
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene oxide nanohybrids for electron transfer-mediated antimicrobial activity†
Nayan Mani
Das
 *ab,
Amit Kumar
Singh
*b,
Debdatta
Ghosh
a and
Dipankar
Bandyopadhyay
*ab
*ab,
Amit Kumar
Singh
*b,
Debdatta
Ghosh
a and
Dipankar
Bandyopadhyay
*ab
aDepartment of Chemical Engineering, Indian Institute of Technology Guwahati, Guwahati – 781039, India. E-mail: nayanmanidas3@gmail.com; dipban@iitg.ac.in
bCentre for Nanotechnology, Indian Institute of Technology Guwahati, Guwahati – 781039, India. E-mail: amitiitg2011@gmail.com
First published on 15th August 2019
Abstract
The rapid increase in the prevalence of antibiotic-resistant bacterial strains poses a global health risk. In this scenario, alternative strategies are needed to combat the alarming rise in multidrug-resistant bacterial populations. For example, metal-incorporated graphene derivatives have emerged as model nanomaterials owing to their intrinsic antibacterial activity together with their biocompatibility. Interestingly, photon-activated phthalocyanine sensitizers have also shown promising physiochemical biocidal effects against pathogenic bacteria populations when conjugated with diverse nanomaterials. Herein, we report the facile synthesis of graphene oxide incorporated zinc phthalocyanine (ZnPc–GO) nanohybrids showing bactericidal activity against Gram-negative Escherichia coli (E. coli) cells, in the absence of any photo-excitation. The ZnPc–GO hybrid nanomaterials were synthesized by the in situ deposition of GO flakes on ZnPc-coated indium tin oxide (ITO) substrates. Two types of morphologically different ZnPc molecules, potato-chip-like α-phase ZnPc, namely ZnPc(A), and nanorod-like β-phase ZnPc(B), were used for the synthesis of the ZnPc(A/B)–GO nanocomposites. The interactions of GO with the underlying ZnPc(A/B) entities in the ZnPc–GO systems were investigated using multiple characterization techniques. It was observed that the GO flakes in the ZnPc(B)–GO nanocomposite possess stronger π–π interactions and thus show a more efficient electron transfer mechanism when compared with the ZnPc(A) counterpart. Furthermore, the E. coli bacterial cells with an electronegative surface demonstrated a profound adherence to the electron-withdrawing ZnPc(B)–GO surface. The death kinetics of bacteria with ZnPc(B)–GO were further investigated using surface potential mapping and Kelvin probe force microscopy (KPFM) analysis. Upon direct contact with ZnPc(B)–GO, the adhered bacterial cells showed outer cell deformation and membrane protein leakage, induced by a proposed charge-transfer mechanism between negatively charged cells and the electron-withdrawing ZnPc(B)–GO surface. These new findings may provide insights into the design of potential ZnPc–GO-based novel antimicrobial nanomaterials or surface coatings.
1. Introduction
The abrupt rise in bacterial mutagenesis has led to widespread antibiotic-resistant infections worldwide, thereby posing a serious threat to public health.1,2 Antibiotics are frequently administered to destroy or inhibit the growth of pathogenic bacteria. However, the exhaustive medical use and misuse of antibiotics has minimized their efficacy, which is attributed to the rapid emergence of antibiotic-resistant strains.3 In order to avert the antibiotic-resistance crisis, researchers are exploring innovative combat strategies to restrain the spread of bacterial pathogens.4–7 In the recent past, the bactericidal and bacteriostatic efficacy of nanomaterials has been widely studied as a promising alternative to conventional antibiotic-based treatment.8–11 In this context, innovative methods like sensors,12,13 bacteriophage-based systems,14 thin film patterns15 and microfluidics16,17 have been employed for deactivation of pathogenic threats in the recent past. Amongst these methods, graphene-based nanomaterials18,19 have been extensively used as antimicrobial agents for numerous biomedical applications in recent years.20–22Graphene derivatives exhibit antimicrobial properties against a wide range of Gram-positive and Gram-negative pathogenic bacterial biofilms.23,24 The ease of surface modification by direct incorporation of functional groups or extrinsic mesoscale materials on the graphene surfaces promotes their usage for antimicrobial therapy.25,26 The antimicrobial efficacies of graphene derivatives can be further enhanced via surface functionalization with metals.23 For example, metallic nanoparticles have been used for decades as antimicrobial agents. Thus, in conjugation with these metallic entities, the antimicrobial activity of graphene derivatives can be further enhanced.27,28 Importantly, previous studies suggest that graphene derivative–metal films may act as efficient electron sinks, which can effectively remove electrons from the bacterial phospholipid membrane.28–31 This results in alteration of the membrane ζ-potential and destabilization of the cell membrane, leading to irreversible bacterial cell damage.30,31 The germicidal efficacy of graphene–metal systems relies on the conductivity of the metallic entity.28 A recent study showed that when E. coli cells were treated with a conducting zinc (Zn)–graphene oxide (GO) composite, the bacterial cells showed more membrane damage in comparison to cells treated with non-metallic or less conductive metallic counterparts present in the GO–metal systems.28 The Zn–GO system induces reactive oxygen species (ROS) generation,32 which disrupts the integrity of the cell membrane, thereby leading to cell lysis.28 These findings inspired us to further investigate Zn–GO metal systems by substituting the conductive Zn metal with more conductive Zn-based entities in the Zn–GO system for further enhancement of its antibacterial activity.
In this direction, we have found that metal phthalocyanines (MPc) are promising substitutes for the Zn metal in Zn–GO systems.33,34 Aromatic phthalocyanine (Pc) molecules are two-dimensional 18π-electron systems, which can be synthesized easily and exhibit non-toxicity, chemical and thermal stability, and substitutional flexibility.33–35 In the Pc molecules, the two central hydrogen atoms can be replaced by metal ions to form metal phthalocyanines (MPc) for various purposes.33,34 Recent studies have shown that MPc molecules containing cationic metal ions are more effective against bacteria than the anionic and neutral metal ion-containing counterparts as the cationic MPc molecules can easily interact with and disrupt the highly anionic bacterial cell wall.35,36 Amongst the reported MPc molecules, zinc phthalocyanine (ZnPc) is considered a model MPc system as it shows the characteristic features of the macrocyclic phthalocyanine (Pc) molecule.37 In the recent past, ZnPc-based derivatives have been employed as promising photosensitizers for sensing,38 optical limiting,39,40 cancer treatment,41 antimicrobial photodynamic therapy,35,42,43 and energy harvesting.44 Thus, it would be very interesting to investigate the activity of graphene derivative–ZnPc conjugates44 as contemporary antimicrobial materials.
In view of this prior art, we report a facile protocol for the synthesis of an antimicrobial zinc phthalocyanine–graphene oxide (ZnPc–GO) nanoconjugate. The synthesis of the ZnPc–GO nanomaterial was evaluated by multiple characterization methods. Following this, we investigated the electron transfer-mediated antibacterial activity of ZnPc–GO on bacterial cells.45 The interaction of the ZnPc molecules with the GO sheets was studied for both α-phase and β-phase orientation of ZnPc molecules. The ZnPc molecules traditionally exhibit α-phase, namely ZnPc(A), at room temperature and β-phase, namely ZnPc(B), at high temperature.37,46 The phase transformation of the ZnPc molecules from the α-phase to the β-phase can be achieved when the ZnPc molecules are heated at temperatures ranging from room temperature of 25 °C to a high temperature of 280 °C.37 It has been observed that upon interaction with the GO substrate, the molecular band gap of the ZnPc molecules is modified owing to charge transfer between the GO substrate and the ZnPc molecules.47,48 As a result of the electronic interaction of the ZnPc molecules with the GO substrate, the ZnPc(B)–GO nanostructure demonstrated significantly enhanced conductivity leading to a regulated charge balance compared to the ZnPc(A)–GO.
Thereafter, we investigated the electron transfer mechanisms47,48 behind the antibacterial effect of the ZnPc–GO nanoconjugates using Kelvin probe force microscopy (KPFM).45 The analysis helps us in measuring the net change in the surface potential of the bacterial cells owing to the ionic transfer of charges between ZnPc–GO nanosheets and the E. coli bacterial cells after their physical interaction with the ZnPc–GO nanosheets. Previous studies of anti-pathogenic MPc–GO nanostructures have solely relied on the photodynamic activation35,42,43 of MPc derivatives to generate ROS for localized antimicrobial activity. In contrast, arguably as the first demonstration, we show the usage of a ZnPc–GO nanocomposite as an antimicrobial agent in the absence of any photonic excitation.
The study uncovers that the destruction of bacterial cells depends on the electron transfer and the surface charge-dependent adherence of bacteria on the ZnPc–GO nanosheets.47,48 Further, based on the characterizations reported, we propose a mechanism for the antibacterial activity of MPc–GO, which has been followed solely through the morphological changes of bacteria in previous reports. Interestingly, we observe that the E. coli cell deformation occurs due to a progressive change in the surface potential of the cell wall within an hour of treatment with the ZnPc–GO nanomaterial. Upon interaction with the ZnPc–GO nanosheets, the temporal change in the bacterial outer membrane potential has been characterized and correlated to the bacterial cell damage with the help of time-dependent KPFM analysis. Unlike antibiotics, the reported electron transfer-mediated antibacterial activity of the ZnPc–GO nanosheets may not result in resistant bacterial strains. The reported results may inspire the design and development of ZnPc–GO-based nanoscale antimicrobial coatings and smart devices49–51 in the near future.
2. Experimental methods
2.1. Materials
Hydrochloric acid (HCl) (37%), hydrogen peroxide (H2O2) (50%), sodium hydroxide (NaOH), sodium chloride (NaCl), potassium permanganate (KMnO4), sulfuric acid (H2SO4) (98%), 125 mm filter paper (grade-1), acetone (CH3COCH3) and ortho-phosphoric acid (H3PO4) (84%) were obtained from Merck (India). Phosphorous pentoxide (P2O5), sodium dodecyl sulfate (SDS) and hydrazine monohydrate (N2H4·H2O) (98%) were obtained from Sigma-Aldrich (India). Zinc phthalocyanine (ZnPc) (MW = 577.91 g mol−1; dye content 97%) was purchased from Sigma-Aldrich (Germany). Graphite flakes (99.99%) were obtained from Alfa Aesar (India). Indium tin oxide (ITO)-coated glass substrates were procured from Macwin (India). The ITO-coated glass substrates were cut into 1 × 1 cm2 pieces and served as the substrate for the ZnPc deposits. Luria–Bertani (LB) medium was purchased from Himedia (India). Parafilm wax paper was obtained from Tarsons (India). The aforementioned chemicals were of analytical grade and were used without further purification. Milli-Q grade water was used in the experiments.2.2. Preparation of ZnPc thin films
The films of ZnPc were deposited over the ITO-coated glass substrates using an organic thermal evaporator (HHV Ltd, India). Prior to deposition, the ITO-coated glass substrates were thoroughly cleaned by dipping in SDS solution for 20 min, followed by gentle brushing and finally repeatedly washing with deionized water and drying. After drying, the ITO-coated glass substrates were transferred into a beaker containing acetone and heated up to boiling temperature for 10 min to remove any organic impurities. Following this, the substrates were again washed thoroughly with jets of deionized water and then dried. The dried substrates were kept in an air-tight container for further use. The cleaned substrates were exposed to oxygen plasma in a UV-ozone chamber for about 10 min and then without any delay the substrates were taken to an organic thermal evaporator chamber for the deposition.The deposition of ZnPc was performed at a high pressure of ∼5.5 × 10−6 bar. The ZnPc powder was placed in a crucible and then placed inside the deposition chamber of the evaporator. The cleaned ITO-coated glass substrates were placed just above the container containing the ZnPc powder. Following this, the crucible was heated to the evaporation temperature of the ZnPc (∼350 °C) within the vacuum system and the amount of deposition was monitored using a thickness-monitoring system. The thickness of the deposited ZnPc thin film was monitored by a thickness-monitoring quartz crystal microbalance fitted inside the evaporator. For our purposes, the optimum thickness of the ZnPc film deposits on the ITO-coated glass substrates was maintained at ∼30 nm. The substrates coated with ZnPc thin films were kept in a clean vacuum environment for further use. The as-synthesized ZnPc thin films on ITO-coated glass were exposed to different temperatures, ranging from room temperature (25 °C) to 280 °C in order to achieve the α-phase to β-phase transformation of ZnPc prior to the deposition of the GO flakes.
2.3. Synthesis of GO flakes
The GO solution was synthesized following the modified Hummer's method.52,53 Initially, 0.5 g of graphite flakes and 3 g of KMnO4 were dispersed in an aqueous mixture consisting of 63 mL of concentrated H2SO4 and 7 mL of H3PO4. Following this, the resulting solution was heated for 12 h at 50 °C. After that, the solution was allowed to cool down and 3 mL of 30% H2O2 along with 50 mL of water were added into the solution. The aforementioned reaction was exothermic in nature and was carried out in an ice environment (∼4 °C), and after completion of the reaction, 100 mL of water was again added into the solution. The resulting solution was then centrifuged at 8000 rpm for 15 min at room temperature and the supernatant liquid was discarded. The precipitate was washed repeatedly with 10% HCl and water until no trace of sulfate was detected by BaCl2 test. Furthermore, the precipitate was thoroughly washed with water to attain a neutral pH. The as-obtained precipitate was then vacuum-dried with P2O5 at room temperature before dispersing it in water and then sonicated for 2 h to obtain the GO solution.The as-synthesized GO was dispersed in water and mechanically stirred for ∼5 min. The resulting solution was then sonicated for 10 min to exfoliate the GO flakes. The exfoliated GO solution was then centrifuged at ∼2000 rpm for 5 min about 2–3 times and the sediment was collected. Thereafter, the sediment was again dispersed in water and centrifuged for 2–3 times at ∼8000 rpm for 15 min. Following this, the exfoliated GO solution was again centrifuged at ∼2000 rpm and ∼8000 rpm for 5 min and 15 min, respectively, about 2–3 times and the sediment was collected. The sediment was again dispersed in water and sonicated for 5 min to obtain a homogeneous dispersion of GO flakes in water. The exfoliated GO solution was then added to methanol with water (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)) and used for the film deposition.
:1 (v/v)) and used for the film deposition.
2.4. Synthesis of ZnPc–GO heterostructures
In order to fabricate the ZnPc–GO nanocomposites, the GO layers were deposited over the ZnPc-coated substrate using our previously reported “funnel” method.54 Briefly, a funnel with a burette with a stopcock was fitted in an upright manner by clamping it to a burette stand. Thereafter, a column of water was created on the funnel, which was covered inside with parafilm wax paper. A ZnPc-coated substrate was kept at the inclined part of the Y shaped funnel on the wax paper. A water sub-phase was maintained inside the funnel on the paper and the ZnPc-coated substrate by closing the stopcock at the bottom of the funnel. Then the GO solution in methanol–water mixture was spread on the water column and kept undisturbed for ∼15 min, which allowed the dispersed GO flakes to orient over the water sub-phase in an ordered manner. Later, the funnel water was allowed to drip out slowly by opening the stopcock. The substrates with ZnPc deposits and coated GO flakes were then dried at 50 °C for 12 h under vacuum. The aforementioned method helped in getting well distributed GO flakes on the ZnPc-coated substrate, thereby, resulting in the ZnPc–GO nanostructures. Furthermore, individual GO flakes were also deposited on blank ITO-coated glass substrates, in the absence of ZnPc deposits, using the aforementioned procedure.2.5. The bacterial adherence on ZnPc–GO nanostructures
The E. coli colonies were grown overnight at 37 °C in Luria–Bertani (LB) medium at 220 rpm for 12 h. The dispersion containing bacteria (grown overnight) was centrifuged at 10000 rpm for 2 min. The pellets thus obtained were washed with de-ionized water and again dispersed in water. The dispersed bacteria were serially diluted with water to obtain bacterial concentrations of 2 × 106 CFU mL−1.55 3 mL of bacterial solution was dispensed in each well of the 12-well microtiter plate and the ZnPc–GO samples were placed in each well, with the coated side of the ITO substrate facing in the upward direction. A few samples were retrieved from the wells after standing for 5 min at ambient conditions and the rest of the samples were left undisturbed in the wells and were incubated for 2 h at 37 °C. Following this, the samples were tilted to remove the excess solution along with the unadhered cells over the surface, prior to atomic force microscopy (AFM) measurements. The antibacterial activity of the ZnPc–GO system was studied for incubated and non-incubated samples using the KPFM56 method.
2.6. Characterization
Field emission scanning electron microscopy (FESEM, Zeiss Sigma, Germany) was used to examine the morphologies of the samples. The samples were vacuum-dried and placed on carbon tape adhered on a stub and platinum-sputtered for FESEM analysis. A laser micro Raman spectrometer (Horiba Jobin Vyon, Model LabRam HR) was used for characterization of the samples. For Raman spectroscopy analysis, the samples were observed at 100× optical microscopic zoom and then excited by 532 nm Nd-YAG laser. The Raman signals were recorded maintaining 1800 grating per mm, 100-hole size and 50% of the ND filter. A constant acquisition time of 5 s and an accumulation time of 2 s were maintained during the procedure. The output power reading of the laser falling on the sample was measured to be ∼50 mW. The samples were also analysed by UV-visible spectrophotometer (Shimadzu, UV-2600). The data were collected after base correction using the blank substrate where the entire deposition was carried out. For the absorption studies, the thin films were deposited on transparent quartz substrates. The film thickness was calculated using an ellipsometer (EP3, Nanofilm, Accurion Scientific Instruments Pvt. Ltd). The X-ray photoelectron spectroscopy (XPS) analysis was performed using a Thermo ESCALAB 250 spectrometer with a monochromatic Al Kα X-ray as the source (15 kV, 150 W).The surface morphologies of the samples were examined using an AFM (Bruker, Innova series) with Si tips. While doing the AFM in tapping mode, a constant cantilever force constant of 0.48 N m−1 was maintained at a set point of ∼2.8 V, whereas, the P and I gains were maintained at 1.0 and 0.5, respectively. For the surface potential (SP) analysis of the samples, the Kelvin probe force microscopy (KPFM) measurements were performed.44 For the KPFM studies, conducting platinum (Pt)/iridium (Ir)-coated tips with an optimum frequency of operation of ∼72 kHz were used. The measurements were carried out in the dual-pass lift mode in order to avoid noise and cross-talk between the topographical and the surface potential measuring images. The images were captured in lift mode and all images were processed in backward lift scanning mode, whereas, the topographies were captured in forward scanning mode. In order to estimate the exact work function of the samples, the images from the KPFM were not further processed by any post-processing software in order to minimize the loss of data. Windows-Scanning-x-Microscope (WSxM) software57 was only used for topography image processing and analysis.
3. Results and discussion
The ZnPc molecules were deposited over the ITO-coated glass substrates by thermal evaporation technique. Later, the 2-D sheets of GO were deposited by the aforementioned funnel-dripping technique to form the ZnPc–GO heterostructures. FESEM, UV-visible spectroscopy, Raman spectroscopy and mapping, AFM, XPS, electrical conductivity measurements and KPFM were used to examine the morphologies and understand the mechanism of interaction between the ZnPc and GO components within the ZnPc–GO heterostructures.3.1. FESEM characterization
Fig. 1 shows the FESEM images of the bare ZnPc deposits and the ZnPc–GO heterostructures over the ITO substrate. Fig. 1(A) shows the surface morphology of the ZnPc deposits at room temperature. The image demonstrates the aggregation of ‘potato-chip-nanoflakes’ throughout the substrate at room temperature, which corresponds to the α-phase of ZnPc molecules, namely ZnPc(A).37,58 The average diameter of the roughly spherical ZnPc(A) nanoflakes was observed to be within 300–500 μm. Previous studies have shown that the π–π stacking interaction between the ZnPc molecules increases at high annealing temperature, thereby resulting in the transition of ZnPc nanoflakes into elongated rod-like structures.37,58 In this study, when the temperature of the ZnPc(A) deposits was increased to 280 °C, the as-obtained nanoflake clusters were transformed into randomly oriented rod-like structures.37 The change in the surface morphology of the ZnPc molecules from nanoflakes to a nanorod-like structure was found to be related to the phase change of ZnPc from the metastable α-phase to the stable β-phase, namely ZnPc(B). | ||
| Fig. 1 FESEM images of (A) ZnPc(A), (B) ZnPc(B), (C) ZnPc(A)–GO and (D) ZnPc(B)–GO. The scale bar is 1 μm in size. | ||
Fig. 1(B) shows the disoriented columnar array of ZnPc(B) nanorods over the substrate, with the size of the nanorods ranging from 250 nm to 400 nm.37,45 The GO nanosheets were deposited on ZnPc molecules to form ZnPc(A/B)–GO nanocomposites. The characterization details of the as-synthesized GO nanosheets are reported in Fig. S1 of the ESI.†Fig. 1(C) and (D) show the morphology of the ZnPc(A)–GO and ZnPc(B)–GO nanocomposites, respectively, which confirmed the stacking of the as-deposited GO flakes over the ZnPc molecules. The FESEM image of the ZnPc(A)–GO hybrid, as shown in Fig. 1(C), demonstrates the characteristic wrinkled texture of GO sheets over the ZnPc(A) nanoflakes. Fig. 1(D) shows the presence of the ZnPc(B) nanorods underneath the wrinkled GO nanosheets.
3.2. UV-visible spectroscopy
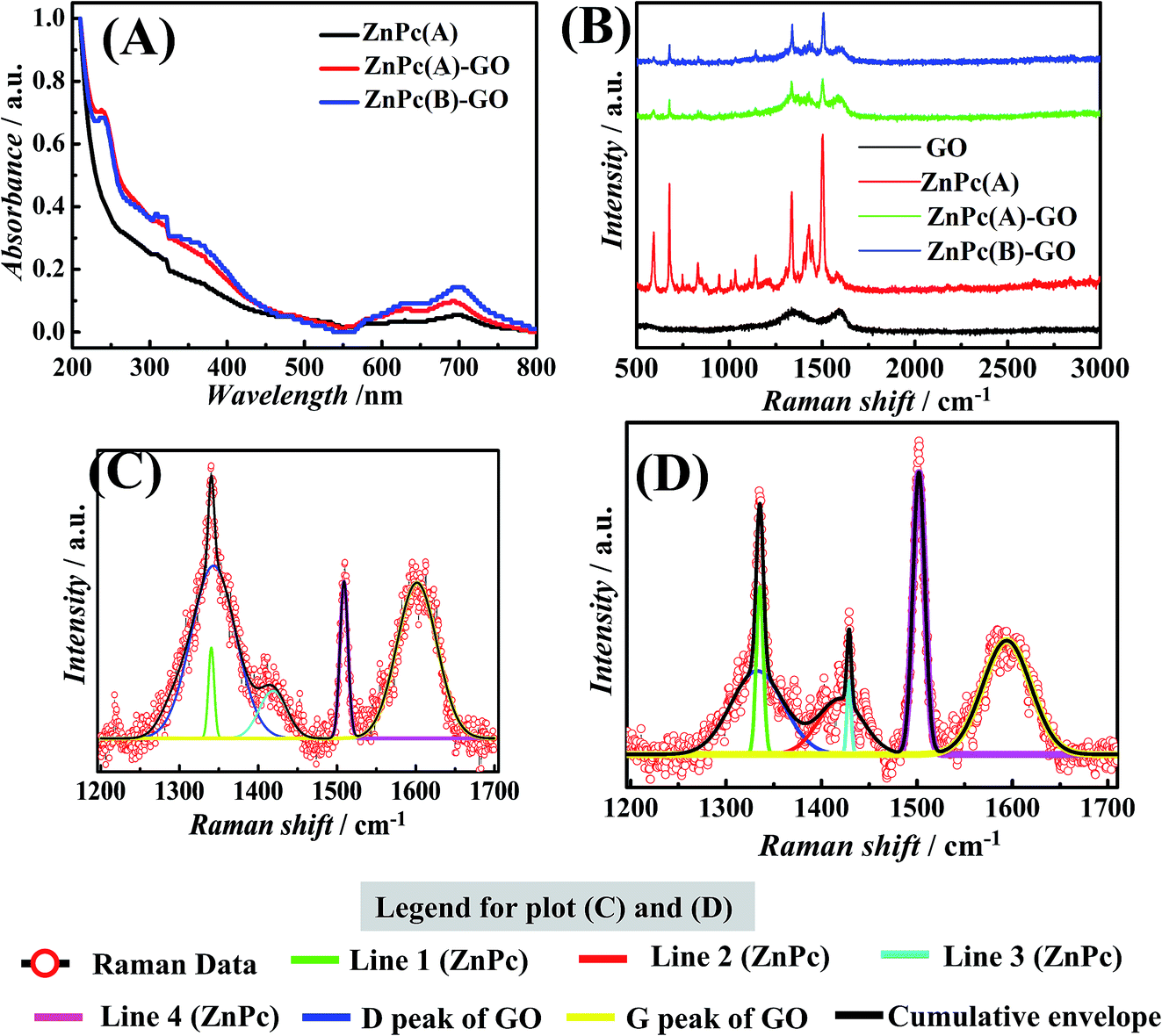
The ZnPc–GO nanohybrids were further investigated by solid-state UV-visible spectroscopy. The UV-vis absorption spectrum of ZnPc(A), as represented by the black line in Fig. 2(A), exhibits a prominent peak centred at 697 nm, followed by a smaller shoulder at 630 nm, representing the characteristic Q-band of metal phthalocyanines (MPcs) associated with the HOMO–LUMO (π–π*) transition of the phthalocyanine rings, and a weak broad Soret band was observed at 315 nm.58,59 In addition to the typical Q-band and Soret band regions of the ZnPc(A) molecules, the absorption spectra of the ZnPc(A)–GO and ZnPc(B)–GO hybrids in Fig. 2(A), represented by red and blue lines, respectively, contain the characteristic absorption peak of the GO moiety at 240 nm, which corresponds to the π–π* transition of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.37,59 The presence of GO absorption peak in the ZnPc–GO hybrids clearly indicates that the GO layers were successfully embedded on the ZnPc thin films. In comparison with the Soret band position of ZnPc(A) molecules at 315 nm, the Soret band absorption peak of ZnPc molecules in the ZnPc(A/B)–GO hybrids was blue-shifted at 310 nm, owing to the π-electron interaction between the GO nanosheets and the ZnPc molecules.59
C bonds.37,59 The presence of GO absorption peak in the ZnPc–GO hybrids clearly indicates that the GO layers were successfully embedded on the ZnPc thin films. In comparison with the Soret band position of ZnPc(A) molecules at 315 nm, the Soret band absorption peak of ZnPc molecules in the ZnPc(A/B)–GO hybrids was blue-shifted at 310 nm, owing to the π-electron interaction between the GO nanosheets and the ZnPc molecules.59
 | ||
| Fig. 2 (A) UV-visible absorption spectra of ZnPc(A) and ZnPc(A)–GO and ZnPc(B)–GO hybrids. (B) Raman spectra of GO flakes, ZnPc(A) deposits, and ZnPc(A)–GO and ZnPc(B)–GO hybrids. The Raman spectra of the ZnPc–GO hybrids in image (B) have been deconvoluted to extract the peak positions of the individual components. Images (C) and (D) represent the deconvolution of the Raman spectra of the ZnPc(A)–GO and ZnPc(B)–GO composites, respectively. | ||
Furthermore, after the deposition of the GO layer on the ZnPc(B) molecules, the Q-band of the ZnPc moiety showed a slight red-shift from 697 nm to 700 nm in the spectrum of the ZnPc(B)–GO hybrid, suggesting that the deposition of the GO flakes onto the ZnPc(B) molecules leads to a strong π–π interaction between the ZnPc(B) and the GO.37,59–61 This strong π–π interaction might have led to the transition of free electrons from the GO sheets to the phthalocyanine group of ZnPc(B), whereas the Q-band of the ZnPc(A) moiety in the spectrum of the ZnPc(A)–GO hybrid remains unaltered.59–61
3.3. Raman spectroscopy and mapping
Further investigations were performed using Raman spectroscopy in order to understand the interaction of the ZnPc molecules and the GO flakes within the nanocomposites. The Raman spectra of the GO flakes, ZnPc(A) thin film, and ZnPc(A)–GO and ZnPc(B)–GO hybrids, as illustrated in Fig. 2(B), were analysed using an excitation wavelength of 532 nm. The Raman spectrum of the GO flakes (denoted by the black line) in Fig. 2(B) shows a prominent D-band around 1350 cm−1 and a characteristic G-band at 1608 cm−1.62 The Raman shift peaks of the ZnPc(A) molecules (denoted by the red line) in Fig. 2(B) consist of primary peaks positioned at 590 cm−1 for benzene ring deformation, 680 cm−1 for macrocycle breathing, 1335 cm−1 and an intense band at 1504 cm−1 for the Zn metal ion linked to the phthalocyanine molecule, along with other secondary peaks.63The presence of all the peaks in the Raman spectrum for ZnPc(A) molecules clearly suggests the formation of a well-organized film over the ITO substrate. The ZnPc(A)–GO and ZnPc(B)–GO hybrids, as denoted by green and blue lines, respectively, in Fig. 2(B), display a characteristic D-band at ∼1347 cm−1 and a G-band at ∼1593 cm−1, which correspond to the GO nanosheets along with the characteristic peaks of the ZnPc molecules.61,62 The G-band shift in the ZnPc(A/B)–GO nanocomposites could be assigned to the extended delocalization of π electrons owing to π–π interaction between the ZnPc and the GO.61,62 The Raman plots of the ZnPc(A/B)–GO nanocomposites were further deconvoluted and re-plotted to gain a better understanding of the interaction between the ZnPc and GO entities.
The deconvolution of the Raman spectra of ZnPc(A)–GO and ZnPc(B)–GO is depicted in Fig. 2(C) and (D), respectively. In Fig. 2(C), the D-band (blue line) and G-band (yellow line) of the GO in the ZnPc(A)–GO hybrids are positioned at wavelengths of 1343 cm−1 and 1601 cm−1, respectively. Further, the characteristic peaks of ZnPc(A) are clearly visible at 1339 cm−1 (line 2) and 1508 cm−1 (line 4) while the secondary peaks (line 1 and 4) of ZnPc(A) is either diffused or not prominent in nature. Furthermore, in the case of the ZnPc(B)–GO nanocomposites, Fig. 2(D) depicts the D-band (blue line) and G-band (yellow line) of GO at 1334 cm−1 and 1594 cm−1, respectively. The two prominent peaks for the ZnPc(B) molecules are positioned at 1335 cm−1 (line 2) and 1501 cm−1 (line 4), comparable to those for the ZnPc(A)–GO composite, while the secondary ZnPc peaks (lines 1 and 4) are prominently visible in the deconvolution plot. The deconvolution suggested three peaks were sufficient for the correct fitting for ZnPc(A) in ZnPc(A)–GO, whereas the occurrence of four convolution peaks led to proper fitting for ZnPc(B) in ZnPc(B)–GO, thereby suggesting that after annealing at 280 °C, the ZnPc molecules had undergone a change in orientation to make itself more pronounced.
The characterizations of the materials corroborated the findings from the FESEM images reported in Fig. 1(B), which depicts the formation of ZnPc nanorods at the high annealing temperature. In the case of the ZnPc(B)–GO composite, the observed shift in the G-band towards lower wavelength could be attributed to the strain in the molecular orientation of GO, resulting in π electron delocalization owing to the resulting π–π interaction between the ZnPc and GO molecules.61,62 The ID/IG ratios for the GO flakes and the ZnPc(A)–GO and ZnPc(B)–GO thin films were calculated to be 0.91, 1.1 and 0.828, respectively.59 The lower ID/IG ratio for ZnPc(B)–GO hybrid suggests that the ZnPc(B) molecules interact more efficiently with the GO flakes as compared to the ZnPc(A) counterparts.61,62 The diminished D peak of GO in the ZnPc(B)–GO composite further confirmed the decrement in defect states in GO flakes leading to an extensive interaction between them.
The change in the physical interaction between the ZnPc and GO moieties with the variation in temperature (morphology of ZnPc molecules) was further investigated using Raman mapping measurements.64Fig. 3(I(A–D)) show the Raman mapping for the site-specific interaction of GO with ZnPc molecules. The site-specific creation of the red hotspots in Raman mapping and the intensification of the red spots clearly indicated the interaction between ZnPc and GO with the increase in temperature from 25 °C [Fig. 3(I(B))] to 280 °C [Fig. 3(I(D))]. The increase in the interaction between ZnPc and GO might be because of the change in morphology from ZnPc(A) to ZnPc(B) in the nanocomposites, as discussed in one of our earlier publications.37
 | ||
| Fig. 3 (I(A)) Optical micrograph (×100 optical zoom) of the ZnPc–GO composite and Raman mapping of (I(B)) ZnPc(A)–GO at 25 °C, I(C) ZnPc(I)–GO at 100 °C and (I(D)) ZnPc(B)–GO at 280 °C. The black arrows and circles show the directions and areas of the image mapping capture, respectively. Image (II(A)) shows the map of the intense peak of the ZnPc(B) molecules (1500 cm−1) and images (II(B)) and (II(C)) depict the maps of the D-band and G-band of the GO layers in the ZnPc(B)–GO clusters at 280 °C. The scale bars in all the images are 2 μm in size. | ||
Fig. 3(I(C)) depicts the intermediate transition state of ZnPc molecules at 100 °C, termed ZnPc(I), present between the ZnPc(A) and ZnPc(B) states while concurrently interacting with the GO layers. The increase in temperature, and thus the morphology change, showed how the ZnPc–GO interaction domains (red spots) increased from Fig. 3(I(B))–(I(D)). The plots suggest that the GO layers more profoundly interacted with the ZnPc(B) molecules than their ZnPc(A) counterparts, which was in agreement with the previous Raman spectra and UV-vis characterizations. The area of the red spot zone was calculated using ImageJ software. The image analysis revealed that ZnPc(B)–GO covered around 52–55% of the total area whereas the interaction zone for ZnPc(A)–GO was found to be nearly 19–22% of the total area. The increase in the zonal percentage values at the elevated temperature complied with the inference of the new surface modification, which broadly originated from the phase change (α to β-phase) of the ZnPc.65,66 In the same Fig. 3(I(A–D)), the faint green-coloured spots suggested the less interactive zones between the ZnPc–GO entities, and the blue indicated the interaction of the ZnPc with the substrate. Furthermore, Fig. 3(II(A–C)) show the detailed analysis of Fig. 3(I(D)) for the ZnPc(B)–GO entities. Fig. 3(II(A–C)) depict the band-specific Raman mapping of ZnPc(B)–GO molecules, at 1500 cm−1 wavelength, for the D-band and G-band of GO, respectively. Further, the dominance of the D-band in Fig. 3(II(B)) and the G-band in Fig. 3(II(C)) in the mapping images denote the availability of the band intensity pattern.
3.4. AFM characterization
AFM analysis was also performed to gain further insights into the interaction between the ZnPc and GO moieties. Fig. 4 shows the AFM topography and phase images of the ZnPc–GO nanocomposites at different temperatures. Fig. 4(I(A)) and (I(B)) show the surface topography and phase contrast AFM images for ZnPc(A)–GO, respectively. Further, Fig. 4(II(A)) and (II(B)) show the surface topography and phase contrast AFM images for ZnPc(I)–GO, respectively. Finally, Fig. 4(III(A)) and (III(B)) show the surface topography and phase contrast AFM images for the ZnPc(B)–GO sample, respectively. | ||
| Fig. 4 AFM topographical and phase contrast images for the ZnPc–GO composites at various temperatures. The surface topology images (I(A)), (II(A)) and (III(A)) correspond to ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO, respectively. The phase contrast images (I(B)), (II(B)) and (III(B)) correspond to ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO, respectively. | ||
AFM phase contrast imaging is very sensitive to numerous mechanical factors, including but not limited to viscoelasticity, adhesion and contact area. Further, the AFM phase contrast images contain the topographic contributions. Thus, an interpretation of the contrast in the phase images provided more information about the interactions between the ZnPc molecules and the GO layers.67 The AFM phase contrast image of ZnPc(B)–GO, as shown in Fig. 4(III(B)), exhibits very distinctive surface edges compared to the ZnPc(A)–GO moieties, as shown in Fig. 4(I(B)). In Fig. 4(I(B)), the ZnPc(A)–GO surface topology shows a significantly corrugated phase structure with many surface irregularities, whereas, as shown in Fig. 4(III(B)), ZnPc(B)–GO exhibits a smooth and planer surface, and this change could result from the charge transfer between the ZnPc and GO interfaces. Previous studies suggested that along with the change in morphology of ZnPc, the phase change in the AFM images could also be attributed to the change in the oxidation state of the individual GO flakes, leading to enhanced conductivity in the ZnPc(B)–GO samples.67 In order to confirm the effect of the varying ZnPc morphologies on the conductivity of the ZnPc–GO samples, further investigations were performed.
3.5. X-Ray photoelectron spectroscopy
The Raman mapping presented a brief overview of the nature of the interaction between the ZnPc and GO entities in the ZnPc–GO nanocomposites. However, in order to gain insight into the interaction at a molecular level, XPS analysis was performed. The XPS spectra of ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO are shown in Fig. S2 (refer to the ESI†). Furthermore, a detailed study of the interaction between the ZnPc and GO was performed by deconvoluting the XPS spectra of ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO for C and O peaks. The interaction of the C and O peaks as one spectral envelope was fitted into four peaks by using Lorentzian peak fitting in XPSPEAK fit software.60
Fig. 5(A–C) depict the Lorentzian deconvoluted peaks of the XPS spectra for the C 1s peak of the ZnPc–GO nanocomposite where the C 1s XPS spectrum of the ZnPc–GO moieties was deconvoluted into four peaks corresponding to different carbon species, namely, CC, C–N, C–O and CO bond interactions.68 The peak positions of the CC, C–N, C–O and CO interactions and the respective curve fitting parameters used for deconvolution of the ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO XPS spectra for C peaks are tabulated in Table S1 (refer to the ESI†). In Fig. 5(A), the peak positions at 284.4 eV, 287.1 eV and 288.6 eV in the C 1s XPS spectrum of ZnPc(A)–GO correspond to the CC, C–O and CO bond interactions, respectively. The peak position at 285.3 eV corresponds to the C–N species of macrocyclic phthalocyanine (Pc) present in the ZnPc–GO system.59
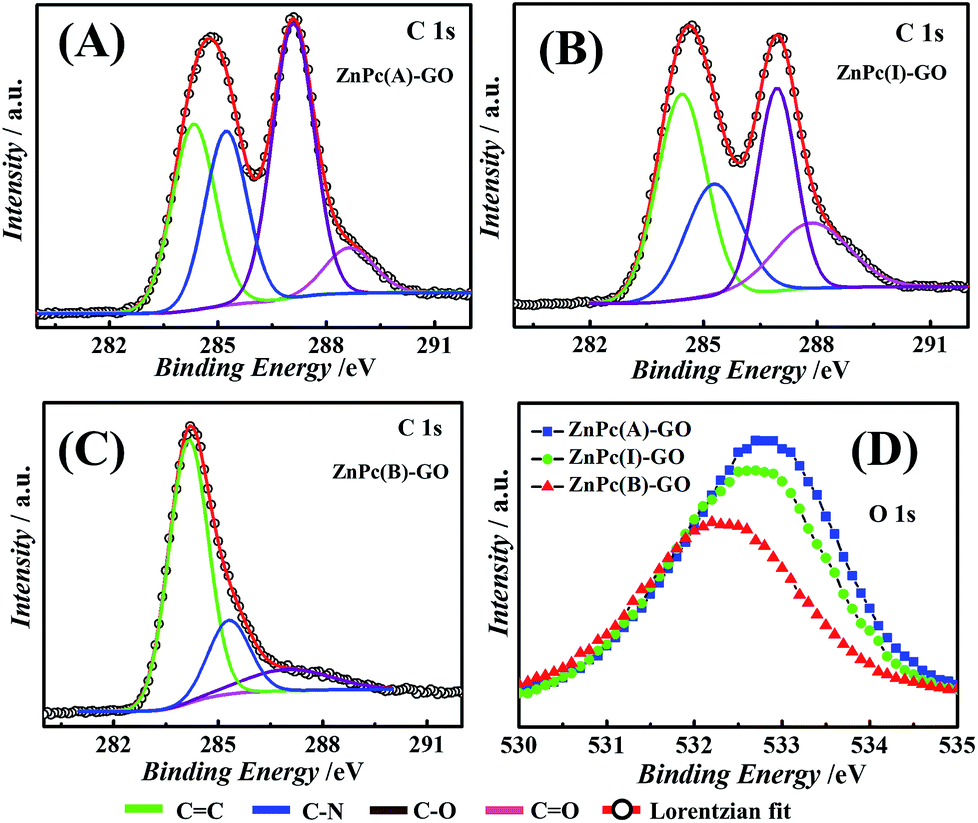 | ||
| Fig. 5 Lorentzian deconvoluted peaks of the XPS spectra for (A) the C peak of ZnPc(A)–GO, (B) the C peak of ZnPc(I)–GO, (C) the C peak of ZnPc(B)–GO and (D) the O peaks of ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO. The symbols ‘C’ and ‘O’ represent elemental carbon and oxygen, respectively. | ||
Fig. 5(B) shows the peaks at 284.4 eV, 285.2 eV, 286.9 eV and 287.9 eV in the C 1s XPS spectrum of ZnPc(I)–GO, which correspond to the CC, C–N, C–O and CO bond interactions, respectively.59 Furthermore, it was observed that the binding energy (eV) and elemental interactions of O and N with C declined gradually from ZnPc(A)–GO to ZnPc(I)–GO. These results indicated a decrease in oxygen with the increment in temperature from 25 °C to 100 °C. In Fig. 5(C), the peaks at 284.2 eV, 285.3 eV, and 287.1 eV in the C 1s XPS spectrum of ZnPc(B)–GO correspond to the CC, C–N, and C–O bond interactions, respectively. These results were also associated with a significant increase in the peak intensity of the CC species. However, the peak for the CO bond interaction was not observed in this case, thereby suggesting a significant depletion in the amount of oxygen molecules.59,69 The depletion in oxygen content could be attributed to partial evolution of reduced GO (rGO) in the ZnPc(B)–GO samples.69
The Lorentzian deconvolution of the XPS spectra of the ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO for O peaks, as shown in Fig. 5(D), clearly suggests a decrement in the oxygen species with the rise in temperature from 25 °C to 280 °C. This observation was in agreement with the results obtained in Fig. 5(A–C). It was evident from the observation that the sp2 carbon present in the graphitic domain of GO interacted with the Pc ring of the ZnPc molecules through a π-stacking mechanism, resulting in the transfer of the electrons from the GO to the Pc moiety. This observation was in agreement with the UV-visible studies, as reported in Fig. 2(A). The depletion of oxygen could be attributed to the fact that, in the presence of ZnPc(B) molecules, all the CO bonds (the epoxy bond or the carboxylic bonds) of the GO flakes might have been disintegrated or destroyed at the high temperature of 280 °C. This phenomenon could also have its origin in the partial conversion of GO to rGO69via thermal reduction, and thus only a very small amount of oxygen in the form of the C–O bond was recorded in Fig. 5(C).
Furthermore, these observations suggest that, unlike previously reported cobalt phthalocyanine (CoPc)–GO systems,47 the interaction between GO and ZnPc was a one-way process. In such a scenario, the electron transfer occurs from the GO sheets to the ZnPc(B) molecules. The ZnPc(B)–GO hybrids lacked ligand-like attacking of the oxygen-functional groups of GO to the central zinc ions of ZnPc(B) molecules owing to the absence of adequate oxygen-functional groups.
