
Cu(II) phenanthroline–phenazine complexes dysregulate mitochondrial function and stimulate apoptosis†
Garret
Rochford
 a,
Zara
Molphy
b,
Kevin
Kavanagh
c,
Malachy
McCann
d,
Michael
Devereux
a,
Andrew
Kellett
b and
Orla
Howe
*a
a,
Zara
Molphy
b,
Kevin
Kavanagh
c,
Malachy
McCann
d,
Michael
Devereux
a,
Andrew
Kellett
b and
Orla
Howe
*a
aFOCAS Research Institute and School of Biological & Health Sciences, Technological University Dublin, City Campus, Dublin 8, Ireland. E-mail: orla.howe@TUDublin.ie
bSchool of Chemical Science and The National Institute for Cellular Biotechnology, Dublin City University, Dublin 9, Ireland
cDepartment of Biology, Maynooth University, Maynooth, Kildare, Ireland
dDepartment of Chemistry, Maynooth University, Maynooth, Kildare, Ireland
First published on 18th October 2019
Abstract
Herein we report an in-depth study on the cytotoxic mechanism of action of four developmental cytotoxic copper(II) complexes: [Cu(phen)2]2+ (Cu-Phen); [Cu(DPQ)(Phen)]2+ (Cu-DPQ-Phen); [Cu(DPPZ)(Phen)]2+; and [Cu(DPPN)(Phen)]2+ (where Phen = 1,10-phenanthroline, DPQ = dipyrido[3,2-f:2′,3′-h]quinoxaline, DPPZ = dipyrido[3,2-a:2′,3′-c]phenazine, and DPPN = benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine). This complex class is known for its DNA intercalative properties and recent evidence—derived from an in vivo proteomic study—supports the potential targeting of mitochondrial function. Therefore, we focused on mitochondrial-mediated apoptosis related to cytotoxic activity and the potential impact these agents have on mitochondrial function. The Cu(II) complexes demonstrated superior activity regardless of aromatic extension within the phenazine ligand to the previously demonstrated activity of cisplatin. Unique toxicity mechanisms were also identified in prior demonstrated cisplatin sensitive and resistant cell lines. Double strand breaks in genomic DNA, quantified by γH2AX foci formation, were then coupled with apoptotic gene expression to elucidate the mechanisms of cell death. These results indicate that while DNA damage-induced apoptosis by BAX, XIAP and caspase-9 and -3 expression is moderate for the Cu(II) complexes when compared to cisplatin, protein targets independent of DNA exert a multimodal mechanistic effect. Significantly, mitochondrial gene expression of oxidative stress, protease expression, and fission/fusion processes—upregulated HMOX, DRP1 and LON, respectively—indicated an increased oxidative damage associated with compromised mitochondrial health upon exposure to these agents. These data support a unique mode of action by these complexes and provide valuable evidence of the developmental potential of these therapeutic inorganic complexes.
Significance to metallomicsThe attachment of copper(II) phenanthroline to three different extended phenazine π-backbones gives novel cationic complexes multimodal biological effects. The DNA-binding and cytotoxic properties of these complexes were probed further to show that the mitochondria are a key player in their biological effects. Our work presents the importance of mitochondrial function and apoptotic cell death as additional mechanisms of action in their multi-modal nature. |
Introduction
Copper(II) complexes are part of a large array of therapeutically active inorganic-based small molecules.1–3 The multimodal cellular activity of Cu(II) complexes reported in the literature includes: association with production of reactive oxygen species,4 and knock-on antioxidant effects, lipid peroxidation,5,6 DNA cleavage,7–9 mitochondrial dysfunction and apoptosis induction.10,11 Cu(II) complexes with different ancillary ligands have shown strong cytotoxicity profiles against a wide spectrum of cancer cell lines.12–16 The Casiopeinas® are a family of Cu(II) ternary complexes that are capable of activation of apoptosis and genotoxicity in a range of medulloblastoma, glioma and colorectal adenocarcinoma cells.17–20 The Cu(II) complexes in this study were synthesised and characterised by Molphy and colleagues.21,22 The Cu(II) complexes presented in this study are Cu(II) phenanthroline (Cu-Phen) (1) (Fig. 1) along with their three phenazine π-backbone extended cationic complexes Cu(II) phenanthroline–phenazine [Cu(DPQ)(Phen)]2+ (where DPQ = dipyrido[3,2-f:2′,3′-h]quinoxaline) (Cu-DPQ-Phen) (2) (Fig. 1), Cu(II) phenanthroline-phenazine [Cu(DPPZ)(Phen)]2+ (where DPPZ = dipyrido[3,2-a:2′,3′-c]phenazine) (Cu-DPPZ-Phen) (3) (Fig. 1) and Cu(II) phenanthroline-phenazine [Cu(DPPN)(Phen)]2+ (where DPPN = benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine) (Cu-DPPN-Phen) (4) (Fig. 1). These Cu(II) complexes were previously biophysically characterised in addition to a preliminary biological screening using the cisplatin resistant SKOV-3 cell line. Both of these studies determined that the complexes induced metal-hydroxo and free hydroxyl species and result in 8-oxo-2′-deoxyguanosine (8-oxo-dG) lesion formation.22 Additionally, the DNA binding constants of the complexes were the highest observed in phenanthrene based complexes and were comparable to the transcription inhibitor actinomycin D. The preliminary biological screening demonstrated cytotoxicity in cisplatin resistant SKOV-3 cells equivalent to adriamycin.21 A more recent study with these complexes in the in vivo model Galleria mellonella details a series of proteomic changes which strongly suggest changes in antioxidant and mitochondrial related responses.23 The in vivo study also determined that the increased DNA binding previously established by DNA binding studies was accompanied by increases in enzymes associated with P450 detoxification, glycolysis, purine metabolism and nitrogen detoxification. Interestingly, the in vivo model demonstrated that superior nuclease action was associated with increased toxicity in the model, while greater DNA binding increased the complexity of the multimodal response mechanism.23 The proteomic study evidenced substantial mitochondrial-related protein involvement in response to the Cu(II) complexes which strongly suggested that mitochondrial-mediated apoptosis was one of the key mechanisms of action. These Cu(II) phenanthroline–phenazine complexes have previously demonstrated strong DNA binding activity, therapeutic activity against cisplatin resistant SKOV-3 cells and compelling evidence of antioxidant and mitochondrial-related dysfunction in G. mellonella. The comparison of these complexes to the activity of the highly successful clinical drug cisplatin is an important comparator for evaluating the therapeutic activity of these complexes.24 The activity of these complexes was compared to cisplatin in MCF-7 and SKOV-3 cells lines.13,14,21 Both MCF-7 and SKOV-3 cell lines have been used in previous studies and have demonstrated different values to those presented in this report. A strong motivation in presenting the isogenic A2780 cell lines along with its therapeutically resistant daughter cell is to address the variability inherent in the use of MCF-7 and SKOV-3 cell lines. | ||
| Fig. 1 (A) Molecular structures of the Cu(II) phenanthroline–phenazine complexes used in this study. (B) Molecular structure of the clinical anticancer drug cisplatin. | ||
Based on previous studies and following from the in vivo investigation of the activity of these Cu(II) complexes, apoptosis and the mitochondria are a central focus of the mechanistic investigation of these complexes in the in vitro mammalian model. Previous studies have investigated the presence of apoptosis as a mechanism of regulated cell death following exposure to a range of Cu(II) complexes.11,25,26 In this study we investigated using gene expression to examine a number of processes in relation to the apoptotic cascade and additionally to genes related to different functions in the mitochondria which are key interrelated processes in apoptosis. Many proteins are involved in the process of apoptosis (Table 1). The B cell lymphoma-2 family (BCL-2) function in both pro- and anti-apoptotic manners. Pro-apoptotic proteins, such as BCL-2 associated X protein (BAX), are activated when mitochondria-mediated apoptosis is signalled, upon which they form monomers of pores that localize to the mitochondrial membrane so that cytochrome c and inhibitors of X-linked Inhibitor of Apoptosis Protein (XIAP), such as second mitochondria-derived activator of caspases (Smac) or serine protease HTRA2/Omi, can potentiate or inhibit the process.27 Anti-apoptotic proteins such as BCL-2 prevent this process occurring by interacting with a BH3 regulatory repressor protein such as Bad. Deficiencies in BCL-2 associated X protein (BAX)/BCL-2 homologous antagonist killer (BAK) have been shown to confer resistance in in vitro models, and over expression in BCL-2 and B-cell Lymphoma extra-large (BCL-XL) is known to increase resistance in vitro.28–31 Caspases are a series of serine proteases with a pivotal role in the initiation (caspase-9) and execution (caspase-3) of apoptosis. The caspase cascade can be initiated in response to DNA damage and mitochondrial dysfunction in addition to other cellular stimuli.32
| Gene | Function | Cellular location |
|---|---|---|
| Caspase-9 | Apoptosis initiator | Cytoplasm |
| Caspase-3 | Apoptosis executioner | |
| BCL-2 | Pro- and anti-apoptotic function | |
| BAX | Pro apoptotic function, associated intrinsic apoptosis | |
| XIAP | Inhibits the activity of caspases | |
| HMOX1 | Reduces oxidative stress through catabolization of haem | Mitochondria |
| TFAM | Mitochondrial related transcription factor, associated with action of TFB1M and TFB2M | |
| TFB1M | Mitochondrial related transcription factors, associated with Nrf-2 action | |
| TFB2M | Mitochondrial related transcription factors, associated with Nrf-2 action | |
| NRF-2 | Transcription element of the ARE complex, activated through oxidative stress | |
| CLPP | Mitochondrial protease, associated with mitochondrial quality control | |
| LON | ||
| SPG7 | ||
| YME1L1 | ||
| DRP1 | Mitochondrial fission regulator | |
| MFN1 | Responsible for mitochondrial fusion and cristae remodelling | |
| MFN2 | ||
| OPA1 | ||
| Actin | Assembly of microfilaments | Cytoplasm (reference genes) |
| β-Tubulin | Polymerization of microtubules | |
Multiple mitochondrial-related genes and associated signalling events that are reported to characterise mitochondrial function were also selected for this study (Table 1). Heme oxygenase (HMOX-1) is a member of the phase II antioxidant enzymes and is primarily involved in the removal of iron based porphyrin, heme from the non-protein moiety of haemoglobin. Nuclear factor (erythroid-derived 2)-like 2 (Nrf-2) is closely involved with the induction of HMOX-1 through the Antioxidant Response Element (ARE) transcription region.33Nrf-2 also forms part of the transcriptional regulation of Transcription factor B1/2, mitochondrial (TFB1M and TFB2M) which in turn regulates the transcription of the general mitochondrial transcription factor, Transcription Factor A (TFAM).34–36 Genes including ATP-dependent Clp protease proteolytic subunit (CLPP), Lon protease homolog 1 (LON1), paraplegin matrix AAA peptidase subunit (SPG7) and ATP-dependent metalloprotease (YME1L1) are involved in the regulation of mitochondrial quality control. LON and CLPP are present in the mitochondrial matrix and are involved in the proteolysis of oxidised proteins from ROS damage and impaired, misfolded protein due to damage respectively.37–39 Dynamin-Related Protein (DRP1), Mitofusin-1/-2 (MFN1), (MFN2) and Dynamin-like 120 kDa protein (OPA1) are involved in the control of mitochondrial fission and fusion. The process of mitochondrial fission is largely controlled by DRP1, which is translocated to the outer membrane of the mitochondria to bring about fission.40 The process of mitochondrial fusion involves OPA1 in concert with MFN1 and MFN2 which regulate mitochondrial fusion from the inner mitochondrial membrane and are essential for cristae remodelling, which also has a regulatory function in oxidative phosphorylation.40,41
This study explores the mitochondrial and apoptotic mechanisms of action of these complexes in in vitro cell models.
Materials and methods
Mammalian cell culture conditions and maintenance
The MCF-7, SKOV-3, A2780 and A2780cis cell lines were obtained from the internal cell bank at Technological University Dublin (TUDublin City Campus) and commercially through the European Collection of Authenticated Cell Cultures (ECACC), Public Health England. Each cell line was grown in Roswell Park Memorial Institute media (RPMI-1640) (Sigma, R8758) supplemented with 12% Foetal Bovine Serum (Sigma, F2442) and 2% L-glutamine (200 mM, Sigma, G7513) in a T75 cell culture flask (Sigma, CLS3276). Cells cultures were incubated at 37 °C in a 5% CO2 atmosphere. Cells were kept in cell culture media until 80% confluent and subcultured using trypsin solution (Thermo, 15090046)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EDTA (Sigma) (1:1).
:EDTA (Sigma) (1:1).
Cytotoxicity assessment by MTT viability
MCF-7, SKOV-3, A2780 and A2780cis cells were subcultured and seeded at 1 × 105 cells in 96-well plates and incubated for 24 h prior to complex exposure. Cell lines were exposed to Cu-Phen (1), Cu-DPQ-Phen (2), Cu-DPPZ-Phen (3) and Cu-DPPN-Phen (4). The cell lines were also treated with cisplatin (Sigma, P4394). The Cu(II) complexes and cisplatin were dissolved in DMSO prior to subsequent dilution with aqueous cell culture media. Cell treatment with complexes 1–4 was performed with an initial stock in DMSO (Sigma, 276855) (5–10 mM) prior to dilution to a range of 0.25–10 μM with aqueous cell culture media. The cells were treated with the Cu(II) complexes (0.25–10 μM) and cisplatin (0.5–200 μM) for 24 h. Drug-treated cell culture media was removed and 100 μl working solution of MTT (methylthiazolyldiphenyl-tetrazolium bromide) (5 mg ml−1) (Sigma, M5655) was added to the cells and incubated for 3 h, as described in the method by Mosmann, 1983.42 The cells were washed (×3) with sterile PBS (Sigma, 806544) and formazan solubilised in 100 μl DMSO. The absorbance was recorded at 595 nm using a 1420 Multilabel Counter Victor3V spectrophotometer (PerkinElmer, USA). Treatments were performed on three separate occasions in triplicate.Immuno-detection of γH2AX foci with flow cytometry and confocal microscopy
A2780 and A2780cis cell lines were selected due to their respective sensitivity and resistance to cisplatin as determined by the MTT viability assay. Additionally, immunostaining analysis was conducted on MCF-7 and SKOV-3 cells and data are included in Supplementary S1 (ESI†). A2780 and A2780cis cells were subcultured and plated at 1 × 105 cells in T25 flasks (Sigma, CLS430639) and incubated for 24 h to allow for attachment and growth. After 24 h the A2780 and A2780cis cells were exposed (at IC25 concentration values as determined by the MTT viability assay) to the Cu(II) complexes and cisplatin for 24 h. The cells were fixed using 2% paraformaldehyde (Sigma, F8775) and permeabilised using 0.25% Triton X-100 (Sigma, T8787). The cells were blocked using 2% Bovine Serum Albumin (Sigma, A9418) and subsequently incubated with the primary antibody, anti-phospho-histone H2A.X (Ser 139), clone JBW301 (Cat# 05-636) (Lot# 2068177) (1:500) (Milipore, Ireland) and fluorescently labelled using Alexa Fluor 488 FITC conjugate (1:200) (ThermoFisher, A-11029). The cells were then washed with PBS and counterstained using propidium iodide (1 μg μl−1) (Sigma, P4864). The Median Fluorescence Intensity (MFI) was calculated from the gated region experimentally determined from controls using the BD Accuri C6 (BD Biosciences, USA) flow cytometer using the whole cell population. The experimental measurements were conducted on three separate occasions. Confocal microscopy was performed using an LSM© 510 Meta Confocal Microscope (Zeiss, Germany) with slides mounted in an inverted configuration. Images were recorded at 620× magnification with controls used to establish the minimum laser gain, amplitude and offset setting in addition to identifying the upper limits of image saturation and fluorophore excitation using argon and helium–neon laser scanning simultaneously. All images were analysed using ImageJ software (open source). A2780cis images were contrast enhanced by 5% using an equalized histogram of the image.
Apoptosis and mitochondrial-related gene expression
The molecular gene targets of apoptosis, inhibition of apoptosis (XIAP) and mitochondrial-related function were analysed by gene expression in real-time (Table 1). These selected candidate genes can characterise the state of the cell specifically related to the mitochondria after exposure to the Cu(II) phenanthroline–phenazine complexes and cisplatin. Well-characterised cisplatin sensitive and resistant cells were adopted to observe mechanistic trends related to sensitivities and resistance, respectively. The primer sequences for each gene of interest and reference gene are presented in Table 2. Primer sequences were designed using NCBI nucleotide sequences (https://www.ncbi.nlm.nih.gov/nucleotide/) and Primer3PLUS and were synthesised by Sigma, Ireland.| Cellular region | Gene | Oligonucleotides (5′–3′) | Gene | Oligonucleotides (5′–3′) |
|---|---|---|---|---|
| Highlighted in bold are the primers of reference genes used to establish the relative expression of the target genes. (F) refers to forward primer while (R) refers to reverse primer. | ||||
| Mitochondrial | HMOX1 | CAGGGCCATGAACTTTGTCC (F) | TFAM | GGGATCTTGCTATGTTGCCC (F) |
| GAGAGGGACACAGTGAGAGG (R) | TTATGGTCAAGCATGGTGGC (R) | |||
| TFB1M | AGTCGCCTCTCTGTTATGGC (F) | TFB2M | ACCAAGTAGACCTCCACACC (F) | |
| ATGGCTGCTCTATCTTGGGC (R) | GTTTTGTCACTTTCGAGCGC (R) | |||
| NRF-2 | ACCAAAACCACCCTGAAAGC (F) | CLPP | CCTTGTTATCGCACAGCTCC (F) | |
| AGGCCAAGTAGTGTGTCTCC (R) | TCGGGTTGAGGATGTACTGC (R) | |||
| LON | GAAGGAAAGTTCGTCTCGCC (F) | SPG7 | ATATCGAGGCCAAGGACAGG (F) | |
| CTCATGGATCTGGGCAAACG (R) | GGCCAGACGGAAAACATACC (R) | |||
| YME1L1 | CTTTTCCGGAGCAGAGTTGG (F) | DRP1 | CCAATTATGCCAGCCAGTCC (F) | |
| CACTTCTTCTTTCAGGCCCC (R) | CTCACAATCTCGCTGTTCCC (R) | |||
| MFN1 | GGAAGAGGCCAGTTTTGCC (F) | MFN2 | CTTGTGTGTGTCCATCTCGC (F) | |
| GCATTTCCACCAGCTCAAGG (R) | AGCATCCTTTCTACCACCCC (R) | |||
| OPA1 | CCCGCTTTATGACAGAACCG (F) | |||
| GTCCTCCGCAAAGTCATTCC (R) | ||||
| Cytoplasmic | Casp 9 | TCCAGATTGACGACAAGTGC (F) | Casp 3 | TTTGTTTGTGTGCTTCTGAG (F) |
| CCTACAAGTCGTGACAGGGA (R) | TATAAGAACCGCTTTAAGT (R) | |||
| BCL-2 | AAGCGGTCCCGTGGATAGA (F) | BAX | AGGATGCGTCCACCAAGAAG (F) | |
| TCCGGTATTCGCAGAAGTCC (R) | CCAGTTGAAGTTGCCGTCAGA (R) | |||
| XIAP | GCACGGATCGTTACTTTTGGACA (F) | |||
| GTGGAAGCACTTCACTTTATCGCC (R) | ||||
| Reference genes | Actin | ACTCTTCCAGCCTTCCTTCC (F) | B-Tubulin | GCTTCTTGGTTTTCCACAGC (F) |
| GTTGGCGTACAGGTCTTTGC (R) | CGTTCTTCAGGTTCGACCTC (R) | |||
A2780 and A2780cis cell lines were seeded at 1 × 104 cells in T25 flasks and allowed 24 h to attach and grow before the cells were treated with the complexes at their IC25 concentration values for 24 h. Similarly, A2780 and A2780cis were treated with cisplatin in the same manner as the Cu(II) complexes. The Cu(II) complexes, cisplatin and untreated controls were incubated in triplicate. The RNA extraction was performed using Tri-Reagent (Sigma, T9424), washed in 75% ethanol and stored in 30 μl of 0.1% diethylpyrocarbonate (DEPC) treated water (Sigma, 40718). Extracted RNA was quantified using a MaestroNano™ spectrophotometer (MaestroGen, USA). Samples were normalized to 150 ng and were converted to cDNA using the qScript cDNA synthesis kit (Quanta Biosciences, 733-1173) employing the following thermal program: 5 min at 22 °C, 30 min at 42 °C, 5 min at 85 °C followed by holding at 4 °C. Synthesised cDNA templates were amplified and quantified using SYBR Green I (Thermo, 4309155) on the 7500 Fast Real-Time PCR System (Applied Biosystems, USA). The final reaction volume of 20 μl was amplified and quantified using the following program: (i) pre-incubation (1 cycle) 95 °C, 5 min, ramp 4.4 °C; (ii) quantification (45 cycles) 95 °C 10 s ramp 4.4 °C, 60 °C 10 s ramp 2.2 °C, 72 °C 10 s ramp 4.4 °C; (iii) melting curve (1 cycle) 95 °C 5 s ramp 4.4 °C, 65 °C 1 min ramp 2.2 °C, 97 °C continuous ramp 0.11 °C acquisition 10 per °C and (iv) cooling (1 cycle) 40 °C 10 s ramp 1.5 °C. Gene amplification of target genes was determined in relation to the reference genes (N = 2–4). Template controls were used to confirm the absence of autofluorescence. Relative expression values were calculated using geometric means of the reference genes and target genes were normalized according to the Livak and Schmittgen formula: 2−ΔCt1(test)−ΔCt2(control).43
Statistical analysis
The MTT assay raw data were normalized to the negative control within each experiment. The statistical package, Prism (ver. 5.0) (GraphPad, USA) was used to establish the 25% inhibitory concentration (IC25) using least squares non-linear regression analysis. The statistical significance of the changes between treatment with the Cu(II) complexes and the cisplatin was determined using one-way ANOVA with Tukey's post hoc test. The flow cytometry data analysis was performed using the BD Accuri C6 software package and Microsoft® Excel (USA) to establish the mean of the MFI and percent positive data. Prism was used to perform one-way ANOVA significance testing with post hoc Bonferroni correction between test complexes and the control. For the analysis of gene expression, the geometric mean Ct values for both actin and tubulin reference genes were used for normalization as per the Vandesompele method.44 Target gene expression values were calculated along with a standard deviation as per the Nordgård, Livak and Schmittgen formula.43,45 Both the positive and negative error bars display the 95% confidence interval around the mean normalized gene expression value.Results
Cytotoxicity assessment by MTT viability
The MTT assay was used to establish the activity of Cu-Phen (1), Cu-DPQ-Phen (2), Cu-DPPZ-Phen (3) and Cu-DPPN-Phen (4) in both cisplatin-sensitive (MCF-7 and A2780) (Table 3) and cisplatin-resistant (SKOV-3, A2780cis) cells relative to the established clinical therapeutic; cisplatin (Table 3). Cisplatin was tested over the concentration range of 0.5–200 μM and Cu-Phen (1), Cu-DPQ-Phen (2), Cu-DPPZ-Phen (3) and Cu-DPPN-Phen (4) were tested over 0.25–10 μM. Cisplatin displayed a varied level of activity across the range of tested cell lines with SKOV-3 cells presenting the indicative profile of platinum resistance.46 Cisplatin displayed differential activity in both A2780 and A2780cis cell lines, with the resistant variant demonstrating a higher concentration in relation to the sensitive variant at 24 h (>200 μM and 63.10 μM, respectively). The Cu(II) complexes (1–4) all showed IC25 values of <10 μM. The A2780 and the cisplatin-resistant counterpart A2780cis demonstrate classic cisplatin resistance, whereas SKOV-3 cells demonstrate cisplatin resistance but to a lower extent. The diminished level of cisplatin resistance observed in SKOV-3 cells has previously been documented in the evaluation of other novel Cu complexes.12,14,16 Cu(II) phenanthroline phenazine complexes demonstrate superior activity (as denoted by *) to cisplatin in all cell lines while the cisplatin sensitive cell lines (MCF-7 and A2780) demonstrate higher toxicity towards the Cu(II) complexes in comparison to both known cisplatin resistant cell lines (SKOV-3 and A2780cis). Exposure to Cu(II) phenanthroline–phenazine complexes demonstrated consistently increased toxicities irrespective of the ligated phenazine structure in comparison to cisplatin with Cu-DPPN-Phen (4) showing slightly lower toxicity possible due to larger size and lipophilicity. Overall the inclusion of cisplatin resistant cell lines demonstrates that cisplatin resistance is not a confounding factor for the activity of the Cu(II) phenanthroline–phenazine complexes.| Treatment | IC25 (μM) ± S.D. | |||
|---|---|---|---|---|
| Cisplatin-sensitive | Cisplatin-resistant | |||
| MCF-7 | A2780 | SKOV-3 | A2780cis | |
| Mean ± standard deviation (N = 3). * denotes p < 0.05 in comparison to cisplatin. ** denotes p < 0.01 in comparison to cisplatin. *** denotes p < 0.001 in comparison to cisplatin. | ||||
| Cisplatin | 0.45 ± 0.07 | 63.10 ± 13.91 | 21.52 ± 4.54 | >200 |
| Cu-Phen (1) | 0.97 ± 0.10 | 0.39 ± 0.07*** | 2.56 ± 0.12*** | 0.83 ± 0.00*** |
| Cu-DPQ-Phen (2) | 1.27 ± 0.40* | 0.34 ± 0.09*** | 2.51 ± 0.09*** | 0.72 ± 0.02*** |
| Cu-DPPZ-Phen (3) | 0.63 ± 0.01 | 0.31 ± 0.04*** | 1.61 ± 0.74*** | 0.39 ± 0.07*** |
| Cu-DPPN-Phen (4) | 1.37 ± 0.32** | 0.36 ± 0.10*** | 5.71 ± 0.92*** | 0.53 ± 0.02*** |
Immunodetection of γH2AX foci with flow cytometry and confocal microscopy
The induction of γH2AX foci was detected using flow cytometry and confocal microscopy after A2780 and A2780cis cells were exposed to the IC25 concentrations of cisplatin and the Cu(II) phenanthroline–phenazine complexes for 24 h and compared to the unexposed control. Foci formation was determined using MFI (Fig. 2).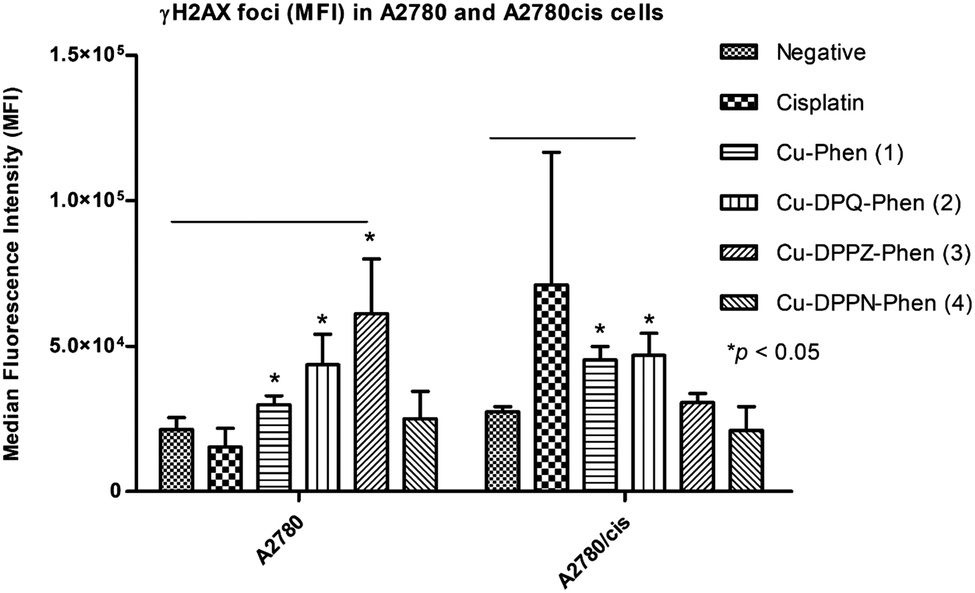 | ||
| Fig. 2 Immunodetection using MFI of γH2AX foci in A2780 and A2780cis cells after 24 h treatment with cisplatin and the Cu(II) complexes. Statistical significance was determined between the negative control and the Cu(II) complexes using one-way ANOVA with Bonferroni correction. | ||
Following the exposure of A2780 to cisplatin and the Cu(II) complexes, a significant increase (p ≤ 0.05) in foci formation was observed, with the exception of Cu-DPPN-Phen (4). Induction of γH2AX foci in A2780 cells occurred to a small extent after exposure to Cu-Phen (1), Cu-DPQ-Phen (2) and Cu-DPPZ-Phen (3) and is absent upon treatment with Cu-DPPN-Phen (4). The induction of γH2AX foci in the A2780cis cells occurs to a larger extent following exposure to Cu-Phen (1) and Cu-DPQ-Phen (2) and is almost entirely absent after exposure to Cu-DPPZ-Phen (3) and Cu-DPPN-Phen (4). The exposure of A2780cis cells to cisplatin induces non-significant variable increases in foci formation. The induction of γH2AX foci was also detected using confocal microscopy imaging to record the incidences of foci formation using the FITC labelled antibody and the nuclear counterstain, propidium iodide (Fig. 3). Overall, the results of confocal microscopy complement the flow cytometry results.
 | ||
| Fig. 3 Confocal microscopy images of γH2AX foci (indicated by yellow arrow and circle) arising from treatment of A2780 and A2780cis cells with cisplatin and the Cu(II) complexes. Red staining indicates nuclear material with green foci representing double stranded DNA break formation (γH2AX). | ||
Apoptosis and mitochondrial-related gene expression
The level of gene expression in the target genes was represented in relation to the geometric mean of the reference genes. The variance for target genes was error propagated and log(2) transformed and a 95% confidence interval was plotted around the ΔΔCt value. Grouped columns in Fig. 4–6 display the log(2) transformed mean expression value relative to the biological negative control, with the broken line at y = 1 indicating lower expression below and higher expression above. | ||
| Fig. 4 A/B Gene expression by A2780 and A2780cis cells following treatment with cisplatin and the Cu(II) complexes. Error bars represent normalized standard deviation. Values below y = 1 (dotted line) represent negative expression. Target genes normalized to the geometric mean of actin and tubulin. N.D. = not detected. | ||
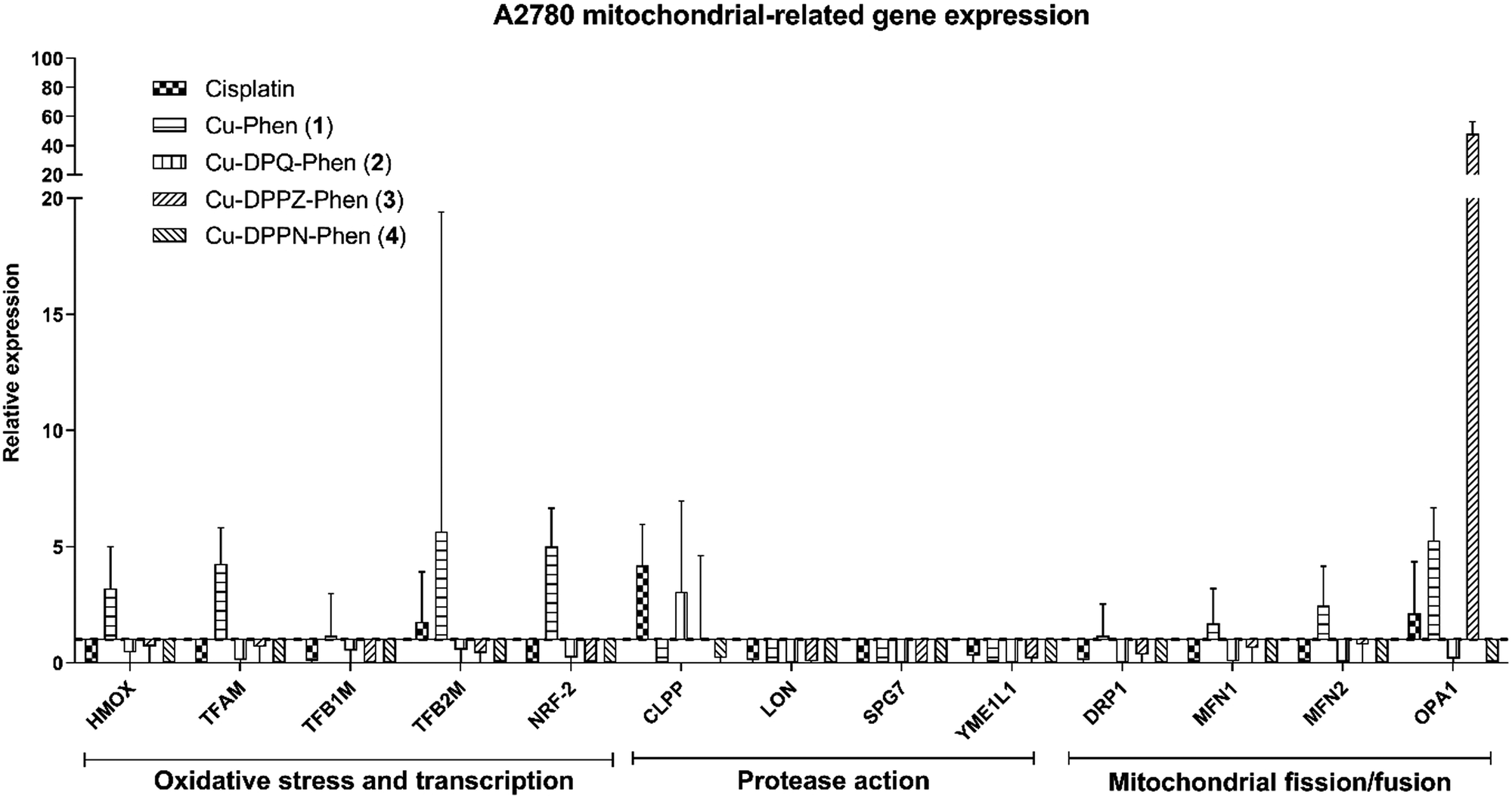 | ||
| Fig. 5 Gene expression by A2780 cells after 24 h exposure to cisplatin and the Cu(II) complexes. Error bars represent normalized standard deviation. Values below y = 1 (dotted line) represent negative expression. Target genes normalized to the geometric mean of actin and tubulin. | ||
 | ||
| Fig. 6 Gene expression by A2780cis cells after 24 h exposure to cisplatin and the Cu(II) complexes. Error bars represent normalized standard deviation. Values below y = 1 (dotted line) represent negative expression. Target genes normalized to the geometric mean of actin and tubulin. N.D. = not detected. | ||
Differential expression of Bax and XIAP expression was observed between A2780 (Fig. 4A) and A2780cis cells (Fig. 4B) when each was exposed to cisplatin. The increase in expression of BCL-2, an anti-apoptotic gene in A2780cis cells, demonstrated a classic mechanism of cisplatin resistance. The apoptotic response following exposure to the Cu(II) complex was markedly different to that of cisplatin. The apoptotic response to the Cu(II) complexes is characterised by increase of BAX and with markedly increased expression in the anti-apoptotic gene, XIAP. Caspase 9, caspase 3, BCL-2 and BAX characterise the response to the Cu(II) complexes, and particularly to exposure to Cu-DPPZ-Phen (3) and Cu-DPPN-Phen (4) in the cisplatin-resistant A2780cis cells. The differential apoptotic response between the Cu(II) complexes and cisplatin in the A2780cis resistant cells demonstrates that the cisplatin resistance is not an impeding factor in the activity of the Cu(II) complexes.
The mitochondrial related gene expression profile produced by the A2780 cells (Fig. 5) demonstrates that the mitochondrial protease gene, CLPP and to a smaller extent the mitochondrial fusion gene, OPA1 are elevated following exposure to cisplatin. This profile is in contrast to that caused by exposure to Cu-Phen (1) where only a small increase in the oxidative stress, transcription, protease and fission/fusion genes was detected. All of the other Cu(II) complexes produce no significant increase except for a strong increase in the mitochondrial fission regulator, OPA1 after exposure to Cu-DPPN-Phen (4). The gene expression profile in the A2780cis cells (Fig. 6) showed significant changes in comparison to the A2780 cells. Cisplatin action features prominently in the mitochondrial fission/fusion regulation in the resistant cells in contrast to the sensitive A2780 cells. In contrast to A2780 cells, exposing A2780cis cells to the Cu(II) complexes produced substantial increases in oxidative stress, transcription, protease and fission/fusion genes. An increase was observed in oxidative stress/transcription and protease genes after exposure of A2780cis cells to Cu-DPQ-Phen (2), which contrasts with the response of the A2780 cells. Considerable increases in the gene groups in A2780cis cells was recorded following treatment with Cu-DPPN-Phen (4), which again differs to the behaviour of the A2780 cells. In contrast to the performance of the A2780 cells, the DRP1 fission regulator has increased expression after exposing the resistant cells to cisplatin and the Cu(II) complexes, demonstrating a similar therapeutic response to both types of metal complex. The use of the cisplatin-resistant A2780cis cells demonstrates that the Cu(II) complexes operate through a different mechanistic mode of action to cisplatin and, furthermore, the Cu(II) complexes have substantially different mechanism(s) of action against cisplatin-resistant cells compared to the cisplatin-sensitive phenotype.
Discussion
Having recently established, the multimodal effects against the in vivo model, G. mellonella in a proteomic study,23 and the previously reported oxidative chemical nuclease and DNA binding properties of the Cu(II) phenanthroline–phenazine complexes,21,22 the aim of this study was to use in vitro models to investigate the mechanism(s) of action of these complexes. Cancer cell lines that confer sensitivity and resistance to the clinical drug, cisplatin, were used so that the mechanisms could be contrasted to cisplatin. Both the MCF-7 (breast) and SKOV-3 (ovarian) cells have been cited in the literature as cisplatin-sensitive and -resistant cell lines, respectively, and are part of the NCI developmental therapeutics program.47–53 Additionally, the use of MCF-7 and SKOV-3 cell lines previously,13,14,21 demonstrated the variable nature of the response to cisplatin in cells derived from different tissues. The A2780 (ovarian) cell line represents the parental lineage of a number of daughter cell lines, which are made resistant to various therapeutics such as doxorubicin (A2780ADR) and cross-resistant to melphalan, doxorubicin and cisplatin (A2780cis). The generation of resistant profiles in the A2780 parent cell lines is brought about through the supplementation of sub-toxic dose of the drug in question in the culture media in order to confer resistance over multiple passages of the cell. While MCF-7 and SKOV-3 cells have previously demonstrated cisplatin-sensitive and -resistant profiles respectively,13,14 the direct comparison between the cell types is limited by their different tissue of origin and subsequently differing gene and protein expression profile. The A2780 and A2780cis cisplatin-resistant daughter cells have transcriptional and protein expression profiles that are comparative, and have been employed in multiple studies examining ovarian cancer and the biological mechanisms that contribute to therapeutic resistance.48–53 The use of the isogenic A2780 parent and resistant A2780cis phenotype has gained popularity as a more homogeneous platform where sub-toxic levels of cisplatin are added to the culture media of A2780cis cells to generate and retain the resistance profile.54 These cells were used to compare the biological effects of the Cu(II) complexes and cisplatin on the MCF-7 and SKOV-3 traditional cells to the effects on the A2780 and A2780cis cells.Overall, the cytotoxicity profile of the Cu(II) phenanthroline–phenazine complexes demonstrated a cytotoxicity profile with the following cadence Cu-DPPZ-Phen (3) (most toxic) > Cu-Phen (1) ∼ Cu-DPQ-Phen (2) > Cu-DPPN-Phen (4) (least toxic). Superior cytotoxicity activity of the Cu(II) phenanthroline–phenazine complexes to the clinical control, cisplatin, was noted in the cisplatin-resistant cell line (A2780cis). A previous study by Molphy et al. established a similar cytotoxic profile against the SKOV-3 cell line which accompanied the highest DNA binding constants and was also comparable to the oxidative DNA damaging agent doxorubicin.21 The development of DNA damage is a key hallmark of cisplatin cytotoxicity and, as such, has been a valuable indicator in the profiling of new therapeutics. γH2AX foci formation is indicative of double stranded breaks (DSBs) in the DNA, and the induction of DSBs by the current Cu(II) complexes was observed. The MCF-7 and A2780 cell lines had less DSBs, which might indicate an alternative mode of action in the resistant cell lines. Additionally, the high levels of DSBs in SKOV-3 and A2780cis cells may be the driver of higher cytotoxicity of the Cu(II) complexes compared to cisplatin, this being despite the development of resistance to cisplatin-induced DNA damage by these two cell lines. This strongly suggests that the Cu(II) complexes are operating through biochemical mechanisms that are distinctly different to those of cisplatin. The identification of alternative mechanisms of action to cisplatin is indeed a developmental objective in metal-based anticancer chemotherapy. Recently our in vivo study in G. mellonella demonstrated increased levels of glutathione S-transferase (GST) in larvae exposed to the Cu(II) complexes.23 The Cu(II) complexes contain polycyclic aromatic rings which have a similar cyclical structure to the DNA damaging agents naphthalene and benzo-[a]-pyrene. Additionally, GST is also upregulated in cells exposed to the chemical nuclease activity of cyclophosphamide and ifosfamide. The generation of oxidative radicals is a key mechanism through which DNA damage occurs. Due to the previously demonstrated nuclease cleavage ability of these Cu(II) complexes22 and structural similarity to the intercalating ruthenium complex Λ-[Ru (phen)2dppz]2+,55 it is likely that the Cu(II) complexes intercalate with the DNA in a similar fashion in addition to the nuclease capability of the complex facilitating DNA strand breakage. The G. mellonella study also determined that enzymes regulating purine synthesis were upregulated along with heat shock protein 90 which are present during increased purine turnover and the formation of the purinosome.56 The high degree of cytotoxicity in SKOV-3 and A2780cis prompted further investigation of apoptosis, which is one of the most important mechanisms of regulated cell death.
Apoptotic cell death is related to loss in cellular viability accompanied by cytotoxicity.27 Transition metal coordinated therapeutics have often been evaluated for their ability to induce apoptosis and other forms of regulated cell death with many evaluative studies on copper complexes using this as a therapeutic endpoint.25,26,57–62 Additionally, cisplatin and other therapeutic compounds have been evaluated for their direct activity against BCL-2.30,63 While these studies have used different markers of apoptosis to demonstrate the induction of this endpoint, the use of gene expression in this study allows multiple pathways in the apoptotic process to be investigated. Mitochondrial-mediated apoptosis can be initiated by the induction of DNA damage through p53 or the action of Bax and BCL-2 regulated by BH3 proteins of the BCL2 family. The upregulation of BCL-2 was observed in the cisplatin-resistant cell lines which is seen as a classic form of drug resistance.64,65 This pattern was in direct contrast to that found when the cells were exposed to the Cu(II) complexes, where increased levels of BAX and XIAP were detected. Additionally, caspase-9 and caspase-3 genes were also expressed with the Cu(II) complexes, indicating a complete apoptotic cascade. The upregulation of BAX is a key progenitor of the formation of the mitochondrial outer membrane pores (MOMP) in mitochondria which then induces the caspase cascade (caspase-9 and caspase-3) for apoptosis.66 The mitochondria are being increasingly recognised as a focal point for a multiple cellular process which proves pivotal in directing the fate of the cell. Critical amongst these are responses to inflammation,67,68 ROS,69–73 and apoptosis,72,74–76 which are closely associated and dependent on the mitochondria. Indeed, substantial effort is being directed towards developing therapeutic targets in the mitochondria or processes associated with its action.77–80
The high toxicity profiles of the Cu(II) complexes with moderate to low in vitro DNA damage recorded by γH2AX foci coupled with mitochondrial-mediated pro-apoptotic gene expression led to further interest in the role and activity of the mitochondria beyond apoptosis. The mitochondrial related gene expression panel was examined through antioxidant enzyme production, HMOX (Table 1), while mitochondrial related transcription was monitored through TFAM, TFB1M, TFB2M and NRF-2 (Table 1). Mitochondrial protease genes were monitored through CLPP, LON, SPG7 and YME1L1 (Table 1). The process of fission/fusion of the mitochondria was examined through DRP1, MFN1, MFN2 and OPA1 (Table 1). The groups of genes representing the mitochondrial, antioxidant, transcription and protease functions demonstrated the highest changes in expression between A2780 and A2780cis resistant cells. Exposure of resistant A2780cis cells to cisplatin induces a characteristic resistance to mitophagy through upregulation of DRP1 and mitochondrial related transcription factors.81,82 The role of DRP1 in mitochondrial remodelling and its relationship to apoptosis has become clearer over the past few years. While DRP1 is involved in mitochondrial membrane impairment and uncoupling of oxidative phosphorylation leading to its identification as a drug resistance factor,83,84 new information has emerged about its direct role in bringing about apoptosis through accelerating cytochrome c release from the mitochondrial membrane,85 and additional functions in the responses to oxidative stress,86 and cellular differentiation.87–89 The activity of the mitochondrial-related transcription factors and the fission/fusion regulator are also known to be involved in the drug resistance response.84,90–92 As determined above, the differences in apoptosis between the Cu(II) complexes and cisplatin centre around the activation of BAX and XIAP, caspase-9 and caspase-3. In contrast to this, the mitochondria fission regulator, DRP1, is commonly upregulated in all exposures. The upregulation of DRP1 has recently been associated with cisplatin resistance81,82,93–95 and apoptosis through cytochrome c release,85 and with a similar upregulation after exposure to the Cu(II) complexes this may indicate the exploitation of a similar mechanism of action. While similarities in mechanisms can be shown with DRP1 activity, the upregulation of all proteases particularly LON, following exposure of A2780cis cells to the Cu(II) complexes may indicate an increased rate of protein turnover due to misfolded, oxidized and otherwise denatured proteins, which may be contributing to the enhanced cytotoxicity of the Cu(II) complexes.96–98 Additionally, the presence of HMOX and transcription factors related to the antioxidant response in the A2780cis cells treated with the Cu(II) complexes, builds on the evidence of upregulated protease factors to demonstrate potential increases in protein dysregulation as a potent factor in the mitochondrial toxicity.36,99 Previously our in vivo G. mellonella study demonstrated increases in protein expression of metabolic enzymes associated with the mitochondria.23 These changes also accompanied increase in pathways associated with protein detoxification and degradation, which complements the gene expression activity observed in this study indicating increased protein turnover potentially due to oxidative damage from mitochondrial dysregulation or the direct action of the Cu(II) complexes.
These Cu(II) complexes have a unique profile of cytotoxicity differing from that of cisplatin. The investigation into the effects of the mitochondria using gene expression has provided a wider context to the mechanistic activity in addition to demonstrating apoptosis. The results presented here demonstrate that Cu(II) phenanthroline–phenazine complexes are a promising therapeutic candidate which are highly potent against cisplatin-resistant cells. The superior activity, together with their multimodal activity against cisplatin-resistant cells offers a potentially novel therapeutic avenue for treating cisplatin-refractory cancers in combination with current chemotherapy.
Conflicts of interest
The authors have no financial conflicts to disclose.Acknowledgements
The authors acknowledge financial support for this study from Technological University Dublin through the ‘Fiosraigh Dean of Graduate Studies award’ (2013 call) for this study. AK and ZM acknowledge support from Science Foundation Ireland (15/CDA/3648).References
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Advances in Copper Complexes as Anticancer Agents, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- L. Ruiz-Azuara and M. E. Bravo-Gómez, Copper Compounds in Cancer Chemotherapy, Curr. Med. Chem., 2010, 17, 3606–3615 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Copper Complexes as Anticancer Agents, Anticancer. Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- C. R. Kowol, P. Heffeter, W. Miklos, L. Gille, R. Trondl, L. Cappellacci, W. Berger and B. K. Keppler, Mechanisms Underlying Reductant-Induced Reactive Oxygen Species Formation by Anticancer Copper(II) Compounds Europe PMC Funders Group, J. Biol. Inorg. Chem., 2012, 17, 409–423 CrossRef CAS PubMed.
- I. B. Afanas’ev, E. A. Ostrachovich and L. G. Korkina, Effect of Rutin and Its Copper Complex on Superoxide Formation and Lipid Peroxidation in Rat Liver Microsomes, FEBS Lett., 1998, 425, 256–258 CrossRef.
- M. Kuzuya, K. Yamada, T. Hayashi, C. Funaki, M. Naito, K. Asai and F. Kuzuya, Role of Lipoprotein-Copper Complex in Copper Catalyzed-Peroxidation of Low-Density Lipoprotein, Biochim. Biophys. Acta, 1992, 1123, 334–341 CrossRef CAS.
- X. Qin, Q. Huang, L. Zhu, H. Xiao, G. Yao, W. Huang, R. Zhu, J. Hu and Y. Zhu, Interaction with Cu 2+ Disrupts the RNA Binding Affinities of RNA Recognition Motif Containing Protein, Biochem. Biophys. Res. Commun., 2014, 444, 116–120 CrossRef CAS.
- A. Marín-Hernández, I. Gracia-Mora, L. Ruiz-Ramírez and R. Moreno-Sánchez, Toxic Effects of Copper-Based Antineoplastic Drugs (Casiopeinas) on Mitochondrial Functions, Biochem. Pharmacol., 2003, 65, 1979–1989 CrossRef.
- J. L. Sagripanti, P. L. Goering and A. Lamanna, Interaction of Copper with DNA and Antagonism by Other Metals, Toxicol. Appl. Pharmacol., 1991, 110, 477–485 CrossRef CAS.
- M. E. Bravo-Gómez, C. Campero-Peredo, D. García-Conde, M. J. Mosqueira-Santillán, J. Serment-Guerrero and L. Ruiz-Azuara, DNA-Binding Mode of Antitumoral Copper Compounds (Casiopeinas Ò) and Analysis of Its Biological Meaning, Polyhedron, 2015, 102, 530–538 CrossRef.
- C. Slator, N. Barron, O. Howe and A. Kellett, [Cu(O-phthalate)(Phenanthroline)] Exhibits Unique Superoxide-Mediated NCI-60 Chemotherapeutic Action through Genomic DNA Damage and Mitochondrial Dysfunction, ACS Chem. Biol., 2016, 11, 159–171 CrossRef CAS.
- A. Kellett, M. O’Connor, M. McCann, O. Howe, A. Casey, P. McCarron, K. Kavanagh, M. McNamara, S. Kennedy, D. D. May, P. S. Skell, D. O’Shea and M. Devereux, Water-Soluble Bis(1,10-Phenanthroline) Octanedioate Cu2+ and Mn2+ Complexes with Unprecedented Nano and Picomolar in Vitro Cytotoxicity: Promising Leads for Chemotherapeutic Drug Development, MedChemComm, 2011, 2, 579–584 RSC.
- A. Kellett, M. O’Connor, M. McCann, M. McNamara, P. Lynch, G. Rosair, V. McKee, B. Creaven, M. Walsh, S. McClean, A. Foltyn, D. O’Shea, O. Howe and M. Devereux, Bis-Phenanthroline Copper(II) Phthalate Complexes Are Potent in Vitro Antitumour Agents with “self-Activating” Metallo-Nuclease and DNA Binding Properties, Dalton Trans., 2011, 40, 1024–1027 RSC.
- A. Kellett, O. Howe, M. O’Connor, M. McCann, B. S. Creaven, S. McClean, A. Foltyn-Arfa Kia, A. Casey and M. Devereux, Radical-Induced DNA Damage by Cytotoxic Square-Planar Copper(II) Complexes Incorporating o-Phthalate and 1,10-Phenanthroline or 2,2’-Dipyridyl, Free Radical Biol. Med., 2012, 53, 564–576 CrossRef CAS.
- A. Prisecaru, M. Devereux, N. Barron, M. McCann, J. Colleran, A. Casey, V. McKee and A. Kellett, Potent Oxidative DNA Cleavage by the Di-Copper Cytotoxin: [Cu2(μ-Terephthalate)(1,10-Phen)4]2+, Chem. Commun., 2012, 48, 6906–6908 RSC.
- A. Prisecaru, V. Mckee, O. Howe, G. Rochford, M. McCann, J. Colleran, M. Pour, N. Barron, N. Gathergood and A. Kellett, Regulating Bioactivity of Cu2+ Bis-1, 10-Phenanthroline Artificial Metallonucleases with Sterically Functionalized Pendant Carboxylates, J. Med. Chem., 2013, 56, 8599–8615 CrossRef CAS PubMed.
- C. Mejia and L. Ruiz-Azuara, Casiopeinas IIgly and IIIia Induce Apoptosis in Medulloblastoma Cells, Pathol. Oncol. Res., 2008, 14, 467–472 CrossRef.
- Ò. J. Serment-Guerrero, P. Cano-Sanchez, E. Reyes-Perez, F. Velazquez-Garcia, M. E. Bravo-Gomez and L. Ruiz-Azuara, Genotoxicity of the Copper Antineoplastic Coordination Complexes Casiopeinas®, Toxicol. In Vitro, 2011, 25, 1376–1384 CrossRef.
- C. Trejo-Solís, G. Palencia, S. Zúñiga, A. Rodríguez-Ropon, L. Osorio-Rico, S. T. Luvia, I. Gracia-Mora, L. Marquez-Rosado, A. Sánchez, M. E. Moreno-García, A. Cruz, E. Bravo-Gó Mez, L. Ruiz-Ramírez, S. Rodríguez-Enriquez and J. Sotelo, Cas IIgly Induces Apoptosis in Glioma C6 Cells In Vitro and In Vivo through Caspase-Dependent and Caspase-Independent Mechanisms 1, Neoplasia, 2005, 7, 563–574 CrossRef.
- F. Carvallo-Chaigneau, C. Trejo-Solís, C. Gómez-Ruiz, E. Rodríguez-Aguilera, L. Macías-Rosales, E. Cortés-Barberena, C. Cedillo-Peláez, I. Gracia-Mora, L. Ruiz-Azuara, V. Madrid-Marina and F. Constantino-Casas, Casiopeina III-Ia Induces Apoptosis in HCT-15 Cells in Vitro through Caspase-Dependent Mechanisms and Has Antitumor Effect in Vivo, Biometals, 2008, 21, 17–28 CrossRef CAS.
- Z. Molphy, A. Prisecaru, C. Slator, N. Barron, M. McCann, J. Colleran, D. Chandran, N. Gathergood and A. Kellett, Copper Phenanthrene Oxidative Chemical Nucleases, Inorg. Chem., 2014, 53, 5392–5404 CrossRef CAS.
- Z. Molphy, C. Slator, C. Chatgilialoglu and A. Kellett, DNA Oxidation Profiles of Copper Phenanthrene Chemical Nucleases, Front. Chem., 2015, 3, 1–9 CAS.
- G. Rochford, Z. Molphy, N. Browne, C. Surlis, M. Devereux, M. McCann, A. Kellett, O. Howe and K. Kavanagh, In-Vivo Evaluation of the Response of Galleria Mellonella Larvae to Novel Copper(II) Phenanthroline-Phenazine Complexes, J. Inorg. Biochem., 2018, 186, 135–146 CrossRef CAS PubMed.
- L. Kelland, Nature Reviews Cancer, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS.
- D. Montagner, V. Gandin, C. Marzano and A. Erxleben, DNA Damage and Induction of Apoptosis in Pancreatic Cancer Cells by a New Dinuclear Bis(Triazacyclonane) Copper Complex, J. Inorg. Biochem., 2015, 145, 101–107 CrossRef CAS.
- L. Fan, M. Tian, Y. Liu, Y. Deng, Z. Liao and J. Xu, Salicylate •Phenanthroline Copper (II) Complex Induces Apoptosis in Triple-Negative Breast Cancer Cells, Oncotarget, 2017, 8, 29823–29832 Search PubMed.
- L. Galluzzi and I. Vitale, Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ., 2018, 25, 486–541 CrossRef.
- G. Kroemer, L. Galluzzi and C. Brenner, Mitochondrial Membrane Permeabilization in Cell Death, Physiol. Rev., 2007, 87, 99–163 CrossRef CAS.
- N. Tajeddine, L. Galluzzi, O. Kepp, E. Hangen, E. Morselli, L. Senovilla, N. Araujo, G. Pinna, N. Larochette, N. Zamzami, N. Modjtahedi, A. Harel-Bellan and G. Kroemer, Hierarchical Involvement of Bak, VDAC1 and Bax in Cisplatin-Induced Cell Death, Oncogene, 2008, 27, 4221–4232 CrossRef CAS.
- W. A. Michaud, A. C. Nichols, E. A. Mroz, W. C. Faquin, J. R. Clark, S. Begum, W. H. Westra, H. Wada, P. M. Busse, L. W. Ellisen and J. W. Rocco, Bcl-2 Blocks Cisplatin-Induced Apoptosis and Predicts Poor Outcome Following Chemoradiation Treatment in Advanced Oropharyngeal Squamous Cell Carcinoma, Clin. Cancer Res., 2009, 15, 1645–1654 CrossRef CAS.
- H. V. Jain and M. Meyer-Hermann, The Molecular Basis of Synergism between Carboplatin and ABT-737 Therapy Targeting Ovarian Carcinomas, Cancer Res., 2011, 71, 705–715 CrossRef CAS.
- G. Ichim and S. W. G. Tait, A Fate Worse than Death: Apoptosis as an Oncogenic Process, Nat. Rev. Cancer, 2016, 16, 539–548 CrossRef CAS.
- L. E. Tebay, H. Robertson, S. T. Durant, S. R. Vitale, T. M. Penning, A. T. Dinkova-Kostova and J. D. Hayes, Mechanisms of Activation of the Transcription Factor Nrf2 by Redox Stressors, Nutrient Cues, and Energy Status and the Pathways through Which It Attenuates Degenerative Disease, Free Radical Biol. Med., 2015, 88, 108–146 CrossRef CAS PubMed.
- N. Gleyzer, K. Vercauteren and R. C. Scarpulla, Control of Mitochondrial Transcription Specificity Factors (TFB1M and TFB2M) by Nuclear Respiratory Factors (NRF-1 and NRF-2) and PGC-1 Family Coactivators, Mol. Cell. Biol., 2005, 25, 1354–1366 CrossRef CAS.
- D. Litonin, M. Sologub, Y. Shi, M. Savkina, M. Anikin, M. Falkenberg, C. M. Gustafsson and D. Temiakov, Human Mitochondrial Transcription Revisited: Only TFAM and TFB2M Are Required for Transcription of the Mitochondrial Genes in Vitro, J. Biol. Chem., 2010, 285, 18129–18133 CrossRef CAS.
- Y. Shi, A. Dierckx, P. H. Wanrooij, S. Wanrooij, N.-G. Larsson, L. M. Wilhelmsson, M. Falkenberg and C. M. Gustafsson, Mammalian Transcription Factor A Is a Core Component of the Mitochondrial Transcription Machinery, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16510–16515 CrossRef CAS.
- A. Cole, Z. Wang, E. Coyaud, V. Voisin, M. Gronda, Y. Jitkova, R. Mattson, R. Hurren, S. Babovic, N. Maclean, I. Restall, X. Wang, D. V. Jeyaraju, M. A. Sukhai, S. Prabha, S. Bashir, A. Ramakrishnan, E. Leung, Y. H. Qia, N. Zhang, K. R. Combes, T. Ketela, F. Lin, W. A. Houry, A. Aman, R. Al-Awar, W. Zheng, E. Wienholds, C. J. Xu, J. Dick, J. C. Y. Wang, J. Moffat, M. D. Minden, C. J. Eaves, G. D. Bader, Z. Hao, S. M. Kornblau, B. Raught and A. D. Schimmer, Inhibition of the Mitochondrial Protease, ClpP, as a Therapeutic Strategy for Human Acute Myeloid Leuekmia HHS Public Access, Cancer Cell, 2015, 27, 864–876 CrossRef CAS.
- P. M. Quirós, T. Langer and C. López-Otín, New Roles for Mitochondrial Proteases in Health, Ageing and Disease, Nat. Rev. Mol. Cell Biol., 2015, 16, 345–359 CrossRef PubMed.
- D. A. Bota and K. J. A. Davies, Mitochondrial Lon Protease in Human Disease and Aging: Including an Etiologic Classification of Lon-Related Diseases and Disorders, Free Radical Biol. Med., 2016, 100, 188–198 CrossRef CAS.
- P. H. Reddy, Inhibitors of Mitochondrial Fission as a Therapeutic Strategy for Diseases with Oxidative Stress and Mitochondrial Dysfunction, J. Alzheimer's Dis., 2014, 40, 245–256 Search PubMed.
- D. A. Patten, J. Wong, M. Khacho, V. Soubannier, R. J. Mailloux, K. Pilon-Larose, J. G. MacLaurin, D. S. Park, H. M. McBride, L. Trinkle-Mulcahy, M.-E. Harper, M. Germain and R. S. Slack, OPA1-Dependent Cristae Modulation Is Essential for Cellular Adaptation to Metabolic Demand, EMBO J., 2014, 33, 2676–2691 CrossRef CAS.
- T. Mosmann, Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- K. J. Livak and T. D. Schmittgen, Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- J. Vandesompele, K. De Preter, B. Pattyn, B. Poppe, N. Van Roy, A. De Paepe and F. Speleman, Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes, Genome Biol., 2002, 3, 34–41 CrossRef.
- O. Nordgård, J. T. Kvaløy, R. K. Farmen and R. Heikkilä, Error Propagation in Relative Real-Time Reverse Transcription Polymerase Chain Reaction Quantification Models: The Balance between Accuracy and Precision, Anal. Biochem., 2006, 356, 182–193 CrossRef.
- C. M. Beaufort, J. C. A. Helmijr, A. M. Piskorz, M. Hoogstraat, K. Ruigrok-Ritstier, N. Besselink, M. Murtaza, W. F. J. van IJcken, A. A. J. Heine, M. Smid, M. J. Koudijs, J. D. Brenton, E. M. J. J. Berns, J. Helleman, R. Pearson, J. C. A. Helmijr, A. M. Piskorz, M. Hoogstraat, K. Ruigrok-Ritstier, N. Besselink, M. Murtaza, W. F. J. Van IJcken, A. A. J. Heine, M. Smid, M. J. Koudijs, J. D. Brenton, E. M. J. J. Berns and J. Helleman, Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of in Vitro Morphological Subtypes, PLoS One, 2014, 9, e103988, DOI:10.1371/journal.pone.0103988.
- R. Shoemaker, The NCI60 Human Tumour Cell Line Anticancer Drug Screen, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, In Vitro and in Vivo Activity and Cross Resistance Profiles of Novel Ruthenium (II) Organometallic Arene Complexes in Human Ovarian Cancer, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS.
- J. C. Hahne, A. Honig, S. R. Meyer, S. Gambaryan, U. Walter, J. Wischhusen, S. F. M. Hausser, S. E. Segerer, N. Fujita, J. Dietl and J. B. Engel, Downregulation of AKT Reverses Platinum Resistance of Human Ovarian Cancers in Vitro, Oncol. Rep., 2012, 28, 2023–2028 CrossRef CAS.
- L. M. McEvoy, S. A. O’Toole, C. D. Spillane, C. M. Martin, M. F. Gallagher, B. Stordal, G. Blackshields, O. Sheils and J. J. O’Leary, Identifying Novel Hypoxia-Associated Markers of Chemoresistance in Ovarian Cancer, BMC Cancer, 2015, 15, 547, DOI:10.1186/s12885-015-1539-8.
- M. Espina, M. Corte-Rodriguez, L. Aguado, M. Montes-Bayin, M. I. Sierra, P. Martinez-Camblor, E. Blanco-Gonzilez and L. M. Sierra, Cisplatin Resistance in Cell Models: Evaluation of Metallomic and Biological Predictive Biomarkers to Address Early Therapy Failure, Metallomics, 2017, 9, 564–574 RSC.
- D. B. Pfankuchen, F. Baltes, T. Batool, J.-P. Li, M. Schlesinger and G. Bendas, Heparin Antagonizes Cisplatin Resistance of A2780 Ovarian Cancer Cells by Affecting the Wnt Signaling Pathway, Oncotarget, 2017, 8, 67553–67566, DOI:10.18632/oncotarget.18738.
- B. Ffrench, C. Gasch, K. Hokamp, C. Spillane, G. Blackshields, T. M. Mahgoub, M. Bates, L. Kehoe, A. Mooney, R. Doyle, B. Doyle, D. O’Donnell, N. Gleeson, B. T. Hennessy, B. Stordal, C. O’Riain, H. Lambkin, S. O’Toole, J. J. O’Leary and M. F. Gallagher, CD10−/ALDH− Cells Are the Sole Cisplatin-Resistant Component of a Novel Ovarian Cancer Stem Cell Hierarchy, Cell Death Dis., 2017, 8, e3128, DOI:10.18632/oncotarget.18738.
- T. A. Ince, A. D. Sousa, M. A. Jones, J. C. Harrell, E. S. Agoston, M. Krohn, L. M. Selfors, W. Liu, K. Chen, M. Yong, P. Buchwald, B. Wang, K. S. Hale, E. Cohick, P. Sergent, A. Witt, Z. Kozhekbaeva, S. Gao, A. T. Agoston, M. A. Merritt, R. Foster, B. R. Rueda, C. P. Crum, J. S. Brugge and G. B. Mills, Characterization of Twenty-Five Ovarian Tumour Cell Lines That Phenocopy Primary Tumours, Nat. Commun., 2015, 6, 7419, DOI:10.1038/ncomms8419.
- C. J. Cardin, J. M. Kelly and S. J. Quinn, Photochemically Active DNA-Intercalating Ruthenium and Related Complexes – Insights by Combining Crystallography and Transient Spectroscopy, Chem. Sci., 2017, 8, 4705–4723 RSC.
- A. M. Pedley and S. J. Benkovic, A New View into the Regulation of Purine Metabolism: The Purinosome, Trends Biochem. Sci., 2017, 42, 141–154 CrossRef CAS.
- J. Zuo, C. Bi, Y. Fan, D. Buac, C. Nardon, K. G. Daniel and Q. Ping Dou, Cellular and Computational Studies of Proteasome Inhibition and Apoptosis Induction in Human Cancer Cells by Amino Acid Schiff Base–Copper Complexes, J. Inorg. Biochem., 2013, 118, 83–93 CrossRef CAS.
- G. Filomeni, S. Piccirillo, I. Graziani, S. Cardaci, A. M. Da, C. Ferreira, G. Rotilio and M. R. Ciriolo, The Isatin-Schiff Base Copper(II) Complex Cu(Isaepy) 2 Acts as Delocalized Lipophilic Cation, Yields Widespread Mitochondrial Oxidative Damage and Induces AMP-Activated Protein Kinase-Dependent Apoptosis, Carcinogenesis, 2009, 30, 1115–1124 CrossRef CAS PubMed.
- S. Tardito, C. Isella, E. Medico, L. Marchiò, E. Bevilacqua, M. Hatzoglou, O. Bussolati and R. Franchi-Gazzola, The Thioxotriazole Copper(II) Complex A0 Induces Endoplasmic Reticulum Stress and Paraptotic Death in Human Cancer Cells, J. Biol. Chem., 2009, 284, 24306–24319 CrossRef CAS PubMed.
- J. Qi, S. Liang, Y. Gou, Z. Zhang, Z. Zhou, F. Yang and H. Liang, Synthesis of Four Binuclear Copper(II) Complexes: Structure, Anticancer Properties and Anticancer Mechanism, Eur. J. Med. Chem., 2015, 96, 360–368 CrossRef CAS PubMed.
- C. Caruso Bavisotto, D. Nikolic, A. Marino Gammazza, R. Barone, F. Lo Cascio, E. Mocciaro, G. Zummo, E. Conway De Macario, A. J. Macario, F. Cappello, V. Giacalone, A. Pace, G. Barone, A. P. Piccionello and C. Campanella, The Dissociation of the Hsp60/pro-Caspase-3 Complex by Bis(Pyridyl)Oxadiazole Copper Complex (CubipyOXA) Leads to Cell Death in NCI-H292 Cancer Cells, J. Inorg. Biochem., 2017, 170, 8–16 CrossRef CAS.
- B. Thati, A. Noble, B. S. Creaven, M. Walsh, K. Kavanagh and D. A. Egan, Apoptotic Cell Death: A Possible Key Event in Mediating the in Vitro Anti-Proliferative Effect of a Novel Copper(II) Complex, [Cu(4-Mecdoa)(Phen)(2)] (Phen = phenanthroline, 4-Mecdoa = 4-Methylcoumarin-6,7-Dioxactetate), in Human Malignant Cancer Cells, Eur. J. Pharmacol., 2007, 168, 16–28 CrossRef.
- K. J. Campbell and S. W. G. Tait, Targeting BCL-2 Regulated Apoptosis in Cancer, Open Biol., 2018, 8, 180002, DOI:10.1098/rsob.180002.
- A. Brozovic, A. Ambriović-Ristov and M. Osmak, The Relationship between Cisplatin-Induced Reactive Oxygen Species, Glutathione, and BCL-2 and Resistance to Cisplatin, Crit. Rev. Toxicol., 2010, 40, 347–359 CrossRef CAS PubMed.
- A.-M. Florea and D. Büsselberg, Cisplatin as an Anti-Tumor Drug: Cellular Mechanisms of Activity, Drug Resistance and Induced Side Effects, Cancers, 2011, 3, 1351–1371 CrossRef CAS.
- L. Grosse, C. A. Wurm, C. Bruser, D. Neumann, D. C. Jans and S. Jakobs, Bax Assembles into Large Ring-like Structures Remodeling the Mitochondrial Outer Membrane in Apoptosis, EMBO J., 2016, 35, 402–413 CrossRef CAS PubMed.
- R. Zhou, A. Yazdi, P. Menu and J. Tschopp, A Role for Mitochondria in NLRP3 Inflammasome Activation, Nature, 2011, 469, 221–225 CrossRef CAS.
- J. Tschopp, Mitochondria: Sovereign of Inflammation?, Eur. J. Immunol., 2011, 41, 1196–1202 CrossRef CAS PubMed.
- L. Bleier, I. Wittig, H. Heide, M. Steger, U. Brandt and S. Dröse, Generator-Specific Targets of Mitochondrial Reactive Oxygen Species, Free Radical Biol. Med., 2015, 78, 1–10 CrossRef CAS PubMed.
- G. S. Shadel and T. L. Horvath, Mitochondrial ROS Signaling in Organismal Homeostasis, Cell, 2015, 163, 560–569 CrossRef CAS PubMed.
- S. I. Okon and M.-H. Zou, Mitochondrial ROS and Cancer Drug Resistance: Implications for Therapy, Pharmacol. Res., 2015, 100, 170–174 CrossRef PubMed.
- H. El-Osta and M. L. Circu, Mitochondrial ROS and Apoptosis, Mitochondrial Mechanisms of Degeneration and Repair in Parkinson's Disease, Springer International Publishing, Cham, 2016, pp. 1–23 Search PubMed.
- S. S. Sabharwal and P. T. Schumacker, Mitochondrial ROS in Cancer: Initiators, Amplifiers or an Achilles’ Heel?, Nat. Rev. Cancer, 2014, 14, 709–721 CrossRef CAS PubMed.
- M. Redza-Dutordoir and D. A. Averill-Bates, Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2977–2992 CrossRef CAS PubMed.
- J. Lopez and S. W. G. Tait, Mitochondrial Apoptosis: Killing Cancer Using the Enemy Within, Br. J. Cancer, 2015, 112, 957–962 CrossRef CAS PubMed.
- S. Marchi, C. Giorgi, J. M. Suski, C. Agnoletto, A. Bononi, M. Bonora, E. De Marchi, S. Missiroli, S. Patergnani, F. Poletti, A. Rimessi, J. Duszynski, M. R. Wieckowski and P. Pinton, Mitochondria-Ros Crosstalk in the Control of Cell Death and Aging, J. Signal Transduction, 2012, 2012, 329635, DOI:10.1155/2012/329635.
- A. S. Heller, Targeting Mitochondria by Mitochondrial Fusion, Mitochondria-Specific Peptides and Nanotechnology, PhD thesis, Universitat Regensburg, 2013 Search PubMed.
- A. Heller, G. Brockhoff and A. Goepferich, Targeting Drugs to Mitochondria, Eur. J. Pharm. Biopharm., 2012, 82, 1–18 CrossRef CAS PubMed.
- C. Bosc, M. A. Selak and J.-E. Sarry, Resistance Is Futile: Targeting Mitochondrial Energetics and Metabolism to Overcome Drug Resistance in Cancer Treatment, Cell Metab., 2017, 26, 705–707 CrossRef CAS PubMed.
- G. Zhang, D. T. Frederick, L. Wu, Z. Wei, C. Krepler, S. Srinivasan, Y. C. Chae, X. Xu, H. Choi, E. Dimwamwa, O. Ope, B. Shannan, D. Basu, D. Zhang, M. Guha, M. Xiao, S. Randell, K. Sproesser, W. Xu, J. Liu, G. C. Karakousis, L. M. Schuchter, T. C. Gangadhar, R. K. Amaravadi, M. Gu, C. Xu, A. Ghosh, W. Xu, T. Tian, J. Zhang, S. Zha, Q. Liu, P. Brafford, A. Weeraratna, M. A. Davies, J. A. Wargo, N. G. Avadhani, Y. Lu, G. B. Mills, D. C. Altieri, K. T. Flaherty and M. Herlyn, Targeting Mitochondrial Biogenesis to Overcome Drug Resistance to MAPK Inhibitors, J. Clin. Invest., 2016, 126, 1834–1856 CrossRef PubMed.
- C. Zhao, Z. Chen, J. Qi, S. Duan, Z. Huang, C. Zhang, L. Wu, M. Zeng, B. Zhang, N. Wang, H. Mao, A. Zhang, C. Xing and Y. Yuan, Drp1-Dependent Mitophagy Protects against Cisplatin-Induced Apoptosis of Renal Tubular Epithelial Cells by Improving Mitochondrial Function, Oncotarget, 2017, 8, 20988, DOI:10.1155/2012/329635.
- X.-J. Han, S.-L. Shi, Y.-F. Wei, L.-P. Jiang, M.-Y. Guo, H.-L. Wu and Y.-Y. Wan, Involvement of Mitochondrial Dynamics in the Antineoplastic Activity of Cisplatin in Murine Leukemia L1210 Cells, Oncol. Rep., 2017, 38, 985–992 CrossRef CAS PubMed.
- M. Bras, V. J. Yuste, G. Roué, S. Barbier, P. Sancho, C. Virely, M. Rubio, S. Baudet, J. E. Esquerda, H. Merle-Béral, M. Sarfati, S. A. Susin, G. Roue, S. Barbier, P. Sancho, C. Virely, M. Rubio, S. Baudet, J. E. Esquerda, H. Merle-Beral, M. Sarfati and S. A. Susin, Drp1 Mediates Caspase-Independent Type III Cell Death in Normal and Leukemic Cells, Mol. Cell. Biol., 2007, 27, 7073–7088 CrossRef CAS PubMed.
- K. J. Thomas and M. R. Jacobson, Defects in Mitochondrial Fission Protein Dynamin-Related Protein 1 Are Linked to Apoptotic Resistance and Autophagy in a Lung Cancer Model, PLoS One, 2012, 7, e45319, DOI:10.1371/journal.pone.0045319.
- B. Oettinghaus, D. D’Alonzo, E. Barbieri, L. M. Restelli, C. Savoia, M. Licci, M. Tolnay, S. Frank and L. Scorrano, DRP1-Dependent Apoptotic Mitochondrial Fission Occurs Independently of BAX, BAK and APAF1 to Amplify Cell Death by BID and Oxidative Stress, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 1267–1276 CrossRef CAS PubMed.
- A. Nishimura, T. Shimauchi, T. Tanaka, K. Shimoda, T. Toyama, N. Kitajima, T. Ishikawa, N. Shindo, T. Numaga-Tomita, S. Yasuda, Y. Sato, K. Kuwahara, Y. Kumagai, T. Akaike, T. Ide, A. Ojida, Y. Mori and M. Nishida, Hypoxia-Induced Interaction of Filamin with Drp1 Causes Mitochondrial Hyperfission-Associated Myocardial Senescence, Sci. Signal., 2018, 11, eaat5185, DOI:10.1155/2012/329635.
- C. Hu, Y. Huang and L. Li, Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals, Int. J. Mol. Sci., 2017, 18, 144, DOI:10.3390/ijms18010144.
- A. Hoque, P. Sivakumaran, S. T. Bond, N. X. Y. Ling, A. M. Kong, J. W. Scott, N. Bandara, D. Hernández, G.-S. Liu, R. C. B. Wong, M. T. Ryan, D. J. Hausenloy, B. E. Kemp, J. S. Oakhill, B. G. Drew, A. Pébay and S. Y. Lim, Mitochondrial Fission Protein Drp1 Inhibition Promotes Cardiac Mesodermal Differentiation of Human Pluripotent Stem Cells, Cell Death Discovery, 2018, 4, 39, DOI:10.1038/s41420-018-0042-9.
- C. Vantaggiato, M. Castelli, M. Giovarelli, G. Orso, M. T. Bassi, E. Clementi and C. De Palma, The Fine Tuning of Drp1-Dependent Mitochondrial Remodeling and Autophagy Controls Neuronal Differentiation, Front. Cell. Neurosci., 2019, 13, 120, DOI:10.1038/s41420-018-0042-9.
- Y.-Y. Chiang, S.-L. Chen, Y.-T. Hsiao, C.-H. Huang, T.-Y. Lin, I.-P. Chiang, W.-H. Hsu and K.-C. Chow, Nuclear Expression of Dynamin-Related Protein 1 in Lung Adenocarcinomas, Mod. Pathol., 2009, 22, 1139–1150 CrossRef CAS PubMed.
- J. A. Kashatus, A. Nascimento, L. J. Myers, A. Sher, F. L. Byrne, K. L. Hoehn, C. M. Counter and D. F. Kashatus, Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth, Mol. Cell, 2015, 57, 537–552 CrossRef CAS PubMed.
- J. Cai, J. Wang, Y. Huang, H. Wu, T. Xia, J. Xiao, X. Chen, H. Li, Y. Qiu, Y. Wang, T. Wang, H. Xia, Q. Zhang and A. P. Xiang, ERK/Drp1-Dependent Mitochondrial Fission Is Involved in the MSC-Induced Drug Resistance of T-Cell Acute Lymphoblastic Leukemia Cells, Cell Death Dis., 2016, 7, e2459, DOI:10.1038/cddis.2016.370.
- K. Zhou, H.-Y. Yang, P.-Y. Tang, W. Liu, Y.-J. Luo, B. Lv, J. Yin, T. Jiang, J. Chen, W.-H. Cai and J. Fan, Mitochondrial Division Inhibitor 1 Protects Cortical Neurons from Excitotoxicity: A Mechanistic Pathway, Neural Regener. Res., 2018, 13, 1552–1560 CrossRef.
- C. Kingnate, K. Charoenkwan, S. Kumfu, N. Chattipakorn and S. C. Chattipakorn, Possible Roles of Mitochondrial Dynamics and the Effects of Pharmacological Interventions in Chemoresistant Ovarian Cancer, EBioMedicine, 2018, 34, 256–266 CrossRef.
- Z. Zhu, Z. Wang, C. Zhang, Y. Wang, H. Zhang, Z. Gan, Z. Guo and X. Wang, Mitochondrion-Targeted Platinum Complexes Suppressing Lung Cancer through Multiple Pathways Involving Energy Metabolism, Chem. Sci., 2019, 10, 3089–3095 RSC.
- L. Gibellini, S. De Biasi, M. Nasi, A. Iannone, A. Cossarizza and M. Pinti, Mitochondrial Proteases as Emerging Pharmacological Targets, Curr. Pharm. Des., 2016, 22, 2679–2688 CrossRef CAS PubMed.
- M. Pinti, L. Gibellini, Y. Liu, S. Xu, B. Lu and A. Cossarizza, Mitochondrial Lon Protease at the Crossroads of Oxidative Stress, Ageing and Cancer, Cell. Mol. Life Sci., 2015, 72, 4807–4824 CrossRef CAS PubMed.
- B. Lu, Mitochondrial Lon Protease and Cancer, Adv. Exp. Med. Biol., 2017, 1038, 173–182 CrossRef CAS PubMed.
- V. Jovaisaite, L. Mouchiroud and J. Auwerx, The Mitochondrial Unfolded Protein Response, a Conserved Stress Response Pathway with Implications in Health and Disease, J. Exp. Biol., 2014, 217, 137–143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mt00187e |
| This journal is © The Royal Society of Chemistry 2020 |