
AgoFISH: cost-effective in situ labelling of genomic loci based on DNA-guided dTtAgo protein†
Lei
Chang‡
 a,
Gang
Sheng‡
b,
Yiwen
Zhang‡
a,
Shipeng
Shao
a,
Yanli
Wang
*bc and
Yujie
Sun
*a
a,
Gang
Sheng‡
b,
Yiwen
Zhang‡
a,
Shipeng
Shao
a,
Yanli
Wang
*bc and
Yujie
Sun
*a
aState Key Laboratory of Membrane Biology, School of Life Sciences, and Biomedical Pioneering Innovation Center (BIOPIC), Peking University, Beijing 100871, China. E-mail: sun_yujie@pku.edu.cn
bKey Laboratory of RNA Biology, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China. E-mail: ylwang@ibp.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 25th March 2019
Abstract
DNA fluorescence in situ hybridization (FISH) has been widely used for visualizing spatial localization of genomic regions in eukaryotic nuclei. Current probe preparation methods for FISH involve high probe synthesis cost and complex experimental procedures, thus limiting their applicability. Here, we report a new FISH method named AgoFISH based on the DNA-guided DNA binding activity of the nuclease-deficient Argonaute protein in Thermus thermophilus (dTtAgo). Fluorescently labelled dTtAgo combined with 5′-phosphorylated single-stranded guide DNA can bring the fluorescent label to the endogenous DNA sequence that is complementary to the guide DNA. We demonstrated that AgoFISH can successfully label repetitive sequences in human and mouse cells, including the centromere, the pericentromere and single-copy tandem repeats. Assembling dTtAgo with a designed pool of single-stranded guide DNAs is able to visualize nonrepetitive sequences within coding genes. Furthermore, AgoFISH can also be applied to dual-color labelling without crosstalk between channels. This cost-effective and simple method provides a convenient and powerful tool for chromatin higher-order structure studies as well as FISH-based clinical molecular diagnosis.
Conceptual insightsWe developed a new method for in situ labelling of genomic loci in fixed cells which introduced a dTtAgo protein (nuclease-deficient Argonaute protein in Thermus thermophilus) with DNA-guided DNA binding activity. Existing approaches for genomic loci labelling involve high costs of DNA synthesis and demanding experimental procedures such as RNA manipulation. AgoFISH can reduce the expense by using short (∼21 nt) single-stranded guide DNA and placing a fluorescent label on dTtAgo, which simplifies the probe preparation procedures. Utilizing AgoFISH, we successfully visualized repetitive sequences and nonrepetitive sequences in human and mouse cells with desirable signal-to-background ratios. Sequential dual-color AgoFISH also enabled simultaneous labelling of two genomic loci. This approach can provide information on the spatial distribution and copy number of genomic regions, which is critical for both basic research and clinical molecular diagnosis. |
FISH is a widely-used method for probing spatial localization of chromatin regions in the eukaryotic nucleus. This method can be used to investigate the chromatin higher-order structure1–3 in basic research and visualize abnormal gene copy number in cancer diagnosis.4 Recent advances in oligonucleotide (oligo)-based probes5,6 have greatly improved the reliability and versatility of FISH as compared to clone-based probes, but have also brought with it the high costs of synthesizing an ∼100 nt oligo library as well as fluorescently labelled oligos. The CASFISH method7 addresses this problem by transferring the fluorescent label onto the nuclease-deficient Cas9 (dCas9) protein, but it still involves complex and demanding probe preparation procedures such as in vitro transcription and RNA purification. Therefore, a simpler and more cost-effective FISH method is needed.
The eukaryotic Argonaute (Ago) protein is an essential component of the RNA-induced silencing complex (RISC) and the major effector in target mRNA cleavage.8 In contrast, the prokaryotic Ago protein preferentially uses single-stranded guide DNA/RNA and cleaves both target RNA and DNA strands.9–12 This DNA/RNA-guided DNA/RNA interference has potential roles in bacterial host defense.10 The Ago protein of the bacterium Marinitoga piezophila has recently been programmed to detect specific RNA species in vitro through guide RNA, demonstrating the potential of applying Ago proteins in DNA/RNA fluorescent labelling.13 For Ago of the bacterium Thermus thermophilus (TtAgo), the optimal guide DNA in vivo is 13–25 nucleotides in length with 5′-phosphorylation, and has strong bias for a 5′ end deoxycytidine.10,14 Binding of TtAgo to guide DNA is able to accelerate target finding as compared with naked guide DNA.15 High-resolution crystal structures of TtAgo have demonstrated that three aspartate residues in the PIWI domain make up the catalytic pocket for target strand cleavage11,16 and a mutation at Asp546 effectively abolishes the cleavage activity but retains normal guide and target binding activity.9,16 Therefore, it is possible to use these properties of dTtAgo to develop a new genome labelling technology.
