
Plasmon enhanced upconverting core@triple-shell nanoparticles as recyclable panchromatic initiators (blue to infrared) for radical polymerization†
Jianming
Zhang‡
 a,
Yue
Huang‡
b,
Xin
Jin‡
b,
Artem
Nazartchouk
b,
Maosong
Liu
a,
Xin
Tong
b,
Yinhua
Jiang
a,
Liang
Ni
a,
Shuhui
Sun
b,
Yuanhua
Sang
c,
Hong
Liu
*cd,
Luca
Razzari
*b,
Fiorenzo
Vetrone
*b and
Jerome
Claverie
*e
a,
Yue
Huang‡
b,
Xin
Jin‡
b,
Artem
Nazartchouk
b,
Maosong
Liu
a,
Xin
Tong
b,
Yinhua
Jiang
a,
Liang
Ni
a,
Shuhui
Sun
b,
Yuanhua
Sang
c,
Hong
Liu
*cd,
Luca
Razzari
*b,
Fiorenzo
Vetrone
*b and
Jerome
Claverie
*e
aSchool of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China
bCentre Énergie, Matériaux et Télécommunications (EMT), Institut National de la Recherche Scientifique (INRS), 1650 Boul. Lionel Boulet, Varennes, Québec J3X 1S2, Canada. E-mail: razzari@emt.inrs.ca; vetrone@emt.inrs.ca
cState Key Laboratory of Crystal Materials, Shandong University, Jinan 250100, China. E-mail: hongliu@sdu.edu.cn
dInstitute for Advanced Interdisciplinary Research (IAIR), University of Jinan, Jinan 250022, China
eDepartment of Chemistry, Université de Sherbrooke, Sherbrooke, Quebec J1K 2R1, Canada. E-mail: jerome.claverie@usherbrooke.ca
First published on 26th March 2019
Abstract
With the rise of stereolithographic processes and advanced fabrication processes, photo-induced polymerization is becoming increasingly popular. The development of panchromatic radical initiators, that is to say initiators which are activated by a wide spectrum, is still in its infancy, with the near-infrared range remaining a challenge. We have reported a rationally-designed nanohybrid which can initiate photo-polymerization from ultraviolet to near-infrared wavelengths. Integration of Ag and Au plasmonic nanoparticles (NPs), upconverting (UC) materials and BiFeO3 (BFO) semiconductors with a rational architecture design results in the formation of an Ag@SiO2@UC@BFO–Au core@triple-shell structure. Compared to the conventional thermal radical polymerization method, this material can initiate polymerization under visible or near-infrared light at room temperature, and the initiator can easily be recycled and reused. The excellent activity of the nanohybrid photoinitiator is attributed to the combination of the optical properties of the UC material and BFO semiconductor and the surface plasmon resonance (SPR) in the core@shell configuration, where the Ag NP core demonstrates dual roles of increasing resonant VIS photon scattering as a “mirror” and improving the upconverting efficiency of the NIR-to-VIS photons as a “resonator”; the Au NPs anchored on the BFO shell further enable an efficient energy transfer of absorbed VIS photon energy from Au to BFO via a plasmon-induced resonance energy transfer (PIRET) process.
Conceptual insightsPhotoinitiation of radical polymerization is widely used in a plethora of technological processes, including in 3D-printing. Known photoinitiators have a limited absorption spectrum which renders the photopolymerization of thick and/or pigmented samples challenging. In this work, we demonstrate how the use of a full solar-spectrum heterogeneous photocatalyst leads to a panchromatic photoinitiator, that is to say a photoinitiator capable of triggering polymerization with visible or near-infrared light, thus covering the entire solar-spectrum. In order to ensure a sufficient radical flux, the heterogeneous photocatalyst includes upconverting nanoparticles, as well as a core acting as an internal resonator and a scattering unit. Thus, it is demonstrated that rationally-designed full solar-spectrum photocatalysts yield promising panchromatic photoinitiators. |
Introduction
Synthetic polymers are one of the most widely used materials in all aspects of our daily life.1 More than 50% of the polymers used worldwide are synthesized via a free-radical polymerization process,1,2 which can be explained by the unique versatility and tolerance for a wide range of functional groups of this kind of reaction. Radical initiation is a critical reaction which affects the overall polymerization rate and molecular weight distribution of the resulting polymeric product.1,2 Light-induced initiation of free radical polymerization is an attractive process because it can be controlled spatially and temporally, leading, inter alia, to the development of 3D-printing and microfabrication of well-defined polymer structures as building blocks in nanotechnology.3–5 Such a process is commercially exploited in an ever-growing number of materials, such as adhesives, inks, nanopatterned surfaces or bio-composites. However, it is mostly restricted to thin films due to the inherent low penetration of light, notably in pigmented samples. Recently, panchromatic photoinitiators were reported to address this issue. Unlike conventional photoinitiating systems, which are wavelength specific, these photoinitiators are able to initiate the polymerization at sensibly the same rate over a wide range of excitation wavelengths.6,7 Reported panchromatic initiators are active under blue (∼400–500 nm) and green light (∼500–600 nm), and exceptionally under red light (∼600–750 nm), but a true panchromatic initiator, which could be activated over the full solar spectrum, is yet to be reported. In particular, the possibility of photopolymerizing with near-infrared (NIR) light is of importance for polymerizing strongly pigmented samples such as those found in biologically relevant environments.Semiconductor nanoparticles (NPs) can generate photoexcited charge carriers upon photon absorption to form radicals, which can trigger a vast number of reactions.8–10 For example, TiO2 and ZnO NPs have been used as photoinitiators for radical polymerization.9,10 However, due to their large bandgap, these NP initiators are only active under ultraviolet (UV) light which only accounts for ∼5% of the solar energy.10 Therefore, photons in the visible (VIS) and NIR range, which account respectively for 49% and 46% of the solar spectrum, are not harvested. To overcome this issue, more efforts are required from the dual aspects of material selection and structural design. In this vein, nanocomposites of core@shell hierarchical structures provide the capacity to multiplex various functionalities within a single unit.11,12 A rationally designed uniform core@multishell structure, with the specialty of harnessing UV, VIS or NIR photons, may lead to a true panchromatic radical photoinitiator for full solar-spectrum photopolymerization.
Bismuth ferrite (BiFeO3, BFO), a multiferroic material, is a promising VIS-light-active photocatalyst with a bandgap of ∼2.0–2.6 eV.13–16 Furthermore, luminescent lanthanide (Ln3+)-doped nanomaterials are known for their ability to convert absorbed NIR light with lower energy to UV or/and VIS photons with higher energy, via a process commonly referred to as upconversion (UC).17 Very recently, UCNP@semiconductor core@shell nanohybrids have been investigated as photocatalysts for the depollution of dyes. In these structures, NIR photons are converted by UCNPs to UV energy which is transferred to the semiconductor shell to generate charge carriers for photocatalysis, revealing a proof of principle that NIR photons can be utilized in photocatalysis via an upconverting process.18–22 We recently also reported UC@BFO NPs as a catalyst for VIS-to-NIR light driven photocatalysis.23 However, in our hands, the activity of these UC@semiconductor nanohybrids as radical photoinitiators is virtually null in the VIS and NIR range.
