
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of ternary sulfide nanomaterials using dithiocarbamate complexes as single source precursors
Anna
Roffey
 ab,
Nathan
Hollingsworth
b and
Graeme
Hogarth
*a
ab,
Nathan
Hollingsworth
b and
Graeme
Hogarth
*a
aDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London SE1 1DB, UK. E-mail: graeme.Hogarth@kcl.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H OAJ, UK
First published on 18th June 2019
Abstract
We report the use of cheap, readily accessible and easy to handle di-isobutyl-dithiocarbamate complexes, [M(S2CNiBu2)n], as single source precursors (SSPs) to ternary sulfides of iron–nickel, iron–copper and nickel–cobalt. Varying decomposition temperature and precursor concentrations has a significant effect on both the phase and size of the nanomaterials, and in some instances meta-stable phases are accessible. Decomposition of [Fe(S2CNiBu2)3]/[Ni(S2CNiBu2)2] at ca. 210–230 °C affords metastable FeNi2S4 (violarite) nanoparticles, while at higher temperatures the thermodynamic product (Fe,Ni)9S8 (pentlandite) results. Addition of tetra-isobutyl-thiuram disulfide to the decomposition mixture can significantly affect the nature of the product at any particular temperature-concentration, being attributed to suppression of the intramolecular Fe(III) to Fe(II) reduction. Attempts to replicate this simple approach to ternary metal sulfides of iron–indium and iron–zinc were unsuccessful, mixtures of binary metal sulfides resulting. Oleylamine is non-innocent in these transformations, and we propose that SSP decomposition occurs via primary–secondary backbone amide-exchange with primary dithiocarbamate complexes, [M(S2CNHoleyl)n], being the active decomposition precursors.
Introduction
Since the properties of nanomaterials are highly dependent upon their size, shape, composition and architecture, then the development of reproducible and controlled synthetic methodologies presents a major challenge.1 Our interest in nanomaterials focuses on the potential role of iron sulfides in prebiotic chemistry, especially the reduction of CO2 to biologically useful molecules believed to have occurred at the interface between hydrothermal fluids and the primordial ocean.2,3 Thus it has been proposed that iron sulfides found in the chimney cavities of hydrothermal vents4 catalysed CO2 reduction forming a primitive acetyl-CoA pathway similar to that in contemporary enzymes.5–7 Greigite is structurally similar to the Fe4S4 cluster sub-units found in ferredoxins8 and its catalytic nature in CO2 activation has been demonstrated,9,10 while iron sulfides have also been shown to catalyse CO2 reduction.11 Thus in a recent communication9 we showed that nanoparticles of greigite can reduce CO2 under ambient conditions into methanol, formic, acetic and pyruvic acid.The greigite used for these experiments was prepared from the controlled hydrothermal decomposition of the dithiocarbamate complex [Fe(S2CNiBu2)3] (1) in oleylamine.12 Dithiocarbamate complexes13 find extensive use as single source precursors (SSPs) towards a range of nanomaterials14–22 their utility stemming from their ease of synthesis, and the ability to tune the solubility, volatility and decomposition properties upon making simple changes to the amine substituents. A further advantage of the dithiocarbamate approach to SSPs is the realisation that this simple ligand forms stable complexes with all the transition metals.12 Thus in seeking to develop our work on the catalytic properties of metal-sulfide nanomaterials we sought to prepare a range of ternary sulfide nanomaterials, primarily containing iron but also nickel. To do this we have used a range of simple air-stable di-isobutyl-dithiocarbamate complexes (Chart 1) and carried out decomposition studies in oleylamine under a range of conditions. This has successfully led to the synthesis of a number of phases of FeNi, FeCu and NiCo sulfides which are described herein, although attempts to prepare FeZn and FeIn sulfides by this method failed.
 | ||
| Chart 1 | ||
Results and discussion
(i) Iron–nickel sulfides
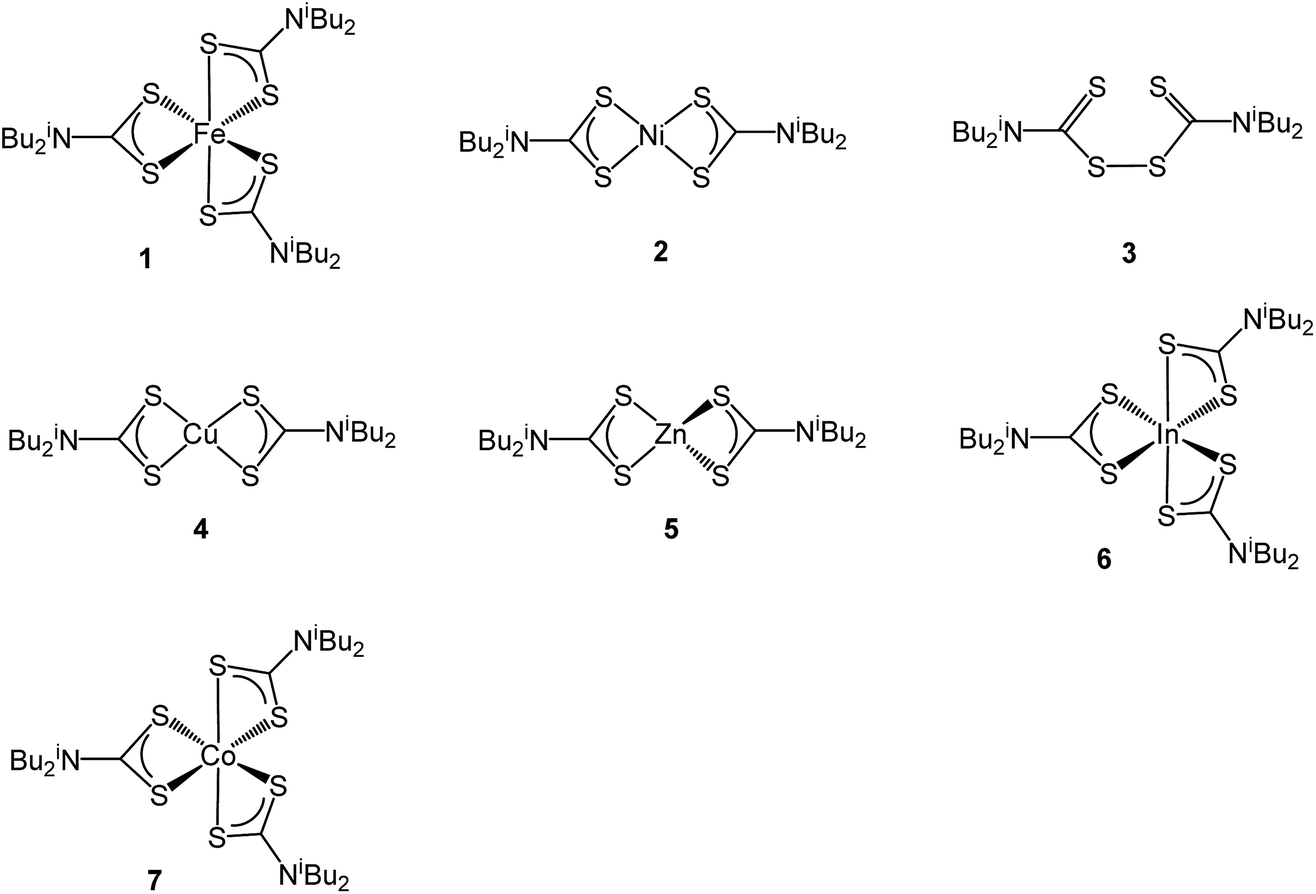
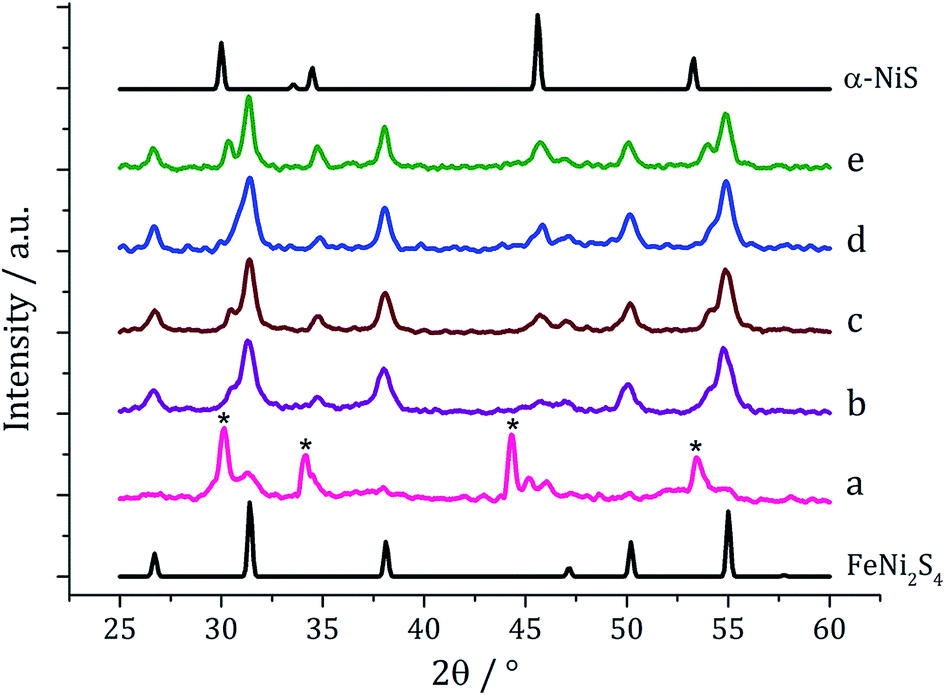
Ternary iron-nickel sulfide materials can take a number of phases,23 most commonly (Fe,Ni)9S8 (pentlandite, pentlandite structure) and FeNi2S3 (violarite, inverse thiospinel structure), although a pyrite type structure (Fe,Ni)S2 (bravoite) has also been reported.24 There are few examples of the synthesis of nanomaterials of these phases. Bezverkhyy and co-workers synthesised pentlandite nanoparticles via a multistep process involving pyrolysis in a H2S/N2 atmosphere of a poorly-defined precursor synthesised from Fe(SO4)·7H2O and Ni(SO4)·7H2O25 and Trávníček reported that a nanoparticulate residue of Fe5Ni4S8 resulted when [Ni(S2CNBziPr)3][FeCl4] was decomposed at 800 °C.26 As far as we are aware, there are no reports of either violarite or bravoite being synthesised in the nanoparticle regime.In our first attempt at preparing ternary iron-nickel sulfide nanomaterials we used [Fe(S2CNiBu2)3] (1) and [Ni(S2CNiBu2)2] (2) as SSPs; decomposition of 2.5 mM quantities of each was carried out in oleylamine at 150, 180, 230, 260 and 280 °C. Upon warming the oleylamine solution a number of temperature-dependent colour changes were observed, being similar to those previously reported in the decomposition of 1 alone, but occurring at ca. 10 °C higher.12 Thus the initially dark brown solution become suddenly clear and pale yellow at 85 °C, then at 90 °C rapidly went black. Samples were heated to the target temperature and held there for 1 h after which the generated nanoparticles were isolated as black powders and analysed by PXRD (Fig. 1). At 150 °C the material generated is mostly amorphous, but by 180 °C peaks for FeNi2S4 begin to emerge and at ca. 230 °C a mixture of FeNi2S4, Fe7S8 and α-NiS is seen. Increasing the temperature to 260 °C sees the emergence of peaks attributed to (Fe,Ni)9S8, and at 280 °C the crystalline phase of the sample appears to be comprised of pure (Fe,Ni)9S8. This suggests that FeNi2S4 (violarite) is metastable, with (Fe,Ni)9S8 (pentlandite) being thermodynamically favourable.
