DOI:
10.1039/C8NH00319J
(Communication)
Nanoscale Horiz., 2019,
4, 464-471
Nano-crater morphology in hybrid electron-collecting buffer layers for high efficiency polymer:nonfullerene solar cells with enhanced stability†
Received
21st September 2018
, Accepted 23rd November 2018
First published on 24th November 2018
Abstract
Organic solar cells based on solution processes have strong advantages over conventional silicon solar cells due to the possible low-cost manufacturing of flexible large-area solar modules at low temperatures. However, the benefit of the low temperature process is diminished by a thermal annealing step at high temperatures (≥200 °C), which cannot be practically applied for typical plastic film substrates with a glass transition temperature lower than 200 °C, for inorganic charge-collecting buffer layers such as zinc oxide (ZnO) in high efficiency inverted-type organic solar cells. Here we demonstrate that novel hybrid electron-collecting buffer layers with a particular nano-crater morphology, which are prepared by a low-temperature (150 °C) thermal annealing process of ZnO precursor films containing poly(2-ethyl-2-oxazoline) (PEOz), can deliver a high efficiency (12.35%) similar to the pristine ZnO layers prepared by the conventional high-temperature process (200 °C) for inverted-type polymer:nonfullerene solar cells. The nano-crater morphology was found to greatly enhance the stability of solar cells due to improved adhesion between the active layers and ZnO:PEOz hybrid buffer layers.
Conceptual insights
Charge-collecting buffer layers play a crucial role in organic solar cells with bulk heterojunction (BHJ) light-absorbing layers because they effectively transfer charges, which are generated by charge separation processes from excitons in the BHJ layers, to the corresponding electrodes. Of various materials, zinc oxide (ZnO) has been intensively studied as an electron-collecting buffer layer for inverted-type organic solar cells because it can be prepared by employing wet-coating processes using precursor solutions. However, to date, thermal annealing at high temperatures (≥200 °C) is still a big hurdle for the ZnO buffer layers to achieve the low-temperature process for organic solar cells. Although a few studies have attempted to reduce the annealing temperature, the device performances couldn’t be desirably improved. In this work, we invented a particular nano-crater structure by the low-temperature (150 °C) thermal annealing of the hybrid buffer layers consisting of ZnO and poly(2-ethyl-2-oxazoline) (PEOz). The low-temperature processed ZnO:PEOz hybrid layers with particular nano-crater morphology delivered one of the highest power conversion efficiencies (PCE = 12.35%), which is very similar to the PCEs in the case of high-temperature (≥200 °C) processed pristine ZnO layers. In addition, the stability of solar cells was noticeably enhanced due to the formation of mechanical interlocking interfaces by the nano-crater morphology in the ZnO:PEOz hybrid buffer layers.
|
Introduction
Charge-collecting buffer layers play a crucial role in organic solar cells with bulk heterojunction (BHJ) light-absorbing layers because they should effectively transfer charges, which are generated by charge separation processes from excitons in the BHJ layers, to their corresponding electrodes.1–3 Most of the inverted-type organic solar cells, the power conversion efficiency (PCE) of which has already exceeded 10% and very recently reached ca. 13–14%, employ metal oxides as charge-collecting buffer layers because of their high charge carrier mobilities and well-matched energy-band structures.4–7
Of various metal oxides, zinc oxide (ZnO) and molybdenum oxide (MoO3) have been intensively employed as an electron-collecting buffer layer and a hole-collecting buffer layer for inverted-type organic solar cells, respectively.8–13 The MoO3 layers, which are just formed on the BHJ layers mainly by the thermal evaporation of MoO3 powders under vacuum, act excellently as a hole-collecting buffer layer without further treatments.14–16 In contrast, the ZnO layers are typically prepared by wet-coating (sol–gel) processes using their precursor solutions, followed by thermal annealing processes at high temperatures (≥200 °C) for securing suitable electron mobility and good film quality.17–21 Note that thermal annealing at low temperatures (ca. 120 °C etc.) was attempted for the sol–gel processed ZnO layers but the PCE was less than 5%.22,23 However, thermal annealing at such a high temperature is a critical drawback because low temperature processes have been recognized as one of the best advantages of organic solar cells over conventional silicon-based inorganic solar cells when it comes to fabrication of flexible plastic film substrates with a typical glass transition temperature of ≪200 °C (see Table S1, ESI†).24–31
To reduce the thermal annealing temperature of ZnO electron-collecting buffer layers for organic solar cells, various wet processes such as aluminum doping, nanoparticle grinding, and hybrid conjugated polymer/inorganic nanocomposite methods have been attempted.32–35 However, the aluminum doping of ZnO using the precursor solutions of zinc acetate and aluminum acetate was found to still require ca. 260 °C because a desirable performance could not be reached by annealing at below 150 °C.32,33 In addition, the nanoparticle grinding method requires a long process time (ca. 500 min), compared to the conventional high temperature process (ca. 60 min), and results in non-uniform ZnO nanoparticles with large aggregates.34 The hybrid conjugated polymer/inorganic nanocomposite method required 110 °C for thermal annealing but the device performance was very poor (PCE = 1.6% only).35
Here we report that hybrid electron-collecting buffer layers with a particular nano-crater morphology, which are prepared by the low-temperature (150 °C) thermal annealing of sol–gel processed ZnO precursor films containing poly(2-ethyl-2-oxazoline) (PEOz), deliver one of the record-high efficiencies (PCE = 12.35%) for inverted-type organic solar cells with the polymer:nonfullerene bulk heterojunction layers based on poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione))] (PBDB-T) and 3,9-bis(2-methylene-(3-(1,1-dicyanomethylene)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene (ITIC) or 3,9-bis(6-methyl-2-methylene-(3-(1,1-dicyanomethylene)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene (IT-M).36–38 The best efficiency was achieved at the PEOz content of 5 wt% which led to nano-craters with a diameter of 100–200 nm. In particular, the stability of the PBDB-T:ITIC solar cells was greatly improved by employing the ZnO:PEOz hybrid electron-collecting buffer layers with the nano-crater morphology due to the formation of mechanically interlocking interfaces.
Results and discussion
Preparation and characteristics of ZnO:PEOz hybrid films
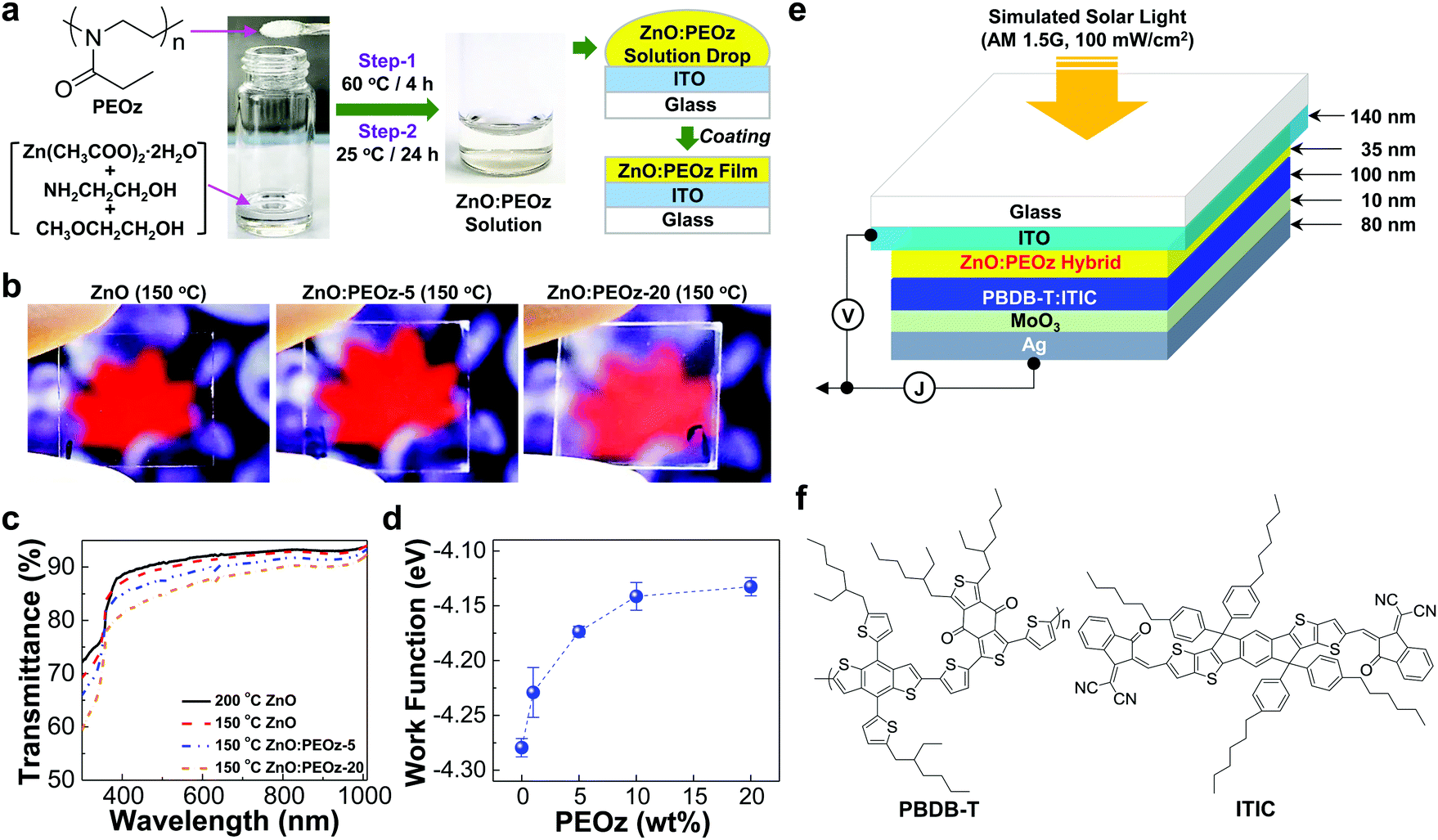
As shown in Fig. 1a, the ZnO precursor solutions including zinc acetate dihydrate (Zn(CH3COO)2·2H2O) and 2-ethanolamine (NH2CH2CH2OH) in 2-methoxyethanol (CH3OCH2CH2OH) were first prepared and then the PEOz polymer was added with various contents, followed by the two step reactions. The resulting solutions after the reactions were optically transparent without any insoluble particles (see Fig. S1a, ESI†), whereas the final films after the thermal annealing at 150 °C showed slightly different optical transparency according to the PEOz content. At a glance, the sol–gel derived ZnO:PEOz hybrid films (optimized thickness = ca. 35 nm) at the PEOz content of 5 wt% seemed to have apparently a similar optical transparency to the pristine ZnO films, but the reduced optical transparency was clearly observed from the hybrid films at 20 wt% (see contrast-enhanced photographs in Fig. 1b). Here it is noted that the optical transparency of all films was sufficiently high that they could not be easily distinguished with the naked eye (see Fig. S1b, ESI†). The detailed difference in the optical transparency can be revealed by the transmittance spectra in Fig. 1c and Fig. S2 (ESI†), disclosing the larger reduction in the optical transparency at the shorter wavelengths (>350 nm) than the longer wavelengths. This can be attributed to the scattering loss of incoming light by the possible presence of phase-separated domains.39,40 Interestingly, however, the work function (an absolute value) of the ZnO:PEOz hybrid films was gradually reduced as the PEOz content increased (Fig. 1d). This lowering of the work function can be attributed to the formation of hybrid states due to the specific interaction between Zn atoms (positive charges) in the ZnO domains and nitrogen atoms (lone pair electrons – negative charges) in the PEOz domains as similarly observed in the case of ultrathin dipole layers.41–50 To examine the performance of the sol–gel processed ZnO:PEOz films, the inverted-type PBDB-T:ITIC solar cells were fabricated by employing MoO3 hole-collecting buffer layers as illustrated in Fig. 1e (see the molecular structure of PBDB-T and ITIC in Fig. 1f).
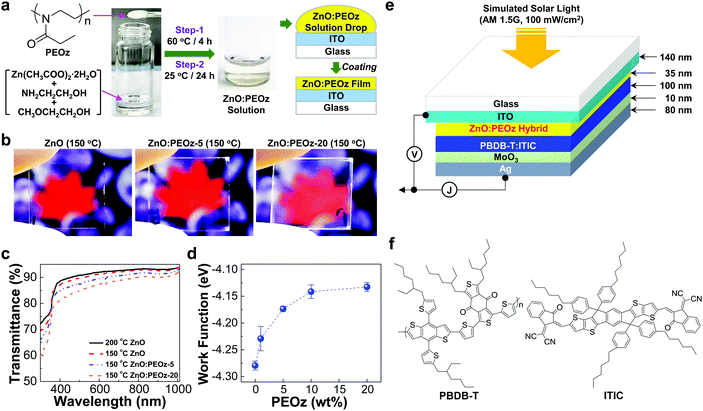 |
| | Fig. 1 Preparation of ZnO:PEOz hybrid films for inverted-type PBDB-T:ITIC solar cells. (a) Procedure for the preparation of ZnO:PEOz hybrid films from precursor solutions via two step reactions. (b) Photographs of the pristine ZnO and ZnO:PEOz hybrid films (layers) coated on the ITO-glass substrates (PEOz-5 and PEOz-20 stand for the PEOz content of 5 wt% and 20 wt%, respectively). (c) Optical transmittance spectra for the pristine ZnO and hybrid films coated on the ITO-glass substrates (note the different annealing temperatures for the pristine ZnO films). (d) Change of the ZnO work function (measured by Kelvin probe) according to the PEOz content in the ZnO:PEOz hybrid films. (e) Device structure for inverted-type solar cells with the BHJ (PBDB-T:ITIC) layers-coated on the ZnO:PEOz hybrid layers. (f) Chemical structure of PBDB-T and ITIC. | |
Performances of solar cells with the ZnO:PEOz hybrid layers
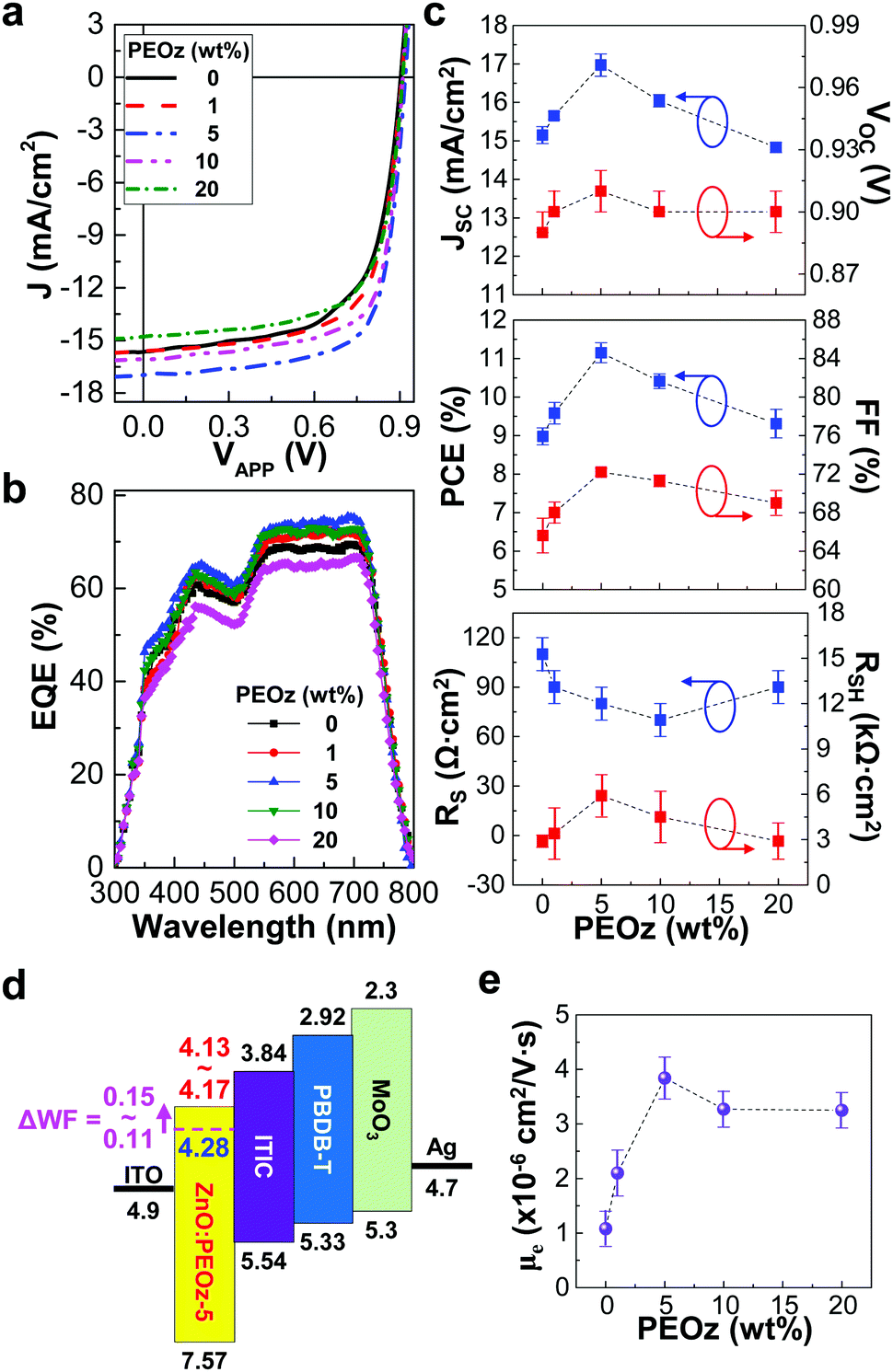
The performance of the inverted-type PBDB-T:ITIC solar cells was measured under simulated solar light (air mass 1.5G, 100 mW cm−2). As shown in Fig. 2a and Fig. S3 (ESI†), the current density–voltage (J–V) curves were pronouncedly changed according to the PEOz content. As the PEOz content increased up to 5 wt%, the J–V curve was shifted towards the increased short circuit current density (JSC) direction. Further increase of the PEOz content (≥10 wt%), however, resulted in relatively poor J–V curves with the reduced JSC values (see the EQE spectra in Fig. 2b). The detailed trend of more than 50 devices is displayed in Fig. 2c, in which the highest JSC value was obtained at the PEOz content of 5 wt%. Similarly, the open circuit voltage (VOC) was increased from 0.90 V to 0.92 V as the PEOz content increased up to 5 wt% and slightly turned down but stayed still higher than that of the control device (0.91 V at 0 wt% PEOz) (see Table 1). This high VOC behavior can be ascribed to the lowered work function by the presence of PEOz as discussed in Fig. 1d (see the work function shift in the flat energy band diagram in Fig. 2d). Here it is worthy to note that a relatively small increase in VOC compared to the work function shift can be attributed to various reasons such as physically imperfect interfacial contacts between ZnO:PEOz hybrid layers and BHJ layers, charge-blocking loss of electrons by the direct contact between ZnO:PEOz hybrid layers and electron-donating components (higher LUMO energy level of PBDB-T) in the BHJ layers, etc., as previously reported (see Table S2, ESI†). In more detail, the imperfect interfacial contacts can give rise to the increased non-ohmic states between the electron-collecting buffer layers and BHJ layers, leading to the lowered VOC.51 In addition, if small portions of donor (PBDB-T) molecules do predominantly contact the electron-collecting buffer layers during the spin-coating process, they can block the electron transfer from the acceptor (ITIC) domains to the electron-collecting buffer layers so that the photovoltage level becomes low accordingly.52
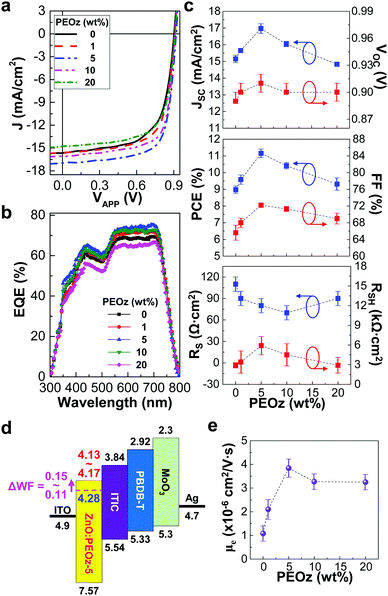 |
| | Fig. 2 Solar cell performances and electron mobility. (a) Light (air mass 1.5G, 100 mW cm−2) J–V curves for the inverted-type PBDB-T:ITIC solar cells with the ZnO:PEOz hybrid layers annealed at 150 °C. (b) EQE spectra for the same solar cells in a. (c) Change of JSC, VOC, PCE, FF, RS and RSH as a function of the PEOz content (note that all data were obtained from the light J–V curves for more than 50 devices). (d) Simplified flat energy band diagram for the inverted-type PBDB-T:ITIC solar cells with the ZnO:PEOz-5 hybrid layers (the shift of ZnO work function is marked with ΔWF in the case of 5 wt% PEOz): note that the minus sign (−) and unit (eV) of energy values were omitted for simplification. (e) Electron mobility (μe) as a function of the PEOz content. | |
Table 1 Summary of solar cell parameters for the inverted-type PBDB-T:ITIC solar cells with the ZnO:PEOz hybrid layers (annealed at 150 °C) under illumination with simulated solar light (air mass 1.5G, 100 mW cm−2)
| Parameters |
PEOz (wt%) |
| 0 |
1 |
5 |
10 |
20 |
|
V
OC (V) |
0.90 (±0.01) |
0.90 (±0.01) |
0.92 (±0.01) |
0.91 (±0.01) |
0.91 (±0.01) |
|
J
SC (mA cm−2) |
15.15 (±0.22) |
15.65 (±0.25) |
16.97 (±0.23) |
16.04 (±0.17) |
14.83 (±0.07) |
| FF (%) |
65.60 (±1.90) |
68.00 (±1.20) |
72.20 (±1.30) |
71.30 (±0.50) |
69.0 (±0.80) |
| PCE (%) |
8.98 (±0.22) |
9.58 (±0.26) |
11.15 (±0.31) |
10.41 (±0.29) |
9.31 (±0.11) |
|
R
S (kΩ cm2) |
0.11 (±0.01) |
0.09 (±0.01) |
0.08 (±0.01) |
0.07 (±0.01) |
0.09 (±0.01) |
|
R
SH (kΩ cm2) |
2.90 (±0.40) |
3.40 (±1.70) |
5.90 (±1.40) |
4.50 (±1.70) |
2.90 (±1.20) |
Interestingly, the fill factor (FF) showed a similar trend to VOC (FF = 69% at 20 wt% PEOz versus FF = 65.6% at 0 wt%), which can be supported by the series resistance (RS) trend that is still lower for 20 wt% than 0 wt% (see the bottom panel in Fig. 2c). As a result, the highest PCE (11.46%), which is very similar to the record high efficiency (11.21%) from the same solar cells with the ZnO layers annealed at 200 °C,36 could be achieved at the PEOz content of 5 wt% by annealing at 150 °C (see the middle panel in Fig. 2c and Table 1). Here the major factors contributing to the highest PCE are regarded as both JSC and FF, which can be explained by the highest electron mobility at the PEOz content of 5 wt% (see Fig. 2e and Fig. S4, ESI†). However, it is considered that the electron mobility is not solely responsible for the performance change of solar cells because the JSC was lower at 20 wt% than 0 wt%, even though the electron mobility was three times higher at 5 wt% than 0 wt% (see Fig. 2e). Here it is worth noting that the performance of polymer:fullerene solar cells, of which the active layers consist of poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene-alt-3-fluorothieno[3,4-b]thiophene-2-carboxylate] (PTB7-Th) and [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM), was also noticeably improved by the ZnO:PEOz hybrid layers (5 wt% PEOz) (see Fig. S5, ESI†). This result confirms the positive effect of the present ZnO:PEOz hybrid layers on the improved performance of organic solar cells.53,54
The morphology of sol–gel processed ZnO:PEOz hybrid layers
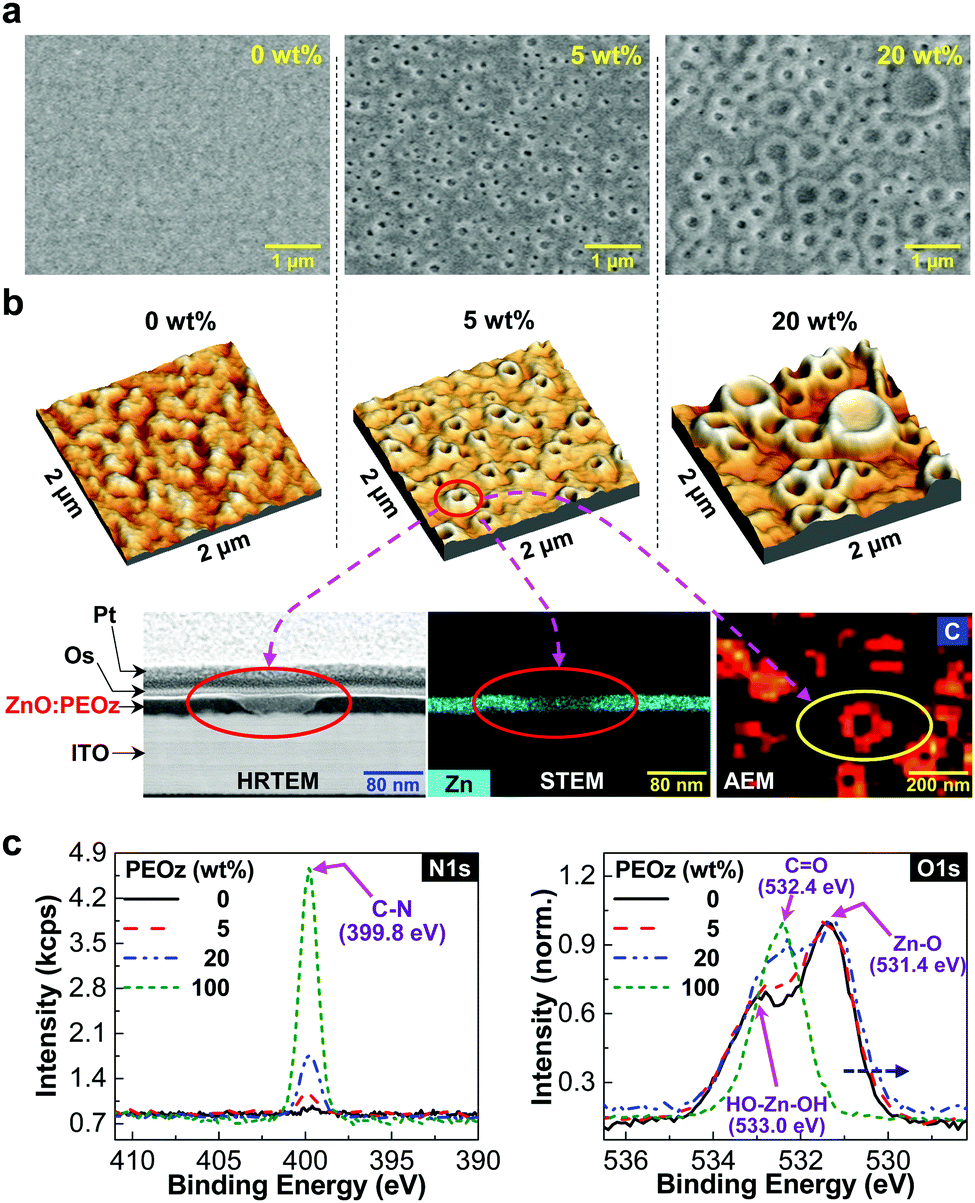
To further understand the discrepancy between the electron mobility and device performance trends according to the PEOz content, the morphology of the ZnO:PEOz hybrid layers was investigated by focusing on the film surface region and cross-section part. The scanning electron microscopic (SEM) images (see Fig. 3a and Fig. S6, ESI†) disclosed that the nanoscale crater-shaped morphology was made on the surface of the ZnO:PEOz hybrid layers with the PEOz content of 5 wt%, even though no particular morphology was observed from the pristine ZnO layer as well as the hybrid layer with a very small PEOz content (1 wt%). Interestingly, as the PEOz content increased from 5 wt% up to 20 wt%, the nano-craters (100–200 nm in diameter at 5 wt%) became larger and their edges were pronouncedly popped up to the front (200–800 nm at 10–20 wt%). These nano-craters with the popped-up edges at the high PEOz content (20 wt%) might increase the interfacial contact resistance with the BHJ layers, as supported by the high RS (see Fig. 2c).
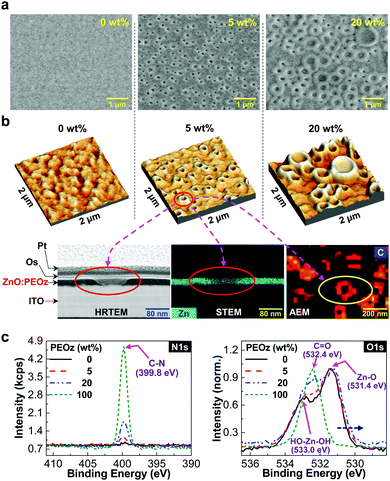 |
| | Fig. 3 Morphology of ZnO:PEOz hybrid films and XPS analysis. (a) SEM images for the ZnO:PEOz hybrid films according to the PEOz content. (b) 3D height-mode AFM images for the ZnO:PEOz hybrid films according to the PEOz content: the nano-craters were further analyzed with HRTEM (imaging for the inner shape of nano-craters from the cross-section part of the hybrid film coated on the ITO-glass substrate), STEM (zinc atom mapping image for the same cross-section part for the HRTEM measurement), and AEM (carbon atom mapping image focusing on the nano-craters in the surface region of the hybrid film). Note that platinum (Pt) and osmium (Os) were coated on the ZnO:PEOz hybrid films for finding the exact location of nano-craters by using SEM before the HRTEM measurement. (c) XPS spectra for the ZnO:PEOz hybrid films coated on the ITO-glass substrates: (left) N1s, (right) O1s. | |
The nano-craters were also measured from the atomic force microscopic (AFM) images, as shown in Fig. 3b (top) and Fig. S7 (ESI†), clarifying that the nano-craters have a volcano-type three-dimensional shape. In particular, the AFM images disclosed that the nano-craters were much rougher and larger at 20 wt% than 5 wt%, which is in good agreement with the SEM images. Note that the surface morphology of the BHJ (PBDB-T:ITIC) layers was also changed owing to the presence of the nano-craters (see Fig. S8, ESI†). As measured with a high-resolution transmission electron microscope (HRTEM) and a scanning transmission electron microscope (STEM) (see the left and middle images in Fig. 3b (bottom) and Fig. S9, ESI†), the cross-section part of the ZnO:PEOz hybrid layer (5 wt%), which was prepared by a focused ion beam (FIB) technique, showed that the inner part of the nano-craters was carved in a round shape with the thin ZnO bottom layer without pinholes as supported by the still high RSH values (see Table 1). The auger electron microscope (AEM) image disclosed that the PEOz component existed at the edge part of the nano-craters (see the right bottom image in Fig. 3b). Considering the detection depth (ca. 5.5 nm from the surface) in the AEM image, the PEOz component might be enriched close to the surface region of the crater edge parts through phase segregation and aggregation processes (see Fig. S10, ESI†).
In order to further confirm the existence and role of the PEOz component, the X-ray photoelectron spectroscopy (XPS) measurement was performed on the ZnO:PEOz hybrid layers. As shown in Fig. 3c (left), the nitrogen peak (N1s) at 399.8 eV was measured for the hybrid layers, which was proportionally increased with the PEOz content. In addition, the oxygen (O1s) peak at around 532.4 eV, which corresponds to oxygen in carbonyl groups in the PEOz polymer, was also gradually increased with the PEOz content (see Fig. 3c right). These XPS results indicate that the PEOz component exists in the hybrid layers. Interestingly, the intensity of the O1s peaks in Fig. 3c (right) was increased by 17.7% and 43.8% for the hybrid layers with the PEOz content of 5 wt% and 20 wt%, respectively, which can confirm the enrichment of the PEOz component toward the surface region of the nano-craters (note that the penetration depth of XPS is ca. 5 nm). Based on the measurement results, the shape of the nano-craters can be illustrated as shown in Fig. S11 (ESI†). Considering the shape of the nano-craters together with the AFM and SEM images, the surface area based on the AFM image with a size of 2 μm × 2 μm can be calculated to be 4.0 × 10−8 cm2 and 5.062 × 10−8 cm2 for 0 wt% and 5 wt% PEOz, respectively. This result indicates that the formation of nano-craters (5 wt% PEOz) can increase the surface area of the ZnO layers by ca. 27%, which could lead to ca. 27%-increased interfacial contact area with the BHJ layers. As a consequence, the capacity of electron collection from the BHJ layers could be as high as ca. 27%, which might be another reason for the PCE improvement in the PBDB-T:ITIC solar cells (5 wt% PEOz).
Stability of solar cells with the ZnO:PEOz hybrid layers
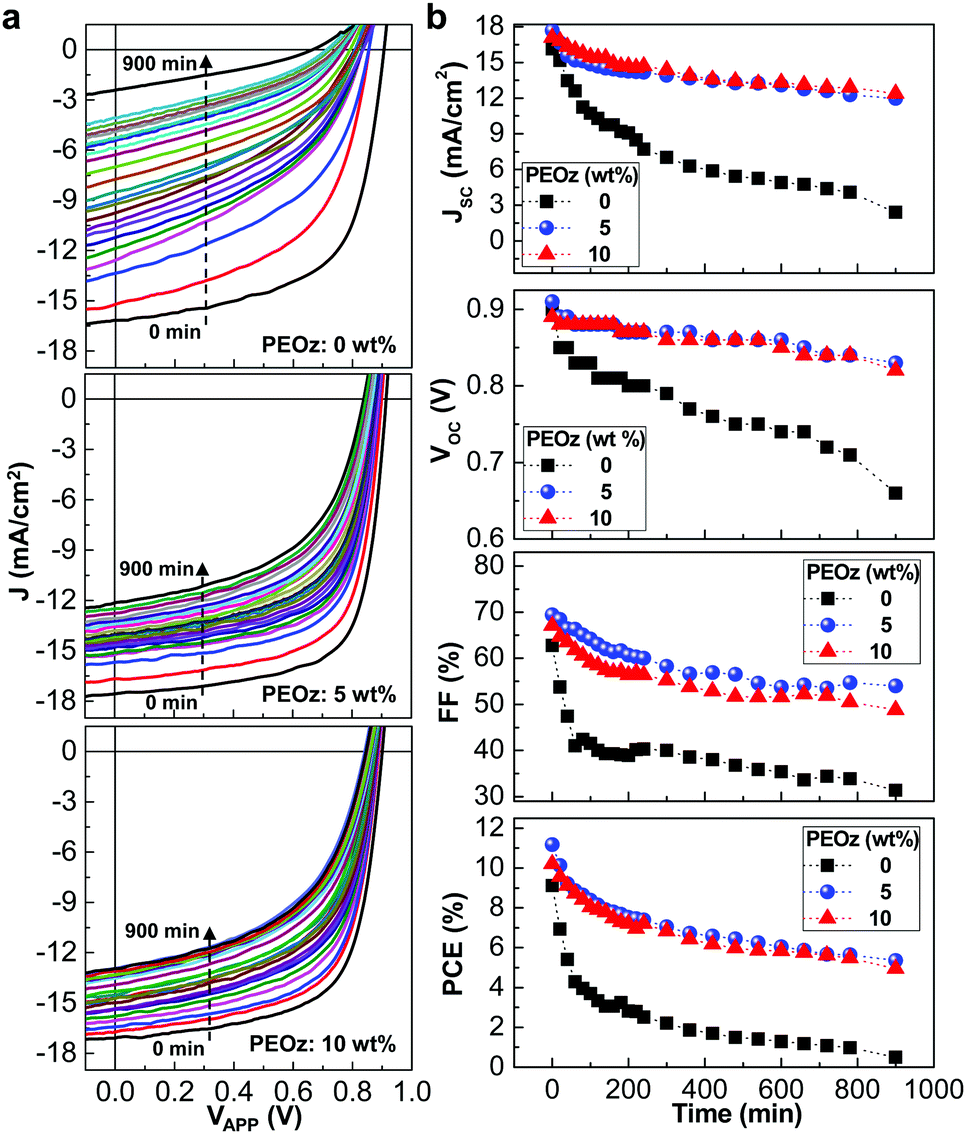
The lifetime of the devices under continuous illumination with simulated solar light (100 mW cm−2) was briefly studied in order to understand the influence of nano-craters on the stability of the present inverted-type PBDB-T:ITIC solar cells with the ZnO:PEOz hybrid layers. As shown in Fig. 4a, the J–V curve was gradually degraded with time for the solar cells with the pristine ZnO layers and became significantly poor after 900 min. In contrast, the solar cells with the ZnO:PEOz hybrid layers exhibited relatively slow degradation in the J–V curve so that the JSC values could be still higher than 12 mA cm−2 even after 900 min. The detailed trend with time disclosed that all solar cell parameters were relatively better for the solar cells with the ZnO:PEOz hybrid layers than those with the pristine ZnO layers (see Fig. 4b). Interestingly, although the JSC decay was slightly slower for the solar cells with the hybrid layers at 10 wt% than 5 wt%, both VOC and FF were relatively well kept at 5 wt%. As a consequence, the slowest PCE decay was achieved for the solar cells with the hybrid layers at the PEOz content of 5 wt%, even though the PCE decay became certainly slower even at 10 wt%. This brief lifetime test supports that the stability of the inverted-type PBDB-T:ITIC solar cells can be remarkably improved by forming the nano-crater morphology in the ZnO electron-collecting buffer layers. One reason behind the improved stability can be attributed to the enhanced adhesion between the BHJ (PBDB-T:ITIC) and ZnO:PEOz hybrid layers due to the formation of mechanical interlocking nanostructures by the presence of the nano-craters (see Fig. S12, ESI†).55,56 The improved adhesion is supported by the dipping test result as shown in Fig. S13 and S14 (ESI†), because the BHJ film dipped in chlorobenzene was almost completely dissolved in the case of the pristine ZnO layer but the dipped part of the BHJ film was considerably maintained in the case of the ZnO:PEOz hybrid layer (5 wt% PEOz).57,58 So the enhanced adhesion might make the contact part of the BHJ layers be relatively stabilized against degradation upon solar light illumination, leading to improved stability (lifetime). Here it is noted that the overall stability of devices might be affected by the photo-degradation of the nonfullerene acceptor (ITIC) because the optical absorption was noticeably decreased after 2 h illumination with solar light but its change was relatively small for the PBDB-T films as reported in our previous work.59
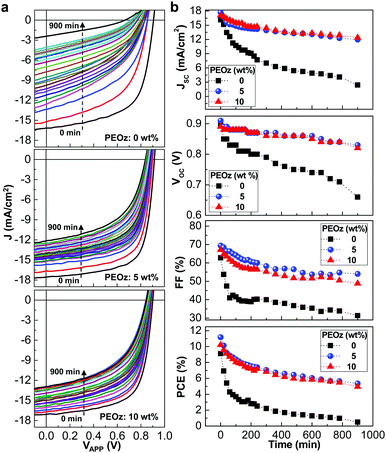 |
| | Fig. 4 Stability of solar cells. (a) Change of light J–V curves for the inverted-type PBDB-T:ITIC solar cells with the ZnO:PEOz hybrid layers (annealed at 150 °C) under continuous illumination with the simulated solar light (air mass 1.5G, 100 mW cm−2) for 900 min. (b) JSC, VOC, FF and PCE as a function of exposure time (data were extracted from the J–V curves in a). | |
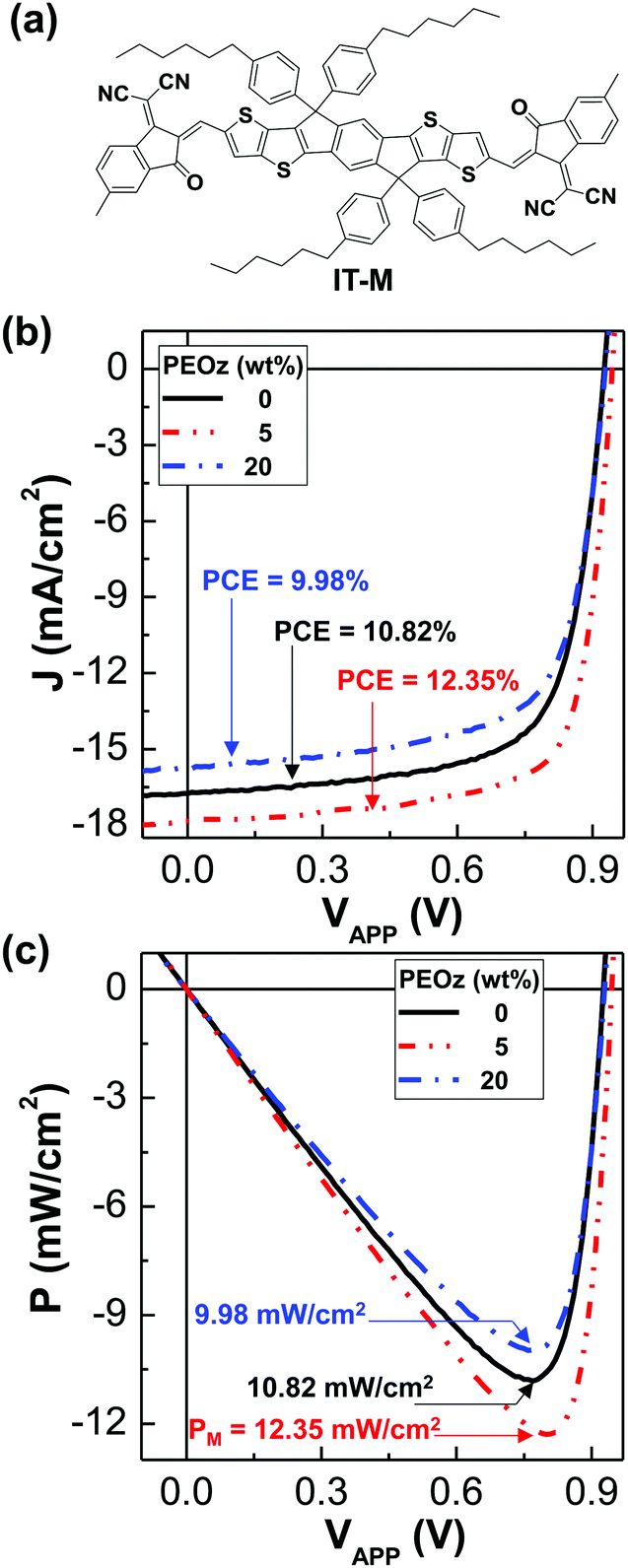
12.35% solar cells with the ZnO:PEOz hybrid layers
Finally, the ZnO:PEOz hybrid layers were applied for another polymer:nonfullerene solar cell with the PBDB-T:IT-M BHJ layers in order to confirm their effects (see Fig. 5). As shown in Fig. 5b, the light J–V curve of the PBDB-T:IT-M solar cells was significantly increased by the presence of the ZnO:PEOz hybrid layers at the PEOz content of 5 wt% which were thermally annealed at 150 °C. The PCE was pronouncedly improved from 10.82% to 12.35%, while the FF increased from 70.2% to 74.2% (see Table 2). However, the PCE was reduced to 9.98% at the PEOz content of 20 wt%, which is in agreement with the trend of the PBDB-T:ITIC solar cells. As a consequence, as shown in Fig. 5c, the maximum output power density was increased by ca. 12% (from 10.82 mW cm−2 to 12.43 mW cm−2) for the ZnO:PEOz hybrid layers at the content of 5 wt% PEOz but it decreased by ca. 10% (from 10.82 mW cm−2 to 9.98 mW cm−2).
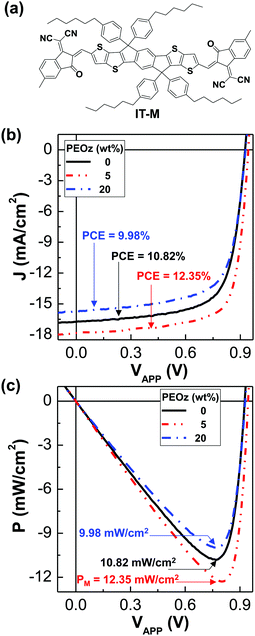 |
| | Fig. 5 PBDB-T:IT-M solar cells. (a) Chemical strcuture of IT-M. (b) Light J–V curves for the PBDB-T:IT-M solar cells with the ZnO:PEOz hybrid layers (annealed at 150 °C) under illumination with the simulated solar light (air mass 1.5G, 100 mW cm−2). (c) Power density–voltage curves from data in b: the maximum power density (PM) is given on each curve. | |
Table 2 Summary of solar cell parameters for the inverted-type PBDB-T:IT-M solar cells with the ZnO:PEOz hybrid layers (annealed at 150 °C) under illumination with simulated solar light (air mass 1.5G, 100 mW cm−2)
| Parameters |
PEOz (wt%) |
| 0 |
5 |
20 |
|
V
OC (V) |
0.92 |
0.94 |
0.92 |
|
J
SC (mA cm−2) |
16.8 |
17.8 |
15.8 |
| FF (%) |
70 |
74 |
69 |
| PCE (%) |
10.8 |
12.4 |
8.0 |
|
R
S (kΩ cm2) |
0.09 |
0.07 |
0.10 |
|
R
SH (kΩ cm2) |
8.50 |
13.40 |
1.50 |
Conclusions
In summary, sol–gel derived ZnO:PEOz hybrid films with a nano-crater morphology were prepared by thermal annealing at a low temperature (150 °C), compared to the conventional high temperature process (200 °C), from corresponding precursor solutions and employed as an electron-collecting buffer layer for inverted-type PBDB-T:ITIC solar cells. The ZnO:PEOz hybrid layers delivered the enhanced PCEs for all solar cells irrespective of PEOz contents, while the highest PCE of 11.46% could be achieved at the PEOz content of 5 wt% in the case of the inverted-type PBDB-T:ITIC solar cells. However, JSC became rather lower at 20 wt% than 0 wt%, even though the electron mobility was still much higher at 20 wt% than 0 wt%. The discrepancy between the higher electron mobility and lower JSC at 20 wt% has been assigned to the poor interfaces with the BHJ layers owing to the relatively much larger and rougher nano-craters at 20 wt% (diameter = ∼800 nm), compared to the smaller and finer nano-craters at 5 wt% (diameter = 100–200 nm) which eventually contributed to the enhanced charge collection due to the increased contact area of the well-made interfaces with the BHJ layers. The brief lifetime test revealed that the stability of the solar cells could be greatly improved by the presence of the present hybrid layers, which has been assigned to the enhanced adhesion between the BHJ and hybrid layers by the formation of mechanically interlocking nanostructures caused by the nano-craters. Finally, the ZnO:PEOz layers (5 wt% PEOz) delivered 12.35% PCE for the inverted PBDB-T:IT-M solar cells. The enhanced efficiency has been assigned to the increased electron transport in the ZnO:PEOz layers which benefits the promoted interfacial contact area with the BHJ layers by the formation of nano-craters. Therefore it is expected that the present ZnO:PEOz hybrid technology can greatly contibute to further advances in organic solar cells when it comes to their one-step coating and low-temperature processes together with a particular nanostructure formation.
Contributions
Y. K. designed and directed overall experiments. J. S. carried out all experiments including device fabrication and measurement. S. N. established the basic experimental conditions for PEOz-based inverted organic solar cell fabrication. D. D. C. B. supervised S. N. during his time in Oxford. H. K. analyzed data including XPS, AEM, TEM, etc. J. S. and Y. K. wrote the first version of the manuscript which was subsequently commented on and edited by S. N., H. K. and D. D. C. B.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Research Foundation of Korea (NRF-Korea) (2016H1D5A1910319, the Basic Science Research Program_2018R1A6A1A03024962, 2018R1D1A3B07046214, 2017M2A2A4A01071010, 2018R1D1A1B07051075). D. D. C. B. thanks the University of Oxford for funding a postdoctoral fellowship for S. N. and for infrastructure support.
References
- H. Cha, S. Wheeler, S. Holliday, S. D. Dimitrov, A. Wadsworth, H. H. Lee, D. Baran, I. McCulloch and J. R. Durrant, Adv. Funct. Mater., 2018, 1704389 CrossRef.
- F.-J. Kahle, C. Saller, S. Olthof, C. Li, J. Lebert, S. Weiß, E. M. Herzig, S. Hüttner, K. Meerholz, P. Strohriegl and A. Köhler, J. Phys. Chem. C, 2018, 122, 21792–21802 CrossRef CAS.
- H. Kim, S. Nam, J. Jeong, S. Lee, J. Seo, H. Han and Y. Kim, Korean J. Chem. Eng., 2014, 31, 1095–1104 CrossRef CAS.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhan, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- W. Zhao, S. Li, S. Zhang, X. Liu and J. Hou, Adv. Mater., 2017, 29, 1604059 CrossRef PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS PubMed.
- K. Zilberberg, S. Trost, H. Schmidt and T. Riedl, Adv. Energy Mater., 2011, 1, 377–381 CrossRef CAS.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. D. Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- M. D. Irwin, D. B. Buchholz, A. W. Hains, R. P. H. Chang and T. J. Marks, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2783–2787 CrossRef CAS.
- V.-H. Tran, R. B. Ambade, S. B. Ambade, S.-H. Lee and I.-H. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 1645–1653 CrossRef CAS.
- J. Jeong, J. Seo, S. Nam, H. Han, H. Kim, T. D. Anthopoulos, D. D. C. Bradley and Y. Kim, Adv. Sci., 2015, 3, 1500269 CrossRef PubMed.
- S. Nam, J. Seo, S. Woo, W. H. Kim, H. Kim, D. D. C. Bradley and Y. Kim, Nat. Commun., 2015, 6, 8929 CrossRef CAS PubMed.
- S. Murase and Y. Yang, Adv. Mater., 2012, 24, 2459–2462 CrossRef CAS.
- J. Liu, S. Shao, G. Fang, B. Meng, Z. Xie and L. Wang, Adv. Mater., 2012, 24, 2774–2779 CrossRef CAS PubMed.
- G. H. Jung, K. Hong, W. J. Dong, S. Kim and J.-L. Lee, Adv. Energy Mater., 2011, 1, 1023–1028 CrossRef CAS.
- B. J. Norris, J. Anderson, J. F. Wager and D. A. Keszler, J. Phys. D: Appl. Phys., 2003, 36, L105–L107 CrossRef CAS.
- P. Jood, R. J. Mehta, Y. Zhang, G. Peleckis, X. Wang, R. W. Siegel, T. Borca-Tasciuc, S. X. Dou and G. Ramanath, Nano Lett., 2011, 11, 4337–4342 CrossRef CAS PubMed.
- B. Sun and H. Sirringhaus, Nano Lett., 2005, 5, 2408–2413 CrossRef CAS PubMed.
- S. Nam, S. G. Hahm, H. Han, J. Seo, C. Kim, H. Kim, S. R. Marder, M. Ree and Y. Kim, ACS Sustainable Chem. Eng., 2016, 4, 767–774 CrossRef CAS.
- Y. Sun, J. H. Seo, C. J. Takacs, J. Seifter and A. J. Heeger, Adv. Mater., 2011, 23, 1679–1683 CrossRef CAS PubMed.
- P. de Bruyn, D. J. D. Moet and P. W. M. Blom, Org. Electron., 2010, 11, 1419–1422 CrossRef CAS.
- J. Czolk, A. Puetz, D. Kutsarov, M. Reinhard, U. Lemmer and A. Colsmann, Adv. Energy Mater., 2013, 3, 386–390 CrossRef CAS.
- P. Docampo, J. M. Ball, M. Darwich, G. E. Eperon and H. J. Snaith, Nat. Commun., 2013, 4, 2761 CrossRef PubMed.
- Y. Zhou, C. Fuentes-Hernandez, T. M. Khan, J.-C. Liu, J. Hsu, J. W. Shim, A. Dindar, J. P. Youngblood, R. J. Moon and B. Kippelen, Sci. Rep., 2013, 3, 1536 CrossRef PubMed.
- L. K. Jagadamma, M. Abdelsamie, A. E. Labban, E. Aresu, G. O. Ndjawa, D. H. Anjum, D. Cha, P. M. Beaujuge and A. Amassian, J. Mater. Chem. A, 2014, 2, 13321–13331 RSC.
- Y.-J. Lee, J. Wang, S. R. Cheng and J. W. P. Hsu, ACS Appl. Mater. Interfaces, 2013, 5, 9128–9133 CrossRef CAS PubMed.
- J. Liu, J. Wu, S. Shao, Y. Deng, B. Meng, Z. Xie, Y. Geng, L. Wang and F. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 8237–8245 CrossRef CAS PubMed.
- H.-C. Chen, S.-W. Lin, J.-M. Jiang, Y.-W. Su and K.-H. Wei, ACS Appl. Mater. Interfaces, 2015, 7, 6273–6281 CrossRef CAS PubMed.
- N. Wu, Q. Luo, Z. Bao, J. Lin, Y.-Q. Li and C.-Q. Ma, Sol. Energy Mater. Sol. Cells, 2015, 141, 248–259 CrossRef CAS.
- X. Jia, N. Wu, J. Wei, L. Zhang, Q. Luo, Z. Bao, Y.-Q. Li, Y. Yang, X. Liu and C.-Q. Ma, Org. Electron., 2016, 38, 150–157 CrossRef CAS.
- H. Oh, J. Krantz, I. Litzov, T. Stubhan, L. Pinna and C. J. Brabec, Sol. Energy Mater. Sol. Cells, 2011, 95, 2194–2199 CrossRef CAS.
- T. Stubhan, I. Litzov, N. Li, M. Salinas, M. Steidl, G. Sauer, K. Forberich, G. J. Matt, M. Halik and C. J. Brabec, J. Mater. Chem. A, 2013, 1, 6004–6009 RSC.
- M. A. Ibrahem, H.-Y. Wei, M.-H. Tsai, K.-C. Ho, J.-J. Shyue and C. W. Chu, Sol. Energy Mater. Sol. Cells, 2013, 108, 156–163 CrossRef CAS.
- W. J. E. Beek, L. H. Slooff, M. M. Wienk, J. M. Kroon and R. A. J. Janssen, Adv. Funct. Mater., 2005, 15, 1703–1707 CrossRef CAS.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- L. Ye, W. Zhao, S. Li, S. Mukherjee, J. H. Carpenter, O. Awartani, X. Jiao, J. Hou and H. Ade, Adv. Energy Mater., 2017, 7, 1602000 CrossRef.
- H. Bin, Z.-G. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657–4664 CrossRef CAS PubMed.
- R. Wahab, S. G. Ansari, Y. S. Kim, M. Song and H.-S. Shin, Appl. Surf. Sci., 2009, 255, 4891–4896 CrossRef CAS.
- G. Iannaccone, A. Bernardi, R. Suriano, C. L. Bianchi, M. Levi, S. Turri and G. Griffini, RSC Adv., 2016, 6, 46915–46924 RSC.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S.-W. Tsang, T.-H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115–120 CrossRef CAS.
- T. Hu, L. Chen, K. Yuan and Y. Chen, Chem. – Eur. J., 2014, 20, 17178–17184 CrossRef CAS PubMed.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS PubMed.
- T. Hu, L. Chen, K. Yuan and Y. Chen, Nanoscale, 2015, 7, 9194–9203 RSC.
- K. Yuan, L. Chen, F. Li and Y. Chen, J. Mater. Chem. C, 2014, 2, 1018–1027 RSC.
- S. Nam, J. Seo, H. Han, H. Kim, S. G. Hahm, M. Ree, Y.-S. Gal, T. D. Anthopoulos, D. D. C. Bradley and Y. Kim, Adv. Mater. Interfaces, 2016, 3, 1600415 CrossRef.
- S. Woo, W. H. Kim, H. Kim, Y. Yi, H.-K. Lyu and Y. Kim, Adv. Energy Mater., 2014, 4, 1301692 CrossRef.
- X. Guan, K. Zhang, F. Huang, G. C. Bazan and Y. Cao, Adv. Funct. Mater., 2012, 22, 2846–2854 CrossRef CAS.
- J. Kesters, T. Ghoos, H. Penxten, J. Drijkoningen, T. Vangerven, D. M. Lyons, B. Verreet, T. Aernouts, L. Lutsen, D. Vanderzande, J. Manca and W. Maes, Adv. Energy Mater., 2013, 3, 1180–1185 CrossRef CAS.
- H.-L. Yip, S. K. Hau, N. S. Baek, H. Ma and A. K.-Y. Jen, Adv. Mater., 2008, 20, 2376–2382 CrossRef CAS.
- A. W. Hains, C. Ramanan, M. D. Irwin, J. Liu, M. R. Wasielewski and T. J. Marks, ACS Appl. Mater. Interfaces, 2010, 2, 175–185 CrossRef CAS.
- W. Tress, K. Leo and M. Riede, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155201 CrossRef.
- A. S. Zoolfakar, R. A. Rani, A. J. Morfa, S. Balendhran, A. P. O’Mullane, S. Zhuiykov and K. Kalantar-zadeh, J. Mater. Chem., 2012, 22, 21767–21775 RSC.
- M.-H. Ham, G. L. C. Paulus, C. Y. Lee, C. Song, K. Kalantar-zadeh, W. Choi, J.-H. Han and M. S. Strano, ACS Nano, 2010, 4, 6251–6259 CrossRef CAS PubMed.
- S. Y. Yang, E. D. O’Cearbhaill, G. C. Sisk, K. M. Park, W. K. Cho, M. Villiger, B. E. Bouma, B. Pomahac and J. M. Karp, Nat. Commun., 2013, 4, 1702 CrossRef PubMed.
- W.-S. Kim, I.-H. Yun, J.-J. Lee and H.-T. Jung, Int. J. Adhes. Adhes., 2010, 30, 408–417 CrossRef CAS.
- M. C. Paiva, I. Ammar, A. R. Campos, R. B. Cheikh and A. M. Cunha, Compos. Sci. Technol., 2007, 67, 1132–1138 CrossRef CAS.
- F. Biscarini, M. Cavallini, D. A. Leigh, S. León, S. J. Teat, J. K. Y. Wong and F. Zerbetto, J. Am. Chem. Soc., 2002, 124, 225–233 CrossRef CAS PubMed.
- E. Park, J. Seo, H. Han, H. Kim and Y. Kim, Adv. Sci., 2018, 1800331 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00319j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b and
Youngkyoo
Kim
b and
Youngkyoo
Kim