Here, we report the rational design of a UC@semiconductor nanohybrid with enhanced photoactivity, which thus behaves as a true panchromatic photo-initiator from blue to NIR light. To achieve this goal, a Ag@SiO2@UC@BFO–Au core@triple-shell plasmonic nanohybrid structure was prepared whereby SiO2, Y2O3:Er3+,Yb3+ (UC) and BFO coatings were deposited in sequence on top of Ag NPs, and Au NPs were finally anchored at the surface of the BFO shell. The addition of each shell on top of each other enhances the photocatalytic activity either in the VIS range or in the NIR range. As a photoinitiator, Ag@SiO2@UC@BFO–Au initiated the radical polymerization of acrylic acid sodium (AAS) at room temperature under VIS and NIR irradiation. Moreover, in contrast to molecular photoinitiators, which are spent during photopolymerization, here the catalyst can be facilely separated/reused with excellent stability. This novel nanohybrid design optimally exploits plasmonics and the UC of NIR photons, thereby considerably favoring photon harvesting for photo-radical polymerization.
Results and discussion
Sample preparation and characterization
Fig. 1a illustrates schematically a multistep procedure to construct the Ag@SiO2@Y2O3:Yb3+,Er3+@BFO–Au nanohybrid. First, Ag NPs with diameter ∼50 nm (D = ∼50 nm) were coated with SiO2 using the classic Stöber method to form uniform Ag@SiO2 structures (step (i), see Fig. S1 in the ESI† for TEM images). By adjusting the reaction time and the silica precursor concentration, the thickness of the SiO2 shell was easily tuned from ∼5 to 30 nm. A second coating of Y,Yb,Er(OH)CO3·H2O with ∼20 nm thickness was deposited on the surface of Ag@SiO2 using a homogeneous precipitation method to generate the Ag@SiO2@Y,Yb,Er(OH)CO3 structure (step (ii)). In the following step, the BFO layer is deposited as the third layer. Conventional BFO synthesis usually involves strongly acidic conditions,13–16,22 whereby Bi(NO3)3·5H2O and Fe(NO3)3·9H2O are first dissolved in acid solution (e.g. pH < 1), followed by precipitating the mixed hydroxide by increasing pH.13–16,22 However, such conditions cannot be employed in our case as the acid would dissolve the Y,Yb,Er(OH)CO3·H2O layer and thus remove the UC material. Unlike nitrate salts, the ethylenediamine tetraacetic acid (EDTA) complexes of both metals (EDTA–Bi/Fe) are soluble at neutral pH.23 Thus the Ag@SiO2@Y,Yb,Er(OH)CO3 particles were suspended in an EDTA solution of Bi/Fe at neutral pH, and ethanol was then added, leading to the precipitation of the Bi/Fe salts at the surface, to yield the Ag@SiO2@Y,Yb,Er(OH)CO3@EDTA–Bi/Fe particle. Afterward, a thermal treatment at 550 °C was conducted to convert Y,Yb,Er(OH)CO3·H2O and EDTA–Bi/Fe into corresponding Y2O3:Er3+,Yb3+ and BFO coatings (step (iii), see the TEM image of Ag@SiO2@UC@BFO in Fig. S2 and characterization details below, ESI†). Finally, the colloidal Au NPs with D = ∼20 nm were anchored on the BFO surface of Ag@SiO2@UC@BFO (step (iv)), leading to the formation of the Ag@SiO2@UC@BFO–Au nanohybrid. | ||
| Fig. 1 (a) Schematic illustration of the construction steps for the Ag@SiO2@UC@BFO–Au core@triple-shell nanoarchitecture. (b and c) TEM images and (d) HR-TEM images of the Ag@SiO2@UC@BFO–Au nanohybrids. (e) STEM image and corresponding EDX elemental maps of the nanohybrids. | ||
Fig. 1b shows the typical TEM image of the Ag@SiO2@Y2O3:Er3+,Yb3+@BFO–Au nanostructures, with size ∼200–300 nm. The rugged outer shell of BFO decorated with Au NPs is visible in the magnified TEM images of Fig. 1c. The Ag core and the two successive coating of SiO2 and Y2O3:Er3+,Yb3+ can be identified due to their distinct contrast. The average thickness of the SiO2 and UC layers is estimated to be ∼30 nm and 10 nm, respectively, while the thickness of the BFO coating varies from 5–15 nm. The HR-TEM image of the surface, as shown in Fig. 1d, reveals the UP–BFO–Au interface, where the lattice fringes of the three components are visible, with a spacing of 0.23 nm corresponding to the {111} planes of face-centered-cubic (FCC) Au and the two larger spacings of 0.28 nm and 0.30 nm corresponding to the {110} crystal planes of BFO and {222} planes of cubic Y2O3, respectively, indicating the formation of oxide crystals after thermal treatment. The core@shell nanostructure was also characterized by energy dispersive X-ray (EDX) spectroscopy, confirming the presence of the main elements (such as Ag, Si, Y, Bi, Fe and Au) as shown in Fig. S2 (ESI†). The elemental composition was also measured via inductively coupled plasma optical emission spectrometry (ICP-OES), and its result is in good agreement with the dosage of precursors (Table S1, ESI†). To further demonstrate the hierarchical configuration of the core@triple-shell architecture, elemental mapping analysis based on EDX measurement was performed. Fig. 1e left shows the High Angle Annular Dark Field Scanning TEM (HAADF-STEM) image corresponding to Fig. 1c, where the hierarchy is again highly visible, and the metal NPs can be clearly identified due to their much higher atomic mass. The elemental maps in Fig. 1e (right side) clearly demonstrate that the distribution of the main elements follows the core@triple-shell architecture perfectly, verifying the successful construction of the Ag@SiO2@UC@BFO–Au nanohybrid.
The crystalline structure of the core@shell NPs was examined by X-ray diffraction (XRD) analysis. Fig. 2a shows the typical XRD patterns of Ag NPs without any coating, where the position of all diffraction peaks can be well indexed to the FCC structure of Ag. After coating with SiO2, the diffraction pattern of the resulting Ag@SiO2 (b) displays a broad amorphous signal corresponding to SiO2 superimposed on the Ag reflections. The XRD pattern of the Ag@SiO2@Y,Yb,Er(OH)CO3 NPs (c) shows a similar reflection feature to that of Ag@SiO2 as the UC layer is amorphous prior to thermal treatment. After thermal treatment and hybridization with Au NPs, the XRD pattern of the Ag@SiO2@UC@BFO–Au structure exhibits the diffraction superimposition of BFO, Y2O3 and metal (Au and Ag) phases (d). On account of the formation of the oxide crystal phases, the intensity of Au and Ag diffractions became relatively weak and the distinction between them in this complex structure by using the XRD technique is not easy due to their very similar diffraction characteristics.
 | ||
| Fig. 2 Characterization of the nanohybrids. XRD patterns of Ag NPs (a), Ag@SiO2 (b), Ag@SiO2@Y(OH)CO3 (c) and the final Ag@SiO2@UC@BFO–Au (d) structures. Standard JCPDS patterns are shown at the bottom. XPS spectra of Fe 2p (e), Au 4f (f), Bi 4f (g) and O 1s (h) of the Ag@SiO2@UC@BFO–Au nanohybrid. Inset of (e) shows the Fe 2p3/2 spectrum. (i) UV-VIS-NIR absorption spectra of the Ag@SiO2@UC@BFO–Au nanohybrid at its different synthetic stages. (j) UC photoluminescence spectra of SiO2@UC, Ag@SiO2@UC and Ag@SiO2@UC@BFO–Au. | ||
The chemical states of the main elements were explored via X-ray photoelectron spectroscopy (XPS). Fig. 2e shows the high resolution XPS spectrum of Fe 2p, which is composed of two main bands at ∼710.0 eV (Fe 2p3/2) and ∼723.5 eV (Fe 2p1/2).24,25 As illustrated in the inset of Fig. 2e, the Fe 2p3/2 band can be deconvoluted into two peaks located at 709.5 (blue curves) and 710.9 eV (red curves), which respectively correspond to Fe2+ and Fe3+ with the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5. This analysis indicates the valence fluctuations of Fe ions in BFO coating, which is unavoidable and common for iron oxide materials.26 The Au 4f spectrum (Fig. 2f) exhibits two typical peaks at 84.0 (Au 4f7/2) and 87.6 eV (Au 4f5/2) that can be assigned to metallic Au.27 The XPS spectrum of another key element, Bi, in the BFO shell, is shown in Fig. 2g, which is composed of two intense peaks at 158.6 and 164.1 eV ascribed to the Bi 4f7/2 and Bi 4f5/2 bands.24,25 The O1s band as shown in Fig. 2h presents an asymmetric feature implying the presence of more than one kind of oxygen species. Spectral deconvolution of the O1s band yields two peaks at 529.3 (red curves) and 531.5 eV (blue curves), corresponding to the typical O2− state in BFO and possible hydroxyl groups (–OH) in oxides,21,23 respectively.
:1.5. This analysis indicates the valence fluctuations of Fe ions in BFO coating, which is unavoidable and common for iron oxide materials.26 The Au 4f spectrum (Fig. 2f) exhibits two typical peaks at 84.0 (Au 4f7/2) and 87.6 eV (Au 4f5/2) that can be assigned to metallic Au.27 The XPS spectrum of another key element, Bi, in the BFO shell, is shown in Fig. 2g, which is composed of two intense peaks at 158.6 and 164.1 eV ascribed to the Bi 4f7/2 and Bi 4f5/2 bands.24,25 The O1s band as shown in Fig. 2h presents an asymmetric feature implying the presence of more than one kind of oxygen species. Spectral deconvolution of the O1s band yields two peaks at 529.3 (red curves) and 531.5 eV (blue curves), corresponding to the typical O2− state in BFO and possible hydroxyl groups (–OH) in oxides,21,23 respectively.
The optical properties for the structures at each synthetic step (Fig. 1a) were characterized by UV-VIS-NIR absorption spectroscopy. Ag NPs show the classic SPR absorption peak at 430 nm (Fig. S3, ESI†). SiO2 and the following Y,Yb,Er(OH)CO3 coating to the Ag NPs lead to the red-shift of the SPR peak position to 437 nm and 440 nm, respectively, with an increase in the peak width as illustrated in Fig. 2i (black and green curves). After forming the Ag@SiO2@UC@BFO structure (blue curves), the sample mainly shows the typical absorption feature of BFO (see Fig. S4 for pure BFO, ESI†), where a strong absorption arises in the UV to VIS spectral region with a sharp absorption cutoff at ∼640 nm corresponding to a bandgap energy of ∼2.1 eV. The anchoring of Au NPs to the surface of Ag@SiO2@UC@BFO (Ag@SiO2@UC@BFO–Au, red solid curves) results in a further considerable increase of absorption from ∼500 to 800 nm, which is ascribed to the SPR band of Au NPs (Fig. S5, ESI†). For both samples with Y2O3:Er3+,Yb3+ layers (blue and red solid curves from Fig. 2i), a relatively weak peak at ∼900 to 1000 nm can be observed, which mostly corresponds to the typical optical feature of UC materials (see below for details).
As the UC material is involved in the nanostructures, the upconverting photoluminescence (PL) property of the samples was examined in a quantitative fashion (Fig. 2j). The sample of SiO2@UC without the Ag core (Fig. S1f for the TEM image, ESI†) exhibits green emission bands located at 510–570 nm, and red ones at 640–680 nm upon excitation with a 980 nm laser. No noticeable emissions with λ < 500 nm can be observed in the PL spectrum (black curves). By involving a Ag core (Ag@SiO2@UC), the intensities of the PL are remarkably amplified with a maximum enhancement of ∼4× for the green emissions, compared with the SiO2@UC NPs (blue curves). By stark contrast, after coating with BFO, a dramatic decrease of the PL is clearly observed for the Ag@SiO2@UC@BFO–Au nanostructures (red curves). This observation is in good agreement with our previous report, in which an efficient Förster resonance energy transfer (FRET) process occurs at the UC–BFO interface,18,23,28,29 as BFO is directly grown on the surface of the UC layer.
Photo-polymerization
Polymerizations of pure acrylic monomers photoinitiated by Ag@SiO2@UC@BFO–Au nanostructures at room temperature were found to be very rapid, leading to large exotherms. In order to carefully assess the influence of each component of the nanostructure on the polymerization kinetics, the photo-polymerization of a slower system, namely 0.2 M sodium acrylate (AAS) aqueous solution, was studied. This choice is justified by the fact that AAS in water has a low reactivity (kp = 9000 L mol−1 s−1 at 20 °C30) in comparison to its neutral counterpart (kp = 95000 L mol−1 s−1 at 20 °C31). It should also be mentioned that the initiation step is the combination of two steps: (a) the generation of free-radicals in solution and (b) the addition of these primary radicals on the first monomer. In most cases, the former is much slower than the latter.31 In our case, hydroxyl radicals are formed in water (see below). Such radicals will react very rapidly with any monomer,32 thus the scope and versatility of Ag@SiO2@UC@BFO–Au as a photoinitiator are not affected by the selection of the monomer for this precise study.
Fig. 3a left shows the photo image of the clear aqueous solution of AAS monomer without any NP photoinitiators. After the addition of Ag@SiO2@UC@BFO–Au NPs under white light irradiation, the viscosity of the reddish suspension increased gradually (middle). The product was characterized by Fourier transform infrared (FTIR) and proton nuclear magnetic resonance (1H-NMR) spectroscopy (Fig. S6, ESI†), to confirm the formation of the PAAS polymer. A clear polymer solution (right) was obtained by separating the NPs via centrifugation. To better understand the reaction mechanisms, the photo-polymerization initiated by Ag@SiO2@UC@BFO–Au was compared with that initiated by Ag@SiO2@UC@BFO, SiO2@UC@BFO, BFO, Y2O3:Er3+,Yb3+ and Au NPs. As shown in Fig. 3b, under white light irradiation at room temperature, without any initiators and with Au and/or Y2O3:Er3+,Yb3+ NPs the polymerization does not proceed, while it was greatly accelerated by introducing BFO and other core@shell structures. The polymerization efficiency upon the formation of various nanostructures was summarized in the order of Ag@SiO2@UC@BFO–Au (Au: ∼2 wt%) > Ag@SiO2@UC@BFO > SiO2@UC@BFO > BFO. In particular, the use of Ag@SiO2@UC@BFO–Au results in ∼60% conversion after 6 h. The effect of Au loading at the surface of Ag@SiO2@UC@BFO on the reaction activity was also investigated. Fig. S7 (ESI†) summarizes the polymerization conversion under VIS and NIR light using Ag@SiO2@UC@BFO samples with Au loadings varying from 0 to ∼2 wt%. Clearly, the highest reaction efficiency is achieved for the samples with ∼2 wt% Au under VIS light and ∼1 wt% Au under NIR light, indicating the presence of appropriate Au contents for the maximum efficiency.
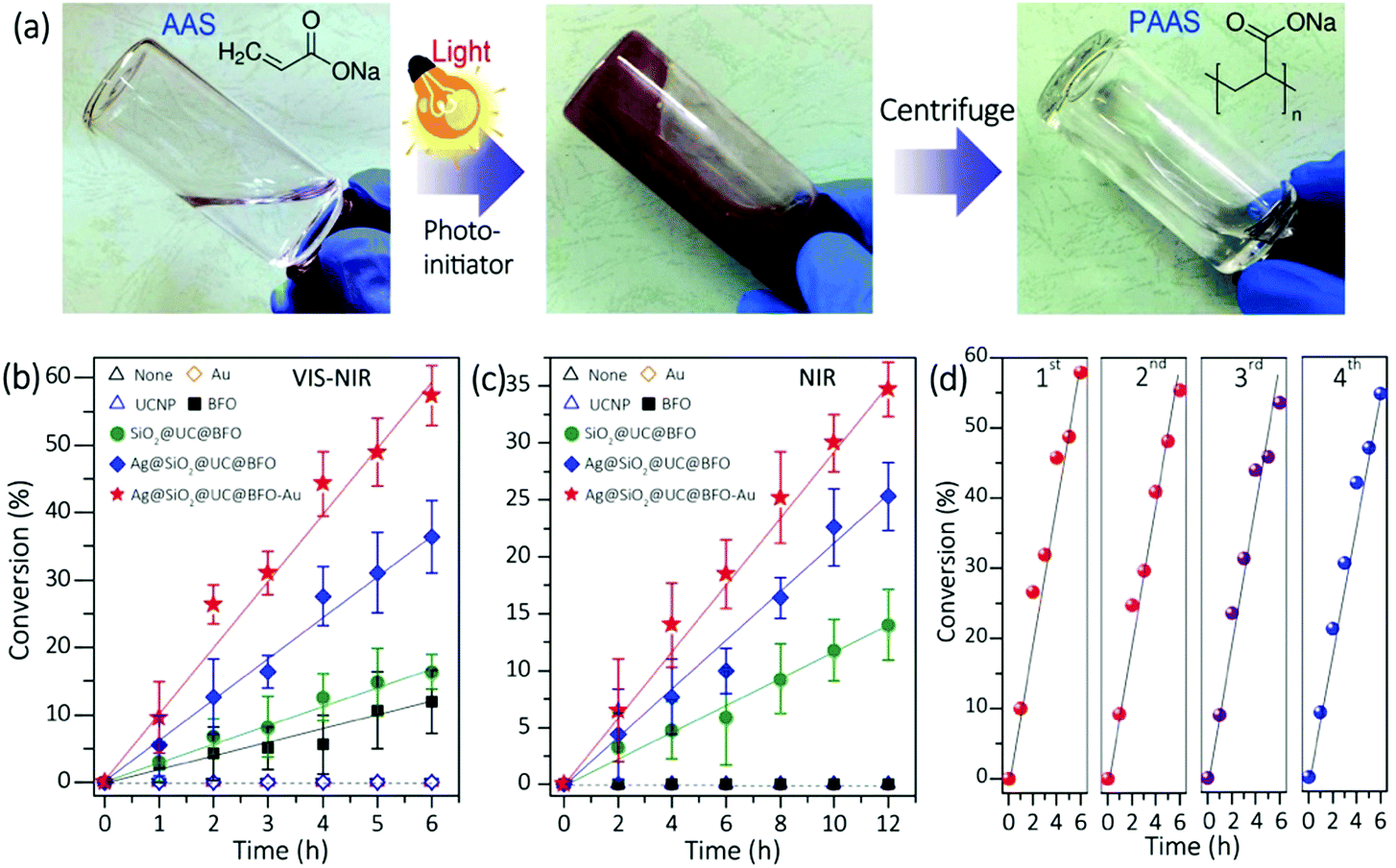 | ||
| Fig. 3 (a) Pictures of AAS aqueous solution without an NP photo-initiator (left) obtained upon polymerization reactions before (middle) and after (right) the removal of the Ag@SiO2@UC@BFO–Au initiator. Conversion of AAS using different NPs as initiators under white light (b) and NIR light (c) (the lines are used to guide the eyes). (d) Repeated runs for the polymerization using Ag@SiO2@UC@BFO–Au under VIS light. | ||
Since UC materials are used in the core@shell hybrids, the NIR photons contained in the white light (Xe lamp) of this study can be upconverted into energies in the VIS wavelength range, which overlaps with the absorption bands of BFO and Au. Thus, it is of high interest to assess the activity of the nanohybrids under NIR irradiation. As shown in Fig. 3c, pure BFO and Au NPs do not show any activity, as they do not absorb NIR photons. As expected, the polymerization took place only in the presence of the nanohybrid initiators where UC materials are involved, with a linear correlation between the conversion ratio and the reaction time. It should be noted that the samples with a Ag NP core show much enhanced activity than that without Ag (e.g., Ag@SiO2@UC@BFO vs. SiO2@UC@BFO).
The activity/stability of the Ag@SiO2@UC@BFO–Au sample was examined repeatedly in four successive polymerization reactions under VIS light. Nanohybrid initiators can easily be collected and reused after each cycle of centrifugation. As illustrated in Fig. 3d, in these continuous cycles the conversion ratio remains nearly unchanged, indicating good reusability/stability of the materials. Thus, Ag@SiO2@UC@BFO–Au works as a true initiator, making this panchromatic photopolymerization a “green” process. This behavior is to be contrasted with a conventional molecular photo-initiator, which is spent during polymerization. In addition, unlike the conventional photocatalysis system for the photo-degradation of small organic molecules, no significant changes in the molecular weight (Mn) and the polydispersity index (PDI) of the photo-synthesized PAAS polymer can be observed. Indeed, in the presence of the Ag@SiO2@UC@BFO–Au NPs, and over the course of the polymerization, the Mn value and the PDI respectively remained at 6000 g mol−1 and around 2, respectively, even after a prolonged white light exposure of 12 hours (83% conversion, Fig. S7, ESI†).
Main active species
To better understand the photo-initiating process, the main active species generated during the photoreaction were identified by scavenging tests.21,23tert-Butyl alcohol (t-BuOH), sodium ethylenediamine tetraacetate (Na2EDTA) and 1,4-benzoquinone (BQ) as scavengers of hydroxyl (OH˙) radicals, holes (h+) and superoxide radical anions (O2˙−),21,23 respectively, were introduced into the aqueous solution for polymerization with the Ag@SiO2@UC@BFO–Au initiator. Fig. 4a displays the photo-polymerization of AAS in the presence of various scavengers under white light illumination. In comparison with the scavenger-free system (black circle), the reaction efficiency for the existence of BQ (O2˙− scavenger, blue triangle) is barely affected. By stark contrast, the reaction in the presence of t-BuOH (OH˙ scavengers, red square) is nearly completely inhibited. The polymer conversion ratio is also largely reduced in the presence of Na2EDTA (hole scavengers, green lozenge), with only ∼7% conversion in 2 h. These results strongly suggest that OH˙ radicals that are conventionally thought to be created by the reaction of water with photogenerated holes, contribute at varying degrees to the radical polymerization. Thus, trapping the holes before they react with water, or trapping the OH˙ radicals both stop the polymerization. The presence of photogenerated OH˙ radicals in our system was also confirmed via electron spin resonance (ESR) spectroscopy (Fig. S8, ESI†). | ||
| Fig. 4 (a) Polymerization initiated using Ag@SiO2@UC@BFO–Au as a photo-initiator under VIS light (λ > 420 nm) irradiation in the presence of different scavengers. (b) Concentration variation of umbelliferone as a function of VIS irradiation upon different nanohybrids. The inset shows the PL spectrum evolution of umbelliferone in coumarin solution at different irradiation times. (c) Simulated absorption (Abs.) spectra of Ag@SiO2@UC@BFO–Au at different synthetic steps. (d) Simulated scattering spectra of Ag@SiO2@UC@BFO–Au (red) and the Ag NP core removed structure in this hybrid (blue). (e) Simulated electric field enhancement spectra of different interfaces of Ag@SiO2@UC@BFO–Au. The 3D view of the electric field enhancement distribution (λ = 550 nm) of the core–shell interfaces (f), surface anchored Au NPs (g) and overall surface anchored Ag@SiO2@UC@BFO–Au (h, for better visual effect, the BFO sphere is set to be semitransparent). | ||
To further verify the generation of OH˙ under irradiation, coumarin was used as a fluorescent probe for the detection of OH˙.23,33,34 Coumarin can react with OH˙ to form 7-OH coumarin (umbelliferone) which is a strong fluorescent emitter.35 The PL of the umbelliferone was measured in the presence of the NP initiator under visible illumination (Fig. 4b).36 Such umbelliferone emission cannot be detected in the absence of the NP initiator or with UCNPs alone. By contrast, OH˙ production with only BFO is rather weak, and this becomes significant by using the nanohybrids; in particular, it is most effective with the Ag@SiO2@UC@BFO–Au hybrid, which is in good agreement with the photo-polymerization tests.
Role of each structural component
In many cases, the generation of charge carriers in VIS-light driven photocatalysis can be enhanced by coupling the semiconductor with plasmonic NPs, such as Au and Ag NPs, either directly or through very thin insulating spacers to form plasmonic heterojunctions. Currently, there are three major mechanisms explaining the dominating plasmonic energy transfer process from a metal to a semiconductor, including (i) light scattering/trapping, (ii) hot electron injection (also called direct electron transfer (DET)) and (iii) plasmon-induced resonance energy transfer (PIRET).37–46 In principle, the exact mechanism chiefly relies on the optical properties of each component along with the configuration of the nanohybrid.38,40–43 Using finite element method (FEM) based numerical simulations, it is possible to investigate the optical responses of the Ag@SiO2@UC@BFO–Au nanohybrid in terms of absorption, scattering and field enhancement. The absorption spectrum of Ag NPs calculated from the FEM simulation shows the characteristic SPR absorption peak at 430 nm (Fig. S3, ESI†). SiO2 and the subsequent UC coating of Ag NPs result in the red-shift of the SPR peak up to 450 nm due to the refractive index increase of the surroundings, as exhibited in Fig. 4c. The formation of a BFO layer induces strong absorption covering the UV-to-VIS spectral range. As expected, the attachment of Au NPs to the BFO surface further increases the VIS absorption with an absorption edge up to 650 nm, due to the superimposition of the SPR band to the BFO absorption spectrum. All these absorptions in the simulation are in good agreement with the experimental observations, suggesting an appropriate model for optical analysis.The red curve shown in Fig. 4d presents the simulated scattering spectrum of the Ag@SiO2@UC@BFO–Au nanohybrid with a strong peak at ∼483 nm, which is due to the presence of the Ag core. For comparison purposes, the same structure was simulated but the Ag core was replaced by water. As can be extracted from Fig. 4d, by taking the integral of the scattering cross sections, the nanohybrid with the Ag core exhibits ∼86% more scattering (orange shadow) compared to the one without the Ag core in the wavelength range from 460 nm to 800 nm. According to the light scattering/trapping mechanism,42,43 light scattering works dominantly in the SPR band for large metal NPs (e.g. Ag NPs with D = ∼50 nm in our case, see Fig. S9 for optical spectra, ESI†), where incident light can be scattered efficiently by the metal NPs to reach the semiconductor, resulting in an improvement in the photon flux in the semiconductor, subsequently improving the photo-charge generation and photocatalytic efficiency.43 Thus, our results strongly indicate a light scattering mechanism working as the dominant process for Ag NP induced plasmonic energy transfer. The Ag core increases light scattering as an “internal mirror”, allowing VIS light to be reused by outer BFO shells and Au NPs or by other neighboring catalytic particles.
The presence of a Ag NP core increases the upconverting capability of the UC component, leading to a more efficient use of NIR photons, as shown experimentally in Fig. 2j. As both Ag and UC layers are physically separated and have no absorption overlap, the observed PL enhancement can be mainly ascribed to a distance-dependent competition between the changes in radiative and non-radiative decay rates of a chromophore in the proximity of a metal surface.47–54 Due to the coupling of Ag SPR and the emission of the UC shell, the radiative decay rate can be increased to achieve a PL enhancement. The non-radiative decay rate will also influence the PL intensity, resulting in PL quenching, because the energy can transfer from the UC shell to the metal NP when the space between the Ag NP and the UC shell is below a certain distance. Thus, an insulating layer of SiO2 with an appropriate thickness plays a vital role in suppressing the non-radiative decay along with preserving the increased radiative decay rate. This distance-dependent PL modulation can quantitatively be described by:51
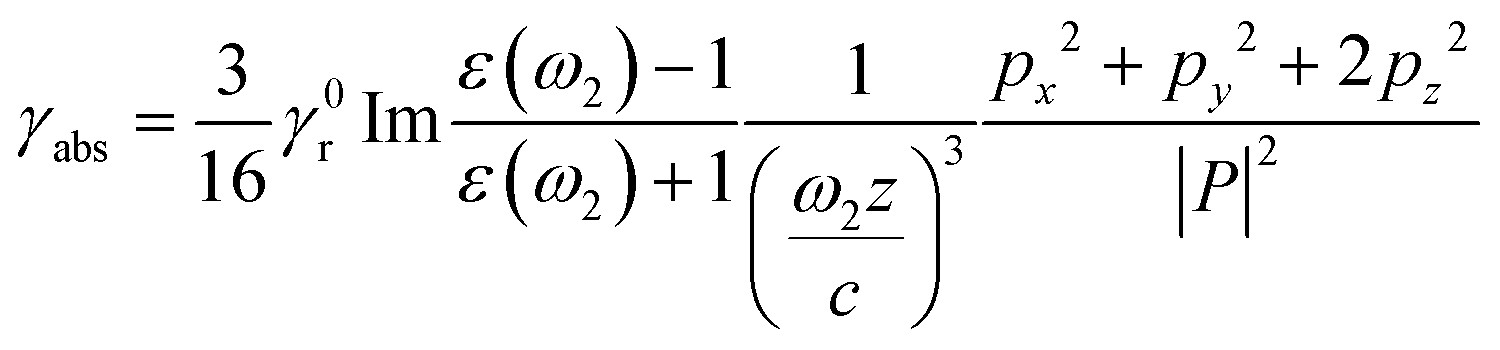 | (1) |
 | (2) |
Numerical simulations were performed again to evaluate the near-field enhancement. As shown in Fig. 4e, the local field at the Au–BFO interface was greatly increased with a maximum enhancement at λ = 550 nm (E/E0 > 37) in a wide VIS region in accordance with the SPR absorption of Au NPs. Fig. 4f–h further display the spatial distribution of the interfacial field intensity of Ag@SiO2@UC@BFO–Au under incident light of 550 nm. The detailed interfacial field distribution is clearly demonstrated in the 3D view of Ag@SiO2@UC@BFO–Au in Fig. 4f, where Au–BFO interfaces show greater field enhancement than Ag–SiO2 and SiO2–UC interfaces (E/E0 ≈ 2). Fig. 4g and h show the near-field distribution of a magnified Au–BFO interface and the outer surface of the Ag@SiO2@UC@BFO–Au structure. The most intense near-field is located at the junctions of the Au NP and BFO surface, which can largely increase the VIS light absorption and boost the transfer of stored plasmonic energy from Au to BFO using a PIRET mode. Wavelength-dependent incident photo-to-electron conversion efficiency (IPCE) measurement was conducted on Ag@SiO2@UC@BFO and Ag@SiO2@UC@BFO–Au hybrids to further confirm this interfacial energy transfer. As illustrated in IPCE spectra of Fig. S10 (ESI†), both samples show good VIS-light activity, due to the presence of BFO. More importantly, in comparison to the sample without Au, Ag@SiO2@UC@BFO–Au demonstrated a distinct photoresponse enhancement in a wide range, indicating a significant energy transfer upon the introduction of Au. Since there is a wide spectral overlap between the SPR band of Au and BFO absorption, and the photoconversion enhancement basically follows the spectral overlap, PIRET is supposed to be the dominating process for plasmonic energy transfer from Au to BFO. In addition, hot electrons may also take part in the interfacial energy transfer due to the intense SPR effect; however, compared with PIRET, this might be nondominant or even negligible as reported in a previous study of plasmonic photocatalysts with similar optical features.46 An investigation to understand the detailed energy transfer process is thus ongoing.
Mechanism
As presented in Scheme 1a, the Y2O3:Er3+,Yb3+ UC layer shows typical upconverted emissions in the VIS range overlapping with the absorption spectrum of BFO–Au, which can result in energy transfer from the UC shell to BFO. Due to the core@shell configuration, the energy transfer is expected to occur through the FRET process and a conventional radiation-reabsorption process, thereby leading to more efficient NIR photon conversion in the visible range. The upconverting mechanism of the Y2O3:Er3+,Yb3+ NPs is illustrated in Scheme 1b. By absorbing NIR photons, Yb3+ ions are excited from the ground state (2F7/2) to the excited state (2F5/2), followed by the subsequent energy transfer to the nearby Er3+ ions, exciting them from the 4I15/2 ground state to the 4I11/2 excited intermediate state. This process is repeated a second time where excited Yb3+ ions again transfer their energy to the Er3+ ions resulting in the population of the higher energy 4F7/2, which further decays to 2H11/2 and 4S3/2 states by non-radiative decay, producing the green emission at ∼525 and 545 nm, respectively. The red emission at ∼640 to 680 nm arises from the 4F9/2 state, which is populated either by non-radiative decay from the upper 2H11/2/4S3/2 states or by direct excitation from the 4I13/2 state after some of the excited Er3+ ions decay non-radiatively from the 4I11/2 intermediate state following the Yb3+ energy transfer (Scheme 1b). Due to the wide VIS spectral responsibility of BFO–Au, photo-charges of BFO can also be excited by energy transfer from the 4S3/2 and 2H11/2 excited states of the Er3+ ions that work as an “internal” VIS light source. | ||
| Scheme 1 (a) Schematic illustration of the spectral overlap of the absorption (red) of the BFO shell and the emission (blue) of the UC core. (b) Energy level diagrams of Yb3+ and Er3+ ions as well as the UC luminescence process upon NIR excitation; the energy transfer from the UC core to the BFO shell. (c) SPR enhanced photoactivity of Ag@SiO2@UC@BFO–Au under VIS and NIR excitation. (d) Schematic process of the AAS photo-polymerization using Ag@SiO2@UC@BFO–Au as a photoinitiator. | ||
Based on the above analysis, the plausible mechanism of the SPR enhanced photon harvesting of Ag@SiO2@UC@BFO–Au can be proposed. As shown in Scheme 1c, under VIS light irradiation (left image), strong near-fields are generated at the Au–BFO interfaces upon the SPR effect of the Au NPs, promoting the plasmonic energy transfer from Au to BFO via a PIRET process to induce efficient charge carrier generation in BFO; the VIS photons that are not absorbed by the BFO shell passing through the shells can be scattered back to the BFO shell by the Ag NP core, which acts as a “nanoscatterer”, resulting in more passes and re-absorption by BFO, which further improves the utilization of VIS light in the hybrids. Under NIR excitation (right image), the NIR light passing through BFO layers is absorbed by the UC layer and upconverted into VIS photons. Due to the presence of the Ag NP core, the UC emission can be greatly enhanced by the SPR induced radiative decay rate increase of the UC emission, which is subsequently transferred to the BFO shell.23
Upon the PIRET from Au to BFO and UC emission, more e−/h+ pairs are generated in BFO, and electrons are excited to the CB of BFO with holes left in the valence band (VB), followed by the migration of photogenerated charges to the surface of BFO to participate in photoredox polymerization initiation. As demonstrated in Scheme 1d, the holes in the VB of the BFO surface can react with H2O to form hydroxyl (OH˙) radicals that work as the main active species to initiate the polymerization. The OH˙ radicals can interact with monomers (AAS) to form the primary free radicals (AAS˙) to trigger the chain propagation.57 Also, the separated electrons can react with surface adsorbed O2 molecules to produce the anion radical of O2˙− as evidenced by the scavenging test. Moreover, the internal electric field resulting from the spontaneous polarization of the BFO shell may also contribute to the separation of the photogenerated e−/h+ pairs, thereby further enhancing photo-activity.58–60
Conclusions
In this work, a rationally designed core@triple-shell nanohybrid composed of plasmonic NPs, UC materials and BFO shells was synthesized for VIS-NIR light driven photo-polymerization. Sodium acrylate monomer can efficiently be polymerized at room temperature using Ag@SiO2@UC@BFO–Au as an initiator under VIS-NIR light irradiation. After the reaction, the polymerized product and the NP initiator can easily be separated/purified by centrifugation, and the NP initiator can be reused, realizing a “green” reaction process. This architectural design of Ag@SiO2@UC@BFO–Au demonstrates unique advantages for photon harvesting: the core@shell morphology can favor the non-radiative energy transfer from the UC shell to BFO, and the plasmonic NPs improve the utilization of VIS and NIR photons. The Ag NP core shows a dual role as a “nanoscatterer” to increase the harvesting of VIS photons in BFO and as a “nanoresonator” to enhance the UC emission. The structural configuration as well as the optical feature of the hybrid greatly favor the plasmonic energy transfer from Au to BFO via a PIRET process. This unique plasmonic core@shell structure significantly improves the solar energy utilization by efficiently capturing VIS and NIR photons. We envision that such panchromatic photoinitiators will be valuable for the development of photopolymerizable pigmented formulations in stereolithographic high-end 3D-printing applications which so far cannot be polymerized due to the light absorption of the pigment at the same wavelength as conventional photoinitiating systems.Experimental
Materials
Ethylene glycol (EG), auric acid, ethanol, NH4OH (30 wt%), Y(NO3)3·6H2O, Yb(NO3)3·6H2O, Er(NO3)3·6H2O, urea, and NaOH were purchased from Alfa Aesar. AgNO3, polyvinylpyrrolidone (PVP, Mw = 55000), tetraethyl orthosilicate (TEOS), Bi(NO3)3·5H2O, Fe(NO3)3·9H2O, coumarin, trisodium citrate, EDTA, HCl, HNO3, CH3COOH, ABV, t-BuOH, disodium ethylenediaminetetraacetate (Na2EDTA) and BQ were purchased from Sigma-Aldrich. Acrylic acid was received from Sinopharm and purified by vacuum distillation. All other chemicals were used without further purification. Water was of nanopure grade (18.2 MΩ cm at 25 °C).
Synthesis of Ag@SiO2 NPs
Ag NPs were synthesized as reported by Xia et al.,61 where a thermal reduction process was performed using AgNO3 dissolved EG with PVP as the protecting agent. The weight ratio was set to be 1:5 between AgNO3 and PVP in order to achieve 50 nm Ag NPs. SiO2 coated Ag NPs were prepared using a Stöber method as reported before. Typically, to obtain the SiO2 shell with a thickness of ∼30 nm, 150 mg of Ag NPs were dispersed in ammonia solution in 200 mL ethanol (4.2 vol% ammonia in ethanol). Under stirring (600 rpm), 0.1 mL of TEOS was quickly injected into the solution.62 By varying the amount of added TEOS, the thickness of the SiO2 shell can be programmed. This mixture was then stirred overnight at room temperature to form the Ag@SiO2 structure.
Synthesis of Ag@SiO2@Y,Yb,Er(OH)CO348
In a typical synthesis, 50 mg of Ag@SiO2 NPs, 1 mL of 1.0 M Y(NO3)3, 5 μL of 0.89 M Yb(NO3)3, 5 μL of 0.89 M Er(NO3)3 and 5.4 g of urea were dissolved in 500 mL of H2O by stirring for 30 min. This mixture was then heated slowly up to 80 °C for 2 h. The pale yellow powder was collected via centrifugation to form the suspension and washed with ethanol twice. The Ag@SiO2@Y,Yb,Er(OH)CO3 powder was dried in a vacuum oven at 60 °C overnight. Ag@SiO2@Y2O3:Er3+,Yb3+ can be prepared by thermally treating part of this powder in a furnace under a nitrogen atmosphere (temperature ramp of 5 °C min−1 from room temperature to 500 °C followed by a 3 h isotherm at 500 °C).Synthesis of Ag@SiO2@UC@BFO
First, to 10 mL of 10% HNO3 solution, 0.242 g (0.5 mmol) of Bi(NO3)3·5H2O and 0.202 g (0.5 mmol) of Fe(NO3)3·9H2O were added to form a clear solution. Subsequently, 0.44 g (1.5 mmol) of EDTA was dissolved to this solution and heated at 80 °C for 1 h under stirring. Water was then boiled-off and the residue was washed with methanol and centrifuged to yield EDTA–Bi/Fe(III) complexes.23 The EDTA–Bi/Fe(III) complex mixture was then dissolved in 5 mL water to form an EDTA–Bi/Fe(III) master-batch solution. Second, 40 mg of the Ag@SiO2@Y,Yb,Er(OH)CO3 powder was dispersed in 5 mL water to form a suspension, and then 200 μL of the Bi/Fe(III) master-batch solution was injected into the suspension and stirred for 2 h. The EDTA–Bi/Fe(III) complexes were then co-precipitated at the surface of Ag@SiO2@Y,Yb,Er(OH)CO3 by adding 10 mL of ethanol dropwise into the mixture solution. The received powder was dried and ground into a fine powder. Finally, the powder was heated at 500 °C in a furnace under a nitrogen atmosphere (the same condition as shown above) to achieve the Ag@SiO2@UC@BFO structure.Synthesis of Ag@SiO2@UC@BFO–Au
The above Ag@SiO2@UC@BFO powders were mixed with colloidal Au NPs with the diameter of ∼20 nm, synthesized using the classic Frens method,63 to form a uniform suspension. Then a HCl solution (1 mM) was added dropwise under strong stirring until the pH value decreased to ∼3–4. The mixture was then stirred at room temperature for another 30 min. Then, the brown precipitate was recovered via centrifugation, followed by washing with distilled water and ethanol and dried in a vacuum oven at 60 °C overnight.Photo-polymerization
Sodium acrylate (AAS) monomer was first synthesized by neutralizing acrylic acid with NaOH. (i) VIS-light-driven photo-polymerization. In a typical process, to an aqueous solution containing 190 mg (2 mmol) of SA and 10 mL of degassed water, 10 mg of NP powder was added and sonicated to form a uniform suspension. The vial was closed tightly and the solution was irradiated using a Xe arc lamp (300 W, CEAUlight, 10 cm distance) with a cut-off filter (λ > 420 nm) in the optical system. (ii) NIR-light-driven photo-polymerization. The sample preparation was the same as that for the VIS light experiments. The only difference is that diode laser beams (λ = 980 nm) were used as the NIR light source. The power of the laser was set to be ∼0.04 W mm−2. At regular intervals, the partial reaction solution was centrifuged to remove the NPs. The polymers in the supernatant were purified/precipitated by mixing with a large amount of warm methanol. The collected polymers were dried under vacuum overnight and used for characterization. (iii) Active species identification. Monomer solutions with 1 mM of tert-butyl alcohol (t-BuOH), Na2EDTA or 1,4-benzoquinone (BQ) were used, in photo-polymerization, as the scavengers of hydroxyl radicals, holes and superoxide radicals, respectively, to identify the possible active species. This test was performed under the same experimental conditions as photo-polymerization under VIS light.Characterization
(i) XRD. The crystal structure of the as-synthesized materials was examined by XRD (D8 Advance; Bruker, Billerica, MA) using CuKα radiation (λ = 1.5418 Å). (ii) TEM and EDS. The morphology of the samples was analyzed using a JEOL-2100F TEM instrument (École Polytechnique de Montréal, Montréal, Canada). Meanwhile, the EDX spectra were taken on the chosen area via TEM for elemental analysis. (iii) UV-Visible absorption spectroscopy. UV-Vis absorption spectra were measured using a PerkinElmer Lambda 750 spectrometer equipped with a 60 mm integrating sphere. (iv) XPS. Samples for XPS measurements were prepared in an identical manner to those for XRD measurements. XPS spectra were acquired using ESCA Escalab 220i XL with a twin Al Kα X-ray source. (v) PL. PL was measured under 980 nm excitation light with 0.08 W mm−2 power density (BWT Beijing Ltd). The emission spectra were recorded by a spectrophotometer (Avaspec-2048L-USB2) using an optical fiber. (vi) GPC. Mn of the polymers was determined using a PL-GPC50 spectrometer using water as the mobile phase. (vii) IPCE measurement is performed using a Zolix SCS-10 IPCE test system. (viii) ICP-OES is conducted using a Spectro Genesis ICP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Z. is grateful for funding from the National Key R&D Program of China (2017YFE0102700) from the Ministry of Science and Technology (MOST) of China, the National Natural Science Foundation of China (NSFC, No. 51272128), and the Jiangsu University (17JDG008). H. L. gratefully acknowledges the NSFC (No. 51732007). J. C., F. V. and L. R. acknowledge funding from the Natural Sciences and Engineering Research Council (NSERC) of Canada. J. C. and F. V. are also grateful for the financial support from Fonds de Recherche du Québec-Nature et Technologies (FRQNT) and NanoQuébec (Major Infrastructures). L. R. further acknowledges the support from PRIMA Quebec. J. C. is grateful to the Canada Research Chair program for supporting this research. X. J. acknowledges FRQNT for a Doctoral Research Scholarship: Bourse d’Excellence pour Étudiants Étrangers (PBEEE – V1). F. V. and J. C. are members of the Quebec Center for Advanced Materials.References
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Cornell, NY, 2010 Search PubMed.
- J. Brydson, Plastics Materials, Butterworth-Heinermann, Oxford, OX, 1999 Search PubMed.
- F. Kettling, B. Vonhören, J. A. Krings, S. Saito and B. J. Ravoo, Chem. Commun., 2015, 51, 1027–1030 RSC.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- B. Kiskan, J. Zhang, X. Wang, M. Antonietti and Y. Yagci, ACS Macro Lett., 2012, 1, 546–549 CrossRef CAS.
- H. Tar, D. Sevinc Esen, M. Aydin, C. Ley, N. Arsu and X. Allonas, Macromolecules, 2013, 46, 3266–3272 CrossRef CAS.
- R. D. Harris, S. Bettis Homan, M. Kodaimati, C. He, A. B. Nepomnyashchii, N. K. Swenson, S. Lian, R. Calzada and E. A. Weiss, Chem. Rev., 2016, 116, 12865–12919 CrossRef CAS PubMed.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 24, 2568–2580 RSC.
- J. Tian, Z. Zhao, A. Kumar, R. I. Boughton and H. Liu, Chem. Soc. Rev., 2014, 43, 6920–6937 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D.-L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- A. Gajović, S. Šturm, B. JanWar, A. Šantić, K. Žagar and M. Čehz, J. Am. Ceram. Soc., 2010, 93, 3173–3179 CrossRef.
- D. Cao, Z. Wang, Nasori, L. Wen, Y. Mi and Y. Lei, Angew. Chem., Int. Ed., 2014, 53, 11027–11031 CrossRef CAS PubMed.
- S. Li, J. Zhang, M. G. Kibria, Z. Mi, M. Chaker, D. Ma, R. Nechache and F. Rosei, Chem. Commun., 2013, 49, 5856–5858 RSC.
- X. Zhang, L. Bourgeois, J. Yao, H. Wang and P. A. Webley, Small, 2007, 3, 1523–1528 CrossRef CAS PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- B. Zheng, Q. Guo, D. Wang, H. Zhang, Y. Zhu and S. Zhou, J. Am. Ceram. Soc., 2015, 98, 136–140 CrossRef CAS.
- Y. Tang, W. Di, X. Zhai, R. Yang and W. Qin, ACS Catal., 2013, 3, 405–412 CrossRef CAS.
- W. Wang, W. Huang, Y. Ni, C. Lu and Z. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 340–348 CrossRef CAS PubMed.
- Z. Xu, M. Quintanilla, F. Vetrone, A. O. Govorov, M. Chaker and D. Ma, Adv. Funct. Mater., 2015, 25, 2950–2960 CrossRef CAS.
- T. Liu, Y. Xu and J. Zhao, J. Am. Ceram. Soc., 2010, 93, 3637–3641 CrossRef CAS.
- J. Zhang, Y. Huang, L. Jin, F. Rosei, F. Vetrone and J. P. Claverie, ACS Appl. Mater. Interfaces, 2017, 9, 8142–8150 CrossRef CAS PubMed.
- A. Jaiswal, R. Das, K. Vivekanand, P. M. Abraham, S. Adyanthaya and P. Poddar, J. Phys. Chem. C, 2010, 114, 2108–2115 CrossRef CAS.
- F. Yan, M. O. Lai, L. Lu and T. J. Zhu, J. Phys. Chem. C, 2010, 114, 6994–6998 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003 Search PubMed.
- J. Zhang, G. Chen, M. Chaker, F. Rosei and D. Ma, Appl. Catal., B, 2013, 132–133, 107–115 CrossRef CAS.
- K. Zhang, S. Zhou, Y. Zhuang, R. Yang and J. Qiu, Opt. Express, 2012, 20, 8675–8680 CrossRef CAS PubMed.
- S. Zhou, N. Jiang, K. Miura, S. Tanabe, M. Shimizu, M. Sakakura, Y. Shimotsuma, M. Nishi, J. Qiu and K. Hirao, J. Am. Chem. Soc., 2010, 132, 17945–17952 CrossRef CAS PubMed.
- I. Lacík, S. Beuermann and M. Buback, Macromol. Chem. Phys., 2004, 205, 1080–1087 CrossRef.
- I. Lacík, S. Beuermann and M. Buback, Macromolecules, 2001, 34, 6224–6228 CrossRef.
- P. Maruthamuthu, Makromol. Chem., Rapid Commun., 1980, 1, 23–25 CrossRef CAS.
- J. Zhang and Y. Nosaka, J. Phys. Chem. C, 2013, 117, 1383–1391 CrossRef CAS.
- J. Zhang and Y. Nosaka, J. Phys. Chem. C, 2014, 118, 10824–10832 CrossRef CAS.
- G. Louit, S. Foley, J. Cabillic, H. Coffigny, F. Taran, A. Valleix, J. P. Renault and S. Pin, Radiat. Phys. Chem., 2005, 72, 119–124 CrossRef CAS.
- Y. Nosaka, S. Komori, K. Yawata, T. Hirakawa and A. Y. Nosaka, Phys. Chem. Chem. Phys., 2003, 5, 4731–4735 RSC.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 865 CrossRef CAS.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- J. Zhang, X. Jin, P. I. Morales-Guzman, X. Yu, H. Liu, H. Zhang, L. Razzari and J. P. Claverie, ACS Nano, 2016, 10, 4496–4503 CrossRef CAS PubMed.
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- S. K. Cushing and N. Wu, J. Phys. Chem. Lett., 2016, 7, 666–675 CrossRef CAS PubMed.
- N. Wu, Nanoscale, 2018, 10, 2679–2696 RSC.
- S. K. Cushing, J. Li, F. Meng, T. R. Senty, S. Suri, M. Zhi, M. Li, A. D. Bristow and N. Wu, J. Am. Chem. Soc., 2012, 134, 15033–15041 CrossRef CAS PubMed.
- J. Li, S. K. Cushing, F. Meng, T. R. Senty, A. D. Bristow and N. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS.
- F. Meng, S. K. Cushing, J. Li, S. Hao and N. Wu, ACS Catal., 2015, 5, 1949–1955 CrossRef CAS.
- P. Anger, P. Bharadwaj and L. Novotny, Phys. Rev. Lett., 2006, 96, 3–6 CrossRef PubMed.
- F. Zhang, G. B. Braun, Y. Shi, Y. Zhang, X. Sun, N. O. Reich, D. Zhao and G. Stucky, J. Am. Chem. Soc., 2010, 132, 2850–2851 CrossRef CAS PubMed.
- J. R. Lakowicz, Anal. Biochem., 2005, 337, 171–194 CrossRef CAS PubMed.
- M. Bauch, K. Toma, M. Toma, Q. Zhang and J. Dostalek, Plasmonics, 2014, 9, 781–799 CrossRef CAS PubMed.
- P. Bharadwaj and L. Novotny, Opt. Express, 2007, 15, 14266–14274 CrossRef CAS PubMed.
- F. W. Kang, J. J. He, T. Y. Sun, Z. Y. Bao, F. Wang and D. Y. Lei, Adv. Funct. Mater., 2017, 27, 1701842 CrossRef.
- J. He, W. Zheng, F. Ligmajer, C.-F. Chan, Z. Bao, K.-L. Wong, X. Chen, J. Hao, J. Dai, S.-F. Yu and D. Y. Lei, Nature, 2017, 6, e16217 CAS.
- P. Yuan, Y. H. Lee, M. K. Gnanasammandhan, Z. Guan, Y. Zhang and Q.-H. Xu, Nanoscale, 2012, 4, 5132–5137 RSC.
- I. Thomann, B. A. Pinaud, Z. Chen, B. M. Clemens, T. F. Jaramillo and M. L. Brongersma, Nano Lett., 2011, 11, 3440–3446 CrossRef CAS PubMed.
- J. Lee, A. O. Govorov, J. Dulka and N. A. Kotov, Nano Lett., 2004, 4, 2323–2330 CrossRef CAS.
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- H. Li, Y. Sang, S. Chang, X. Huang, Y. Zhang, R. Yang, H. Jiang, H. Liu and Z. L. Wang, Nano Lett., 2015, 15, 2372–2379 CrossRef CAS PubMed.
- Y. Cui, J. Briscoe and S. Dunn, Chem. Mater., 2013, 25, 4215–4223 CrossRef CAS.
- S. Li, B. Alotaibi, W. Huang, Z. Mi, N. Serpone, R. Nechache and F. Rosei, Small, 2015, 11, 4018–4026 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS PubMed.
- C. Graf, D. L. J. Vossen, A. Imhof and A. Blaaderen, Langmuir, 2003, 19, 6693–6700 CrossRef CAS.
- N. Nath and A. Chilkoti, Anal. Chem., 2004, 76, 5370–5378 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization and simulation details. See DOI: 10.1039/c9nh00026g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |