
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmino acid-based ionic liquids as precursors for the synthesis of chiral nanoporous carbons†
Sapir Shekef
Aloni
a,
Milena
Perovic
b,
Michal
Weitman
a,
Reut
Cohen
a,
Martin
Oschatz
 bc and
Yitzhak
Mastai
*a
bc and
Yitzhak
Mastai
*a
aDepartment of Chemistry, The Institute of Nanotechnology, Bar-Ilan University, Ramat-Gan 5290002, Israel. E-mail: mastai@biu.ac.il
bMax Planck Institute of Colloids and Interfaces, Potsdam-Golm Science Park, Am Mühlenberg 1 OT Golm, Potsdam 14476, Germany
cInstitute of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, D-14476 Potsdam, Germany
First published on 16th November 2019
Abstract
The synthesis of chiral nanoporous carbons based on chiral ionic liquids (CILs) of amino acids as precursors is described. Such unique precursors for the carbonization of CILs yield chiral carbonaceous materials with high surface area (≈620 m2 g−1). The enantioselectivities of the porous carbons are examined by advanced techniques such as selective adsorption of enantiomers using cyclic voltammetry, isothermal titration calorimetry, and mass spectrometry. These techniques demonstrate the chiral nature and high enantioselectivity of the chiral carbon materials. Overall, we believe that the novel approach presented here can contribute significantly to the development of new chiral carbon materials that will find important applications in chiral chemistry, such as in chiral catalysis and separation and in chiral sensors. From a scientific point of view, the approach and results reported here can significantly deepen our understanding of chirality at the nanoscale and of the structure and nature of chiral nonporous materials and surfaces.
Introduction
Chirality is a key factor in almost all biological processes that involve molecular recognition and has great importance in numerous chemical and biological applications.1,2 In recent years, due to tremendous developments in nanotechnology, chirality at the nanoscale has become of huge importance. Many studies were conducted in the field of chiral nanotechnology with the aim of using chiral nanosystems in various applications, mainly asymmetric synthesis, chiral analysis, and chiral separations. Over the last years, several insightful reviews were published on chiral nanotechnology and the chirality of nanostructures.3–8 In general, chirality plays an important role in the properties of nanomaterials, including chiral supramolecules, chiral nanoparticles, and DNA nanotechnology.Chiral nanoparticles,9–14 chiral surfaces,15–20 and chiral mesoporous materials,20–28 are attracting great attention owing to their significant advantages such as high surface area and diverse chiral functionalities that confer many advantages for catalysis, bio-recognition, chiral separation processes, and other applications. Over the years, various synthetic methods were developed for preparing chiral mesoporous materials with a strong focus on chiral mesoporous silica.21,24,26,29–43 One approach for the preparation of chiral mesoporous materials is based on molecular imprinting which is very similar to chiral imprinting of polymers.44 In other studies, various types of chiral templates led to the formation of chiral silica. For example, Álvaro et al.45 proved that combining binaphthyl and cyclohexadiyl organic template groups with a tetraethyl orthosilicate precursor induces chiral porous silica formation. The group of Avnir contributed greatly to this study of chiral mesoporous silica. In several papers, it was shown that various chiral template molecules, such as propranolol, 2,2,2-trifluoro-1-(9-anthryl) ethanol, or tyrosine, can be used for the synthesis of chiral mesoporous silica using sol–gel matrix production.32,33,35,36 Our group also reported on the use of various chiral block copolymers as templates for the formation of chiral silica.12,30,43,46 Furthermore, Oda et al.41,47,48 investigated the self-assembly organization of chiral amphiphilic molecules by controlling several parameters and studied the mechanism of formation of chiral mesoscopic molecular assemblies. Finally, chiral anionic surfactant-templated mesoporous silica materials yielded particularly chiral well-ordered structures, and in some cases even generated complex helical rod-like structures.26,37,49 Another important class of porous materials where chirality can be easily incorporated are metal–organic frameworks (MOFs).50 The mild conditions for the synthesis of these materials allow direct incorporation of various functionalities directly into the framework.
Although nanoporous substrates have proven to be very useful in the area of chiral nanomaterials, most of those nanoporous materials are still based on silica. However, carbon-based porous materials have advantages over silica such as higher thermal and chemical stability, electric conductivity, tuneable porosity, diverse ways of syntheses, as well as a wide range of possibilities for the choice of precursors. On the other side, the significant disadvantage of porous carbon materials over silica is their less defined surface chemistry and surface energy which makes the controlled functionalization of their surface with chiral functional groups rather difficult.
Recently we have introduced a new type of chiral mesoporous carbon materials based on chiral ionic liquids (CILs). CILs, mostly based on amino acids, were used as precursors and after a carbonization process, enantioselective nanoporous carbon has been formed. In recent years, the field of CILs51–55 experienced rapid growth and new synthetic methods were established with many new applications, such as asymmetric organic synthesis, chromatography, and chiral separation but their conversion to high-surface-area chiral porous carbons remained poorly investigated.
In this article, we extend our research on enantioselective microporous carbon in view of our previous article. In this article, we describe the synthesis of a variety of CILs based on amino acids and the synthesis chiral nanoporous carbonaceous materials that were not reported in our previous article. Moreover, we apply several new techniques to characterize the chirality of chiral nanoporous carbonaceous materials such as isothermal titration calorimetry (ITC) and cyclic voltammetry (CV). Furthermore in our previous article the mechanism for the formation of chirality and the origin of enantioselective nature of the nanoporous carbonaceous materials was unclear therefrom in the article we focused on exploration of the mechanism of chirality in our nanoporous carbonaceous materials using technique of thermogravimetric analysis combined with gas chromatography and mass spectrometry (TGA-GC-MS) to investigate and confirm the chiral nature of the nanoporous carbons.
Methods
Chemicals
The following analytical-grade chemicals were purchased from Aldrich-Sigma: L-tyrosine (>99%), D-tyrosine (>98%), iodomethane (>99.8%), potassium hydrogen carbonate, chloroform, acetone, L-tartaric acid (>98%), D-tartaric acid (>98%) and sodium tetrafluoroborate.Synthesis of CILs
In Scheme 1 the overall route for the synthesis of chiral ionic liquids is shown. CILs were synthesized based on enantiomers of amino acids. L- or D-tyrosine methyl ester hydrochloride (5.00 g, 21.6 mmol) was placed in a 250 mL round bottom flask, 120 mL acetone was added, and the mixture was stirred. Iodomethane (18.44 g, 129.9 mmol, 6 eq.) was added, followed by the addition of potassium hydrogen carbonate (10.80 g, 108.0 mmol, 5 eq.), and the suspension was vigorously stirred. After 16–22 h, the solvent was evaporated. Chloroform (30 mL) was added to the residue and the mixture was stirred for 30 min. The impurities in the solvent were removed by suction filtration and 33 mL of water were added to the solid. The mixture was stirred and heated to 90 °C, giving a clear solution. It was then cooled to room temperature, and further with an ice bath, leading to the formation of a solid. The solid was filtered, washed with cold water and dried to obtain N,N,N-trimethyl-L-tyrosine methyl ester iodide. Ion exchange was performed by the following method. A saturated solution of sodium tetrafluoroborate in water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 g g−1) was prepared, the methyl ester iodide was added, the mixture was heated while stirring at 50–55 °C for 1–3 h, and then cooled to room temperature. The solid was filtered, washed with cold water, and dried to give a white solid. The purity of the as-synthesized Tyr-based CILs, which we term L- and D-CIL(Tyr), was verified by 1H and 13C NMR spectroscopy and mass spectrometry (see the ESI†).
:1 g g−1) was prepared, the methyl ester iodide was added, the mixture was heated while stirring at 50–55 °C for 1–3 h, and then cooled to room temperature. The solid was filtered, washed with cold water, and dried to give a white solid. The purity of the as-synthesized Tyr-based CILs, which we term L- and D-CIL(Tyr), was verified by 1H and 13C NMR spectroscopy and mass spectrometry (see the ESI†).
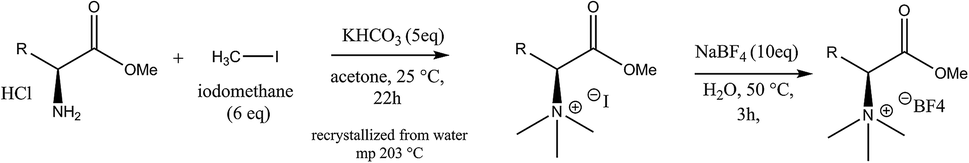 | ||
| Scheme 1 The overall schematic route for the synthesis of chiral ionic liquids (CILs). | ||
Carbonization process
Each CIL, based on a specific amino acid, was mixed and carbonized with an equal part of a eutectic mixture of NaCl and ZnCl2 salts (1:3 molar ratio). In a closed chamber furnace, 1 g of the mixture was placed in an oven under N2 flow at 25 °C for 1 h, and the furnace was heated to 450 °C with a rate of 2.5 °C min−1, maintained for 1 h, followed by gradual cooling to room temperature. The solid carbonization products were washed extensively with water to extract remaining salts, resulting in carbonized CIL material, termed CIL-C.
Characterization
The CIL morphology was studied using a FEI Quanta FEG 250 scanning electron microscope (SEM) and a JEOL JEM-1400 (LaB6, 120 kV) transmission electron microscope (TEM). TEM samples were prepared by dipping a drop of aqueous CIL dispersion on a 400-mesh carbon-coated copper grid. The CILs were tested for the presence of inorganic salt residue by X-ray diffraction (XRD) using a Bruker AXS D8 Advance diffractometer equipped with Cu Kα (λ = 1.5418 Å) applying 2θ step size of 0.02° and counting time of 1 s per step. Low-pressure physisorption measurements were performed on a Quantachrome Autosorb IQ apparatus using Ar at 87 K as the adsorptive (sample weight ∼20 mg). Prior to a physisorption measurement, the sample was degassed under vacuum at 150 °C for 20 h. The pore size distributions were calculated using quenched solid density functional theory (QSDFT) method (adsorption branch kernel) for Ar adsorbed on carbon with cylindrical/sphere pore shape at 87 K, integrated into the ASiQwin 3.0 analysis software (Quantachrome). The specific surface area (SSA) of the material was calculated using the multipoint Brunauer–Emmett–Teller (BET) model in the relative pressure range of 0.006–0.078. The total pore volume was determined at a relative pressure of 0.99. Micropore volume has been calculated from the cumulative pore volume at a diameter of 2 nm. The Raman spectrum was recorded using a Witec (focus innovations) Raman Microscope operating with an objective (Nikon, 10×/0.25, ∞/−WD 6.1) and an excitation wavelength of 532 nm with an intensity of 3.5 mW. Thermogravimetric analysis (TGA) was performed using a thermo microbalance TG 209 F1 Libra (Netzsch, Selb, Germany). A platinum crucible was used for the measurement of 10 mg of a sample in synthetic air flow of 10 mL min−1. The sample was heated to 1000 °C with a heating rate of 10 K min−1. The data was recorded and analyzed by the Proteus (6.0.0) software package. Absorbance measurements were performed with Chirascan circular dichroism (CD) spectrometer (Applied Photophysics, UK) using a bandwidth of 3 nm, from 320 to 200 nm, with step size and duration of 1 nm and 3 s, respectively. Isothermal titration calorimetry (ITC) measurements were performed with a VP-ITC calorimeter (Northampton, MA). The volume of the reservoir cell was 1.442 mL, and it was thermostated at 30 °C. Before measurements, solutions were degassed under vacuum for 5 min to eliminate air bubbles. The sample cell was loaded with 1.442 mL of a 0.2 wt% suspension of chiral carbon nanoparticles in water, and the 300 μL syringe was filled with 200 mM of D- or L-tartaric acid (TA) solution. A titration run consisted of consecutive injections of 5 μL, each with a duration of 8.5 s, separated by 300 s intervals. The cell was constantly stirred at a 300 rpm. To eliminate the nonspecific heat effects and the heat of the dilution of the chiral solutions, we carried out control experiments in which L- and D-TA stock solutions (5 μL) were injected into double distilled water at 30 °C. Thermogravimetric analysis coupled with gas chromatography and mass spectrometry (TGA-GC/MS) experiments were performed with PerkinElmer ‘Pyris 1’ TGA, ‘Clarus 680’ GC and ‘Clarus SQ 8 C’ MS instrument. 2.09 mg of L-CIL(Tyr)-C and 4.03 mg of L-CIL(Tyr) were subjected to TGA oven, with a heating rate of 20 K min−1from 25 to 700 °C under a nitrogen atmosphere (balance purge 80 mL min−1; sample purge 20 mL min−1) on alumina crucibles. TGA was connected to a transfer-line (TL9000) of ‘Red Shift’ heated to 280 °C, which is connected to a 100 μL loop. The gas mixture was then separated over 5 MS (30 m × 0.25 mm) column using helium gas carrier at 1.0 mL min−1 flow. The column temperature was raised from 40 °C to 300 °C at 20 K min−1 and then held at 300 °C for 5 min. The mass spectrometer conditions were as follows: electronic impact ionization at 70 eV, source temperature 220 °C and GC transfer line 250 °C. Mass range 35–300 Da with 0.2 s dwell time. The data were collected and analyzed by GCMS PerkinElmer TurboMass software.Results
HR-SEM and HR-TEM images of carbons L- and D-CIL(Tyr)-C are shown in Fig. 1. As can be seen in the HR-SEM images, the CIL(Tyr)-C particles present a spherical shape with an average size of 1 μm in diameter however the carbons show broad particle size distributions (Fig. 1a) and a porous structure and high surface roughness (Fig. 1b). The nanoscale structure of the porous carbon was characterized using HR-TEM. Typical micrographs of the porous carbon are shown in Fig. 1c. The micrographs clearly show that the structure consists of spherical pores of ca. 2 nm in diameter, arranged in a random fashion. | ||
| Fig. 1 Electron microscopy images of CIL(Tyr)-C obtained by HR-SEM (a and b), and HR-TEM (c). | ||
The XRD pattern of L-CIL(Tyr)-C shows an amorphous carbon structure that is typical for templated carbon materials with narrow pores, together with several distinct peaks that can be ascribed to the inorganic salts used for the synthesis of carbonaceous material (Fig. S1–S3†). This is in agreement with the results of thermogravimetric analysis (TGA), performed under the flow of synthetic air (Fig. S4†). The residual mass of approximately 24% at 1000 °C confirms the presence of remaining inorganic salts and due to organic residues that are originated from the chiral ionic liquids (see below at description of TGA-GC/MS results). The minor weight loss at low temperatures is likely due to adsorbed water molecules.
Low-pressure Ar physisorption experiments were carried out to analyze the pore structure of the chiral carbonaceous materials (Fig. 2 and Table S1†).
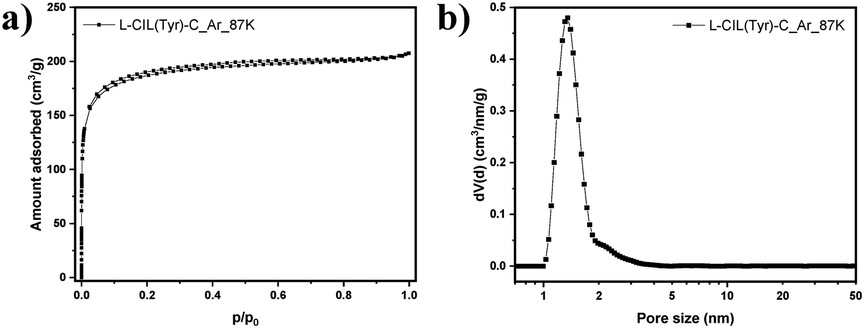 | ||
| Fig. 2 Argon physisorption isotherm (87 K) (a), and corresponding differential pore size distribution calculated with QSDFT (argon on carbons with cylindrical/sphere pores at 87 K, adsorption branch kernel) (b). | ||
The use of argon as an adsorptive at 87 K offers advantages over nitrogen at 77 K in particular for micropore analysis. Namely, the absence of quadrupole moment in the case of Ar prevents the specific interactions with surface functional groups, allowing to establish a more straightforward correlation between the pressure at which adsorption occurs and the pore size. This is of particular importance for the highly polar IL-based carbons discussed here with a high density of functional groups. L-CIL(Tyr)-C exhibits a type I(b) isotherm, indicating its mainly microporous structure, with a small amount of narrow mesopores.56 The SSA of 625 m2 g−1 is significantly higher than for previously reported CIL-derived carbon materials.57 The PSD analysis (Fig. 2b) shows that the pores are centered at a diameter of 1.69 nm. The sample shows a micropore volume of 0.22 cm3 g−1 and a total pore volume of 0.26 cm3 g−1. This indicates that the salt melt acts as a template in this synthesis and is responsible for the development of significant porosity.
Raman spectroscopy is another useful tool to characterize sp2-based carbon structures by their so-called disordered (D) and graphite (G)-like bands with varying intensity, position, and width.58 In the Raman spectrum of L-CIL(Tyr)-C (Fig. S3†), the D band near 1350 cm−1 originates from the breathing modes of the six-fold sp2-hybridized carbon rings in the presence of defects and disorder. The G band near 1590 cm−1 is caused by bond stretching of sp2-hybridized carbon in either rings or chains. The peak height ratio of the D- and G-band (ID/IG) is proportional to the amount of six-membered sp2 carbon rings, which is commonly employed to evaluate the level of carbon ordering in porous carbons. After fitting the spectra with a 4-band model, the ID/IG ratio for L-CIL(Tyr)-C is determined to be 1.87, which is a typical value for porous carbon materials with high local disorder.58
In summary, it was shown that micro-sized particles of porous carbon with high surface area can be synthesized using chiral ionic liquids based on different amino acids, combined with a salt melt carbonization process. Apart from CILs based on tyrosine CIL(Tyr), phenylalanine CIL(Phe) and proline and CIL(Pro) other CILs were also successfully synthesized and carbonized.
Afterward, we studied the chiral recognition capability of the chiral nanoporous carbon. In the first set of experiments, we examined the chiral recognition of CIL(Tyr)-C. We chose adsorption of L-phenylalanine from solution on the CIL(Tyr)-C as a representative case to demonstrate the chiral recognition ability of the nanoporous carbon using circular dichroism (CD) spectroscopy. We performed selective chiral adsorption measurements of 5 mM enantiomeric solutions of L-phenylalanine. Solutions were added to different amounts of nanoporous carbon (20, 30 and 40 mg L- or D-CIL(Tyr)-C), and CD signals were measured. The optical activity, namely the CD signals, of the chiral solutions was measured after adsorption equilibrium was achieved for approximately one day. The results of L-phenylalanine adsorption onto L- or D-CIL(Tyr)-C can be seen in Fig. S8.† A comparison of the adsorption measurements clearly shows the stereoselective uptake of the nanoporous carbon, displaying a significant difference in the adsorption of the two enantiomers. For example, approximately 2.90 mmol of L-Phe were adsorbed on 40 mg of D-CIL(Tyr)-C, while only 2.28 mmol of L-Phe was adsorbed on 40 mg of L-CIL(Tyr)-C. The CD results confirm the preference of L-phenylalanine for chiral adsorption on the D enantiomer of CIL(Tyr)-C. Based on the equilibrium concentrations of L-Phe, an equilibrium chiral discrimination ratio of 1.34 was calculated. This figure is relatively high for nanoporous materials with chiral functionalization and is sufficient for carrying out a successful enantiomeric separation on the chiral nanoporous carbon.
In order to support these findings and further demonstrate the enantioselective nature of CIL(Tyr)-C, isothermal titration calorimetry (ITC) was performed to measure the adsorption enthalpy of enantiomers onto CIL(Tyr)-C. In recent years it was shown that the ITC method can be used for detecting and measuring the chirality of nanomaterials;59–62 the enantioselective factor is obtained by calculating the difference between the adsorption enthalpies of the two enantiomers.
In a typical chiral ITC experiment, a solution of a chiral probe (e.g., enantiomers of amino acids) is titrated into the sample cell with the nanoporous carbon powders. The heat (ΔH) released due to molecular interactions is monitored as a function of time. Each peak represents the change in heat associated with the injections of the chiral probe solutions into the nanoporous carbon in the ITC reaction cell. The total heat of interaction is determined by the area under the peaks. The main contribution to the total change in enthalpy (ΔH) is due to the enthalpy of chiral binding between the D and L enantiomers and the chiral carbon. However, other enthalpy changes can result from the heat of dilution of the chiral solutions and from nonspecific heat effects, and these are calculated and corrected using a number of control experiments. In our ITC experiments, we used enantiomers of L- and D-tartaric acid (TA) as the chiral probe, and the ITC experiments were carried out in the following manner: a 1 mg mL−1 sample of L- or D-CIL(Tyr)-C was placed in the ITC cell, and 5 μL of a 10 mM D- or L-TA solution was injected into the suspension of the microporous carbon. The distinct difference between the D and L microporous carbon is shown in Fig. 4. It is evident that when L-TA is injected into L-CIL(Tyr)-C microporous carbon, the maximum heat flow for the first ITC peak is ca. 1.79 μcal s−1, whereas when D-TA is injected, the maximum heat flow in the first peak reduces significantly to ca. 1.56 μcal s−1. From the integration of the ITC peaks vs. injection number, the heat of adsorption for tartaric acid on L-CIL(Tyr)-C can be calculated, as shown in Fig. 3c. Moreover, the integration of the ITC curves in Fig. 3c represents the total heat of adsorption, of each enantiomer (ΔHads). We calculated ΔHabs values of 5.82 cal mol−1 for L-TA and 4.94 cal mol−1 for D-TA. These results prove the chiral nature of CIL(Tyr)-C and show an enantioselectivity value of about 1.17, similar to the results obtained in the CD chiral adsorption measurements. A similar ITC experiment was performed with L- and D-TA solutions injected into D-CIL(Tyr)-C nanoporous carbon, and the results of these experiments are presented in Fig. 3b and d. In this case, the maximum heat flows for the first ITC peak are about 1.8 and 1.66 μcal for D- and L-TA, respectively, with corresponding enthalpies of chiral binding of 5.82 and 4.94 cal mol−1, the exact opposite values with respect to L-CIL(Tyr)-C, confirming the enantioselectivity value of 1.18. Hence, ITC nicely shows the chiral recognition of CIL(Tyr)-C. The chirality of the nanoporous carbon is shown to be dependent on the chirality of the tyrosine; nanoporous carbon prepared from L-tyrosine shows a preference for adsorption of L enantiomers, while the nanoporous carbon of D-tyrosine shows a preference for adsorption of D enantiomers. It should be noted that our results from the CD selective chiral adsorption and ITC measurements are in line with results reported for chiral nanoporous materials, for example, silica, that were prepared by molecular imprinting methods.36,43 In other words, the chirality of the nanoporous materials is determined by the chirality of the precursor molecules. In our case, nanoporous tyrosine-derived carbon, which originated from the chiral ionic liquid of L-tyrosine, had L chiral recognition, while carbon synthesized from the CIL of D-tyrosine had D chiral recognition.
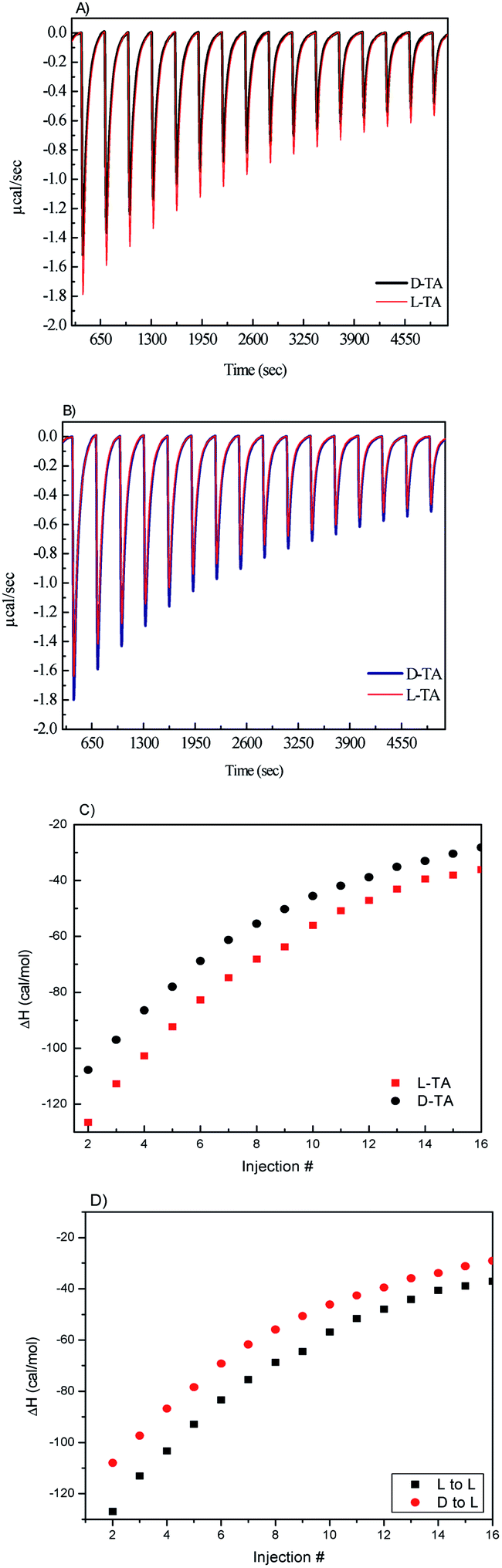 | ||
| Fig. 3 ITC heat of adsorption histograms and integrated enthalpies for the chiral nanoporous carbons. Histograms for injection of D- and L-TA solutions into L-CIL(Tyr)-C (a), and D-CIL(Tyr)-C (b), and heat of adsorption for the probe molecules on L-CIL(Tyr)-C (c), and D-CIL(Tyr)-C (d), obtained by integration of the peaks in (a) and (b) after each injection. | ||
Next electrochemical techniques such as cyclic voltammetry (CV) can be used to investigate the chirality of surfaces.63,64 In a set of CV experiments, we examined the chiral recognition ability of CIL(Tyr)-C by using it as an electrode. We prepared carbon electrodes for the CV experiments from CIL(Tyr)-C as follows: 6 mg of the mesoporous carbon was mixed with 4 mg of carbon black and 0.2% NAFION wt% to give the optimal combination for preparing the carbon electrode. The chiral solutions for the CV measurements were L- and D-TA (5 mM), with Na2SO4 (0.1 mM) as supporting electrolyte. Cyclic voltammetry was performed with D and L carbon electrodes, and their electrochemical activities were measured. From the electrochemistry literature, it is known that oxidation of TA occurs at about 0.6–0.7 V vs. RHE on the carbon electrodes. The results of the CV measurements are shown in Fig. 4; the oxidation peaks for solutions of the L and D enantiomers of TA show a different shape, depending on the chiral nature of the mesoporous carbon electrodes. In order to quantify the different electrochemical behavior, the total electrochemical current for the oxidation process, Q, can be calculated from the areas below the oxidation of the CV curves. The ratio between the total current for oxidation of D and L-CIL(Tyr)-C carbons (QD/QL) can give an indication of the chiral selectively of the carbon electrodes. The results show that the D-CIL(Tyr)-C electrode selectively oxidizes more of the L enantiomer of TA, while the L-CIL(Tyr)-C electrode selectively oxidizes more of the D enantiomer. The chiral recognition of tartaric acid enantiomers can be calculated by the total current ratios: for the D-CIL(Tyr)-C electrode the enantioselectivity factor is QD/QL = 1.068, while for the L-CIL(Tyr)-C electrode the enantioselectivity factor is much greater: QL/QD = 1.393.
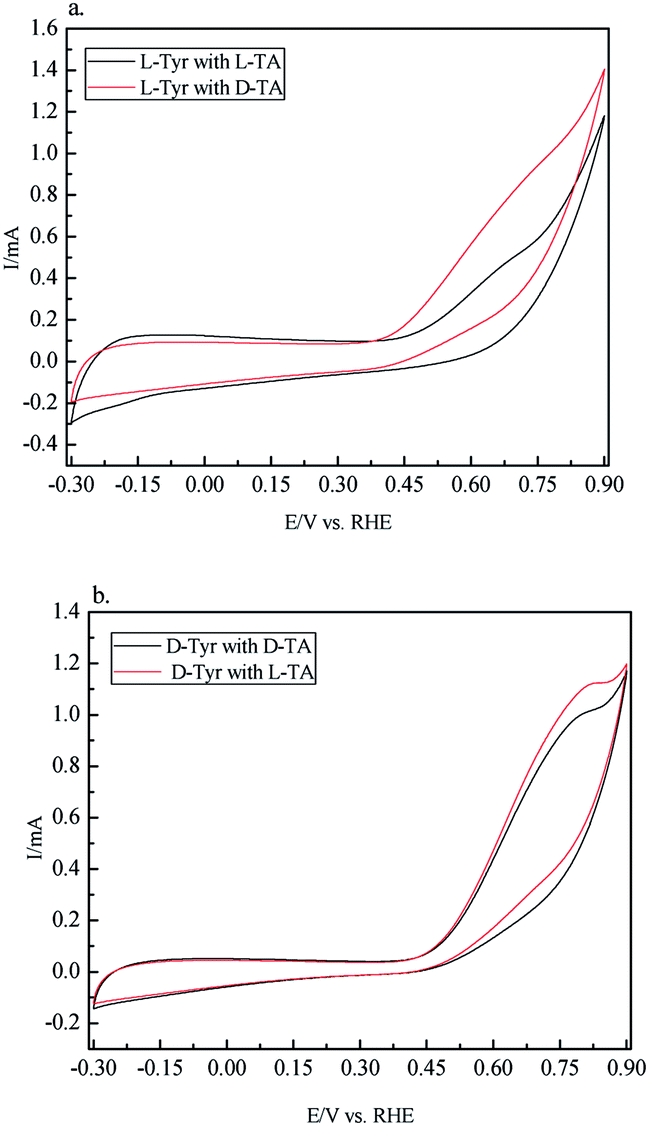 | ||
| Fig. 4 Chiral recognition of Tyr-based CIL-C electrodes: (a) L-CIL(Tyr)-C electrode and (b) D-CIL(Tyr)-C electrode with L/D-TA. CV measurements were taken at room temperature in solutions of 5 mM L- or D-TA in 0.1 M Na2SO4. | ||
It should be emphasized that the CV measurements display a chiral behavior that is inverse compared to the CD and ITC measurements. The electrochemical measurements demonstrate chiral recognition that is inverse with respect to the chirality of the mesoporous carbon, i.e. the electrode based on L-CIL(Tyr)-C shows high chiral recognition for the D enantiomer of TA, while the other electrode, based on D-CIL(Tyr)-C, shows high chiral recognition for the L enantiomer. It is still unclear to us what might be the reason for the observed difference in chiral recognition. One possible explanation is based on the nature of the CV measurements which differs from that of the chiral adsorption from CD and ITC measurements. In the latter, we measured the chiral recognition at chemical equilibrium, while in the electrochemical measurements we probed the chiral recognition under dynamic and diffusion conditions of electrochemical reactions. This explanation is supported by other studies that reported the preferred formation of homochiral domains of TA enantiomers on metallic surfaces due to weak interactions between TA and the metallic surface. Nevertheless, the electrochemical studies have clearly demonstrated the chiral nature and enantioselective properties of the carbon electrodes.
In summary, all the techniques used in this work clearly indicate the enantioselective nature of the carbons, however, still the main question regarding the origin of the chirality in our carbon materials is unclear. Overall, in chiral nanoporous materials such as silica, the molecular mechanism leading to the formation of chirality is based on the chiral templating mechanism. Nevertheless, in our case, we assume that the chiral nature of our carbon is due to the presence of chiral residues of the ionic liquids used as the source of carbon in our synthesis method. It is known from previous studies on nanoporous carbons prepared with zinc ions that zinc ions are able to coordinate and stabilize polar organic functional groups even at elevated temperatures65,66 In our previous work57 we speculated that chiral organic motifs remain in the carbonized scaffold after the carbonization process. This hypothesis was supported by thermogravimetric analysis combined with gas chromatography and mass spectrometry (TGA-GC-MS).
In order to verify our assumption regarding the mechanism for the chirality in our carbon, we decided to perform a series of TGA-GC-MS measurements of the pure chiral ionic liquids and of the carbons. TGA-GC-MS is a powerful analytical methodology for composite materials. In these experiments, we can analyze the volatile compounds that are released from the carbons so that a combination of GC/MS and TGA can give another dimension of understanding of the carbon structure and the origin of chirality. All the TGA-GC/MS measurements were carried out under nitrogen atmosphere. First, we measured the TGA-GC/MS profile for the pure L-CIL(Tyr) as shown in Fig. 5a(i) and b(i). From the TGA curve is can be seen that the pure L-CIL(Tyr) decomposition from ca.120 °C up to 550 °C and the maximum decomposition rate is observed at 310 °C. The pyrolysis at 310 °C of L-CIL(Tyr) gave three main products: I, II and III with designate mass (and their corresponding base fragment peak) characterized each compound: 178 (119), 192 (161), 178 (147) for I, II, III respectively. The analysis of L-CIL(Tyr)-C in TGA-GC/MS is not a trivial one since it consists of a mixture of organic and inorganic compounds and where the organic compounds are found only in trace amounts (12% weight loss). Consequently, the TGA thermogram feature is of a moderate slope, indicating a slow release of the organic components. The emitted gases from the L-CIL(Tyr)-C at 400 °C were collected and accumulated in a 100 μL loop and the results are shown in Fig. 5a(ii) and b(ii). Then the mixture was injected and separated using a capillary GC column. Since the extent of the analysts is extremely low, in the total ion chromatogram (TIC) we could not detect the presence of organic compounds that are related to chiral liquid L-CIL(Tyr). However, when we examined the extracted ion chromatograms (EICs) of the designated masses, the chromatogram contained the same three major components as in the L-CIL(Tyr). All three major thermal products are related to L-CIL(Tyr) that releases its quaternary ammonium cation. The mass spectrum of the peak at retention time (RT) 7.88 minutes closely matches to methyl 2,3-dihydro-1-benzofuran-3-carboxylate, I, that seems to be a thermal rearrangement product. The peaks at RTs 8.40 and 8.86 are the degradation products methyl p-methoxycinnamate II and methyl p-hydroxycinnamate III that may form by elimination reactions.
 | ||
| Fig. 5 Gas chromatography and mass spectrometry, (RT and MS chromatograms) and the TGAs profiles of L-L-CIL(Tyr) and L-CIL(Tyr)-C. | ||
In conclusion, the above have showed two important findings first that the carbon prepared at 450 °C still contain organic residues and that those organic residues are originated from the chiral ionic liquids. In general, these results prove our claims that chiral organic motifs remain attached to the carbonized scaffold in the final carbon, and those chiral functional groups provide the chiral recognition observed in our carbon.
Conclusions
Over the last years, there have been substantial achievements in the synthesis and applications of new chiral nanostructures and chiral nanoporous materials. In this paper, we described a new synthesis process developed for preparing enantioselectivity nanoporous carbon-based on chiral ionic liquids (CILs) as source material. We showed that the CILs based on amino acids could be used for the fabrication of chiral nanoporous carbon materials with high surface areas. We demonstrated the enantioselective property of those chiral nanoporous carbons by a set of unique analytical methods, i.e. selective adsorption CD and ITC. It is obvious that our new chiral nanoporous carbon can find many important applications in chiral chemistry, ranging from enantiomeric separation, chiral catalysis and chiral sensors. In addition, we studied our chiral carbons with thermogravimetric analysis combined with gas chromatography and mass spectrometry (TGA-GC-MS) measurements and shown that their enantioselectivity is due to are originated from the chiral ionic liquids. In general, these results prove our claims that chiral organic residues that are attached to the carbonized material. It is clear that in the near future new approaches for the preparation and use of chiral nanoporous materials will be developed, and our work is part of this general trend. However, further detailed and systematic experimental studies are needed to attain a better understanding of the molecular mechanism for the formation of chiral properties and of the chiral behavior of these new chiral carbons in order to develop appropriate applications. Nearly all aspects of chiral nanomaterials including synthesis, separation, and analysis can benefit greatly from the use of chiral porous carbon materials. We believe that the approach presented in this article is significant for the development of novel chiral porous materials for enantioselective applications and can contribute significantly to our understanding of the structure and behavior of chiral nanomaterials and nano-surfaces.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Shekef-Aloni would like to acknowledge the Bar-Ilan President's Ph.D. Scholarship Foundation. We would like to thank Mr Bar Gavriel for assistance with CV measurements. This research was supported by the German-Israeli Foundation for Scientific Research and Development (GIF, Grant No. I-87-302.10-2015).Notes and references
- T. J. Maher and D. A. Johnson, Drug Dev. Res., 1991, 24, 149–156 CrossRef CAS.
- N. M. Maier, P. Franco and W. Lindner, J. Chromatogr. A, 2001, 906, 3–33 CrossRef CAS.
- D. B. Amabilino, Chem. Soc. Rev., 2009, 38, 669–670 RSC.
- D. S. Bag, T. C. Shami and K. U. Rao, Def. Sci. J., 2008, 58, 626–635 CrossRef.
- J. Zhang, M. T. Albelda, Y. Liu and J. W. Canary, Chirality, 2005, 17, 404–420 CrossRef CAS PubMed.
- W. Ma, L. Xu, A. F. de Moura, X. Wu, H. Kuang, C. Xu and N. A. Kotov, Chem. Rev., 2017, 117, 8041–8093 CrossRef CAS PubMed.
- C. Gautier and T. Bürgi, ChemPhysChem, 2009, 10, 483–492 CrossRef CAS PubMed.
- Y. Mastai, Chem. Soc. Rev., 2009, 38, 772–780 RSC.
- C. Gautier and T. Bürgi, J. Am. Chem. Soc., 2006, 128, 11079–11087 CrossRef CAS PubMed.
- Y. Xia, Y. Zhou and Z. Tang, Nanoscale, 2011, 3, 1374–1382 RSC.
- B. Chen, J. Deng and W. Yang, Adv. Funct. Mater., 2011, 21, 2345–2350 CrossRef CAS.
- P. Paik, A. Gedanken and Y. Mastai, J. Mater. Chem., 2010, 20, 4085–4093 RSC.
- L. C. Preiss, L. Werber, V. Fischer, S. Hanif, K. Landfester, Y. Mastai and R. Muñoz-Espí, Adv. Mater., 2015, 27, 2728–2732 CrossRef CAS PubMed.
- H. Alhassen, V. Antony, A. Ghanem, M. M. A. Yajadda, Z. J. Han and K. Ostrikov, Chirality, 2014, 26, 683–691 CrossRef CAS PubMed.
- T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed.
- R. M. Hazen and D. S. Sholl, Nat. Mater., 2003, 2, 367–374 CrossRef CAS PubMed.
- R. Raval, Chem. Soc. Rev., 2009, 38, 707–721 RSC.
- F. Zaera, J. Phys. Chem. C, 2008, 112, 16196–16203 CrossRef CAS.
- J. C. Lang and D. W. Armstrong, Curr. Opin. Colloid Interface Sci., 2017, 32, 94–107 CrossRef CAS.
- A. J. Gellman, ACS Nano, 2010, 4, 5–10 CrossRef CAS PubMed.
- H. Qiu and S. Che, Chem. Soc. Rev., 2011, 40, 1259–1268 RSC.
- S. Che, J. Nanosci. Nanotechnol., 2006, 6, 1557–1564 CrossRef CAS PubMed.
- D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273–282 CrossRef CAS PubMed.
- K. E. Shopsowitz, H. Qi, W. Y. Hamad and M. J. MacLachlan, Nature, 2010, 468, 422–425 CrossRef CAS PubMed.
- C. Li, H. Zhang, D. Jiang and Q. Yang, Chem. Commun., 2007, 547–558 RSC.
- H. Jin, Z. Liu, T. Ohsuna, O. Terasaki, Y. Inoue, K. Sakamoto, T. Nakanishi, K. Ariga and S. Che, Adv. Mater., 2006, 18, 593–596 CrossRef CAS.
- J. A. Kelly, M. Giese, K. E. Shopsowitz, W. Y. Hamad and M. J. MacLachlan, Acc. Chem. Res., 2014, 47, 1088–1096 CrossRef CAS PubMed.
- T. Cheng, Q. Zhao, D. Zhang and G. Liu, Green Chem., 2015, 17, 2100–2122 RSC.
- P. Paik, A. Gedanken and Y. Mastai, Microporous Mesoporous Mater., 2010, 129, 82–89 CrossRef CAS.
- P. Paik, A. Gedanken and Y. Mastai, ACS Appl. Mater. Interfaces, 2009, 1, 1834–1842 CrossRef CAS PubMed.
- C. Casado, J. Castán, I. Gracia, M. Yus, A. Mayoral, V. Sebastián, P. López-Ram-de-Viu, S. Uriel and J. Coronas, Langmuir, 2012, 28, 6638–6644 CrossRef CAS PubMed.
- S. Fireman-Shoresh, D. Avnir and S. Marx, Chem. Mater., 2003, 15, 3607–3613 CrossRef CAS.
- S. Fireman-Shoresh, S. Marx and D. Avnir, Adv. Mater., 2007, 19, 2145–2150 CrossRef CAS.
- S. Fireman-Shoresh, S. Marx and D. Avnir, J. Mater. Chem., 2007, 17, 536–544 RSC.
- S. Fireman-Shoresh, I. Popov, D. Avnir and S. Marx, J. Am. Chem. Soc., 2005, 127, 2650–2655 CrossRef CAS PubMed.
- S. Marx and D. Avnir, Acc. Chem. Res., 2007, 40, 768–776 CrossRef CAS PubMed.
- S. Che, Z. Liu, T. Ohsuna, K. Sakamoto, O. Terasaki and T. Tatsumi, Nature, 2004, 429, 281–284 CrossRef CAS PubMed.
- X. Meng, T. Yokoi, D. Lu and T. Tatsumi, Angew. Chem., 2007, 119, 7942–7944 CrossRef.
- D. Berthier, T. Buffeteau, J.-M. Léger, R. Oda and I. Huc, J. Am. Chem. Soc., 2002, 124, 13486–13494 CrossRef CAS PubMed.
- R. Oda, F. Artzner, M. Laguerre and I. Huc, J. Am. Chem. Soc., 2008, 130, 14705–14712 CrossRef CAS PubMed.
- R. Oda, I. Huc, M. Schmutz, S. J. Candau and F. C. MacKintosh, Nature, 1999, 399, 566–569 CrossRef CAS PubMed.
- K. Sugiyasu, S. Tamaru, M. Takeuchi, D. Berthier, I. Huc, R. Oda and S. Shinkai, Chem. Commun., 2002, 1212–1213 RSC.
- A. Gabashvili, D. D. Medina, A. Gedanken and Y. Mastai, J. Phys. Chem. B, 2007, 111, 11105–11110 CrossRef CAS PubMed.
- J. Kupai, E. Rojik, P. Huszthy and G. Szekely, ACS Appl. Mater. Interfaces, 2015, 7, 9516–9525 CrossRef CAS PubMed.
- M. Álvaro, M. Benitez, D. Das, B. Ferrer and H. García, Chem. Mater., 2004, 16, 2222–2228 CrossRef.
- G. Levi, Y. Scolnik and Y. Mastai, ACS Appl. Mater. Interfaces, 2016, 8, 23356–23361 CrossRef CAS PubMed.
- Y. Okazaki, T. Buffeteau, E. Siurdyban, D. Talaga, N. Ryu, R. Yagi, E. Pouget, M. Takafuji, H. Ihara and R. Oda, Nano Lett., 2016, 16, 6411–6415 CrossRef CAS PubMed.
- N. Ryu, Y. Okazaki, K. Hirai, M. Takafuji, S. Nagaoka, E. Pouget, H. Ihara and R. Oda, Chem. Commun., 2016, 52, 5800–5803 RSC.
- T. Ohsuna, Z. Liu, S. Che and O. Terasaki, Small, 2005, 1, 233–237 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- J. Ding and D. W. Armstrong, Chirality, 2005, 17, 281–292 CrossRef CAS PubMed.
- K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 2008, 3235–3250 CrossRef.
- A. Winkel, P. V. G. Reddy and R. Wilhelm, Synthesis, 2008, 2008, 999–1016 CrossRef.
- C. P. Kapnissi-Christodoulou, I. J. Stavrou and M. C. Mavroudi, J. Chromatogr. A, 2014, 1363, 2–10 CrossRef CAS PubMed.
- L. González, B. Altava, M. Bolte, M. I. Burguete, E. García-Verdugo and S. V Luis, Eur. J. Org. Chem., 2012, 2012, 4996–5009 CrossRef.
- M. Thommes, K. Kaneko, A. V Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- I. Fuchs, N. Fechler, M. Antonietti and Y. Mastai, Angew. Chem., Int. Ed., 2016, 55, 408–412 CrossRef CAS PubMed.
- M. Oschatz, P. Pré, S. Dörfler, W. Nickel, P. Beaunier, J.-N. Rouzaud, C. Fischer, E. Brunner and S. Kaskel, Carbon, 2016, 105, 314–322 CrossRef CAS.
- A. Shval and Y. Mastai, Chem. Commun., 2011, 47, 5735–5737 RSC.
- L. Werber, L. C. Preiss, K. Landfester, R. Muñoz-Espí and Y. Mastai, Chirality, 2015, 27, 613–618 CrossRef CAS PubMed.
- R. Dimova, R. Lipowsky, Y. Mastai and M. Antonietti, Langmuir, 2003, 19, 6097–6103 CrossRef CAS.
- L. Werber and Y. Mastai, Chirality, 2018, 30, 619–631 CrossRef CAS PubMed.
- G. A. Attard, J. Phys. Chem. B, 2001, 105, 3158–3167 CrossRef CAS.
- S. P. Mathew, P. C. Mondal, H. Moshe, Y. Mastai and R. Naaman, Appl. Phys. Lett., 2014, 105, 242408 CrossRef.
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed.
- S. J. Yang, M. Antonietti and N. Fechler, J. Am. Chem. Soc., 2015, 137, 8269–8273 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00520j |
| This journal is © The Royal Society of Chemistry 2019 |