
Vinyl sulfonamide synthesis for irreversible tethering via a novel α-selenoether protection strategy†
Gregory B.
Craven
 a,
Dominic P.
Affron‡
a,
Philip N.
Raymond
a,
David J.
Mann
b and
Alan
Armstrong
*a
a,
Dominic P.
Affron‡
a,
Philip N.
Raymond
a,
David J.
Mann
b and
Alan
Armstrong
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London W12 0BZ, UK. E-mail: a.armstrong@imperial.ac.uk
bDepartment of Life Sciences, Imperial College London, South Kensington Campus, London SW7 2AZ, UK
First published on 13th December 2018
Abstract
Vinyl sulfonamides are valuable electrophiles for targeted protein modification and inhibition. We describe a novel approach to the synthesis of terminal vinyl sulfonamides which uses mild oxidative conditions to induce elimination of an α-selenoether masking group. The method complements traditional synthetic approaches and typically yields vinyl sulfonamides in high purity after aqueous work-up without requiring column chromatography of the final electrophilic product. The methodology is applied to the synthesis of covalent fragments for use in irreversible protein tethering and crucially enables the attachment of diverse fragments to the vinyl sulfonamide warhead via a chemical linker. Using thymidylate synthase as a model system, ethylene glycol is identified as an effective linker for irreversible protein tethering.
Introduction
Protein-reactive small molecules are being widely employed to probe and modulate biological systems.1–3 These covalent probes contain a reactive warhead that modifies the protein and a specificity element, which selectively directs the warhead to the target protein. To facilitate the design of such probes, covalent fragments can be screened against recombinant proteins using mass spectrometry and fluorescence-based tethering methodologies as well as in biochemical assays to identify chemical starting points for ligand optimisation.4–9 Alternatively, covalent fragments can be directly applied to cell-based disease models to identify new protein targets through activity-based protein profiling strategies.10–12 To maximise the efficiency of these approaches, libraries of covalent fragments are required which contain chemical diversity of both the warhead and specificity element. As such, methodologies that enable the synthesis of new covalent fragments are of great importance.The choice of reactive warhead is crucial to the success of any covalent ligand approach and Michael acceptors, such as vinyl sulfonamides,13 vinyl sulfones14 and acrylamides,15 can target cysteine with high specificity. Terminal vinyl sulfonamides form stable protein adducts and are found in covalent inhibitors of multiple protein classes including deubiquitylating enzymes (USP7 inhibitor),16 GTPases (KRas(G12C) inhibitor)17 and kinases (ERK2 and BTK inhibitors)18,19 (Fig. 1). Relative to acrylamides, vinyl sulfonamides are more electrophilic and therefore display a higher degree of reactivity towards nucleophilic amino acids, making them desirable warheads for targeting non-catalytic cysteine residues.20 Indeed, it has recently been shown that vinyl sulfonamides can undergo selective reaction with lysine residues via an aza-Michael addition, offering the potential to target a wide range of protein sites.21 Apart from their potential in covalent therapeutics, vinyl sulfonamides have also emerged as attractive reagents for chemical biology applications, being used in the preparation of antibody–drug conjugates (ADCs),22 in disulfide stapling,21 as chemical genetics probes23 and for the specific introduction of fluorophores and enrichment tags into proteins and peptides.24,25
