
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReconfigurable assembly of charged polymer-modified Janus and non-Janus particles: from half-raspberries to colloidal clusters and chains†
Claudia
Marschelke
ab,
Olga
Diring
ab and
Alla
Synytska
 *ab
*ab
aLeibniz Institute of Polymer Research Dresden e. V., Hohe Straße 6, 01069 Dresden, Germany. E-mail: synytska@ipfdd.de
bDresden University of Technology, Faculty of Mathematics and Science, Institute of Physical Chemistry and Polymer Physics, 01062, Dresden, Germany
First published on 22nd August 2019
Abstract
Understanding the dynamic and reversible assembly of colloids and particles into complex constructs, inspired by natural phenomena, is of fundamental significance for the fabrication of multi-scale responsive and reconfigurable materials. In this work, we investigate the pH-triggered and reconfigurable assembly of structures composed of binary mixtures of oppositely charged polyacrylic acid (PAA)-modified non-Janus and poly(2-dimethylamino)ethyl methacrylate (PDMAEMA)/poly(N-isopropylacrylamide) (PNIPAM)-modified Janus particles driven by electrostatic interactions. Three different target structures are visible both in dispersions and in dry state: half-raspberry structures, colloidal clusters and colloidal chains depending on the mass, numerical and particle size ratio. All formed structures are well-defined and stable in a certain pH range. Half-raspberry-like structures are obtained at pH 6 and numerical ratios NJP/PAA-HP of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :500 (for 200-PAA-HP), 1:44 (for 450-PAA-HP) and 1:15 (for 650-PAA-HP), respectively, due to electrostatic interactions between the central JP and the excessive PAA-HP. Colloidal chains and cluster-like structures are generated at numerical ratios NJP/PAA-HP of 4:5 (for 200-PAA-HP), 4:3 (for 450-PAA-HP), and 4:1 (for 650-PAA-HP). Moreover, the smaller the size of a “connecting” PAA colloid, the larger is the average length of a colloidal chain. Depending on the particle size ratio SJP/PAA-HP, some of the observed structures can be disassembled on demand by changing the pH value either close to the IEP of the PDMAEMA (for half-raspberries) or PAA (for colloidal clusters and chains) and then reassembled into new stable structures many times. The obtained results open a pathway to pH-controlled reconfigurable assembly of a binary mixture composed of polymeric-modified non-Janus and Janus particles, which allow the reuse of particle building blocks.
:500 (for 200-PAA-HP), 1:44 (for 450-PAA-HP) and 1:15 (for 650-PAA-HP), respectively, due to electrostatic interactions between the central JP and the excessive PAA-HP. Colloidal chains and cluster-like structures are generated at numerical ratios NJP/PAA-HP of 4:5 (for 200-PAA-HP), 4:3 (for 450-PAA-HP), and 4:1 (for 650-PAA-HP). Moreover, the smaller the size of a “connecting” PAA colloid, the larger is the average length of a colloidal chain. Depending on the particle size ratio SJP/PAA-HP, some of the observed structures can be disassembled on demand by changing the pH value either close to the IEP of the PDMAEMA (for half-raspberries) or PAA (for colloidal clusters and chains) and then reassembled into new stable structures many times. The obtained results open a pathway to pH-controlled reconfigurable assembly of a binary mixture composed of polymeric-modified non-Janus and Janus particles, which allow the reuse of particle building blocks.
Introduction
Self-assembly is the basic concept whereby complex structures in nature are formed and it comprises all dimensions, starting from the atomic or molecular level, to cellular processes, phenomena of ethology, and cosmic formations such as solar systems and galaxies. In this context, the internal dynamics in nature are a source of inspiration to form controllable and reversible assembly structures using artificial building blocks such as colloidal particles with a rich functionality of the individual particles, and can serve the development of novel materials for the application in micromotors,1–3 photonics/plasmonics,4 drug delivery,5 catalysis,6 solar cells,7 sensors,8 and electronic ink technology.9The assembly process can be triggered, on the one hand, by the intrinsic properties of the colloids such as charges,10–14 amphiphilicity15 and depletion interactions16,17 forming supraparticular assemblies. On the other hand, assembly can be provoked field-assisted by an electric,1,18–20 magnetic21–25 or flow field.26,27 Assemblies of chemically and geometrically isotropic building blocks typically lead to symmetric structures, while colloids with chemical and/or geometrical anisotropies, such as Janus particles, can assemble into more sophisticated, asymmetric structures depending on the mutual orientation.10,11 Taking advantage of their chemically anisotropic structure, the assembly of Janus particles can lead to a variety of structures which are inaccessible to their homogeneous counterparts.
Static self-assembly, which was already intensively studied for non-Janus20,28 and Janus particles,10,11,26,29,30 comprises thermodynamically stable systems in an equilibrium state.31,32 Once these static assemblies are formed, the structures are irreversible.
In contrast to that, dynamic systems include non-equilibrium structures which can be reversibly changed by energy input or an external stimulus including pH, light, and temperature.31,32 Thus, the variation of a chosen parameter or stimulus accomplishes a programmable adaption of the system. This adaptation is reversible upon the removal of the stimulus. Programmed and especially dynamic assemblies of particles and colloids are of importance for fundamental research and offer great potential for understanding of natural sensory processes and their application-oriented imitation.
Nevertheless, there are only few examples of reversible self-assembly of non-Janus12,14,27,33–44 and Janus particles15,25,34,45 discussed in the literature. The reported colloidal building blocks exhibit either a purely soft (organic),14 purely hard (inorganic), or hybrid (soft/hard) character.43 An example for dynamic assemblies of soft colloids was shown by Crassous and coworkers who employed oppositely charged temperature-responsive core–shell particles with complementary shapes for a reversible lock-and-key assembly mechanism.14 Comprehensive studies towards the dynamic self-assembly of soft15,46 as well as hard Janus particles1,2,23,25,29,47,48 were performed by Granick et al. Already in 2011, they studied the kinetic pathways of self-assembly of sulfate polystyrene Janus particles that were made hydrophobic on one hemisphere through successive deposition of titanium and gold thin films, onto which monolayers of n-octadecanethiol were formed.15 They showed that at very low salt concentrations (3.8 mM NaCl), particles repel each other due to electrostatic forces, whereas an increase of the ionic strength weakens the repulsive interactions causing triple helix growth by face-sharing tetrahedral. Later, the group of Granick also investigated the impact of magnetic fields on the assembly of hard half-nickel-coated silica Janus particles.25 By applying a rotating magnetic field of moderate strength, the Janus particles arrange perpendicularly to the field and form hexagonal rotating crystals. Increase of the magnetic field strength causes disaggregation of the crystals into dumbbell-like particle assemblies. In 2016, Velev and co-workers described a thermoresponsive system consisting of polystyrene/iron oxide latex Janus particles.45 The iron oxide-coated patches are selectively wetted with liquid lipids, driving the particle assembly into colloidal clusters above the phase transition temperature via inter-particle capillary bridges. Temperature decrease causes the fluid-to-gel transition of the liquid lipids and subsequent the disassembly of the clusters due to the loss of the capillary forces. Dynamic interactions can also originate from UV-induced switches, as shown by Ren et al. who designed SiO2–Pt Janus catalytic micromotors functionalized with spiropyran moieties49 self-assemble into multiple motors due to electrostatic interactions and π–π stacking induced by UV light irradiation, and disassemble into single motors when the light is switched to green. Recently, Kraft et al. demonstrated the potential of non-Janus colloidal joints with controlled stiffness and motion range by assembling them into flexible colloidal chains (called “colloidal polymers”) and clusters (called “colloidal molecules”).43
The aforementioned examples concern the utilization of either soft latex particles or hard/stiff silica particles, which were partly coated with metals or low-molecular compounds. However, to the best of our knowledge, no reports on the programmable and reconfigurable assembly of organic–inorganic polymer-modified colloidal systems are presented in the literature. These systems are especially attractive due to their broadened diversity of functionalities coming from the polymeric shells in terms of their chemical design and responsiveness to pH, ionic strength, UV light or temperature12 as well as adaptivity. The polymer coating may also impact the kinetics of reversibility of transitions between different structures due to interdiffusion and interpenetration of the polymeric chains. However, stimuli-responsiveness of hairy Janus particles was solely used for the initiation of self-assembly processes,10,11 while switchability of these assemblies caused by changes of ambient parameters remained challenging and has not been investigated yet.
Here, we report on the reconfigurable assembly of structures composed of binary mixtures of poly(2-dimethylamino)ethyl methacrylate (PDMAEMA)/poly(N-isopropylacrylamide) (PNIPAM) Janus and polyacrylic acid (PAA)-decorated non-Janus particles. The particles assemble either in half-raspberry-like structures, colloidal clusters or colloidal chains depending on the particle size ratio SJP/PAA-HP and mass ratio MJP/PAA-HP as well as numerical ratio NJP/PAA-HP. The formed structures are well-defined and stable in a certain pH range. Some of the structures also have the advantage that the particle interactions are bistable. They can be disassembled on demand by changing the pH value, and then reassembled into new stable structures many times.
Experimental section
Materials and methods
Synthesis of spherical SiO2-based non-Janus and Janus particles
A similar procedure was used for the grafting of PtBA on 200 nm, 450 nm, and 650 nm initiator-modified silica particles. For the polymerization, 70 μL PMDTA (0.5 M in DMF), 3 mL tBA, 70 μL CuBr2 (0.1 M solution in DMF), and 0.15 μL ethyl α-bromoisobutyrate (EBiB) were added to the red-fluorescent particles. The mixture was sonicated and purged with Ar, followed by the injection of 150 μL of Sn(II)-2-ethylhexanoate. The polymerization was performed under continuous stirring at 110 °C for 15 minutes. After the particles were purified by centrifugation and redispersion cycles in toluene and dried, hydrolysis was performed to yield polyacrylic acid (PAA). Briefly, PtBA-covered particles were suspended in 10 mL chloroform in a Teflon centrifuge vial, accompanied by adding 4 mL of methanesulfonic acid and rapidly stirring the mixture for five minutes. The modified particles were collected by diluting the mixture with diethyl ether, purified by centrifugation and dried.
Preparation of colloidosomes and grafting of the PDMAEMA. Colloidosomes with 1000 nm large APTES-modified silica spheres were prepared by a wax–water Pickering emulsion approach described elsewhere.50,51 The ATRP-initiator (α-bromoisobutyric acid) was then immobilized onto the exposed particle surface.10 Subsequently, wax was dissolved in hexane, and partly initiator-covered particles were used for polymerization. PDMAEMA was grafted on the modified particles in the same manner as described above for the homogeneously coated particles at 70 °C for 120 min.
Grafting of PNIPAM. The “grafting to” approach was utilized to graft the second polymer onto the particles modified in the previous step (PDMAEMA/NH2-JP). For this purpose, silica particles with the grafted first polymer were dispersed in 25 mL of a 1 wt% carboxy terminated poly(N-isopropylacrylamide) (PNIPAM) solution in ethanol, and stirred for 2 hours. Next, the solvent was evaporated and the particles were annealed at 150 °C overnight. The ungrafted polymer chains were removed by multiple redispersion cycles of particles in appropriate solvents and subsequent centrifugation. As a result, bi-component 1000 nm-PDMAEMA/PNIPAM JPs were obtained.
Assembly experiments
Self-assembly experiments were carried out by mixing different mass ratios MJP/PAA-HP and numerical ratios NJP/PAA-HP of 1000-PDMAEMA/PNIPAM Janus particles (JP) with homogeneously PAA-covered particles (PAA-HP) (Table 1).| PAA-HP diameter [nm] | Particle size ratio SJP/PAA-HP | 1000-PDMAEMA/PNIPAM-JP:PAA-HP |
|||
|---|---|---|---|---|---|
| Mass ratio MJP/PAA-HP | Numerical ratio NJP/PAA-HP | ||||
| 200 | 5 | 1:4 |
100:1 |
1:500 |
4:5 |
| 450 | 2.2 | 1:4 |
15:1 |
1:44 |
4:3 |
| 650 | 1.5 | 1:4 |
15:1 |
1:15 |
4:1 |
For a typical experiment, 1 mg mL−1 dispersions of particles of all sizes were prepared in filtered, aqueous KCl solution (1 or 10 mmol L−1); pH 2, 6 and 9 were adjusted by addition of 0.1 M HCl or KOH. All dispersions were sonicated and stirred at 700 rpm for several hours under cooling to avoid crosslinking of the PDMAEMA shells. Then the particles were mixed in appropriate ratios by taking aliquots from the initial 1 mg mL−1 dispersions depending on the target structures. For the half-raspberry-like structures, the dispersion with 1000-PDMAEMA/PNIPAM-JP was dropwise slowly added to the dispersion of PAA-HP under continuous stirring. For the colloidal chains and clusters, vice versa, the dispersion of PAA-HP was dropwise slowly added to the dispersion of 1000-PDMAEMA/PNIPAM-JP. The resulting mixtures were diluted to 0.5 mg mL−1, stirred at 700 rpm for one hour, and sonicated for 20 minutes. Samples for SEM and FLM imaging were taken at this point. In order to obtain individual micro-clusters in SEM and FLM, the mixtures were diluted five times and sonicated for 20 more minutes. For SEM, 5 μL droplets of the samples were placed on a silicon wafer and left to dry. For investigation of the pH-triggered reversibility, variation of the pH value was carried out under potentiometric control by addition of 0.1 M HCl or KOH. After adjustment of the pH value, the dispersions were stirred at 700 rpm for one hour and sonicated for 20 minutes before samples for SEM and FLM were taken again. For statistical analysis, 250 clusters taken from five samples at each pH value were counted via FLM. SEM images were always acquired to investigate the stability of the formed constructs in dry state.
A complete overview of the prepared samples using different particle size ratios SJP/PAA-HP is displayed in Table 1. The mass ratio MJP/PAA-HP was used as parameter for the preparation of the binary particle mixtures and describes the applied mass of JP per mass of PAA-HP. The numerical ratio NJP/PAA-HP describes the applied number of 1000-PDMAEMA/PNIPAM-JP per PAA-HP and was estimated using eqn (1):
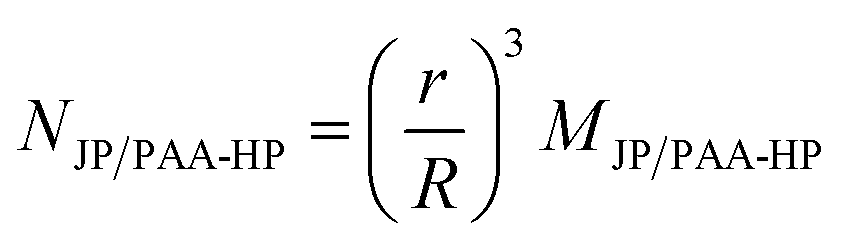 | (1) |
Results and discussion
Synthesis and characterization of homogeneous and Janus particles
:1 (PDMAEMA:PNIPAM), which corresponds with the depth of the particle penetration into the wax phase during the colloidosome preparation process as described elsewhere (Fig. 2b and c).52
| Sample ID | D core (SEM) [nm] | D core+shellH | H dry (TGA) [nm] | IEP |
|---|---|---|---|---|
| 200-PAA-HP | 208 ± 11 | 734 | 28 | <2 |
| 450-PAA-HP | 446 ± 18 | 940 | 14 | <2 |
| 650-PAA-HP | 639 ± 15 | 1203 | 19 | <2 |
| 1000-PDMAEMA-HP | 990 ± 23 | 1392 | 75 | 9.5 |
| 1000-PDMAEMA/PNIPAM-JP | 990 ± 23 | 1385 | 42 | 8.3 |
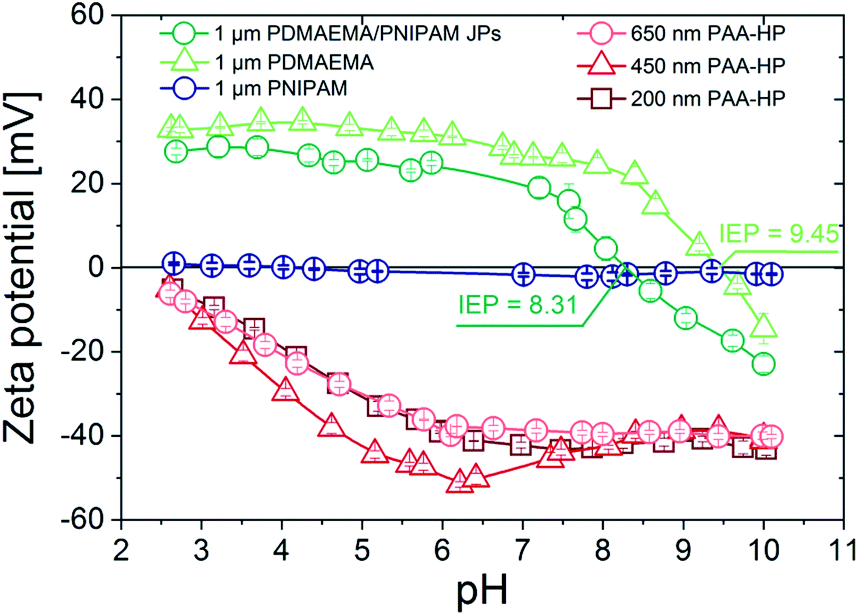 | ||
| Fig. 1 pH-dependent zeta potential of the synthesized PAA-HP (red) and PDMAEMA-HP and JP (green), as well as PNIPAM-HP as reference. | ||
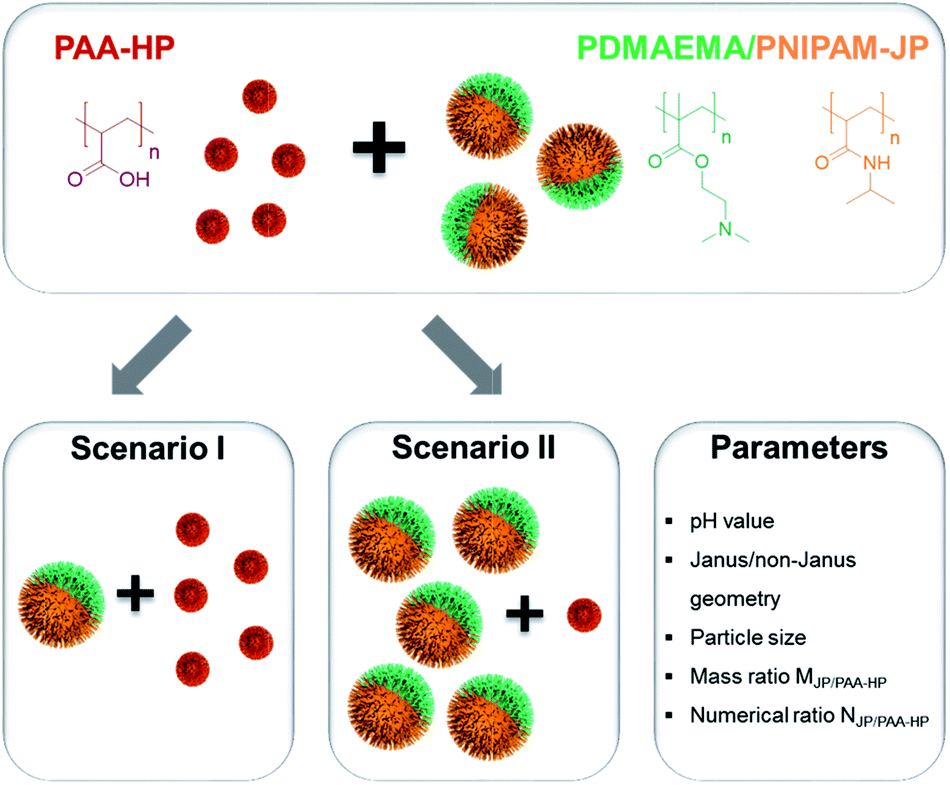 | ||
| Fig. 2 Two proposed scenarios for the assembly of binary mixtures of oppositely charged PAA-HP and 1000-PDMAEMA/PNIPAM-JP in dispersions. Varied parameters are: pH value, particle geometry (Janus and non-Janus), particle size, mass ratio MJP/PAA-HP and numerical ratio NJP/PAA-HP. | ||
The pH-responsive swelling behavior of PAA and PDMAEMA polymers grafted on silica particles was intensively studied in ref. 11. Briefly, the both used polymers exhibit an opposite swelling behavior (Fig. S3†). The PAA-modified particles are highly swollen at pH 9 due to the high negative charge density of PAA chains (swelling ratio is around 8), intermediately swollen at pH 6, and collapsed at pH 2 due to the absence of charges. Contrary, PDMAEMA polymer chains are protonated, highly charged and thus highly swollen at pH 2 (swelling ratio is around 5), intermediately stretched out at pH 6, and collapsed at pH 9. At pH 6 both polymers are in a charged and swollen state, which was set as starting pH value for all self-assembly experiments.
Starting from pH 6, the pH value was then adjusted to either pH 9 or pH 2, and backwards during five cycles in order to investigate the impact of the pH on the reversibility of self-assembled structures. To avoid interferences based on the increase of the ionic strength du, the ionic strength for all self-assembly experiments was adjusted with potassium chloride either to 1 (for half-raspberry-like structures) or 10 mmol L−1 (for cluster- and chain-like structures).
Analysis of formed structures and their reversibility
Two main scenarios were pursued for the self-assembly of binary mixtures of oppositely charged PAA-HP and 1000-PDMAEMA/PNIPAM-JP via electrostatic interactions to understand the shape of formed structures and their pH-triggered reversibility. In scenario I, an excess of smaller homogeneously modified particles was mixed with Janus particles of a larger size (Fig. 2, scenario I). Scenario II comprises the opposite situation, in which an excess of Janus particles with a larger diameter was combined with smaller homogeneous particles (Fig. 2, scenario II). For the implementation of these two scenarios, we synthesized three types of particles: 200 nm, 450 nm and 650 nm homogeneously decorated with PAA (PAA-HP), 1000 nm large particles homogeneously decorated with PDMAEMA (PDMAEMA-HP, for reference purposes), and 1000 nm large Janus particles decorated with PDMAEMA and PNIPAM shells at their opposite sides (JP). The particles size ratio SJP/PAA-HP = DJP/DPAA-HP with D corresponding to the diameter of the silica core, the mass ratio MJP/PAA-HP = MJP/MPAA-HP, and the numerical ratio NJP/PAA-HP = NJP/NPAA-HP ultimately determine the shape of the formed structure.:4 (corresponds to numerical ratios NJP/PAA-HP of 1:500, 1:44 and 1:15 for 200-, 450- and 650-PAA-HP, respectively) and the appropriate particle size ratio SJP/PAA-HP was varied between 5.0 (for 200-PAA-HP), 2.2 (for 450-PAA-HP) and 1.5 (for 650-PAA-HP). In order to visualize the structures in dispersion, PAA-HP were modified with a red fluorescent dye (rhodamine) and PDMAEMA/PNIPAM-JPs with a green fluorescent dye (fluorescein) in the silica core during the Stöber process.
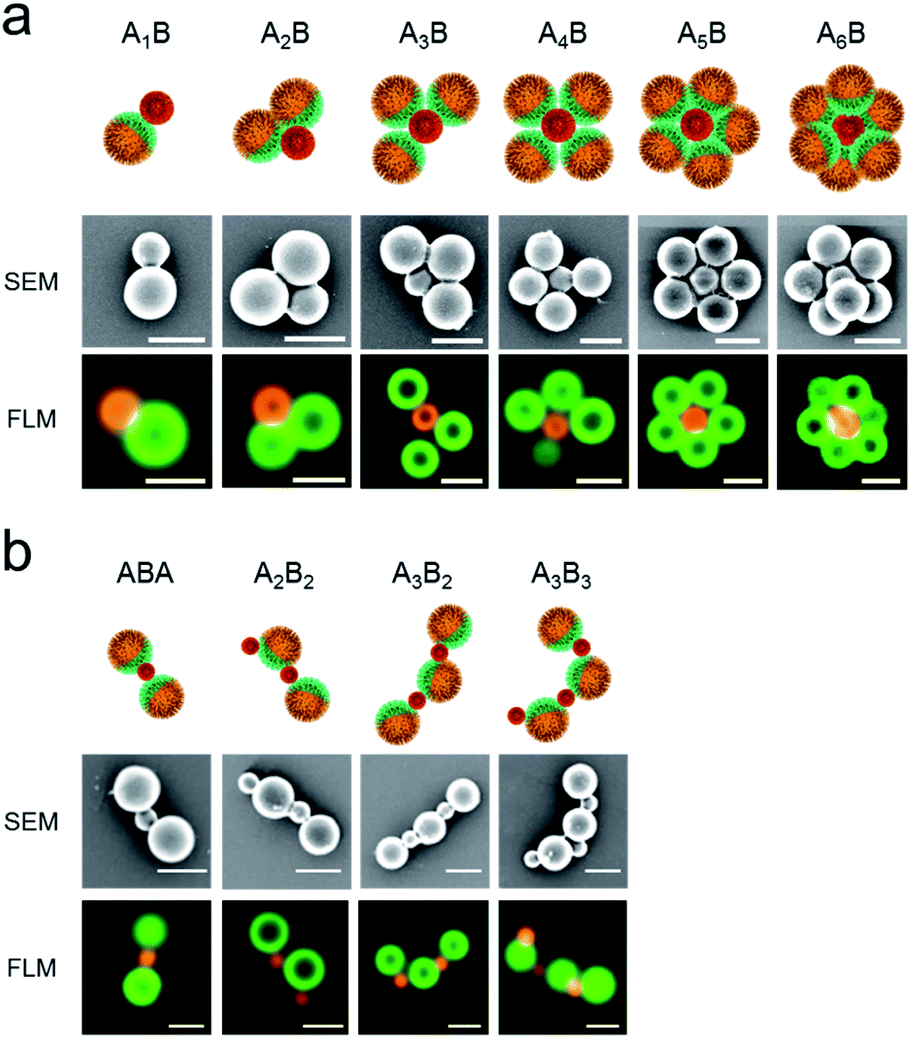
The formation of a wide range of ABn-like structures by different numbers n of smaller PAA-HP (named as B) surrounding one larger PDMAEMA-decorated JP (named as A) was observed and visualized via FLM and SEM.
