
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2D MXene-containing polymer electrolytes for all-solid-state lithium metal batteries†
Qiwei
Pan
*ab,
Yongwei
Zheng
a,
Sankalp
Kota
a,
Weichun
Huang
ac,
Shijun
Wang
a,
Hao
Qi
a,
Seyong
Kim
a,
Yingfeng
Tu
 c,
Michel W.
Barsoum
a and
Christopher Y.
Li
*a
c,
Michel W.
Barsoum
a and
Christopher Y.
Li
*a
aDepartment of Materials Science and Engineering, Drexel University, Philadelphia, PA 19104, USA. E-mail: chrisli@drexel.edu
bCollege of Materials Science and Engineering, South China University of Technology, Guangzhou, 510640, China. E-mail: panqw@scut.edu.cn
cCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China
First published on 14th September 2018
Abstract
Nanocomposite polymer electrolytes (CPEs) are promising materials for all-solid-state lithium metal batteries (LMBs) due to their enhanced ionic conductivities and stability to the lithium anode. MXenes are a new two-dimensional, 2D, family of early transition metal carbides and nitrides, which have a high aspect ratio and a hydrophilic surface. Herein, using a green, facile aqueous solution blending method, we uniformly dispersed small amounts of Ti3C2Tx into a poly(ethylene oxide)/LiTFSI complex (PEO20-LiTFSI) to fabricate MXene-based CPEs (MCPEs). The addition of the 2D flakes to PEO simultaneously retards PEO crystallization and enhances its segmental motion. Compared to the 0D and 1D nanofillers, MXenes show higher efficiency in ionic conductivity enhancement and improvement in the performance of LMBs. The CPE with 3.6 wt% MXene shows the highest ionic conductivity at room temperature (2.2 × 10−5 S m−1 at 28 °C). An LMB using MCPE with only 1.5 wt% MXene shows rate capability and stability comparable with that of the state-of-the-art CPELMBs. We attribute the excellent performance to the 2D geometry of the filler, the good dispersion of the flakes in the polymer matrix, and the functional group-rich surface.
Introduction
Rechargeable lithium ion batteries (LIBs) have been widely used in applications such as portable electronics, electric vehicles and large-scale energy storage. To solve the inherent safety issue of LIBs and further enhance their energy density, all-solid-state lithium metal batteries (LMBs) have been proposed.1–3 In these LMBs, solid-state electrolytes are utilized to replace the toxic, volatile, and flammable liquid electrolytes, with the high capacity lithium metal (3860 mA h g−1) anode to replace the low capacity graphite (372 mA h g−1) in LIBs. However, active lithium metal anodes typically lead to faster lithium dendrite formation and sequential short-circuit in the cells. Solid-state electrolytes with good mechanical properties are therefore of crucial importance since they are anticipated to inhibit dendritic growth.3–8 Moreover, lithium metal can act as the lithium source in the battery to enable the application of nonlithiated materials, such as sulfur or oxygen as the cathode to greatly improve the energy density of the battery.9Solid-state electrolytes with high ionic conductivities, wide electrochemical windows, and long-term stability are desirable for LMBs. Both inorganic electrolytes, such as Li7La3Zr2O12 (LLZO),10 Li7La2.75Ca0.25Zr1.75Nb0.25O12 (LLCZN),1 and Li2S–P2S5,11 and solid polymer electrolytes (SPEs) based on poly(ethylene oxide) (PEO),12 poly(propylene carbonate),13 and poly(methacrylate) (PMA),14 have been reported. Inorganic electrolytes typically have high ionic conductivities at room temperature (RT). However, brittleness and reactions with the lithium metal anodes have hindered their application in LMBs. SPEs are flexible and light-weight, whereas they suffer from relatively low RT ionic conductivities. To take advantage of both inorganic and polymeric materials, Weston and Steele fabricated the first composite electrolyte (CPE), PEO-LiClO4-Al2O3 in 1982.15,16 Croce et al. later reported that the RT ionic conductivities of the SPEs of PEO-LiClO4 were greatly enhanced (1000 times) by adding ceramic nanoparticles such as Al2O3 or TiO2.17 Numerous ceramic materials have since been introduced into polymer electrolytes to form CPEs. These fillers can be classified on the basis of their dimensions. Zero-dimensional (0D) fillers include SiO2,17–19 ZrO2,20 TiO2,21 MgAl2O4,22 Li6.4La3Zr1.4Ta0.6O12 (LLZTO),23,24etc. One-dimensional (1D) Li0.5La0.5TiO3 (LLTO) nanowires,25,26 halloysite nanoclay,27 2D graphene oxide (GO),28,29 and clay30 have also been used in CPEs. The general mechanistic reasons for increased ionic conductivities in CPEs is three-fold: (1) the fillers act as plasticizers to lower the crystallinity of the polymers and enhance the motion of the polymer segments; (2) ion transfer pathways can be formed on the filler surfaces; (3) dissociation of the lithium salts is facilitated because of the interaction between selective ions and filler surface functional groups. Therefore, fillers with large surface areas and rich surface functional groups are suitable to prepare CPEs, as demonstrated by the success of in situ nanosilica and porous nano-Al2O3.18,31
2D materials have higher specific surface areas compared to 0D or 1D materials, and therefore are considered as promising candidates for CPEs. The unique 2D feature also could render anisotropic properties of the CPEs, as demonstrated in clay, GO, phase separated polymers, and even polymer single crystals.32–36 The in-plane conductivity along the 2D filler surface can be two orders of magnitude higher than that along the normal direction of the filler.34 MXenes are a new family of 2D transition metal carbides and/or nitrides, which are best described as Mn+1XnTx, where M is an early transition metal, X is carbon and/or nitrogen, T is the terminating group (O, OH or F), x is the number of T, and n is the number of X (varies from 1 to 3).37,38 Different from graphene, MXenes are hydrophilic due to their terminal groups. This hydrophilicity is critical in applications such as capacitors,39,40 LIB anodes,41,42 electromagnetic interference (EMI) shielding,43etc. For CPEs, this hydrophilic surface can enhance the interaction between MXene and the polymer chain, leading to reduced PEO crystallinity and enhanced ionic conductivity. In addition, compared with GO, a typical 2D flake used in CPEs, over 20 types of MXenes with rich layers and surface chemistry can be prepared under relatively mild conditions, offering an unprecedented opportunity for solid state battery research.
In this study polymer/MXene composites have been fabricated and characterized. The first work mixes Ti3C2Tx with polyvinyl alcohol (PVA) and polydiallyldimethylammonium chloride.44 Liu et al. incorporated MXene with acidic Nafion, sulfonated poly(etherether ketone) and basic chitosan to prepare polymer composites for proton transport in fuel cells.45 We have recently demonstrated that in a salt-free MXene polymer nanocomposite, the MXene fillers have an intriguing effect on PEO crystallization.46 In this work, we hypothesized that because of its large surface area and hydrophilic surface with rich functional groups, MXene could be an excellent nanofiller for CPE. Here we report the first study on MXene-containing CPEs (MCPEs). We show that MXene inhibit PEO crystallization, enhance the ionic conductivities and accelerate polymer chain dynamics. MCPE-based LMBs have also been fabricated. Our tests demonstrate that state-of-the-art rate capabilities and stability are achieved at a much lower nanofiller content compared with other CPE systems. We therefore envisage that MCPEs could be a new class of materials for all-solid-state LMBs.
Experimental section
Materials
Poly(ethylene oxide) (PEO, Mn =300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and bis(trifluoromethane)sulfonimide Li salt (LiTFSI, 99.95%) were purchased from Sigma-Aldrich and used as received. Nitrogen gas was bubbled through deionized, DI, water (Ricca Chemical Company), for 0.5 h to remove the dissolved oxygen. Commercially available Ti2AlC powders were purchased from Kanthal in Sweden. Lithium foil (99.9%) was purchased from Alfa Aesar. 6 M hydrochloric acid (HCl) was purchased from Fisher Scientific. Polypropylene membranes – with a 0.22 μm pore size – were purchased from Celgard LLC.
000 g mol−1) and bis(trifluoromethane)sulfonimide Li salt (LiTFSI, 99.95%) were purchased from Sigma-Aldrich and used as received. Nitrogen gas was bubbled through deionized, DI, water (Ricca Chemical Company), for 0.5 h to remove the dissolved oxygen. Commercially available Ti2AlC powders were purchased from Kanthal in Sweden. Lithium foil (99.9%) was purchased from Alfa Aesar. 6 M hydrochloric acid (HCl) was purchased from Fisher Scientific. Polypropylene membranes – with a 0.22 μm pore size – were purchased from Celgard LLC.
Preparation of composite polymer electrolytes
A Ti3C2Tx colloidal suspension comprised of single and few layered flakes was prepared as previously reported.46Fig. 1a shows the green, one-pot MCPE fabrication process. PEO and LiTFSI with a [EO]/[Li+] ratio of 20 were dissolved in de-aerated DI water. Colloidal Ti3C2Tx suspension was then added to the mixture. The flask was sealed, and the suspension was stirred at RT for 24 h, sonicated for 10 min and then cast onto a PTFE Petri dish to evaporate the solvent at RT. Membranes were obtained after further drying at 70 °C for 72 h, and then at 120 °C for 4 h under vacuum. MCPEs with 0, 0.5:100, 2:100, 5:100, and 10:100 MXene to PEO mass ratios were prepared. All membranes were stored in an MBraun glove box (H2O < 1 ppm, O2 < 1 ppm) for at least one week before being tested.
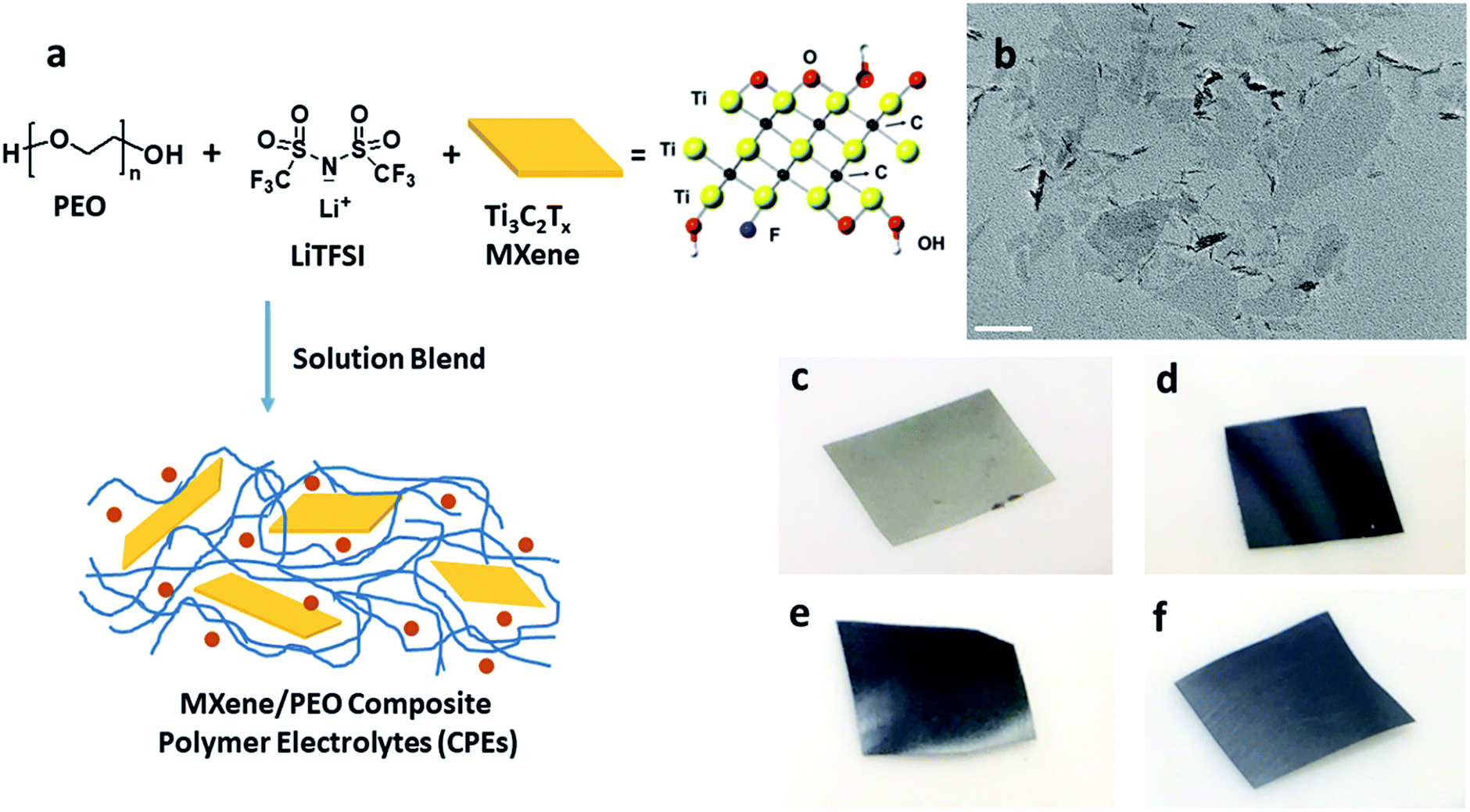 | ||
| Fig. 1 Fabrication of MCPEs. (a) Preparation procedure of MCPEs. (b) TEM micrograph of the as-prepared few-layer MXene, scale bar is 100 nm. (c–f) Photographs of the MCPE membranes, (c) PEO20-LiTFSI-MXene0.005, (d) PEO20-LiTFSI-MXene0.02, (e) PEO20-LiTFSI-MXene0.05 and (f) PEO20-LiTFSI-MXene0.1. | ||
LiFePO4|MCPE|Li batteries fabrication and testing
A composite LiFePO4 cathode was prepared as reported elsewhere.6 The loading of active material was approximately 2.88 mg cm−2, corresponding to a current density of 0.49 mA cm−2 at 1C. 2032-type coin-cell batteries were assembled in the glove box using a lamination method. The MCPE membrane was used as the separator in-between the cathode and lithium metal anode. The potential window used was between 4.0 V and 2.5 V. All LMBs were galvanostatically cycled at 60 °C and tested with an Arbin battery tester.Characterization
Differential scanning calorimetry (DSC) experiments were conducted using a DSC Q2000 (TA Instruments) with Tzero pans. Transmission electron microscopy (TEM) experiments were performed on a JEOL JEM2100. The MCPEs were embedded in an epoxy resin, and microtomed into ∼100 nm thin sections using a Leica UC6 ultramicrotome. Scanning electron microscopy (SEM) images were obtained with a Zeiss Supra 50VP SEM. The SEM specimens were prepared by sectioning the MCPE membranes in the glove box.The ionic conductivity was measured in the glove box using a custom-made cell32 connected to a Princeton Applied Research Parstat 2273 Potentiostat, with POWERSUITE software. The MCPE membrane and a 120 μm thick PTFE ring spacer were sandwiched in-between the two stainless steel blocking electrodes in the cell. Specimens with 5 mm in diameter of fixed thickness were then obtained by hot-pressing the cell at 110 °C for 2 h in the glove box. After cooling to 28 °C, temperature scan of the ionic conductivity of the CPE with 10 °C increments was conducted using a hot-stage with temperature accuracy of ±1 °C in the glove box. A 20 mV ac perturbation and a frequency range of 1 MHz to 0.1 Hz were used for each measurement. The ionic conductivity (σ) was calculated assuming
| σ = L/(A × R) | (1) |
The Li ion transference numbers, tLi+, of all the CPEs were measured at 60 °C using the method proposed by Bruce et al.47 and by Appetecchi et al.48 The impedance of the Li symmetric cell was measured before and after polarization with a DC voltage pulse (ΔV = 10–30 mV) with a Gamry Interface 1000 Potentiostat. tLi+ was calculated assuming
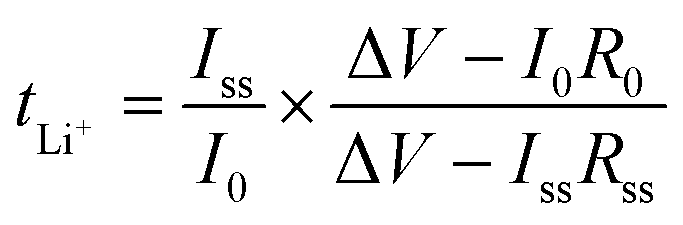 | (2) |
Linear sweep voltammetry (LSV) measurements were performed at 60 °C using a Gamry Interface 1000 Potentiostat. Li metal and stainless steel were used as the reference and the working electrodes, respectively. A linear sweep was conducted from 2 V to 5 V at a scan rate of 0.1 mV s−1. The electronic conductivity of the MCPE was measured by placing it between two stainless steel electrodes and applying a potential ΔV = 1 V until a steady state current was obtained. The membrane's resistance was calculated from the steady state current. The electronic conductivity was then calculated using eqn (1).
Results and discussion
As noted above, LiF/HCl solution was used to etch the Al layers to yield Ti3C2Tx.46 The chemical structure of Ti3C2Tx is shown in Fig. 1a. Terminal functional groups, such as O, OH, and F, are on the surface of the flakes. Fig. 1b is a TEM bright field micrograph of the obtained few-layer MXene flakes with lateral sizes up to 400 nm. The MCPEs were fabricated using solution blending of PEO/LiTFSI and MXene aqueous solution/suspension (Fig. 1a). The [EO]/[Li+] ratio was fixed to 20, since PEO/LiTFSI SPE shows the highest ionic conductivity at this ratio.49 Four samples were prepared, denoted as PEO20-LiTFSI-MXenem, where the superscript m is the weight ratio of MXene to PEO, controlled to be 0.005, 0.02, 0.05, and 0.1. The mass contents of MXene in these four samples are therefore calculated to be 0.38%, 1.5%, 3.6% and 7.0%, respectively (Table 1). The subscript 20 is the molar ratio of EO to Li+. Table 1 summarizes the characteristics of the four samples. The MXene-free SPE, PEO20-LiTFSI, is used as the control. Photographs of MCPEs are also shown in Fig. 1c–f. At low MXene content, PEO20-LiTFSI-MXene0.005, the membrane is light grey (Fig. 1c). Dark membranes were obtained with higher MXene contents (Fig. 1d to f).| CPE | MXene content in SPE, wt% | T g (°C) | T m (°C) | Crystallinitya | T c,onset (°C) | T c,peak (°C) | σ electronic,60°C (×10−10 S cm−1) | t Li+ |
|---|---|---|---|---|---|---|---|---|
| a Based on DSC second heating thermograms. | ||||||||
| PEO20-LiTFSI | 0 | −39.1 | 50.2 | 29.5% | 31.3 | 27.2 | 1.40 | 0.18 |
| PEO20-LiTFSI-MXene0.005 | 0.38 | −40.4 | 51.7 | 34.4% | 36.1 | 32.1 | 1.51 | 0.18 |
| PEO20-LiTFSI-MXene0.02 | 1.5 | −40.8 | 44.9 | 23.1% | 30.0 | 24.5 | 5.00 | 0.18 |
| PEO20-LiTFSI-MXene0.05 | 3.6 | −45.8 | 45.5 | 27.3% | 30.7 | 23.1 | 7.02 | 0.17 |
| PEO20-LiTFSI-MXene0.1 | 7.0 | −40.8 | 50.5 | 27.5% | 36.3 | 32.8 | 2.67 | 0.16 |
To study the morphology of MCPE films, ∼100 nm thin sections of the MCPE film were obtained using ultramicrotomy for TEM observation. Due to its hydrophilic surface, MXene is compatible with PEO20-LiTFSI. The TEM image in Fig. 2a shows that the MXene flakes are well dispersed in the PEO matrix. It is also evident that MXenes are exfoliated and the single-layer platelets have a thickness of ∼1 nm (Fig. 2b). Similar single-layer MXene morphology can be found in the previously reported PVA/MXene composites.44 The cross-section of MCPEs was also examined using SEM, and the image shows that the films are dense without obvious voids (Fig. 2c). EDS elemental mapping (Fig. 2d–g) confirms the uniform incorporation of Ti3C2Tx in the MCPE membranes.
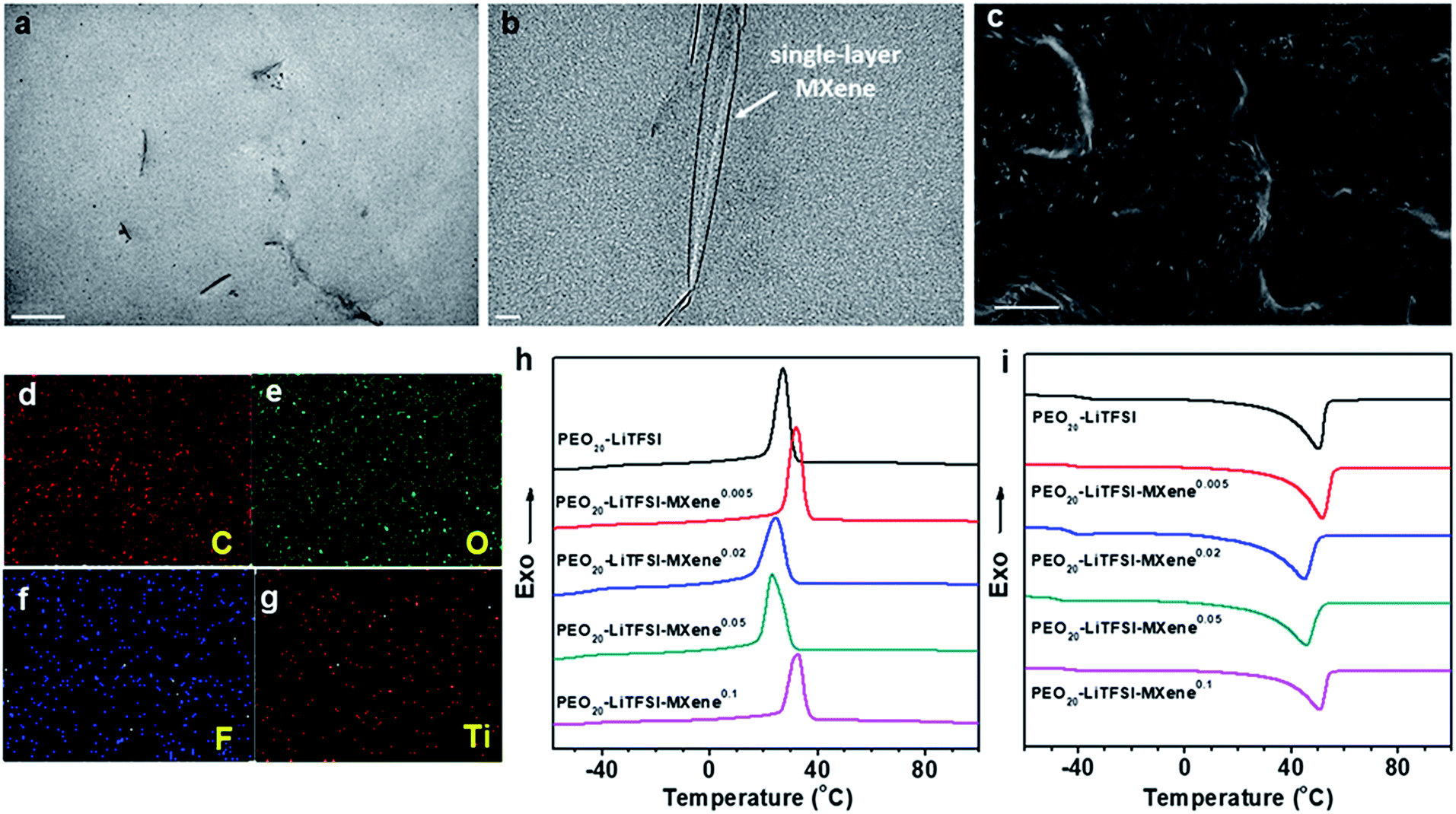 | ||
| Fig. 2 TEM micrographs of the cross-section of PEO20-LiTFSI-MXene0.02 ((a), scale bar is 500 nm; (b), scale bar is 20 nm). SEM image ((c), scale bar is 3 μm) and SEM-EDS surface scan of C (d), O (e), F (f) and Ti elements (g) in the cross-section of PEO20-LiTFSI-MXene0.05. DSC thermograms obtained at a rate of 10 °C min−1 during first cooling (h) and second heating (i). | ||
The phase behavior of the MCPEs was studied using DSC. Fig. 2h and i show the first cooling and subsequent heating thermograms in the DSC experiment obtained at a rate of 10 °C min−1. Crystallization/melting and a glass transition can be observed in each of the thermogram. Table 1 also lists the transition temperatures of each sample. Incorporating MXenes into PEO20-LTFSI clearly affects the phase transition of PEO. Its glass transition temperature (Tg) is reduced. It suggests enhanced polymer chain dynamics, which is crucial for ion transport in SPEs.
Since crystalline PEO reduces ion transport,32 decreased crystallinity is desirable in SPE design. Interestingly, for the MCPEs, the crystallization temperature first increases from 27.2 °C to 32.1 °C at the lowest MXene loading (m = 0.005), then decreases to 24.5 and 23.1 °C when m is 0.02 and 0.05, respectively. Further increasing m to 0.1 increases the crystallization temperature to 32.8 °C. In our recent publication,46 we reported a similar observation in PEO/MXene nanocomposites without the Li salt. We showed that this intriguing behavior of PEO crystallization can be attributed to the competing nucleation and nano-confining effects of the 2D filler on PEO crystallization. At low concentrations, nucleation is dominant since there are few MXene nanoflakes in the composites to confine and slow down crystal growth. As the MXene content increases to m = 0.02 and 0.05, while nucleation is fast, the abundance of the 2D flakes inhibits growth into large crystals. When m further increases to 0.1, most of the polymer is in the vicinity of the filler surface, and nucleation effects therefore dominate again, leading to the increased crystallization temperature for PEO20-LiTFSI-MXene0.1. Similar trends are observed in cooling on-set temperatures, melting peak temperatures, and crystallinity, as shown in Table 1.
XRD patterns of PEO20-LiTFSI and the MCPEs at RT confirm their crystalline structure (Fig. S1†). For all the samples, two major diffraction peaks are observed. The one at 2θ = 19.15° corresponds to the (120) plane of the PEO monoclinic crystal structure; the other at 2θ = 23.32° belongs to the (032) plane.
Fig. 3a compares the AC ionic conductivities of the MCPEs fabricated here. Three specimens were measured for each sample and the standard deviation was less than 22%. The plot shows a change of slope at ∼45–50 °C, which is attributed to the melting of the PEO crystals, consistent with the DSC results. The temperature-dependent conductivity data were fitted using the modified Vogel–Tammann–Fulcher (VTF) equation (ESI, Fig. S3†). Fitting results show that at high temperature, the ion transport follows a VTF mechanism. The divergence from the VTF behavior at low temperature is due to PEO crystallization. The filler effect on the SPE conductivity can be more clearly revealed when plotting the conductivity vs. MXene content (Fig. 3b). At 60 °C, the filler-free PEO20-LiTFSI shows an ionic conductivity of 0.36 mS m−1. The ionic conductivity gradually increases with the addition of the MXene, reaching a maximum of 0.69 mS cm−1 for PEO20-LiTFSI-MXene0.05. Further increase in the MXene content reduces the conductivity to 0.54 mS cm−1. Similar trends can be observed at 28 °C (Fig. 3b): the conductivity increases from 6.4 × 10−6 S cm−1 for the filler-free sample, to 2.2 × 10−5 S cm−1 for PEO20-LiTFSI-MXene0.05, and then decreases to 9.47 × 10−6 S cm−1 for the PEO20-LiTFSI-MXene0.1 composition. Note that the ionic conductivity of 2.2 × 10−5 S cm−1 at 28 °C is comparable to state-of-the-art CPE values such as PEO8-LiClO4-10% in situ SiO2,18 PEO8-LiClO4-10%TiO2,17 PEO18-LiTFSI-10%SiO2,19 and PEO8-LiClO4-40 vol%LATP,50etc. Notably, the filler loading needed to obtain these ionic conductivities is significantly lower compared with the aforementioned systems, implying that Ti3C2Tx is more effective in enhancing the conductivities of CPEs. We attributed this to a combination of the 2D geometry of the MXene and strong interaction between the PEO chain and the hydrophilic surface. The decrease in conductivity at the highest MXene loading in Fig. 3b can be attributed to the higher crystallinity and the tortuous ion pathways associated with 2D nanofillers, similar to the morphological effects on ionic transport observed in other SPE systems.33,34,36,51,52
 | ||
| Fig. 3 (a) Temperature scan of the ionic conductivities of the MCPEs. (b) Ionic conductivities vs. MXene content at 28 °C and 60 °C. (c) Lithium transference number measurement of the PEO20-LiTFSI-MXene0.02. (d) Linear sweep voltammetry of the MCPEs (scan rate 0.1 mV s−1). Curves are shifted along the current axis for clarity. | ||
As noted above, MXenes possess high electron conductivities and indeed MXene/polymer composites with high electronic conductivities have been achieved when the MXene loadings are high.43,44 For SPE application, the electrolyte membrane has to be ionically conductive but electronically insulating to avoid short circuiting of the cells. At low enough loadings, however, the electronic conductivity should be negligible when the filler particles are unable to form a percolation pathway. The reported DC polarization method was used to measure the electronic conductivities at 60 °C of filler-free SPE and the MCPEs.23,53 Fig. S2† shows that there is not much difference between the DC polarization curves of the samples. The electronic conductivities calculated from the steady state currents are listed in Table 1 and at about ≈10−10 S cm−1, which are six orders of magnitude lower than the MCPE ionic conductivities. This observation implies that the Ti3C2Tx flakes are fully surrounded by the PEO matrix at low loadings, unable to form a percolation pathway for electronic conduction, and thus suitable as SPEs for LMBs.
The lithium ion transference number (tLi+) is measured by DC polarization and AC electrochemical impedance spectroscopy29 before and after polarization. A typical time dependence of the ionic current of a PEO20-LiTFSI-MXene0.02 is shown in Fig. 3c. The inset in Fig. 3c shows Nyquist plots of the MCPE-based lithium symmetric cells before and after polarization, confirming the stability of the interface and a relatively low interface resistance between the MCPE and lithium metal. From these results we calculated the values of tLi+ of the MCPEs at 60 °C and listed them in Table 1. Typical values of approximately 0.18 are found for all electrolytes. Note that filler-dependent tLi+ has been reported in CPEs,27 which is typically attributed to the filler effect on polymer chain dynamics as well as the selective interaction of the filler with the cations and anions of the lithium salts used.
Individual Ti3C2Tx sheets have a zeta potential of −20 mV (in aqueous solutions with pH = 6), which implies that the surfaces are negatively charged.54 These negative charges can attract lithium cations and have them preferentially interact with the filler surfaces, similar to the case of basic Al2O3.31 This type of interaction accelerates ion transport as it provides an alternative pathway for cation transport. The constancy of the tLi+ values with filler content suggests that neither the cations nor anions are strongly immobilized on the Ti3C2Tx surfaces.
The electrochemical stability of the filler-free SPE and MCPEs was investigated using linear sweep voltammetry from 2 to 5 V at a scan rate of 0.1 mV s−1 at 60 °C, and the results are shown in Fig. 3d. PEO20-LiTFSI is stable up to 4.7 V. For MCPEs, the electrochemical stability remains constant at ≈4.7 V. It should be noted that enhanced electrochemical stability has been reported in some CPE systems, such as PEO8-LiClO4-10% in situ SiO2.18 Since the electrochemical instability of the PEO/LiTFSI complex arises from the decomposition of the anion,55 it's reported that the enhancement comes from the interaction between the filler and TFSI−. In the present case, the MXenes with a negatively charged surface do not provide strong preferential adsorption of anions in the MCPEs, leading to the similar observed working voltage compared with the MXene-free sample.
To study the electrodeposition of lithium with the MCPEs, a lithium symmetric cell with PEO20-LiTFSI-MXene0.02 as the electrolyte and separator was assembled and galvanostatically cycled using a current density of 0.3 mA cm−2 at 60 °C (Fig. S4†). One hour charge/discharge cycling was conducted. Stable voltage is obtained after cycling for approximately 60 hours as shown in Fig. S4,† which confirms that a stable MCPE/Li interface was formed in the symmetric cell.
Considering the high ionic conductivities, wide electrochemical windows, and stability to Li metal of our MCPEs at 60 °C, LMBs were fabricated to evaluate their potential as separators in SPEs. From a LMB fabrication standpoint, MCPEs with less nanofillers are more desirable, due to the high cost and mass density of the nanofillers compared with polymers. To this end, PEO20-LiTFSI-MXene0.02 was chosen to fabricate LMBs. Composite cathodes were prepared using LiFePO4 as the active material. The previously reported polyhedral oligomeric silsesquioxane (POSS)-based cross-linked hybrid SPE (POSS-2PEG6K) was used as the binder,6,56 and carbon black as the conductive phase. An active material to binder to conductive phase weight ratio of 60:32:8 was chosen. Coin cells (2032 type) were fabricated in the glove box and galvanostatically cycled at 60 °C. The voltage profiles at different rates are shown in Fig. 4a, where C/x denotes the charge/discharge of the theoretical cathode capacity (C, 170 mAh g−1) in x h. A typical potential plateau is observed for all rates studied (Fig. 4a). Charge/discharge capacities above 150 mAh g−1 were obtained at C/10 and C/5. Increasing the charge/discharge rates to C/3 and C/2 slightly decreases the capacities to 140 and 130 mAh g−1, respectively. A capacity of 92 mAh g−1 is delivered at 1C. The LMB was also galvanostatically cycled at a C/3 rate for 100 cycles. The plot of discharge capacity and coulombic efficiency vs. cycle number is shown in Fig. 4b. The LMB retained 91.4% of its original capacity after 100 cycles, and the coulombic efficiency was >97% during cycling. EIS was used to track the resistance change of the battery. The Nyquist plots of the LiFePO4|PEO20-LiTFSI-MXene0.02|Li battery before and after 100 cycles are shown in Fig. S5.† The intercept of the spectra with the real axis reflects the bulk resistance of MCPE. The semicircle denotes the resistance of the MCPE/Li interface, which slightly increased after cycling.
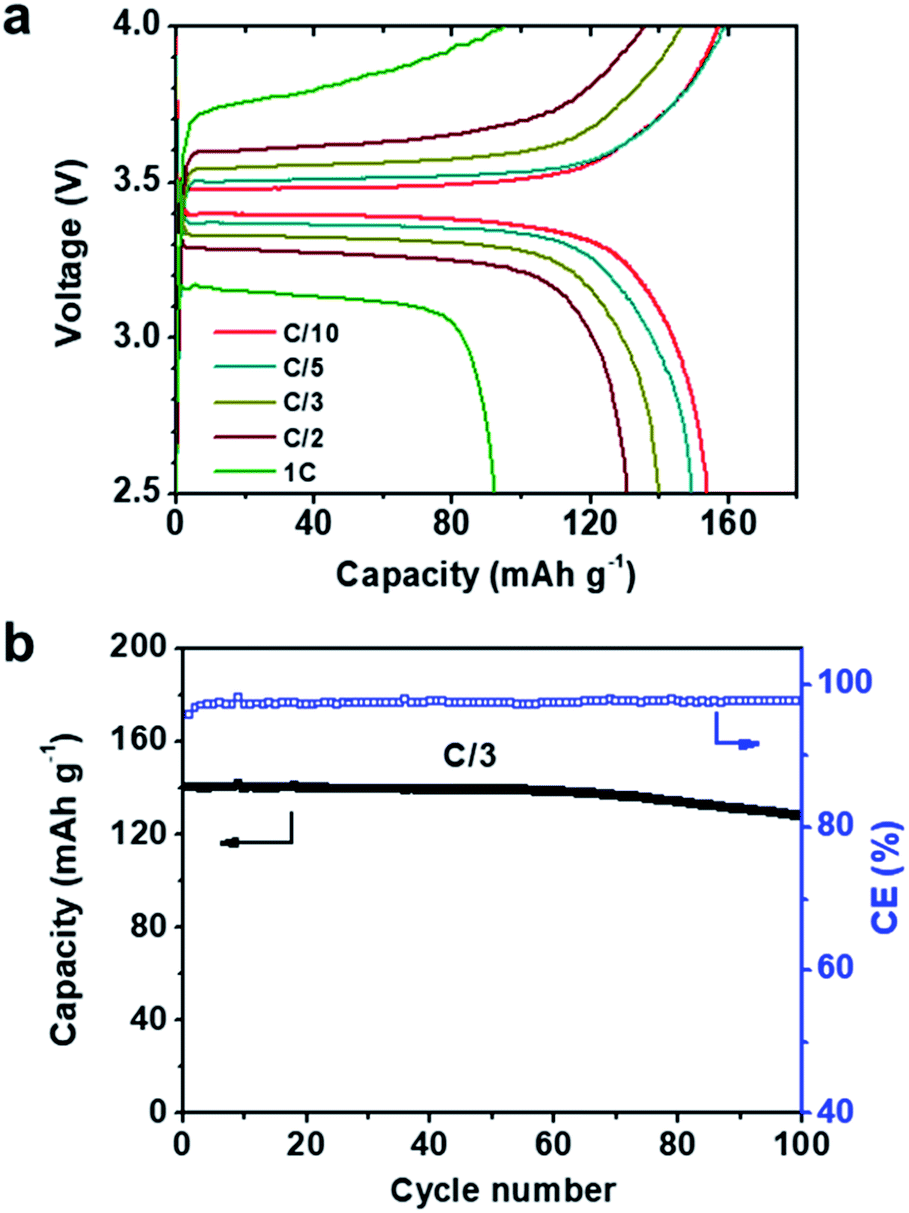 | ||
| Fig. 4 Performance of the LiFePO4|PEO20-LiTFSI-MXene0.02|Li battery at 60 °C. (a) Voltage profile at different C rates. (b) Capacity and coulombic efficiency vs. cycle number at C/3. The LiFePO4 loading is ≈2.88 mg cm−2. | ||
Notably, the rate capabilities and stabilities of these LMBs are comparable with some of the best reported LMBs to date such as PEO8-LiClO4-10% in situ SiO218 and PEO-12.7 vol% nano LLZTO.23 It is important to note that much less filler is used in our MCPE-based LMBs. We attribute this excellent performance to the good dispersion of MXene, which in turn results in high surface areas and the richness of the surface functional groups.
Conclusions
A novel 2D material, few-layer Ti3C2TxMXene is used as the nanofiller to incorporate with PEO-LiTFSI to prepare CPEs by aqueous solution blending. The TEM and SEM results show that MXene is well-dispersed in the polymer matrix due to its hydrophilic surface. The 2D filler enhances PEO chain dynamics and retards its crystallization. Furthermore, this 2D filler is more efficient in enhancing ionic conductivity and improving LMB performance than 0D and 1D nano fillers due to its large surface area and hydrophilic surface. The CPE with 3.6 wt% MXene shows the highest ionic conductivity at room temperature (2.2 × 10−5 S m−1 at 28 °C). The CPEs show lithium transference numbers of around 0.18, and electrochemical stability up to 4.7 V. A LiFePO4/Li battery with CPE containing 1.5wt% MXene as the electrolyte tested at 60 °C for 50 cycles at C/3 (C = 170 mAh g−1) yields a stable capacity of ≈140 mAh g−1. The facility and green production method by which these electrolytes can be made, together with the small loadings needed to enhance their properties, suggest that MXenes are a promising 2D material to prepare CPEs for all-solid-state LMB applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation through grants CBET-1510092, CBET-1603520 and DMR-1740795 as well as ACS-PRF (54945-ND7).References
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- P. Hovington, M. Lagacé, A. Guerfi, P. Bouchard, A. Mauger, C. M. Julien, M. Armand and K. Zaghib, Nano Lett., 2015, 15, 2671–2678 CrossRef CAS PubMed.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- C. X. Zu and H. Li, Energy Environ. Sci., 2011, 4, 2614–2624 RSC.
- D. T. Hallinan, S. A. Mullin, G. M. Stone and N. P. Balsara, J. Electrochem.Soc., 2013, 160, A464–A470 CrossRef CAS.
- Q. Pan, D. M. Smith, H. Qi, S. Wang and C. Y. Li, Adv. Mater., 2015, 27, 5995–6001 CrossRef CAS PubMed.
- R. Khurana, J. L. Schaefer, L. A. Archer and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7395–7402 CrossRef CAS PubMed.
- C. Wang, Y. Yang, X. Liu, H. Zhong, H. Xu, Z. Xu, H. Shao and F. Ding, ACS Appl. Mater. Interfaces, 2017, 9, 13694–13702 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- A. Sakuda, A. Hayashi, Y. Takigawa, K. Higashi and M. Tatsumisago, J. Ceram. Soc. Jpn., 2013, 121, 946–949 CrossRef CAS.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- J. Zhang, J. Zhao, L. Yue, Q. Wang, J. Chai, Z. Liu, X. Zhou, H. Li, Y. Guo, G. Cui and L. Chen, Adv. Energy Mater., 2015, 5, 1501082 CrossRef.
- Z. Zhu, M. Hong, D. Guo, J. Shi, Z. Tao and J. Chen, J. Am. Chem. Soc., 2014, 136, 16461–16464 CrossRef CAS PubMed.
- J. E. Weston and B. C. H. Steele, Solid State Ionics, 1982, 7, 75–79 CrossRef CAS.
- B. Kumar and L. G. Scanlon, J. Power Sources, 1994, 52, 261–268 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- D. Lin, W. Liu, Y. Liu, H. R. Lee, P.-C. Hsu, K. Liu and Y. Cui, Nano Lett., 2016, 16, 459–465 CrossRef CAS PubMed.
- S. Liu, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda, O. Yamamoto and J. Yang, J. Power Sources, 2010, 195, 6847–6853 CrossRef CAS.
- L. Damen, J. Hassoun, M. Mastragostino and B. Scrosati, J. Power Sources, 2010, 195, 6902–6904 CrossRef CAS.
- I. Gurevitch, R. Buonsanti, A. A. Teran, B. Gludovatz, R. O. Ritchie, J. Cabana and N. P. Balsara, J. Electrochem. Soc., 2013, 160, A1611–A1617 CrossRef CAS.
- N. Angulakshmi, K. S. Nahm, J. R. Nair, C. Gerbaldi, R. Bongiovanni, N. Penazzi and A. M. Stephan, Electrochim. Acta, 2013, 90, 179–185 CrossRef CAS.
- J. Zhang, N. Zhao, M. Zhang, Y. Li, P. K. Chu, X. Guo, Z. Di, X. Wang and H. Li, Nano Energy, 2016, 28, 447–454 CrossRef CAS.
- J. Zhang, X. Zang, H. Wen, T. Dong, J. Chai, Y. Li, B. Chen, J. Zhao, S. Dong, J. Ma, L. Yue, Z. Liu, X. Guo, G. Cui and L. Chen, J. Mater. Chem. A, 2017, 5, 4940–4948 RSC.
- W. Liu, N. Liu, J. Sun, P.-C. Hsu, Y. Li, H.-W. Lee and Y. Cui, Nano Lett., 2015, 15, 2740–2745 CrossRef CAS PubMed.
- W. Liu, S. W. Lee, D. Lin, F. Shi, S. Wang, A. D. Sendek and Y. Cui, Nat. Energy, 2017, 2, 17035 CrossRef CAS.
- Y. Lin, X. Wang, J. Liu and J. D. Miller, Nano Energy, 2017, 31, 478–485 CrossRef CAS.
- S. Gao, J. Zhong, G. Xue and B. Wang, J. Membr. Sci., 2014, 470, 316–322 CrossRef CAS.
- W. Jia, Z. Li, Z. Wu, L. Wang, B. Wu, Y. Wang, Y. Cao and J. Li, Solid State Ionics, 2018, 315, 7–13 CrossRef CAS.
- Y. W. Chen-yang, Y. T. Chen, H. C. Chen, W. T. Lin and C. H. Tsai, Polymer, 2009, 50, 2856–2862 CrossRef CAS.
- P. A. R. D. Jayathilaka, M. A. K. L. Dissanayake, I. Albinsson and B. E. Mellander, Electrochim. Acta, 2002, 47, 3257–3268 CrossRef CAS.
- S. Cheng, D. M. Smith and C. Y. Li, Macromolecules, 2014, 47, 3978–3986 CrossRef CAS.
- S. Cheng, D. M. Smith and C. Y. Li, Macromolecules, 2015, 48, 4503–4510 CrossRef CAS.
- S. Cheng, D. M. Smith, Q. Pan, S. Wang and C. Y. Li, RSC Adv., 2015, 5, 48793–48810 RSC.
- D. M. Smith, B. Dong, R. W. Marron, M. J. Birnkrant, Y. A. Elabd, L. V. Natarajan, V. P. Tondiglia, T. J. Bunning and C. Y. Li, Nano Lett., 2012, 12, 310–314 CrossRef CAS PubMed.
- D. M. Smith, Q. Pan, S. Cheng, W. Wang, T. J. Bunning and C. Y. Li, Adv. Mater. Interfaces, 2017, 1700861 Search PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CAS.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- M.-Q. Zhao, M. Torelli, C. E. Ren, M. Ghidiu, Z. Ling, B. Anasori, M. W. Barsoum and Y. Gogotsi, Nano Energy, 2016, 30, 603–613 CrossRef CAS.
- M.-Q. Zhao, M. Sedran, Z. Ling, M. R. Lukatskaya, O. Mashtalir, M. Ghidiu, B. Dyatkin, D. J. Tallman, T. Djenizian, M. W. Barsoum and Y. Gogotsi, Angew. Chem., Int. Ed., 2015, 54, 4810–4814 CrossRef CAS PubMed.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. Man Hong, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137–1140 CrossRef CAS PubMed.
- Z. Ling, C. E. Ren, M.-Q. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676–16681 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, X. Zhang, Y. Li and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 20352–20363 CrossRef CAS PubMed.
- Z. Huang, S. Wang, S. Kota, Q. Pan, M. W. Barsoum and C. Y. Li, Polymer, 2016, 102, 119–126 CrossRef CAS.
- P. G. Bruce, J. Evans and C. A. Vincent, Solid State Ionics, 1988, 28, 918–922 CrossRef.
- G. B. Appetecchi, G. Dautzenberg and B. Scrosati, J. Electrochem. Soc., 1996, 143, 6–12 CrossRef CAS.
- K. Hayamizu, E. Akiba, T. Bando and Y. Aihara, J. Chem. Phys., 2002, 117, 5929 CrossRef CAS.
- H. Zhai, P. Xu, M. Ning, Q. Cheng, J. Mandal and Y. Yang, Nano Lett., 2017, 17, 3182–3187 CrossRef CAS PubMed.
- W. Huang, Q. Pan, H. Qi, X. Li, Y. Tu and C. Y. Li, Polymer, 2017, 128, 188–199 CrossRef CAS.
- D. M. Smith, S. Cheng, W. D. Wang, T. J. Bunning and C. Y. Li, J. Power Sources, 2014, 271, 597–603 CrossRef CAS.
- S. N. Patel, A. E. Javier, G. M. Stone, S. A. Mullin and N. P. Balsara, ACS Nano, 2012, 6, 1589–1600 CrossRef CAS PubMed.
- Y. Ying, Y. Liu, X. Wang, Y. Mao, W. Cao, P. Hu and X. Peng, ACS Appl. Mater. Interfaces, 2015, 7, 1795–1803 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- Q. Pan, D. Barbash, D. M. Smith, H. Qi, S. E. Gleeson and C. Y. Li, Adv. Energy Mater., 2017, 1701231 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00206a |
| This journal is © The Royal Society of Chemistry 2019 |