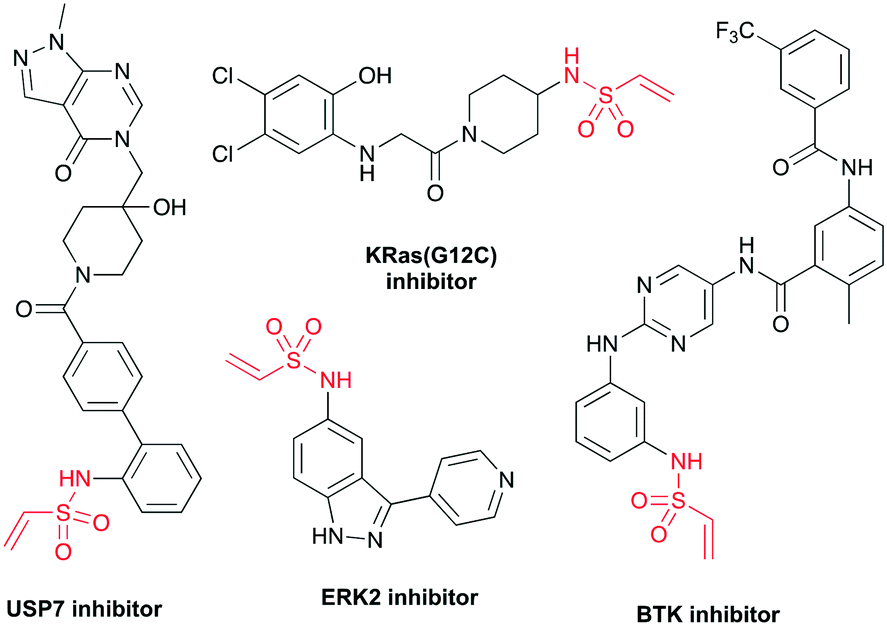 | ||
| Fig. 1 Structures of terminal vinyl sulfonamide containing protein inhibitors. | ||
We have previously reported methodology for synthesising acrylamide fragment libraries26 but the synthesis of terminal vinyl sulfonamides presents distinct challenges. The relatively high electrophilicity of vinyl sulfonamides makes them incompatible with many reaction conditions and nucleophilic functional groups and therefore late stage installation is generally required. However, vinyl sulfonamide formation is often characterised by poor substrate scope and low yields because the vinyl sulfonylation reagents (2-haloethane-1-sulfonyl chloride or vinyl sulfonyl chloride) can undergo extensive side reactions (Scheme 1).27 Unfortunately, the vinyl sulfonamide products themselves are often prone to polymerization during silica gel chromatography which makes the removal of such side-products challenging.28 We therefore investigated an alternative route that would enable vinyl sulfonamide formation under mild conditions and in sufficient purity for use in biological assays without the need for column chromatography. To achieve this, we looked to explore whether vinyl sulfonamides could be masked as α-selenoether ethyl sulfonamides and then be liberated under mild oxidative conditions by in situ syn-elimination of the resultant selenoxide, a strategy which has been successful in acrylamide synthesis.29,30
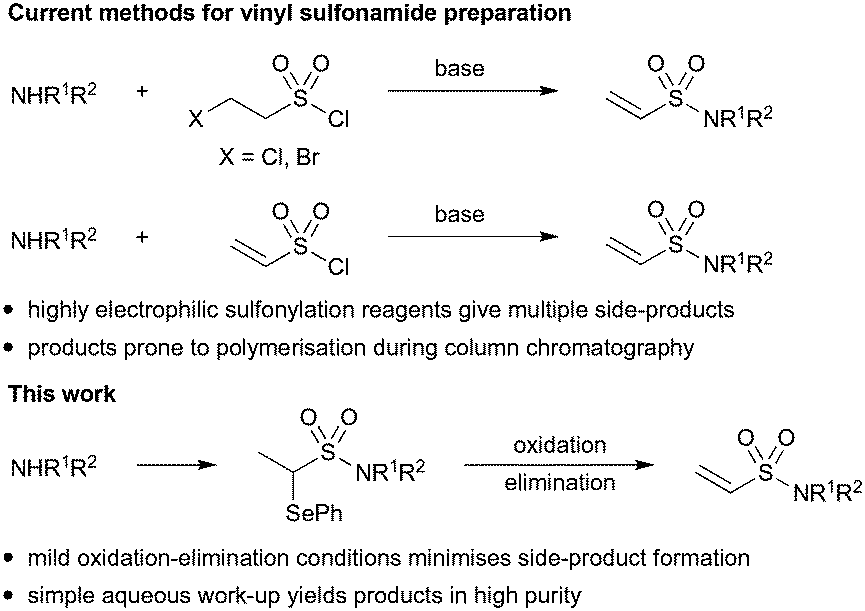 | ||
| Scheme 1 Strategies for the preparation of terminal vinyl sulfonamides. | ||
Results and discussion
Synthesis of vinyl sulfonamides via α-selenoether oxidation
It was anticipated that the most direct route of preparing α-selenoether ethyl sulfonamides precursors would be through direct amine sulfonylation with the proposed sulfonyl chloride reagent 1 (Scheme 2). Unfortunately, all efforts to generate this key reagent from its parent sulfonate salt 2 were unsuccessful: desulfonylation to give alkyl chloride 3 was observed even under cryogenic chlorination conditions, a phenomenon which has previously been documented for similar thioether analogues.31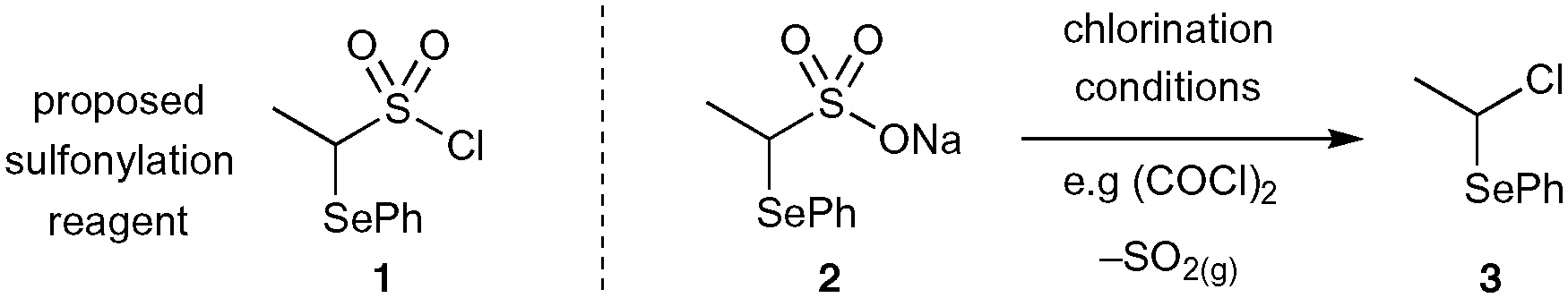 | ||
| Scheme 2 Proposed sulfonylation reagent 1 is inaccessible via chlorination of sulfonate salt 2 due to rapid SO2 elimination. | ||
To circumvent this, a stepwise strategy was implemented in which the amine substrate 4 is first sulfonylated with 1-bromoethane-1-sulfonyl chloride and then the bromide displaced with phenyl selenide (Scheme 3a). The resulting selenoether 6 is stable towards column chromatography or further functionalisation and can be readily converted into the corresponding vinyl sulfonamide 7 by treatment with the mild oxidant sodium metaperiodate. As a proof of concept, 4-aminopiperidine 4a was taken through the reaction sequence (Scheme 3b): sulfonylation gave sulfonamide 5a in high yield which successfully underwent selenide displacement to give selenoether 6a. Pleasingly, oxidation with sodium metaperiodate gave vinyl sulfonamide 7a in excellent purity (<95%) after a simple aqueous work up to remove the water soluble by-products.
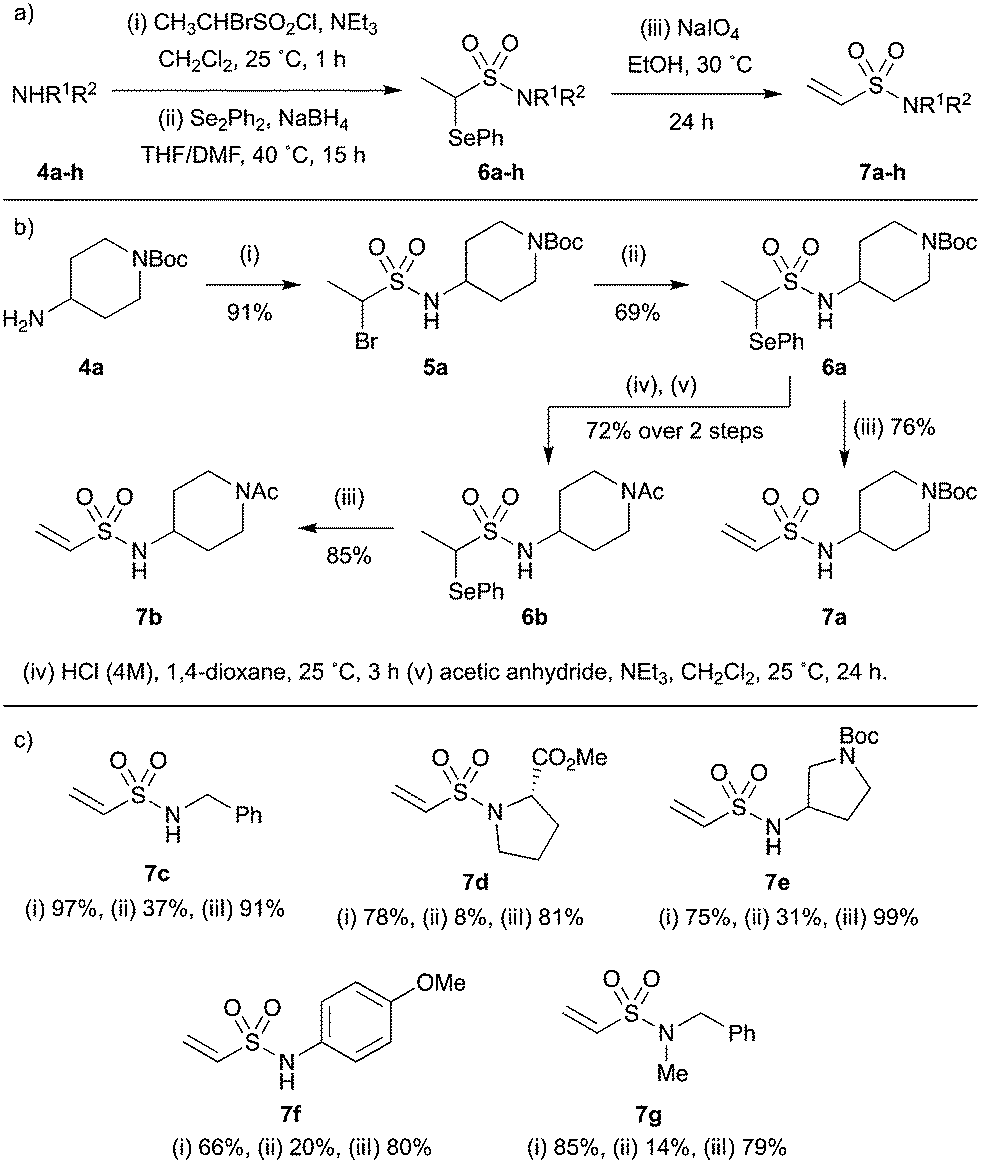 | ||
| Scheme 3 Synthesis of terminal vinyl sulfonamides. (a) General scheme and reaction conditions; (b) proof-of-concept study using 4-aminopiperidine 4a and (c) substrate scope. | ||
To investigate the synthetic utility of the method, attempts were made to further functionalise intermediate 6a before unmasking the vinyl moiety. The selenoether protecting group was found to be compatible with t-butyl carbamate deprotection and acetylation conditions, yielding N-acetyl sulfonamide 6b which was successfully converted into the corresponding vinyl sulfonamide 7b, all in high yields.
Next, the scope of the main reaction sequence was evaluated (Scheme 3c). Unfortunately, low yields were generally observed for the selenide displacement step (ii), where an unwanted proto-debrominated side product was typically observed. However, both the sulfonylation (i) and oxidation (iii) reactions proceeded well with aryl, alkyl and benzyl substituted amines, yielding a diverse collection of terminal vinyl sulfonamides with desirable fragment-like properties (7c–g).
It should be noted that for certain substrates, such as indoline 5h, proto-debromination during the selenide displacement step appeared to dominate the reaction and the desired product (6h) could not be isolated (Scheme 4). This unreliable transformation was expected to limit the synthetic utility of the methodology with regards to library development. However, we postulated that incorporation a chemical linker between the fragment and vinyl sulfonamide functionalities would allow the selenoether protecting group to be installed prior to the fragment, enabling the synthesis of each library member in two robust steps.
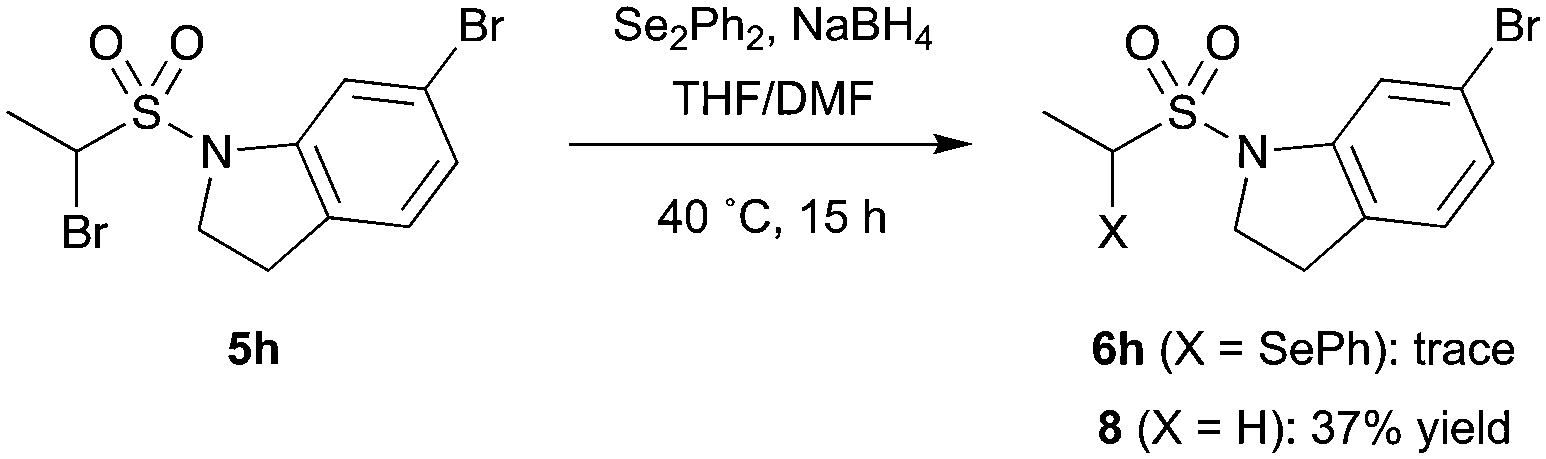 | ||
| Scheme 4 Attempted selenide displacement resulted in proto-debrominated product. | ||
Synthesis of PEG-linked vinyl sulfonamide fragments
Irreversible tethering has emerged as an attractive method for screening irreversible cysteine reactive-fragments against recombinant proteins.32–34 Typically, libraries of electrophilic fragments are incubated with the target protein and the degree of protein modification is monitored by intact-protein mass spectrometry (Fig. 2a). It was anticipated that incorporation of a flexible chemical linker between the vinyl sulfonamide warhead and the fragment could also enable effective irreversible tethering if the linker allows the warhead to adopt a cysteine-reactive conformation in the protein–ligand complex (Fig. 2b). An appropriately designed chemical linker would (1) make the library synthesis more efficient by incorporating the α-selenoether functionality prior to fragment installation, (2) facilitate efficient template-guided reactivity by allowing access to a large number of reactive conformations upon fragment coordination and (3) normalize the reactivity of the vinyl sulfonamide warhead by insulating it from steric and electronic influence of the fragment. Poly(ethylene)glycol (PEG) was selected as a suitable linker because it is flexible, resistant to nonspecific protein binding and confers enhanced aqueous solubility relative to alkyl linkers.35 A carboxylic acid functionalised scaffold was desired so that amine fragments could be easily incorporated by amide bond coupling.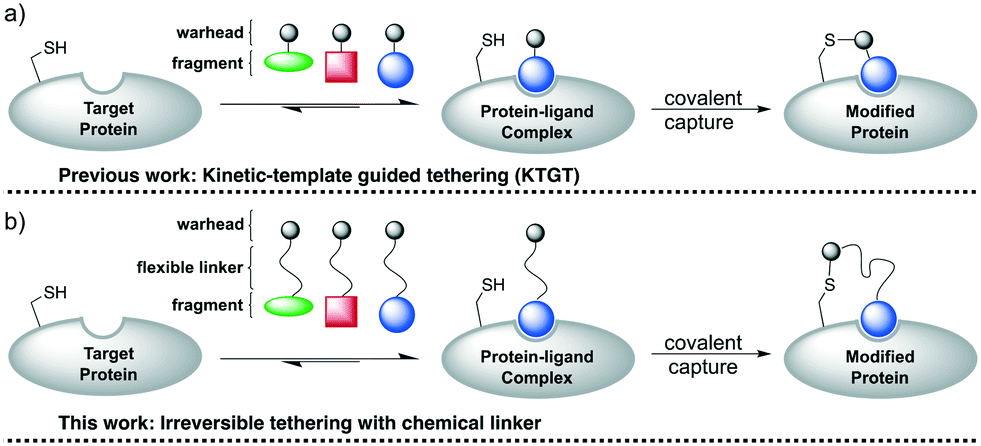 | ||
| Fig. 2 Chemical linker approach for irreversible tethering. | ||
To investigate the effect of linker length, two PEG-linked scaffolds 13 (n = 1) and 14 (n = 2) were examined (Scheme 5). N-Terminal functionalisation of the PEG linkers was achieved by sequential sulfonylation and selenide displacement of amines 9 and 10 to give alcohols 11 and 12 in reasonable yields. Then the O-termini were capped with the versatile carboxylic acid functionality by alkylation of alcohols 11 and 12 with bromoacetic acid in excellent yields.
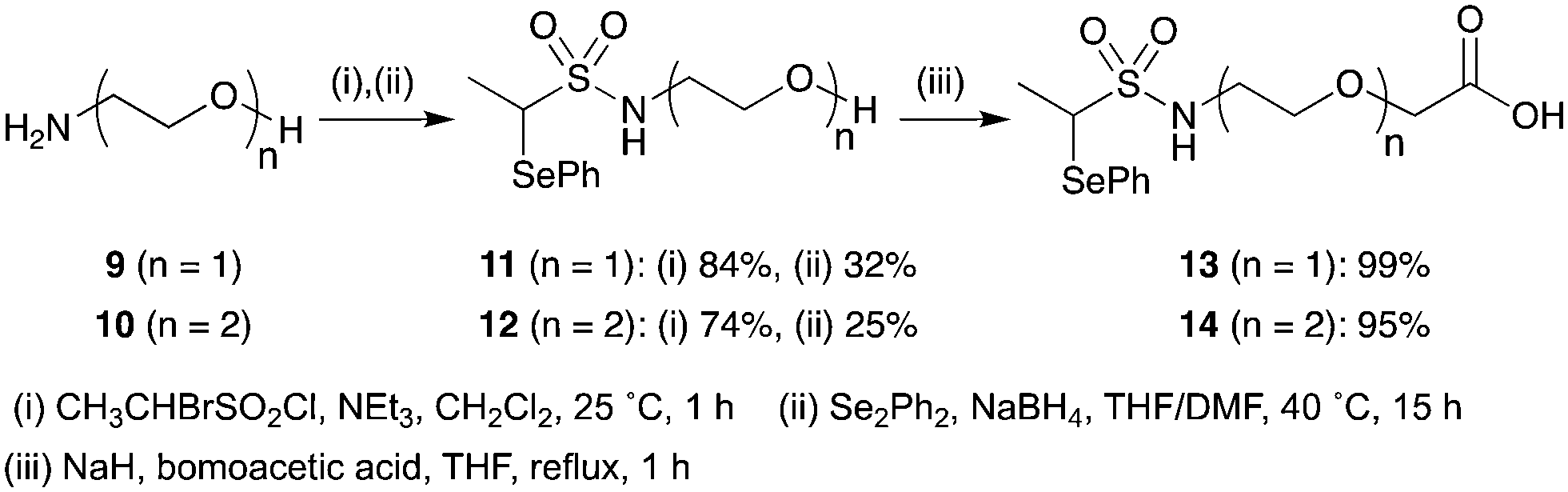 | ||
| Scheme 5 Preparation of carboxylic acid functionalised PEG linkers. | ||
With the key intermediates 13 and 14 in hand, a variety of amine fragments were converted into PEG-linked vinyl sulfonamides using a simple two step procedure. First, amide bond formation was effected using either Lewis acid catalysis with ZrCp2Cl2 (15a–c and 16a–b) or using stoichiometric activation with HATU (16d–f) (Scheme 6). Secondly, oxidation with NaIO4 generated the vinyl sulfonamide, typically in sufficient purity after aqueous work up that chromatography was not required. Pleasingly, the methodology proved successful for both PEG-linked scaffolds across a range of amine substrates, including alkyl and aryl examples. Crucially, by pre-installing the α-selenoether functionality into the linker, this strategy generates each product in two reliable steps.
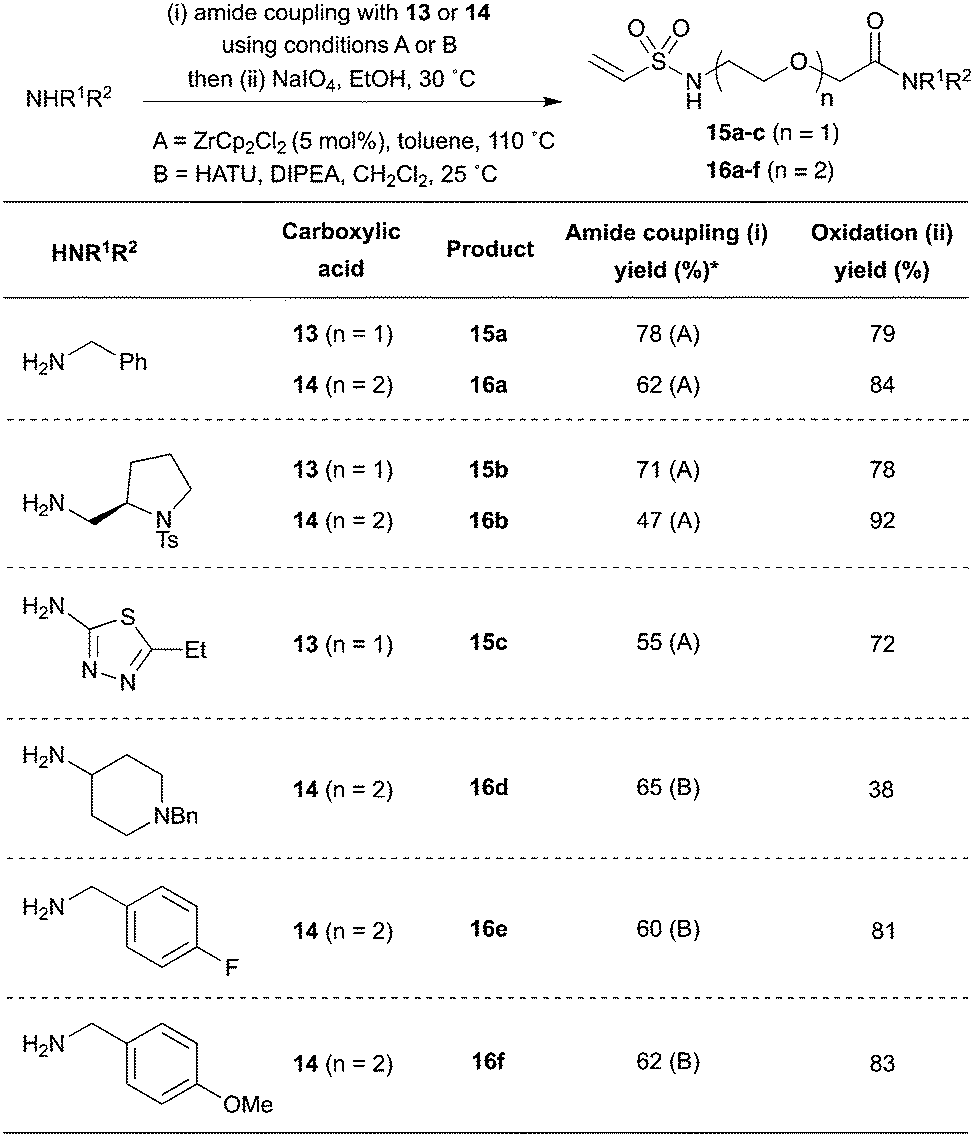 | ||
| Scheme 6 Two step synthesis of PEG-linked vinyl sulfonamides. *Amide coupling by method A or B. | ||
Applying the chemical linker strategy to irreversible tethering
To investigate whether fragment binding can induce kinetic-template guided tethering in the context of these PEG-linked vinyl sulfonamide scaffolds, proof of concept experiments were performed on thymidylate synthase (TS). Fragments derived from N-tosyl D-proline are well characterised binders of TS.36 In previous work, we have demonstrated that the catalytic cysteine of TS reacts selectively with an acrylamide functionalised N-tosyl D-proline fragment in the presence of other acrylamides including benzyl acrylamide.33 Therefore for this study, N-tosyl D-proline linked vinyl sulfonamides 15b and 16b would serve as positive controls while benzyl linked vinyl sulfonamides 15a and 16a as negatives.Therefore thymidylate synthase was incubated separately with binary mixtures of the positive and negative PEG-linked vinyl sulfonamides and the reactions analyzed by mass spectrometry (Fig. 3). After thirty minutes thorough modification of thymidylate synthase had been achieved in both cases, indicated by a low intensity peak for the native protein. In the case of the longer linker (n = 2), the adduct corresponding to labeling by negative control 16a (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 752 Da) had equal intensity to the positive control 16b adduct (30899 Da), which indicates no kinetic template rate acceleration and this modification probably corresponds to background reactivity. Pleasingly, however, for the shorter linker (n = 1) tethering by the positive control 15b occurred at a faster rate than the negative control 15a, demonstrating that significant template-guided rate enhancement was in operation.
752 Da) had equal intensity to the positive control 16b adduct (30899 Da), which indicates no kinetic template rate acceleration and this modification probably corresponds to background reactivity. Pleasingly, however, for the shorter linker (n = 1) tethering by the positive control 15b occurred at a faster rate than the negative control 15a, demonstrating that significant template-guided rate enhancement was in operation.
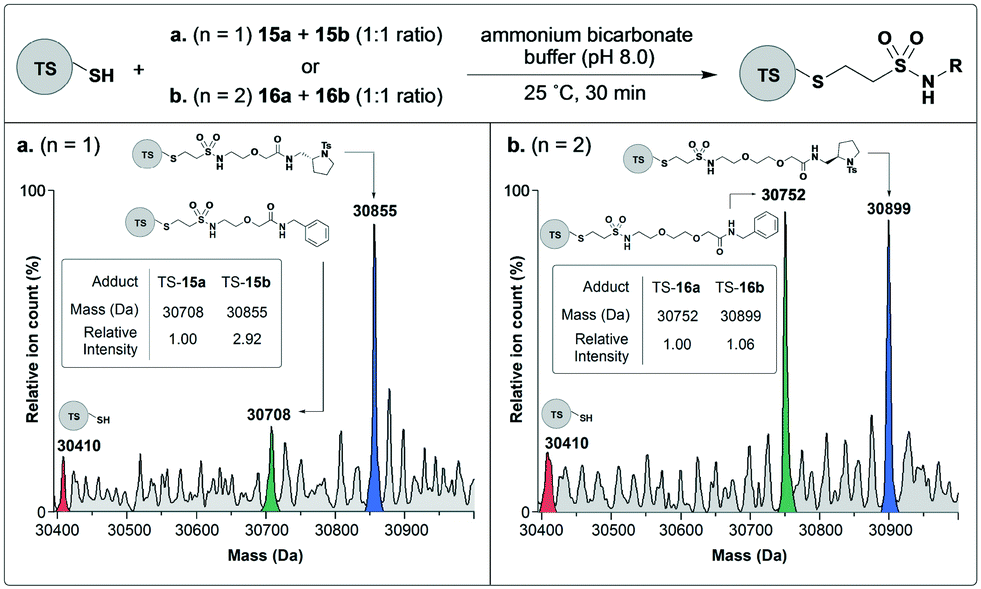 | ||
| Fig. 3 Testing PEG-linked vinyl sulfonamides against TS. TS (10 μM) was incubated with a mixture of positive control and negative control ligands (each at 100 μM) and analyzed by intact protein mass spectrometry after 30 minutes. (a) Testing PEG(n = 1) ligands 15a and 15a (b) testing PEG(n = 2) ligands 16a and 16b. | ||
For the positive controls 15a and 16a, the spacing between the terminal vinyl carbon and the tosyl sulfonamide functionalities are 13.4 Å and 16.0 Å respectively. Analysis of the previously published crystal structure of the N-tosyl D-proline fragment in complex with TS (PDB: 1F4E) reveals that there is only a 7.1 Å spacing between the reactive thiol of Cys146 and the tosyl sulfonamide functionality of the bound fragment and we attribute the observed selectivity for the shorter linker to its closer match to this separation. We hypothesise that the PEG(n = 2) linker is too long to enable an efficient reaction once the fragment has bound into the active site of TS which would account for the lack of positive/negative control selectivity. However, we note that for proteins for which the reactive cysteine residue lies further outside of the binding pocket, such a long scaffold may be required for efficient labelling.
Since the PEG(n = 1) scaffold gave encouraging results in the KTGT assay, a kinetic investigation was carried out to determine whether the PEG(n = 1) linker is effective at normalizing the intrinsic reactivity of the vinyl sulfonamide warhead. When investigating acrylamide reactivity for our previously published study on KTGT, 1,3,4-thiadiazole-based acrylamide 17 was identified as highly reactive and was therefore removed from the screening library.33 It was anticipated that the PEG-linker would insulate the warhead from the electronic properties of the fragment such that vinyl sulfonamides 15a, 15b and 15c would have similar intrinsic reactivities.
In order to determine the intrinsic reactivity of electrophiles towards conjugate thiol addition, an NMR-based rate study was carried out, using a published procedure.32 Accordingly, vinyl sulfonamides 15a, 15b and 15c were each reacted with excess N-acetyl cysteine methyl ester in a deuterated buffer system and the concentration of the electrophile was monitored over time by 1H NMR spectroscopy. As the electrophile is the limiting reagent, the pseudo-first-order rate constant (kobs) was extracted by plotting the natural logarithm of the electrophile concentration over time (Scheme 7 and Fig. S1†).
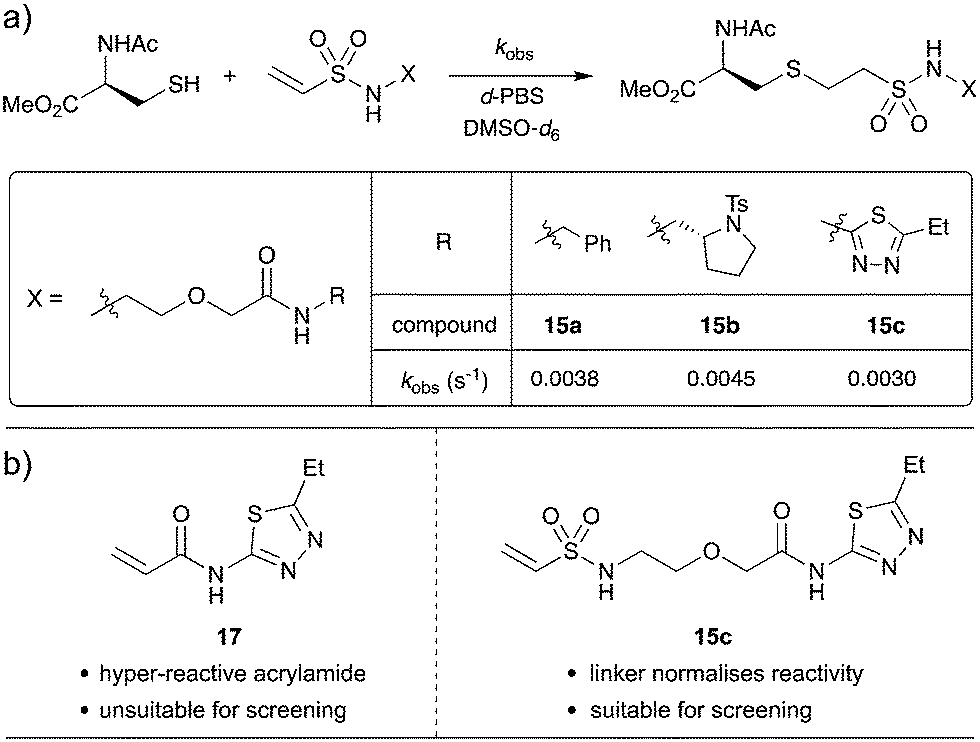 | ||
| Scheme 7 (a) NMR rate study. Vinyl sulfonamides (10 mM) were reacted with N-acetyl cysteine methyl ester (78 mM) and the rate of reaction monitored by 1H NMR spectroscopy. (b) Comparison of unlinked and linker thiadiazole fragments for irreversible tethering. | ||
The effectiveness of the linker is evident as only a 1.5 fold difference in reactivity was observed between least reactive and most reactive fragments, with an average pseudo-first-order rate constant of 3.77 × 10−3 s−1. Indeed, 1,3,4-thiadiazole 15c reacted most slowly of the three electrophiles, which highlights that this linker will allow screening of fragments that were previously thought of as hyper reactive.
Conclusions
Incorporation of a linker into the KTGT construct is necessary to provide a more robust screening assay and is expected to advance this methodology by allowing fragments greater flexibility for protein binding. Herein we have described a novel methodology for the synthesis of terminal vinyl sulfonamide fragments and demonstrated its application to the synthesis of unlinked and linked electrophilic fragments. Ethylene glycol linked vinyl sulfonamides were found to have homogeneous intrinsic reactivity and provide positive/negative selectivity in the KTGT assay. In addition, the linker was able to modulate the reactivity of electrophiles that were previously considered unusable in libraries. The developed synthetic route will allow the creation of large vinyl sulfonamide linked fragment libraries, as incorporation of amine fragments can be achieved in two robust steps.Experimental
Synthesis
All synthetic procedures and characterisation data are found in the ESI.†Protein labelling experiments
Thymidylate synthase (recombinantly expressed, 10 μM) in ammonium bicarbonate (10 mM) and DTT (1 mM) solution was treated with binary mixtures of ligands (15a + 15b) or (16a + 16b) each at 100 μM and incubated at rt. The reactions were analyzed by intact protein ESI mass spectrometry after 30 minutes: to a 25 μL aliquot of the reaction, 0.5% formic acid (50 μL) and MeOH (50 μL) were added and the mixture injected onto a Micromass LCT Premier mass spectrometer at 30 μL min−1. Data was collected in the m/z range 500–2500 in positive ion mode using electrospray ionization and the spectra were deconvoluted with maximum entropy software from Waters. Maximum entropy deconvolutions were performed with a mass step of 1 over a minimum of 4 charge states.NMR rate study
To a 550 μL solution of N-acetyl cysteine methyl ester (94 mM) and internal standard dichloromethane (12 mM) in 4:1 deuterated PBS:DMSO-d6 was added 110 μL of electrophile (60 mM) in DMSO-d6. The resulting solution was analyzed immediately by 1H NMR, capturing spectra every 20 seconds for 5 minutes. All spectra were phase and baseline corrected using MestReNova before analysis. The integration of the vinyl peaks relative to the dichloromethane peak was used to monitor the electrophile concentration. The pseudo-first-order rate constant was calculated from the linear gradient of the natural logarithm of the electrophile concentration over time. Deuterated PBS recipe: 20 mM Na3PO4, 50 mM NaCl in D2O was adjusted to pD 8 with DCl solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Institute of Chemical Biology (Imperial College London), the UK Engineering and Physical Sciences Research Council (Studentship award EP/F500416/1), Biotechnology and Biological Sciences Research Council (Studentship award BB/J014575/1).References
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed
.
- D. S. Johnson, E. Weerapana and B. F. Cravatt, Future Med. Chem., 2010, 2, 949–964 CrossRef CAS PubMed
- R. Schulz, A. Atef, D. Becker, F. Gottschalk, C. Tauber, S. Wagner, C. Arkona, A. A. Abdel-Hafez, H. H. Farag, J. Rademann and G. Wolber, J. Med. Chem., 2018, 61, 1218–1230 CrossRef CAS PubMed
- S. G. Kathman and A. V. Statsyuk, MedChemComm, 2016, 7, 576–585 RSC
- G. B. Craven, D. P. Affron, C. E. Allen, S. Matthies, J. G. Greener, R. M. L. Morgan, E. W. Tate, A. Armstrong and D. J. Mann, Angew. Chem., Int. Ed., 2018, 57, 5257–5261 CrossRef CAS PubMed
- D. Becker, Z. Kaczmarska, C. Arkona, R. Schulz, C. Tauber, G. Wolber, R. Hilgenfeld, M. Coll and J. Rademann, Nat. Commun., 2016, 7, 12761 CrossRef CAS PubMed
- R. M. Miller, V. O. Paavilainen, S. Krishnan, I. M. Serafimova and J. Taunton, J. Am. Chem. Soc., 2013, 135, 5298–5301 CrossRef CAS PubMed
- D. A. Erlanson, J. A. Wells and A. C. Braisted, Annu. Rev. Biophys. Biomol. Struct., 2004, 33, 199–223 CrossRef CAS PubMed
- D. Becker, Z. Kaczmarska, C. Arkona, R. Schulz, C. Tauber, G. Wolber, R. Hilgenfeld, M. Coll and J. Rademann, Nat. Commun., 2016, 7, 12761 CrossRef CAS PubMed
- L. A. Bateman, T. B. Nguyen, A. M. Roberts, D. K. Miyamoto, W.-M. Ku, T. R. Huffman, Y. Petri, M. J. Heslin, C. M. Contreras, C. F. Skibola, J. A. Olzmann and D. K. Nomura, Chem. Commun., 2017, 53, 7234–7237 RSC
- L. Bar-Peled, E. K. Kemper, R. M. Suciu, E. V. Vinogradova, K. M. Backus, B. D. Horning, T. A. Paul, T.-A. Ichu, R. U. Svensson, J. Olucha, M. W. Chang, B. P. Kok, Z. Zhu, N. T. Ihle, M. M. Dix, P. Jiang, M. M. Hayward, E. Saez, R. J. Shaw and B. F. Cravatt, Cell, 2017, 171, 696–709 CrossRef CAS PubMed
- J. L. Counihan, A. L. Wiggenhorn, K. E. Anderson and D. K. Nomura, ACS Chem. Biol., 2018, 13, 1970–1977 CrossRef CAS PubMed
- W. R. Roush, J. Cheng, B. Knapp-Reed, A. Alvarez-Hernandez, J. H. McKerrow, E. Hansell and J. C. Engel, Bioorg. Med. Chem. Lett., 2001, 11, 2759–2762 CrossRef CAS PubMed
- S. Y. Woo, J. H. Kim, M. K. Moon, S.-H. Han, S. K. Yeon, J. W. Choi, B. K. Jang, H. J. Song, Y. G. Kang, J. W. Kim, J. Lee, D. J. Kim, O. Hwang and K. D. Park, J. Med. Chem., 2014, 57, 1473–1487 CrossRef CAS PubMed
- P. A. Jackson, J. C. Widen, D. A. Harki and K. M. Brummond, J. Med. Chem., 2017, 60, 839–885 CrossRef CAS PubMed
- A. P. Turnbull, S. Ioannidis, W. W. Krajewski, A. Pinto-Fernandez, C. Heride, A. C. L. Martin, L. M. Tonkin, E. C. Townsend, S. M. Buker, D. R. Lancia, J. A. Caravella, A. V. Toms, T. M. Charlton, J. Lahdenranta, E. Wilker, B. C. Follows, N. J. Evans, L. Stead, C. Alli, V. V. Zarayskiy, A. C. Talbot, A. J. Buckmelter, M. Wang, C. L. McKinnon, F. Saab, J. F. McGouran, H. Century, M. Gersch, M. S. Pittman, C. G. Marshall, T. M. Raynham, M. Simcox, L. M. D. Stewart, S. B. McLoughlin, J. A. Escobedo, K. W. Bair, C. J. Dinsmore, T. R. Hammonds, S. Kim, S. Urbé, M. J. Clague, B. M. Kessler and D. Komander, Nature, 2017, 550, 481–486 CrossRef CAS PubMed
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed
- R. A. Ward, N. Colclough, M. Challinor, J. E. Debreczeni, K. Eckersley, G. Fairley, L. Feron, V. Flemington, M. A. Graham, R. Greenwood, P. Hopcroft, T. D. Howard, M. James, C. D. Jones, C. R. Jones, J. Renshaw, K. Roberts, L. Snow, M. Tonge and K. Yeung, J. Med. Chem., 2015, 58, 4790–4801 CrossRef CAS PubMed
- X. Li, Y. Zuo, G. Tang, Y. Wang, Y. Zhou, X. Wang, T. Guo, M. Xia, N. Ding and Z. Pan, J. Med. Chem., 2014, 57, 5112–5128 CrossRef CAS PubMed
- M. E. Flanagan, J. A. Abramite, D. P. Anderson, A. Aulabaugh, U. P. Dahal, A. M. Gilbert, C. Li, J. Montgomery, S. R. Oppenheimer, T. Ryder, B. P. Schuff, D. P. Uccello, G. S. Walker, Y. Wu, M. F. Brown, J. M. Chen, M. M. Hayward, M. C. Noe, R. S. Obach, L. Philippe, V. Shanmugasundaram, M. J. Shapiro, J. Starr, J. Stroh and Y. Che, J. Med. Chem., 2014, 57, 10072–10079 CrossRef CAS PubMed
- Z. Li, R. Huang, H. Xu, J. Chen, Y. Zhan, X. Zhou, H. Chen and B. Jiang, Org. Lett., 2017, 19, 4972–4975 CrossRef CAS PubMed
- R. Huang, Z. Li, Y. Sheng, J. Yu, Y. Wu, Y. Zhan, H. Chen and B. Jiang, Org. Lett., 2018, 20, 6526–6529 CrossRef CAS PubMed
- A. L. Garske, U. Peters, A. T. Cortesi, J. L. Perez and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15046–15052 CrossRef CAS PubMed
- R. Huang, Z. Li, P. Ren, W. Chen, Y. Kuang, J. Chen, Y. Zhan, H. Chen and B. Jiang, Eur. J. Org. Chem., 2018, 2018, 829–836 CrossRef CAS
- H. Chen, R. Huang, Z. Li, W. Zhu, J. Chen, Y. Zhan and B. Jiang, Org. Biomol. Chem., 2017, 15, 7339–7345 RSC
- C. E. Allen, P. R. Curran, A. S. Brearley, V. Boissel, L. Sviridenko, N. J. Press, J. P. Stonehouse and A. Armstrong, Org. Lett., 2015, 17, 458–460 CrossRef CAS PubMed
- C. S. Rondestvedt, J. Am. Chem. Soc., 1954, 76, 1926–1929 CrossRef CAS
- J. Sinha, M. Podgórski, S. Huang and C. N. Bowman, Chem. Commun., 2018, 54, 3034–3037 RSC
- K. B. Sharpless, R. F. Lauer and A. Y. Teranishi, J. Am. Chem. Soc., 1973, 95, 6137–6139 CrossRef CAS
- S.-R. Sheng, X.-C. Wang, X.-L. Liu and C.-S. Song, Synth. Commun., 2003, 33, 2867–2872 CrossRef CAS
- J. F. King and K. C. Khemani, Can. J. Chem., 1985, 63, 619–622 CrossRef CAS
- S. G. Kathman, Z. Xu and A. V. Statsyuk, J. Med. Chem., 2014, 57, 4969–4974 CrossRef CAS PubMed
- R. H. Nonoo, A. Armstrong and D. J. Mann, ChemMedChem, 2012, 7, 2082–2086 CrossRef CAS PubMed
- R. Cardoso, R. Love, C. L. Nilsson, S. Bergqvist, D. Nowlin, J. Yan, K. K.-C. Liu, J. Zhu, P. Chen, Y.-L. Deng, H. J. Dyson, M. J. Greig and A. Brooun, Protein Sci., 2012, 21, 1885–1896 CrossRef CAS PubMed
- K. L. Prime and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 10714–10721 CrossRef CAS
- D. A. Erlanson, A. C. Braisted, D. R. Raphael, M. Randal, R. M. Stroud, E. M. Gordon and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9367–9372 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00566d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2019 |