3.6. Electrical conductivity and surface potential measurements
Fig. 6(A) shows the current (I)–voltage (V) measurements for the ZnPc–GO hybrids performed using a 6217B source meter. The conductivity measurements were done on the ZnPc–GO hybrids fabricated over the ITO-coated glass surface with a work function value of about 4.7 eV.70 Previous studies have shown that the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of ZnPc molecules are situated at ∼5.3 eV and ∼3.3 eV, respectively.71 As the energy band gap between the HOMO band of the ZnPc and the ITO electrode was very small (∼0.3 eV), the I–V characteristics exhibited the rectifying characteristics of a Schottky junction.72 In this work, the incorporation of GO (with a work function of ∼4.3 eV) created a metastable entity within the HOMO–LUMO band of the ZnPc–GO heterostructure, which helped in increasing the diffusion of the separated charged species, owing to the fact that GO can be used as an efficient hole transport material.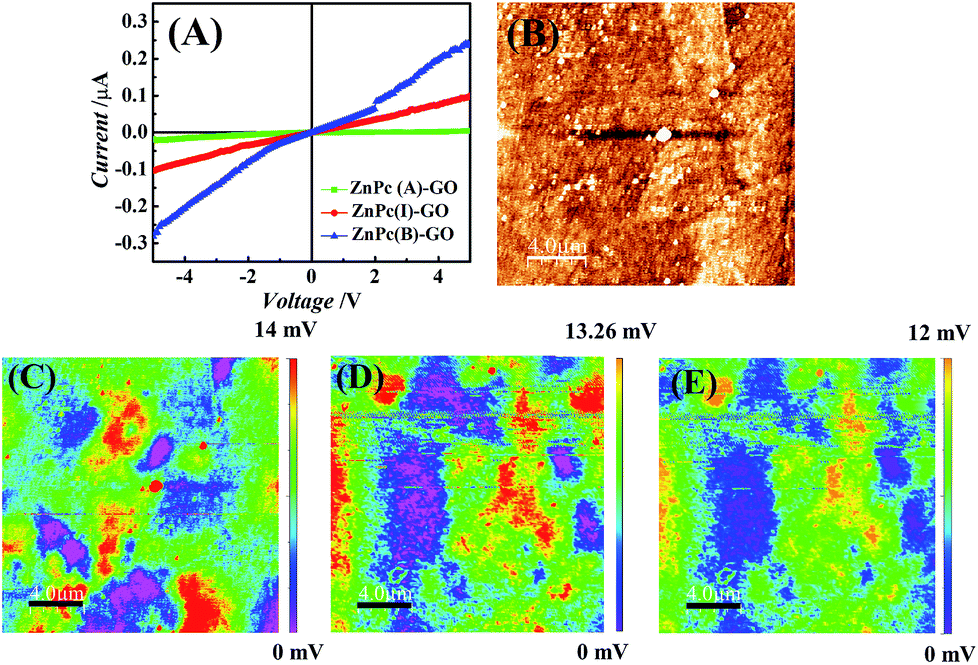 | ||
| Fig. 6 (A) I–V curve measurements of the ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO samples. (B) AFM topography image of ZnPc–GO over an ITO-coated glass substrate. The KPFM images or surface potential mapping of the (C) ZnPc(A)–GO, (D) ZnPc(I)–GO and (E) ZnPc(B)–GO samples. The scale bar in all the images is 4 μm in size. The colour gradient bars in images (C) and (D) represent the changes in the surface potential (SP) of the analysed samples. | ||
Fig. 6(A) also shows an increase in the conductivity for ZnPc(B)–GO samples as compared to its ZnPc(A) counterparts. A 7.4% increase in the conductivity was recorded for ZnPc(B)–GO molecules with respect to ZnPc(A)–GO samples. The temperature-dependent increment in conductivity was in good agreement with the results reported by Chunder et al. for the interaction between the GO sheets and the tetrasulfonate salt of copper phthalocyanine (TSCuPc).73 The exciton separation to form electrons and holes at the interface of ZnPc(B)–GO was found to be very efficient, which favoured a well suited electron–hole separation at the interface. The GO moiety could also play a dual role of electron–hole separation and hole transportation agent owing to good efficiency in hole transportation.74,75 The heating of ZnPc at 280 °C led to the evolution of more ordered and extended structures of ZnPc(B) molecules in the ZnPc(B)–GO hybrids, thereby leading to the decrease in the interplanar distance of the stacking molecules. Furthermore, the partial evolution of rGO in the ZnPc(B)–GO hybrid reduced the grain boundary, leading to improvement in the charge transport properties.
The electrical conductivity enhancements were further extended in terms of the measuring of the work function on the ZnPc(A/B)–GO hybrids with the help of the AFM Pt/Ir conducting tips. The tip was given an appropriate bias (0.5 V) to map the SP of the surfaces with variation in temperature using KPFM or surface potential microscopy (SPoM) measurements.56Fig. 6(B) shows the AFM topography of the ZnPc(A)–GO hybrid over the ITO-coated glass substrate and the SPoM measurements of the ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO hybrids are reported in Fig. 6(C)–(E), respectively. The figures suggest that the SP of the ZnPc–GO hybrids altered with the rise in temperature from 25 °C to 280 °C.
Fig. 6(C) shows the SPoM images of ZnPc(A)–GO consisting of several localized red spots, thereby suggesting the presence of higher SP zones over the sample surface. Conversely, the same localized red spots were not observed in the case of ZnPc(I/B)–GO, as shown in Fig. 6(D) and (E). These observations clearly suggest that the SP decreased with the increase in the heating temperature, as seen in the case of the ZnPc(B)–GO hybrid. The overall work function values for the ZnPc(A)–GO, ZnPc(I)–GO and ZnPc(B)–GO samples were calculated to be 4.504 eV, 4.506 eV and 4.508 eV, respectively. The details of the work function (ϕs) calculations for various experimental samples are tabulated in Table S2 (refer to the ESI†). The decrease in the work function over the ZnPc(B)–GO surface indicated a significant increase in the electron–hole separation, thereby resulting in the increment of the overall electrical conductivity. The presence of lower surface potential domains in the ZnPc(B)–GO sample at 280 °C, compared to the ZnPc(A)–GO counterpart at 25 °C, led to the increment in the electrical conductivity of the ZnPc(B)–GO sample, which was in agreement with the I–V curve measurement, as shown in Fig. 6(A).
3.7. Interaction of bacterial cells with ZnPc–GO
It may be noted here that a Gram-negative bacterium cell has a supplemental outer membrane composed of phospholipids and lipopolysaccharides (LPS). The presence of carboxylate and phosphate groups in the peptidoglycan and LPS layers imparts a net negative charge to the surface of these cells.76 The net negative surface charge of the bacterial cells helps them to adhere and proliferate as biofilms on positively charged abiotic surfaces.77 Thus, the facilitation of an electron transfer interaction between the negatively charged bacterial membrane to an electron-withdrawing or electropositive surface may disrupt the surface potential of the bacterial cell membrane, thereby resulting in cell death.29In this line, we propose a novel GO-based antibacterial material, namely ZnPc–GO, which relies on electronic charge transfer from the negatively charged bacterial cell membrane to the ZnPc–GO entities for inactivation of bacterial cells. Furthermore, we attempt to uncover the underlying mechanism behind the ZnPc–GO antibacterial activity using KPFM or surface potential microscopy (SPoM). E. coli, a Gram-negative bacterium, was used as a model prokaryotic organism for all the experiments performed in this work. In order to uncover the antibacterial activity of the ZnPc–GO entities and mechanism of electrostatic interaction between the ZnPc–GO and E. coli cells, the diluted bacteria culture solution was introduced over the ZnPc–GO surface, as mentioned in the experimental section. The spatiotemporal variation in the bacterial cell morphology was investigated, in order to understand the influence of ZnPc–GO on cell integrity.
The low bacterial cell count in Fig. 7(A) and (B) clearly suggests that the bacterial cells exhibited very minimal adherence on the GO nanosheets and the ZnPc(A)–GO surface, respectively. However, the ZnPc(B)–GO surface possesses favourable surface conditions for the adherence of bacterial cells. Fig. 7(C) shows that the maximum number of cells were found to adhere on the ZnPc(B)–GO surfaces, which suggests that the negatively charged E. coli interacts preferentially with the ZnPc(B)–GO surface via electrostatic interaction. The previous characterization results already confirmed that the GO nanoflakes showed a strong π–π interaction with ZnPc(B). This strong π–π interaction led to the transition of free electrons from GO sheets to the ZnPc(B),60 whereas, the ZnPc(A) molecules failed to show a profound electronic interaction with the GO nanoflakes. The maximal bacterial cell adherence on the ZnPc(B)–GO surface might be owing to the electron-charge transfers from the GO surface to the ZnPc(B) molecules, which rendered the GO surface more electron-deficient. Subsequently, the GO surface assumed a partial electropositive charge. Thus, owing to the high count of adhered bacterial cells, the ZnPc(B)–GO nanocomposite was considered to be the best configuration for bacterial cell capture and antibacterial studies. Henceforth, all the KPFM measurements were performed using ZnPc(B)–GO. Before performing the KPFM study, we carried out an antibacterial efficacy test with ZnPc(B)–GO with the help of confocal microscopy. The details of the results are incorporated in Fig. S4 in Section 5 of the ESI.†78
 | ||
| Fig. 7 AFM micrographs showing the adherence of E. coli bacterial cells on (A) GO nanosheets (control) and (B) ZnPc(A)–GO and (C) ZnPc(B)–GO samples. The scale bar in all the images is 10 μm in size. | ||
Previous studies have shown that after cell death, the bacterial outer membrane disintegrates and the internal cellular components leak out from the dead bacterial cells.29 LPS molecules, which provide the cell surface potential to the live bacteria, also leak from the cell environment. Subsequently, the process diminishes the overall negative cell potential of the dead bacteria.29 This provided us with the hint that the measurement of the surface potential across a bacterial cell could be one approach to differentiate a live bacterium from a dead one. Importantly, the surface potential measurement technique could be implemented using the KPFM mode of AFM. Thus, we employed this method to determine the fate of the bacterial cells in the presence of ZnPc(B)–GO.
The work function of the sample (ϕs, in eV) was calculated by AFM by using Pt/Ir tips in KPFM mode. The measurements were carried out in dual-pass lift mode in order to avoid cross-talk between the surface topography and SP measurements. The work function of the Pt/Ir-coated AFM tip was calibrated using a highly ordered pyrolytic graphite (HOPG) sample as standard. The work function of the tip (ϕt, in eV) was calibrated to be ∼4.52 eV after three repetitions. The KPFM measurements provided the overall contact potential difference (CPD, or VCPD in V) values for the measured sample. The contact potential difference or CPD (VCPD) was measured by KPFM method and the work function of the tip (ϕt) was determined by calibration. Further, the surface potential (ϕs) of the sample was calculated as ϕs = ϕt − eVCPD, where e is the unit electronic charge. The details of the surface potential (ϕs) calculations for various experimental samples are tabulated in Table S2 (refer to the ESI†). The interaction between the ZnPc(B)–GO and the bacterial cell provided the resultant CPD and thus the work function of the sample (ϕs) was estimated from the calculations.
The ZnPc(B)–GO sample was retrieved from the well after a period of 5 min (without incubation) and immediately mounted on the AFM sample-holder stage then analysed in KPFM mode. All the KPFM analysis in this work was performed without chemically or heat fixing the cells on to the sample or substrate in order to keep the cell nourished and alive for real-time monitoring of SP variation using the SP mapping technique. Out of the large distribution, a typical E. coli cell was selected and the length of the bacterial cell was found to be ∼2.5 μm with a typical height of ∼0.3 μm.
Fig. 8(A) shows the captured AFM phase image of an E. coli population adhered on a ZnPc(B)–GO sample. Fig. 8(B) shows the surface height topology of a single bacterial cell. The SP mapping was performed on the bacterial cell, as shown in Fig. 8(C), and the resultant CPD value was recorded to be ∼82 mV and the work function of the sample (ϕs) was estimated to be ∼4.438 eV. Fig. 8(C) clearly depicts that most of the electronic charge was confined within the intact bacterial cell at this point in time.
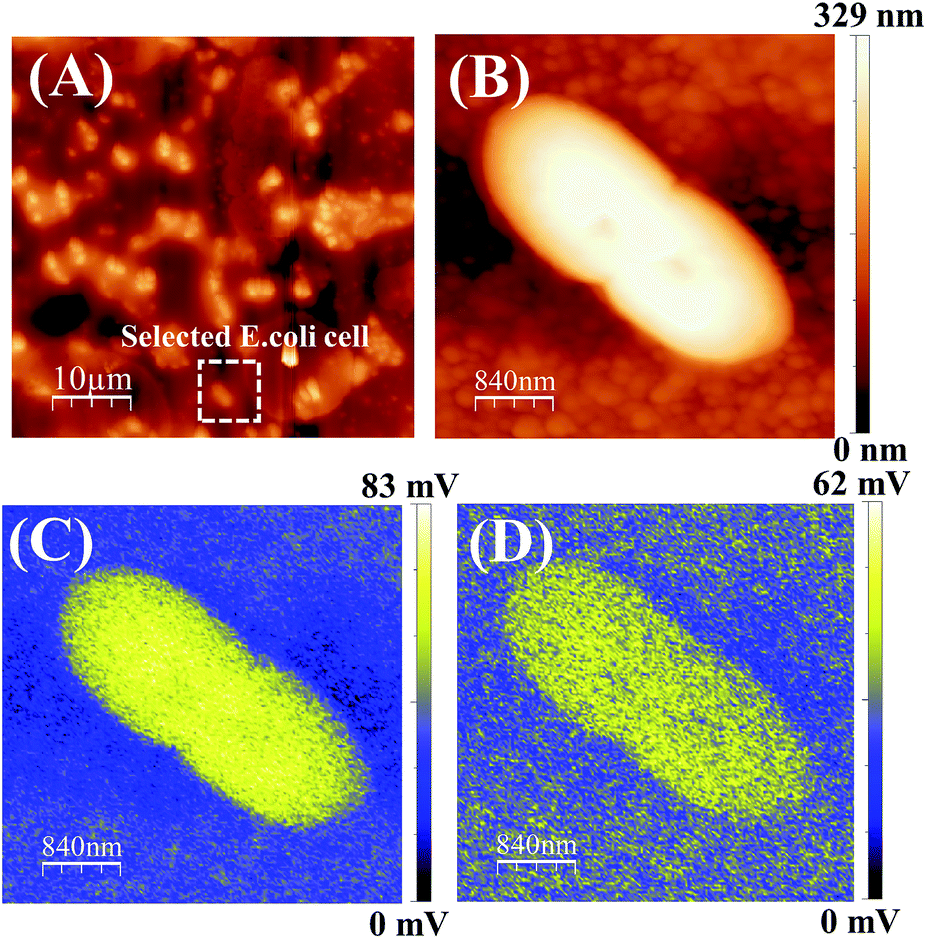 | ||
| Fig. 8 (A) AFM phase micrograph of an E. coli population adhered on the ZnPc(B)–GO sample. The sample was kept at room temperature for 5 min, without incubation. The white square in the image represents the bacterial cell selected for KPFM analysis. (B) Surface height topology of the selected bacterial cell. Surface potential (SP) mapping of the selected bacterial cell after (C) 0 min and (D) a 30 min stand-by time period. The scale bars in images (A) and (B–D) are 10 μm and 840 nm, respectively. | ||
The sample was left on the stage for another 30 min and the KPFM analysis was again performed on the same bacterial cell and its corresponding SP mapped image is shown in Fig. 8(D). The net CPD value was recorded to be ∼62 mV and work function of the sample (ϕs) was estimated to be ∼4.459 eV in this case. Interestingly, a variation in the charge distribution area was observed across the cell, as shown in Fig. 8(D). This experiment indicated a change in the cell surface potential of the bacteria with a noticeable increment in the work function of the sample (ϕs) from ∼4.438 eV to ∼4.459 eV. Although the cell structure did not show any signs of structural deformation, however, a variation of the surface potential (SP) was clearly visible from the work function measurement. The increment in the work function and the electrostatic surface potential of the bacterial cell surface clearly indicated that the bacterium was losing free electrons from its outer membrane. Subsequently, the electrons were transferred from the cell to the ZnPc(B)–GO sample. The surface potential gradient diffused out of the cell, suggesting probable minor leakage of LPS molecules from the bacterium to the surrounding environment.79,80 A change in the SP or work function was not sufficient to conclude that the bacterial cells under consideration were not viable. Thus, we further extended the study to include the results after incubating for longer time durations.
For further investigations, we incubated the ZnPc(B)–GO sample with the bacterial cell structure for 2 h, and KPFM analysis was performed. Fig. 9(A) shows the surface height topology of two bacterial cells. The surface height of the E. coli was found to decrease from ∼0.3 μm to ∼0.16 μm, probably owing to the outer membrane thinning and loss in cellular integrity. Fig. S3 (refer to the ESI†) presents the FESEM images of an E. coli cell prior to and after incubation with the ZnPc(B)–GO nanocomposite for 2 h. The FESEM images show the morphological changes in the E. coli bacterial cell after 2 h incubation, thereby indicating the onset of cell membrane degradation.
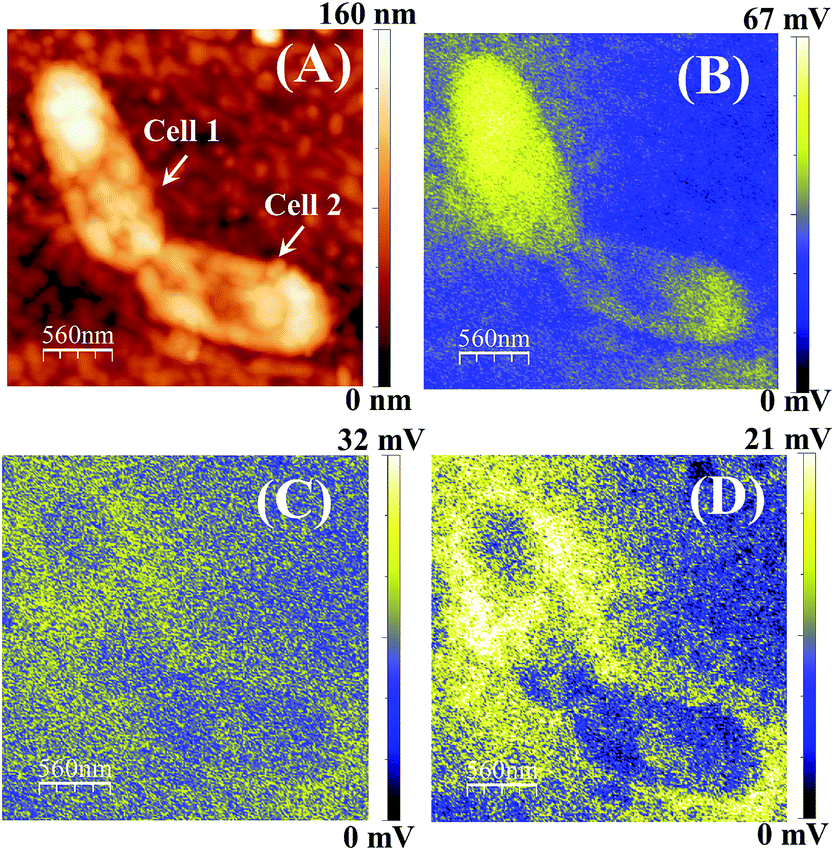 | ||
| Fig. 9 (A) Surface height topology of E. coli cells adhered on the ZnPc(B)–GO sample captured after 2 h of incubation. Surface potential (SP) mapping of the bacterial cells analysed at (B) 0 min, (C) 30 min and (D) after a 1 h stand-by time period, after 2 h of incubation. The scale bar in all images is 560 nm. | ||
The SP mapping was performed on the bacterial cell, as shown in Fig. 9(B), and the resultant CPD value was recorded to be ∼67 mV. Subsequently, the work function of the sample (ϕs) was estimated to be ∼4.453 eV. The SP mapping image of bacterial cells on the ZnPc(B)–GO samples in Fig. 9(B) clearly depicts that the majority of the electronic charge was confined within the bacterial membrane of the cell. However, minor leakage of charged species from the bacterial cell was also observed in this case. The sample was left on the stage for another 30 min and the KPFM analysis was again performed on the same bacterial cell, as shown in Fig. 9(C). The net CPD value was recorded to be ∼32 mV and the work function of the sample (ϕs) was estimated to be ∼4.488 eV in this case. After a stand-by time period of 30 min, the increment in the yellow region indicates profound LPS leakage with visible structural deformation in the bacterial cells under investigation.
The SP of the sample was further mapped in KPFM mode after 1 h time lapse. Fig. 9(D) shows the apparent scattering of charged membrane proteins from the cell, depicted as a yellow coloured area. The prevalent change in SP across the bacteria cell membrane is also shown, which indicates membrane damage and cytoplasmic leakage.
The net CPD value and work function of the sample (ϕs) were recorded to be ∼21 mV and ∼4.499 eV, respectively, in this case. The observations indicated that with the increase of the stand-by time period after 2 h of incubation, the work function of the ZnPc(B)–GO–E. coli system also increased. This increment of the work function was intensely correlated with the change in the electrostatic surface potential gradient of the cell surface owing to the cytoplasmic leakage and cell damage.29Fig. 9(B–D) also suggest that the leaked LPS molecules did not bleed quickly after the rupture of the outer cellular membrane. However, the bleeding was rather progressive for a certain span after the charge-transfer-induced cell damage occurred.
3.8. Proposed mechanism for E. coli and ZnPc(B)–GO interaction
Fig. 10(A) depicts the physical interaction of the Gram-negative E. coli cells with the ZnPc(B)–GO system in which the GO nanoflakes were positioned on a random array of ZnPc(B) nanorods. A schematic is shown in Fig. 10(B) in order to illustrate the proposed mechanism of the interaction observed in Fig. 10(A). The scheme also helps in gaining a brief understanding of the bacterial cell response to the GO flakes film on the ZnPc(B) molecule.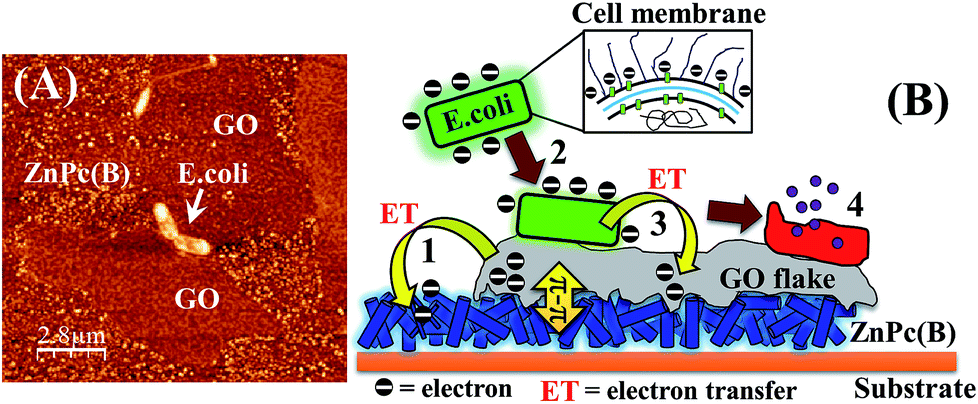 | ||
| Fig. 10 (A) An AFM phase micrograph of E. coli cells adhered on ZnPc(B)–GO after 2 h incubation and a stand-by time period of 1 h under ambient conditions. (B) Schematic diagram illustrating the proposed mechanism for the interaction between the E. coli cells and ZnPc(B)–GO. | ||
According to Fig. 10(B), the mechanism of the interaction between the E. coli cells and the ZnPc(B)–GO along with the antibacterial activity of the ZnPc(B)–GO can be explained as follows:
(1) The deposition of GO nanoflakes on the array of ZnPc(B) nanorods facilitated an electronic interaction of the GO flakes with the underlying ZnPc(B) molecules via the strong π–π interaction between ZnPc(B) and the graphitic domain of the GO flakes.60 The strong π–π interaction between the components of the ZnPc(B)–GO system resulted in the relocation of the electrons from the GO sheets to the Pc group of the underlying ZnPc(B) molecules.
(2) The electron-charge transfer from the GO flakes to the ZnPc(B) molecules renders the GO surface more electron-deficient. This imparted partial electropositive surface potential to the GO surface. Passive evidence for the change of the electronic charge surrounding each component was investigated by measuring the ζ-potential, as tabulated in Table S3 (refer to the ESI†). The peptidoglycan and LPS layers present in the outer membrane of the bacterial cell imparted a net negative charge to the surface of these cells. The electropositive GO surface facilitated the adherence to these negatively charged E. coli cells.
(3) The physical interaction of the adhered E. coli cells with the GO layer of the ZnPc(B)–GO nanocomposite was resulted in the formation of a Schottky barrier owing to Fermi level alignment. This arrangement promoted electron transfer from the outer membrane of the bacterial cell to the acceptor GO flakes.29 The negative surface potential of the bacterial cells was measured to be in the range of 32–48 mV.81 The bacterial cell membrane started progressively losing electrons to the GO flakes, which transferred these acquired electrons to the underlying ZnPc molecules to form a circuit for electron transfer.
(4) The ZnPc(B)–GO system acted as an electron pump, which progressively transferred the electrons away from the bacterial cell membrane. The continuous electron extraction from the cells results in the development of ROS-dependent oxidative stress in the bacteria,29 which eventually caused the destruction of the membrane integrity. Fig. S6 in the ESI† shows the progressive generation of ROS when the ZnPc(B)–GO interacts with E. coli bacteria and might be one of the causes of the rupture of the bacterial cell walls.29,82,83 Subsequently, the membrane might have bled the cytoplasmic proteins, resulting in the deformation of the cell before death. A test carried out with the help of UV-visible spectroscopy provided proof of cytoplasmic leakage. The details are provided in Fig. S7 of Section 8 in the ESI.†84,85
(5) It was also observed that upon physical interaction with the ZnPc(B) nanorods, the ultrathin GO basal sheets attained the barb-like shape of the underlying randomly arranged ZnPc(B) nanorods. Upon direct interaction with the protective outer membrane of the bacterial cell, there was a chance that the sharp protruding edges of the ZnPc(B) nanorods–GO could penetrate into the membrane, resulting in mechanical rupture of the phospholipids of the cell membrane, effecting irreversible damage to the cell membrane integrity.
In a nutshell, aided by state-of-art Kelvin probe force microscopy (KPFM), we have shown the dissipation of charge from a single bacterial cell during the interaction with ZnPc–GO nanohybrids, which leads to the death of E. coli cells. The time-dependent cellular damage and leakage of intrinsic proteins and nucleic acids from the bacterial cell have been captured by employing KPFM mapping and UV-visible spectroscopy. The mapping directly showed that the surrounding space of a single bacterial cell might be populated with progressive leaking of the lipopolysaccharide (LPS) resulting in the change in the contact potential (VCPD) with respect to the AFM conducting tips.
In order to implement the protocol, we chose a ZnPc–GO hybrid nanomaterial. The electropositive ZnPc (established by ζ-potential measurement) was found to tune the electronegativity of the GO surface, which was again established by ζ-potential measurement. In order to confirm the interaction of ZnPc in its two different α- and β-phases, structural and optical characterization was carried out with AFM, FESEM, Raman and Raman mapping. The experiments uncovered that there is a progressive physical interaction between ZnPc and GO with the former in its two different phase formats. The experiments also corroborated the variation of the surface charge with the structural variation owing to the interaction of the ZnPc and GO moieties.
The main objective of the phase variant ZnPc interaction with GO was to fine-tune the surface charge of GO targeting increased adherence of Gram-negative E. coli bacteria on its surface. This might have led to the bleeding of the charge from the bacterial cell while maintaining charge equilibrium at the bacteria–composite interface. Thus, the ZnPc tuned, electron-deficient GO flakes were found to scavenge free charge from the bacterial surface, which led to the sudden change in the surface potential on the bacterial cell, causing its death. The experiments on the charge assimilation-induced bacterial death unearthed an unprecedented pathway of antimicrobial activity of 2D nanohybrids, which could be useful for therapeutics without the use of drugs.
4. Conclusions
A 2D ZnPc–GO nanocomposite was prepared to study the interaction of ZnPc with GO sheets for α-ZnPc(A) and β-ZnPc(B) phase orientations. The phase transformation from α-ZnPc(A) to β-ZnPc(B) was done by heating from room temperature to 280 °C. The ZnPc(B) molecules demonstrated better interaction with the GO nanosheets while the ZnPc(B)–GO nanostructure showed significantly enhanced conductivity and a regulated charge balance. The GO nanoflakes showed strong π–π interactions with ZnPc(B) owing to the transition of free electrons from the GO sheets to ZnPc(B).The 2D ZnPc(B)–GO nanohybrids thus obtained were tested for antimicrobial activity, which was explored through physical electrostatic interaction rather than chemical interaction. The long-prevailing notions of bacterial interaction with graphene derivatives, such as cutting (leading to leakage of cytoplasm), wrapping (leading to stress generation) and trapping (generation of reactive oxygen species; ROS), were extended from electrostatic interaction experiments between ZnPc(B)–GO nanohybrids and bacterial cells with the help of KPFM. The experiments uncovered the death of E. coli through dissipation of charge from the surface. The time-dependent cellular damage and progressive leakage of intrinsic LPS from the bacterial cell membrane were captured using KPFM mapping. The electron-deficient GO flakes after their attachment with ZnPc(B) were found to scavenge free charge from the bacterial surface, which led to rapid changes in the surface potential on the bacterial cell, causing its death.
The results reported here may lead to a very new pathway of charge-transfer-mediated antibacterial activity using 2D nanohybrid materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank MeitY Grant no. 5(9)/2012-NANO, IMPRINT 8058, and DST SERB Grant no. EMR/2016/001824, Government of India, for financial support. Nayan Mani Das thanks IIT Guwahati for support through the IPDF grant. The authors also acknowledge support from CIF, IIT Guwahati. We thank Dr Dhrubojyoti Roy and Prof. Manabendra Mukherjee for helping us with the XPS characterization facility at SINP, Kolkata India.Notes and references
- N. Woodford and M. J. Ellington, Clin. Microbiol. Infect., 2007, 13, 5–18 CrossRef CAS PubMed.
- M. Frieri, K. Kumar and A. Boutin, Journal of Infection and Public Health, 2017, 10, 369–378 CrossRef PubMed.
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef CAS PubMed.
- M. Chen, Q. Yu and H. Sun, Int. J. Mol. Sci., 2013, 14, 18488–18501 CrossRef PubMed.
- A. C. Rios, C. G. Moutinho, F. C. Pinto, F. S. Del Fiol, A. Jozala, M. V. Chaud, M. M. D. C. Vila, J. A. Teixeira and V. M. Balcão, Microbiol. Res., 2016, 191, 51–80 CrossRef CAS PubMed.
- M. P. Illa, M. Khandelwal and C. S. Sharma, Emergent Materials, 2018, 1, 105–120 CrossRef.
- S. Hosseini, F. Ibrahim, I. Djordjevic and L. H. Koole, Analyst, 2014, 139, 2933–2943 RSC.
- H. A. Hemeg, Int. J. Nanomed., 2017, 12, 8211–8225 CrossRef CAS PubMed.
- J. T. Seil and T. J. Webster, Int. J. Nanomed., 2012, 7, 2767–2781 CAS.
- L. Wang, C. Hu and L. Shao, Int. J. Nanomed., 2017, 12, 1227–1249 CrossRef CAS.
- K. Kaviyarasu, C. M. Magdalane, K. Kanimozhi, J. Kennedy, B. Siddhardha, E. S. Reddy, N. K. Rotte, C. S. Sharma, F. T. Thema, D. Letsholathebe, G. T. Mola and M. Maaza, J. Photochem. Photobiol., B, 2017, 173, 466–475 CrossRef CAS.
- A. Thiha, F. Ibrahim, S. Muniandy, I. J. Dinshaw, S. J. Teh, K. L. Thong, B. F. Leo and M. Madou, Biosens. Bioelectron., 2018, 107, 145–152 CrossRef CAS PubMed.
- J. Mokili, D. Wibowo, S. Kassegne, V. Ramesh, J. Chi, A. Khosla and A. Narenji, IET Nanobiotechnol., 2015, 9, 153–163 CrossRef PubMed.
- J. J. Barr, R. Auro, N. Sam-Soon, S. Kassegne, G. Peters, N. Bonilla, M. Hatay, S. Mourtada, B. Bailey, M. Youle, B. Felts, A. Baljon, J. Nulton, P. Salamon and F. Rohwer, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13675–13680 CrossRef CAS PubMed.
- T. Yaguchi, M. Dwidar, C. K. Byun, B. Leung, S. Lee, Y. K. Cho, R. J. Mitchell and S. Takayama, Biomacromolecules, 2012, 13, 2655–2661 CrossRef CAS PubMed.
- F. I. Uba, S. R. Pullagurla, N. Sirasunthorn, J. Wu, S. Park, R. Chantiwas, Y. K. Cho, H. Shin and S. A. Soper, Analyst, 2015, 140, 113–126 RSC.
- E. Roy, G. Stewart, M. Mounier, L. Malic, R. Peytavi, L. Clime, M. Madou, M. Bossinot, M. G. Bergeron and T. Veres, Lab Chip, 2015, 15, 406–416 RSC.
- V. B. Mohan, K. Lau, D. Hui and D. Bhattacharyya, Compos. B Eng., 2018, 142, 200–220 CrossRef CAS.
- V. Singh, D. Joung, L. Zhai, S. Das, S. I. Khondaker and S. Seal, Prog. Mater. Sci., 2011, 56, 1178–1271 CrossRef CAS.
- H. Ji, H. Sun and X. Qu, Adv. Drug Delivery Rev., 2016, 105, 176–189 CrossRef CAS PubMed.
- F. Perreault, A. F. De Faria, S. Nejati and M. Elimelech, ACS Nano, 2015, 9, 7226–7236 CrossRef CAS PubMed.
- A. N. Banerjee, Interface Focus, 2018, 8, 20170056 CrossRef PubMed.
- M. D. Rojas-Andrade, G. Chata, D. Rouholiman, J. Liu, C. Saltikov and S. Chen, Nanoscale, 2017, 9, 994–1006 RSC.
- S. Szunerits and R. Boukherroub, J. Mater. Chem. B, 2016, 4, 6892–6912 RSC.
- J. Liu, J. Tang and J. J. Gooding, J. Mater. Chem., 2012, 22, 12435–12452 RSC.
- S. Shi, F. Chen, E. B. Ehlerding and W. Cai, Bioconjugate Chem., 2014, 25, 1609–1619 CrossRef CAS PubMed.
- K. A. Whitehead, M. Vaidya, C. M. Liauw, D. A. C. Brownson, P. Ramalingam, J. Kamieniak, S. J. Rowley-Neale, L. A. Tetlow, J. S. T. Wilson-Nieuwenhuis, D. Brown, A. J. McBain, J. Kulandaivel and C. E. Banks, Int. Biodeterior. Biodegrad., 2017, 123, 182–190 CrossRef CAS.
- S. Panda, T. K. Rout, A. D. Prusty, P. M. Ajayan and S. Nayak, Adv. Mater., 2018, 30, 1702149 CrossRef PubMed.
- J. Li, G. Wang, H. Zhu, M. Zhang, X. Zheng, Z. Di, X. Liu and X. Wang, Sci. Rep., 2014, 4, 4359 CrossRef PubMed.
- X. Zou, L. Zhang, Z. Wang and Y. Luo, J. Am. Chem. Soc., 2016, 138, 2064–2077 CrossRef CAS.
- L. Shi, J. Chen, L. Teng, L. Wang, G. Zhu, S. Liu, Z. Luo, X. Shi, Y. Wang and L. Ren, Small, 2016, 12, 4165–4184 CrossRef CAS.
- V. Palmieri, M. C. Lauriola, G. Ciasca, C. Conti, M. D. Spirito and M. Papi, Nanotechnology, 2017, 28, 1–18 CrossRef.
- S. N. Nyamu, L. Ombaka, E. Masika and M. Ng’ang’a, Adv. Chem., 2018, 2598062, 1–8 Search PubMed.
- D. Mondal and S. Bera, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2014, 5, 033002 Search PubMed.
- Y. Gao, B. Mai, A. Wang, M. Li, X. Wang, K. Zhang, Q. Liu, S. Wei and P. Wang, Photodiagn. Photodyn. Ther., 2018, 21, 316–326 CrossRef CAS PubMed.
- P. Mikula, L. Kalhotka, D. Jancula, S. Zezulka, R. Korinkova, J. Cerny, B. Marsalek and P. Toman, J. Photochem. Photobiol., B, 2014, 138, 230–239 CrossRef CAS PubMed.
- D. Roy, N. M. Das, N. Shakti and P. S. Gupta, RSC Adv., 2014, 4, 42514–42522 RSC.
- S. Eken Korkut, D. Akyüz, K. Özdogan, Y. Yerli, A. Koca and M. K. Şener, Dalton Trans., 2016, 45, 3086–3092 RSC.
- O. M. Bankole and T. Nyokong, New J. Chem., 2016, 40, 10016–10027 RSC.
- C. Nitschke, S. M. O'Flaherty, M. Kröll and W. J. Blau, J. Phys. Chem. B, 2004, 108, 1287–1295 CrossRef CAS.
- A. R. N. Zamani, M. R. Mashayekhi, M. F. S. Jadid, Y. Faridvand, H. Tajalli and R. Rahbarghazi, Lasers Med. Sci., 2018, 33, 1969–1978 CrossRef.
- P. Hu, Z. Chen, L. Li, J. Chen, S. Chen, M. Huang and S. Zhou, J. Lumin., 2013, 152, 103–107 Search PubMed.
- M. R. Ke, J. M. Eastel, K. L. K. Ngai, Y.-Y. Cheung, P. K. S. Chan, M. Hui, D. K. P. Ng and P.-C. Lo, Eur. J. Med. Chem., 2014, 84, 278–283 CrossRef CAS.
- N. Karousis, J. Ortiz, K. Ohkubo, T. Hasobe, S. Fukuzumi, Á. Sastre-Santos and N. Tagmatarchis, J. Phys. Chem. C, 2012, 116, 20564–20573 CrossRef CAS.
- J. J. Bang, S. R. Russell, K. K. Rupp and S. A. Claridge, Anal. Methods, 2015, 7, 7106–7127 RSC.
- S. Senthilarasu, S.-J. Baek, S. D. Chavhan, J. Lee and S.-H. Lee, J. Nanosci. Nanotechnol., 2008, 8, 5414–5417 CrossRef CAS.
- J. H. Yang, Y. Gao, W. Zhang, P. Tang, J. Tan, A. H. Lu and D. Ma, J. Phys. Chem. C, 2013, 117, 3785–3788 CrossRef CAS.
- G. de la Torre, T. Torres, D. M. Guldi, G. Katsukis, A. Roth, J. Malig and M.-E. Ragoussi, J. Am. Chem. Soc., 2014, 136, 4593–4598 CrossRef PubMed.
- A. K. Singh, K. K. Dey, A. Chattopadhyay, T. K. Mandal and D. Bandyopadhyay, Nanoscale, 2014, 6, 1398–1405 RSC.
- A. K. Singh, S. Rarotra, V. Pasumarthi, T. K. Mandal and D. Bandyopadhyay, J. Mater. Chem. A, 2018, 6, 9209–9219 RSC.
- T. Bhuyan, A. K. Singh, D. Dutta, A. Unal, S. S. Ghosh and D. Bandyopadhyay, ACS Biomater. Sci. Eng., 2017, 3, 1627–1640 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- S. Kumar, A. K. Singh, A. K. Dasmahapatra, T. K. Mandal and D. Bandyopadhyay, Carbon, 2015, 89, 31–40 CrossRef CAS.
- N. M. Das, S. Kumar and D. Bandyopadhyay, Carbon, 2017, 121, 612–624 CrossRef CAS.
- U. Goswami, S. Basu, A. Paul, S. S. Ghosh and A. Chattopadhyay, J. Mater. Chem. C, 2017, 5, 12360–12364 RSC.
- W. Melitz, J. Shen, A. C. Kummel and S. Lee, Surf. Sci. Rep., 2011, 66, 1–27 CrossRef CAS.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- A. Chowdhury, B. Biswas and B. Mallik, Sci. Adv. Mater., 2013, 5, 1297–1306 CrossRef CAS.
- W. Song, C. He, W. Zhang, Y. Gao, Y. Yang, Y. Wu, Z. Chen, X. Li and Y. Dong, Carbon, 2014, 77, 1020–1030 CrossRef CAS.
- Z. Wang, C. He, W. Song, Y. Gao, Z. Chen, Y. Dong, C. Zhao, Z. Li and Y. Wu, RSC Adv., 2015, 5, 94144–94154 RSC.
- M. Mukherjee, M. Samanta, U. K. Ghorai, S. Murmu, G. P. Das and K. K. Chattopadhyay, Appl. Surf. Sci., 2018, 449, 144–151 CrossRef CAS.
- J. Zhu, Y. Li, Y. Chen, J. Wang, B. Zhang, J. Zhang and W. J. Blau, Carbon, 2011, 49, 1900–1905 CrossRef CAS.
- M. Szybowicz, W. Bała, K. Fabisiak, K. Paprocki and M. Drozdowski, J. Mater. Sci., 2011, 46, 6589–6595 CrossRef CAS.
- P. N. Nirmalraj, T. Lutz, S. Kumar, G. S. Duesberg and J. J. Boland, Nano Lett., 2011, 11, 16–22 CrossRef CAS.
- M. Szybowicz, W. Bała, K. Fabisiak, K. Paprocki and M. Drozdowski, J. Mater. Sci., 2011, 46, 6589–6595 CrossRef CAS.
- Y. Zhu, Q.-K. Yu, G.-Q. Ding, X.-G. Xu, T.-R. Wu, Q. Gong, N.-Y. Yuan, J.-N. Ding, S.-M. Wang, X.-M. Xie and M.-H. Jiang, Nanoscale Res. Lett., 2014, 9(1–6), 48 CrossRef.
- J. P. Froning, P. Lazar, M. Pykal, Q. Li, M. Dong, R. Zbořil and M. Otyepka, Nanoscale, 2017, 9, 119–127 RSC.
- Y. Yu, M. Zhou, W. Shen, H. Zhang, Q. Cao and H. Cui, Carbon, 2012, 50, 2539–2545 CrossRef CAS.
- M. Lee, J. Lee, S. Y. Park, B. Min, B. Kim and I. In, Sci. Rep., 2015, 5, 11707 CrossRef PubMed.
- M. G. Helander, Z. B. Wang, J. Qiu, M. T. Greiner, D. P. Puzzo, Z. W. Liu and H. Lu, Science, 2011, 332, 944–947 CrossRef CAS PubMed.
- D. O. Grynko, O. M. Fedoryak, P. S. Smertenko, N. A. Ogurtsov, A. A. Pud, Y. V. Noskov and O. P. Dimitriev, J. Phys. D: Appl. Phys., 2013, 46, 495114 CrossRef.
- G. D. Sharma and D. Saxena, J. Mater. Sci.: Mater. Electron., 1999, 10, 539–544 CrossRef CAS.
- A. Chunder, T. Pal, S. I. Khondaker and L. Zhai, J. Phys. Chem. C, 2010, 114, 15129–15135 CrossRef CAS.
- S. Li, Ḱ. K. Tu, Ḱ. C. Lin, C. Chen and M. Chhowalla, ACS Nano, 2010, 4, 3169–3174 CrossRef CAS PubMed.
- C. T. G. Smith, R. W. Rhodes, M. J. Beliatis, K. D. G. Imalka Jayawardena, L. J. Rozanski, C. A. Mills and S. R. P. Silva, Appl. Phys. Lett., 2014, 105, 073304 CrossRef.
- I. M. Helander, I. Kilpeläinen and M. Vaara, FEBS Lett., 1997, 409, 457–460 CrossRef CAS.
- A. Terada, A. Yuasa, T. Kushimoto, S. Tsuneda, A. Katakai and M. Tamada, Microbiology, 2006, 152, 3575–3583 CrossRef CAS.
- S. K. Gogoi, P. Gopinath, A. Paul, A. Ramesh, S. S. Ghosh and A. Chattopadhyay, Langmuir, 2006, 22, 9322–9328 CrossRef CAS.
- I. Bononi, V. Balatti, S. Gaeta and M. Tognon, Appl. Environ. Microbiol., 2008, 74(20), 6470–6472 CrossRef CAS.
- D. B. Kell and E. Pretorius, Integr. Biol., 2015, 7, 1339–1377 CrossRef CAS.
- S. Halder, K. K. Yadav, R. Sarkar, S. Mukherjee, P. Saha, S. Haldar, S. Karmakar and T. Sen, SpringerPlus, 2015, 4, 672 CrossRef PubMed.
- J. F. Woolley, J. Stanicka and T. G. Cotter, Trends Biochem. Sci., 2013, 38(11), 556–565 CrossRef CAS PubMed.
- H. Choi, Z. Yang and J. C. Weisshaar, Proc. Natl. Acad. Sci. U. S. A., 2015, 5, E303–E310 CrossRef PubMed.
- W. Harm, Biological Effects of Ultraviolet Radiation, 1980, ISBN: 9780521293624 Search PubMed.
- Z. Yang, H. Choi and J. C. Weisshaar, Biophys. J., 2018, 114, 368–379 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of GO, XPS measurement data of samples, work function calculations, and FESEM images of bacteria. See DOI: 10.1039/c9na00272c |
| This journal is © The Royal Society of Chemistry 2019 |