
The role of metal ion binding in the antioxidant mechanisms of reduced and oxidized glutathione in metal-mediated oxidative DNA damage
Elias O. U.
Eteshola
 a,
Devin A.
Haupt
b,
Stephen I.
Koos
c,
Lee A.
Siemer
d and
Daniel L.
Morris
Jr
*c
a,
Devin A.
Haupt
b,
Stephen I.
Koos
c,
Lee A.
Siemer
d and
Daniel L.
Morris
Jr
*c
aDepartment of Pharmacology & Cancer Biology/Department of Surgery, Duke University, Durham, NC 27710, USA
bCornerstone Controls, Indianapolis, IN 46278, USA
cDepartment of Chemistry and Biochemistry, Rose-Hulman Institute of Technology, Terre Haute, IN 47803, USA. E-mail: morris@rose-hulman.edu; Fax: +1 812 872 6019; Tel: +1 812 877 8314
dCovance Laboratories, Indianapolis, IN 46214, USA
First published on 14th November 2019
Abstract
The antioxidant activity of glutathione in its reduced (GSH) and oxidized (GSSG) forms against metal-mediated oxidative DNA damage was studied by monitoring production of 8-hydroxy-2′-deoxyguanosine (8-OH-dG) from calf-thymus DNA. GSH and GSSG were combined with Fe(II) and Cu(II) before and after addition of DNA to investigate the role of metal coordination in the antioxidant mechanism. The antioxidant behavior of GSH and GSSG was also compared to the known radical scavenger DMSO. GSH and GSSG lower oxidative DNA damage for Fe(II) and Cu(II) reactions. GSH only exhibited appreciable antioxidant behavior when combined with Fe(II) prior to adding DNA, and GSH and GSSG were slightly more effective against Cu(II)-mediated damage when combined with Cu(II) prior to adding DNA. Raman spectra of GSH in the presence of Cu(II) indicate that Cu(II) oxidizes GSH and raises the possibility that the antioxidant activity of GSH against Cu(II) reactions may be attributed to its ability to form GSSG. No evidence of GSH oxidation in the presence of Fe(II) was observed. The fluorescent probe dichlorofluorescein diacetate (DCF-DA) shows that the presence of GSH (for Cu(II) reactions) and GSSG (for Fe(II) and Cu(II) reactions) lowers levels of reactive oxygen species (ROS) in bulk solution. Overall, the results suggest that the mechanism of antioxidant activity for GSH and GSSG against Fe(II) and Cu(II)-mediated oxidative damage involves metal coordination, and isothermal titration calorimetry (ITC) studies of the Cu(II)–GSSG system show an enthalpically favored complexation reaction with an apparent 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry.
:1 stoichiometry.
Significance to metallomicsTransition metals are crucial to the function of biochemical processes in living organisms. However, certain metal ions, including Fe(II) and Cu(II), react with H2O2 (a normal byproduct of aerobic cell metabolism) to produce harmful reactive oxygen species (ROS). Oxidative damage by ROS is associated with numerous diseases and clinical conditions. Glutathione (GSH) and glutathione disulfide (GSSG) appear to function as antioxidants against metal-mediated oxidative damage by coordinating Fe(II) and Cu(II). Understanding the abilities of these and other antioxidants to form coordination complexes and characterizing their behavior has important implications in preventing or controlling disorders related to metal-mediated oxidative damage. |
Introduction
Reactive oxygen species (ROS); such as the hydroxyl radical (˙OH), superoxide radical (˙O2−) and peroxide ions (O22−); are natural byproducts of cellular respiration. While ROS are essential in various cellular processes, including signal transduction cascades and homeostasis, an imbalance in endogenous ROS levels can lead to aberrant and destructive oxidation of lipids and membranes as well as RNA and DNA lesions, strand breaks and cross-links.1–10 Oxidative DNA and RNA damage can interfere with cellular replication, transcription, and lead to genetic mutations and chromosomal instability that can result in tumorigenesis if not repaired by the host DNA damage machinery or aborted via apoptotic fail-safes.11,12Metal ions and protonated amines can help with the charge stabilization required for the large poly-anionic charge distribution of nucleic acids such as DNA.13–15 Metal ions can interact with DNA via coordination with the phosphate groups, sugar oxygen atoms, nitrogenous base atoms, or a combination thereof.13 Some metal ions interact with purine N7 and pyrimidine N3 atoms to perturb the double helical structure of DNA. An important reaction in free radical biology and chemistry is the Fenton reaction in which Fe(II) reacts with H2O2 to form ˙OH.16–19 Other transition metal ions, including Cu(I) and Cu(II), undergo Fenton-like reactions to produce ROS.20–22 This leads to various adducts and lesions in DNA, especially at the G–C sites where coordinated metal ions can more readily generate ROS.13,15,23–25 These radicals can increase the frequency and accumulation of mutagenic events in nuclear and mitochondrial DNA – a hallmark of many solid tumors.2 These mutations can be quantified via measurement of site-specific DNA damage markers, such as 8-hydroxy-2′-deoxyguanosine (8-OH-dG), its tautomeric form 8-oxo-2′-deoxyguanosine (8-oxo-dG) or protein expression profiles of implicated signaling proteins and transcription factors such as AP-1 (activator protein 1) and NF-κB (nuclear factor kappa-light-chain enhancer of activated B cells).2,26–33
A variety of mechanisms exists to combat and intercept chemical carcinogens and mutagens including endogenous antioxidants, which can sequester reactive oxygen species and prevent them from propagating radical chain reactions.34 Antioxidants include a variety of enzymes, thiols, selones, and polyphenols such as superoxide dismutase (SOD), catalase, glutathione, vitamin C (L-ascorbic acid), and vitamin E (α-tocopherol).35,36 Antioxidants have been studied in pharmacology as potential therapeutics for strokes and neurodegenerative diseases such as Alzheimer's and Parkinson's,18,36–38 and their potential roles as anti-cancer therapeutics has started attracting attention, especially with chalcogenic (oxygen family) agents including selenium and sulfur compounds.39–45 Reduced glutathione (γ-L-glutamyl-L-cysteinylglycine or GSH) is a sulfur-containing tripeptide that plays an important role in cellular protection from oxidative damage of lipids, proteins and nucleic acids.37,46,47 A better understanding of how sulfur-based antioxidants, such as glutathione, work to ameliorate the harmful effects of metal-mediated oxidative DNA damage would enhance near-term therapeutic breakthroughs against cancer, inflammatory, and neurodegenerative diseases.29,37
Antioxidants have been viewed traditionally as radical scavengers. However, recent work suggests that many sulfur and selenium compounds exhibit antioxidant activity against metal-mediated production of ROS through a mechanism that involves metal ion coordination.41,48–53 Glutathione in both its reduced (GSH) and oxidized (GSSG) forms is of particular interest. GSH has been shown to lower 8-OH-dG formation for reactions involving Fe(II) and H2O2, and its behavior as an antioxidant was attributed to formation of an Fe(II)–GSH complex with radical scavenging properties.54 GSH is also reported to decrease 8-OH-dG formation in reactions involving Cu(I) and Cu(II).55 GSSG appears to be more effective than GSH at mitigating DNA strand nicking arising from Cu(I)-mediated ROS production, and this was attributed to the ability of GSSG to coordinate Cu(I). However, the role of Fe(II) coordination in the abilities of GSH and GSSG to protect against DNA nicking is unclear.48 Cu(I) and Cu(II) have been reported to form complexes with both GSH and GSSG, and the interrelated redox behavior of reported Cu(I)–GSH and Cu(II)–GSSG complexes has been reviewed.56
The purpose of this study is to gain further insight into the role that metal ion coordination plays in the antioxidant activities of GSH and GSSG. Fe(II) and Cu(II) mediated oxidative DNA damage was initiated by adding H2O2 and probed by measuring production of the site-specific DNA damage marker 8-OH-dG. Reactions in which calf thymus DNA was pre-incubated with metal ions prior to addition of GSH or GSSG (condition 1 reactions) and reactions where the two compounds were pre-incubated with metal ions prior to adding DNA (condition 2 reactions) were performed to determine if the antioxidant activity of GSH and GSSG is limited when metal ions are able to interact with DNA initially (condition 1) as opposed to being free in solution (condition 2). For comparison, condition 1 and 2 reactions were also performed in the presence of the known radical scavenger DMSO. Standard redox potentials indicate that oxidation of GSH to GSSG by Cu(II) and Fe(III) (produced by air oxidation of Fe(II)) is favorable, and this raises the possibility that antioxidant activity under these conditions may involve GSSG rather than, or in addition to, GSH. Conversion of GSH to GSSG in the presence of Cu(II) and Fe(II) was probed using Raman spectroscopy by monitoring the intensity of the 2586 cm−1 thiol band. Dichlorofluorescein diacetate (DCF-DA) is a commonly used probe for ROS production in cells, and it was used in this study to determine if decreases in the site-specific oxidative DNA damage marker 8-OH-dG are associated with lower overall ROS production or if a more site-specific protective effect is responsible. Lastly, isothermal titration calorimetry (ITC) was used to study the redox-stable Cu(II)/GSSG system and gain thermodynamic data on formation of a Cu(II)–GSSG coordination complex.
Experimental
Materials and methods
Calf thymus DNA (sodium salt, type 1 fibers), Catalase (bovine, crystalline suspension in H2O/0.1% thymol, 21600 units per mg protein), P1 nuclease, alkaline phosphatase (bovine, 500 DEA units per mg protein) and DCF-DA (2′,7′-dichlorofluorescin diacetate) were purchased from Sigma Aldrich. Tris (free base) was obtained from J. T. Baker. Imidazole, glutathione (GSH, reduced form, 98%) and glutathione disulfide (GSSG, oxidized form, 95%) were obtained from Acros Organics. DMSO was purchased from BDH, Fe(NH4)2(SO4)2·6H2O (FAS) was obtained from Fisher and CuSO4·5H2O was from Alfa Aesar. All reagents were used without further purification, and all solutions were prepared using ultrapure water.
Oxidative DNA damage studies
The protocol for performing the DNA oxidation reactions in the presence of metal ions has been published previously.57 Reaction mixtures contained 0.5 mg mL−1 calf thymus DNA in 120 mM NaCl and 20 mM Tris (pH = 7.0). For condition 1 reactions metal ions were added to the DNA solutions to achieve a final concentration of 25 μM, and the solutions were allowed to pre-incubate at 37 °C for 30 min. A metal-mediated oxidative DNA damage study involving 6 μM Cu(I) and 2 μM Fe(II) has been reported previously in which the concentrations of GSH and GSSG ranged from 1 μM up to 10 mM. Maximum DNA damage inhibition was achieved with concentrations ranging from 10 μM up to 10 mM, depending on the metal ion and antioxidant.48 In the present study with 25 μM Fe(II) and Cu(II), a 2:1 ratio of GSH or GSSG to metal ion provided a detectable protective effect without reaching maximum DNA damage inhibition. However, DMSO had to be present at a concentration of 250 mM to provide a comparable protective effect.47 Therefore, antioxidants were added to the DNA reactions to achieve final concentrations of 50 μM for GSH and GSSG and 250 mM for DMSO. For condition 2 reactions the antioxidants were combined with metals and allowed to pre-incubate at 37 °C for 30 min before adding DNA. Addition of H2O2 (final concentration of 35 mM) initiated the oxidative damage reactions, and the reactions mixtures were incubated for 1 h at 37 °C. Five replicate samples were prepared for each set of reactions.
Catalase (600 units) was added to the reaction mixtures to decompose excess H2O2 and quench the reactions. Sodium acetate buffer (pH = 5.0) was added to the reaction mixtures (final concentration of 170 mM) followed by heating at 95 °C for 5 min to denature the DNA and rapid cooling in an ice bath. P1 nuclease (1 unit) was added, and the reactions were incubated overnight at 37 °C. The reactions were adjusted to pH of 8.0 using Tris buffer (final concentration of 60 mM) followed by addition of alkaline phosphatase (55 units) and incubated for 8–24 h at 37 °C. Reaction mixtures were frozen prior to HPLC analysis.
Reversed-phase high-pressure liquid chromatography (HPLC) was used to monitor the degree of oxidative DNA damage (in the form of the modified base 8-OH-dG). The reaction mixtures were filtered using Whatman® Mini-UniprepTM 0.2 μm syringeless, self-filtering propylene autosampler vials. All chromatographic separations were performed using a Shimadzu VP Series liquid chromatograph (LC-10AT dual pump, FCV-10AL quaternary low-pressure solvent mixer, SIL-20A HT autosampler, and SPD-10A dual channel variable wavelength absorption detector at 254 and 297 nm). An ESA Coulochem III ECD at a potential of 280 mV was used to provide on-line electrochemical detection of the 8-OH-dG DNA damage marker. The column used in this study was a Beckman CoulterTM Ultrasphere octadecylsilane (ODS) (45 × 4.6 mm, 5.0 μm particle diameter) with an ODS guard cartridge (4.0 mm × 3.0 mm). Injection volumes were 50 μL with a mobile phase flowrate of 1.0 mL min−1 and a 24.5 min solvent program. The initial mobile phase composition was 100% water for 30 seconds to focus the sample at the head of the column followed by a step gradient to 85 mM ammonium acetate containing 3% acetonitrile. After 7.5 min the acetonitrile component was increased to 100% linearly over a 5 min time period and held at 100% acetonitrile for 4.5 min. Shimadzu VP Series EZStart software version 7.4 was used for peak integration. The mole ratio of 8-OH-dG to unmodified dG in each individual DNA digest was calculated. Computing mole ratios for each individual reaction has the advantage of normalizing for any differences in digestion efficiency among the various trials. This ratio was set to 100% damage for Fe(II) and Cu(II) reactions in the absence of sulfur compounds (positive control), and the mole ratios of 8-OH-dG to unmodified dG in the reactions containing sulfur compounds were divided by the 100% damage ratios to calculate a percent damage value for these reactions. The average ± standard deviation of the 8-OH-dG:unmodified dG mole ratio of five independently prepared samples for each set of reaction conditions was used to calculate percent damage values unless otherwise noted. An F test was employed to determine if the variances between condition 1 and 2 reactions was equal, and a two-tailed t-test at a 95% confidence level was applied to determine if differences between the conditions were significant.
GSH oxidation studies
Raman spectroscopy was used to study conversion of GSH to GSSG in the presence of Fe(II) and Cu(II) by monitoring the change in intensity of the 2586 cm−1 thiol band. Raman spectra were acquired using a Kaiser RamanRxn1 spectrometer with a 400 mW 785 nm laser source. Fe(II) and Cu (II) were added to GSH solutions to achieve final concentrations of 12 mM GSH and 2 mM metal ion in 100 mM Tris buffer (pH = 7.0). Spectra were acquired 20 min after the metal ion was added to the GSH solution and at 30 min intervals afterward. The integration time was 60 s, and five spectra were averaged for each solution at each time interval. The solutions were maintained at room temperature during the experiment.DCF-DA studies
DCF-DA (2′,7′-dichlorofluorescin diacetate) is an accepted probe for detecting intracellular ROS.58,59 DCF-DA is nonfluorescent, but upon oxidation by ROS it is converted to fluorescent DCF. It was used in this study as a probe for ROS generated in Fe(II) and Cu(II)-mediated oxidative DNA damage reactions according to a previously published procedure.57 Condition 1 reactions were prepared by combining DNA and metals (at the same concentrations as the oxidative DNA damage studies) and incubating at 37 °C for 30 min before the addition of the sulfur antioxidants, DCF-DA and H2O2. Condition 2 reactions involved combining metal ions and the sulfur antioxidants and incubating at 37 °C for 30 min followed by addition of DNA, DCF-DA and H2O2. The antioxidant concentrations were 500 mM for DMSO and 50 μM for GSH and GSSG. A stock solution of DCF-DA in DMSO (5.0 mg mL−1) was diluted by a factor of two with 0.02 M NaOH prior to each experiment and added to the reactions to achieve a final DFC-DA concentration of 5 μg mL−1. The H2O2 concentration in the reaction mixtures was 0.88 mM. After addition of H2O2, reactions were allowed to proceed for 1 h at 37 °C before adding 600 units of catalase to decompose any remaining H2O2. Positive controls were reactions between the metal ions and H2O2 in the presence of DNA and DCF-DA. Samples were prepared in triplicate for each set of reactions. All solutions were diluted by a factor of 10 with 20 mM Tris buffer (pH 7.0) containing 120 mL NaCl, and fluorescence was measured immediately using a PerkinElmer LS50B spectrofluorometer (λex = 503 nm, λem = 512 nm, spectral bandwidth = 2.5 nm).Cu(II)/GSSG ITC studies
ITC studies of the Cu(II)/GSSG system were carried out using a TA Instruments Nano ITC Low Volume Isothermal Titration Calorimeter with a gold cell of active volume of 175 μL and TA Instruments ITC Run® acquisition software. Data was analyzed using TA Instruments NanoAnalyze® software. Titrations were performed at 25 °C at a stir rate of 350 rpm. Titrant was added in 20 steps (each separated by 5 min) using 2.5 μL increments from a 50 μL syringe. The sample cell contained 300 μL of 0.4 mM GSSG prepared in either 100 mM Tris or 25 mM imidazole buffer (pH = 7.4 and ionic strength (μ) = 0.1). Identifying an appropriate buffer for ITC measurements involving bioinorganic systems can be challenging because of metal–buffer interactions that may lead to precipitate formation and metal–buffer complexes stronger than the desired metal–ligand complex. HEPES is a useful biological buffer at pH 7.4, but it forms a precipitate with Cu(II) at the concentrations used in this study. Therefore, the present study was limited to Tris (the buffer used for the oxidative DNA damage studies) and imidazole buffers. A pH of 7.4 was chosen for the ITC studies so that comparisons with literature values can be made readily. The 50 μL syringe (buret) was filled with 3.2 mM CuSO4 in the same buffer as the GSSG solution. The reference cell contained 350 μL of ultrapure water. All solutions, including the water for the reference cell were degassed by applying a vacuum of 413 mm Hg for 10 min. When ITC is used to study binding reactions involving metal ions the thermodynamics of metal–solution interactions (including metal ion binding with the buffer) must be accounted for. This information is often not available in tables of standard thermodynamic data and must be determined experimentally by performing a titration reaction for which the binding constant between the metal and ligand is known so that the metal–buffer interaction can be determined. The Cu(II)/EDTA system, for which thermodynamic data is readily available, was used for this purpose. For these studies the sample cell was filled with 300 μL of a 0.46 mM Na2EDTA solution prepared in either 100 mM Tris or 25 mM imidazole buffer at pH 7.4 and 0.1 ionic strength. The 50 μL syringe (buret) was filled with 3.2 mM CuSO4 in the corresponding buffer. At least three replicate titrations were performed in each buffer for the GSH, GSSG and EDTA systems. Heat of dilution for the addition of Cu(II) solution was accounted for by titrating Cu(II) solution into 300 μL of each buffer under conditions identical to the other titrations.Results and discussion
Oxidative DNA damage
The 8-OH-dG:unmodified dG mole ratios for the Fe(II) and Cu(II) reactions in the absence (positive control) and presence of the antioxidants GSH, GSSG and DMSO are reported in Table 1 along with the corresponding percent damage values. The concentrations and conditions used in this study yielded anywhere from 3 to 9 nmol of unmodified dG and 0.1 to 0.8 nmol of 8-OH-dG, depending on the specific metal and reaction type. Fig. 1 shows the percent damage levels for reactions involving Fe(II) (left panel) in the presence of GSH, GSSG and DMSO under conditions 1 and 2, and the results for the Cu(II) reactions are shown in the right panel.
:nmol unmodified dG ratio is reported for each set of Fe(II) and Cu(II) reactions in the absence (positive control) and presence of the antioxidants GSH, GSSG and DMSO. The reported ratios are the average nmol 8-OH-dG:nmol unmodified dG ratios calculated for each individual DNA digest (five replicates unless otherwise noted) in a given set (positive control, condition 1 and condition 2). Percent damage levels were calculated by dividing the average nmol 8-OH-dG:nmol unmodified dG ratios of the digests containing antioxidants by the average mole ratio of the positive control (i.e. equating the mole ratio of the positive control to 100% damage). (S↓) indicates a statistically significant smaller percent damage level when comparing condition 1 and 2 reactions for a given metal ion and antioxidant. An F test was employed to determine if the variances between condition 1 and 2 reactions were equal, and a two-tailed t-test at a 95% confidence level was applied to determine if differences between the conditions were significant
| Antioxidant | Fe(II) reactions | nmol 8-OH-dG/nmol unmodified dG | Percent damage |
|---|---|---|---|
| a Average of four trials. b Average of three trials. | |||
| GSH | Positive control | 0.0189 ± 0.0031a | 100 |
| Condition 1 | 0.0170 ± 0.0023a | 90.1 ± 19.3 | |
| Condition 2 | 0.0130 ± 0.0021a | 68.9 ± 16.1 | |
| GSSG | Positive control | 0.0676 ± 0.0061a | 100 |
| Condition 1 | 0.0280 ± 0.0074b | 41.5 ± 11.6 | |
| Condition 2 | 0.0261 ± 0.0045 | 38.6 ± 7.5 | |
| DMSO | Positive control | 0.0519 ± 0.0047 | 100 |
| Condition 1 | 0.0330 ± 0.0010a | 63.5 ± 6.1 | |
| Condition 2 | 0.0344 ± 0.0022 | 66.3 ± 7.3 | |
| Antioxidant | Cu(II) reactions | nmol 8-OH-dG/nmol unmodified dG | Percent damage |
|---|---|---|---|
| GSH | Positive control | 0.126 ± 0.006 | 100 |
| Condition 1 | 0.0854 ± 0.0039 | 67.7 ± 4.3 | |
| Condition 2 | 0.0720 ± 0.0022 | 57.1 ± 3.1 (S↓) | |
| GSSG | Positive control | 0.141 ± 0.008 | 100 |
| Condition 1 | 0.0559 ± 0.0046 | 39.6 ± 3.9 | |
| Condition 2 | 0.0480 ± 0.0029 | 34.0 ± 2.8 (S↓) | |
| DMSO | Positive control | 0.137 ± 0.006a | 100 |
| Condition 1 | 0.105 ± 0.004 | 76.5 ± 4.3 | |
| Condition 2 | 0.103 ± 0.003a | 75.2 ± 4.0 |
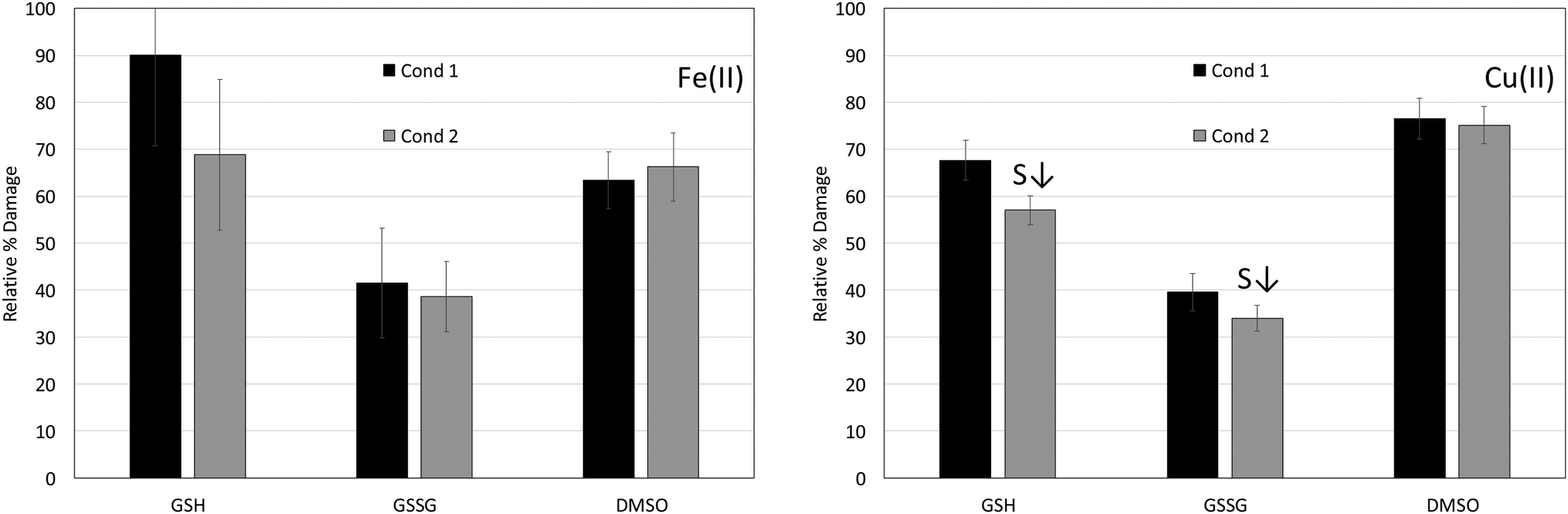 | ||
| Fig. 1 Relative percent damage levels (from Table 1) for Fe(II) and Cu(II) condition 1 and 2 reactions in the presence of GSH, GSSG and DMSO. Table 1 provides details on the number of replicate trials for each set of reactions and describes how the percent damage levels were calculated. (S↓) indicates a statistically significant smaller percent damage level when comparing condition 1 and 2 reactions. An F test was employed to determine if the variances between condition 1 and 2 reactions were equal, and a two-tailed t-test at a 95% confidence level was applied to determine if differences between the conditions were significant. | ||
Fe(II) reactions
The results in Table 1 and Fig. 1 (left panel) show that GSH lowers the percent damage level of Fe(II)-mediated reactions to 68.9 ± 16.1% for condition 2 reactions, but it is virtually ineffective at decreasing oxidative damage for condition 1 reactions (90.1 ± 19.3% damage). Percent damage levels for reactions in the presence of GSSG are 41.5 ± 11.6% for condition 1 reactions and 38.6 ± 7.5 for condition 2 reactions, and DMSO lowers the damage levels to 63.5 ± 6.1% and 66.3 ± 7.3% for condition 1 and 2 reactions, respectively. GSSG is the most effective antioxidant for the Fe(II) reactions under both conditions 1 and 2, and neither GSSG nor DMSO exhibit a statistically significant difference in percent damage levels between conditions 1 and 2. The results for the Fe(II) reactions are summarized in Table 1.GSH has been shown to protect DNA from oxidative damage when a solution containing Fe(II) and GSH was added to DNA and H2O2. Hydroxyl radical (˙OH) and thiyl radical were detected in the presence of GSH indicating that GSH does not inhibit ˙OH formation, and it was suggested that the protective effect of GSH is due to an Fe(II)–GSH complex that is capable of ˙OH scavenging.48,54
GSH only exhibits a measurable protective effect for the Fe(II) condition 2 reactions in which Fe(II) is allowed to interact with GSH before DNA is added. Lower percent damage levels for condition 2 reactions suggest an affinity between Fe(II) and GSH that is hindered when metal ions are allowed to interact with DNA prior to GSH addition. Due to the short lifetime of radical species, the effectiveness of a radical scavenger (like DMSO) is limited, and high concentrations are required to exhibit noticeable antioxidant behavior.60,61 However, the percent damage levels for the condition 2 GSH reactions are comparable to those in the presence of DMSO even though DMSO is present at a much larger concentration (250 mM compared to 50 μM for GSH). The percent damage levels in the presence of DMSO also do not depend on the order in which metal ions and DNA are combined (condition 1 or condition 2), which is expected for an antioxidant that functions by a radical scavenging mechanism rather than metal ion binding. These results suggest that the antioxidant mechanism of Fe(II) in the presence of GSH cannot be attributed solely to a radical scavenging mechanism and may involve metal ion coordination.
The lack of a significant difference in percent damage levels between conditions 1 and 2 in the presence of GSSG could be interpreted to mean that GSSG functions as a radical scavenger in the Fe(II) reactions. However, GSSG inhibits production of the site-specific oxidative damage marker 8-OH-dG significantly more than DMSO (despite the lower concentration of GSSG (25 μM) relative to DMSO (250 mM)). This also indicates that GSSG is exhibiting more than radical scavenging activity against Fe(II)-mediated production of ROS, supporting metal ion coordination.
Cu(II) reactions
The results for Cu(II) in Table 1 and Fig. 1 (right panel) show percent damage levels of 67.7 ± 4.3% and 57.1 ± 3.1% when GSH is present under conditions 1 and 2, respectively. The difference in condition 1 and 2 damage levels is statistically significant. The percent damage levels when GSSG is present under conditions 1 and 2 exhibit a borderline significant difference (t = 2.6 with t critical = 2.3 for a two-tail test assuming equal variances) with 39.6 ± 3.9% damage for condition 1 and 34.0 ± 2.8% damage for condition 2. Consistent with its radical scavenging behaviour, DMSO shows no dependence on the order in which it is combined with metal ions and DNA with 76.5 ± 4.3% damage for condition 1 reactions and 75.2 ± 4.0% damage for condition 2 reactions. Similar to the Fe(II) results, GSSG is also the most effective antioxidant for the Cu(II) reactions. However, GSH exhibits lower percent damage levels than DMSO. Copper-peroxide complexes or singlet oxygen have been proposed as the primary ROS produced from Cu-mediated reactions, and while DMSO is an effective ˙OH scavenger it is not as effective at quenching other ROS.62,63 However, DMSO is present at a high concentration (250 mM) in these reactions and still exhibits some antioxidant activity in the Cu(II) reactions. The percent damage levels for the reactions involving Cu(II) in the presence of GSH, GSSG and DMSO are presented in Table 1.GSH has been shown to lower 8-OH-dG production for reactions involving Cu(II) when present in a 3:1 excess over the metal ion, and this was attributed to reduction of Cu (II) by GSH to form a Cu(I)–GSH complex that stabilized Cu(I) and prevented it from forming ROS. GSH was also effective against oxidative damage in the presence of Cu(I) and H2O2 until the H2O2 concentration was increased to the point where it oxidized GSH stoichiometrically (1:2 ratio of H2O2:GSH). When the H2O2 concentration was increased to the point where it oxidized the available GSH, Cu(I) and H2O2 reacted to produce ROS.55 A Cu(II)–GSSG complex has been reported, characterized spectroscopically and shown to react directly with H2O2 to produce molecular oxygen with subsequent regeneration of Cu(II)–GSSG,64 and the antioxidant behavior of GSSG against Cu(I) mediated damage has been attributed to a mechanism involving coordination of Cu with GSSG.48 The 2:1 GSH:Cu(II) ratio and large excess of H2O2 used in the present study do not map directly with previous work, but the lower percent damage levels observed when GSH is added to Cu(II) prior to DNA (condition 2) and the fact that GSH is more effective at lowering percent damage than the radical scavenger DMSO (despite the large concentration difference) are consistent with an antioxidant mechanism that involves Cu(II) coordination. GSSG also shows slightly lower percent damage levels when added to Cu(II) prior to DNA (condition 2), and the damage levels are substantially lower than those with DMSO. These results also indicate an antioxidant mechanism that involves Cu(II) coordination. The differences between condition 1 and 2 percent damage levels are small, albeit statistically significant, and suggest that GSH and GSSG have only a slight affinity for Cu(II) (small formation constant) when combined prior to adding DNA.
GSH oxidation by H2O2 and metal ions
GSH is oxidized by H2O2 (even in the absence of glutathione peroxidase (GPX)). The kinetics and mechanism of this reaction have been studied.65 In the absence of GPX the reaction between GSH and H2O2 is reported to involve an initial fast decay of H2O2 (50%) that is attributed to formation of a compound between H2O2 and GSH. The second part of this process is reported take up to 6 h to remove the remaining H2O2 when H2O2:GSH = 1.66 A high concentration of H2O2 (35 mM) was employed in the present study (giving a H2O2:GSH ratio of 700) so that it is able to permeate the DNA structure and react with DNA-bound metal ions. Depending on the kinetics of GSH oxidation compared to those of the metal/H2O2 reaction that forms ROS, it is possible that much of the antioxidant activity observed for GSH is actually due to GSSG that results from oxidation of GSH by the large excess of H2O2. However, if antioxidant activity was solely dependent on oxidation of GSH to GSSG by H2O2, differences in percent damage levels between condition 1 and 2 reactions would not be anticipated. The increased effectiveness of GSH in the Cu(II) condition 2 reactions suggest that other factors may contribute.
An aspect of GSH (the reduced form of glutathione) that must be considered in order to understand its behavior as an antioxidant against metal-mediated oxidative damage is its ability to be oxidized by the metal ions used in this study. The standard reduction potential of the GSSG/GSH couple is reported to be in the range of −160 to −270 mV vs. SHE at pH 7.0.67 Reduction of Fe(III) to Fe(II) occurs at a standard potential of 770 mV vs. SHE, and the standard reduction potential of Cu(II) to Cu(I) is 153 mV vs. SHE. These standard potentials are favorable for GSH to reduce Fe(III) (formed by air oxidation of Fe(II) and/or reaction with H2O2) and Cu(II) to form GSSG, and this raises the possibility that the increased effectiveness of GSH in condition 2 reactions (in which the metal ions and GSH are pre-incubated for 30 min) is due to GSH being converted to GSSG prior to H2O2 addition.
In the presence of excess GSH (1:3 mole ratio of Cu(II):GSH or greater) Cu(II) is reportedly reduced to Cu(I), and Cu(I) is coordinated by GSH to form the Cu(I)–(GSH)2 complex. This complex has been shown to react with molecular oxygen in a reversible reaction to produce superoxide anions followed by regeneration of molecular oxygen and reduced Cu(I)–(GSH)2.68,69 However, dismutation of superoxide anion to form H2O2 and Cu(II)–GSSG is favored at low pH values (6.8), long preincubation times (30 min) and high temperatures (37 °C).70 With the exception of the 1:3 mole ratio of Cu(II):GSH, these conditions are very similar to the current study.
NMR spectrometry has been used previously to show the oxidation of GSH by Cu(II). A white precipitate was formed upon addition of 0.5 equivalents of Cu(II) to GSH in D2O, and the NMR spectrum of the resultant solution indicated conversion to GSSG.71 Raman spectroscopy was employed in the present study to probe metal-mediated oxidation of GSH to GSSG by monitoring the decrease in the intensity of the 2586 cm−1 band that corresponds to the thiol (–S–H) group in GSH. Monitoring GSH oxidation using Raman spectroscopy instead of NMR allows the use of lower GSH concentrations (12 mM) that are closer to physiological intracellular concentrations (1 to 6 mM).72 Raman spectroscopy can also be used to probe GSH oxidation in the presence of Fe(II)/Fe(III), which cannot be studied using NMR due to the paramagnetic character of Fe.
Raman spectra showing the 2586 cm−1 thiol band of 12 mM GSH solutions at varying times after addition of Cu(II) and Fe(II) (2 mM) are presented in Fig. 2. The intensity of the 2586 cm−1 thiol band in a solution containing only GSH in Tris buffer does not change, indicating that air oxidation of GSH to GSSG is not significant during the time frame of the experiment (Fig. 2a). The intensity of the 2586 cm−1 thiol band in the presence of Cu(II) decreases noticeably in the first 20 min after metal addition and stays about the same at 50 min after addition (Fig. 2b). While the Cu(II) and GSH concentrations (2 mM and 12 mM, respectively) and temperature (room temperature) in the Raman study are different than those in the oxidative damage studies (25 μM metal and 50 μM GSH at 37 °C), these results support the possibility that GSH could be converted to GSSG in the presence of Cu(II), especially during the 30 min pre-incubation at 37 °C under condition 2. The intensity of the 2586 cm−1 thiol band did not change perceptibly in the GSH solution up to 70 min after addition of Fe(II) (Fig. 2c). These results indicate that GSH is not oxidized to GSSG significantly in the presence of Fe(II) during the time frame of the oxidative damage studies.
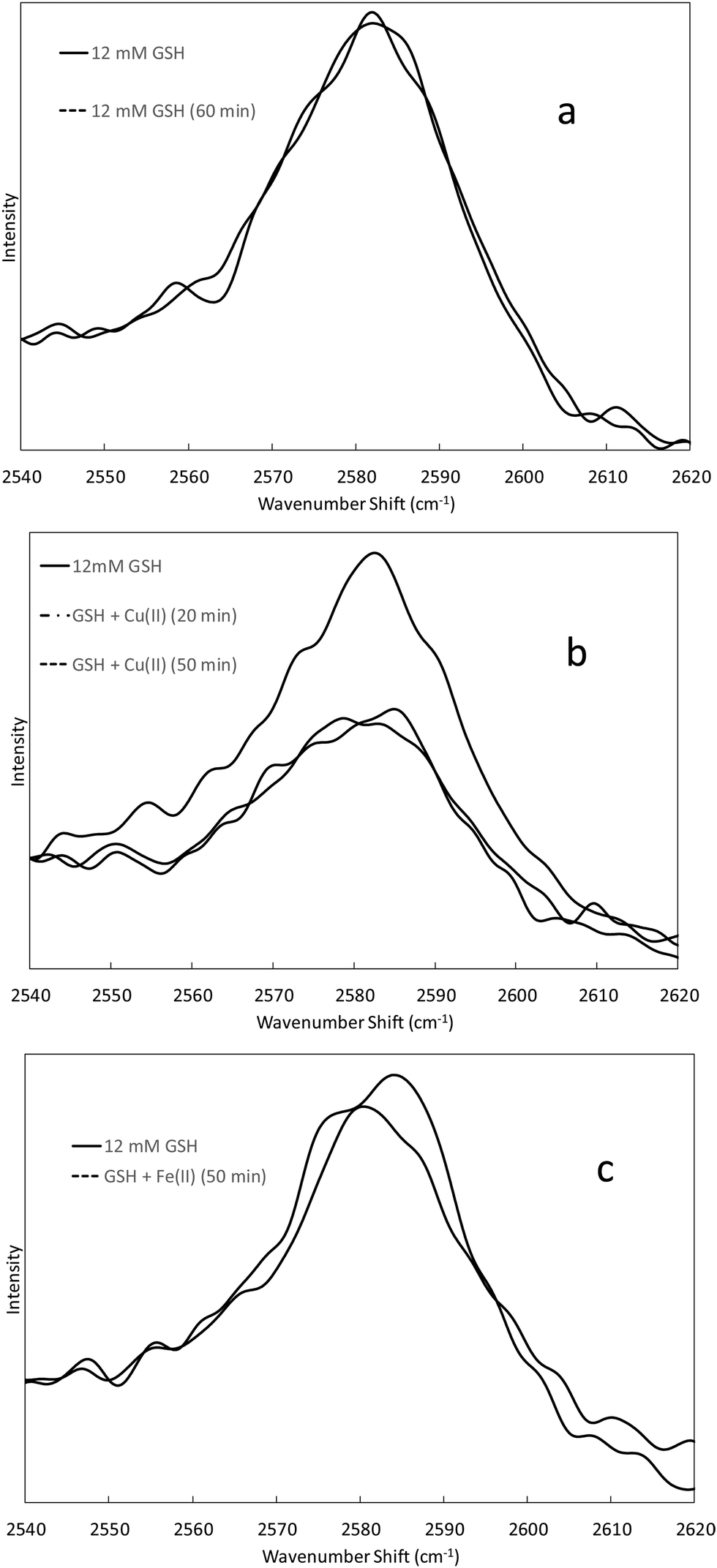 | ||
| Fig. 2 Raman spectra of the 2586 cm−1 thiol band of GSH at various times. Panel (a) corresponds to 12 mM GSH in 100 mM Tris at pH 7.0. Panel (b) corresponds to 12 mM GSH in 100 mM Tris at pH 7.0 before and after addition of Cu(II) (2 mM). Panel (c) corresponds to 12 mM GSH in 100 mM Tris at pH 7.0 before and after addition of Fe(II) (2 mM). | ||
 | ||
| Fig. 3 DCF fluorescence intensities from condition 1 and 2 Fe(II) and Cu(II) reactions with H2O2 in the presence of GSH, GSSG and DMSO. The average ± standard deviation of three independently prepared samples for each set of reactions is reported. The “Metal” reaction corresponds to a positive control in which the metal and H2O2 were combined in the absence of any antioxidant. | ||
DCF-DA fluorescence
Fig. 3 shows relative DCF fluorescence intensities (RFI) observed in Fe(II) and Cu(II) reactions in the presence of GSH, GSSG and DMSO for condition 1 and 2 DNA reactions. The Fe(II) reactions exhibit lower overall fluorescence intensities compared to the Cu(II) reactions. This may be due in part to air oxidation of Fe(II) to Fe(III), which will lower the concentration of Fe(II) available for reaction with H2O2. Redox cycling between Cu(I) and Cu(II) can allow ROS production until the H2O2 is depleted. The presence of GSH increases RFI in the condition 1 and 2 Fe(II) reactions above the Fe(II) + H2O2 positive control (metal + H2O2). However, GSH lowers RFI when present in the reactions between Cu(II) and H2O2. GSSG lowers RFI significantly for both the Fe(II) and Cu(II) reactions compared to the positive metal control reactions, and DMSO lowers RFI for the Fe(II) reactions to levels similar to those of GSSG. DMSO was not as effective at lowering RFI for the Cu(II) reactions, but its presence still achieved RFI's similar to GSH. Values of RFI for the reactions are presented in Table 2. There is no statistically significant difference in the fluorescence intensities between the condition 1 and 2 reactions.| Metal ion\antioxidant | Metal | GSH | GSSG | DMSO | |||
|---|---|---|---|---|---|---|---|
| Cond. 1 | Cond. 2 | Cond. 1 | Cond. 2 | Cond. 1 | Cond. 2 | ||
| Fe(II) | 156.2 ± 9.4 | 176.7 ± 6.3 | 183.3 ± 8.2 | 85.4 ± 4.1 | 80.0 ± 10.8 | 71.9 ± 7.2 | 76.4 ± 8.5 |
| Cu(II) | 535.0 ± 21.7 | 481.3 ± 10.5 | 462.6 ± 22.1 | 320.3 ± 8.3 | 299.5 ± 31.7 | 465.1 ± 12.3 | 475.0 ± 13.6 |
When comparing the DCF fluorescence results with percent damage levels from the site-specific oxidative damage studies (Fig. 1 and Table 1), GSH decreases percent damage levels for site-specific damage (8-OH-dG formation) for condition 2 Fe(II) reactions, but DCF fluorescence indicates that GSH increases ROS production in bulk solution above that of the Fe(II) metal positive control. In the Cu(II) reactions GSH provides lower percent damage levels than DMSO for site-specific oxidative damage involving Cu(II), but DCF fluorescence indicates similar levels of ROS in bulk solution for these two antioxidants. GSSG was the most effective antioxidant in the site-specific oxidative damage studies, and its presence results in some of the lowest fluorescence intensities observed among the Fe(II) and Cu(II) reactions. At a high enough concentration, a radical scavenger, like DMSO, is expected to be effective at quenching ROS in bulk solution, and GSSG lowers RFI to the same levels as that of DMSO in the Fe(II) reactions. However, DMSO does not lower RFI to the same levels as GSSG in the Cu(II) reactions. This is consistent with the fact that the reaction between Cu(II) and H2O2 is reported to produce copper-peroxide complexes or singlet oxygen while DMSO is more effective as a ˙OH scavenger.62,63 Since the reaction between Fe(II) and H2O2 produces ˙OH, DMSO lowers the RFI for the Fe(II) reactions much more effectively than for the Cu(II) reactions.
Comparing the effectiveness of GSH, GSSG and DMSO in the oxidative damage and DCF fluorescence results, GSH appears to be more effective at minimizing site-specific oxidative damage than lowering ROS levels in bulk solution for the Fe(II) and Cu(II) reactions. However, the DCF results for GSSG map well with the trend observed in the oxidative damage studies and demonstrate that the presence of GSSG lowers site-specific oxidative damage and ROS in bulk solution. This is consistent with formation of a coordination complex with GSSG that suppresses the abilities of Fe(II) and Cu(II) to react with H2O2. Again, it is important to consider the concentrations of GSH and GSSG compared to DMSO when assessing their abilities to lower ROS production. GSH and GSSG are present in the DCF-DA study at a concentration of 50 μM while the DMSO concentration is 500 mM. Given the significant concentration difference, the abilities of GSH and GSSG to lower RFI's similar to or below those achieved from DMSO are not consistent with a radical scavenging mechanism. Unlike the oxidative damage studies, the DCF fluorescence results show no significant differences between condition 1 and 2 reactions, indicating that the order in which metal ions, antioxidants and DNA are combined does not affect the population of metal ions in bulk solution significantly enough to see a difference in RFI's. This is consistent with a small formation constant for any metal-antioxidant complex that is formed.
ITC studies of Cu(II) + GSSG
ITC can yield valuable thermodynamic information about chemical reactions (e.g. ΔH, ΔS, ΔG, and equilibrium constants) as well as reaction stoichiometry. However, significant time is required for the calorimeter and solutions to reach thermal equilibrium prior to the start of a titration, and sufficient time must be allowed for the system to re-establish equilibrium after each incremental addition of titrant. This makes ITC studies difficult for chemical systems that are not stable in solution or oxidized readily by air. It is also difficult to interpret ITC results when several different reactions take place upon addition of titrant. Given the effectiveness of GSSG against Cu(II)-mediated oxidative damage, the fact that Fe(II) is readily air oxidized and the possibility that GSH is be oxidized by Cu(II), ITC studies were performed on the redox-stable Cu(II)–GSSG system. Representative titration curves of 3.2 mM Cu(II) into 0.4 mM GSSG in 100 mM Tris and 25 mM imidazole buffers (pH = 7.4, μ = 0.1) are presented in Fig. 4. The ITC data have been corrected for heat of dilution by subtracting a background titration in which Cu(II) was added to the respective buffer with no GSSG present. The background titration data were fit using a linear model, and the Cu(II)/GSSG titration curves were fit using an independent model to determine Cu(II):GSSG stoichiometry (n) and the overall change in enthalpy measured in the system (ΔHITC). The ITC curves indicate a reaction between Cu(II) and GSSG with a 1:1 stoichiometry the system (ΔHITC). The ITC curves indicate a reaction between Cu(II) and GSSG with a 1:1 stoichiometry.
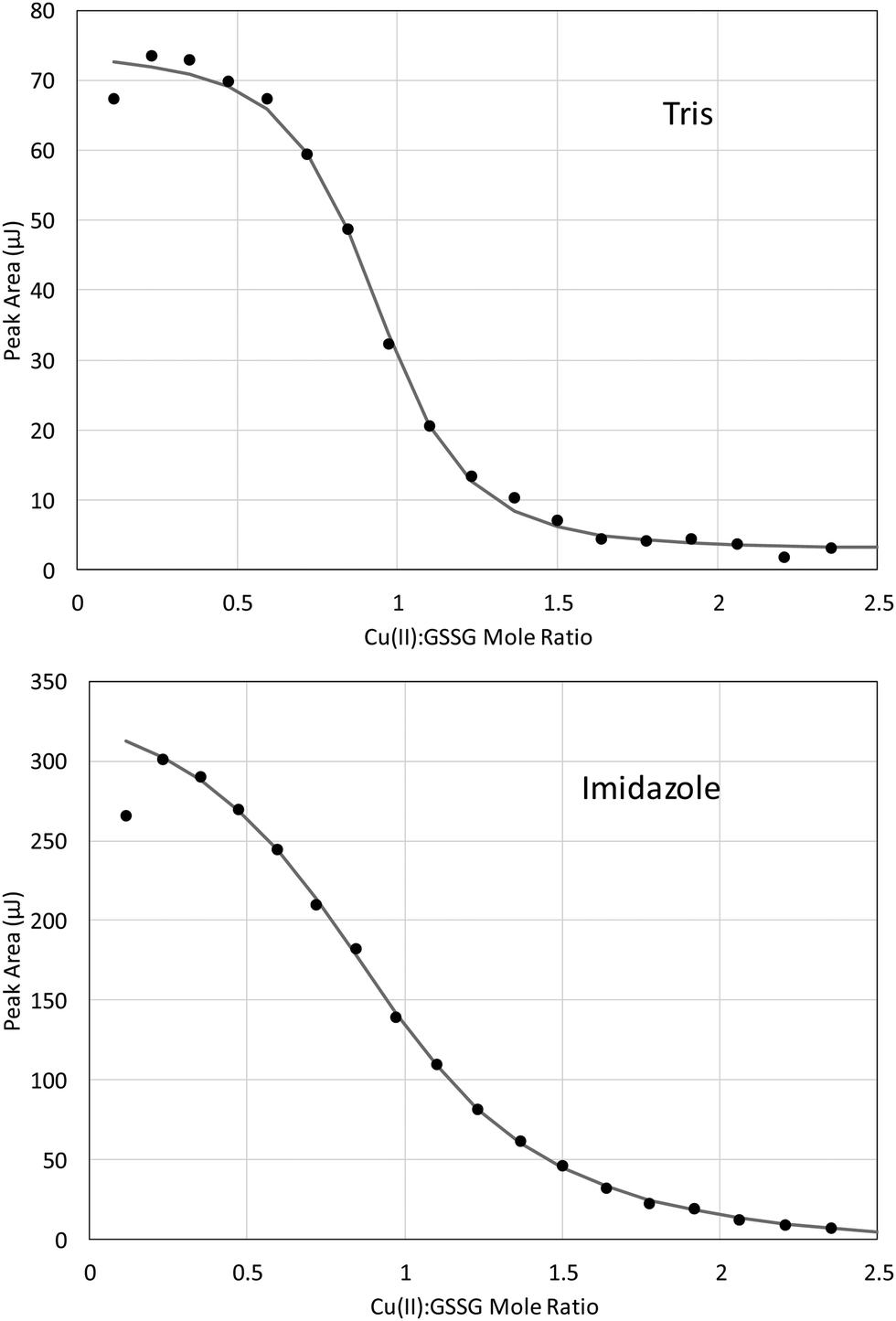 | ||
| Fig. 4 ITC titration curves of 3.2 mM Cu(II) titrated into 0.4 mM GSSG in 100 mM Tris (top panel) and 25 mM imidazole (bottom panel) buffer (pH = 7.4, μ = 0.1) at 25 °C. Background titrations (to correct for heat of dilution) were fit to a linear model and subtracted from the data. The remaining curve was fit to an independent model. | ||
The heat of reaction between Cu(II) and GSSG (ΔHCu–GSSG) can be determined from ΔHITC as long as the stoichiometry of the complex (n) and the number of any protons transferred during the binding event (m) are known. Eqn (1) shows how ΔHITC is related to ΔHCu–GSSG, the enthalpy associated with metal–buffer interaction(s) (ΔHCu–B), the heat of buffer protonation (ΔHH–B) and heat of proton transfer from GSSG (ΔHH–GSSG) upon binding Cu(II).
| ΔHITC = n(ΔHCu–GSSG − ΔHCu–B) + m(ΔHH–B − ΔHH–GSSG) | (1) |
Evaluating ITC data for metal binding interactions has been reviewed.73,74 Values for ΔHH–B and ΔHH–GSSG are available in the literature, but values of ΔHCu–B must often be determined experimentally.75,76 Thermodynamic data for the well-characterized Cu(II)–EDTA reaction is reported in standard tables, so values of ΔHCu–B for the Tris and imidazole systems were determined experimentally by titrating EDTA with Cu(II) in the Tris and imidazole buffers under the same conditions as the Cu(II)/GSSG titrations.77 The various thermodynamic data derived from the experimental data are included in Table 3.
:GSSG stoichiometry. ΔHITC comes from a fit of the raw ITC data (corrected for heat of dilution by subtracting a background titration of Cu(II) into the specified buffer). ΔHCu–B corresponds to the enthalpy change associated with the interaction of Cu(II) with the specified buffer, and these values were determined using Cu(II) into EDTA titrations. ΔHCu–GSSG is the enthalpy change for formation of the Cu(II)–GSSG complex, and it was calculated using eqn XYZ with a = 0.13 and b = 1.95
| Buffer | n | ΔHITC (kJ mol−1) | ΔHCu–B (kJ mol−1) | ΔHCu–GSSG (kJ mol−1) (2 proton transfer) | ΔHCu–GSSG (kJ mol−1) (4 proton transfer) |
|---|---|---|---|---|---|
| a Average of three trials. b Average of four trials. | |||||
| 100 mM Tris | 0.90 ± 0.01a | 8.9 ± 0.3a | −66.0 ± 0.9a | −34.3 ± 0.3a | −10.7 ± 0.6a |
| 25 mM imidazole | 0.94 ± 0.02b | 46.9 ± 1.7b | −56.3 ± 0.2a | −6.4 ± 1.9b | −10.2 ± 1.0b |
GSSG has been reported to coordinate with Cu(II) through the amino and carboxyl groups of the glutamic acid residue in the pH range of 5–10.78 GSSG possesses 6 acidic protons with approximate pKa values of 2.0, 2.6, 3.3, 4.0, 8.7 and 9.6.79,80 At pH 7.4 the amino groups on the glutamic acid residues should be protonated, and GSSG should exist as H2GSSG2− (H2L). Assuming that Cu(II) coordination involves the amino groups, m (the total number of protons transferred during metal binding) in eqn (1) should equal a + 2b where a and b are the fractions of GSSG that exist as HGSSG3− (HL) and H2GSSG2− (H2L), respectively at pH 7.4. Making this substitution and rearranging eqn (1) gives:
| nΔHCu–GSSG = ΔHITC + (a + b)ΔHHL + bΔHH2L − (a + 2b)ΔHH–B + nΔHCu–B | (2) |
Based on pKa5 = 8.7, a = 0.05 and b = 0.95 at pH 7.4, and assuming that two protons are transferred from GSSG upon Cu(II) binding the average ΔHCu–GSSG values are −34.3 ± 0.3 kJ mol−1 in Tris buffer and −6.4 ± 1.9 kJ mol−1 in imidazole buffer (Table 3). This lack of agreement between the average ΔHCu–GSSG values acquired in the two different buffers is concerning and indicates one or more flawed assumptions.
Eqn (1) can be rearranged to give:
| ΔHITC + nΔHCu–B = mΔHH–B − mΔHH–GSSG + nΔHCu–GSSG | (3) |
According to eqn (3), a plot of ΔHITC + nΔHCu–B as a function of ΔHH–B for two or more buffers with significantly different heats of protonation will yield a straight line with a slope m. These data are plotted in Fig. 5 for the Cu(II)–GSSG reaction in Tris and imidazole buffers. The plot has a slope of 4.04 with a standard error of ±0.05, indicating that 4 protons are transferred from GSSG upon coordination with Cu(II). This is contrary to the previous assumption that a total of two protons are transferred from the amino groups of the glutamic acid residues. However, a previous study of the Cu(II)–GSSG complex in solution suggests the possibility of a binuclear complex (at high metal to ligand ratio) of the form Cu(GSSG)2Cu where a Cu(II) ion coordinates through each glutamyl residue on two GSSG molecules.81 Such a complex would exhibit an apparent 1:1 stoichiometry and involve the transfer of 4 protons because each amine group on the two glutamyl residues of each GSSG (4 amine groups total) would lose a proton upon Cu(II) coordination.
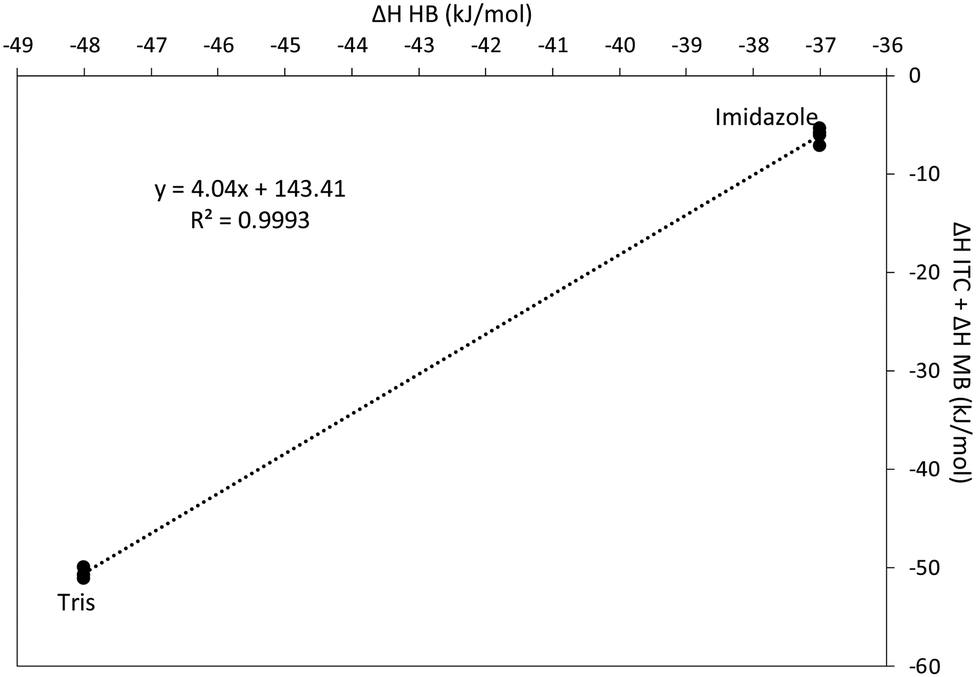 | ||
| Fig. 5 Determination of the number of acidic protons displaced upon coordination of Cu(II) with GSSG. According to eqn (3) a plot of this type generated from two or more buffers with different heats of protonation (Tris and imidazole) will yield a straight line. | ||
Note from eqn (3) that the intercept (Int) of the plot in Fig. 5 can be used to calculate ΔHCu–GSSG. Combining eqn (2) and (3) gives:
| ΔHCu–GSSG = (Int + (a + b)ΔHHL + bΔHH2L)/n | (4) |
Conclusions
The antioxidant activity of reduced and oxidized glutathione was studied and compared to that of the known radical scavenger DMSO to ascertain the role that metal ion coordination and/or radical scavenging play in the abilities of GSH and GSSG to decrease metal-mediated oxidative DNA damage.GSSG was found to be the most effective antioxidant against Fe(II) and Cu(II)-induced damage. GSH and GSSG exhibit equivalent or lower percent damage levels than the known radical scavenger DMSO, despite being present at significantly lower concentrations than DMSO, and these results suggest that GSH and GSSG exhibit more than radical scavenging activity in the Fe(II) and Cu(II) reactions. An antioxidant mechanism that involves metal coordination is especially supported in the case of Cu(II) given the lower percent damage levels observed for condition 2 reactions in which Cu(II) is combined with GSH or GSSG prior to adding DNA.
The large excess of H2O2 used in the reactions in this study are capable of oxidizing GSH to GSSG, but this does not explain the lower percent damage levels for condition 2 Cu(II) reactions (in which metal ions and GSH are preincubated for 30 min prior to adding DNA and H2O2). Decreasing intensity of the thiol Raman band of GSH at 2586 cm−1 in the presence of Cu(II) suggests that GSH is oxidized by Cu(II) to form GSSG and Cu(I). Dissolved oxygen in the solution can oxidize Cu(I) back to Cu(II), which can oxidize additional GSH and/or form a Cu(II)–GSSG coordination complex. Therefore, the antioxidant activity of GSH in the Cu(II) reactions may actually involve a Cu(II)–GSSG coordination complex, the formation of which is favored when Cu(II) and GSH are allowed to preincubate prior to adding H2O2. Oxidation of GSH to GSSG in the presence of Fe(II) was not observed.
The fluorescent probe DCF-DA was employed to determine if decreased site-specific DNA damage correlated with decreased ROS levels in bulk solution for condition 1 and 2 Fe(II) and Cu(II) reactions. GSH lowers site-specific damage for condition 2 Fe(II) reactions, but ROS production in bulk solution is observed to increase above the positive metal control reaction. Site-specific damage is lowered for condition 1 and 2 Cu(II) reactions, and the presence of GSH exhibits a slight decrease in ROS levels compared to the positive metal control. GSSG lowers site-specific damage and ROS levels in bulk solution substantially for Fe(II) and Cu(II) reactions, and DMSO is also very effective at lowering ROS levels in bulk solution for Fe(II) reactions (which produce ˙OH). DMSO is less effective at lowering ROS levels for the Cu(II) reactions, which are believed to produce copper-peroxide complexes or singlet oxygen instead of ˙OH. Considering the relative concentrations of GSH and GSSG compared to DMSO, the fluorescence studies also indicate that the relative effectiveness of GSH and GSSG as antioxidants compared to DMSO is not consistent with a radical scavenging mechanism and also lends support for an antioxidant mechanism involving metal coordination.
Isothermal titration calorimetry (ITC) confirms formation of a Cu(II)–GSSG coordination complex exhibiting apparent 1:1 stoichiometry. However, studies of this system in two separate buffers show that 4 protons are transferred during the complexation reaction, and this could be explained by formation of a binuclear complex (Cu(GSSG)2Cu) where the two glutamyl residues on two GSSG molecules coordinate with two Cu(II) ions. The value of ΔHCu–GSSG was determined to −10.4 ± 0.8 kJ mol−1 and reveals that the complexation reaction is enthalpically favored. ITC experiments are capable of yielding values of formation constants, but determining an accurate value for the Cu(II)–GSSG species requires further, carefully-designed ITC experiments that are optimized for this purpose.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
DAH and SIK were supported by the RSURP/Weaver Summer Research Program. The support of the Department of Chemistry and Biochemistry at Rose-Hulman Institute of Technology (RHIT) is acknowledged along with the help of James “Lou” Johnson and Cyndi Erwin from RHIT.References
- K. J. Davies, Oxidative stress: the paradox of aerobic life, Biochem. Soc. Symp., 1995, 61, 1–31 CrossRef CAS.
- M. Valko, D. Leibfritz, J. Moncol, M. T. Cronin, M. Mazur and J. Telser, Free radicals and antioxidants in normal physiological functions and human disease, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS.
- T. P. Devasagayam, J. C. Tilak, K. K. Boloor, K. S. Sane, S. S. Ghaskadbi and R. D. Lele, Free radicals and antioxidants in human health: current status and future prospects, J. Assoc. Physicians India, 2004, 52, 794–804 CAS.
- J. A. Imlay and S. Linn, DNA damage and oxygen radical toxicity, Science, 1988, 240, 1302–1309 CrossRef CAS.
- B. Halliwell and O. I. Aruoma, DNA and Free Radicals, Ellis Horwood, Chichester, 1993 Search PubMed.
- D. R. Lloyd, D. H. Phillips and P. L. Carmichael, Generation of putative intrastrand cross-links and strand breaks in DNA by transition metal ion-mediated oxygen radical attack, Chem. Res. Toxicol., 1997, 10, 393–400 Search PubMed.
- C. J. Burrows and J. G. Muller, Oxidative nucleobase modifications leading to strand scission, Chem. Rev., 1998, 98, 1109–1151 CrossRef CAS.
- J. Cadet, T. Douki, D. Gasparutto and J. L. Ravanat, Oxidative damage to DNA: formation, measurement and biochemical features, Mutat. Res., 2003, 531, 5–23 CrossRef CAS.
- M. Podhorecka, A. Skladanowski and P. Bozko, H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy, J. Nucleic Acids, 2010, 2010, 920161 Search PubMed.
- B. Halliwell and J. M. C. Gutteridge, Role of free radicals and catalytic metal ions in human disease: an overview, Methods Enzymol., 1990, 186, 1–85 CAS.
- T. Paz-Elizur, D. E. Brenner and Z. Livneh, Interrogating DNA repair in cancer risk assessment, Cancer Epidemiol., Biomarkers Prev., 2005, 14, 1585–1587 CrossRef.
- C. Lengauer, K. W. Kinzler and B. Vogelstein, Genetic instability in colorectal cancers, Nature, 1997, 386, 623–627 CrossRef CAS.
- J. Anastassopoulou, Metal-DNA interactions, J. Mol. Struct., 2003, 651–653, 19–26 CrossRef CAS.
- B. Lippert, Multiplicity of metal ion binding patterns to nucleobases, Coord. Chem. Rev., 2000, 200–202, 487–516 CrossRef CAS.
- D. L. Morris, DNA-bound metal ions: recent developments, Biomol. Concepts, 2014, 5, 397–407 CAS.
- C. Walling, Fenton's reagent revisited, Acc. Chem. Res., 1975, 8, 125–131 CrossRef CAS.
- M. Valko, H. Morris and M. T. D. Cronin, Metals, toxicity and oxidative stress, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef CAS.
- K. Jomova and M. Valko, Advances in metal-induced oxidative stress and human disease, Toxicology, 2011, 283, 65–87 CrossRef CAS PubMed.
- K. Barbusinski, Fenton reaction - Controversy concerning the chemistry, Ecol. Chem. Eng. S, 2009, 16, 347–358 CAS.
- J. L. Sagripanti and K. H. Kraemer, Site-specific oxidative DNA damage at polyguanosines produced by copper plus hydrogen peroxide, J. Biol. Chem., 1989, 264, 1729–1734 CAS.
- D. R. Lloyd and D. H. Phillips, Oxidative DNA damage mediated by copper(II), iron(II) and nickel(II) Fenton reactions: evidence for site-specific mechanisms in the formation of double-strand breaks, 8-hydroxydeoxyguanosine and putative intrastrand cross-links, Mutat. Res., 1999, 424, 23–36 CrossRef CAS.
- A. M. Fleming, J. G. Muller, I. Ji and C. J. Burrows, Characterization of 2'-deoxyguanosine oxidation products observed in the Fenton-like system Cu(II)/H2O2/reductant in nucleoside and oligodeoxynucleotide contexts, Org. Biomol. Chem., 2011, 9, 3338–3348 RSC.
- V. Tereshko, C. J. Wilds, G. Minasov, T. P. Prakash, M. A. Maier, A. Howard, Z. Wawrzak, M. Manoharan and M. Egli, Detection of alkali metal ions in DNA crystals using state-of-the-art X-ray diffraction experiments, Nucleic Acids Res., 2001, 29, 1208–1215 CrossRef CAS.
- H. Rosemeyer, The chemodiversity of purine as a constituent of natural products, Chem. Biodiversity, 2004, 1, 361–401 CrossRef CAS.
- C. Angele-Martinez, C. Goodman and J. Brumaghim, Metal-mediated DNA damage and cell death: mechanisms, detection methods, and cellular consequences, Metallomics, 2014, 1358–1381 RSC.
- M. R. Shigenaga, C. J. Gemeno and B. N. Ames, Urinary 8-hydroxy-2′-deoxyguanosine as a biological marker of in vivo oxidative DNA damage, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9679–9701 CrossRef PubMed.
- J. Cadet, T. Delatour, T. Douki, D. Gasparutto, J. P. Pouget, J. L. Ravanat and S. Sauvaigo, Hydroxyl radicals and DNA base damage, Mutat. Res., 1999, 424, 9–21 CrossRef CAS.
- F. Taddei, H. Hayakawa, M. Bouton, A. Cirinesi, I. Matic, M. Sekiguchi and M. Radman, Counteraction by MutT protein of transcriptional errors caused by oxidative damage, Science, 1997, 278, 128–130 CrossRef CAS.
- M. Valko, C. J. Rhodes, J. Moncola, M. Izakovic and M. Mazura, Free radicals, metals and antioxidants in oxidative stress-induced cancer, Chem.-Biol. Interact., 2006, 160, 1–40 CrossRef CAS.
- M. Ameyar, M. Wisniewska and J. B. Weitzman, A role for AP-1 in apoptosis: the case for and against, Biochimie, 2003, 85, 747–752 CrossRef CAS PubMed.
- T. D. Gilmore, Introduction to NF-kappaB: players, pathways, perspectives, Oncogene, 2006, 25, 6680–6684 CrossRef CAS.
- N. D. Perkins, Integrating cell-signalling pathways with NF-kappaB and IKK function, Nat. Rev. Mol. Cell Biol., 2007, 8, 49–62 CrossRef CAS.
- A. R. Brasier, The NF-kappaB regulatory network, Cardiovasc. Toxicol., 2006, 6, 111–130 CrossRef CAS PubMed.
- R. A. Weinberg, The Biology of Cancer, Norton, W. W. & Company, Inc., New York, 2nd edn, 2014 Search PubMed.
- C. Balsano and A. Alisi, Antioxidant effects of natural bioactive compounds, Curr. Pharm. Des., 2009, 15, 3063–3073 CrossRef CAS.
- H. Sies, Oxidative stress: oxidants and antioxidants, Exp. Physiol., 1997, 82, 291–295 CrossRef CAS.
- J. B. Schulz, J. Lindenau, J. Seyfried and J. Dichgans, Glutathione, oxidative stress and neurodegeneration, Eur. J. Biochem., 2000, 267, 4904–4911 CrossRef CAS PubMed.
- P. G. Shekelle, S. C. Morton, L. K. Jungvig, J. Udani, M. Spar, W. Tu, M. J. Suttorp, I. Coulter, S. J. Newberry and M. Hardy, Effect of supplemental vitamin E for the prevention and treatment of cardiovascular disease, J. Gen. Intern. Med., 2004, 19, 380–389 CrossRef.
- G. F. Combs and W. P. Gray, Chemopreventive agents: selenium, Pharmacol. Ther., 1998, 79, 179–192 CrossRef CAS.
- L. Letavayova, V. Vickova and J. Brozmanova, Selenium: from cancer prevention to DNA damage, Toxicology, 2006, 227, 1–14 CrossRef CAS PubMed.
- R. R. Ramoutar and J. L. Brumaghim, Antioxidant and anticancer properties and mechanisms of inorganic selenium, oxo-sulfur, and oxo-selenium compounds, Cell Biochem. Biophys., 2010, 58, 1–23 CrossRef CAS.
- Z. Zhu and W. Jiang, Selenium in prevention of cancer: evidence and mechanism, Biomed. Res. Trace Elem., 2008, 19, 282–289 CAS.
- J. Brozmanova, D. Manikova, V. Vlckova and M. Chovanec, Selenium: a double-edged sword for defense and offence in cancer, Arch. Toxicol., 2010, 84, 919–938 CrossRef CAS.
- L. Jiang, K. H. Yang, J. H. Tian, Q. L. Guan, N. Yao, N. Cao, D. H. Mi, J. Wu, B. Ma and S. H. Yang, Efficacy of antioxidant vitamins and selenium supplement in prostate cancer prevention: a meta-analysis of randomized controlled trials, Nutr. Cancer, 2010, 62, 719–727 CrossRef CAS.
- M. Vinceti, G. Dennert, C. M. Crespi, M. Zwahlen, M. Brinkman, M. P. Zeegers, M. Horneber, R. D'Amico and C. Del Giovane, Selenium for preventing cancer, Cochrane Database Syst. Rev., 2014, CD005195 Search PubMed.
- D. Gérard-Monnier and J. Chaudiere, Metabolism and antioxidant function of glutathione, Pathol. Biol., 1996, 44, 77–85 Search PubMed.
- E. O. U. Eteshola, MS thesis, Rose-Hulman Institute of Technology, 2015.
- E. E. Battin and J. L. Brumaghim, Metal specificity in DNA damage prevention by sulfur antioxidants, J. Inorg. Biochem., 2008, 102, 2036–2042 CrossRef CAS PubMed.
- E. E. Battin and J. L. Brumaghim, Antioxidant activity of sulfur and selenium: A review of reactive oxygen species scavenging, glutathione peroxidase, and metal-binding antioxidant mechanisms, Cell Biochem. Biophys., 2009, 55, 1–23 CrossRef CAS PubMed.
- R. R. Ramoutar and J. L. Brumaghim, Investigating the antioxidant properties of oxo-sulfur compounds on metal-mediated DNA damage, Main Group Chem., 2007, 6, 143–153 CrossRef CAS.
- E. E. Battin, M. T. Zimmerman, R. R. Ramoutar, C. E. Quarles and J. L. Brumaghim, Preventing metal-mediated oxidative DNA damage with selenium compounds, Metallomics, 2011, 3, 503–512 RSC.
- E. E. Battin, N. R. Perron and J. L. Brumaghim, The central role of metal coordination in selenium antioxidant activity, Inorg. Chem., 2006, 45, 499–501 CrossRef CAS PubMed.
- M. T. Zimmerman, C. A. Bayse, R. R. Ramoutar and J. L. Brumaghim, Sulfur and selenium antioxidants: challenging radical scavenging mechanisms and developing structure-activity relationships based on metal binding, J. Inorg. Biochem., 2015, 145, 30–40 CrossRef CAS.
- N. Spear and S. D. Aust, Effects of glutathione on Fenton reagent-dependent radical production and DNA oxidation, Arch. Biochem. Biophys., 1995, 324, 111–116 CrossRef CAS.
- N. Spear and S. D. Aust, Hydroxylation of deoxyguanosine in DNA by copper and thiols, Arch. Biochem. Biophys., 1995, 317, 142–148 CrossRef CAS.
- M. E. Aliaga, C. López-Alarcón, R. Bridi and H. Speisky, Redox-implications associated with the formation of complexes between copper ions and reduced or oxidized glutathione, J. Inorg. Biochem., 2016, 154, 78–88 CrossRef CAS PubMed.
- W. E. Hart, S. P. Marczak, A. R. Kneller, R. A. French and D. L. Morris, The abilities of selenium dioxide and selenite ion to coordinate DNA-bound metal ions and decrease oxidative DNA damage, J. Inorg. Biochem., 2013, 125, 1–8 CrossRef CAS PubMed.
- B. Halliwell and M. Whiteman, Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean?, Br. J. Pharmacol., 2004, 142, 231–255 CrossRef CAS PubMed.
- M. Karlsson, T. Kurz, Ulf T. Brunk, S. E. Nilsson and C. I. Frennesson, What does the commonly used DCF test for oxidative stress really show?, Biochem. J., 2010, 428, 183–190 CrossRef CAS PubMed.
- J. E. Repine, O. W. Pfenninger, D. W. Talmage, E. M. Berger and D. E. Pettijohn, Dimethyl sulfoxide prevents DNA nicking mediated by ionizing radiation or iron/hydrogen peroxide-generated hydroxyl radical, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 1001 CrossRef CAS.
- Z. W. Yu and P. J. Quinn, Dimethyl sulphoxide: A review of its applications in cell biology, Biosci. Rep., 1994, 14, 259 CrossRef CAS PubMed.
- K. Yamamoto and S. Kawanishi, Hydroxyl free radical is not the main active species in site-specific DNA damage induced by copper(II) ion and hydrogen peroxide, J. Biol. Chem., 1989, 264, 15435–15440 CAS.
- S. Frelon, T. Douki, A. Favier and J. Cadet, Hydroxyl radical is not the main reactive species involved in the degradation of DNA bases by copper in the presence of hydrogen peroxide, Chem. Res. Toxicol., 2003, 16, 191–197 Search PubMed.
- M. E. Aliaga, D. Andrade-Acuña, C. López-Alarcón, C. Sandoval-Acuña and H. Speisky, Cu(II)-disulfide complexes display simultaneous superoxide dismutase- and catalase-like activities, J. Inorg. Biochem., 2013, 129, 119–126 CrossRef CAS PubMed.
- Z. Abedinzadeh, M. Gardes-Albert and C. Ferradini, Kinetic study of the oxidation mechanism of glutathione by hydrogen peroxide in neutral aqueous medium, Can. J. Chem., 1989, 67, 1247–1255 CrossRef CAS.
- J. W. Finley, E. L. Wheeler and S. C. Witt, Oxidation of glutathione by hydrogen peroxide and other oxidizing agents, J. Agric. Food Chem., 1981, 29, 404–407 CrossRef CAS PubMed.
- K. K. Millis, K. H. Weaver and D. L. Rabenstein, Oxidation/reduction potential of glutathione, J. Org. Chem., 1993, 58, 4144–4146 CrossRef CAS.
- H. Speisky, M. Gómez, C. Carrasco-Pozo, E. Pastene, C. Lopez-Alarcón and C. Olea-Azar, Cu(I)-glutathione complex: a potential source of superoxide radicals generation, Bioorg. Med. Chem., 2008, 16, 6568–6574 CrossRef CAS PubMed.
- H. Speisky, M. Gómez, F. Burgos-Bravo, C. López-Alarcón, C. Jullian, C. Olea-Azar and M. E. Aliaga, Generation of superoxide radicals by copper–glutathione complexes: Redox-consequences associated with their interaction with reduced glutathione, Bioorg. Med. Chem., 2009, 17, 1803–1810 CrossRef CAS PubMed.
- M. E. Aliaga, C. Carrasco-Pozo, C. López-Alarcón and H. Speisky, The Cu(I)–glutathione complex: factors affecting its formation and capacity to generate reactive oxygen species, Transition Met. Chem., 2010, 35, 321–329 CrossRef CAS.
- N. Kato, M. Nakamura and T. Uchiyama, 1H NMR studies of the reactions of copper(I) and copper(II) with D-penicillamine and glutathione, J. Inorg. Biochem., 1999, 75, 117–121 CrossRef CAS PubMed.
- F. Tietze, Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues, Anal. Biochem., 1969, 27, 502–522 CrossRef CAS PubMed.
- N. E. Grossoehme, A. M. Spuches and D. E. Wilcox, Application of isothermal titration calorimetry in bioinorganic chemistry, J. Biol. Inorg. Chem., 2010, 15, 1183–1191 CrossRef CAS PubMed.
- R. A. Johnson, O. M. Manley, A. M. Spuches and N. E. Grossoehme, Dissecting ITC data of metal ions binding to ligands and proteins, Biochim. Biophys. Acta, 2016, 1860, 892–901 CrossRef CAS PubMed.
- R. Goldberg, N. Kishore and R. Lennen, Thermodynamic Quantities for the Ionization Reactions of Buffers, J. Phys. Chem. Ref. Data, 2002, 31, 231–370 CrossRef CAS.
- P. Crea, C. De Stefano, M. Kambarami, F. J. Millero and V. K. Sharma, Effect of Ionic Strength and Temperature on the Protonation of Oxidized Glutathione, J. Solution Chem., 2008, 37, 1245–1259 CrossRef CAS.
- R. M. Smith and A. E. Martell, Critical Stability Constants, Springer, Boston, MA, vol. 6, suppl. 2, 1989 Search PubMed.
- G. Formicka-Kozłowska, H. Kozłowski and B. Jezowska-Trzebiatowska, Metal-glutathione interaction in aqueous solution. Nickel(II), cobalt(II) and copper(II) complexes with oxidized glutathione, Acta Biochim. Pol., 1979, 26, 239–248 Search PubMed.
- D. Vila-Viçosa, V. H. Teixeira, H. A. Santos and M. Machuqueiro, Conformational study of GSH and GSSG using constant-pH molecular dynamics simulations, J. Phys. Chem. B, 2013, 117, 7507–7517 CrossRef PubMed.
- N. W. Pirie and K. G. Pinhey, The titration curve of glutathione, J. Biol. Chem., 1929, 84, 321–333 CAS.
- J. Z. Pedersen, C. Steinkühler, U. Weser and G. Rotilio, Copper-glutathione complexes under physiological conditions: structures in solution different from the solid state coordination, Biometals, 1996, 9, 3–9 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |