
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic control of a galvanic replacement reaction on cuprous oxide†
James M.
Lowe
and
Robert H.
Coridan
 *
*
Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville, AR 72701, USA. E-mail: rcoridan@uark.edu; Tel: +1-479-575-5077
First published on 15th January 2019
Abstract
Galvanic replacement (GR) reactions are a versatile approach to fabricating hierarchically structured functional nanomaterials for catalytic, plasmonic, and sensing applications. Most research efforts aim to identify chemical strategies to control the resultant morphology of GR deposition on metallic nanoparticle seeds. Recently, GR has become a method of interest for fabricating heterogeneous interfaces for these applications. Here, we study the chemical mechanism for the GR reaction of AuCl4− on Cu-based thin films. X-ray photoelectron spectroscopy and structural characterization show that, while the GR reaction proceeds through the direct dissolution of Cu and reduction of AuCl4− on Cu, the reaction on Cu2O results in the solid-state formation of CuO at the interface which passivates the interface from further Au deposition. As a result, the chemistry and morphology of Au deposited on Cu2O is limited by the rate of CuO dissolution in the background acidic electrolyte. To explain the observed differences between the GR reaction of AuCl4− on Cu and Cu2O interfaces, we propose a new mechanism for the GR reaction on Cu2O surfaces where disproportionation is the limiting intermediate reaction which can be mediated by AuCl4− concentration and by the photoelectrochemical generation of Cu nanoparticles throughout the bulk of the Cu2O. Consequently, the hierarchical structure of the GR deposition of Au can be chemically controlled on Cu2O films. More generally, this highlights how the details of the chemical kinetics at the reaction interface can be exploited to tailor the resulting nanostructure of metals deposited via GR reactions.
Introduction
Galvanic replacement (GR) is the electroless redox reaction that occurs at the interface of a substrate and a solution containing metal ions.1,2 The difference in the reduction potentials between the substrate and ions drives the spontaneous oxidation of the substrate and deposition of metal atoms via the reduction of ions. GR reactions are sensitive to the chemistry of the metal ions and surface chemistry of the metal substrate.3–5 As a result, the morphology and composition of the GR-deposited metal can be controlled by parameters such as the faceting of the starting substrate, the ligand structure of the ions, the concentration of each component, and the incorporation of other reducing agents.6–13 GR reactions are used to synthesize a wide range of multi-metallic hierarchical nanostructures of interest to a wide range of optical, plasmonic, catalytic, and sensing applications.14–17Copper is a particularly interesting substrate for GR reactions due to the wide range of nanostructures, nanocrystals, and films that can be chemically synthesized as a starting material. The mechanism for the GR-driven deposition of Au (from AuCl4−, for example) on Cu metal substrates through the reduction of Au salt and the oxidation/dissolution of Cu in acidic electrolytes:4,18
| 3Cu(s) + 2AuCl4−(aq) → 3Cu2+(aq) + 2Au(s) + 8Cl−(aq) | (1) |
This mechanism describes the GR reaction on both nanoparticle Cu substrates and on thin films. While it is not as common as metallic Cu, Cu2O is also of interest as a GR substrate. The reduction potentials of Cu(0) (Cu ⇌ Cu2+ + 2e−, E0 = −0.3419 V) and Cu2O (Cu2O + 2H2O + 2e− ⇌ 2Cu2+ + 2OH−, E0 = −0.360 V) are roughly equal and, naively, should demonstrate roughly the same reactivity.19 Cu2O is useful because the shape and faceting of nanoparticles and thin-films, and therefore the resulting noble metal structures from GR, can be controlled during chemical synthesis.20,21 The GR reaction of AuCl4− also occurs spontaneously on Cu2O, though less is known about the mechanism and kinetics. The overall reaction on Cu2O has been proposed to follow a similar stoichiometric mechanism to the one observed on Cu metal in acid:20,22
| 3Cu2O(s) + 2AuCl4−(aq) + 6H+(aq) → 6Cu2+(aq) + 2Au(s) + 8Cl−(aq) + 3H2O | (2) |
In this work, we elucidate the mechanism for the GR reaction of AuCl4− on Cu and Cu2O thin film substrates by extensive characterization of the surface and bulk transformations of the substrates. Contrary to previously proposed stoichiometric mechanisms for GR on Cu2O (Reaction 2), we observe that the reaction proceeds through the solid-state formation of a passivating layer of CuO at the interface, which is not observed for the same GR reaction on Cu. As a result, the kinetics and overall morphology of the Au deposition on Cu2O are dependent on the rate of dissolution of the GR-inactive CuO. We show that the morphology and fidelity of the deposited Au can be controlled by the concentration of the noble metal salts and by the presence of Cu nanoinclusions throughout the Cu2O film that form during photoelectrodeposition. Based on the experiments described here, we propose a mechanism for the GR-based deposition of Au on Cu2O interfaces that is initiated by disproportionation of the Cu2O surface.
Experimental section
Copper(II) sulfate hydrate (98%; Sigma-Aldrich), lactic acid solution (reagent grade, ≥85%; Sigma-Aldrich), sodium hydroxide solution (50% w/w; VWR), sodium tetrachloroaurate(III) dihydrate (99%; Sigma-Aldrich), sulfuric acid (95–98% ACS grade; VWR), and water (HPLC grade; VWR) were used as received. Fluorine-doped tin oxide (FTO)-coated substrates (TEC-15, 12–14 Ω sq; MTI Corp) were diced and rinsed in acetone, methanol, and isopropanol, then dried under nitrogen prior to use.Materials characterization
X-ray diffraction (XRD) measurements (Mini-Flex II, Rigaku) were performed using Cu-Kα radiation (λ = 1.54 Å). Scanning electron micrographs (SEM) were measured with a FEI Nova Nanolab SEM. X-ray photoelectron spectroscopy (XPS) measurements (Phi Versaprobe) were performed with a monochromated Al Kα source (1486.6 eV).Preparation of copper-based thin films
Electrodeposited and photoelectrodeposited Cu2O films were prepared identically to previously reported methods.23,24 The electrodeposition solution was prepared by dissolving CuSO4 in a 3.0 M lactic acid solution to a final CuSO4 concentration of 40.0 mM. The pH of the electrodeposition solution was adjusted to 10.0 by the slow addition of NaOH solution. Electrodeposition onto FTO electrodes was carried out in a three-electrode configuration with a Pt mesh counter electrode and a Ag/AgCl (saturated KCl) reference electrode (Bioanalytical Systems) with a potentiostat (Bio-Logic SP-240). The electrodeposition bath was heated to 60 °C by partially submerging it into a heated water bath. The films were synthesized by potentiostatic electrodeposition at −0.4 V vs. Ag/AgCl until a charge density of 0.36 C cm−2 had passed. Photoelectrodeposited Cu2O films were prepared under identical conditions to electrodeposited ones, though deposition was stimulated by a 455 nm LED (350 mW cm−2). The resultant electrodeposited or photoelectrodeposited Cu2O films were roughly 600 nm thick based on previously published measurements.24 CuO films were prepared by annealing electrodeposited Cu2O films at 500 °C in air for 1 h (10 °C min−1 ramp rate). The complete transformation to CuO was confirmed by XRD before proceeding. 600 nm Cu films were deposited onto FTO substrates by electron beam evaporation (10 Å s−1 deposition rate).Galvanic replacement (GR) reactions on Cu-based films
Au GR reaction solutions of specified concentrations were prepared by dissolving NaAuCl4 into 0.01 M sulfuric acid. NaOH solution was added slowly to adjust the solution to pH 2.7. The GR reaction was performed by submerging the substrate in GR solution for the desired time. After submersion, the films were rinsed in water and dried under nitrogen.Results
We characterized the surface chemistry of the evaporated Cu, electrodeposited Cu2O, and air-annealed CuO films exposed to 5 mM AuCl4− GR solution. Under exposures of up to 60 s, the Cu 2p XPS spectra for the Cu substrate remained constant, indicating that the oxidation state of the Cu interface was maintained during the reaction (Fig. 1a). Cu 2p XPS spectra of CuO showed the broad features and satellite peaks indicative of Cu(II) for GR solution exposures of up 3600 s (Fig. 1b). For Cu 2p XPS measurements on Cu2O (Fig. 1c), we observed the rapid oxidation of the surface for 2 s of GR solution exposure. The film was completely transformed from Cu(I) to Cu(II) for the resolvable depth of the XPS measurement. Cu2O exposed to a null solution (0 mM AuCl4− in H2SO4 adjusted to pH 2.7) showed no change in the oxidation state of the film after 60 s in the solution. Cu2O films were dissolved on electrodes exposed to the null solution for 30 min or greater, though a slight amount of metallic Cu was formed on the surface. This is an observation of the disproportionation of Cu2O followed by the dissolution of Cu2O (Fig. S1 and S2†), which is known to occur on Cu2O in dilute sulfuric acid.25,26 Au 4f XPS measurements on each substrate (Fig. 1d) showed Au deposition on Cu and Cu2O surfaces within 2 s, but no deposition on CuO for any exposure duration. The null solution slowly dissolved each of the oxide substrates (Fig. S1†). | ||
| Fig. 1 The Cu 2p XPS spectra for the GR reaction of 5 mM AuCl4− on copper-based thin film substrates. The oxidation state Cu on the (a) Cu substrate and on the (b) CuO substrate remains constant for all exposure times. In each plot, the 0 s scan indicates the measurement of the unexposed electrode. On Cu2O (c), the Cu 2p spectra indicates that the surface oxidizes to Cu(II) within the first 2 s of exposure. Oxidation was not observed for the 0 mM null solution, indicating that the GR reaction transforms the interface rather than the sulfuric acid. (d) The Au 4f XPS spectra for the GR reaction of 5 mM AuCl4− on Cu, Cu2O and CuO. Au is observed on Cu and Cu2O substrates within 2 s of exposure, while no Au is observed on CuO, even at 3600 s exposure. | ||
We measured the dynamics of the surface oxidation on Cu2O films by the GR reaction as a function of AuCl4− concentration by Cu 2p XPS (Fig. 2a). The surface was fully oxidized within 2 s of exposure to the 5.0 mM and 50.0 mM solutions. A small amount of oxidation was observed in the 0.2 mM solutions after 300 s, though not much more than the native CuO formed on as-prepared Cu2O. A significant amount of oxidation was observed for the films exposed to the 1.0 mM GR solution. The intensity of the Cu(I) and Cu(II) peaks was roughly equal after 300 s.
 | ||
| Fig. 2 The Cu 2p XPS spectra for Cu2O thin film substrates exposed to varying concentration AuCl4− GR solutions. The substrate was observed to oxidize within 2 s to CuO for 5.0 mM (green) and 50.0 mM AuCl4− (purple) solutions. The oxidation of the Cu2O substrate progresses more slowly a lower AuCl4− concentrations. Little oxidation is observed for 0.2 mM AuCl4−, and a pronounced CuO peak is observed for 1.0 mM AuCl4− after 300 s exposure. | ||
XRD measurements (Fig. 3a) for the time series of GR deposition on Cu shows only a small peak (2θ = 28.0°) after 60 s that corresponds to the (120) Bragg reflection of the orthorhombic Pbam CuAu intermetallic phase.27 On Cu2O, a weak feature was observed at the angle corresponding to the Au(111) Bragg reflection (2θ = 38.1°). Peaks with stronger intensities were observed in XRD measurements with increasing AuCl4− concentration after 60 s exposure (Fig. 3b). At 50.0 mM, the intensity of the (120) orthorhombic CuAu Bragg reflection was significantly higher than for the 5 mM exposure. Other diffraction peaks were observed for the 50.0 mM exposure, including one corresponding to the Au(111) Bragg reflection and others that indicate significant alloying between Cu and Au. The peak positions not attributable to the substrate or pure metal deposition (2θ = 32°; 2θ = 38–41°) can be assigned to a number of Au–Cu intermetallic phases.28 Additionally, the diffraction intensity for the Cu substrate was significantly less intense at 50.0 mM than for other exposures, showing that the substrate film was being rapidly consumed in the GR reaction. On Cu2O, only the Au(111) and Au(200) Bragg reflections were observed, with only a slight intensity increase compared to the 5 mM case (Fig. 3c). No XRD features indicating large scale crystalline Au deposition was observed at lower concentrations for either Cu or Cu2O films. Energy-dispersive X-ray spectroscopy showed that a significant amount of Au was deposited on Cu2O surfaces exposed to 1 mM AuCl4− (Fig. S3†).
 | ||
| Fig. 3 (a) XRD measurements of a time series of 5 mM AuCl4− GR reactions on Cu (blue) and Cu2O (red) thin films. No significant change is observed in either film for 2 s and 10 s exposures. A small peak (blue arrow; 2θ = 28.0°) was observed on the Cu substrate at an angle corresponding to the (120) Bragg reflection for an orthorhombic CuAu alloy.27 After 60 s, Au was observed on the Cu2O substrate (red arrow; 2θ = 38.1°, corresponding to the Au (111) Bragg reflection). (b) XRD measurements of GR deposition on Cu and Cu2O substrates after 60 s from varying AuCl4− concentration. At low concentrations (0.2 mM, 1.0 mM), no significant features were observed beyond the peaks for the substrate on either the Cu or Cu2O films. The (120) Cu–Au alloy Bragg reflection was significantly more intense on Cu exposed to 50 mM AuCl4−. Other features (purple arrows) indicated significant alloying and solid solution formation between Cu and Au at this high concentration, though the peak positions can be assigned to a number of Au–Cu intermetallic phases.28 On Cu2O, the Bragg intensity for Au was slightly more intense for 50 mM AuCl4− than for 5 mM, but no XRD features indicating alloy formation were observed. (c) An isolated region of the XRD measurements in (b), emphasizing the concentration-dependent observation of the Au (111) Bragg reflection. For reference, the calculated diffraction patterns for the known phases of the Au–Cu alloy system are shown in Fig. S4.† | ||
We imaged the surface morphology of the deposited Au on Cu after a 60 s exposure to the GR solution (Fig. 4). At concentrations of up to 5 mM AuCl4−, the GR reaction resulted in a uniform, homogenous deposition of Au. For comparison, the Cu substrate and the Au-coated substrate are shown in Fig. S5.† At 50 mM AuCl4−, the surface became porous by the deposition, though the Au was homogenously distributed over the roughened interface. On Cu2O, a 60 s exposure to the GR solution resulted in the deposition of Au nanoparticles with a distribution that varied with concentration. A sparse distribution of Au nanoparticles was observed for 0.2 mM AuCl4−. A roughly uniform distribution of 50–100 nm Au nanoparticles was observed for the 1 mM solution. The particle size and the heterogeneity of the distribution increased with increasing AuCl4− concentration (5 mM and 50 mM). Additionally, the oriented, pyramidal surface characteristic of electrodeposited Cu2O was etched flat at these high concentrations.
 | ||
| Fig. 4 Scanning electron micrographs of GR deposition after a 60 s exposure on (left) Cu substrates and (right) Cu2O substrates for varying AuCl4− concentration. On Cu, conformal deposition was observed for concentrations of (a) 5.0 mM and lower. At 50.0 mM (b), the film was etched by the GR reaction to form a microporous surface, though the deposition was uniform otherwise. A sparse distribution of very small (<50 nm) Au nanoparticles were observed on the Cu2O substrates exposed to 0.2 mM GR solution (c). At higher AuCl4− concentrations, GR-deposited Au formed nanoparticles on the surface of Cu2O (d–f), with increasing characteristic size and interparticle distance with increasing AuCl4− concentration. The characteristic pyramidal morphology of electrodeposited Cu2O films was etched flat at 5.0 mM and 50.0 mM AuCl4−. The dark regions in the images of the interface are Cu2O (from XRD measurements) with a thin layer of CuO (from XPS). SEM images with larger fields of view are shown in Fig. S6.† | ||
We used photoelectrodeposited Cu2O films to study the effects of excess Cu on surface oxidation during the GR reaction. At sufficient illumination intensity, the photoelectrodeposition of Cu2O results in a film that is doped by a homogeneous distribution of Cu nanoparticles.24 This is indicated by the emergence of a Scherrer-broadened Cu(111) peak in an XRD measurement of Cu2O electrodeposited under 455 nm LED illumination at an intensity of 350 mW cm−2 (Fig. 5a). Cu 2p XPS measurements of the surface oxidation dynamics on Cu-doped Cu2O films showed that the rate of oxidation was reduced at low concentrations (Fig. 5b). Virtually no oxide formation was observed on films exposed to 0.2 mM or 1.0 mM films for 300 s, indicating that any initial CuO at the interface had been etched away in the solution. The surfaces were almost totally transformed to Cu(II) at 5.0 mM and 50.0 mM within 2 s of GR solution exposure. We observed Au deposition at all concentrations by Au 4f XPS (Fig. 5c), with the signal increasing with exposure time. As a result, the intensity in XRD measurements (Fig. 6) for the Au(111) and Au(200) Bragg reflections was higher on the photoelectrodeposited Cu2O compared to ordinary Cu2O exposed to identical GR conditions (Fig. 3b). No diffraction peaks were observed for Au for 0.2 mM or 1.0 mM GR exposure even after 300 s. While Au is evident on these surfaces from XPS, the deposition is too thin to generate measurable Bragg reflections.
 | ||
| Fig. 5 (a) XRD measurements of electrodeposited (green) and photoelectrodeposited (black) Cu2O films. Measurements of the photoelectrodeposited films are distinguished by broadened Cu2O Bragg reflections and a Cu (111) Bragg reflection indicating the presence of metallic Cu nanoinclusions throughout the bulk of the film. (b) Cu 2p XPS of photoelectrodeposited Cu2O exposed to the GR reaction. At high concentrations (5.0 mM, 50 mM) the interface was fully oxidized within 2 s of exposure, as observed on ordinary Cu2O. Virtually no surface oxidation was observed for the film exposed to 1.0 mM or 0.2 mM AuCl4− for exposures of up to 300 s, compared to the significant oxidation observed on ordinary Cu2O (Fig. 2). (c) Au 4f XPS indicating Au deposition at each concentration and exposure time, with increasing XPS intensity for longer exposure times. | ||
 | ||
| Fig. 6 (a) XRD of the Au deposition on photoelectrodeposited Cu2O after a 60 s exposure to GR solution. No significant XRD features were observed for the low concentration samples (0.2 mM, 1.0 mM). The Au (111) and Au (200) Bragg reflections were observed for 5.0 mM and 50.0 mM GR reactions with higher intensities than the corresponding peaks on ordinary Cu2O (Fig. 3b). (b) An isolated region of the XRD measurements in (b), emphasizing the concentration-dependent observation of the Au (111) Bragg reflection. | ||
The SEM-imaged structure of GR-deposited Au on Cu-doped Cu2O after a 60 s exposure is shown in Fig. 7. At 1.0 mM, the resultant deposition consisted of small nanoparticles (Fig. 7a) distributed more homogeneously (Fig. 7b) than for the ordinary Cu2O (Fig. 4). The Au deposition formed larger, more heterogeneously dispersed nanoparticles with increasing AuCl4− concentration for 5.0 mM and 50.0 mM solutions (Fig. 7c,d).
 | ||
| Fig. 7 Scanning electron micrographs of photoelectrodeposited Cu2O substrates exposed to GR solutions with varying AuCl4− concentration. (a) The Au deposition was more conformal for 1.0 mM AuCl4− than the one observed on ordinary Cu2O substrate (Fig. 4). (b) The deposition at the GR solution level for 1.0 mM AuCl4− deposition. At higher concentrations of AuCl4− ((c) 5.0 mM; (d) 50.0 mM), sparse films of Au nanoparticles were deposited, resembling those observed on ordinary Cu2O. | ||
Discussion
The previously proposed mechanism (Reaction 2) for the AuCl4− GR reaction on Cu2O closely follows the one for the same reaction on Cu. The results of the experiments described in this work show that there are distinct mechanisms for the GR reaction of AuCl4− on Cu and Cu2O substrates. The oxidation state of a Cu substrate remains unchanged during the GR reaction, regardless of the duration of the exposure or concentration of AuCl4−. The Au deposition is relatively homogeneous over the exposed substrate, with some induced roughness occurring at higher concentrations of AuCl4−. The formation of intermetallic Cu–Au alloys and the continuous presence of Cu at the substrate surface even after significant Au deposition suggests that the GR reaction on Cu metal depends on the mobility of Cu atoms through the solid deposition to maintain the reaction. The chemical parameters that limit the GR reaction are the concentration of AuCl4− and solution access to the Cu surface. These observations are consistent with and expand on the GR mechanism proposed by other researchers for Cu substrates (Reaction 1).For Cu2O, exposure to AuCl4− forms a CuO layer at the interface that is inactive for the GR reaction, but dissolves under exposure to sulfuric acid. The heterogeneity of the Au deposition on Cu2O and the rate of surface oxidation increases with increasing AuCl4− concentration. Doping the Cu2O substrate with Cu nanoparticles via photoelectrodeposition increases the resistance to the formation of CuO and the uniformity of the Au deposition. If Cu2O was the direct reactant, as suggested by the previously proposed mechanism (Reaction 2), the surface would maintain the Cu(I) oxidation state throughout the reaction. However, after sufficient exposure to AuCl4− in H2SO4, the surface is transformed to Cu(II) (in the form of CuO) to such a complete degree that only Cu(II) is observable by XPS. The formation of a passivating surface oxide rather than the direct dissolution of substrate is also known for electroless noble metal deposition on Si in aqueous solutions. Similar to the GR reaction on Cu2O, the electroless deposition reaction on Si requires an active oxide etchant like hydrofluoric acid to proceed.29,30
The difference in the GR reaction for the two substrates suggests a more complex mechanism for Cu2O substrates. Cu2O is known to disproportionate under acidic conditions based on the standard reduction potentials for Cu(II) and Cu(I) in aqueous, acidic environments.25,26 Cu formation was observed in these studies for the dissolution of electrodeposited Cu2O films in the null, sulfuric acid-only solution (Fig. S1 and S2†). We propose that disproportionation is the initial step for the GR-driven deposition of Au on Cu2O (Fig. 8). To initiate the reaction, the surface of the Cu2O film disproportionates upon exposure to the acidic AuCl4− deposition solution,
| Cu2O(s) → CuO(s) + Cu(s) | (3) |
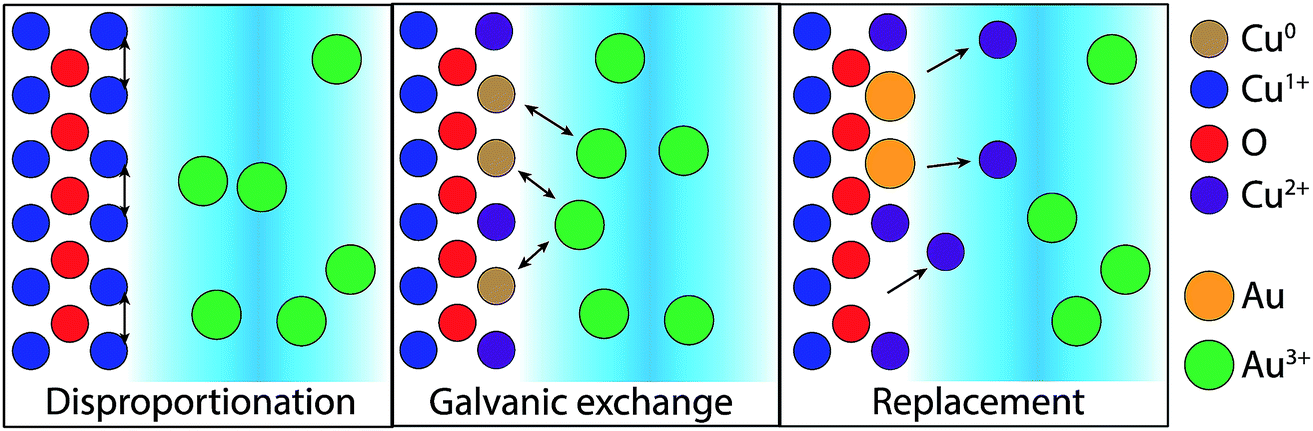 | ||
| Fig. 8 The proposed mechanism for the AuCl4− GR reaction on Cu2O. To initiate the GR reaction, the surface must first disproportionate, forming Cu and CuO on the surface. The Cu and dissolved AuCl4− participate in the galvanic exchange, resulting in a surface of Au and CuO. The rate of the disproportionation intermediate reaction and the recruitment of electrons from the near-surface bulk to form surface Cu(0) limits further deposition until the CuO is etched by the acid in solution. | ||
A mechanism for the GR reaction on Cu2O that depends on disproportionation as an intermediate step explains the AuCl4− concentration-dependent kinetics of CuO formation and heterogeneity of Au deposition on Cu2O. Similarly, the doping of photoelectrodeposited Cu2O by Cu nanoinclusions throughout a substrate mitigates the formation of a reaction-limiting amount of CuO at the surface at low AuCl4−. On undoped Cu2O, the consumption of Cu from the surface by the GR reaction traps the equilibrium Reaction 3 at the products. The depletion of interfacial Cu2O extends into the near-surface bulk of the substrate, which transforms entirely into CuO as observed from XPS measurements. The excess of Cu(0) throughout the photoelectrodeposited film contributes to the disproportionation reaction by LeChatlier's principle, providing a reservoir of Cu to maintain the equilibrium presence of Cu2O near the interface. The resulting photoelectrodeposited Cu2O is more resistant to the build-up of GR-limiting CuO at the interface, particularly at low AuCl4− concentrations, and thus can maintain the reaction to form a more homogeneous deposition. The complex energetics of the Au/Cu2O/CuO interface may promote the collection of electrons from the bulk on surface Au(0) to drive further reduction of AuCl4− to catalyze the observed nanoparticle formation rather than a homogenous layer, though further experiments are necessary to explore this. As disproportionation is characteristic of the interfacial chemistry between the acidic media and the Cu2O surface, it potentially mediates the GR reaction observed for other noble metal salts, such as Ag+, PtCl62− or PdCl42−, which also have appropriate reduction potentials for driving GR reactions on Cu and Cu2O.31–34
Conclusion
In conclusion, we have characterized the interfacial chemistry of Cu and Cu2O thin films after the deposition of Au via galvanic replacement reaction of AuCl4− in H2SO4. The conventional reaction mechanism is an apt description of GR on Cu, where surface Cu(0) is the direct reactant. We have outlined experiments that show that the disproportionation mediates the same reaction on Cu2O, resulting in Au deposition that is limited by the formation of CuO on the interface. Further deposition depends on the kinetics of CuO dissolution and electron drift from the near-surface bulk at the interface. The passivation of the surface and further reduction of Au on deposited Au provides a chemical handle for controlling the size and distribution of Au nanoparticles at the interface. As a result, this new mechanistic understanding can lead to new methods for engineering the morphology-dependent functional interfaces.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Center for Advanced Surface Engineering, under the National Science Foundation (grant number IIA-1457888). We acknowledge engaging discussions on this topic with Prof. Mita Dasog (Dalhousie University) and Prof. Jingyi Chen (University of Arkansas).References
- L. Magagnin, R. Maboudian and C. Carraro, J. Phys. Chem. B, 2002, 106, 401–407 CrossRef CAS.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- L. Au, X. Lu and Y. Xia, Adv. Mater., 2008, 20, 2517–2522 CrossRef CAS PubMed.
- A. P. O'Mullane, S. J. Ippolito, A. M. Bond and S. K. Bhargava, Electrochem. Commun., 2010, 12, 611–615 CrossRef.
- A.-A. El Mel, M. Chettab, E. Gautron, A. Chauvin, B. Humbert, J.-Y. Mevellec, C. Delacote, D. Thiry, N. Stephant, J. Ding, K. Du, C.-H. Choi and P.-Y. Tessier, J. Phys. Chem. C, 2016, 120, 17652–17659 CrossRef CAS.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS PubMed.
- D. K. Smith and B. A. Korgel, Langmuir, 2008, 24, 644–649 CrossRef CAS PubMed.
- C. J. DeSantis, A. C. Sue, M. M. Bower and S. E. Skrabalak, ACS Nano, 2012, 6, 2617–2628 CrossRef CAS PubMed.
- K. D. Gilroy, P. Farzinpour, A. Sundar, R. A. Hughes and S. Neretina, Chem. Mater., 2014, 26, 3340–3347 CrossRef CAS.
- B. Goris, L. Polavarapu, S. Bals, G. Van Tendeloo and L. M. Liz-Marzán, Nano Lett., 2014, 14, 3220–3226 CrossRef CAS PubMed.
- F. Hoshyargar, J. Crawford and A. P. O'Mullane, J. Am. Chem. Soc., 2017, 139, 1464–1471 CrossRef CAS PubMed.
- E. A. Sutter and P. W. Sutter, Nanoscale, 2017, 9, 1271–1278 RSC.
- V. Bansal, H. Jani, J. Du Plessis, P. J. Coloe and S. K. Bhargava, Adv. Mater., 2008, 20, 717–723 CrossRef CAS.
- B. Zhang, P. Xu, X. Xie, H. Wei, Z. Li, N. H. Mack, X. Han, H. Xu and H.-L. Wang, J. Mater. Chem., 2011, 21, 2495–2501 RSC.
- C. Li, Y. Su, X. Lv, H. Shi, X. Yang and Y. Wang, Mater. Lett., 2012, 69, 92–95 CrossRef CAS.
- H. Rasouli, S. H. Tabaian and M. Rezaei, RSC Adv., 2016, 6, 22500–22510 RSC.
- I. E. Stewart, S. Ye, Z. Chen, P. F. Flowers and B. J. Wiley, Chem. Mater., 2015, 27, 7788–7794 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, 95th edn, 2014 Search PubMed.
- L. Xiong, S. Li, B. Zhang, Y. Du, P. Miao, Y. Ma, Y. Han, H. Zhao and P. Xu, RSC Adv., 2015, 5, 76101–76106 RSC.
- Y. Shang and L. Guo, Adv. Sci., 2015, 2, 1500140 CrossRef PubMed.
- X. Liu, RSC Adv., 2011, 1, 1119–1125 RSC.
- T. D. Golden, M. G. Shumsky, Y. Zhou, R. A. VanderWerf, R. A. Van Leeuwen and J. A. Switzer, Chem. Mater., 1996, 8, 2499–2504 CrossRef CAS.
- J. M. Lowe, Q. Yan, M. Benamara and R. H. Coridan, J. Mater. Chem. A, 2017, 5, 21765–21772 RSC.
- D. Nicholls, Complexes and First-Row Transition Elements, Palgrave Macmillan, London, 1975 Search PubMed.
- F. A. Cotton, G. Wilkinson, M. Bochmann and R. N. Grimes, Advanced Inorganic Chemistry: A Comprehensive Text, Wiley-Interscience, New York, 5th edn, 1988 Search PubMed.
- P. Villars, L. D. Calvert and W. B. Pearson, Pearson's Handbook of Crystallographic Data for Intermetallic Phases, American Society for Metals, Metals Park, Ohio, USA, 1985, Vol. 2 Search PubMed.
- H. Okamoto, D. J. Chakrabarti, D. E. Laughlin and T. B. Massalski, J. Phase Equilib., 1987, 8, 454 CrossRef CAS.
- G. V. Kuznetsov, V. A. Skryshevsky, T. A. Vdovenkova, A. I. Tsyganova, P. Gorostiza and F. Sanz, J. Electrochem. Soc., 2001, 148, C528–C532 CrossRef CAS.
- J. Jiang, Z. Huang, C. Xiang, R. Poddar, H.-J. Lewerenz, K. M. Papadantonakis, N. S. Lewis and B. S. Brunschwig, ChemSusChem, 2017, 10, 4657–4663 CrossRef CAS PubMed.
- S.-H. Ye, X.-J. He, L.-X. Ding, Z.-W. Pan, Y.-X. Tong, M. Wu and G.-R. Li, Chem. Commun., 2014, 50, 12337–12340 RSC.
- M. D. Susman, R. Popovitz-Biro, A. Vaskevich and I. Rubinstein, Small, 2015, 11, 3942–3953 CrossRef CAS PubMed.
- H. Luo, J. Zhou, H. Zhong, L. Zhou, Z. Jia and X. Tan, RSC Adv., 2016, 6, 99105–99113 RSC.
- P. Liu, Z. Cheng, L. Ma, M. Zhang, Y. Qiu, M. Chen and F. Cheng, RSC Adv., 2016, 6, 76684–76690 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00396c |
| This journal is © The Royal Society of Chemistry 2019 |