 | ||
| Fig. 1 PXRD patterns for samples prepared from 1 with 2 at (a) 150 °C, (b) 180 °C, (c) 230 °C, (d) 260 °C and (e) 280 °C, with reference patterns for FeNi2S4 (ICDD card no. 47-1740) and (Fe,Ni)9S8 (ICDD card no. 75-2024). ‘*’ indicates Fe7S8 peaks and ‘+’ indicates α-NiS peaks. | ||
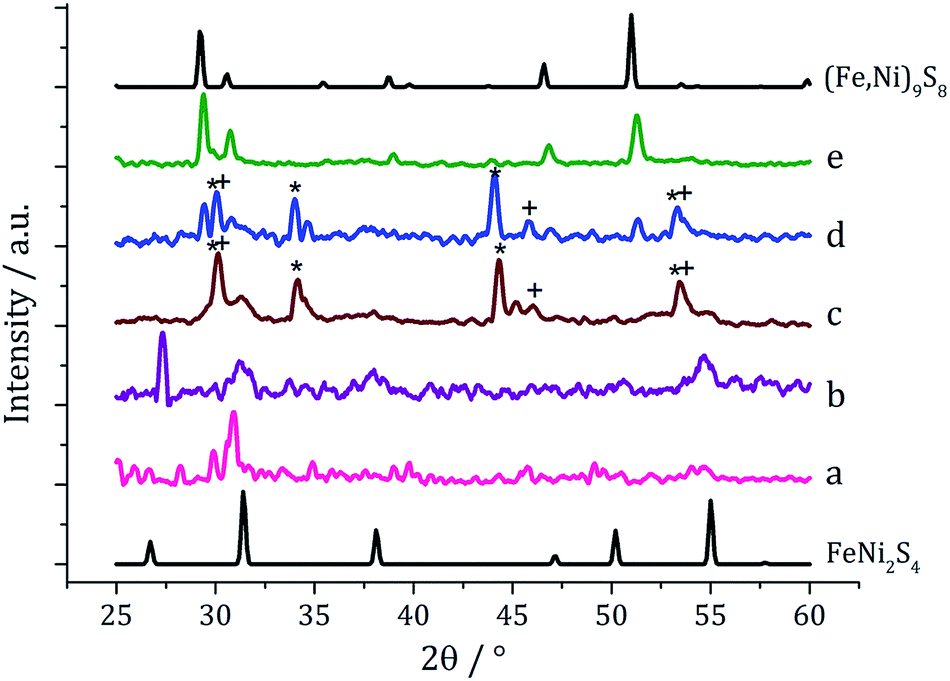
The pure (Fe,Ni)9S8 produced at 280 °C was analysed by TEM and HRTEM being comprised of roughly hexagonal nanoparticles of 53 nm (SD 18 nm) average diameter (Fig. 2). These are significantly smaller than nanoparticulate pentlandite prepared by Bezverkhyy (Av. 240 nm).25 HRTEM analysis reveals a d-spacing of 3.55 Å corresponding to the [220] lattice plane (3.57 Å).
 | ||
| Fig. 2 Particle size histogram (left) and TEM image with HRTEM insert (right) of the (Fe,Ni)9S8 sample prepared from 1 with 2 at 280 °C. | ||
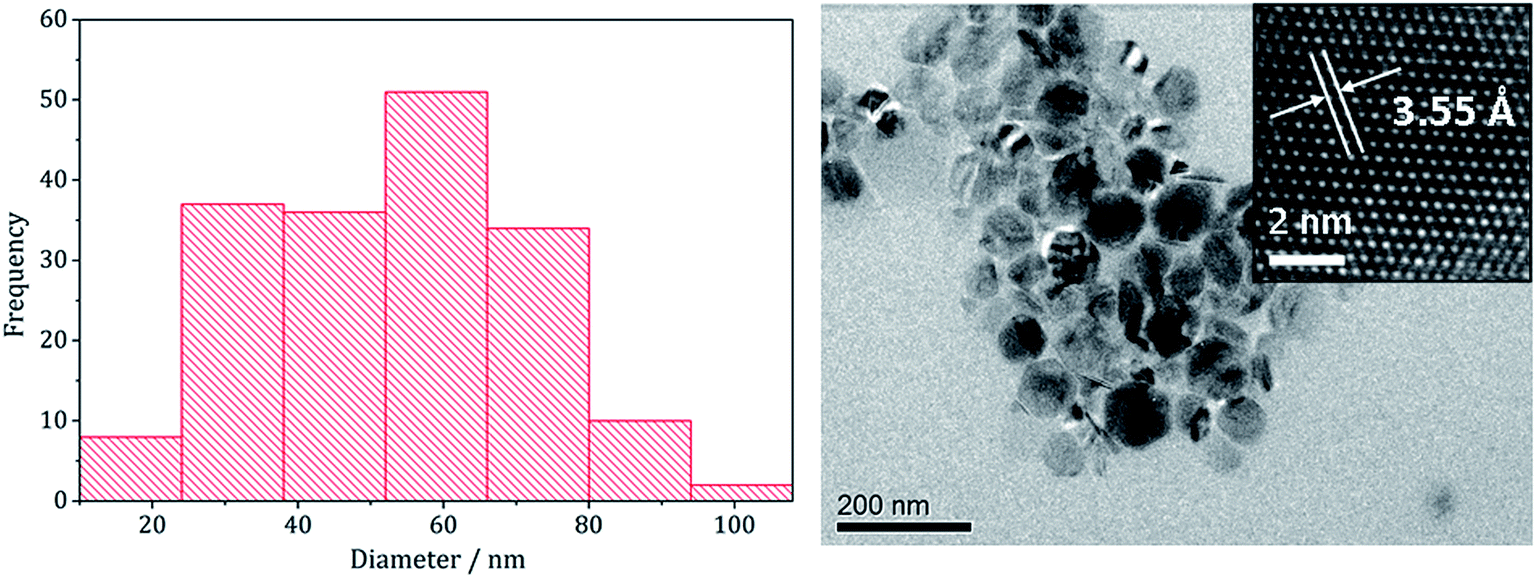
In recent work we found that decomposition of 1 and 2 alone were significantly influenced by addition of tetra-isobutylthiuram disulfide (3) leading to the increased stability of metastable phases and allowing access to the thiospinel phases Fe3S4 and Ni3S4.22 For 1 this was related to an intramolecular ligand–metal electron transfer leading to reduction to an Fe(II) product and extrusion of 3, the addition of which moves the equilibrium towards the Fe(III) species.12 We thus investigated how the addition of 3 would affect the decomposition of mixtures of 1 and 2. Consequently all reactions were repeated with addition of ca. 4 equivalents of 3 (i.e. 10 mM ca. 2 eq. to each precursor). PXRD analysis revealed a similar trend to the samples prepared in the absence of thiuram disulfide but with some notable differences (Fig. 3). Thus, while spectra are similar at low temperatures, at 230 °C seemingly pure FeNi2S4 is formed, that is without Fe7S8 and α-NiS impurities seen in the absence of 3. This indicates that addition of 3 stabilises the thiospinel, FeNi2S4, phase possibly preventing formation of binary metal sulfides. At 260 °C the peaks attributed to FeNi2S4 broaden and those of Fe7S8 and α-NiS begin to appear, while at 280 °C the sample is mainly Fe7S8 and α-NiS, with some FeNi2S4 remaining.
 | ||
| Fig. 3 PXRD patterns for samples prepared from mixtures of 1, 2 and 3 at (a) 150 °C, (b) 180 °C, (c) 230 °C, (d) 260 °C, (e) 280 °C, and (f) 300 °C, with reference patterns for FeNi2S4 (ICDD card no. 47-1740) and (Fe,Ni)9S8 (ICDD card no. 75-2024). ‘*’ indicates Fe7S8 peaks and ‘+’ indicates α-NiS peaks. | ||
To probe whether the thermodynamically stable phase, (Fe,Ni)9S8, could be accessed in the presence of 3 but at higher temperatures, a decomposition was performed at 300 °C with addition of only 2 eq. of 3. PXRD of the resulting material showed the crystalline phase to be a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of Fe7S8 and α-NiS, with neither FeNi2S4 or (Fe,Ni)9S8 being present. This suggests addition of 3 prevents formation of (Fe,Ni)9S8, making binary metal sulfide phases more thermodynamically favourable.
:1 mix of Fe7S8 and α-NiS, with neither FeNi2S4 or (Fe,Ni)9S8 being present. This suggests addition of 3 prevents formation of (Fe,Ni)9S8, making binary metal sulfide phases more thermodynamically favourable.
Analysing the FeNi2S4 particles produced at 230 °C by TEM (Fig. 4) showed them to be short rods embedded in hexagonal interlocking sheets with an average diameter of 36.5 nm (SD 21 nm), being slightly smaller than (Fe,Ni)9S4 nanoparticles formed in the absence of 3 (average diameter 53.1 nm). HRTEM analysis of the nanoparticles reveals d-spacings of 3.29 Å, corresponding to the [220] lattice plane in FeNi2S4 (3.35 Å).
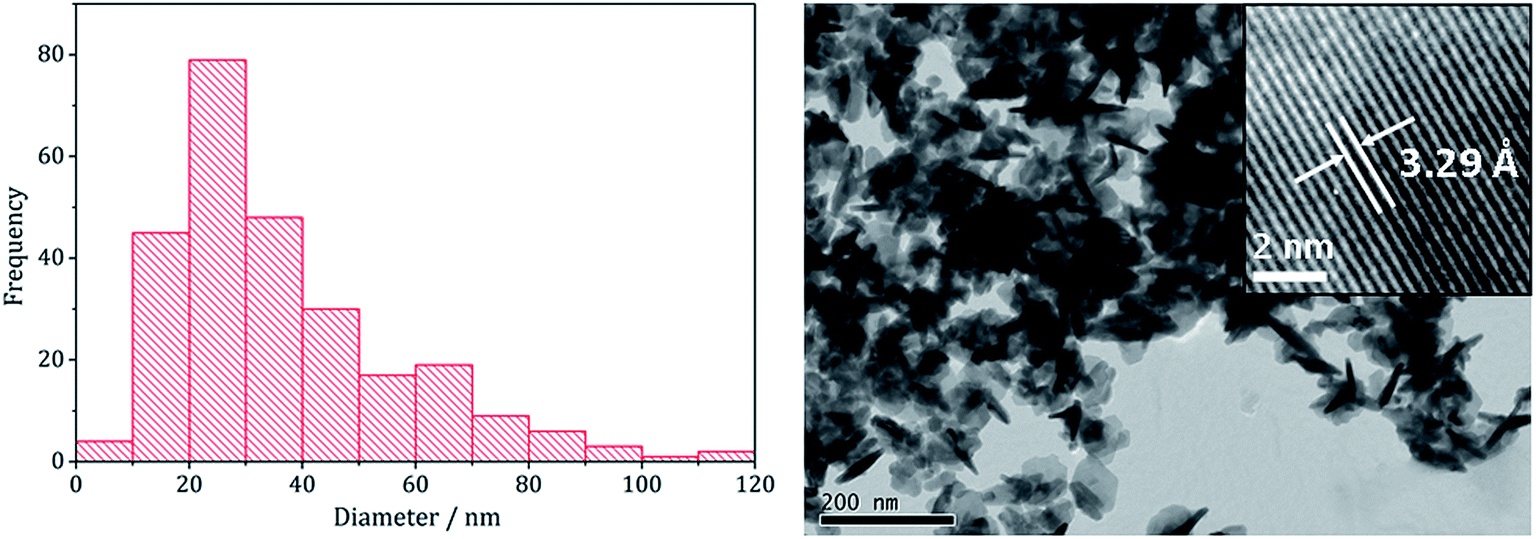 | ||
| Fig. 4 Particle size histogram (left) and TEM image with HRTEM insert (right) of the FeNi2S4 sample prepared from mixtures of 1–3 at 230 °C. | ||
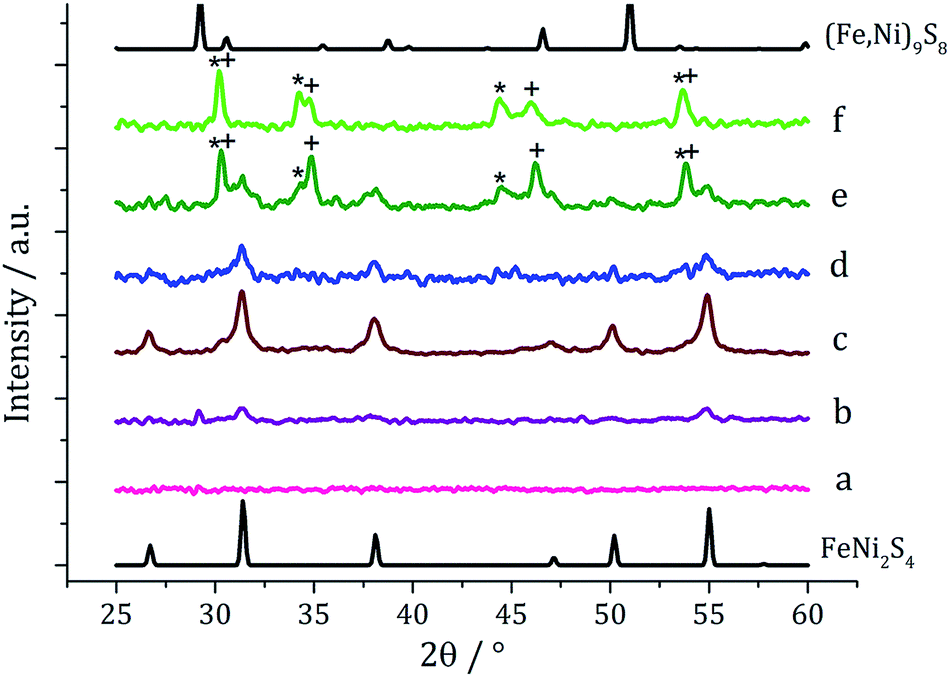
We next varied precursor concentrations, equimolar amounts of 1 and 2 at 2.5, 5, 10, 20 and 25 mM were decomposed in oleylamine at 230 °C. PXRD analysis reveals a trend in the phase of metal sulfide produced with increasing concentration of precursors (Fig. 5). At 2.5 mM the majority of the crystalline sample corresponds to Fe7S8, with some FeNi2S4 and α-NiS, while at higher concentrations the crystalline phase is predominantly the thiospinel FeNi2S4, with some α-NiS impurity. This is consistent with the concentration study performed on 112 where at low precursor concentrations (5 mM) Fe7S8 was formed, and as the concentration was increased (up to 50 mM) the phase changed to the thiospinel, Fe3S4. When a similar study was performed on 2, changing concentration had no effect on nickel sulfide phase; α-NiS being formed at all concentrations.
 | ||
| Fig. 5 PXRD patterns for samples prepared from 1–2 at concentrations of (a) 2.5 mM, (b) 5 mM, (c) 10 mM, (d) 20 mM and (e) 25 mM, with reference patterns for FeNi2S4 (ICDD card no. 47-1740) and α-NiS (ICDD card no. 02-1273). ‘*’ indicates peaks for Fe7S8. | ||
Experiments were repeated upon addition of four equivalents of 3 and PXRD analysis again shows a concentration-dependent phase change (Fig. 6). At low concentrations pure violarite is formed, but upon increasing the majority of the crystalline material matches the pattern for (Fe,Ni)S2 (bravoite, a nickelian pyrite) and for the 1:2:3 = 20:20:80 mM sample the pattern matches pure (Fe,Ni)S2. Peaks are, broad suggesting that the nanoparticles are small, being consistent with the concentration study on the nickel complex 2 in the presence of 3.22 In the ternary system, when the concentration is increased further, additional peaks at slightly higher 2θ angles appear, indicative of the presence of FeS2 (pyrite). This is interesting since in the binary iron sulfide system, pyrite was not accessible at high concentrations, even after addition of 3. It should also be noted that at higher concentrations, low angle (10–20°) broad peaks are observed, possibly resulting from excess sulfur.
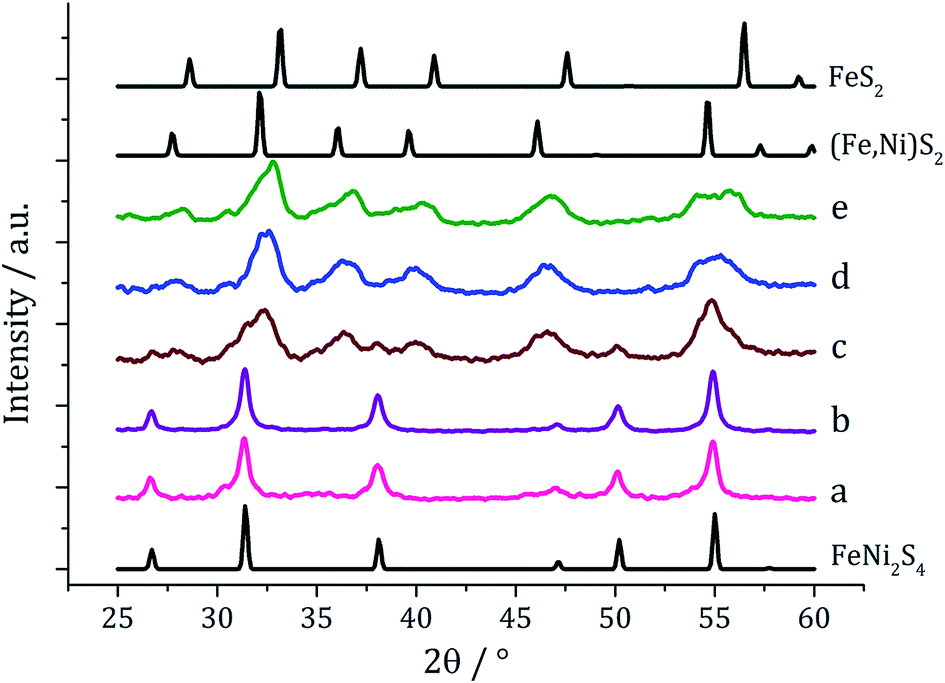 | ||
| Fig. 6 PXRD patterns for samples prepared from mixtures of 1–3 (1:1:2) at concentrations of 1–2 of (a) 2.5 mM, (b) 5 mM, (c) 10 mM, (d) 20 mM and (e) 25 mM, with reference patterns for FeNi2S4 (ICDD card no. 47-1740), (Fe,Ni)S2 (ICDD card no. 88-1710) and FeS2 (ICDD card no. 42-1340). | ||
Nanoparticles produced at 20 mM 1–2 were analysed by TEM, being approximately spherical and with small crystallite size (average diameter 6.7 nm, SD = 1.9 nm), as indicated by the broad peaks in the PXRD pattern. They closely resemble NiS2 nanoparticles formed at high concentration of 1 decomposed with 3 (1:3 = 20:40 mM) which had an average diameter of 4.2 nm (SD = 1.3 nm).22 HRTEM analysis (Fig. 7) of the (Fe,Ni)S2 nanoparticles reveals d-spacings of 3.10 and 2.45 Å, which match the [111] and [200] planes of (Fe,Ni)S2 (3.21 and 2.49 Å respectively).
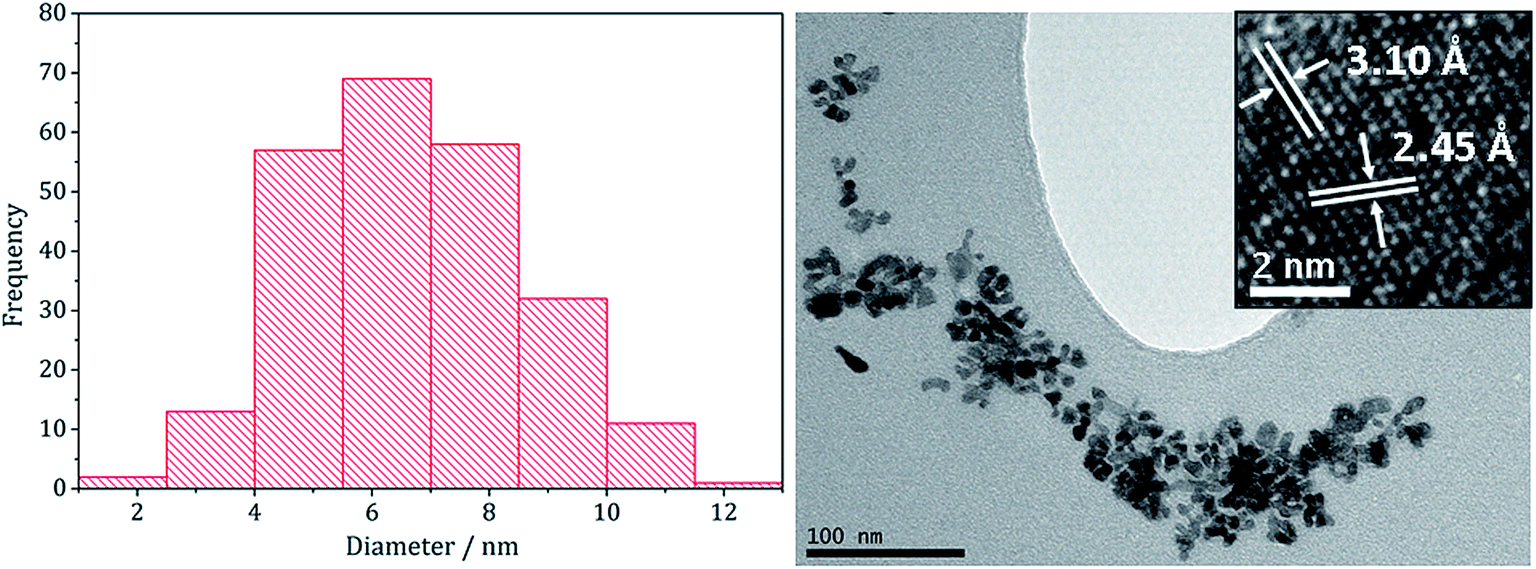 | ||
| Fig. 7 Particle size histogram (left) and TEM image with HRTEM insert (right) of the (Fe,Ni)S2 sample prepared from 1–3 at concentrations 20:20:80 mM. | ||
In summary, products of the mixed iron and nickel ternary sulfide system were found to be highly dependent upon both the decomposition temperature and precursor concentration, with similarities to both the iron sulfide and nickel sulfide binary systems. The pentlandite phase (Fe,Ni)9S8, thiospinel phase FeNi2S4 and pyrite phase (Fe,Ni)S2 could be selectively produced by varying reaction temperature, precursor concentration and employing the tetra-isobutylthiuram disulfide additive 3. We believe that this is the first reported synthesis of the latter two phases in nanoparticulate form.
(ii) Iron–copper sulfides
Copper–iron sulfide has many known phases including Cu5FeS4 (bornite), CuFeS2 (chalcopyrite) and CuFe2S3 (cubanite)27 and there are numerous reports on their synthesis, although only a few involving the decomposition of dithiocarbamate complexes. Thus, methods to synthesise nanoparticulate copper–iron sulfides include; solvothermal and microwave-assisted decomposition of metal salts with sulfur sources, colloidal synthesis and laser ablation of bulk CuFeS2.28 Two reports have previously detailed the use of dithiocarbamate SSPs for the synthesis of copper–iron sulfide nanoparticles. Gupta utilised a ‘hot-injection’ method to prepare CuFeS2 nanoparticles of ca. 12 nm diameter, injecting a mixture of [Cu(S2CNEt2)2] and [Fe(S2CNEt2)3] in oleylamine/dichlorobenzene into a solution of sulfur in oleylamine/trioctylphosphine at 180 °C,29 while Pang and co-workers synthesised CuFeS2 nanoparticles of ca. 6 nm (ref. 30) upon injecting solution of NaS2CNEt2 in dodecanethiol into a hot (140 °C) solution of FeCl3 and CuCl2 oleic acid/dodecanethiol; dithiocarbamate complexes presumably being formed in situ.30Following on from our methodology described above, we decomposed equimolar amounts (2.5 mM) of brown [Cu(S2CNiBu2)2] (4) and [Fe(S2CNiBu2)3] (1) in oleylamine at 230 °C. The reaction was then repeated with the addition 4 eq. of thiuram disulfide 3, and the resulting brown powders were analysed. No discernible colour changes occurred during these reactions, clear purple dispersions resulting in chlorinated solvents. PXRD analysis (Fig. 8) showed that the crystalline phase produced in both cases corresponded to CuFeS2 (chalcopyrite). This is in accordance with previous literature.29,30 TEM analysis revealed that addition of 3 had little effect on the size or morphology of the nanoparticles, which were roughly spherical (Fig. 9). Average particle diameters were 11 nm (SD = 4 nm) without 3, and 10 nm (SD = 4 nm) with 3. HRTEM analysis (Fig. 9) revealed a d-spacing of 2.99 Å, which matches the [112] plane (3.21 Å, CuFeS2).
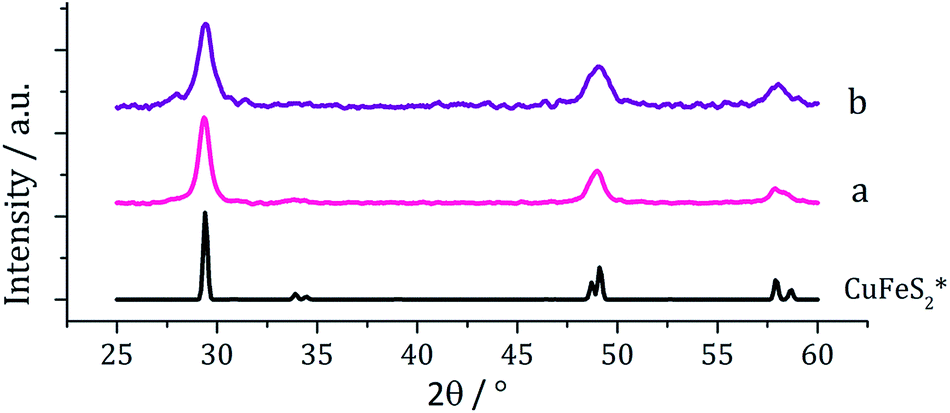 | ||
| Fig. 8 PXRD patterns for samples prepared from (a) 1 and 4 and (b) 1 and 4 with added 3, with reference pattern for bulk CuFeS2 (ICDD card no. 37-0471). | ||
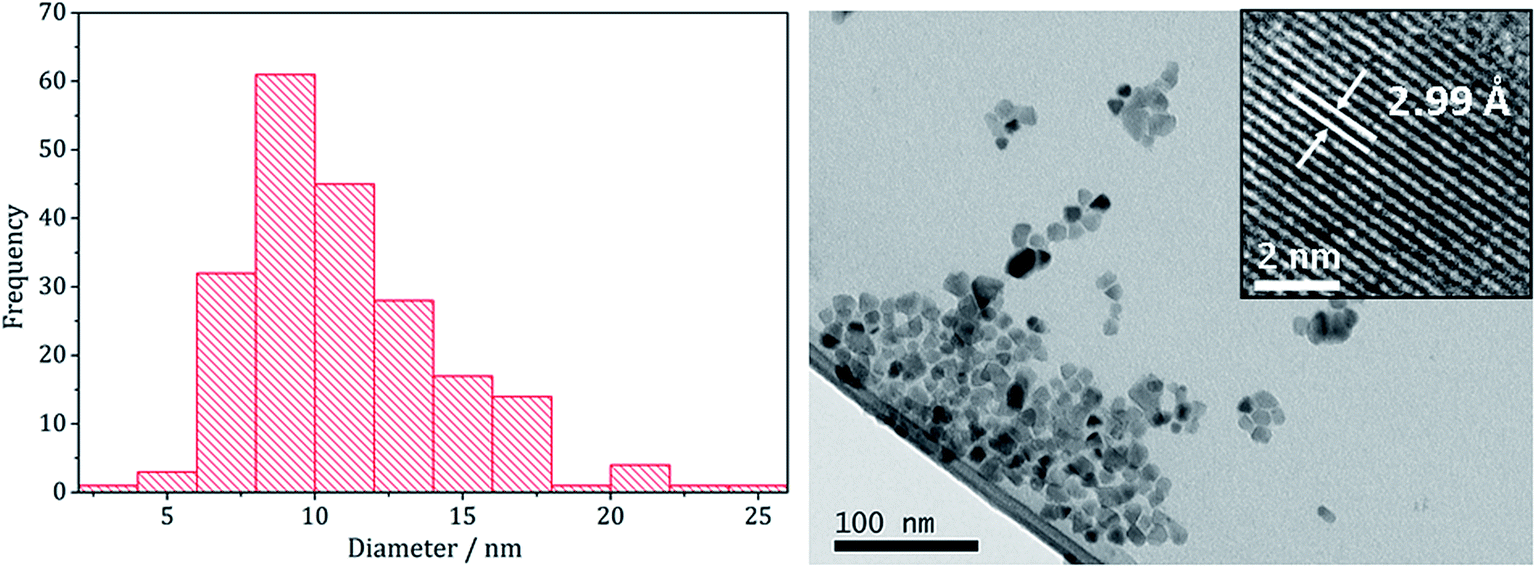 | ||
| Fig. 9 Particle size histogram (left) and TEM image with HRTEM insert (right) of the sample prepared from 1 and 4. | ||
These results are in accord with those of Gupta and co-workers29 who generated similar nanomaterials at 180 °C. Even upon increasing the temperature by 50 °C we saw no significant change in the nanoparticle size suggesting that during nanoparticle formation the duration of burst nucleation and the number of nucleation sites is not affected by temperature.
(iii) Attempts to prepare iron–zinc sulfides
There are several known phases of the ternary sulfide of iron and zinc; (Zn,Fe)S wurtzite ({ZnFe}S(W), an iron doped wurtzite), (Zn,Fe)S marmatite ({ZnFe}S(S), an iron rich sphalerite) and ZnFe2S4, a thiospinel31 but few reports on nanoparticulate synthesis and none involving a dithiocarbamate SSP. Feng and co-workers prepared thin films of (Zn,Fe)S(W) by MOCVD at 350 °C from [Fe(CO)5], Me2Zn and H2S32 and the same material was synthesised in nanoparticulate form upon solvothermal decomposition of [Zn(Aftscz)2] and [Zn(AftsczH)2Cl2] (AftsczH = monoacetylferrocene thiosemicarbazole) in ethylene glycol.33 Nanoparticles of (Zn,Fe)S(S) have also been synthesised; Chen et al. prepared Zn0.9Fe0.1S upon annealing nanoparticles produced from a micro-emulsion of [Fe(CO)5], [Zn(O2CCH3)2] and CH3(CH2)11OSO3Na (sodium dodecyl sulphate) in water with added thioacetamide.34We used equimolar amounts (2.5 mM) of [Zn(S2CNiBu2)2] (5) and [Fe(S2CNiBu2)3] (1) in oleylamine at 230 °C in the absence and presence of 4 eq. of thiuram disulfide 3. As with previous decompositions involving 1, dark brown solutions suddenly became clear and pale yellow at ca. 85 °C, and at 100 °C quickly turned black. Dark brown powders were isolated but PXRD analysis revealed that the crystalline phase corresponded to a mixture of iron sulfide and zinc sulfide phases (Fig. 10). Interestingly, in both the zinc sulfide phase matched to the pattern for the sphalerite rather than the wurtzite phase. Notably the characteristic peak for ZnS(W) at 39.8° 2θ is absent. Peaks match well for ZnS(S), and are not shifted to lower angles, as previously observed for mixed iron and zinc sulfide systems32 suggesting that no ternary mixed-metal sulfide has been formed. The reason for this is unclear, but possibly the precursors decompose at quite different temperatures, giving rise to separate nucleation events and forming binary metal sulfide nanoparticles. The sample prepared in the absence of 3 appears to also have peaks for the iron sulfide Fe7S8 and the sample prepared with 3 contains peaks which match well to Fe3S4, the thiospinel phase of iron sulfide. This is consistent with previous results reported on the iron sulfide binary phase system.
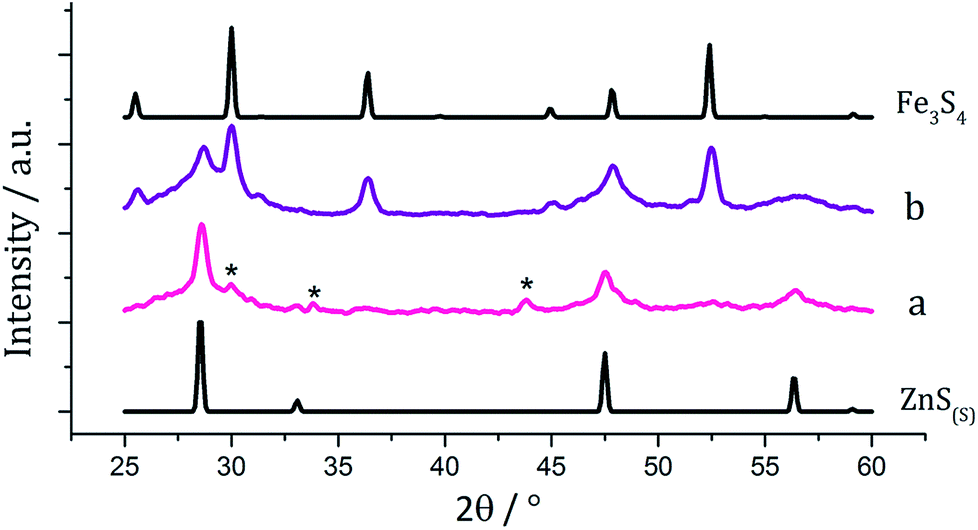 | ||
| Fig. 10 PXRD patterns for samples prepared from (a) 1 with 5 and (b) 1 with 5 and 3, with reference patterns for bulk ZnS(S) (ICDD card no. 77-2100) and Fe3S4 (ICDD card no. 16-0713). ‘*’ indicates peaks for Fe7S8. | ||
TEM images support the PXRD analysis, clearly showing a two phase system (Fig. 11). Samples comprised large hexagonal particles (average diameter 32 nm, SD 16 nm) which resemble the iron sulfide phases observed in the binary system, and thin nanowires (length = 16 nm, SD 15 nm, width = 2.9 nm, SD 0.8 nm) resembling zinc sulfide material produced in the binary system.
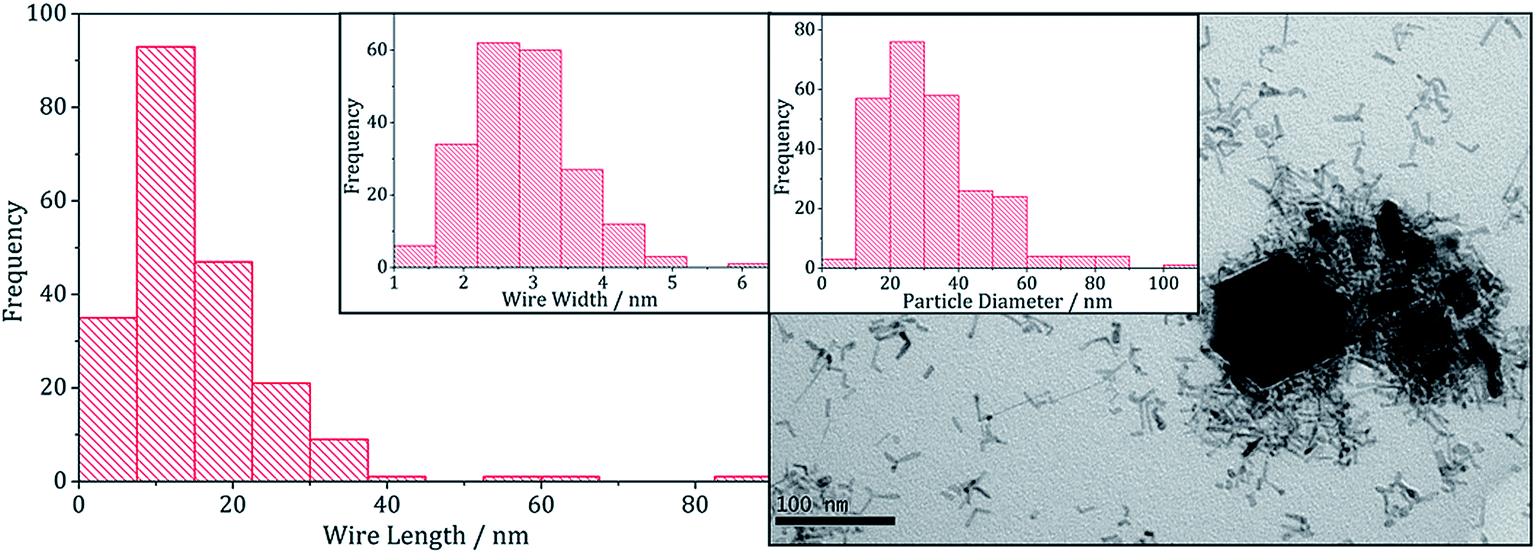 | ||
| Fig. 11 Wire-length histogram with wire-width histogram insert (left) and TEM image with particle diameter histogram insert (right), of the sample prepared from 1 with 5. | ||
(iv) Attempts to prepare iron–indium sulfides
There have been few reported syntheses of nanoparticulate FeIn2S4, and none using solvothermal methods. Related to this work, Wu et al. recently synthesised FeIn2S4 microspheres using an indium dithiocarbamate precursor.35 Equimolar amounts (2.5 mM) of pale yellow [In(S2CNiBu2)3] (6) and dark brown [Fe(S2CNiBu2)3] (3) were decomposed in oleylamine, being repeated upon addition of 3, and the resulting brown powders were analysed. PXRD patterns (Fig. 12) did not match that of the mixed iron indium sulfide, FeIn2S4, being broad and indicative of low crystallinity. Peaks for Fe3S4 were detectable in both, as well as for β-In2S3 in the sample prepared with added 3. As with the iron-zinc sulfide chemistry ternary mixed-metal sulfides were absent. Interestingly, unlike the iron zinc sulfide samples, peaks for pyrrhotite are not observed in the sample prepared without 3, but peaks matching Fe3S4 are seen in both. This might suggest that the presence of the indium precursor 6 or indium sulfide product β-In2S3 helps to stabilise the iron sulfide thiospinel. | ||
| Fig. 12 PXRD patterns for samples prepared from (a) 1 with 6, (b) 1 with 6 and 3, with reference patterns for β-In2S3 (ICDD card no. 25-0390) and Fe3S4 (ICDD card no. 16-0713). | ||
(v) Nickel–cobalt sulfides
Although the focus of our work with ternary chalcogenides is on mixed-metal iron sulfides, many other combinations of mixed-metal sulfide materials are possible. The nickel–cobalt sulfide system has several possible phases including; NiCo8S8 (pentlandite structure), NiCo2S4 and CoNi2Co4 (both thiospinel structure). Nanoparticles of NiCo8S8 have been synthesised by Bezverkhyy et al. in a two-step process; firstly Co(NO3)2 and Ni(NO3)2 were reacted with Na2S in water and then the dried precipitate was heated with H2S/H2 for 3 h at 300 °C.25 Urchin-like nanostructures of NiCo2S4 were synthesised by Jiang and co-workers in a two-step process involving the hydrothermal decomposition of NiCl2 and CoCl2 with urea in an autoclave at 140 °C, followed by a further reaction with Na2S at 160 °C (ref. 36) and nanoparticles of CoNi2S4 were formed in an autoclave by Pang et al., from the slow (24 h) decomposition of [Co(O2CMe)2] and [Ni(O2CMe)2] mixtures with sulfur dissolved in an oleylamine/anisole mixture.37Equimolar amounts (2.5 mM) of [Ni(S2CNiBu2)2] (2) and [Co(S2CNiBu2)3] (7) were decomposed in oleylamine at 230 °C, being repeated upon addition of thiuram disulfide 3, and the resulting brown powders analysed. PXRD analysis (Fig. 13) revealed no significant shift in peaks between the two samples, indicating that 3 has no observable effect on the phase of ternary sulfide produced. In addition, to elucidate whether the nickel or cobalt rich thiospinel structures had been formed (CoNi2S4 or NiCo2S4 respectively) both samples were analysed by energy-dispersive X-ray spectroscopy (EDX) and found to have atomic compositions of cobalt, nickel and sulfur of 1.76:1.60:4 and 1.52:1.32:4, respectively. Although the ratios of metal to sulfur vary slightly, they remain approximately 1.1:1 Co:Ni, indicating an intermediate (Ni,Co)3S4 phase. The difference between the PXRD patterns for CoNi2S4 and NiCo2S4 is small; nickel and cobalt having very similar ionic radii and electronegativity, and the patterns obtained for the samples synthesised here closely match both.
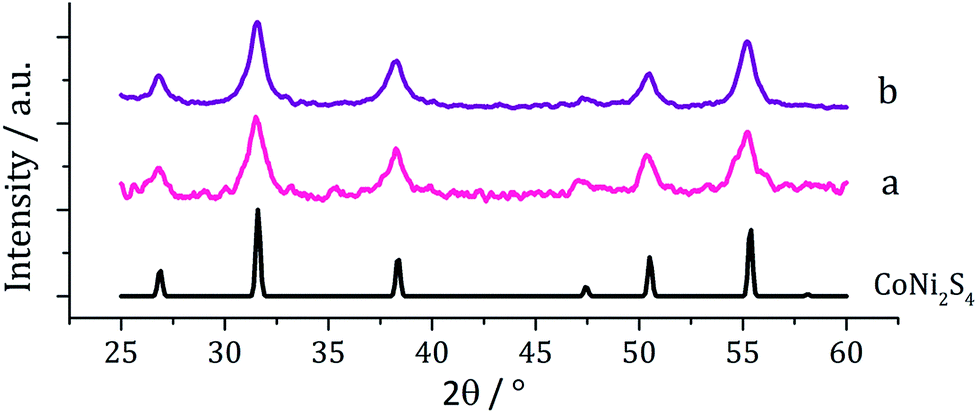 | ||
| Fig. 13 PXRD patterns for samples prepared from (a) 2 with 7 and (b) 2 with 7 and 3, with reference pattern for bulk CoNi2S4 (ICDD card no. 22-0334). | ||
TEM analysis revealed the addition 3 also had little effect on the size or morphology of the nanoparticles formed, which were roughly spherical (Fig. 14). The average particle diameters were 10 nm (SD = 5 nm) for the sample prepared without 3, and 10 nm (SD = 4 nm) with 3. Nanoparticles of CoNi2S4 previously reported by Pang et al.37 were reported to have ‘quasi-spherical’ morphology with an average diameter of approximately 8–15 nm, similar to those produced in the current study. HRTEM analysis (Fig. 13) of the current (Ni,Co)3S4 nanoparticles reveals a d-spacing of 2.67 Å, which matches the [222] plane (2.72 Å, CoNi2S4).
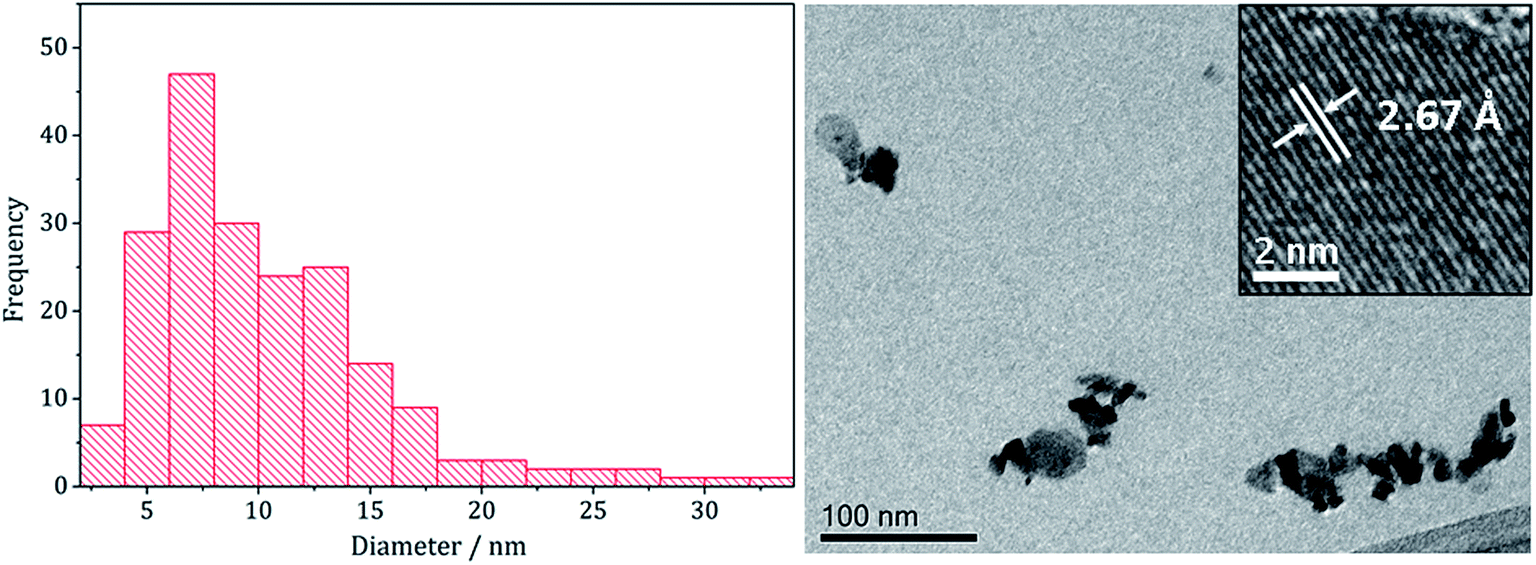 | ||
| Fig. 14 Particle size histogram (left) and TEM image with HRTEM insert (right) of the (Ni,Co)3S4 sample prepared from 2 with 7 and 3. | ||
Summary and conclusions
Nanomaterials of iron–nickel sulfide, iron–copper sulfide and nickel–cobalt sulfide were successfully prepared via the decomposition of mixtures of simple metal–dithiocarbamate complexes in oleylamine, and for iron–nickel mixtures, various sulfide phases were accessible upon varying decomposition conditions. In contrast, attempts to synthesise iron–zinc sulfide and iron–indium sulfide via this method were unsuccessful, producing only binary metal sulfides. This may be due to the precursor complexes decomposing at different temperatures, although this has not been fully established. We have previously shown that [Fe(S2CNiBu2)3] (1) undergoes an intramolecular electron-transfer process at ca. 80 °C in oleylamine to generate [Fe(S2CNiBu2)2] and thiuram disulfide (S2CNiBu2)2 (3).12 This also occurs during decompositions carried out here as shown by the sudden colour change from brown to yellow at ca. 80–90 °C. Thus the decomposition mixtures contain an Fe(II) and not an Fe(III) dithiocarbamate complex. Consistent with this supposition, addition of 3 to the decomposition mixture has a significant effect on the precise nature of the nanomaterials formed. We have also previously shown that decomposition of [Ni(S2CNiBu2)2] (2) in oleylamine occurs via an initial amide-exchange process to generate the primary amine derivative [Ni(S2CNHoleyl)2]21,22 and we have evidence that [Fe(S2CNiBu2)2] behaves in the same way.12 Thus, while it appears that iron–nickel sulfides result from decomposition of [Fe(S2CNiBu2)3] (1) and [Ni(S2CNiBu2)2] (2), most likely it is [Ni(S2CNHoleyl)2] and [Fe(S2CNHoleyl)2] that are actually decomposing via extrusion of oleylNCS. We have less information about the precise nature of 4–7 in oleylamine but cannot rule out similar amide-exchange reactions being important. Hence, a general comment about the decomposition of dithiocarbamate SSPs in oleylamine (and other amines) is that, while changes to the alkyl-substituents may appear to lead to different nanomaterial sizes and shapes, this is most likely related to the rate of the amide-exchange reaction rather than the relative strengths of the C–S bonds that are cleaved. This may account for the failed attempts to prepare iron–zinc and iron–indium sulfides, in which we obtained only mixtures of binary sulfides. Thus if amide-exchange (and subsequent decomposition) is much faster for [Fe(S2CNiBu2)2] than either [Zn(S2CNiBu2)2] or [In(S2CNiBu2)3] then it is easy to see how iron–sulfides would be generated first followed by a slower process to afford zinc or indium sulfide. Consequently we are focusing our current efforts on the synthesis (and subsequent decomposition) of primary-amine dithiocarbamate derivatives, [M(S2CNHR)n], since this should allow for far better control over the decomposition process (e.g. lower temperatures, non-amine solvents etc.) potentially providing access to a wider range of binary and ternary metal sulfide nanomaterials.Experimental section
General procedures
Unless otherwise stated, manipulations were performed under a dry, oxygen-free dinitrogen atmosphere using standard Schlenk techniques or in a MBRAUN Unilab glovebox. All solvents used were stored in alumina columns and dried with anhydrous engineering equipment, such that the water concentration was 5–10 ppm. All other reagents were procured commercially from Aldrich and used without further purification.Physical measurements
1H and 13C{1H} NMR spectra were obtained on either a Bruker Avance III 400 or Avance 600 spectrometers. All spectra were recorded using CDCl3 which was dried and degassed over molecular sieves prior to use; 1H and 13C{1H} chemical shifts are reported relative to SiMe4. Mass spectra were obtained using either Micromass 70-SE spectrometer using Electron Ionisation (EI) or a Thermo Finnigan MAT900xp spectrometer using Fast Atom Bombardment (FAB) ionisation. Elemental analysis was carried using Elemental Analyser (CE-440) (Exeter Analytical Inc). PXRD were measured on a Bruker AXS D4 diffractometer using CuKα1 radiation, patterns obtained being compared to database standards. For TEM characterisation a 4 μl droplet of nanoparticle suspension (CHCl3) was placed on a holey carbon-coated copper TEM grid and allowed to evaporate in air under ambient laboratory conditions for several minutes. TEM images were obtained using a JEOL-1010 microscope at 100 kV equipped with a Gatan digital camera. HRTEM measurements were collected using a Jeol 2100 (high resolution) TEM with a LaB6 source operating at an acceleration voltage of 200 kv. Micrographs were taken on a Gatan Orius Charge-coupled device (CCD).Synthesis and characterisation of molecular precursors
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C], 992 (s), 1244 (s) [CS], 1145 (s) [C2N].
C], 981 (s) [CS].
C], 992 (s), 1244 (s) [CS], 1145 (s) [C2N].
C], 981 (s) [CS].
Decomposition studies
In a typical synthesis the dithiocarbamate complexes (2.5 mM) were added to oleylamine (20 mL) in a three-neck round bottom flask attached to a water condenser and evacuated and refilled with nitrogen repeatedly for ca. 15 minutes. The solution was heated to 230 °C for 1 h. The mixture was slowly cooled to room temperature, whereupon methanol (80 mL) was added with stirring. The mixture was centrifuged and the solution decanted leaving behind the resultant nanoparticles. This procedure was repeated three times and then the material was dried under vacuum.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the EPSRC for funding; PhD studentship to AR and postdoctoral fellowship to NH.References
- See for example; (a) A. K Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef PubMed; (b) A.-H. Lu, E. L. Salabas and F. Schueth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed; (c) T. Trinade, P. O'Brien and N. L. Pickett, Chem. Mater., 2001, 13, 3843 CrossRef; (d) T. Hyeon, Chem. Commun., 2003, 927 RSC; (e) A. L. Rogach, D. V. Talapin, E. V. Shevchenko, A. Kornowski, M. Haase and H. Weller, Adv. Funct. Mater., 2002, 12, 653 CrossRef CAS; (f) M. A. Malik, M. Afzaal and P. O'Brien, Chem. Rev., 2010, 110, 4417 CrossRef CAS PubMed.
- W. Martin and M. J. Russell, Philos. Trans. R. Soc., B, 2003, 358, 59 CrossRef CAS PubMed.
- M. J. Russell and A. J. Hall, J. Geol. Soc., 1997, 154, 377 CrossRef CAS PubMed.
- M. J. Russell, R. M. Daniel, A. J. Hall and J. A. Sherringham, J. Mol. Evol., 1994, 39, 231 CrossRef CAS.
- M. J. Russell and W. Martin, Trends Biochem. Sci., 2004, 29, 358 CrossRef CAS PubMed.
- G. Wachtershauser, Chem. Biodiversity, 2007, 4, 584 CrossRef PubMed.
- N. Lane and W. Martin, Cell, 2012, 151, 1406 CrossRef CAS PubMed.
- U. P. Apfel and W. Weigand, Angew. Chem., Int. Ed., 2011, 50, 4262 CrossRef CAS PubMed.
- A. Roldan, N. Hollingsworth, A. Roffey, H.-U. Islam, J. B. M. Goodall, C. R. A. Catlow, J. A. Darr, W. Bras, G. Sankar, K. B. Holt, G. Hogarth and N. H. de Leeuw, Chem. Commun., 2015, 51, 7501 RSC.
- A. Yamaguchi, M. Yamamoto, K. Takai, T. Ishii, K. Hashimoto and R. Nakamura, Electrochim. Acta, 2014, 141, 311 CrossRef CAS.
- M. G. Vladimirov, Y. F. Ryzhkov, V. A. Alekseev, V. A. Bogdanovskaya, V. A. Otroshchenko and M. S. Kritsky, Orig. Life Evol. Biospheres, 2004, 34, 347 CrossRef CAS.
- A. Roffey, N. Hollingsworth, H.-U. Islam, W. Bras, G. Sankar, N. De Leeuw and G. Hogarth, Nanoscale Adv. Search PubMed , submitted.
- G. Hogarth, Prog. Inorg. Chem., 2005, 53, 71 CrossRef CAS.
- M. Afzaal, M. A. Malik and P. O'Brien, J. Mater. Chem., 2010, 20, 4031 RSC.
- (a) A. Saunders, A. Vecht and G. Tyrell, Chem. Abs., 1988, 108, 66226h Search PubMed; (b) A. Birri, B. Harvey, G. Hogarth, E. Subasi and F. Ugar, J. Organomet. Chem., 2007, 692, 2448 CrossRef CAS; (c) R. Nomura, K. Miyawaki, T. Toyosaki and H. Matsuda, Chem. Vap. Deposition, 1996, 2, 174 CrossRef CAS; (d) M. Kemmler, M. Lazell, P. O'Brien, D. J. Otway, J.-H. Park and J. R. Walsh, J. Mater. Sci.: Mater. Electron., 2002, 13, 531 CrossRef CAS; (e) M. J. Almond, H. Redman and D. A. Rice, J. Mater. Chem., 2000, 10, 2842 RSC; (f) F. Srouji, M. Afzaal, J. Waters and P. O'Brien, Chem. Vap. Deposition, 2005, 11, 91 CrossRef CAS; (g) P. O'Brien, D. J. Otway and J.-H. Park, Mater. Res. Soc. Symp. Proc., 2000, 606, 133 CrossRef; (h) J. M. Soon, L. Y. Goh, K. P. Loh, Y. L. Foo, L. Ming and J. Ding, Appl. Phys. Lett., 2007, 91, 084105 CrossRef; (i) O. C. Monteiro, H. I. S. Nogueira and T. Trindade, Chem. Mater., 2001, 13, 2103 CrossRef CAS; (j) P. O'Brien, D. J. Otway and J. R. Walsh, Thin Solid Films, 1998, 315, 57 CrossRef.
- (a) B. L. Druz, Y. N. Eutukhov and M. Y. Rakhlin, Organomet. Chem., 1988, 1, 357 Search PubMed; (b) P. O'Brien, J. R. Walsh, I. M. Watson, M. Motevalli and L. Henrikson, J. Chem. Soc., Dalton Trans., 1996, 2491 RSC; (c) R. D. Pike, H. Cui, R. Kershaw, K. Dwight, A. Wold, T. N. Blanton, A. A. Wernberg and H. J. Gysling, Thin Solid Films, 1993, 224, 221 CrossRef; (d) D. M. Frifo, O. F. Z. Khan and P. O'Brien, J. Cryst. Growth, 1989, 96, 989 CrossRef; (e) J. Wang, J. R. Hyde, J. W. Wilson, K. Mallik, P. J. Sazio, P. O'Brien, M. A. Malik, M. Afzaal, C. Q. Nguyen, M. W. George, S. M. Howdle and D. C. Smith, Adv. Mater., 2009, 21, 4115 CrossRef; (f) Y. M. Rumyantsev, N. I. Fainer, M. L. Kosinova, B. M. Ayupov and N. P. Sysoeva, J. Phys. IV, 1999, 9, 777 CAS; (g) N. I. Fainer, M. L. Kosinova, Y. M. Rumyantsev, E. G. Salman and F. A. Kuznetsov, Thin Solid Films, 1996, 280, 16 CrossRef CAS; (h) N. I. Fainer, Y. M. Rumyantsev, M. L. Kosinova and F. A. Kuznetsov, Electrochemical Society, 1997, 25–97, 1437 Search PubMed; (i) D. Zeng, M. J. Hamden-Smith, T. M. Alam and A. L. Rheingold, Polyhedron, 1994, 13, 2715 CrossRef CAS; (j) B. Ludolph, M. A. Malik, P. O'Brien and N. Revaprasadu, Chem. Commun., 1998, 1849 RSC; (k) N. Revaprasadu, M. A. Malik, J. Carstens and P. O. Brien, J. Mater. Chem., 1999, 9, 2885 RSC.
- (a) M. A. Malik, P. O'Brien and N. Revaprasadu, Chem. Mater., 2002, 14, 2004 CrossRef CAS; (b) M. A. Malik, N. Revaprasadu and P. O'Brien, Chem. Mater., 2001, 13, 913 CrossRef CAS; (c) M. Lazell and P. O'Brien, J. Chem. Soc., Chem. Commun., 1999, 2041 RSC; (d) P. Sreekumari Nair, N. Revaprasadu, T. Radbakrishnan and G. A. Kolawole, J. Mater. Chem., 2001, 11, 1555 RSC; (e) T. Trindade, P. O'Brien and X.-M. Zhang, Chem. Mater., 1997, 9, 523 CrossRef CAS; (f) D. C. Onwudiwe and P. A. Ajibade, Int. J. Mol. Sci., 2011, 12, 5538 CrossRef CAS PubMed; (g) D. C. Onwudiwe and P. A. Ajibade, Mater. Lett., 2011, 65, 3258 CrossRef CAS; (h) P. A. Ajibade, D. C. Onwudiwe and M. J. Moloto, Polyhedron, 2011, 30, 246 CrossRef CAS; (i) V. S. R. Pullabhotla, S. M. Rajasekhar and N. Revaprasadu, J. Nanosci. Nanotechnol., 2011, 11, 1201 CrossRef CAS PubMed; (j) R. Marx Nirmal, K. Pandian and K. Sivakumar, Appl. Surf. Sci., 2011, 257, 2745 CrossRef CAS; (k) L. D. Nyamen, P. R. S. Viswanadha, A. A. Nejo, P. Ndifon and N. Revaprasadu, New J. Chem., 2011, 35, 1133 RSC; (l) E. Y. M. Lee, N. H. Tran and R. N. Lamb, Appl. Surf. Sci., 2005, 241, 493 CrossRef CAS; (m) M. Motevalli, P. O'Brien, J. R. Walsh and I. M. Watson, Polyhedron, 1996, 15, 2801 CrossRef CAS; (n) P. O'Brien, J. R. Walsh, I. M. Watson, L. Hart and S. R. P. Silva, J. Cryst. Growth, 1996, 167, 133 CrossRef; (o) L. D. Nyamen, A. A. Nejo, V. S. R. Pullabhotla, P. T. Ndifon, M. A. Malik, J. Akhtar, P. O'Brien and N. Revaprasadu, Polyhedron, 2014, 67, 129 CrossRef CAS; (p) L. D. Nyamen, N. Revaprasadu, R. V. S. R. Pullabhotla, A. A. Nejo, P. T. Ndifon, M. A. Malik and P. O'Brien, Polyhedron, 2013, 56, 62 CrossRef CAS; (q) A. L. Abdelhady, M. Afzaal, M. A. Malik and P. O'Brien, J. Mater. Chem., 2011, 21, 18768 RSC.
- (a) W. Han and M. Y. Gao, Cryst. Growth Des., 2008, 8, 1023 CrossRef CAS; (b) Y. Zhang, Y. Du and Q. Wang, CrystEngComm, 2010, 12, 3658 RSC; (c) M. Akhtar, J. Akhter, M. A. Malik, P. O'Brien, F. Tuna, J. Raftery and M. Helliwell, J. Mater. Chem., 2011, 21, 9737 RSC; (d) M. Akhtar, A. L. Abdelhady, M. A. Malik and P. O'Brien, J. Cryst. Growth, 2012, 346, 106 CrossRef CAS.
- (a) A. Adeogun, M. Afzaal and P. O'Brien, Chem. Vap. Deposition, 2006, 12, 597 CrossRef CAS; (b) O. C. Monteiro and T. Trindade, Mater. Res. Bull., 2004, 39, 357 CrossRef CAS; (c) Y. Lou, A. C. S. Samia, J. Cowen, K. Banger, X. Chen, H. Lee and C. Burda, Phys. Chem. Chem. Phys., 2003, 5, 1091 RSC; (d) Z. Liu, D. Xu, J. Liang, J. Shen, S. Zhang and Y. Qian, J. Phys. Chem. B, 2005, 109, 10699 CrossRef CAS PubMed; (e) J. C. Bear, N. Hollingsworth, P. D. McNaughter, A. G. Mayes, M. B. Ward, T. Nann, G. Hogarth and I. P. Parkin, Angew. Chem., Int. Ed., 2014, 53, 1598 CrossRef CAS PubMed; (f) K. Ramasamy, V. L. Kuznetsov, K. Gopal, M. A. Malik, J. Raftery, P. P. Edwards and P. O'Brien, Chem. Mater., 2013, 25, 266 CrossRef CAS; (g) K. Ramasamy, M. A. Malik and P. O'Brien, Chem. Commun., 2012, 48, 5703 RSC; (h) M. Afzaal, C. L. Rosenberg, M. A. Malik, A. J. P. White and P. O'Brien, New J. Chem., 2011, 35, 2773 RSC; (i) K. Ramasamy, M. A. Malik and P. O'Brien, Chem. Sci., 2011, 2, 1170 RSC.
- (a) P. O'Brien and J. Waters, Chem. Vap. Deposition, 2006, 12, 620 CrossRef; (b) H. Cui, R. D. Pike, R. Kershaw, K. Dwight and A. Wold, J. Solid State Chem., 1992, 101, 115 CrossRef CAS; (c) P. O'Brien, J.-H. Park and J. Waters, Thin Solid Films, 2003, 431, 502 CrossRef; (d) G. H. Singhal, R. I. Botto, L. D. Brown and K. S. Colle, J. Solid State Chem., 1994, 109, 166 CrossRef CAS; (e) R. Nomura and N. Nakai, Trans. Mater. Res. Soc. Jpn., 2003, 28, 1287 Search PubMed; (f) X. Chen, Z. Wang, X. Wang, J. Wan, J. Liu and Y. Qian, Chem. Lett., 2004, 33, 1294 CrossRef CAS; (g) R. Chauhan, M. Trivedi, J. Singh, K. C. Molloy, G. Kociok-Köhn, U. P. Mulik, D. P. Amalnerkar and A. Kumar, Inorg. Chim. Acta, 2014, 415, 69 CrossRef CAS.
- N. Hollingsworth, A. Roffey, H.-U. Islam, M. Mercy, A. Roldan, W. Bras, M. Wolthers, C. R. A. Catlow, G. Sankar, G. Hogarth and N. H. de Leeuw, Chem. Mater., 2014, 26, 6281 CrossRef CAS.
- A. Roffey, N. Hollingsworth, H.-U. Islam, M. Mercy, C. R. A. Catlow, G. Sankar, N. H. de Leeuw and G. Hogarth, Nanoscale, 2016, 8, 11067 RSC.
- A. Wold and K. Dwight, Solid State Chemistry: Synthesis, Structure, and Properties of Selected Oxides and Sulfides, Chapman and Hall, 1993 Search PubMed.
- (a) D. J. Vaughan and J. R. Craig, Am. Mineral., 1947, 70, 1036 Search PubMed; (b) G. Kullerud, Can. Mineral., 1963, 7, 353 CAS; (c) V. Raghavan, J. Phase Equilib. Diffus., 2004, 25, 373 CrossRef CAS; (d) M. R. Thornber, J. Appl. Electrochem., 1983, 13, 253 CrossRef CAS; (e) P. Bayliss, Am. Mineral., 1989, 74, 1168 CAS.
- I. Bezverkhyy, P. Afanasiev and M. Danot, J. Phys. Chem. B, 2004, 108, 7709 CrossRef CAS.
- R. Pastorek, P. Štarha, T. Peterek and Z. Trávníček, Polyhedron, 2011, 30, 2795 CrossRef CAS.
- M. E. Fleet, Rev. Mineral. Geochem., 2006, 61, 365 CrossRef CAS.
- (a) A. M. Wiltrout, N. J. Freymeyer, T. Machani, D. P. Rossi and K. E. Plass, J. Mater. Chem., 2011, 21, 19286 RSC; (b) V. S. Gurin, Colloids Surf., A, 1998, 142, 35 CrossRef CAS; (c) A. M. Malyarevich, K. V Yumashev, N. N. Posnov, V. P. Mikhailov and V. S. Gurin, Appl. Phys. B, 2000, 70, 111 CrossRef CAS; (d) A. M. Malvarevich, K. V Yumashev, N. N. Posnov, V. P. Mikhailov and V. S. Gurin, J. Appl. Spectrosc., 2000, 67, 359 Search PubMed; (e) E. A. Nevar, N. A. Savastenko, V. Bruser, D. A. Lopatik, F. May, A. V Butsen, N. V Tarasenko and V. S. Burakov, J. Appl. Spectrosc., 2010, 77, 136 CrossRef; (f) K. Aup-Ngoen, T. Thongtem, S. Thongtem and A. Phuruangrat, Mater. Lett., 2013, 101, 9 CrossRef CAS.
- Y.-H. A. Wang, N. Bao and A. Gupta, Solid State Sci., 2010, 12, 387 CrossRef CAS.
- D. Liang, R. Ma, S. Jiao, G. Pang and S. Feng, Nanoscale, 2012, 4, 6265 RSC.
- (a) V. Blagojević, G. A. Gledhill, A. Hamilton, S. B. Upadhyay, P. M. Nikolić, M. B. Pavlović and D. I. Raković, Infrared Phys., 1991, 31, 387 CrossRef; (b) A. A. Vaipolin, Y. A. Nikolaev, I. K. Polushina, V. Y. Rud', Y. V. Rud', E. I. Terukov and N. Fernelius, Semiconductors, 2003, 37, 681 CrossRef.
- Q. J. Feng, D. Z. Shen, J. Y. Zhang, Y. M. Lu, Y. C. Liu and X. W. Fan, Mater. Chem. Phys., 2006, 96, 158 CrossRef CAS.
- D. Disale and S. S. Garje, J. Organomet. Chem., 2011, 696, 3328 CrossRef.
- Y. Li, C. Cao and Z. Chen, Mater. Lett., 2011, 65, 2432 CrossRef CAS.
- Y. Shi, Y. Wang and L. Wu, J. Phys. Chem. C, 2013, 117, 20054 CrossRef CAS.
- H. Chen, J. Jiang, L. Zhang, H. Wan, T. Qi and D. Xia, Nanoscale, 2013, 5, 8879 RSC.
- W. Du, Z. Zhu, Y. Wang, J. Liu, W. Yang, X. Qian and H. Pang, RSC Adv., 2014, 4, 6998 RSC.
- A. H. White, R. Roper, E. Kokot, H. Waterman and R. L. Martin, Aust. J. Chem., 1964, 17, 294 CrossRef CAS.
- S. C. Ngo, K. K. Banger, M. J. DelaRosa, P. J. Toscano and J. T. Welch, Polyhedron, 2003, 22, 1575 CrossRef CAS.
- A. M. Bond and A. F. Hollenkamp, Inorg. Chem., 1990, 29, 284 CrossRef CAS.
- M. L. Riekkola and O. Makitie, J. Therm. Anal., 1982, 25, 89 CrossRef CAS.
- J. E. J. Schmitz and J. G. M. Van der Linden, Inorg. Chem., 1984, 23, 3298 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |