
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of naturally occurring serine/cysteine variations on the structure and function of Pseudomonas metallothioneins†
Jelena
Habjanič
 a,
Serge
Chesnov
b,
Oliver
Zerbe
a and
Eva
Freisinger
*a
a,
Serge
Chesnov
b,
Oliver
Zerbe
a and
Eva
Freisinger
*a
aDepartment of Chemistry, University of Zurich, Zurich, Switzerland. E-mail: freisinger@chem.uzh.ch
bUniversity of Zurich/ETH Zurich, Functional Genomics Centre Zurich, Zurich, Switzerland
First published on 15th November 2019
Abstract
Metallothioneins (MTs), small cysteine-rich metal-binding proteins, support the viability of organisms under normal physiological conditions and help them to respond to different environmental stressors. Upon metal coordination (e.g. ZnII, CdII, CuI) they form characteristic polynuclear metal–thiolate clusters that are known for their high thermodynamic stability and kinetic lability. However, despite numerous studies, it is still not understood how MTs modulate their metal-binding properties. Pseudomonas MTs are an emerging subclass of bacterial MTs, distinct for their high number of His residues and for several unique features such as an intrinsically disordered long C-terminal tail and multiple variations in the number and nature of coordinating amino acids. These variations might provide the bacteria with a functional advantage derived from evolutionary adaptation to heterogeneous environments. Nearly 90% of the known Pseudomonas MT sequences feature a central YC![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) xxC motif, that is altered to YC
xxC motif, that is altered to YC![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) xxC in the rest. We demonstrate that the additional Cys residue serves as a coordinating ligand without influencing the metal-binding capacity, the overall metal-binding stability or the structure. However, the additional ligand changes intra-cluster dynamics and, as a consequence, modulates metal transfer reactions that could be functionally advantageous in vivo.
xxC in the rest. We demonstrate that the additional Cys residue serves as a coordinating ligand without influencing the metal-binding capacity, the overall metal-binding stability or the structure. However, the additional ligand changes intra-cluster dynamics and, as a consequence, modulates metal transfer reactions that could be functionally advantageous in vivo.
Significance to metallomicsThe metal clusters of metallothioneins (MTs) are known for their unique properties such as a high thermodynamic stability that is nevertheless combined with a kinetic lability. Both are crucial for their biological functions, among others, in metal ion homeostasis and detoxification. However, insights into the modulation of metal-binding properties by amino acid sequence variations is scarce. In this work we demonstrate how altering the number of potentially coordinating ligands in form of naturally occurring Ser/Cys variations help modulating metal transfer reactions in Pseudomonas MTs. These variations do not influence the overall protein structure but significantly change intra-cluster dynamics. |
Introduction
Metallothioneins (MTs) are unique, low-molecular-weight (6–20 kDa) proteins, characterised by an extraordinary high cysteine content (15–30%). The Cys residues are organized in specific Cys motifs (e.g. CxC, CxxC, CC, CCxCC, CxCC) that allow the formation of diverse metal–thiolate clusters. Depending on the biological nature of the bound transition metal ion, MTs are involved in a wide variety of vital physiological processes, which include metal homeostasis and detoxification, as well as a defence against nitrative and oxidative stress owing to the redox properties of the Cys residues.1–3 The main differences between bacterial and vertebrate MTs are the involvement of His residues in metal ion coordination, the greater sequence diversity now also featuring aromatic residues, and a higher percentage of secondary structural elements.4,5
Pseudomonas metallothionein (PsdMT) sequences were identified only recently5,6 and the encoded proteins form a highly conserved subfamily of bacterial metallothioneins (bacMTs). PsdMTs share a conserved Cys distribution pattern with cyanobacterial MTs, the most intensively investigated bacMTs, but the crucial difference is the replacement of one of the two coordinating His ligands in cyanobacterial MTs by a non-coordinating residue (e.g. Ala, Gly, Ser) in PsdMTs. Furthermore, the His content in PsdMTs is generally unusually high, some sequences contain a long C-terminal Cys-free amino acid stretch with a highly conserved sequence,7 and in almost 90% of the sequences identified so far an additional Cys instead of a Ser residue can be found in one of the central motifs, i.e. YCxxC instead of YCxxC. This additional Cys residue is also present in 30% of cyanobacterial MTs (Fig. 1).
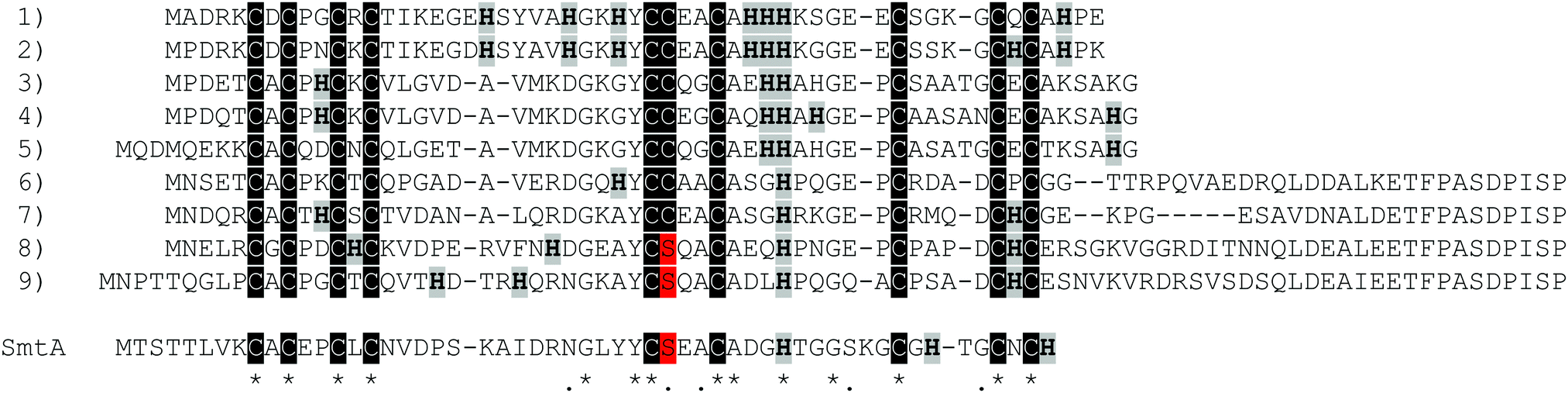 | ||
| Fig. 1 Sequence alignment of different Pseudomonas metallothioneins. (1) P. fluorescens strain Pf0-1 (primary accession number: Q3K9Q2), (2) P. fluorescens R124 (K0X1F5), (3) P. fluorescens SBW25 (C3K8N4), (4) P. fluorescens BRIP34879 (L7HBL7), (5) P. fluorescens BBc6R8 (V7E9L0), (6) P. aeruginosa PAO1 (Q9I1X5), (7) P. putida KT2440 (Q88HU1), (8) P. fluorescens Q2-87 (J2EKT7), (9) P. fluorescens BTR19 (A0A1Q5X1W4), as well as the cyanobacterial SmtA MT from Synechococcus elongatus PCC 7942 (P30331) for comparison. Cys residues are highlighted in black, His residues in grey, Cys/Ser variations in red. | ||
In our previous work, we have structurally and functionally characterized some of these novel features using an MT from Pseudomonas fluorescens Q2-87 (PflQ2 MT, sequence 8 in Fig. 1).7 Our studies revealed that the long C-terminal tail is intrinsically disordered and neither influencing the metal-binding properties nor the structure of the rest of the protein. However, it might potentially serve as a biologically relevant binding site for globular proteins or other interacting molecules. Surprisingly, PsdMTs display differences in their maximum metal-binding capacity towards ZnII and CdII, metal ions that are often considered isostructurally replaceable, as reflected in formation of a Cd4versus a Zn3 cluster. The maximum binding capacity for ZnII was restored to four by introducing a second coordinating His residue as found in cyanobacterial MTs but is accompanied by a decrease in kinetic lability. The investigation of the additional non-coordinating His residues present in the sequence revealed no significant impact on stability, structure or metal-binding properties of the protein, and the biological relevance remains unclear.
In the present study, we address another major feature of PsdMTs, that is a naturally occurring Ser/Cys variation in the central stretch of the Cys distribution pattern (YC/xxC). The change of Ser to Cys implies altering only a single atom, that is oxygen to sulphur. However, for MTs, this change potentially introduces an additional metal binding ligand and therefore might significantly affect the metal-binding properties, and hence function and structure. Upon metal binding, the Cys thiol group is deprotonated, reducing its apparent pKa value from approx. 8.5 in the free amino acid8,9 by 4–5 units, depending on the metal ion. In contrast, the higher electronegativity of the oxygen (χ = 3.44) compared to the sulphur (χ = 2.58)10 prevents deprotonation of the hydroxymethyl group of Ser under physiological conditions. In fact, Ser residues have never been observed to coordinate to metal ions in MTs, despite their high abundance. For example, in vertebrate MTs they are the second most abundant residue after Cys.11,12 Strikingly, Ser residues are often located in the direct vicinity of Cys residues, and mutagenesis studies showed that conserved Ser residues contribute to the strength and overall stability of metal binding.12
The previously studied PflQ2 MT has a Ser residue at the specified position (Ser29, YCxxC). The S29C mutant of the protein will be used here to evaluate if the additional Cys residue is participating in metal ion coordination and whether it changes metal-binding properties, function and structure in comparison to the wild-type protein. In addition, an MT with a naturally occurring Cys residue at this position (Cys28), Pseudomonas putida KT2440 (PpKT MT, sequence 7 in Fig. 1), is studied together with its C28S mutant.
Materials and methods
Chemicals and solutions
The enzymes and purification systems used for plasmid construction were purchased from Fermentas (Le Mont-sur-Lausanne, Switzerland) and Sigma-Aldrich (Buchs, Switzerland). All buffers and chemicals were ACS grade or comparable from Sigma-Aldrich (Buchs, Switzerland), Merck (Darmstadt, Germany), Chemie Brunschwig (Basel, Switzerland), and Roth AG (Arlesheim, Switzerland). Chelex® 100 resin was from Bio-Rad (Reinach, Switzerland). Isotopically labelled compounds (tris(hydroxymethyl)aminomethane, Tris, (D11, 98%), D-glucose (U-13C6, 99%), ammonium chloride (15N, 99%), and cadmium chloride (113Cd, 95.24%)) were purchased from Cambridge Isotope Laboratories (ReseaChem, Burgdorf, Switzerland), while D2O (99.8%) was purchased from Armar Chemicals (Döttingen, Switzerland). Plasmid pRK793 for the production of tobacco etch virus (TEV) protease in Escherichia coli was a gift from David Waugh (Addgene plasmid #8827).13 All solutions were prepared using Millipore water and were degassed under vacuum or saturated with argon. To exclude oxygen, experiments were performed in a Type B Vinyl Anaerobic Chamber (Coy Lab, USA) equipped with a palladium catalyst and operated with a 5% hydrogen/95% nitrogen gas mix wherever possible.Protein expression and purification
The coding sequence of the PpKT MT (residues 1–73) was optimised for the expression in E. coli (Fig. S1, ESI†) and constructed from commercially obtained oligonucleotides (Microsynth AG, Balgach, Switzerland) (Table S1, ESI†). A tobacco etch virus (TEV) protease cleavage site was introduced at the 5′end of the protein-encoding sequence. Using the upstream BamHI and the downstream XmaI restriction sites, the coding sequence was cloned into a pGEX-4T-1 expression vector, which encodes an N-terminal glutathione S-transferase (GST) tag (Fig. S2, ESI†). The shortened version of the protein lacking the C-terminal tail, sh_PpKT MT (residues 1–51) was constructed in the same manner. Both newly constructed plasmids, as well as previously constructed plasmids of the full-length PflQ2 MT and its shortened version sh_PflQ2 MT,7 were used as a DNA template for the site-directed mutagenesis to construct the C28S and S29C mutants, respectively. Oligonucleotides used for the site-directed mutagenesis are listed in Table S1 (ESI†).All proteins were expressed and purified as previously described.7 The identity of proteins was confirmed by electrospray ionization mass spectrometry (ESI-MS) (Fig. S3, ESI†).
Determination of metal ion binding capacity
To determine the maximum metal-binding capacity, an excess of the respective metal ion was added to the apo-protein and the pH increased to 7.4. To remove the metal ion excess, the samples were either purified by size exclusion chromatography (SEC; Superdex Peptide 10/300 GL column; GE Healthcare, Glattbrugg, Switzerland) or by incubation with Chelex resin, both pre-equilibrated with 50 mM Tris HCl, 50 mM NaCl, pH 7.4. Samples of the holo-protein forms obtained in this way were analysed by flame atomic absorption spectroscopy (F-AAS) on an AA240FS spectrometer (Agilent Technologies AG, Basel, Switzerland) to determine the metal ion concentration and by the 2,2′-dithiopyridine (2-PDS) assay to determine the protein concentration via thiol quantification.14 Additionally, each sample was analysed by ESI-MS.CdII titrations of apo-MTs
For each titration, 800 μL of a 10–15 μM protein solution in 10 mM Tris HCl, 50 mM NaCl, pH 7.4 in a septum-sealed spectrophotometric cuvette (1.4 mL, 1 cm optical path length; Hellma AG, Zumikon, Switzerland) was used. Increasing amounts of a CdCl2 solution (1 mM) were added with a microsyringe (Hamilton, Bonaduz, Switzerland). After each addition, a UV spectrum was collected over a spectral range of 210 nm to 350 nm at a scan speed of 480 nm min−1 with 1 nm data intervals. The buffer solution was used as a baseline and automatically subtracted. Measurements were performed on an Agilent Cary 60 UV-Vis spectrometer with Cary WinUV 5.1 software. After the titration experiment, each protein solution was analysed for oxidation by thiolate group quantification with the 2-PDS assay.Determination of average metal-binding stability
The overall apparent stability constants of the CdII forms were determined from competition experiments with the potentially octadentate chelator 1,2-bis-(2-amino-5-fluorophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (5F-BAPTA, Biotium, USA).15 To this end, a 150 μM solution of the respective metallated protein in 50 mM Tris HCl, 300 mM NaCl, pH 7.4, was incubated with 2.5 mM 5F-BAPTA overnight at room temperature. 19F{1H} spectra were acquired using a Bruker AV2-400 spectrometer equipped with a 5 mm QNP probehead at 300 K using a spectral width of 237 ppm, a relaxation delay of 1 s, and 500 scans for each experiment. Calculations and value corrections were performed as described previously.7Mass spectrometry
ESI-MS spectra were acquired using a Synapt G2 quadrupole time-of-flight spectrometer (Waters, UK) with a capillary voltage of 2.8 V, a cone voltage of 40 V, and a source temperature of 80 °C. The apo-forms were measured in MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.15% aqueous formic acid (1:1) and the holo-forms in MeOH:30 mM aqueous ammonium acetate (1:1). The m/z data were deconvoluted using the MaxEnt1 software with a resolution of the output mass of 0.1 Da per channel and a Uniform Gaussian Damage Model at the half-height of 0.1 Da.
:0.15% aqueous formic acid (1:1) and the holo-forms in MeOH:30 mM aqueous ammonium acetate (1:1). The m/z data were deconvoluted using the MaxEnt1 software with a resolution of the output mass of 0.1 Da per channel and a Uniform Gaussian Damage Model at the half-height of 0.1 Da.
NMR studies
To prepare the different metallated species, an exact amount of cadmium ions in the form of a CdCl2 solution was added dropwise to the freshly prepared apo-protein solution and the pH was increased to 7.4 with 1 M Tris-D11 and/or small amounts of NaOH. Samples were lyophilized and resuspended in a buffer containing 50 mM Tris-D11 HCl, 50 mM NaCl, pH 7.4 (90% H2O/10% D2O) just prior to the measurements. The average concentration of 15N (13C,15N) labelled samples and those used for 113Cd measurements was around 0.5 mM and 1–2 mM, respectively. Referencing of 1H chemical shifts was done with the external standard 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS, 0.2% pH 7.5), while 15N and 13C frequencies were referenced indirectly. The 113Cd chemical shifts were referenced to an external 0.1 M Cd(ClO4)2 solution. Experiments were recorded on a Bruker 600 MHz spectrometer equipped with a CRYO TCI triple-resonance probe (1H, 13C, 15N) if not stated differently. The 1D 113Cd and [113Cd,1H]-HSQC-TOCSY spectra were recorded on a Bruker 500 MHz spectrometer equipped with a CRYO 5 mm QNP probe in the temperature range from 275 to 320 K.Assignments and structure calculations were performed in the same way as described previously.7
15N relaxation measurements
The backbone amide dynamics were studied by measuring 15N longitudinal and transverse relaxation rates (R1, R2) as well as 15N{1H}-NOE experiments. Delays were set to 0.01, 0.015, 0.025, 0.05, 0.1, 0.15, 0.2, 0.5, 1.0, 1.5, 2.0, and 2.5 s for the longitudinal (T1) relaxation measurement and to 0, 5, 15, 25, 50, 75, 100, 125, 150, 250, 300, 400, 450, and 500 ms for the transverse (T2) relaxation measurement. The peaks were integrated with SPSCAN, and the data were analysed using in-house written scripts. In order to extract the relaxation rate, the signal intensities were least square fitted to an exponential decay:| I = I0e−t/Ti |
Thermal denaturation
NMR samples of 13C,15N labelled Cd4sh_PflQ2 MT and its S29C mutant were lyophilized and resuspended in 100% D2O. [13C,1H]-HSQC spectra were measured from 290 K to 323 K in 3 K intervals. Spectra were calibrated against an internal standard (tetramethylsilane), analysed using CARA,16 and peak positions adjusted manually at each temperature. 1H and 13C chemical shifts differences against the standard at 290 K were computed for the different temperatures and tabulated in the form of their absolute values. The absolute values of the chemical shift differences were computed by subtracting corresponding chemical shifts: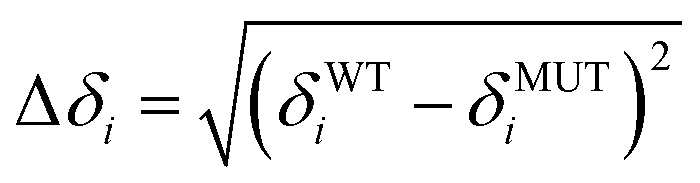
CdII transfer reactions to EDTA
The Cd4 species of sh_PflQ2 MT and sh_PpKT MT as well as of their respective mutants, S29C-sh_PflQ2 MT and C28S-sh_PpKT MT, were prepared by addition of exactly four equivalents of cadmium ions to the respective apo-forms. Subsequently, the pH was increased to 7.4 with small amounts of aqueous ammonia solutions (5–20%). For each transfer reaction, 100 μL of a solution containing 10 μM of the respective Cd4 species and 40 μM ethylenediaminetetraacetic acid (EDTA) were prepared and incubated at room temperature. The stability constant for the [Cd(EDTA)]2− complex is 1016.5 M−1 (25 °C, 0.1 M ionic strength).17At different time intervals, 3 μL aliquots of each solution were desalted using C18 ZipTips (Merck Millipore, Darmstadt, Germany). The samples were eluted with 10 mM ammonium acetate in 30% MeOH and analysed with the Synapt G2 quadrupole time-of-flight spectrometer operating in the positive ion mode with settings as described above. Data were acquired for 2 min in the m/z range between 50 and 5000 Da. Relative quantification of the detected species was performed based on the sum of signal intensities of +3 and +4 ions.
Results and discussion
Similar to PflQ2 MT, the PpKT MT contains a 32-residue long C-terminal tail. For PflQ2 MT, it was shown that this tail is intrinsically disordered and does neither influence the metal-binding properties nor the structure of the rest of the protein.7 The latter was also confirmed for PpKT MT by comparing the [15N,1H]-HSQC spectra of the full-length protein and its shortened version (residues 1–51) (data not shown). Hence, we will focus in this study only on the data obtained with the truncated constructs of the proteins to simplify assignments. Furthermore, while PsdMTs can bind both zinc and cadmium ions, we limit our investigations here to CdII because ZnII ions are spectroscopically silent in most of the experiments presented.Impact of the Ser/Cys variation on the metal-binding properties
To assess the effect of the Ser/Cys variation, first, its influence on the metal ion binding capacity was analysed. Based on F-AAS and the 2-PDS assay all four constructs can bind up to four CdII ions (PflQ2 MT (3.65 ± 0.16), S29C-PflQ2 MT (3.79 ± 0.14), PpKT MT (3.62 ± 0.20), C28S-PpKT MT (3.78 ± 0.16)). This was confirmed by ESI-MS (Fig. S4, ESI†) showing a major signal for the Cd4 species and a minor for the Cd3 species, the latter being an indication for a certain lability of the 4th binding site for all four constructs. The overall apparent metal binding stabilities of the Cd3 and Cd4 species were analysed using 19F NMR spectroscopy and the metal chelator 5,5′-difluoro-BAPTA and show only minor differences between the four constructs, the most apparent being a higher stability of Cd3PpKT MT by 0.75 log units compared to its C28S mutant (Fig. S5, ESI†). Hence, while obviously the Ser/Cys variation does not influence the CdII binding capacity and has a very minor influence on the thermodynamic stability, this does not exclude the possibility that the additional 10th Cys residue might be involved in CdII binding. To answer this question, the four different apo-proteins were titrated with CdII ions, and the absorptivity increase of the thiolate-to-cadmium charge transfer band at 250 nm was monitored by UV spectroscopy (Fig. 2). As a result, the maximum molar absorptivity between wild-type and mutant protein differs by approx. 7000 M−1 cm−1 for both MTs, that is 42700 M−1 cm−1versus 35900 M−1 cm−1 for PpKT MT and its C28S mutant, and 45000 M−1 cm−1versus 38200 M−1 cm−1 for PflQ2 MT and its S29C mutant. Hence the 10th Cys residue clearly increases the maximum molar absorptivity, confirming its involvement in CdII binding.
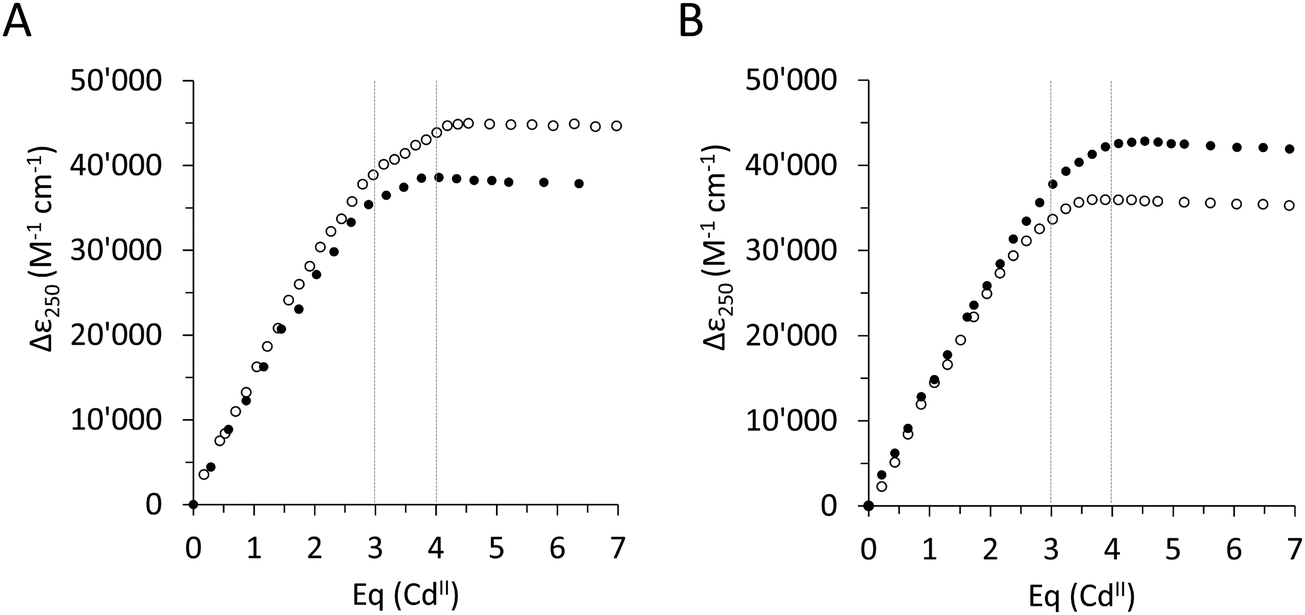 | ||
| Fig. 2 Coordination of cadmium ions. Plot of thiolate-to-cadmium charge transfer band absorptivity at 250 nm for the CdII titrations of (A) apo-PflQ2 MT (black dots) and its S29C mutant (open circles), and (B) apo-PpKT MT (black dots) and its C28S mutant (open circles). | ||
Impact on the structure, backbone dynamics and stability of the protein fold
In the structure of Cd4PflQ2 MT, Ser29 has an essential role as an N-cap for a short α-helix, forming two hydrogen bonds, one from its carbonyl oxygen (O) to the amide proton (HN) of Ala33 and a second one from the side-chain oxygen (Oγ) to the amide proton (HN) of Ala31.7 Since the CdII binding studies showed that upon mutation of Ser29, the additional 10th Cys serves as an additional ligand, the observation of some conformational changes seems likely. However, analysis of the Cα and Cβ chemical shift differences of both Cd3 and Cd4 species of PflQ2 MT and its S29C mutant (Fig. S6 and S7, ESI†) reveals that significant changes only occur in the vicinity to the mutation. Indeed, the NMR solution structure of Cd4S29C-PflQ2 MT is highly similar to the wild-type (Fig. 3) and hence, any significant impact of the Ser-to-Cys mutation on the protein structure can be excluded.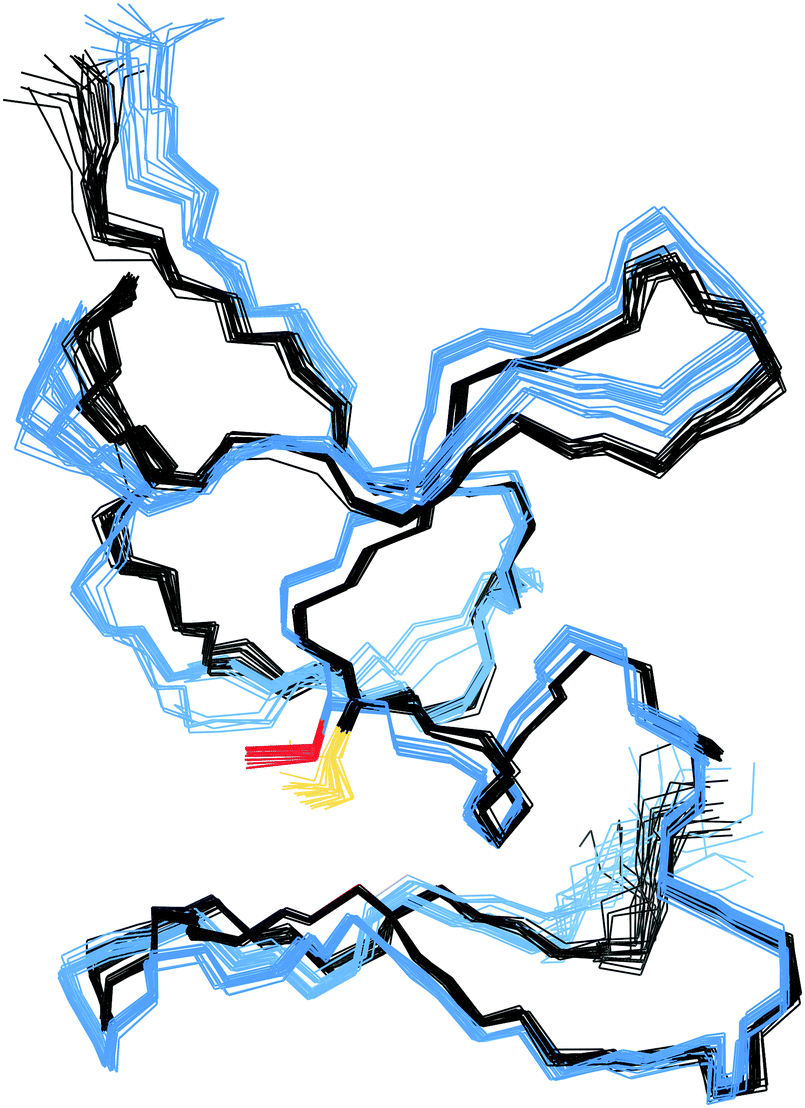 | ||
| Fig. 3 NMR solution structures of Cd4PflQ2 MT and Cd4S29C-PflQ2 MT. The protein backbone of Cd4PflQ2 MT and Cd4S29C-PflQ2 MT is depicted in black and blue, respectively, and the corresponding Ser29 or Cys29 are highlighted in dark yellow or red. | ||
The comparison of backbone amide dynamics between PflQ2 MT and its S29C mutant reveals a moderate perturbation in 15N{1H}-NOE values and longitudinal relaxation rates (R1). The most prominent differences are observed for Cys residues (Fig. S8 and S9, ESI†). More pronounced difference between wild-type and mutant protein are observed for the transverse relaxation rates (R2) (Fig. S10, ESI†). The considerably higher R2 rates for the S29C mutant, again mostly for Cys residues, suggest the presence of exchange processes driven by Cys residues. Therefore, although the two protein folds are highly similar, the Ser-to-Cys mutation triggers a certain degree of change in the dynamics.
The impact on the protein fold stability was investigated with NMR melting studies, monitoring Cα and Cβ chemical shift changes for the Cd4 species of PflQ2 MT and its S29C mutant upon temperature increase from 290 K to 323 K. The results demonstrate an overall destabilisation of the S29C mutant compared to the wild-type (Fig. S11, ESI†). Surprisingly, the most significant differences are not in the direct vicinity of the mutation but predominantly localized in the amino acid stretch from Pro8 to Tyr27 with several other significant changes in polar or charged residues distributed along the entire protein sequence. In contrast to most other proteins, whose structures are to a large extend stabilized by hydrophobic interactions, MTs feature a rather high percentage of polar and charged amino acids that contribute to protein structure and stability via electrostatic interactions. Hence the observed changes in the S29C mutant are well in line with observed reduced protein stability. In order to further investigate if protein sequences are naturally optimised to adapt to either Ser or Cys at this specific position, we also investigated the PpKT MT, that contains a 10th Cys residue and the corresponding C28S mutant by NMR. The [15N,1H]-HSQC spectrum of Cd4PpKT MT shows pronounced line broadening (Fig. 4A), which is significantly smaller in Cd4C28S-PpKT MT (Fig. 4B) and even more in Cd3PpKT MT (Fig. 4C) as both spectra display sharper peaks. It seems thus that the additional Cys residue introduces exchange processes, possibly indicating that interconversion between different metal-coordination modes takes place, which is more evident when the fourth CdII ion is bound.
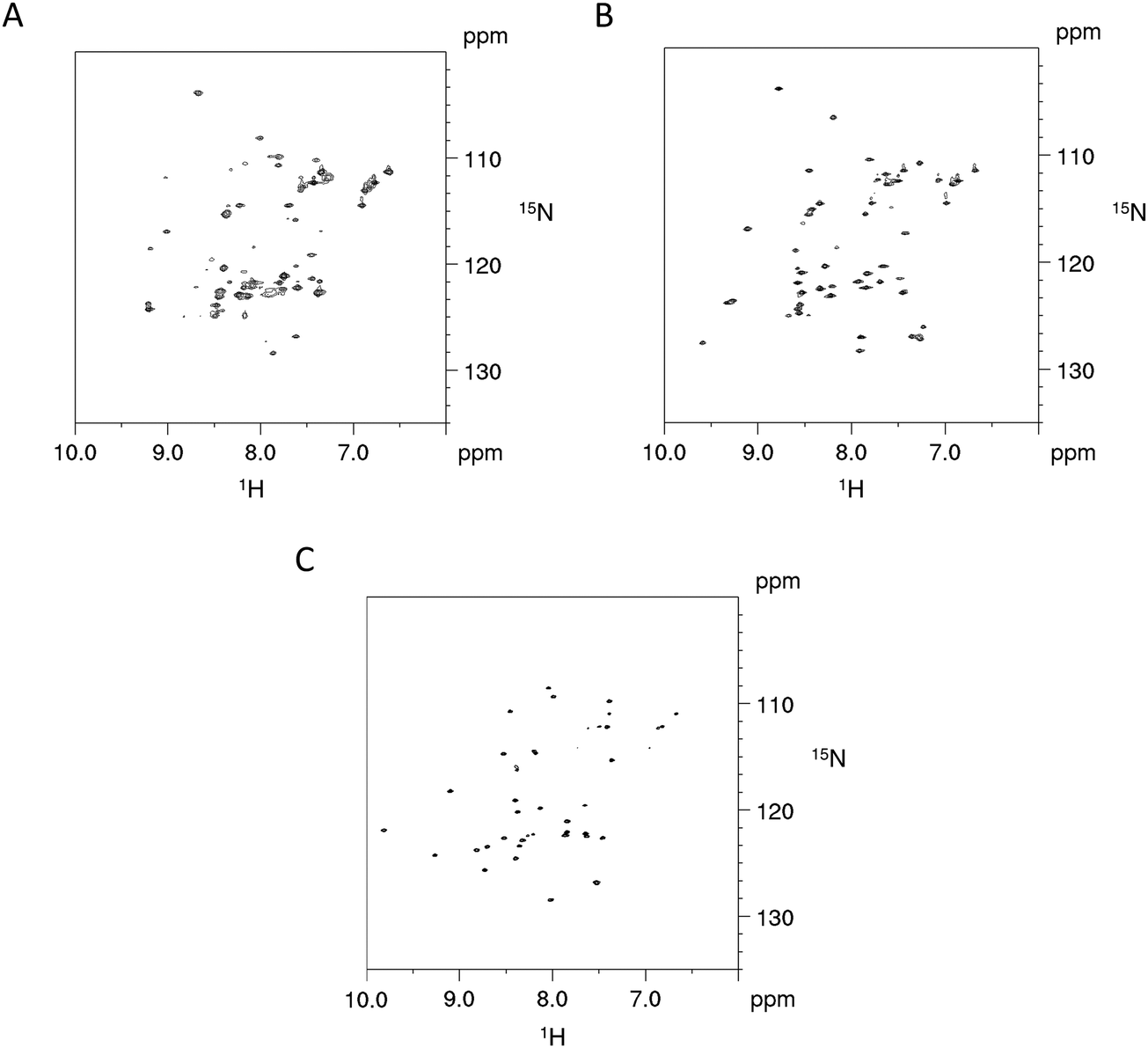 | ||
| Fig. 4 NMR studies of PpKT MT and its C28S mutant. 600 MHz [15N,1H]-HSQC spectra of Cd4PpKT MT (A) and the C28S mutant (B) (T = 300 K) as well as of Cd3PpKT MT (C). | ||
This line broadening prohibits proper determination of the PpKT MT structure. However, assignment of backbone chemical shifts for Cd3PpKT MT reveals the characteristic “zinc-finger fold” pattern seen in PflQ2 MT7 and SmtA18 (low-field shift of Cys27 HN) (9.702 ppm due to hydrogen bonding of Cys5 Sγ; high-field shift of Ala32 Hα (1.886 ppm) due to interaction with the aromatic ring of Tyr26), indicating a highly similar protein fold.
Impact on the metal ion coordination environment and the cluster structure
Considering the contribution of the additional Cys residue in PpKT and S29C-PflQ2 MT to CdII binding, its influence on the metal ion coordination environment and cluster structure was further investigated. The 1D 113Cd spectrum of Cd4PflQ2 MT displays four peaks representing four different CdII coordination sites (Fig. 5A). Three of the peaks are sharp, while the fourth peak at 535 ppm with H36, C7, C32, C49 as coordinating residues is significantly broader and only visible at higher temperature.7 This broadening is attributed to an exchange process on the intermediate time scale for this site compared to the other three sites, which share Cys10 as a common residue.7 Interestingly, the integral of that resonance is also significantly reduced, beyond the error of integration. In studies of human MT-3 (also referred to as “human neuronal growth inhibitory factor” (GIF)), a similar observations was made in that some of the 113Cd resonances were very broad at ambient temperature and became more sharp at higher temperature, while their integral was still much smaller than the integral of the other signals.19 This was interpreted as a coupled slow-configurational exchange process, due to Pro-Xaa cis/trans isomerization, with a fast conformational exchange that likely takes place in those Pro-Xaa isomer subpopulations that cannot coordinate to the metal ions.15 The investigated MTs contain a very large number of Pro residues, and this is the case not only for residues spacing the Cys/His residues that coordinate to the signal at 535 ppm (Cd-D). Moreover, this effect is much less visible in the Cd3 species, indicating a non-obvious contribution to this effect from the additional Cd2+ ion.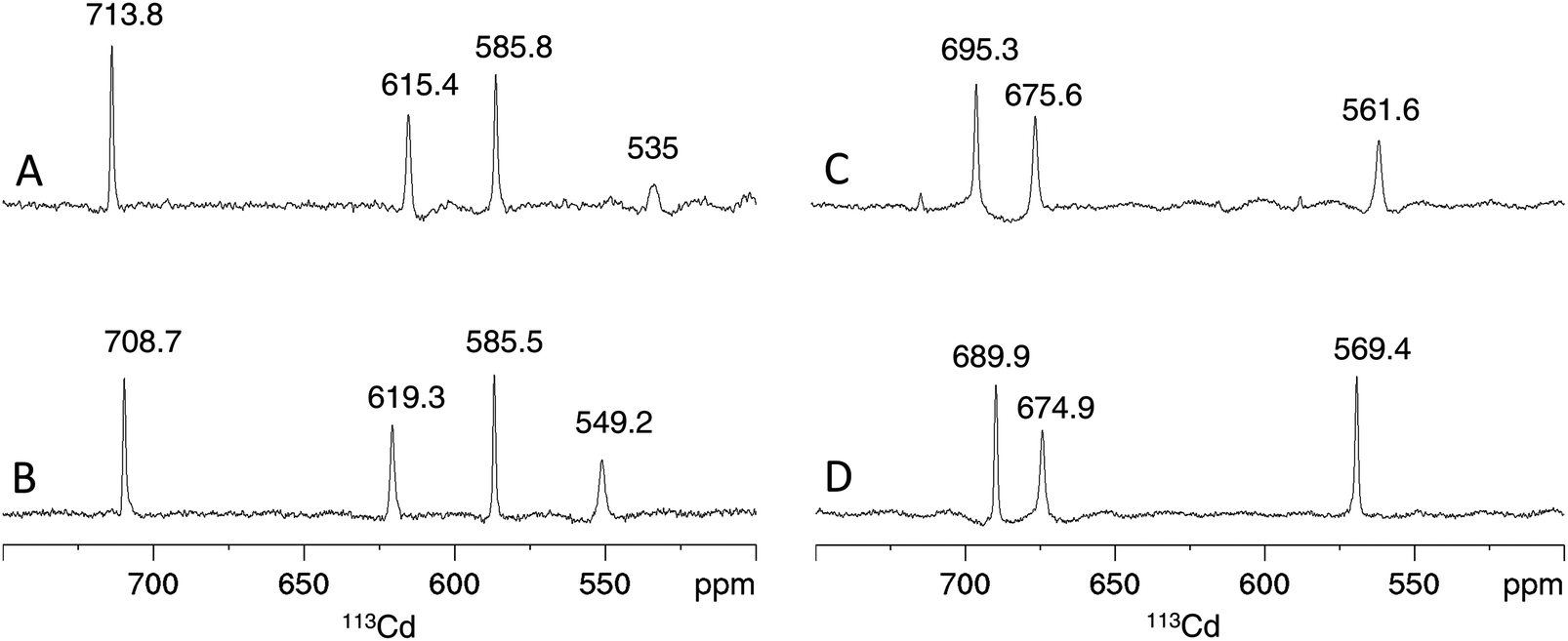 | ||
| Fig. 5 Cadmium coordination environment in PflQ2 MT and the S29C mutant. Proton-decoupled 1D 113Cd spectra at 320 K of the Cd4 (A and B) and Cd3 (C and D) species of PflQ2 MT (A and C) and S29C-PflQ2 MT (B and D). Peak integrals are listed in Table S2 (ESI†). | ||
Also the 1D 113Cd spectrum of the S29C mutant shows four distinct peaks, but here the fourth peak (at 549.2 ppm) is sharper and downfield shifted, while chemical shifts changes of the other three peaks are minor (Fig. 5B). Considering the extraordinary high sensitivity of the 113Cd chemical shift even to small differences in the coordination environment as well as to exchange processes,20 the coordination mode for the three sites (708.7, 619.3, 585.8 ppm) can be assumed to be virtually identical in wild-type and S29C mutant. In contrast, the more significant difference for the fourth site (549.2 ppm) points to a change in the coordination environment and/or an alteration in the timescale of the exchange process observed in the wild-type. For the Cd3 species of both PflQ2 MTs and its S29C mutant (Fig. 5C and D), the 1D 113Cd spectra contain three well-defined peaks corresponding to three distinct cadmium binding sites. As the chemical shifts minimally differ between the two spectra, it is clear that changes in the chemical environment of the CdII ions are minor.
In analogy to the [15N,1H]-HSQC spectrum (Fig. 4A), the 1D 113Cd spectrum of Cd4PpKT MT at 320 K shows various broad, low intensity and ill-defined peaks in the range from 550 ppm to 750 ppm (Fig. 6A). When the temperature is lowered to 280 K many low-intensity peaks, some of which are sharp, occur indicating that the exchange process is now in the slow-exchange NMR regime. In contrast and in analogy to the [15N,1H]-HSQC spectrum (Fig. 4B), the 1D 113Cd spectrum of Cd4C28S-PpKT MT is better defined and shows three peaks of higher intensity (704.9, 624.2, and 606 ppm), one of which is sharp (Fig. 6B). Apparently, there is a change in the timescale of the exchange processes between PpKT MT and its C28S mutant that is reflected in the number and broadness of peaks in the 113Cd spectrum. Again, Cd3PpKT MT is even less dynamic, and its 1D 113Cd spectrum shows three well-defined peaks (683.6, 673.9, and 602.6 ppm) at a lower temperature (280 K; Fig. 6C), similar to the spectra of Cd3PflQ2 MT and its S29C mutant. In contrast, in the spectrum of Cd3C28S-PpKT MT only two peaks (691.8 and 676.5 ppm; Fig. 6D) are observed and only at a higher temperature (320 K). Hence, these results corroborate the view that the Ser/Cys variation influences the exchange processes by altering the timescale of intra-cluster dynamics while retaining the overall chemical environment.
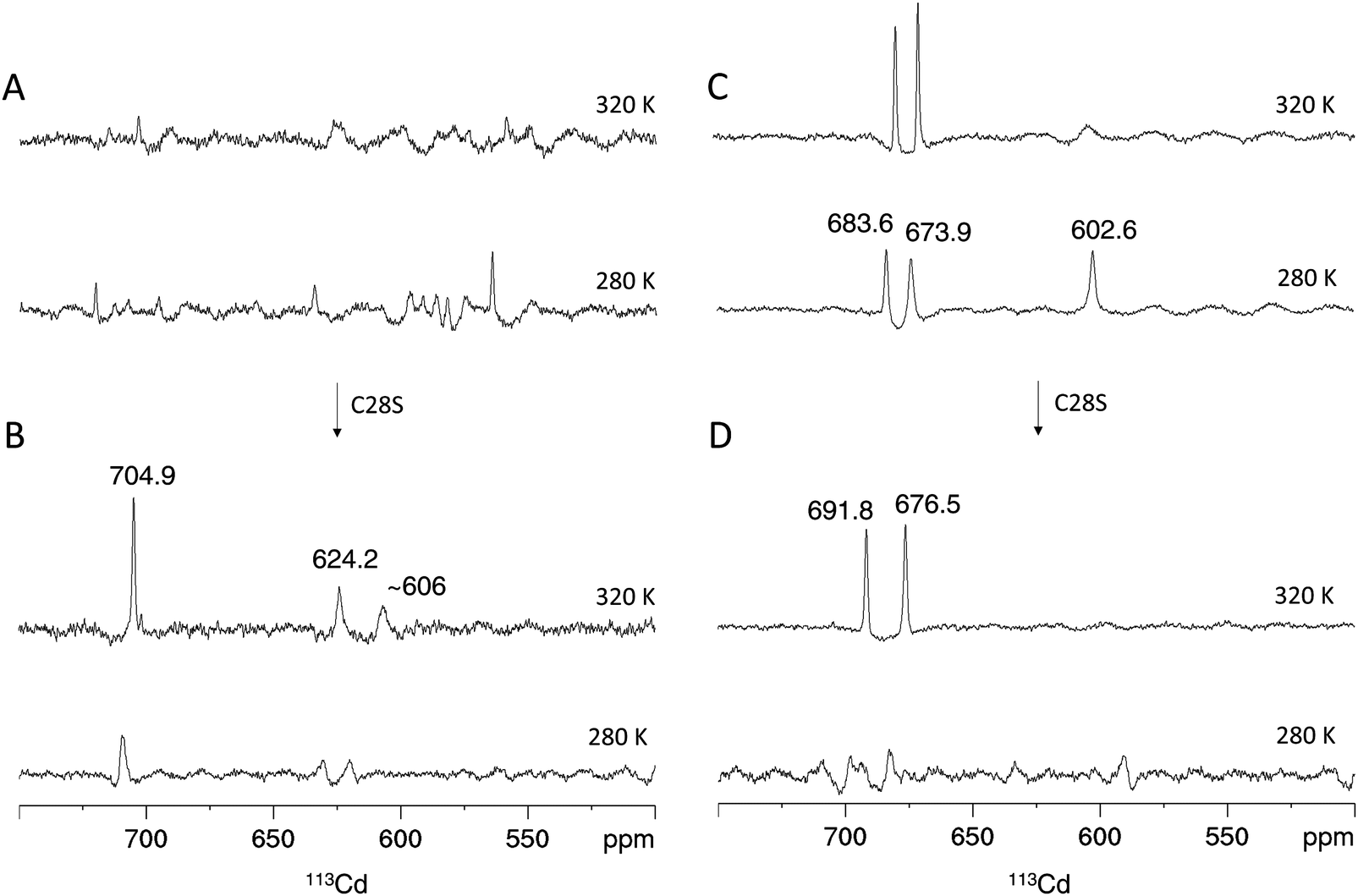 | ||
| Fig. 6 113Cd NMR of PpKT and its C28S mutant. Proton-decoupled 1D 113Cd spectra of the Cd4 species of PpKT MT (A) and the C28S mutant (B). Cd3 species of PpKT MT (C) and its C28S mutant (D). Peak intregrals are listed in Table S2 (ESI†). | ||
Due to the well-resolved NMR spectra, we were able to further address the impact of the additional Cys ligand on the Cd4 cluster structure in S29C-PflQ2 MT. As shown above, NMR studies of Cd4S29C-PflQ2 MT reveal that the Cd4 cluster has four distinct CdII coordination sites reflected by the four defined peaks in the 1D 113Cd spectrum (Fig. 5B). As in the previously published wild-type structure, the chemical shifts of two peaks (708.7 ppm and 619.3 ppm) are in the range of Cys4 polynuclear CdII coordination while the other two peaks (585.5 ppm and 549.2 ppm) are indicative for either the involvement of one His residues in coordination, a deviation from ideal tetrahedral tetrathiolate geometry, or a transient coordination.7,21,22
Like in wild-type Cd4PflQ2 MT, His36 coordinates to one of the CdII ions as detected by the characteristic scalar coupling of 113Cd to Nε2 of His36 (Fig. S12, ESI†) in the long-range [15N,1H]-HSQC spectrum. In analogy to the wild-type protein, the 113Cd peak at 549.2 ppm was assigned to the Cys3His site. This was additionally confirmed during structure calculation since assigning the Cys3His site to peak at 585.5 ppm resulted in significant violations in NMR distances. Therefore, we assume that the signal at 585.5 ppm is upfield-shifted due to a deviation from ideal tetrahedral tetrathiolate coordination geometry accompanied by transient coordination of the fourth ligand as was previously discussed for the wild-type.7 Since a total of 11 ligands (10 Cys and 1 His) in Cd4S29C-PflQ2 MT are involved in CdII coordination, a typical α-type cluster as characteristic for vertebrate and echinoderm MTs23,24 and also found in cyanobacterial MTs (where two terminal Cys residues are replaced by His)24,25 would be possible in principle (Fig. 7A). However, the additional Cys ligand is located at position 29 in the sequence rather than at position 44 as expected from sequence alignments of cyanobacterial MTs. Furthermore, if Cys29 would indeed restore an α-type cluster, the zinc-binding capacity should be increased from three to four as seen for A44H-PflQ2 MT.7 However, neither the zinc-binding capacity nor overall binding stability was affected by introducing the additional Cys residue (Fig. S13, ESI†).
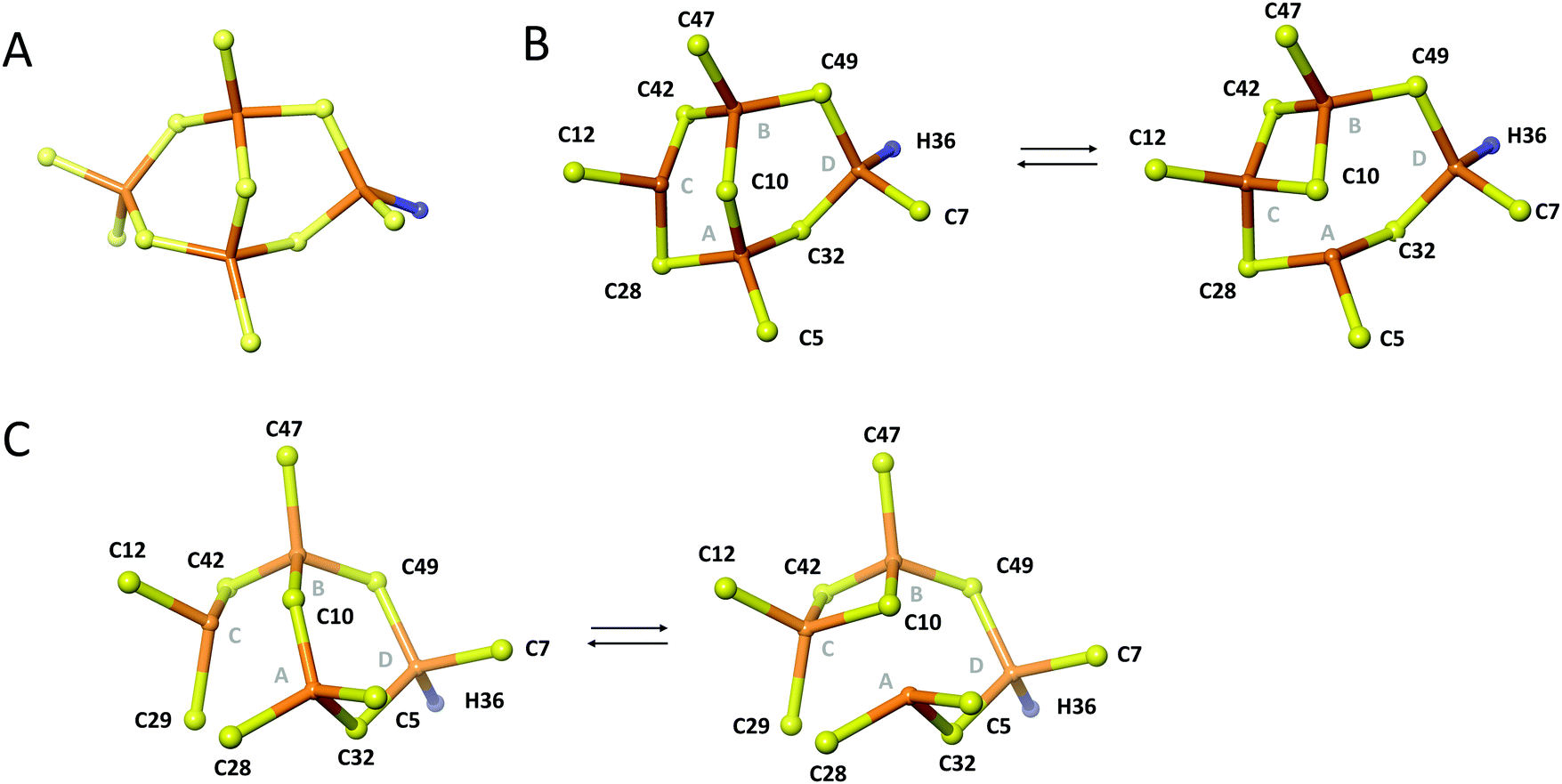 | ||
| Fig. 7 Topology of the metal cluster in Cd4S29C-PflQ2 MT. (A) Theoretical α-type cluster with Cd4Cys10His topology. Rearrangements of the Cd4 cluster of the PflQ2 MT (B) and its S29C mutant (C) showing two bridging modes of residue Cys10. Part B is Reproduced from ref. 7 with permission from The Royal Society of Chemistry. | ||
Due to the low number of experimentally observed metal–ligand connectivities (Fig. S14 and Table S3, ESI†), it is not possible to directly determine the cluster structure. However, Cys-Cβ chemical shifts are highly sensitive indicators to changes in metal binding, and the differences are small between the spectra of Cd4PflQ2 MT and its S29C mutant. Therefore, it can be excluded that any of the Cys residues (except for C28 that is in the direct vicinity of the mutation) changes from bridging to terminal binding mode (or vice versa), and hence the two clusters are most likely highly similar. When these restrictions are added as restraints to the structure calculation in addition to the experimentally observed connectivities, the Cd4 cluster topology as depicted in Fig. 7C is obtained for the S29C mutant while all other topologies cause a larger number of violations of experimental restraints. In the calculated structure, the additional Cys29 acts as a terminal ligand at site C, converting Cys28, that bridges sites A and C in the wild-type structure, into a terminal ligand at site A. Interestingly, Cys10 still serves as a ligand between three metal sites (A, B, C), as observed in the wild-type (Fig. 7B), forming two interchanging arrangements. The resulting cluster structure is also in agreement with the 113Cd NMR data: The peak at 549.2 ppm belongs to cadmium site D that has the same Cys3His coordination as in the wild-type. Since this peak was the most affected one (see above), we attribute the observed decreased linewidth to a change in the rate of exchange from the intermediate to the fast-regime, while the shift to higher ppm values indicates also a change in the relative populations of the interconverting states: Since the shift occurs to higher ppm, this suggests a higher population of regular tetrahedral tetrathiolate coordination. Interestingly, cadmium site C, that is the most affected by the newly coordinating ligand, does experience very little change in the 113Cd chemical shift (585.8 vs. 585.5 ppm), likely because the additional coordination is only transient and because changing coordination from Cys28 to the neighboring Cys29 requires little structural changes.
Overall, the Cd4 cluster structure of the S29C mutant shows that the additional Cys ligand does not drastically change the cluster topology, but increases ligand crowding that promotes the change in coordination and increases dynamics.
The long-range [15N,1H]-HSQC spectrum of Cd3S29C-PflQ2 MT again confirms the involvement of His36 in the coordination (Fig. S15, ESI†), and the 113Cd peak at 569.4 ppm in the 1D 113Cd spectrum is assigned to the Cys3His site (Fig. 5D). The 113Cd chemical shifts of the other two peaks (689.9 ppm, 674.9 ppm) are again indicative for polynuclear CdCys4 coordination sites. While for Cd3PflQ2 MT His36, Cys7, Cys32, and Cys49 complete the CdCys3His site,7 the [113Cd,1H]-HSQC-TOCSY spectrum of Cd3S29C-PflQ2 MT shows five CdII-ligand correlations, that is to His36, Cys7, Cys29, Cys32, and Cys49 (Fig. S16, ESI†). This observation is, at least at first glance, surprising: We observed in the CdII titration experiments (Fig. 2) that the molar absorptivity for S29C-PflQ2 MT further increases after the addition of three equivalents of CdII, and much more than observed for PflQ2 MT. For PflQ2 MT, data showed that both in the Cd3 and in the Cd4 cluster all ligands (one His and nine Cys) coordinate. The slight increase in absorptivity upon addition of the fourth CdII was attributed to the transition of some of the Cys ligands from terminal to bridging coordination mode. However, the much more significant increase for S29C-PflQ2 MT suggests that the coordination of the fourth CdII involves an additional Cys residue, and obviously Cys29 would be the likely candidate for this. An argument against this assumption is that all Cys-Cβ resonances in the [13C,1H]-HSQC spectrum of Cd3S29C-PflQ2 MT, including the one of Cys29, are in the chemical shift range (29–33 ppm) typical for deprotonated thiol groups, and deprotonation of Cys residues at or near physiological pH should only occur upon metal coordination (Fig. S17, ESI†). Since a pentacoordinated CdII ion is highly unlikely and in addition not in-line with the observed chemical shift in the 1D 113Cd spectrum, the five correlations observed in the [113Cd,1H]-HSQC-TOCSY spectrum most likely represent an equilibrium between two interchanging CdCys3His coordination modes. Hence, while Cys29 is not significantly changing the coordination environment in the Cd3 species, it promotes faster ligand exchange.
Possible functional implications – metal transfer reactions
In order to test the potential functional impact of altered intra-cluster dynamics, the CdII ion transfer from the four different MTs to EDTA was probed using ESI-MS. The transfer reaction is completed within 1–10 min for all MTs as no more changes in speciation are observed (Fig. 8 and Fig. S18–S20, ESI†). Generally, there is apparently a very fast transfer of the first CdII equivalent taking place during the time period needed to collect the first ESI-MS spectra after EDTA addition (time point “1 min” in Fig. S20, ESI†). The predominant difference between the behaviour of PpKT and PflQ2 MTs is that for the entire duration of the experiment, no completely demetallated forms are observed for the two PpKT constructs, while for PflQ2 as well as S29C_PflQ2 MT already after 1 min small amounts of the respective apo-species can be detected. Comparing the amount of detected Cdx species for wild-type and mutant constructs of PflQ2 MT after equilibrium is reached, rather small but peculiar differences are observed (Fig. 8). While generally ESI-MS is considered to be a qualitative method, the similarity of wild-type and respective mutant protein, as well as the identical sample and measurement conditions, allow for some more quantitative interpretations: Mutating Ser to Cys increases the amount of the Cd3 species, while the fraction of the apo and Cd1 species is significantly reduced. Hence, while the fourth CdII is clearly less tightly bound in the S29C mutant (see also Fig. S4, ESI†), the other binding sites are apparently stabilized in the form with the additional Cys residue. The difference between PpKT MT and its C28S mutant is also striking: Again, the amounts of Cd3 species detected is higher in the form that contains 10 Cys residues, that is wild-type PpKT MT, while for the C28S mutant the Cd2 species dominates. Hence, while the additional Cys residue seems to cause weaker binding of the fourth CdII in Cd4S29C-PflQ2 MT, it stabilizes the Cd3 species considerably both for S29C-PflQ2 and PpKT MT.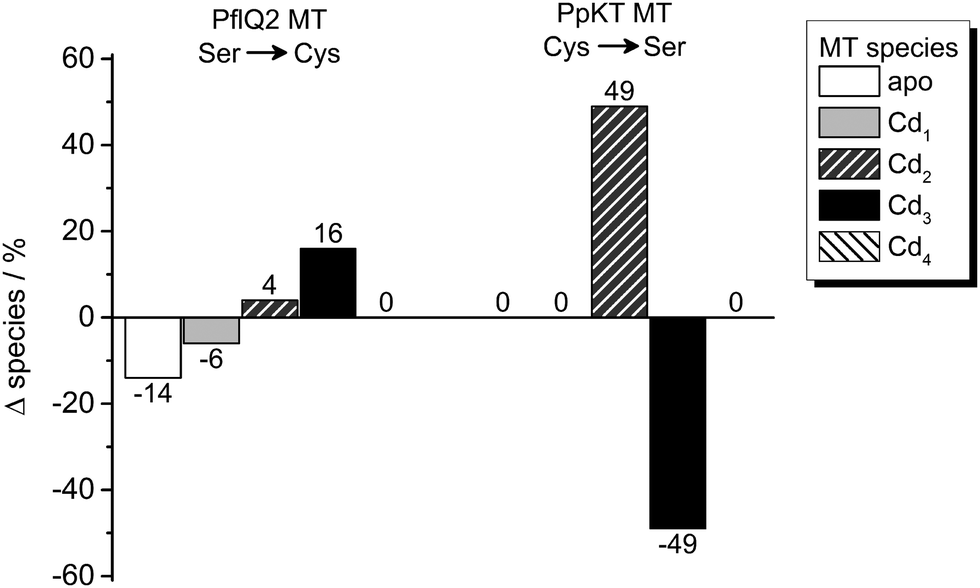 | ||
| Fig. 8 Cadmium transfer from CdIIMTs to EDTA. Percentage difference of species observed by ESI-MS (Fig. S20, ESI†) for Ser or Cys containing PflQ2 as well as Cys or Ser containing PpKT after incubation with four equiv. of EDTA relative to CdII ions for 60 min. The CdIIMT forms used were prepared by addition of precisely four equiv. of CdII ions to the apo proteins without further purification. | ||
The fact that our structural studies show the same protein fold for the Cd3 and Cd4 species of PflQ2 MT as well as its S29C mutant argues against a structural role of the additional Cys residue and implies a functional relevance instead. As shown before, the additional Cys residue increases cluster dynamics by providing an additional ligand for metal ion coordination. Hence, it might modulate metal transfer reactions by transiently coordinating metal ions and enabling metal ion transfer without protein unfolding.
Conclusions
MT sequences from different Pseudomonas species exhibit features previously not observed for any other MT. These unique properties have the potential of modulating metal-binding properties and functions of MTs in vivo and presumably reflect the environmental adaptation of Pseudomonas species. One of those characteristics is the presence of a Cys residue in the YC/xxC motif in almost 90% of PsdMT sequences identified so far. A comparative study of four different constructs was performed including sequences with a naturally occurring Ser (PflQ2 MT) or Cys (PpKT MT) residue at the indicated position as well as the corresponding S29C and C28S mutants. Results show that the additional 10th Cys residue is involved in the binding of CdII ions but is influencing neither the metal-binding capacity nor the overall metal-binding strength. Interestingly, the three-dimensional structure of Cd4S29C-PflQ2 MT reveals no significant structural influence evoked by the additional Cys residue. Furthermore, although binding of an additional Cys residue now provides enough ligands (11) to build a typical MT α-cluster, the additional ligand instead participates in transient metal-binding resulting in an even more dynamic cluster that interchanges between different coordination modes.
These alterations may be correlated with the type and amount of sub-metallated species formed during the metal transfer reaction to EDTA. For MT forms that feature such an additional Cys residue, the predominant formation of the Cd3 species is observed, and the amount of sub-metallated species is significantly reduced. Therefore, we hypothesise that the Ser/Cys variation modulates important metal transfer reactions that might be favourable for the overall function of PsdMTs in vivo.
Abbreviations
| MT | Metallothionein |
| bacMTs | Bacterial metallothioneins |
| PsdMTs | Metallothioneins from Pseudomonas species |
| PflQ2 MT | Metallothionein from Pseudomonas fluorescens Q2-87 |
| sh_PflQ2 MT | C-terminal truncated form of metallothionein from Pseudomonas fluorescens Q2-87 |
| PpKT MT | Metallothionein from Pseudomonas putida KT2440 |
| sh_PpKT MT | C-terminal truncated version of metallothionein from Pseudomonas putida KT2440 |
| SEC | Size-exclusion chromatography |
| ESI-MS | Electrospray ionization mass spectrometry |
| F-AAS | Flame atomic absorption spectroscopy |
| Tris | Tris(hydroxymethyl)aminomethane |
| TCEP | Tris(2-carboxyethyl)phosphine |
| 5F-BAPTA | 1,2-Bis-(2-amino-5-fluorophenoxy)ethane-N,N,N′,N′-tetra-acetic acid |
| 2-PDS | 2,2′-Dithiopyridine |
| GST | Glutathione S-transferase |
| TEV | Tobacco etch virus |
| EDTA | Ethylenediaminetetraacetic acid. |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Simon Jurt (Department of Chemistry, UZH) for help with analysis of 15N relaxation data and express overflowing gratefulness to Prof. em. Milan Vašák († 2019) for continuous discussions. The work was financially supported by the Forschungskredit of the University of Zurich, grant no. [FK-15-085] and [FK-17-091] to JH.References
- C. A. Blindauer, in Binding, Transport and Storage of Metal Ions in Biological Cells, ed. W. Maret and A. G. Wedd, The Royal Society of Chemistry, 2014, pp. 606–665 Search PubMed.
- P. Coyle, J. C. Philcox, L. C. Carey and A. M. Rofe, Metallothionein: the multipurpose protein, Cell. Mol. Life Sci., 2002, 59, 627–647 CrossRef CAS PubMed.
- B. Ruttkay-Nedecky, L. Nejdl, J. Gumulec, O. Zitka, M. Masarik, T. Eckschlager, M. Stiborova, V. Adam and R. Kizek, The role of metallothionein in oxidative stress, Int. J. Mol. Sci., 2013, 14, 6044–6066 CrossRef CAS PubMed.
- C. A. Blindauer, Bacterial metallothioneins, Met. Ions Life Sci., 2009, 5, 51–81 CAS.
- C. A. Blindauer, Bacterial metallothioneins: past, present, and questions for the future, J. Biol. Inorg. Chem., 2011, 16, 1011–1024 CrossRef CAS PubMed.
- G. L. Winsor, E. J. Griffiths, R. Lo, B. K. Dhillon, J. A. Shay and F. S. L. Brinkman, Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database, Nucleic Acids Res., 2016, 44, D646–D653 CrossRef CAS PubMed.
- J. Habjanič, O. Zerbe and E. Freisinger, A histidine-rich Pseudomonas metallothionein with a disordered tail displays higher binding capacity for cadmium than zinc, Metallomics, 2018, 10, 1415–1429 RSC.
- J. Kyte, Structure in Protein Chemistry, Garland Science, New York, 2nd edn, 2007 Search PubMed.
- G. Roos, N. Foloppe and J. Messens, Understanding the pKa of redox cysteines: The key role of hydrogen bonding, Antioxid. Redox Signaling, 2013, 18, 94–127 CrossRef CAS PubMed.
- L. C. Allen, Electronegativity is the average one-electron energy of the valence-shell electrons in ground-state free atoms, J. Am. Chem. Soc., 1989, 111, 9003–9014 CrossRef CAS.
- J. H. R. Kägi, Y. Kojima, M. M. Kissling and K. Lerch, Metallothioneins: An exceptional metal thiolate protein, Ciba Found. Symp., 1979, 72, 223–237 Search PubMed.
- T. Emoto, M. Kurasaki, S. Oikawa, M. Suzuki-Kurasaki, M. Okabe, F. Yamasaki and Y. Kojima, Roles of the conserved serines of metallothionein in cadmium binding, Biochem. Genet., 1996, 34, 239–251 CrossRef CAS PubMed.
- R. B. Kapust, J. Tözser, J. D. Fox, D. E. Anderson, S. Cherry, T. D. Copeland and D. S. Waugh, Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency, Protein Eng., 2001, 14, 993–1000 CrossRef CAS.
- A. O. Pedersen and J. Jacobsen, Reactivity of the thiol-group in human and bovine albumin at pH 3-9, as measured by exchange with 2,2′-dithiodipyridine, Eur. J. Biochem., 1980, 106, 291–295 CrossRef CAS.
- D. W. Hasler, L. T. Jensen, O. Zerbe, D. R. Winge and M. Vašák, Effect of the two conserved prolines of human growth inhibitory factor (metallothionein-3) on its biological activity and structure fluctuation: comparison with a mutant protein, Biochemistry, 2000, 39, 14567–14575 CrossRef CAS PubMed.
- R. L. J. Keller, Computer aided resonance assignment tutorial, Cantina Verlag, Goldau, 2004 Search PubMed.
- D. C. Harris, Quantitative chemical analysis, W. H. Freeman and Co, New York, 8th edn, 2010 Search PubMed.
- C. A. Blindauer, M. D. Harrison, A. K. Robinson, J. A. Parkinson, P. W. Bowness, P. J. Sadler and N. J. Robinson, Multiple bacteria encode metallothioneins and SmtA-like zinc fingers, Mol. Microbiol., 2002, 45, 1421–1432 CrossRef CAS PubMed.
- P. Faller, D. W. Hasler, O. Zerbe, S. Klauser, D. R. Winge and M. Vašák, Evidence for a dynamic structure of human neuronal growth inhibitory factor and for major rearrangements of its metal–thiolate clusters, Biochemistry, 1999, 38, 10158–10167 CrossRef CAS.
- M. Vašák, Application of 113Cd NMR to metallothioneins, Biodegradation, 1998, 9, 501–512 CrossRef.
- L. Hemmingsen, L. Olsen, J. Antony and S. P. A. Sauer, First principle calculations of 113Cd chemical shifts for proteins and model systems, J. Biol. Inorg. Chem., 2004, 9, 591–599 CrossRef CAS.
- G. L. Öz, D. L. Pountney and I. M. Armitage, NMR spectroscopic studies of I = 1/2 metal ions in biological systems, Biochem. Cell Biol., 1998, 76, 223–234 CrossRef.
- J. Hidalgo, R. S. Chung, M. Penkowa and M. Vašák, Structure and function of vertebrate metallothioneins, Met. Ions Life Sci., 2009, 5, 279–317 CAS.
- L. Vergani, Metallothioneins in aquatic organisms: Fish, crustaceans, molluscs, and echinoderms, Met. Ions Life Sci., 2009, 5, 199–237 CAS.
- C. A. Blindauer, M. D. Harrison, J. A. Parkinson, A. K. Robinson, J. S. Cavet, N. J. Robinson and P. J. Sadler, A metallothionein containing a zinc finger within a four-metal cluster protects a bacterium from zinc toxicity, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9593–9598 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mt00213h |
| This journal is © The Royal Society of Chemistry 2020 |