We observed raspberry-like (for non-Janus PDMAEMA-decorated particles, Fig. S4–S6†) and half-raspberry structures (for JP, exemplarily shown for 650-PAA-HP particles in Fig. 3). It should be noted that these structures were found both in situ in dispersion (FLM images, lower row in Fig. 3), and in dry state (SEM images, middle row in Fig. 3). Thus, drying effects did not seem to have any effect on the formation of the micro-structures. As anticipated, the degree of coverage is higher for non-Janus PDMAEMA particles compared to the Janus ones due to the spatially separated and, thus smaller, PDMAEMA areas on the Janus particles. Furthermore, we always observed a distribution of all ABn-like structures (Fig. 3 and 4 left column), comprising dumbbell-like AB1 up to half-raspberry AB6 structures. AB4-like assemblies were found to be the predominant structure for this system with 650-PAA-HP (≈35% yield, Fig. 4 lowermost row).
 | ||
| Fig. 3 Observed half-raspberry-like structures: schematic cartoons and representative SEM and FLM images obtained in dispersion of a mixture of non-Janus 650-PAA-HP and 1000-PDMAEMA/PNIPAM-JP (mass ratio MJP/PAA-HP 1:4, numerical ratio NJP/PAA-HP 1:15). FLM images were obtained with FITC and TRITC filters and the combined colors. The ionic strength was adjusted to 1 mmol L−1 with KCl. Scale bars: 1 μm. | ||
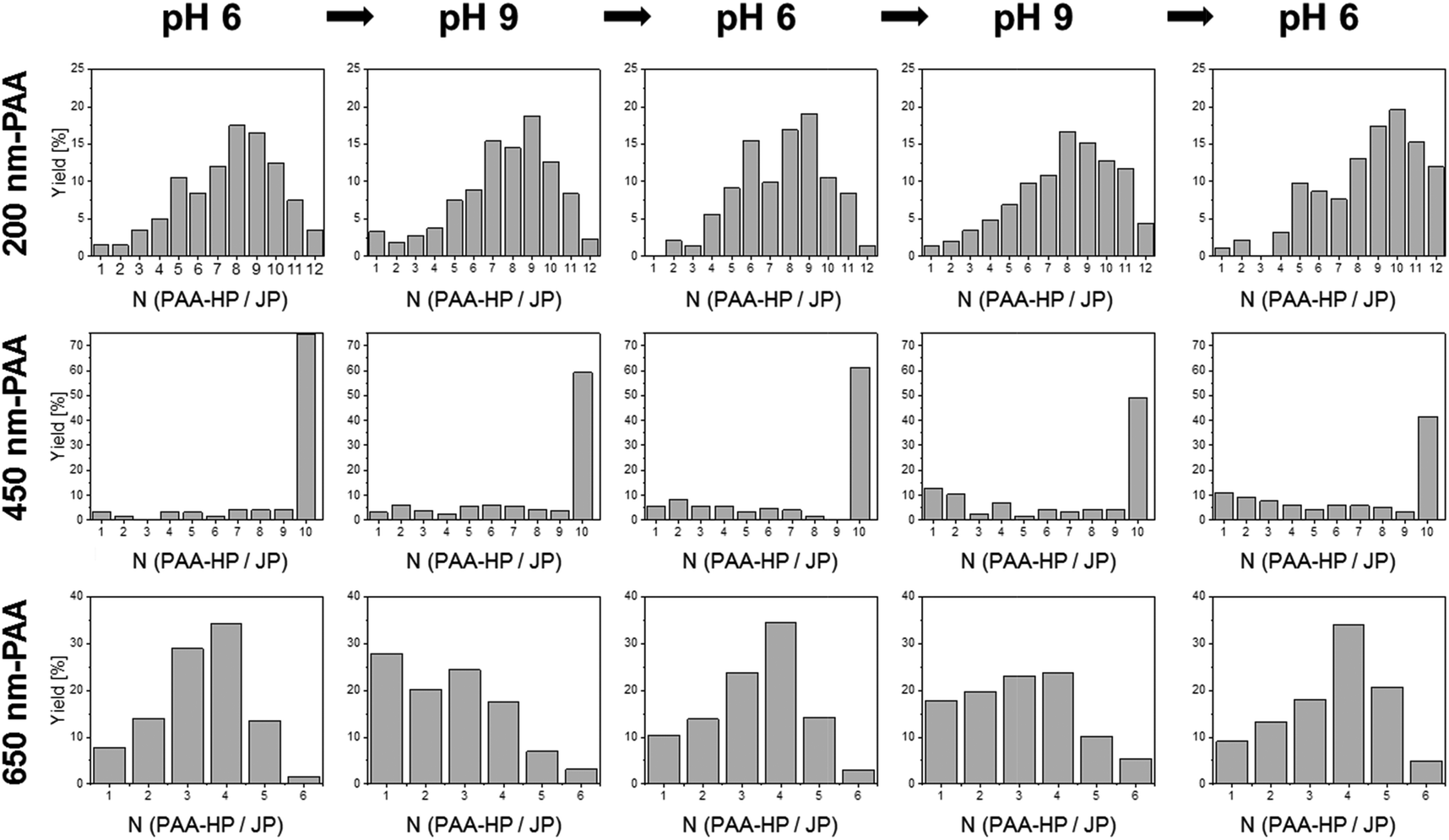 | ||
| Fig. 4 Reversible self-assembly experiments: statistical analysis of the number of PAA-HP attaching to a single 1000-PDMAEMA/PNIPAM-JP (mass ratio MJP/PAA-HP 1:4; numerical ratios 1:500 for 200-PAA-HP, 1:44 for 450-PAA-HP, and 1:15 for 650-PAA-HP) forming a half-raspberry-like structure obtained from reversible pH switch from pH 6 to pH 9 and backwards. The ionic strength was adjusted to 1 mmol L−1 with KCl. 250 clusters were counted for each pH value, based on FLM images of five independent experiments. | ||
ABn-type half-raspberry constructs were also observed for higher particle size ratios SJP/PAA-HP of 2.2 (for 450-PAA-HP) and 5.0 (for 200-PAA-HP), which was realized using PAA-HP with larger diameters of 450 and 200 nm at a constant mass ratio MJP/PAA-HP of 1:4 (corresponds to NJP/PAA-HP of 1:44 for 450-PAA-HP and 1:500 for 200-PAA-HP). As expected, the maximum found number of PAA-HP attached to a single central JP increased with increasing particle size ratio (Fig. 4, left column).
For a more detailed quantification, the theoretically maximum number Nmax of smaller PAA-HP in a hexagonal closed packing pattern on a single large JP was calculated as previously described in ref. 11 and 53 using eqn (2):
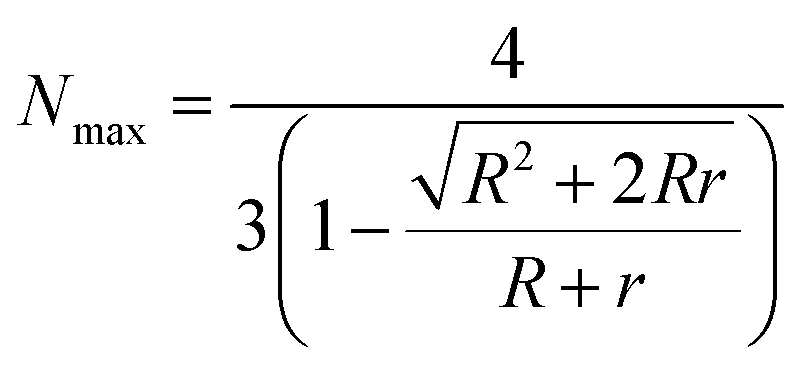 | (2) |
For this purpose, the effective radii of the particles were calculated either based on the polymer shell thicknesses H in dry state and the swelling ratio of the respective polymer via cryo-TEM11 (rcryo-TEM and Rcryo-TEM), or via DLS measurements (rDLS and RDLS). RDLS and rDLS were calculated by 0.5 times Dcore+shellH(DLS) (Table 3; see Table 2, third column for Dcore+shellH(DLS)). The effective radius rcryo-TEM is based on the swelling ratio (SR), which was determined via cryo-TEM to be 3 for PAA and 4 for PDMAEMA at neutral pH.11 Thus, rcryo-TEM is calculated as follows:
| rcryo-TEM = 0.5.(Hdry(TGA)SR + dcoreH) | (3) |
| PAA system | Effective radius r [nm] | N Theorymax | N Experimentmax | ||
|---|---|---|---|---|---|
| Cryo-TEM | DLS | Cryo-TEM | DLS | ||
| 200-PAA-HP | 142 | 367 | 69 | 22 | 12 |
| 450-PAA-HP | 246 | 470 | 30 | 16 | 10 |
| 650-PAA-HP | 354 | 602 | 18 | 12 | 6 |
| 1000-PDMAEMA/PNIPAM-JP | 584 | 693 | — | — | — |
H dry(TGA) can be taken from Table 2, fourth column.
The experimentally obtained number of small HP on the surface of a large JP was compared with the maximum theoretical number (Table 3, Fig. 5).
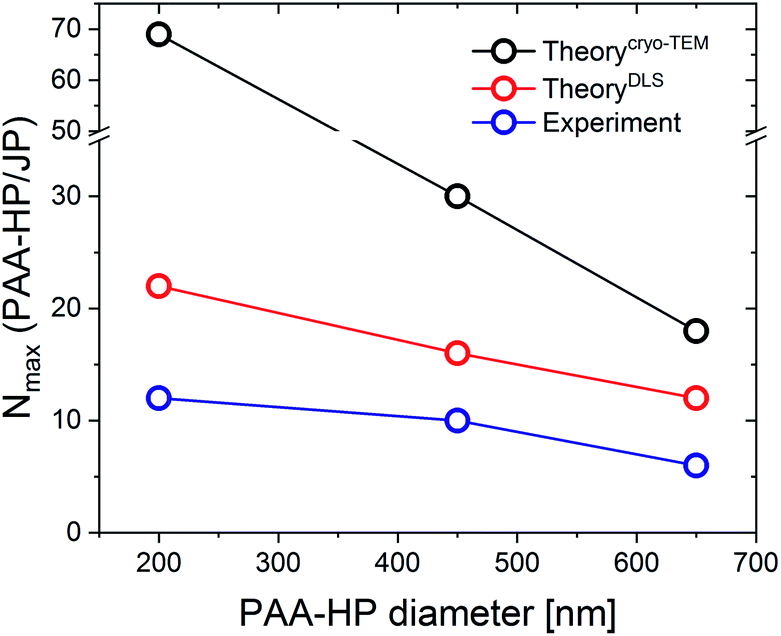 | ||
| Fig. 5 Comparison of the theoretical and experimental numbers N of PAA-HP on a single JP depending on the PAA particle size. | ||
The theoretical results show that the number of PAA-HP on a single JP decreases when increasing the PAA-particle size (Fig. 5), which is in reasonable agreement with the results from ref. 11. However, much lower numbers of PAA-HP attached to one single JP were found in the DLS and cryo-TEM experiments compared to the theoretical results in accordance with further investigations.11 In the experiments, hexagonal close packing could be hindered by the repulsive forces between two neighboring PAA-HP carrying similar negative charges, which leads to empty spaces between the PAA-HP.11 On the contrary, the theoretical model does not consider charges of the colloids and resulting attraction or repulsion between them at all. Generally, theoretical results determined with effective radii from DLS measurements (Fig. 5, red line) correspond better with the experimental findings (Fig. 5, blue line) than the theoretical results obtained via cryo-TEM (Fig. 5, black line). We assume that the hydrodynamic radius obtained from DLS measurements represents a more realistic value for the determination of the size of a polyelectrolyte-decorated particle.
 | ||
| Fig. 6 (a) Average number of 200-, 450- and 650-PAA-HP on a single 1000-PDMAEMA/PNIPAM-JP forming a half-raspberry-like assembly at pH 6 and pH 9. (b) Average number of particles (200-, 450- or 650-PAA-HP and 1000-PDMAEMA/PNIPAM-JP) forming a cluster or chain-like structure at pH 6 and pH 2. | ||
Contrary, reversibility of structures made of 200-PAA-HP or 450-PAA-HP and 1000-PDMAEMA/PNIPAM-JP could not be observed at all (Fig. 4 and 6a). We assume that the found tendencies are caused by geometrical and electrostatic effects. Indeed, all sizes of PAA-modified are attached to the PDMAEMA shell of the central 1000-PDMAEMA/PNIPAM-JP due to their opposite charges. But when the PAA-particles are small compared to the central JP (<650 nm), they are spatially enclosed and locked by electrostatics and/or entanglements (“caging”) with the PDMAEMA polymer chains to a larger extent. The extensive three-dimensional enveloping of small PAA-particles in the PDMAEMA shell of the JPs hinders their release from the central 1000-PDMAEMA/PNIPAM-JP even after pH increase. Thus, the dense packing of the smaller particles is irreversible. The stability of assembled structures can be compared with polyelectrolyte multilayers fabricated e.g. via layer-by-layer processes,54,55 which are thermodynamically stable structures. Molecular-dynamics simulation also showed that the polyelectrolyte multilayers are “fuzzy” and the molecules of one layer interpenetrate other layers.56 Spruijt and coworkers reported a similar effect on polymer brush-modified colloidal particles. They presented systems consisting of oppositely charged colloidal particles, which assembled into large clusters, and disassembled when increasing the salt concentration. They found that interparticle interactions are governed by a time-dependent attractive electrostatic complexation that strengthens in time due to interpenetration of the polymer brushes.12 Indeed, the average value of 200-PAA-HP per 1000-PDMAEMA/PNIPAM-JP forming half-raspberry-like assemblies slightly increases with proceeding cycle number (Fig. 6a, black curve), which seemed to hint at strengthening electrostatic complexation in time as described by Spruijt et al. However, an opposite tendency was found for 450-PAA-HP: less 450-PAA-HP attached to a single Janus particle with increasing cycle number (Fig. 6a, red curve). Therefore, we assume that not only electrostatic, but particularly geometrical “caging” effects may have an effect on the irreversibility of assemblies. Small 200-PAA-HP could be easily and irreversibly caged in the shell of the Janus particles, while medium-sized 450-PAA-HP were released in time despite almost constant ionic strength because of weaker “caging” effects. Only 650-PAA-HP are large enough that neither “caging” nor ionic effects occurred, which enabled the reversibility of the system.
Maas et al. investigated the influence of the particle size on the assembled constructs, and reported that the use of small particles in assembly processes of binary particle mixtures leads to uncontrolled aggregation, bridging of the particles and high polydispersity of the formed structures.57 They hypothesize that this phenomenon is not governed by kinetics, but rather by aggregation of the small particles within the ion cloud of the central particles, when the adsorbing particle is small compared to the diameter of the ion cloud.57 Based on this hypothesis, we cannot disprove that the effect of aggregation of small particles within the ion cloud of the central particles also occurs for 200-PAA-HP due to the broad distribution of observed ABn structures. 450-PAA-HP show a more narrow distribution of the ABn structures probably due to the more controlled aggregation within the ion cloud of the 1000-PDMAEMA/PNIPAM-JP. However, all PAA-HP appeared to randomly attach at the PDMAEMA-hemisphere of the 1000-PDMAEMA/PNIPAM-JP and rather not with the maximum distance to each other because of the neutralization of the PAA chains in the PDMAEMA ion cloud.
:1 for 450- and 650-PAA-HP and 100:1 for 200-PAA-HP (corresponds to numerical ratios NJP/PAA-HP to 4:5 for 200-PAA-HP, 4:3 for 450-PAA-HP, and 4:1 for 650-PAA-HP) and are exemplarily shown for 650-PAA-HP and JPs in Fig. 7a. In order to quantify the self-assembly result, we counted the number of particles forming AnB structures consisting of different numbers n of larger JP A and smaller PAA-HP B (obtained from FLM images). The scope of structures ranges from dumbbell-like structures, such as A1B, to flower-like constructs, such as A5B or A6B.
 | ||
| Fig. 7 Observed colloidal clusters and colloidal chains. (a) Schematic and representative SEM and FLM images of colloidal clusters, formed in dispersion from 1000-PDMAEMA-PNIPAM-JP and 650-PAA-HP (mass ratio MJP/PAA-HP 15:1, numerical ratio NJP/PAA-HP 4:1). (b) Schematic and representative SEM and fluorescence microscopy images of colloidal chains, formed in dispersion from 1000-PDMAEMA-PNIPAM-JP and 450-PAA-HP (mass ratio MJP/PAA-HP 15:1, numerical ratio NJP/PAA-HP 4:3). FLM images were obtained with different filters and the combined colors. The ionic strength was adjusted to 10 mmol L−1 with KCl. Scale bars: 1 μm. | ||
Furthermore, the particle size ratio SJP/PAA-HP seemed to have an impact on the size of the clusters. An increase of the particle size ratio SJP/PAA-HP resulted in a decrease of the occurring cluster size (Fig. 8). A small central 200-PAA-HP enables the formation of clusters consisting of maximum four particles due to the steric hindrance and fast charge compensation. The small surface of the 200-PAA-HP leads also to small distances between the partly positively charged JP, which leads to repulsive electrostatic forces between them, and hinders the attachment of further JP to the central 200-PAA-HP. Larger central PAA-HP provide a larger surface and a higher number of negative charges per PAA particle, thus more partly positively charged JPs can be included in a cluster.
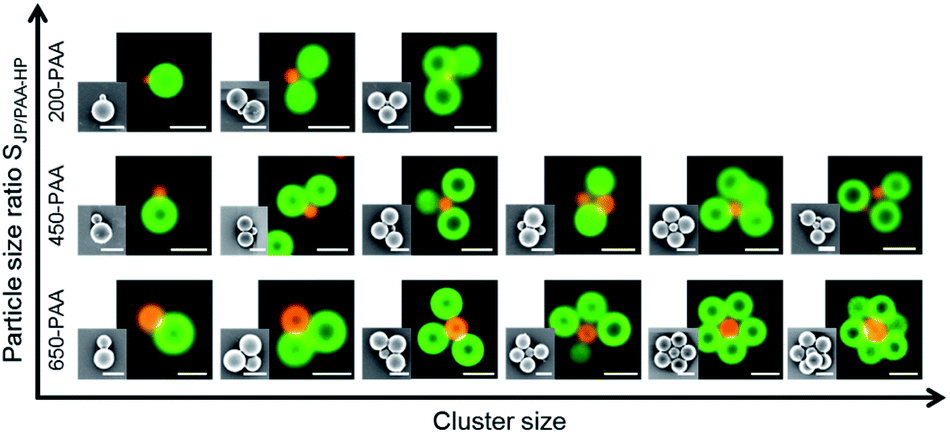 | ||
| Fig. 8 Phase diagram with representative SEM and FLM images of colloidal clusters formed during the self-assembly of PAA-HP (diameter varied between 200 nm, 450 nm and 650 nm) and 1000-PDMAEMA/PNIPAM-JP (constant diameter of 1000 nm; mass ratio MJP/PAA-HP 15:1, numerical ratios NJP/PAA-HP 4:5, 4:3 and 4:1, respectively); the cluster size decreases with increasing particle size ratio SJP/PAA-HP. Scale bar: 1 μm. x-axis: geometry/shape and size of formed clusters; y-axis: particle size ratio SJP/PAA-HP. | ||
:1 (corresponds to numerical ratios NJP/PAA-HP to 4:5 for 200-PAA-HP, 4:3 for 450-PAA-HP, and 4:1 for 650-PAA-HP) and are exemplarily summarized for 450-PAA-HP and JP in Fig. 7b and 9. Although colloidal clusters and colloidal chains emerge simultaneously at this mass ratio, the formation of colloidal clusters is favored over colloidal chains in general (Fig. 10). Similar structures and tendencies were shown by Kraft et al., who also obtained a mixture of chain-like (colloidal polymer) and cluster structures (colloidal molecules) made of DNA linker-modified particles at particle size ratio of 2:1. The percentage of the formed structures could be also programmed by controlling the mixing ratio.43
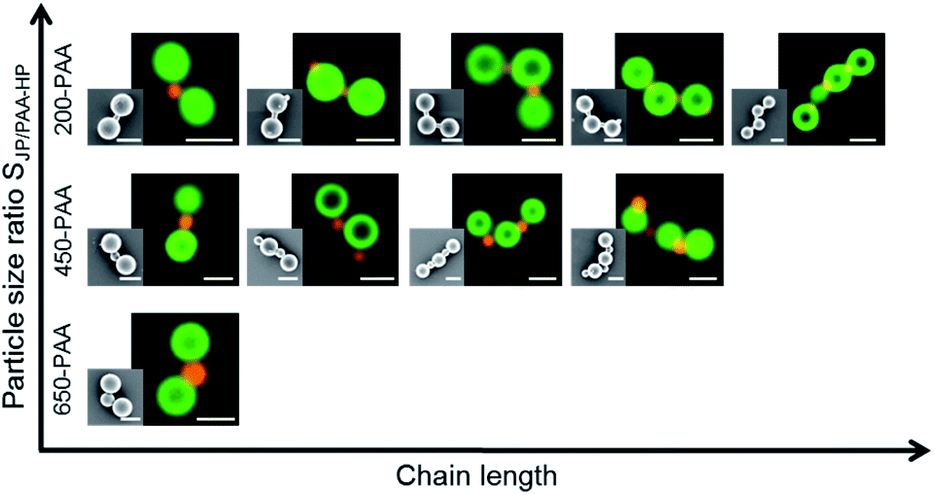 | ||
| Fig. 9 Diagram with representative SEM and FLM images of the colloidal chains formed during the self-assembly of PAA-HP and 1000-PDMAEMA/PNIPAM-JP (mass ratio MJP/PAA-HP 15:1, numerical ratios NJP/PAA-HP 4:5, 4:3 and 4:1, respectively); the chain length increases with increasing particle size ratio SJP/PAA-HP. Scale bar: 1 μm. x-axis: geometry/shape and size of formed chains; y-axis: particle size ratio SJP/PAA-HP. | ||
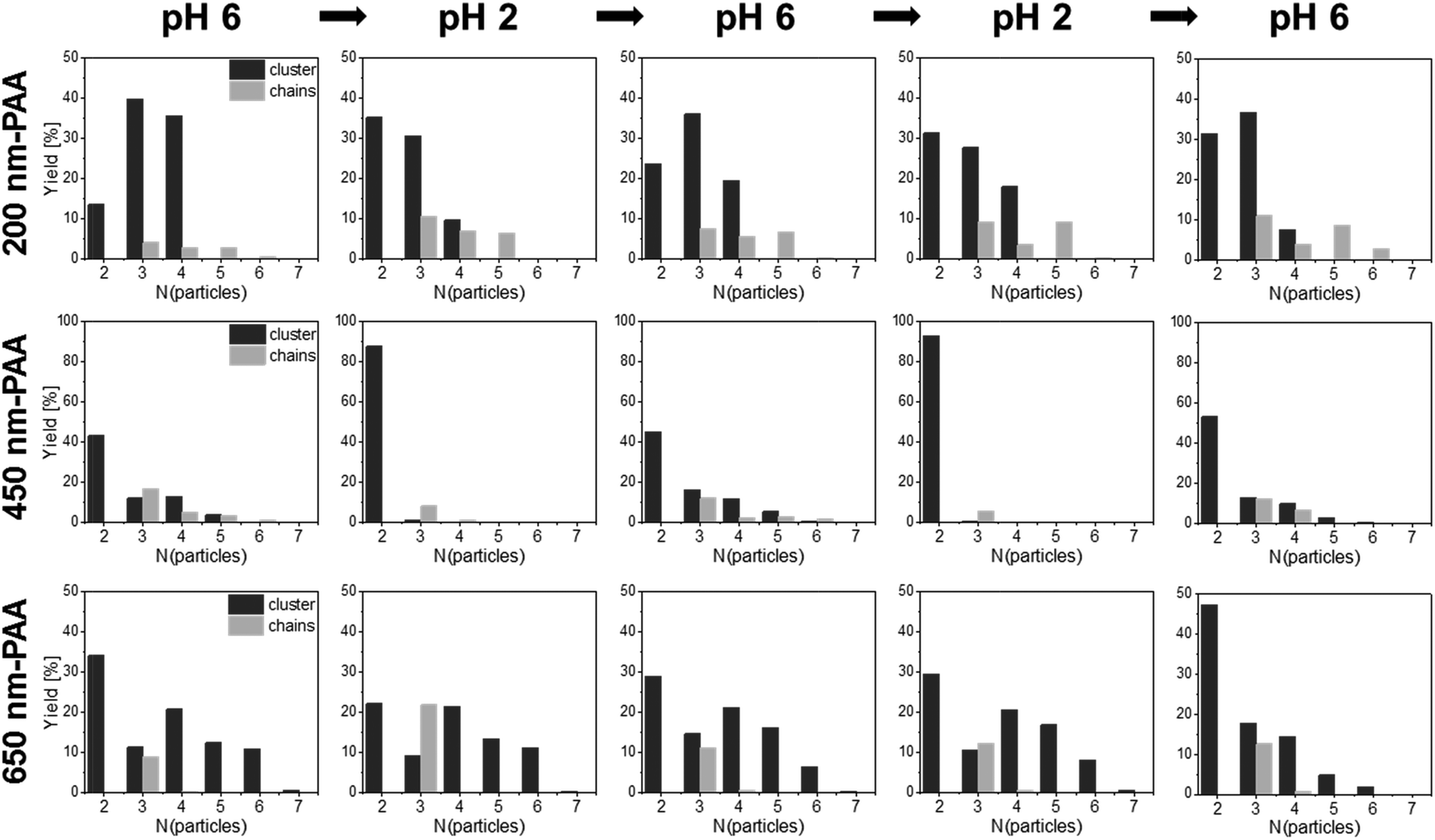 | ||
| Fig. 10 Reversible self-assembly experiments: statistical analysis of the number of PAA-HP and 1000-PDMAEMA-PNIPAM JP (mass ratio MJP/PAA-HP 15:1, numerical ratios NJP/PAA-HP 4:5, 4:3 and 4:1, respectively) forming a colloidal cluster or a colloidal chain obtained from reversible pH switch from pH 6 to pH 2 and backwards. The ionic strength was adjusted to 10 mmol L−1 with KCl. 250 clusters were counted for each pH value, based on FLM images of five independent experiments. | ||
The length of AnBm-like chains exhibits a dependency on the particle size ratio SJP/PAA-HP (Fig. 9), which is, interestingly, opposite to the tendency found for clusters (cf.Fig. 8): the higher the particle size ratio SJP/PAA-HP, the higher the length of the formed chains (Fig. 9). The formation of colloidal chains is preferred for smaller PAA-HP (200 nm and 450 nm), while the formation of clusters is preferred for larger connecting PAA-HP. It means the smaller the size of the “connecting” PAA-HP, the higher is the probability to form longer chains. This tendency can be explained with the increasing numerical ratio NJP/PAA-HP when increasing the particle size ratio SJP/PAA-HP (Table 1). That means that with increasing diameter of the PAA-HP the number of available PAA-HP per 1000-PDMAEMA/PNIPAM-JP decreases, which reduces the probability to form longer colloidal chains declines. Furthermore, the observed trend is probably supported by steric matters: smaller PAA-HP hamper the attachment of more than two large JP to form a cluster, which facilitates the formation of colloidal chains.
The most pronounced reversibility was observed for 450-PAA-HP and 1000-PDMAEMA/PNIPAM-JPs for both clusters and colloidal chains (Fig. 6b, Tables S2–S4†). At pH 6, larger clusters and chains comprising three, four or five particles were present (due to electrostatic interactions between oppositely charged PAA-HP and JP) although the number of A1B clusters was predominant (average number of particles forming a cluster or chain is 2.9; Fig. 6b, Table S4†). A decrease of the pH value down to pH 2 (close to the IEP of PAA) caused a loss of negative charges in the PAA polymer chains resulting in a weak electrostatic attraction to the JPs. Thus, the yield of longer chains and larger clusters is reduced at pH 2, and the average number of particles forming a cluster or chain decreases by 28% from 2.9 to 2.1 (Fig. 6b, Table S4†). This tendency can be inverted multiple times by increasing and re-decreasing the pH value (Fig. 6b). This dynamic reversibility determined by pH is also discernible for 200-PAA-HP and 650-PAA-HP even though the effect was not as pronounced as for 450-PAA (Fig. 6b, Tables S2–S4†).
Conclusions
In summary, we investigated the pH-triggered reconfigurable assembly of structures composed of binary mixtures of poly(2-dimethylamino)ethyl methacrylate (PDMAEMA)/poly(N-isopropylacrylamide) (PNIPAM) Janus and polyacrylic acid (PAA)-decorated non-Janus particles. We found that the particle size ratio SJP/PAA-HP and the numerical ratio NJP/PAA-HP strongly determine the geometry of the obtained constructs as well as the quality of the reversibility of the assembly process. The formed structures are well-defined and stable in a certain pH range. The structures also have the advantage that the particle interactions are bistable. They can be disassembled on demand by changing the pH value, and then reassembled into new stable structures many times. Specifically, half-raspberry-like assemblies with a higher number of smaller PAA-particles attached to the larger central JPs are formed at pH 6 due to strong electrostatic interactions and at a mass ratio MJP/PAA-HP of 1:4 (corresponding numerical ratios NJP/PAA-HP of 1:500 for 200-PAA-HP, 1:44 for 450-PAA-HP, and 1:15 for 650-PAA-HP). However, only binary particle mixtures with a particle size ratio SJP/PAA-HP of 2.2 could be reversibly disassembled when the pH is increased close to the IEP of the PDMAEMA polymer chains causing weakened attraction between the particles.
Both colloidal clusters and chains are formed at a mass ratio MJP/PAA-HP of 15:1 (corresponding numerical ratios NJP/PAA-HP of 4:5 for 200-PAA-HP, 4:3 for 450-PAA-HP, and 4:1 for 650-PAA-HP). The formation of clusters was generally preferred over the formation of colloidal chains for all sizes of PAA-HP and all pH values. The particle size ratio SJP/PAA-HP strongly determined the size of the formed colloidal clusters: an increase of SJP/PAA-HP decreased the size of the formed clusters. Colloidal clusters could also be reversibly reassembled to chain-like structures (preferably of A1B-type) by decreasing the pH to 2 which is close to the IEP of the PAA-decorated particles. However, colloidal chains, although they were observed at the same mass ratio MJP/PAA-HP and numerical ratio NJP/PAA-HP like colloidal clusters, exhibited the opposite dependency on the particle size ratio SJP/PAA-HP compared to colloidal clusters: by increasing the particle size ratio SJP/PAA-HP, the length of the colloidal chains could be increased.
The presented approach provides new insight into the topic of reconfigurable self-assembly of particles with surface polymer patches into colloidal superstructures with controlled shapes. This may open up a variety of self-assembly-based applications towards miniaturization of machines, catalysis, photonics/plasmonics, photovoltaics, or nanoscale electronics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the German Research Foundation (DFG, Grant SY 125/4-1) and the Leibniz Institute of Polymer Research Dresden (IPF) for generous financial support.References
- J. Zhang, J. Yan and S. Granick, Angew. Chem., Int. Ed. Engl., 2016, 55, 5166–5169 CrossRef CAS PubMed.
- J. Yan, M. Han, J. Zhang, C. Xu, E. Luijten and S. Granick, Nat. Mater., 2016, 15, 1095–1099 CrossRef CAS PubMed.
- W. Gao, A. Pei, X. Feng, C. Hennessy and J. Wang, J. Am. Chem. Soc., 2013, 135, 998–1001 CrossRef CAS.
- F. Li, D. P. Josephson and A. Stein, Angew. Chem., Int. Ed. Engl., 2011, 50, 360–388 CrossRef CAS PubMed.
- M. Vallet-Regi, Chemistry, 2006, 12, 5934–5943 CrossRef CAS.
- J. A. Melero, R. van Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS PubMed.
- K. Hou, B. Tian, F. Li, Z. Bian, D. Zhao and C. Huang, J. Mater. Chem., 2005, 15, 2414–2420 RSC.
- Y. Gao and Y. Yu, J. Am. Chem. Soc., 2013, 135, 19091–19094 CrossRef CAS PubMed.
- P. Johal and S. Chaudhary, International Journal of Advances in Engineering Sciences and Applied Mathematics, 2013, 2, 106–110 Search PubMed.
- A. Kirillova, G. Stoychev, L. Ionov and A. Synytska, Langmuir, 2014, 30, 12765–12774 CrossRef CAS PubMed.
- A. Kirillova, G. Stoychev and A. Synytska, Faraday Discuss., 2016, 191, 89–104 RSC.
- E. Spruijt, H. E. Bakker, T. E. Kodger, J. Sprakel, M. A. Cohen Stuart and J. van der Gucht, Soft Matter, 2011, 7, 8281–8290 RSC.
- M. E. Leunissen, C. G. Christova, A. P. Hynninen, C. P. Royall, A. I. Campbell, A. Imhof, M. Dijkstra, R. van Roij and A. van Blaaderen, Nature, 2005, 437, 235–240 CrossRef CAS.
- A. M. Mihut, B. Stenqvist, M. Lund, P. Schurtenberger and J. J. Crassous, Sci. Adv., 2017, 3, e1700321 CrossRef.
- Q. Chen, J. K. Whitmer, S. Jiang, S. C. Bae, E. Luijten and S. Granick, Science, 2011, 331, 199–202 CrossRef CAS.
- D. J. Kraft, R. Ni, F. Smallenburg, M. Hermes, K. Yoon, D. A. Weitz, A. van Blaaderen, J. Groenewold, M. Dijkstra and W. K. Kegel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10787–10792 CrossRef CAS PubMed.
- S. Asakura and F. Oosawa, J. Polym. Sci., 1958, 33, 183–192 CrossRef CAS.
- S. Gangwal, O. J. Cayre and O. D. Velev, Langmuir, 2008, 24, 13312–13320 CrossRef CAS.
- S. Gangwal, A. Pawar, I. Kretzschmar and O. D. Velev, Soft Matter, 2010, 6, 1413–1418 RSC.
- K. L. Heatley, F. Ma and N. Wu, Soft Matter, 2017, 13, 436–444 RSC.
- Q. Chen, E. Diesel, J. K. Whitmer, S. C. Bae, E. Luijten and S. Granick, J. Am. Chem. Soc., 2011, 133, 7725–7727 CrossRef CAS PubMed.
- S. K. Smoukov, S. Gangwal, M. Marquez and O. D. Velev, Soft Matter, 2009, 5, 1285–1292 RSC.
- J. Yan, M. Bloom, S. C. Bae, E. Luijten and S. Granick, Nature, 2012, 491, 578–581 CrossRef CAS PubMed.
- K. P. Yuet, D. K. Hwang, R. Haghgooie and P. S. Doyle, Langmuir, 2010, 26, 4281–4287 CrossRef CAS PubMed.
- J. Yan, S. C. Bae and S. Granick, Adv. Mater., 2015, 27, 874–879 CrossRef CAS.
- A. Nikoubashman, Soft Matter, 2016, 13, 222–229 RSC.
- Y. Abe, B. Zhang, L. Gordillo, A. M. Karim, L. F. Francis and X. Cheng, Soft Matter, 2017, 13, 1681–1692 RSC.
- A. F. Demirors, P. P. Pillai, B. Kowalczyk and B. A. Grzybowski, Nature, 2013, 503, 99–103 CrossRef PubMed.
- J. Zhang and S. Granick, Faraday Discuss., 2016, 191, 35–46 RSC.
- A. M. Percebom, J. J. Giner-Casares, N. Claes, S. Bals, W. Loh and L. M. Liz-Marzan, Chem. Commun., 2016, 52, 4278–4281 RSC.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- N. Krasnogor, S. Gustafson, D. A. Pelta and J. L. Verdegay, Systems Self-Assembly: Multidisciplinary Snapshots, Elsevier, Amsterdam, 2008 Search PubMed.
- Y. Wang, G. Chen, M. Yang, G. Silber, S. Xing, L. H. Tan, F. Wang, Y. Feng, X. Liu, S. Li and H. Chen, Nat. Commun., 2010, 1, 87 CrossRef CAS.
- Y. Liu, X. Han, L. He and Y. Yin, Angew. Chem., Int. Ed. Engl., 2012, 51, 6373–6377 CrossRef CAS.
- C. Rossner, O. Glatter and P. Vana, Macromolecules, 2017, 50, 7344–7350 CrossRef CAS.
- C. Rossner, Q. Tang, O. Glatter, M. Muller and P. Vana, Langmuir, 2017, 33, 2017–2026 CrossRef CAS.
- Z. Tian, C. Yang, W. Wang and Z. Yuan, ACS Appl. Mater. Interfaces, 2014, 6, 17865–17876 CrossRef CAS.
- Y. Guo, B. G. P. van Ravensteijn, C. H. J. Evers and W. K. Kegel, Langmuir, 2017, 33, 4551–4558 CrossRef CAS.
- Z. Sun, W. Ni, Z. Yang, X. Kou, L. Li and J. Wang, Small, 2008, 4, 1287–1292 CrossRef CAS.
- M. Grzelczak and L. M. Liz-Marzan, J. Phys. Chem. Lett., 2014, 5, 2455–2463 CrossRef CAS PubMed.
- T. Lee, K. Gizynski and B. A. Grzybowski, Adv. Mater., 2017, 29, 1704274 CrossRef.
- B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647–5678 RSC.
- I. Chakraborty, V. Meester, C. van der Wel and D. J. Kraft, Nanoscale, 2017, 9, 7814–7821 RSC.
- S. Wintzheimer, T. Granath, M. Oppmann, T. Kister, T. Thai, T. Kraus, N. Vogel and K. Mandel, ACS Nano, 2018, 12, 5093–5120 CrossRef CAS PubMed.
- B. Bharti, D. Rutkowski, K. Han, A. U. Kumar, C. K. Hall and O. D. Velev, J. Am. Chem. Soc., 2016, 138, 14948–14953 CrossRef CAS.
- Q. Chen, S. C. Bae and S. Granick, J. Am. Chem. Soc., 2012, 134, 11080–11083 CrossRef CAS.
- C. Yu, J. Zhang and S. Granick, Angew. Chem., Int. Ed. Engl., 2014, 53, 4364–4367 CrossRef CAS PubMed.
- J. Yan, S. C. Bae and S. Granick, Soft Matter, 2015, 11, 147–153 RSC.
- Q. Zhang, R. Dong, X. Chang, B. Ren and Z. Tong, ACS Appl. Mater. Interfaces, 2015, 7, 24585–24591 CrossRef CAS PubMed.
- S. Berger, A. Synytska, L. Ionov, K.-J. Eichhorn and M. Stamm, Macromolecules, 2008, 41, 9669–9676 CrossRef CAS.
- S. Berger, L. Ionov and A. Synytska, Adv. Funct. Mater., 2011, 21, 2338–2344 CrossRef CAS.
- A. Synytska, A. Kirillova and L. Isa, ChemPlusChem, 2014, 79, 656–661 CrossRef CAS.
- H. Xing, Z. Wang, Z. Xu, N. Y. Wong, Y. Xiang, G. L. Liu and Y. Lu, ACS Nano, 2012, 6, 802–809 CrossRef CAS PubMed.
- M. Müller, B. Urban and S. Schwarz, Langmuir, 2018, 34, 8129–8144 CrossRef.
- J. J. Richardson, J. Cui, M. Bjornmalm, J. A. Braunger, H. Ejima and F. Caruso, Chem. Rev., 2016, 116, 14828–14867 CrossRef CAS PubMed.
- B. Abu-Sharkh, J. Chem. Phys., 2005, 123, 114907 CrossRef PubMed.
- M. Maas, C. C. Silverio, J. Laube and K. Rezwan, J. Colloid Interface Sci., 2017, 501, 256–266 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00522f |
| This journal is © The Royal Society of Chemistry 2019 |