Thus, we develop a new fluorescence in situ hybridization method, named AgoFISH, based on the DNA-guided DNA binding activity of dTtAgo. In design, fluorescently labelled dTtAgo is premixed with 5′-phosphorylated single-stranded guide DNA (ssDNA), and then applied to fixed cells to bring the fluorescent label to the endogenous DNA sequence that is complementary to the guide DNA, thus visualizing any region-of-interest on the chromosome (Fig. 1A). Compared with conventional FISH techniques for which synthesis of fluorescently labelled ssDNA probes is the most costly step, AgoFISH has the following two advantages that greatly reduce the cost, especially when labelling long non-repetitive sequences. Firstly, AgoFISH uses guide ssDNA of 21 nt, 1/3–1/5 the length of ssDNA used in conventional FISH methods. Secondly, AgoFISH places the fluorescent label on dTtAgo as a fusion protein which can be expressed and purified in Escherichia coli, and does not involve complex and demanding probe preparation procedures. Thus this technology has promising cost-effectiveness and simplicity.
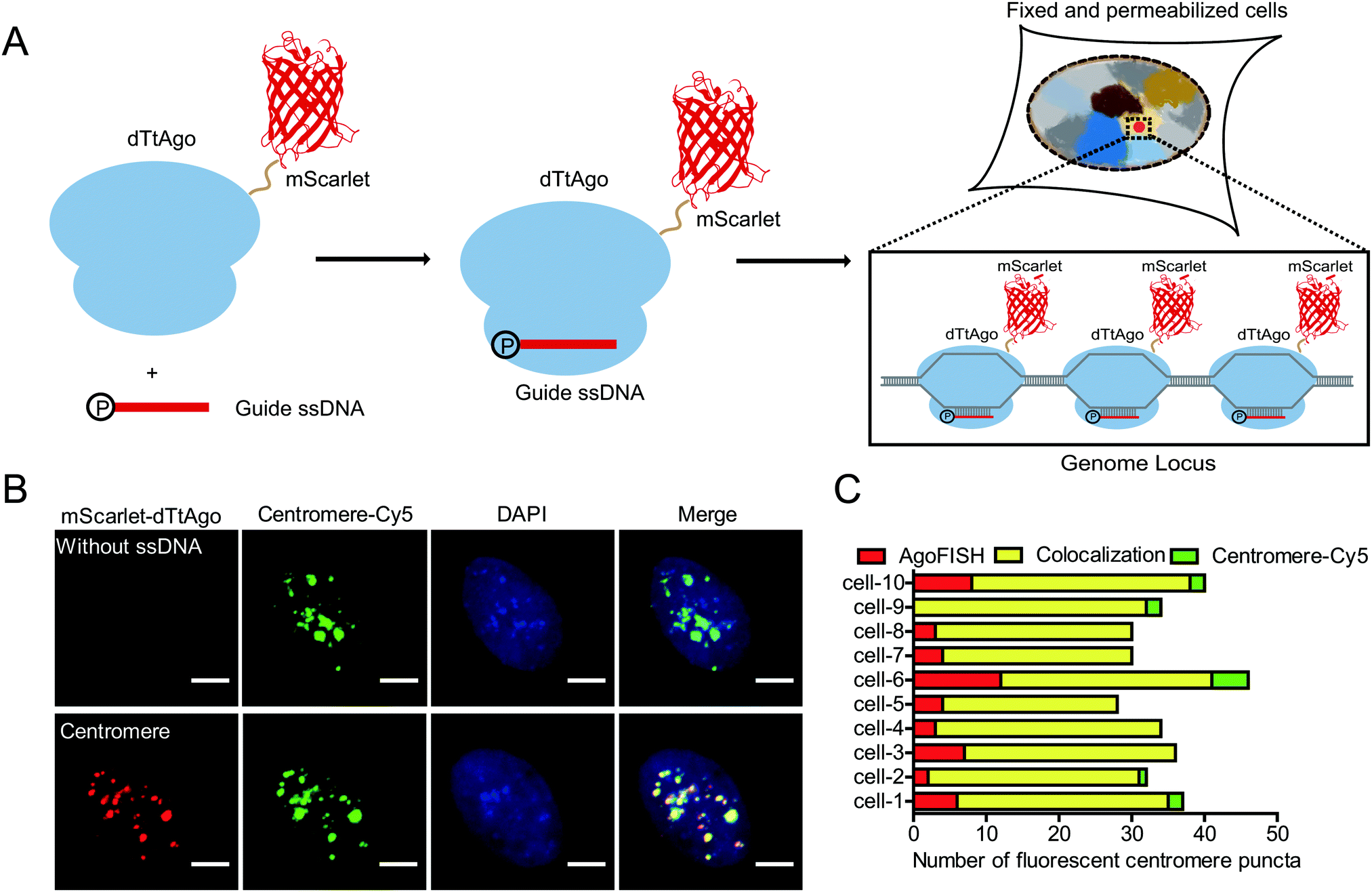 | ||
| Fig. 1 Construction of the AgoFISH labelling system for fluorescent imaging of genomic sequences: (A) Schematic of the AgoFISH strategy. (B) Co-labelling of centromeres using AgoFISH (red) and traditional DNA FISH (green) in U2OS cells. Puncta were shown under the same intensity threshold in the AgoFISH channel for two samples, while under two-fold maximum intensity threshold in the traditional DNA FISH channel for the control sample without ssDNA. Scale bar, 5 μm. Maximum projections of z-stacks are shown, z-stack step size, 0.5 μm. (C) Quantitative results of the sample shown in (B). Quantification of centromere targeting specificity based on the three colocalization scenarios in 10 cells. Yellow: apparent colocalization between AgoFISH labelled puncta and traditional FISH probe labelled puncta; red: AgoFISH labelled puncta without apparent traditional FISH probe labelled puncta; green: traditional FISH probe labelled puncta without apparent AgoFISH labelled puncta. | ||
To realize such purpose, we first fluorescently labelled dTtAgo by constructing a dTtAgo fusion protein that contains a fluorescent protein mScarlet or EGFP at the N terminus (mScarlet-dTtAgo/EGFP-dTtAgo), and then purified the recombinant dTtAgo fusion protein expressed in Escherichia coli. As a proof-of-concept experiment, we first chose to label the highly repetitive centromeric regions in human cells. Commercially synthesized, 5′-phosphorylated 21 nt ssDNA (ssCentromere) that is complementary to the centromeric repeats was mixed with mScarlet-dTtAgo at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The probe mixture was then applied to denatured human U2OS cells and hybridized overnight. Images from the mScarlet channel showed distinct puncta of centromeres with a high signal-to-background ratio (Fig. S1A, ESI†). The number of detected centromeres was in agreement with the previously reported number of chromosomes in human cell lines17 (Fig. S1B, ESI†). In contrast, mScarlet-dTtAgo alone showed a low and evenly dispersed signal, suggesting that without the guide ssDNA, dTtAgo exhibits little specific binding to genomic sequences (Fig. S1A, ESI†).
:1. The probe mixture was then applied to denatured human U2OS cells and hybridized overnight. Images from the mScarlet channel showed distinct puncta of centromeres with a high signal-to-background ratio (Fig. S1A, ESI†). The number of detected centromeres was in agreement with the previously reported number of chromosomes in human cell lines17 (Fig. S1B, ESI†). In contrast, mScarlet-dTtAgo alone showed a low and evenly dispersed signal, suggesting that without the guide ssDNA, dTtAgo exhibits little specific binding to genomic sequences (Fig. S1A, ESI†).
A secondary probe binding sequence was added to the 3′ end of ssDNA and colocalization between the secondary probe and dTtAgo was observed, suggesting that dTtAgo indeed binds to ssDNA (Fig. S2, ESI†). To further demonstrate the specificity and efficiency of AgoFISH labelling, we simultaneously labelled centromeres using AgoFISH and traditional DNA FISH probes with Cy5 labelling. The DNA FISH probe has the same sequence as ssCentromere and is conjugated to a Cy5 dye at the 5′ end. Centromeres detected in the two channels were well colocalized (∼80%) and similar in number (Fig. 1B and C). Also, the maximum intensity threshold of puncta in the Cy5 channel was two times as high in the control sample without ssDNA as that in the other sample with ssDNA, suggesting that the AgoFISH probes have similar affinity for target sequences as compared with traditional DNA FISH probes. We also measured the cell total fluorescence intensity of the Cy5 channel in samples with and without ssDNA (Fig. S3, ESI†). We found that the total fluorescence intensity in the Cy5-labelled classical FISH probe channel was also two times as high in the control sample without ssDNA for AgoFISH as that in the other sample with ssDNA. Thus, AgoFISH and classical FISH would show equivalent performance using the same probe sequences and number.
To promote wide application of AgoFISH, we optimized the protocol to further improve the cost-effectiveness and labelling efficiency. The 5′ phosphorylation of guide ssDNA is essential for dTtAgo binding and also greatly adds to the cost of commercial synthesis. Alternatively, ssDNA can be phosphorylated with T4 polynucleotide kinase (PNK) to lower the experimental expense. We then tested the efficiency of T4 PNK-mediated phosphorylation of guide ssDNA. As expected, unphosphorylated ssDNA was unable to guide dTtAgo to specific genomic loci (Fig. 2A). In contrast, T4 PNK-mediated customized phosphorylation of the ssCentromere had similar labelling results to commercial phosphorylation (Fig. 2A). Therefore, by doing phosphorylation via T4 PNK for synthesized unmodified ssDNA, we were able to further reduce the cost of AgoFISH.
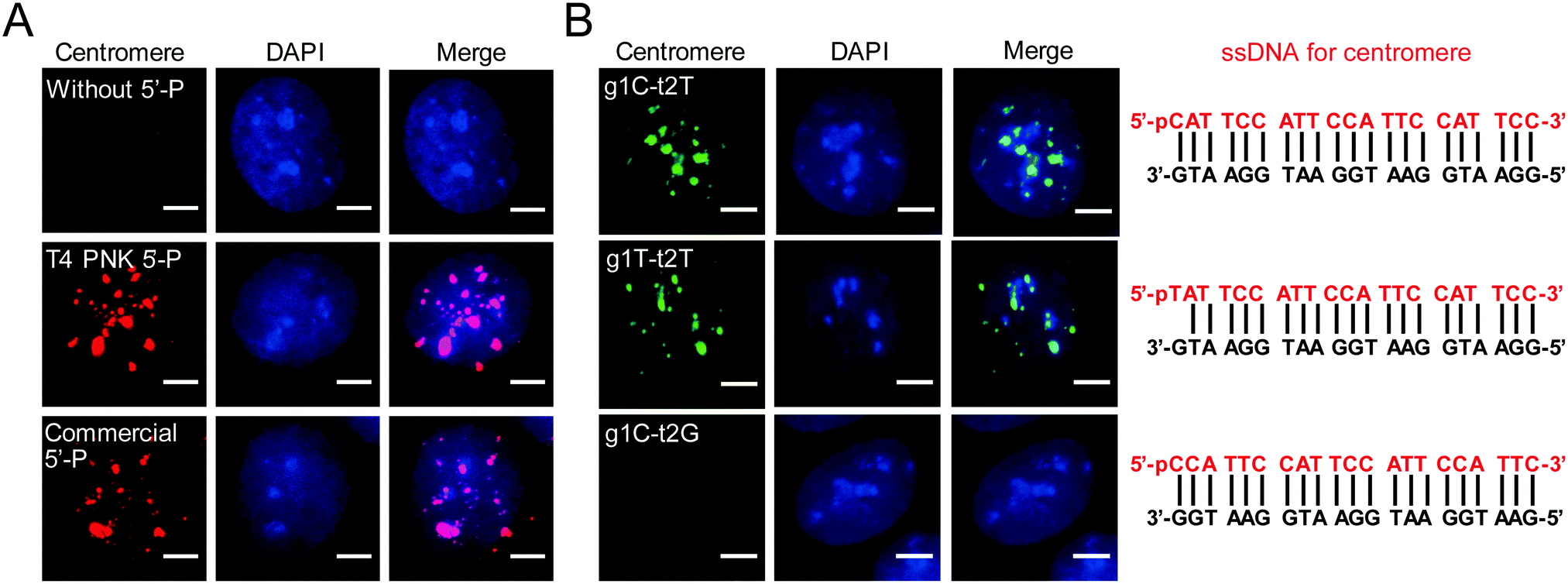 | ||
| Fig. 2 Optimization of probe design and synthesis: (A) AgoFISH labelling of centromeres in U2OS cells with different phosphorylation methods of ssDNA. (B) AgoFISH labelling of centromeres in U2OS cells using ssDNA beginning with different nucleotides (left). Design of ssDNA sequences are indicated on the right. Puncta were shown under the same intensity threshold in the AgoFISH channel for three samples in (A) and (B). Maximum projections of z-stacks are shown. The nucleus was stained by DAPI (blue). z-stack step size, 0.4 μm; scale bar, 5 μm. | ||
Based on previous in vivo,10in vitro14 and structural studies,9,11 dTtAgo preferentially binds to deoxyguanosine at the first position at the end of the target sequence (t1G) and does not have preference for a specific 5′ end nucleotide on the guide DNA. However, unpaired deoxythymidine as the first nucleotide at the 5′ end of guide DNA (g1T) is always used in research, and a paired deoxycytidine (g1C) is more commonly used in nature. Thus, we designed different guide DNA sequences for centromeres and discovered that t1G, g1C and g1T showed similar labelling results (Fig. 2B). Besides, we found that the second nucleotide at the end of the target DNA was also important for dTtAgo binding (Fig. 2B), which is consistent with the finding that 72% of guide ssDNAs found in the complex with TtAgo in vivo have deoxyadenosine at the second position (g2A).10 Designing probes according to these rules can greatly improve the labelling efficiency of AgoFISH.
We then performed AgoFISH in mouse embryonic stem cells (mESCs) to show its versatility and applicability in cells of different species. Labelling of the highly repetitive pericentromeric sequences (major satellite repeats) showed well correlated clusters between the mScarlet channel that represents dTtAgo and the DAPI channel (Fig. 3A), as shown previously.7 In human cells, the mucin 4 (MUC4) gene on chromosome 3 encodes for a cell surface glycoprotein that is implicated in cancer development and metastasis. AgoFISH could robustly label exon 2 of the MUC4 gene that contains a repetitive sequence with ∼400 copies18 in human U2OS cells (Fig. 3B). In some cases, we detected two spatially adjacent loci, representing the MUC4 gene undergoing replication (Fig. 3B). We also successfully labelled a non-coding tandem repeat region located at 242 Mb on chromosome 2 with ∼68 copies. These results demonstrated the capability of AgoFISH to visualize endogenous coding genes and non-coding sequences with a low copy number. The number of fluorescent puncta detected by AgoFISH was in agreement with the number of chromosomes in U2OS cells determined by karyotype analysis (Fig. S4, ESI†).
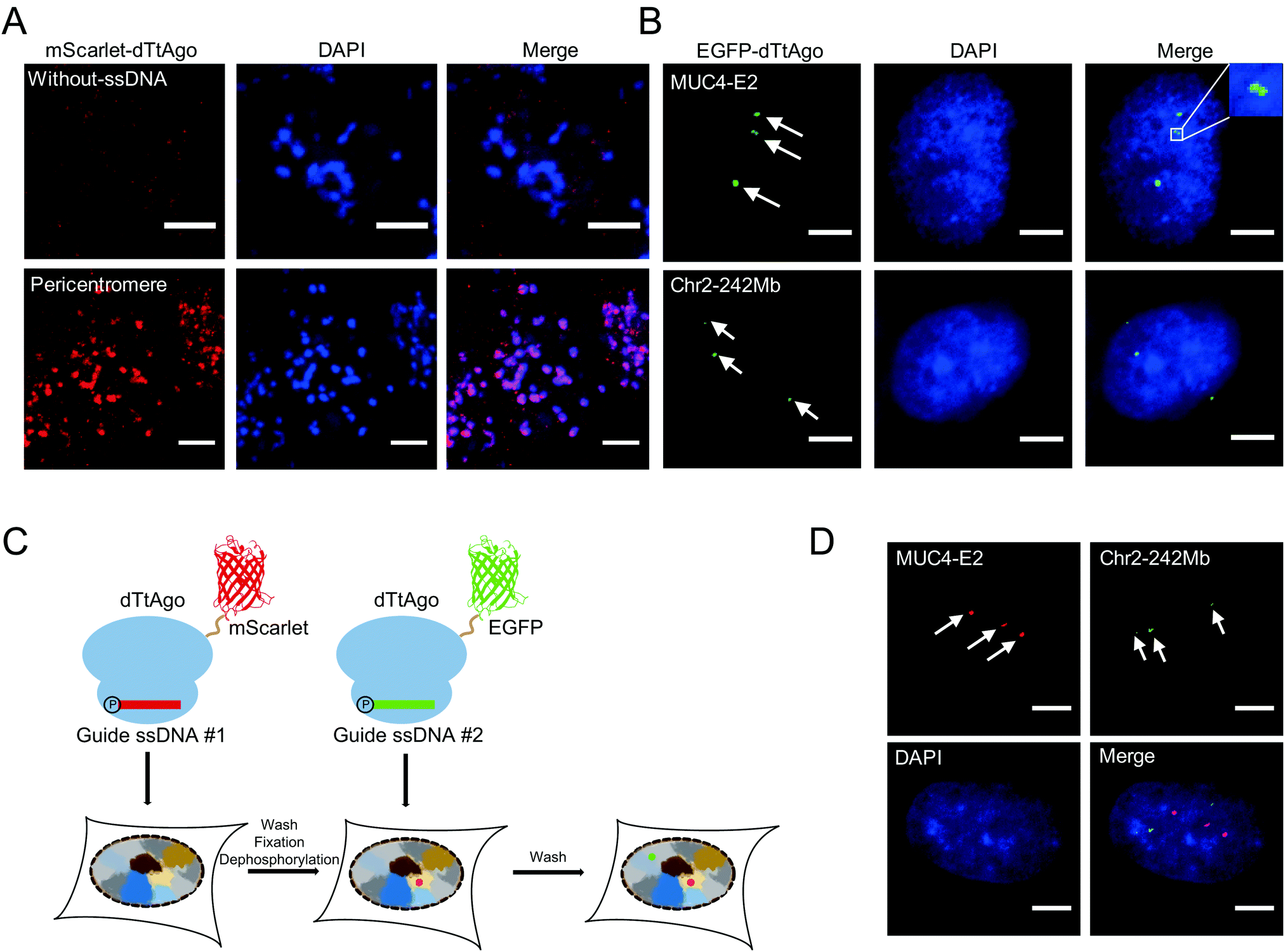 | ||
| Fig. 3 Applications of AgoFISH: (A) AgoFISH labelling of pericentromeres (major satellite repeats) in mESCs. Puncta were shown under the same intensity threshold in the AgoFISH channel for each sample. Maximum projections of z-stacks are shown; step size, 0.5 μm. (B) AgoFISH labelling of two tandem repeat sequences in exon 2 of the MUC4 gene (MUC4-E2) and at 242 Mb on chromosome 2 (Chr2-242 Mb) in U2OS cells, separately. (C) Schematic of the dual-color sequential AgoFISH method. (D) Dual-color sequential AgoFISH labelling of MUC4-E2 and Chr2-242 Mb in U2OS cells. The nucleus was stained by DAPI (blue). Maximum projections of z-stacks are shown, z-stack step size, 0.5 μm. Scale bar, 5 μm. | ||
Simultaneous imaging of multiple genomic loci can provide valuable information on the spatial relationship between sequences of interest. We first tried a one-step dual-color AgoFISH procedure in which two sets of ssDNA respectively targeting two different genomic regions were separately mixed and incubated with dTtAgo labelled with mScarlet and EGFP. However, we observed severe crosstalk between the two channels, which was likely due to unstable binding between dTtAgo and ssDNA. Furthermore, sequential dual-color AgoFISH without additional treatment also showed cross-reactivity between the two channels (Fig. S5, ESI†), probably resulting from the dissociation of round 1 dTtAgo/ssDNA from target sequences and the excess of round 1 dTtAgo or ssDNA that could not be removed. These results indicated that the guide ssDNA-mediated binding between fluorescently-labelled dTtAgo and target DNA sequences is not absolutely stable and undergoes exchange during hybridization. In order to make a quantitative evaluation, we measured the on–off kinetics of dTtAgo-EGFP on centromeres using the fluorescence recovery after photobleaching (FRAP) assay (Fig. S6, ESI†). The recovery rate of dTtAgo-EGFP (t1/2) is 28 s. The immobile fraction of dTtAgo-EGFP is 95.6% (Fi = 0.956), while the mobile fraction is 4.4% (Fm = 0.044). The large immobile fraction of dTtAgo-EGFP guarantees that fluorescently-labelled dTtAgo binding to the target genomic loci can be detected in the image. In summary, most of the loaded fluorescently labelled dTtAgo binds stably to target sequences, but there also exists a small fraction with a fast exchange rate which may lead to crosstalk in dual-color labelling.
To address this problem, we introduced the following modifications to the sequential dual-color AgoFISH protocol: after the first round of hybridization and washing, the cells were fixed with paraformaldehyde (PFA) to stabilize the dTtAgo/ssDNA/target DNA complex, and then treated with calf-intestinal alkaline phosphatase (CIP) to prevent round 1 ssDNA from binding to round 2 dTtAgo (Fig. 3C). Following this modified protocol, we sequentially labelled MUC4-E2 and the tandem repeat sequence on chromosome 2 in the same cell. The results showed distinct locations of the two sequences with no detectable cross-reactivity (Fig. 3D). The sample without CIP treatment between two rounds of hybridization showed colocalization events, demonstrating that CIP treatment is crucial for preventing cross-reactivity between two channels (Fig. S5, ESI†). Thus, the modified sequential dual-color AgoFISH protocol effectively eliminates crosstalk between two rounds of hybridization and reflects the spatial relationship between genomic loci.
To demonstrate the capability of AgoFISH to label nonrepetitive genomic sequences, we designed 406 ssDNAs tiling the ∼24 kb nonrepetitive region in the first intron of the MUC4 gene (Fig. 4A). The designing process was based on OligoArray5,19 and took into account the special requirements of AgoFISH guide ssDNA such as length, GC content and beginning nucleotides. This process can be easily applied to other customized genomic regions of interest. The labelling results in U2OS cells showed distinct puncta that were clearly separable from background fluorescence (Fig. 4B). Therefore, the results provided evidence that AgoFISH can robustly label endogenous nonrepetitive genes in human cells. Furthermore, we chose another two tandem repeat sequences and one non-repetitive sequence to demonstrate the versatility of AgoFISH (Fig. S7, ESI†).
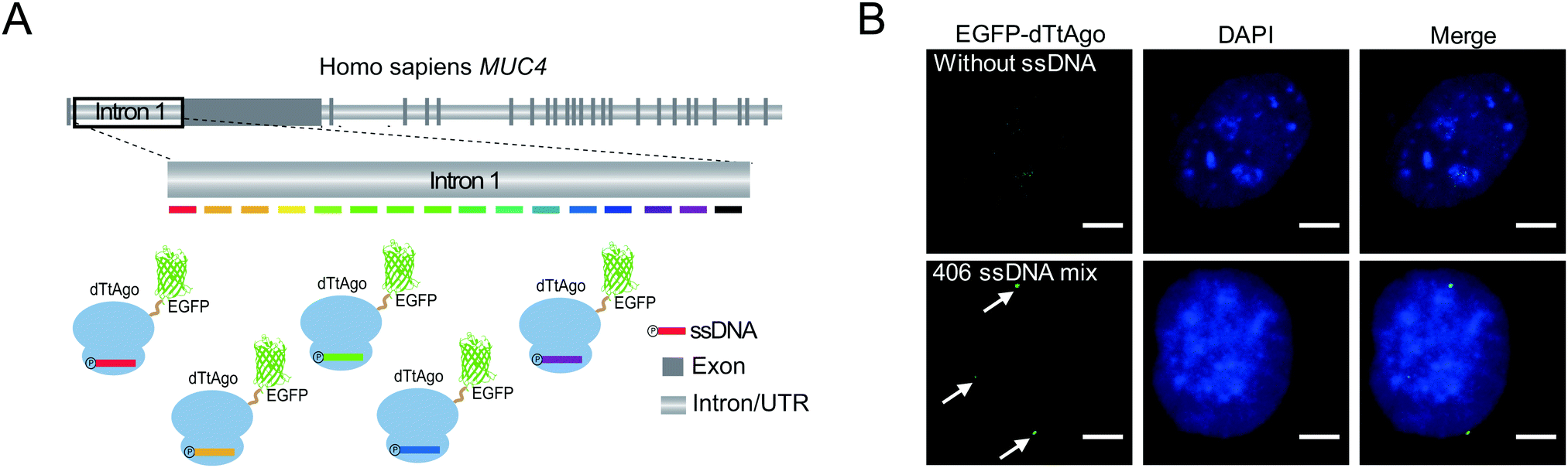 | ||
| Fig. 4 AgoFISH labelling of non-repetitive sequences: (A) Schematic of labelling a non-repetitive sequence by AgoFISH. (B) AgoFISH labelling of non-repetitive sequences in intron 1 of the MUC4 gene (MUC4-I1) in U2OS cells. Puncta were shown under the same intensity threshold in the AgoFISH channel. The nucleus was stained by DAPI (blue). Maximum projections of z-stacks are shown, z-stack step size, 0.5 μm. Scale bar, 5 μm. | ||
Conclusions
In summary, we developed a new simple and cost-effective FISH method, named AgoFISH, based on the DNA-guided DNA binding property of nuclease-deficient Ago protein. Optimizations of the beginning sequence and phosphorylation method of guide DNA further improved the labelling efficiency and lowered the experimental costs. Utilizing AgoFISH, we successfully visualized repetitive sequences in human cells and mESCs. Sequential dual-color AgoFISH enabled simultaneous labelling of two genomic loci with virtually no crosstalk between channels. We were also able to label nonrepetitive sequences within coding genes with a relatively high signal-to-background ratio (Table S1, ESI†). We anticipate AgoFISH to be a new tool with wide applicability and high commercial prospects, especially for three-dimensional structure studies of the genome and FISH-based clinical molecular diagnosis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Ge Zhan from the laboratory of Dr Xiaohua Shen in Tsinghua University for providing fixed mESCs. We also thank Dr Hongxia Lv at the core imaging facility of the School of Life Sciences, Peking University for imaging support. This work was supported by grants from the National Key R&D Program of China, No. 2017YFA0505302, and the National Natural Science Foundation of China, 21573013 and 21825401 for Y. S., 31725008 for Y. W. and 31571335 for G. S.Notes and references
- A. N. Boettiger, B. Bintu, J. R. Moffitt, S. Wang, B. J. Beliveau, G. Fudenberg, M. Imakaev, L. A. Mirny, C. T. Wu and X. Zhuang, Nature, 2016, 529, 418–422 CrossRef CAS PubMed.
- S. Wang, J. H. Su, B. J. Beliveau, B. Bintu, J. R. Moffitt, C. T. Wu and X. Zhuang, Science, 2016, 353, 598–602 CrossRef CAS PubMed.
- B. Bintu, L. J. Mateo, J. H. Su, N. A. Sinnott-Armstrong, M. Parker, S. Kinrot, K. Yamaya, A. N. Boettiger and X. Zhuang, Science, 2018, 362, eaau1783 CrossRef PubMed.
- C. M. Ellis, M. J. Dyson, T. J. Stephenson and E. L. Maltby, J. Clin. Pathol., 2005, 58, 710–714 CrossRef CAS PubMed.
- B. J. Beliveau, E. F. Joyce, N. Apostolopoulos, F. Yilmaz, C. Y. Fonseka, R. B. McCole, Y. Chang, J. B. Li, T. N. Senaratne, B. R. Williams, J. M. Rouillard and C. T. Wu, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 21301–21306 CrossRef CAS PubMed.
- B. J. Beliveau, A. N. Boettiger, M. S. Avendano, R. Jungmann, R. B. McCole, E. F. Joyce, C. Kim-Kiselak, F. Bantignies, C. Y. Fonseka, J. Erceg, M. A. Hannan, H. G. Hoang, D. Colognori, J. T. Lee, W. M. Shih, P. Yin, X. Zhuang and C. T. Wu, Nat. Commun., 2015, 6, 7147 CrossRef CAS PubMed.
- W. Deng, X. Shi, R. Tjian, T. Lionnet and R. H. Singer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11870–11875 CrossRef CAS PubMed.
- L.-A. MacFarlane and P. R. Murphy, Curr. Genomics, 2010, 11, 537–561 CrossRef CAS PubMed.
- Y. Wang, S. Juranek, H. Li, G. Sheng, G. S. Wardle, T. Tuschl and D. J. Patel, Nature, 2009, 461, 754–761 CrossRef CAS PubMed.
- D. C. Swarts, M. M. Jore, E. R. Westra, Y. Zhu, J. H. Janssen, A. P. Snijders, Y. Wang, D. J. Patel, J. Berenguer, S. J. J. Brouns and J. van der Oost, Nature, 2014, 507, 258–261 CrossRef CAS PubMed.
- G. Sheng, H. Zhao, J. Wang, Y. Rao, W. Tian, D. C. Swarts, J. van der Oost, D. J. Patel and Y. Wang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 652–657 CrossRef CAS PubMed.
- Y.-R. Yuan, Y. Pei, J.-B. Ma, V. Kuryavyi, M. Zhadina, G. Meister, H.-Y. Chen, Z. Dauter, T. Tuschl and D. J. Patel, Mol. Cell, 2005, 19, 405–419 CrossRef CAS PubMed.
- A. Lapinaite, J. A. Doudna and J. H. D. Cate, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3368–3373 CrossRef CAS PubMed.
- D. C. Swarts, M. Szczepaniak, G. Sheng, S. D. Chandradoss, Y. Zhu, E. M. Timmers, Y. Zhang, H. Zhao, J. Lou, Y. Wang, C. Joo and J. van der Oost, Mol. Cell, 2017, 65, 985–998 CrossRef CAS PubMed.
- W. E. Salomon, S. M. Jolly, M. J. Moore, P. D. Zamore and V. Serebrov, Cell, 2015, 162, 84–95 CrossRef CAS PubMed.
- Y. Wang, G. Sheng, S. Juranek, T. Tuschl and D. J. Patel, Nature, 2008, 456, 209–213 CrossRef CAS PubMed.
- S. Shao, W. Zhang, H. Hu, B. Xue, J. Qin, C. Sun, Y. Sun, W. Wei and Y. Sun, Nucleic Acids Res., 2016, 44, e86 CrossRef PubMed.
- B. Chen, L. A. Gilbert, B. A. Cimini, J. Schnitzbauer, W. Zhang, G. W. Li, J. Park, E. H. Blackburn, J. S. Weissman, L. S. Qi and B. Huang, Cell, 2013, 155, 1479–1491 CrossRef CAS PubMed.
- J. M. Rouillard, M. Zuker and E. Gulari, Nucleic Acids Res., 2003, 31, 3057–3062 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00028c |
| ‡ These three authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |