
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNano-SiO2 coating enabled uniform Na stripping/plating for dendrite-free and long-life sodium metal batteries†
Fuyi
Jiang
a,
Tianjiao
Li
a,
Peng
Ju
b,
Jianchao
Sun
 *a,
Chuang
Liu
a,
Yiwei
Li
a,
Xueqin
Sun
*a and
Chengcheng
Chen
c
*a,
Chuang
Liu
a,
Yiwei
Li
a,
Xueqin
Sun
*a and
Chengcheng
Chen
c
aSchool of Environment and Materials Engineering, Yantai University, Yantai, Shandong 264005, China. E-mail: jianchaoabc@163.com; sxq@ytu.edu.cn
bKey Laboratory of Marine Bioactive Substances and Analytical Technology, Marine Ecology Center, First Institute of Oceanography, Ministry of Natural Resources (MNR), Qingdao, Shandong 266061, China
cKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China
First published on 18th November 2019
Abstract
Metallic sodium, which has a suitable redox potential and high theoretical capacity, is regarded as an ideal anode material for rechargeable Na metal batteries. However, dendrite growth on sodium metal during cycling has seriously restricted its practical applications. Herein, we employed a low-cost and facile brushing method to fabricate a porous nano-SiO2 coating, which can induce a relatively uniform distribution of Na+ flux and suppress the growth of Na dendrites. The nano-SiO2 coating with high porosity can decrease the Na stripping/plating overpotential (<50 mV) over 400 cycles at 5 mA cm−2. Moreover, when coupled with a Na3V2(PO4)3 (NVP) cathode, the Na with SiO2 coating (Na@SiO2) composite anode shows a favorable suitability in a full cell. Compared with the one with a bare Na anode, the full cell with the Na@SiO2 anode delivers a 27.8% higher discharge capacity (94.6 vs. 74 mA h g−1 at 1C) after 1000 cycles.
Introduction
Nowadays, as fossil fuel-related energy is consumed excessively and environmental awareness boosts, the demand for alternative clean energy is becoming more and more urgent.1–4 Over the past few decades, rechargeable lithium-ion batteries (LIBs) have dominated portable electronics, medical microelectronics, and electric vehicles (EVs) as one of the most promising energy storage systems.5–9 However, with the increasing demand for LIBs and shortage of lithium resources, the cost of lithium raw materials has increased rapidly.10–12 Therefore, researching and exploring substitutes for LIBs has become one of the significant directions in the development of rechargeable batteries.13–15 Because of the abundant reserves, low cost, and suitable electrochemical potential of Na, rechargeable Na batteries have received widespread attention and are regarded as the most promising substitute to LIBs.16–20 However, some serious problems have hampered the practical application of rechargeable Na batteries.21 The charge and Na+ distribution on the surface of sodium metal are non-uniform during continuous charge–discharge cycling, resulting in the formation of sodium dendrites.22,23 The dendrites not only affect the capacity and life of batteries, but cause safety problems such as short-circuits and explosions, hindering the practical application of sodium metal batteries (SMBs).24,25 Therefore, solving the problem of Na metal dendrites generated during charge–discharge cycling is the key to improving the cycling stability and accelerating the commercial application of Na metal anodes.26Recently, many methods have been adopted to restrain dendritic Na formation, including (1) the stabilization of the SEI layer by electrolyte modification;27–29 (2) engineering artificial SEI layers by surface modification;30–32 and (3) engineering nanostructured current collectors and hosts by artificial manufacture.33–35 For instance, Wang and co-workers designed potassium bis(trifluoromethylsulfonyl)-imide (KTFSI) as a bifunctional electrolyte additive to stabilize the Na metal electrode.36 In the work reported by Sun's group, an ultrathin protective coating was constructed as a protective layer on a Na metal anode through the ALD technique.21 Luo' group stabilized Na metal anodes by employing a porous Al current collector as the plating substrate.37 Hu and co-workers manufactured a stable Na-carbonized wood composite anode as a 3D host for Na metal.38 These strategies provide insights for Na metal protection and achieve excellent electrochemical performance. Unfortunately, the expensive and complex preparation process poses substantial obstacles to the practical application of these strategies. Recently, Chen and Kang's group have designed nano-Super P and nano-CaCO3 coatings respectively, which restrain the growth of Li and Zn dendrites.39,40 Inspired by this, we explored a low-cost, simple, safe and scalable strategy to inhibit the growth of non-uniform dendrites in the Na metal anode.
Herein, we report a simple method to construct a porous nano-SiO2 coating as an auxiliary layer for inhibiting the growth of Na dendrites. We found that the nano-SiO2 coating with a high porosity can be easily permeated by electrolyte. Compared with the bare Na anode (Fig. 1a), the Na anode with the SiO2 coating induces a relatively uniform distribution of Na+ flux (Fig. 1b).
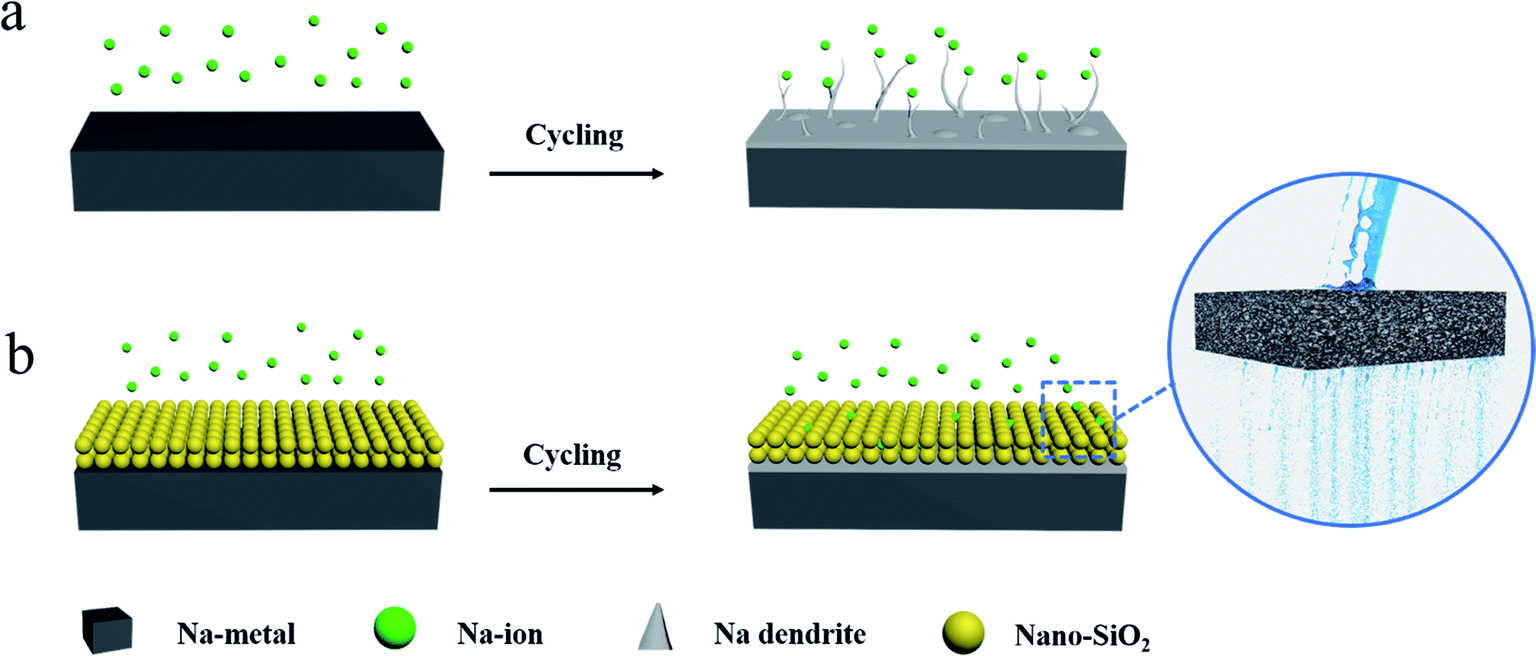 | ||
| Fig. 1 Schematic illustrations of the Na stripping/plating behavior on (a) bare and (b) nano SiO2 coated Na electrodes. | ||
The SiO2 coating on the surface of Na acted as an electrolyte transport channel (like pores in water-permeable bricks), effectively dispersing Na+ flux and achieving the purpose of suppressing dendrites. Consequently, the Na@SiO2 composite electrode used in symmetrical cells exhibited excellent cycling stability with significantly reduced polarization. Furthermore, Na dendrites were found to be absent in the Na@SiO2 electrode even at a current density of 5 mA cm−2 after 100 cycles. Moreover, we also tested a Na metal electrode with Super P coating, clearly finding that insulation coating (SiO2) has advantages over conductive coating (Super P). Finally, we successfully constructed a full cell with an NVP cathode and a Na@SiO2 anode, proving the benefits of this coating strategy for potential applications.
Experimental
Fabrication of Na@SiO2 and Na@Super P electrodes
Firstly, a certain amount of nano SiO2 or Super P was dried in a vacuum oven at 60 °C for 10 hours. Then, a Na disk (12 mm in diameter, 0.3 mm in thickness) was evenly coated with a SiO2 or Super P layer (∼100 μm) via brushing and rolling in an Ar-filled glovebox, and the mass loading of the layer is about 4.4 mg cm−2.Structural characterization
A scanning electron microscope (SEM, JSM-7610F) with an EDX attachment was used for obtaining SEM images and the corresponding EDX mapping images. For observing the morphology of electrodes after cycling, batteries were first disassembled in a glovebox and then the electrodes were rinsed with diglyme to remove residual Na salts. XRD patterns were obtained using a Rigaku D/MAX 2500/PC diffractometer with Cu Kα radiation (λ = 0.154 nm). Noticeably, the Na samples for SEM tests were loaded in a sealing device and covered with Kapton tape to avoid direct contact with air.Electrochemical measurements
To investigate the electrochemical properties of Na stripping/plating, symmetrical 2032-type coin cells were assembled with two identical electrodes, which were either Na@SiO2 or bare Na electrodes. For the preparation of the NVP cathode, NVP powder was mixed with carbon black and polyvinylidene fluoride (weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) using N-methyl-2-pyrrolidone as the solvent. The mass loading of the NVP cathode is about 1 mg. The electrolyte solution was 1 M sodium hexafluorophosphate (NaPF6) in diglyme. All batteries were assembled in an argon-filled glovebox using the same dose of electrolyte (50 μL), and Celgard membrane was used as the separator. The batteries were first left to stand for four hours to stabilize the interface before tests. Galvanostatic cycling was carried out on a standard battery tester (Neware CT-4008-5 V/10 mA). EIS and CV were conducted on an electrochemical workstation. CV was performed in the potential window from 2.5 to 3.8 V at a scan rate of 0.1 mV s−1.
:1:1) using N-methyl-2-pyrrolidone as the solvent. The mass loading of the NVP cathode is about 1 mg. The electrolyte solution was 1 M sodium hexafluorophosphate (NaPF6) in diglyme. All batteries were assembled in an argon-filled glovebox using the same dose of electrolyte (50 μL), and Celgard membrane was used as the separator. The batteries were first left to stand for four hours to stabilize the interface before tests. Galvanostatic cycling was carried out on a standard battery tester (Neware CT-4008-5 V/10 mA). EIS and CV were conducted on an electrochemical workstation. CV was performed in the potential window from 2.5 to 3.8 V at a scan rate of 0.1 mV s−1.
Results and discussion
We employed a low-cost and facile brushing method to fabricate the Na@SiO2 composite electrode. The hydrophobic SiO2 used in this experiment is amorphous and the particle size is about 15 nm (Fig. S1†). Fig. 2a shows an optical photo and SEM image of a bare Na electrode, revealing a smooth, flat and shiny metallic surface. After brushing the nano SiO2, the surface of Na was evenly coated with a white layer of spherical nano-SiO2 particles (inset in Fig. 2b). Clearly, the fresh nano-SiO2 coating is highly porous (Fig. 2b), which promotes electrolyte diffusion and ion migration in nano-channel pores.41–45 Due to the high viscosity of Na, the nano-SiO2 particles are tightly attached to the metal surface and will not fall off when the Na@SiO2 electrode is inverted with tweezers (insets in Fig. 2b). As shown in Fig. 2c, the thickness of the nano-SiO2 coating is about 100 μm, which can be confirmed by EDX elemental mapping. | ||
| Fig. 2 The characterization of Na and Na@SiO2 electrodes. (a) Top-view SEM image of a bare Na electrode and the corresponding digital image (inset). (b) Top-view SEM image of the Na@SiO2 electrode, and the corresponding top and side view digital images (insets). (c) Cross-sectional SEM image of the Na@SiO2 electrode and the corresponding EDX elemental mapping images of Si and Na (insets). | ||
To assess the electrochemical performance of the Na@SiO2 electrode, symmetric cells with two identical Na@SiO2 electrodes were assembled in 1 M NaPF6/diglyme electrolyte. For comparison, symmetric cells with two identical bare Na electrodes (thickness of ∼300 μm) were assembled.
Fig. 3a shows the galvanostatic cycling performance of symmetric cells with Na@SiO2 (yellow lines) and bare Na electrodes (green lines), respectively, for 400 cycles with a capacity of 1 mA h cm−2 at 1, 3 and 5 mA cm−2. The symmetric cell with Na@SiO2 electrodes exhibited stable voltage profiles and much lower hysteresis (5 mV) than the one with bare Na electrodes at 1 mA cm−2. The bare Na electrode displayed a gradual increase in hysteresis (>25 mV), where the overpotential increases first, and then irregularly fluctuates, indicating a micro-short circuit.46 When the current density was increased to 3 mA cm−2, the Na@SiO2 electrode still exhibited a small overpotential (15 mV) and very stable voltage profiles. In comparison, the symmetric cells with bare Na electrodes showed an anomalistic and apparently fluctuating voltage (>140 mV). As the current density was further increased to 5 mA cm−2, the symmetrical cell with the bare Na electrode had large voltage polarization (>180 mV) and obviously poor stability, which is due to the uneven plating/stripping of Na metal, leading to the formation of dendrites. In contrast, the cell with the Na@SiO2 electrode not only exhibited a much lower polarization (50 mV) but also realized very stable cycling under the same conditions. Furthermore, the rate behavior of the symmetric cells with bare Na and Na@SiO2 electrodes is presented in Fig. 3b. As the current density increased from 0.5 to 10 mA cm−2, the voltage polarization of the bare Na electrode increased greatly and tended to be unstable, which was attributed to irregular growth of Na dendrites on the electrode surface. Unfortunately, when the current density returned to 0.5 mA cm−2, the battery no longer returned to its previous state, indicating poor cycling stability. In contrast, the cell using the Na@SiO2 electrode continued to cycle steadily to 10 mA cm−2. It is worth mentioning that when the current density returned to 0.5 mA cm−2, the voltage profile was as stable as it was initially. To further demonstrate the superiority of the Na@SiO2 electrode, higher capacity cycling of the symmetrical cell was performed and the results are presented in Fig. S2.† The result demonstrated that the stability of the Na@SiO2 electrode is significantly improved in virtue of the nano-SiO2 coating, which could induce a relatively uniform distribution of Na+ flux over the entire Na surface.26,45,47
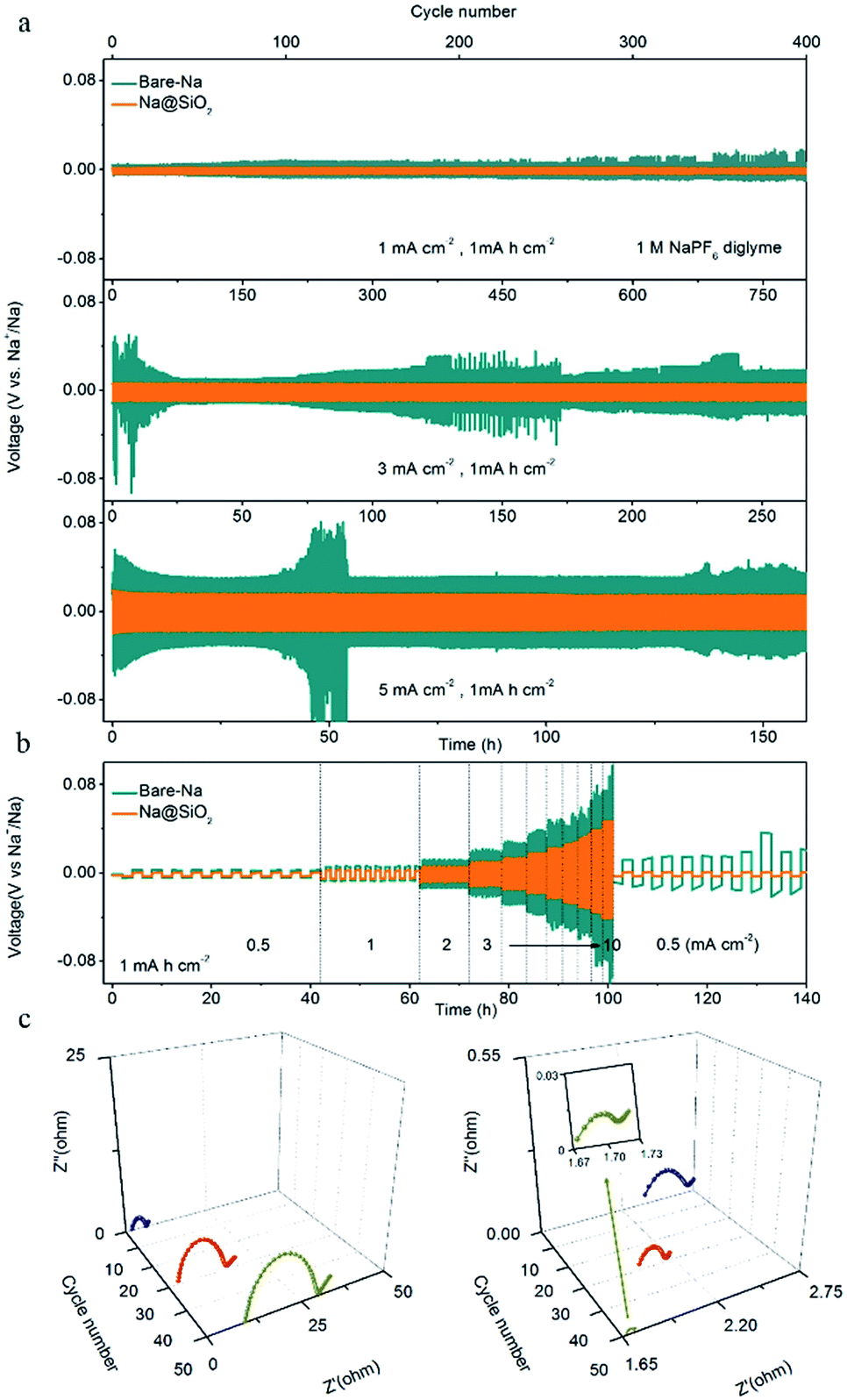 | ||
| Fig. 3 Electrochemical performance of the Na@SiO2 electrode. (a) Comparison of cycling stability of bare Na (green) and Na@SiO2 electrodes (orange) in symmetrical cells at current densities of 1, 3 and 5 mA cm−2 with a capacity of 1 mA h cm−2. (b) Rate performance. (c) EIS plots of bare Na (left) and Na@SiO2 electrodes (right) in symmetrical cells after different cycles at a current density of 1 mA cm−2 with a capacity of 1 mA h cm−2. | ||
Moreover, electrochemical impedance spectroscopy (EIS) was performed after 1, 30, and 50 cycles at a current density of 1 mA cm−2. The interfacial resistance of the SEI and surface charge transfer impedance of the electrode were estimated according to the diameter of the semicircle in the high frequency range.26,48 As shown in Fig. 3c, the Nyquist plots of the bare Na electrode show that the interface resistance increases after cycling (19 Ω in the 1st cycle, 25 Ω in the 30th cycle, and 35 Ω in the 50th cycle). This phenomenon is explained by the morphological transformation of the bare Na surface, indicating the occurrence of fractures in the SEI layer and the obvious growth of Na dendrites,26 which is proven in the subsequent SEM images. In addition, the ontological impedance of the symmetric cells with bare Na increases as the number of cycles increases, which is due to the generation of a large amount of non-conductive material during repeated repair of the SEI layer process, thereby degrading the overall conductivity of the battery.49 The Na@SiO2 electrode exhibited a smaller semicircle after cycling, indicating that the charge transfer resistance of the Na@SiO2 composite electrode is much lower than that of bare Na. This could be attributed to the fact that the porous nano-SiO2 coating induced a relatively uniform distribution of Na+ flux and a stable SEI layer was formed on the Na metal electrode surface, thereby achieving the purpose of suppressing the generation of Na dendrites.50
The morphological evolution of bare Na and Na@SiO2 electrodes after 100 cycles is shown in Fig. 4. The bare Na electrode exhibited a rough surface with rugged and mossy Na deposition at a current density of 1 mA cm−2 (Fig. 4a), which differed from the fresh Na electrode morphology. When the current density was increased to 3 and 5 mA cm−2, the bare Na electrode showed an extremely inhomogeneous surface with excessive Na dendrites and dead Na (Fig. 4b and c). The morphological variation is mainly caused by the non-uniform Na plating/stripping and high reaction kinetics on the surface of sodium metal. In contrast, the morphology of the Na@SiO2 electrode retained a flat surface without any detectable protrusions (Fig. 4d–f), which is attributed to the porous nano-SiO2 coating for homogenizing the distribution of Na+ on the Na surface. The above morphological evolution results demonstrate the role of nano-SiO2 coating in restraining Na dendritic growth.
 | ||
| Fig. 4 Morphological evolution of bare Na and Na@SiO2 electrodes after cycling. SEM images of (a–c) bare Na electrodes and (d–f) Na@SiO2 electrodes after 100 stripping/plating cycles at different current densities, respectively. The current density is marked at the top of the figure and the stripping/plating capacity was 1 mA h cm−2. | ||
In order to further study the deposition behavior of Na on the Na@SiO2 electrode, we further tested the Na metal electrode with conductive Super P coating (Na@Super P). The Super P used in this experiment is amorphous and the particle size is about 30 nm (Fig. S3†). The thickness of the Super P coating is about 100 μm (Fig. S4†). The electrodes with SiO2 and Super P coatings all present small wetting angles (Fig. S5†), demonstrating excellent electrolyte permeability. Although both the SiO2 layer and Super P layer are porous coatings, in terms of electrochemical properties, the latter has lower cycling stability than the former (Fig. 5a). The symmetric cells with the Na@Super P electrodes exhibited larger voltage hysteresis after 55 h. As shown in Fig. 5b, no Na metal was found on the surface of the SiO2 coating after 50 cycles. We further proved that sodium metal was deposited at the bottom of the nano-SiO2 coating layer after battery cycling by a cross-sectional SEM test (Fig. S6†). However, Na metal appeared on the surface of the Na@Super P electrodes after 50 cycles (Fig. 5c). The surface of the bare Na electrode is no longer flat after 100 cycles (Fig. S7a†), which is due to the formation of dendrites. The Na@SiO2 electrode is almost unchanged (Fig. S7b†), while the Na@Super P electrode has changed from black to a metallic luster (Fig. S7c†). The high conductivity of the Super P layer may weaken the position limiting ability on Na electrodeposition, leading to preferential deposition of Na above the conductive coating rather than below it.40 In contrast, owing to its electrically insulating properties, there should be a large variation of potential across the nano-SiO2 coating.51 Therefore, only the potential close to the surface of Na was low, and it was low enough for Na+ to undergo a reduction reaction, forming a position-selective, bottom-up Na deposition process, instead of preferential deposition on Na protrusions/dendrite tips.52
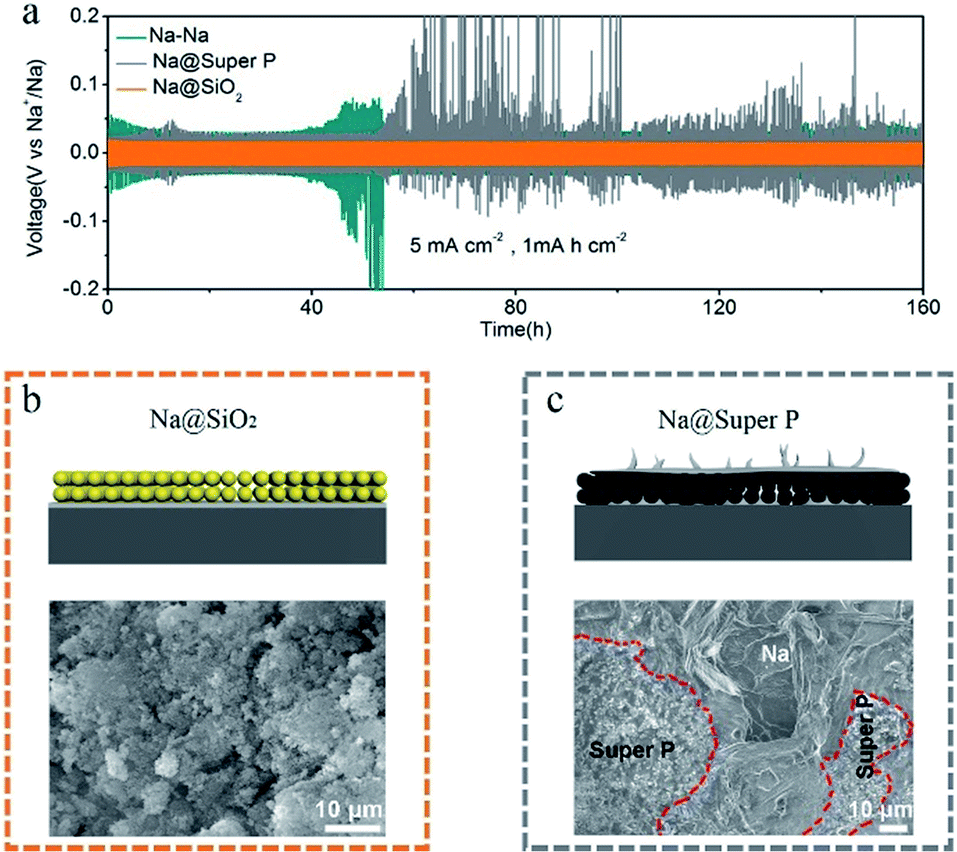 | ||
| Fig. 5 (a) Comparison of the cycling stability of bare Na (green), Na@SiO2 (orange) and Na@Super P (gray) electrodes at a current density of 5 mA cm−2 with a capacity of 1 mA h cm−2. Schematic illustrations and SEM images of (b) Na@SiO2 and (c) Na@Super P electrodes after 50 stripping/plating cycles at a current density of 5 mA cm−2 with a capacity of 1 mA h cm−2. | ||
To demonstrate the practical application of the Na@SiO2 electrode, we further assembled full cells with bare Na or Na@SiO2 as the anode and NVP as the cathode, respectively. The XRD profiles of NVP were consistent with PDF no. 70-3613 (Fig. S8a†). The SEM image indicates that the NVP particles are cross-linked with CNTs (Fig. S8b†).
A much higher NVP capacity was obtained with the Na@SiO2 anodes during rate cycling (Fig. 6a). The NVP–Na@SiO2 full cell exhibits superior capacities of 107, 99, 93, 89, 84, 80, 78, 76, 74 and 72.5 mA h g−1 with the current density increasing from 1 to 10C (1C = 118 mA g−1), respectively. After 150 consecutive cycles, a capacity of 106 mA h g−1 at 1C and an excellent coulombic efficiency of 99.4% are obtained. In contrast, the cells with bare Na anodes offer much lower capacities of 101, 89, 83, 80, 77, 72, 67, 64, 61 and 59 mA h g−1 with pronounced instability. Fig. 6b shows the cyclic voltammograms (CVs) of the NVP electrode, in which there are evident redox pairs at ∼3.4 V. Furthermore, when galvanostatically cycled at 1C, the battery with the Na@SiO2 anodes presented much better cycling stability than the one with bare Na anodes. As shown in Fig. 6c, it is observed that the cell with the Na@SiO2 anode shows stable electrochemical performance (94.6 mA h g−1 after 1000 cycles, a capacity retention of 86.4%, and a coulombic efficiency of 99.8%). In sharp contrast, the cell with bare Na anodes exhibited a smaller initial discharge capacity of 100 mA h g−1, and only retained 74 mA h g−1 after 1000 cycles with extremely unstable coulombic efficiency. The corresponding charge/discharge voltage–capacity profiles at the 1st and 500th cycles are shown in Fig. S9,† respectively. These results demonstrate the good adaptability of the Na@SiO2 anode in full cells.
 | ||
| Fig. 6 Electrochemical performance of the full cells with bare Na or Na@SiO2 as the anode and NVP as the cathode. (a) Rate capability of the NVP–Na and NVP–Na@SiO2 cells at different rates from 1 to 10C (1C = 118 mA g−1). (b) CVs of the NVP cathode at 0.1 mV s−1 coupled with the Na@SiO2 anode. (c) Cycling stability of full cells with the bare Na or Na@SiO2 anode at 1C, respectively. | ||
Conclusions
In conclusion, we developed a low-cost and simple brushing method to improve the stripping/plating stability of Na metal anodes. The porous nano-SiO2 coating induces a relatively uniform distribution of Na+ flux, resulting in uniform and dendrite-free deposition of metallic sodium. High cycling stability with low hysteresis was achieved in the Na@SiO2 electrode even at a very high current density of 5 mA cm−2. Furthermore, the Na@SiO2|NVP full cell exhibits high cycling stability and superior suitability, making it far better than the full cell with bare Na as the anode. Our work provides an alternative route for the fabrication of safe Na metal anodes, which will be of great significance in the practical application of Na metal anodes for high-energy Na-based batteries.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51772257, 51702328 and 21805171), Major Basic Research Project of Shandong Natural Science Foundation (ZR2018ZC1459) and Shandong Natural Science Foundation (ZR2018BB038 and ZR2017BD002).Notes and references
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- Y. Zhao, K. R. Adair and X. Sun, Energy Environ. Sci., 2018, 11, 2673–2695 RSC.
- J. Sun, Y. Lu, H. Yang, M. Han, L. Shao and J. Chen, Research, 2018, 2018, 6914626 Search PubMed.
- S. J. Tan, X. X. Zeng, Q. Ma, X. W. Wu and Y. G. Guo, Electrochem. Energy Rev., 2018, 1, 113–138 CrossRef CAS.
- J. Lu, Z. Chen, F. Pan, Y. Cui and K. Amine, Electrochem. Energy Rev., 2018, 1, 35–53 CrossRef CAS.
- G. Yang, H. Wang, B. Zhang, S. Foo, M. Ma, X. Cao, J. Liu, S. Ni, M. Srinivasan and Y. Huang, Nanoscale, 2019, 11, 9556–9562 RSC.
- Y. He, C. Lu, S. Liu, W. Zheng and J. Luo, Adv. Energy Mater., 2019, 9, 1901810 CrossRef.
- S. Liu, L. Deng, W. Guo, C. Zhang, X. Liu and J. Luo, Adv. Mater., 2019, 31, 1807585 CrossRef PubMed.
- A. Ponrouch, D. Monti, A. Boschin, B. Steen, P. Johansson and M. R. Palacín, J. Mater. Chem. A, 2015, 3, 22–42 RSC.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3431–3448 CrossRef CAS PubMed.
- X. Wang, L. Ma and J. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 41297–41303 CrossRef CAS PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- Y. Liu, H. Kang, L. Jiao, C. Chen, K. Cao, Y. Wang and H. Yuan, Nanoscale, 2015, 7, 1325–1332 RSC.
- C. Chen, T. Li, H. Tian, Y. Zou and J. Sun, J. Mater. Chem. A, 2019, 7, 18451–18457 RSC.
- A. P. Cohn, N. Muralidharan, R. Carter, K. Share and C. L. Pint, Nano Lett., 2017, 17, 1296–1301 CrossRef CAS PubMed.
- K. Kretschmer, B. Sun, J. Zhang, X. Xie, H. Liu and G. Wang, Small, 2017, 13, 1603318 CrossRef PubMed.
- B. Xiao, H. Liu, J. Liu, Q. Sun, B. Wang, K. Kaliyappan, Y. Zhao, M. N. Banis, Y. Liu, R. Li, T.-K. Sham, G. A. Botton, M. Cai and X. Sun, Adv. Mater., 2017, 29, 1703764 CrossRef PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- R. Zhang, Y. Cui, W. Fan, G. He and X. Liu, Electrochim. Acta, 2019, 295, 181–186 CrossRef CAS.
- Y. Zhao, L. V. Goncharova, A. Lushington, Q. Sun, H. Yadegari, B. Wang, W. Xiao, R. Li and X. Sun, Adv. Mater., 2017, 29, 1606663 CrossRef PubMed.
- H. Tian, Z. W. Seh, K. Yan, Z. Fu, P. Tang, Y. Lu, R. Zhang, D. Legut, Y. Cui and Q. Zhang, Adv. Energy Mater., 2017, 7, 1602528 CrossRef.
- W. Luo and L. Hu, ACS Cent. Sci., 2015, 1, 420–422 CrossRef CAS PubMed.
- R. D. D. I. Iermakova, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2015, 166, 7060–7066 CrossRef.
- X. Li, L. Zhao, P. Li, Q. Zhang and M. Wang, Nano Energy, 2017, 42, 122–128 CrossRef CAS.
- S. Chi, X. Qi, Y. Hu and L. Fan, Adv. Energy Mater., 2018, 8, 1702764 CrossRef.
- N. Ingersoll, Z. Karimi, D. Patel, R. Underwood and R. Warren, Electrochim. Acta, 2019, 297, 129–136 CrossRef CAS.
- J. Lee, Y. Lee, J. Lee, S. M. Lee, J. H. Choi, H. Kim, M. S. Kwon, K. Kang, K. T. Lee and N. S. Choi, ACS Appl. Mater. Interfaces, 2017, 9, 3723–3732 CrossRef CAS PubMed.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- Q. Liu, J. Xu, S. Yuan, Z. Chang, D. Xu, Y. Yin, L. Li, H. Zhong, Y. Jiang, J. Yan and X. Zhang, Adv. Mater., 2015, 27, 5241–5247 CrossRef CAS PubMed.
- W. Luo, C. Lin, O. Zhao, M. Noked, Y. Zhang, G. W. Rubloff and L. Hu, Adv. Energy Mater., 2017, 7, 1601526 CrossRef.
- Y. Zhao, L. V. Goncharova, Q. Zhang, P. Kaghazchi, Q. Sun, A. Lushington, B. Wang, R. Li and X. Sun, Nano Lett., 2017, 17, 5653–5659 CrossRef CAS PubMed.
- J. Sun, C. Guo, Y. Cai, J. Li, X. Sun, W. Shi, S. Ai, C. Chen and F. Jiang, Electrochim. Acta, 2019, 309, 18–24 CrossRef CAS.
- Y. Lu, Q. Zhang, M. Han and J. Chen, Chem. Commun., 2017, 53, 12910–12913 RSC.
- H. J. Yoon, N. R. Kim, H. J. Jin and Y. S. Yun, Adv. Energy Mater., 2018, 8, 1701261 CrossRef.
- Q. Shi, Y. Zhong, M. Wu, H. Wang and H. Wang, Angew. Chem., 2018, 57, 9069–9072 CrossRef CAS PubMed.
- S. Liu, S. Tang, X. Zhang, A. Wang, Q. Yang and J. Luo, Nano Lett., 2017, 17, 5862–5868 CrossRef CAS PubMed.
- W. Luo, Y. Zhang, S. Xu, J. Dai, E. Hitz, Y. Li, C. Yang, C. Chen, B. Liu and L. Hu, Nano Lett., 2017, 17, 3792–3797 CrossRef CAS PubMed.
- J. Wang, J. Liu, Y. Cai, F. Cheng, Z. Niu and J. Chen, ChemElectroChem, 2018, 5, 1702–1707 CrossRef CAS.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- H. Bian, J. Zhang, M. F. Yuen, W. Kang, Y. Zhan, D. Y. W. Yu, Z. Xu and Y. Y. Li, J. Power Sources, 2016, 307, 634–640 CrossRef CAS.
- S. J. R. Prabakar, J. Jeong and M. Pyo, Electrochim. Acta, 2015, 161, 23–31 CrossRef CAS.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294–1305 RSC.
- C. Wang, A. Wang, L. Ren, X. Guan, D. Wang, A. Dong, C. Zhang, G. Li and J. Luo, Adv. Funct. Mater., 2019, 29, 1905940 CrossRef.
- C. Wei, R. Zhang, X. Zheng, Q. Ru, Q. Chen, C. Cui, G. Li and D. Zhang, Inorg. Chem. Front., 2018, 5, 3126–3134 RSC.
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2016, 16, 572 CrossRef PubMed.
- N. Muralidharan, A. P. Cohn, R. Carter, K. Share and C. L. Pint, Nano Lett., 2017, 17, 1296–1301 CrossRef PubMed.
- D. Lin, Y. Liu, Z. Liang, H. W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626 CrossRef CAS PubMed.
- J. Meng, F. Chu, J. Hu and C. Li, Adv. Funct. Mater., 2019, 29, 1902220 CrossRef.
- X. Q. Zhang, X. Chen, X. B. Cheng, B. Q. Li, X. Shen, C. Yan, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 5301–5305 CrossRef CAS PubMed.
- K. N. Wood, E. Kazyak, A. F. Chadwick, K. H. Chen, J. G. Zhang, K. Thornton and N. P. Dasgupta, ACS Cent. Sci., 2016, 2, 790–801 CrossRef CAS PubMed.
- D. D. Joshua, W. Gallaway, A. Gaikwad, C. Corredor, S. Banerjee and D. Steingart, J. Electrochem. Soc., 2010, 157, 1279–1286 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00658c |
| This journal is © The Royal Society of Chemistry 2019 |