
Crystallographic and SAR analyses reveal the high requirements needed to selectively and potently inhibit SIRT2 deacetylase and decanoylase†
Ling-Ling
Yang
a,
Wei
Xu
a,
Jie
Yan
a,
Hui-Lin
Su
a,
Chen
Yuan
a,
Chao
Li
a,
Xing
Zhang
a,
Zhu-Jun
Yu
b,
Yu-Hang
Yan
b,
Yamei
Yu
b,
Qiang
Chen
b,
Zhouyu
Wang
a,
Lin
Li
a,
Shan
Qian
*a and
Guo-Bo
Li
 *b
*b
aCollege of Food and Bioengineering, Xihua University, Sichuan 610039, China. E-mail: qians33@163.com
bKey Laboratory of Drug Targeting and Drug Delivery System of Ministry of Education, Department of Medicinal Chemistry, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: liguobo@scu.edu.cn
First published on 7th December 2018
Abstract
A high-quality X-ray crystal structure reveals the mechanism of compound 1a inhibiting SIRT2 deacetylase and decanoylase. Structure–activity relationship (SAR) analysis of the synthesized derivatives of 1a reveals the high requirements needed for selective inhibitors to bind with the induced hydrophobic pocket and potently inhibit sirtuin 2 deacetylase.
Human sirtuins are nicotinamide adenine dinucleotide (NAD+)-dependent deacylases, which are key regulators of metabolism, stress, and immune and inflammatory responses, influencing a range of diseases, including cancer.1–3 There are seven isotypes of sirtuins (SIRT1–7), which differ in their deacylase activity and subcellular localization. Sirtuins can remove various acyl groups (e.g., acetyl, palmitoyl, myristoyl, crotonyl, succinyl, glutaryl, and decanoyl) from histones and other protein substrates,1,4–6 but each isoform has specific preference for acyl-lysine substrates. SIRT2, mainly located in the cytoplasm, shows deacetylase and defatty-acylase activities to various protein substrates, including metabolic enzymes (e.g., S6K1, PEPCK1, G6PD, LDH, and PGAM), differentiation proteins (e.g., tubulin, PAR3, keratin 8, and PRLR), and transcription factors (e.g., p300, p53, Foxo1, HIF1α, and NFκB).1,7,8 The abnormal activity of SIRT2, found in disease models,3,9,10 promotes the development of selective small-molecule inhibitors to investigate associated molecular mechanisms and to exploit treatments for relevant diseases.
There are several reports of structurally diverse small-molecule SIRT2 inhibitors,11–20 some of which exhibit high selectivity for SIRT2 over other sirtuin isoforms, such as aminothiazoles,21–23 2-anilinobenzamide,24 thienopyrimidinones,25 and 1,2,4-oxadiazoles.19 The common inhibition mechanism of these selective inhibitors involves their binding to the specific hydrophobic pocket.19,21–23,25 Through structure-guided molecular designs, we recently identified N-(3-(phenoxymethyl)phenyl)acetamide derivatives as highly selective SIRT2 inhibitors, which show inhibitory activities against SIRT2 highly-expressed human breast cancer cells and non-small cell lung cancer cells;26,27 similar to other reported selective inhibitors, they bind with the induced hydrophobic pocket as observed by crystallographic analyses.27
Previous structure–activity relationship (SAR) analyses of the derivatives with different moieties at the 3- or 4-position of the phenyl of the N-(3-(phenoxymethyl)phenyl)acetamide scaffold revealed that the 3-substituted derivatives are likely more potent than the 4-substituted derivatives.26 Nevertheless, we lately found that compound 1a (IC50 = 1.32 μM), which bears a benzenethiol motif at the 4 position, shows more potent inhibition to SIRT2 deacetylase than 3-substituted compound 1b (IC50 = 6.65 μM, Fig. 1a and S1†). Since SIRT2 has defatty-acylase activity in addition to its deacetylase activity, and particularly, one compound (NPD11033) was reported recently by Kudo et al. to have the ability to inhibit deacetylase but not the defatty-acylase reaction catalysed by SIRT2,28 we hence tested whether 1a and 1b inhibit SIRT2 decanoylase (as a representative of defatty-acylases). We observed that 1a could inhibit SIRT2 decanoylase with an IC50 value of 11.6 μM, more potently than 1b (IC50 = 62.1 μM, Fig. 1a and S2†). These results reveal that 1a has different inhibitory potency against SIRT2 deacetylase and decanoylase, which is, consistently, more potent than 1b, reflecting that 1a binds with SIRT2 via a manner which blocks the binding of acetylated and canoylated substrates. We report the single-crystal X-ray diffraction structure of the SIRT2:1a complex, and the SAR analyses of new derivatives of 1a with SIRT2 deacetylase and decanoylase in the following sections.
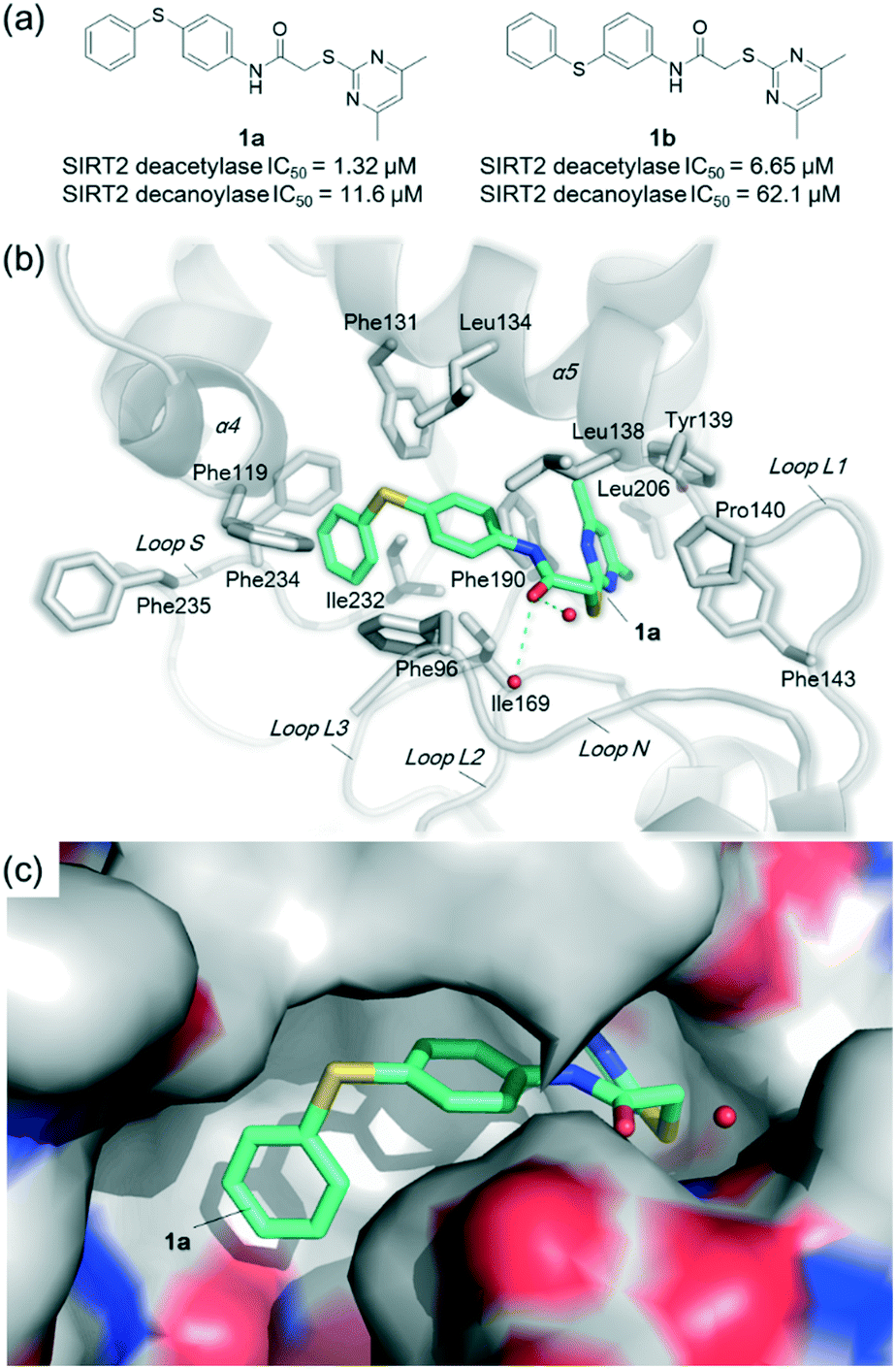 | ||
| Fig. 1 Crystallographic analysis reveals the binding mode of 1a with SIRT2. (a) Chemical structures of 1a/1b and their inhibitory activities to SIRT2 deacetylase and decanoylase. (b) View from the SIRT2:1a complex structure (PDB ID 5YQM) showing that 1a is positioned to form hydrophobic interactions with residues Phe234, Ile232, Phe119, Phe131, Leu134, Leu138, Tyr139, Phe96, Pro140, Phe143, Ile169 and Phe190, and π–π stacking interactions with Phe190 and Phe96. (c) View from the molecular surface representation of the hydrophobic pocket with 1a. | ||
Co-crystallization experiments yielded a high-quality structure of SIRT2 in complex with 1a, which was solved to 1.75 Å (PDB ID 5YQM). The crystallization conditions, data collection and refinement statistics are given in Tables S1 and S2.† SIRT2 crystallized in the previously described space group P1211 with one molecule per asymmetric unit (Table S2†), and one molecule of 1a identified in the hydrophobic pocket at the interface of the Rossmann fold domain and the zinc-binding domain; 1a could be confidently modelled into the clear Fo–Fc density map (B-factors 1.2 times greater than the main chain) (Fig. S3†).
The crystal structure reveals the binding mode of 1a with SIRT2, which is similar to that of 2-((4,6-dimethylpyrimidin-2-yl)thio)-N-(3-(phenoxymethyl)phenyl)acetamide (A1) with SIRT2 (PDB ID 5YQL)27 (r.m.s.d. of 0.15 Å over 236 Cα atoms, Fig. S4†). 1a is positioned to form hydrophobic interactions with the residues in the hydrophobic site, including Phe234 (loop S), Ile232 (loop S), Phe119 (α4), Phe131 (α5), Leu134 (α5), Leu138 (α5), Tyr139 (α5), Phe96 (loop N), Pro140 (loop L1), Phe143 (loop L1), Ile169 (loop L2), and Phe190 (loop L3) (Fig. 1b). The 4,6-dimethylpyrimidine motif of 1a forms π–π stacking interactions with Phe190 (loop L3) and Phe96 (loop N) (Fig. 1b). Notably, two water molecules are observed in the hydrophobic pocket, which are likely important for the binding of 1a (Fig. 1b); equivalent water molecules are also observed in the complex structures of SIRT2:A1 (PDB ID 5YQL),27 SIRT2:SirReal2 (PDB ID 4RMG),22 and SIRT2:SirReal probes (PDB ID 5DY5).23
Superimposition of SIRT2:1a (PDB ID 5YQM) with SIRT2:HKK(Ac)LRF (PDB ID 3JR3)29 reveals that the binding of 1a induces substantial rearrangements of loop S and α4, e.g., the positional change of Phe235 that is important for HKK(Ac)LRF binding (Fig. 1b and S5†), which hence block the binding of the acetylated substrates. We also observe that 1a occupies the myristoyl binding pocket (Fig. S6†), which hence can disturb the myristoylated or other fatty-acylated substrates for binding. These results clearly reveal the mechanism of 1a inhibiting both the SIRT2 deacetylase and decanoylase.
A comparison of the structures of SIRT2:1a (PDB ID 5YQM), SIRT2:A1 (PDB ID 5YQL),27 SIRT2:SirReal2 (PDB ID 4RMG),22 SIRT2:thienopyrimidinone (PDB ID 5MAT),25 and SIRT2:2-anilinobenzamide (PDB ID 5Y5N)24 reveals the common pharmacophore features of these selective inhibitors and evidence of the flexibility in the conformation of loop N, suggesting that this loop (particularly Phe96 and Arg97) may be important in inhibitor capture (Fig. S7†). Notably, 1a and SirReal2 are positioned to perfectly bind with the hydrophobic pocket around residues Phe119, Phe234, Phe131, Leu134, and Ile232 (Fig. 1b and c and S8†); compared with 1a, the diphenylsulfane moiety of 1b may not fit perfectly with this hydrophobic pocket as indicated by molecular docking analyses (Fig. S9†). These results reflect the high requirements needed for selective inhibitors to bind with the induced hydrophobic pocket and potently inhibit SIRT2 catalytic activities.
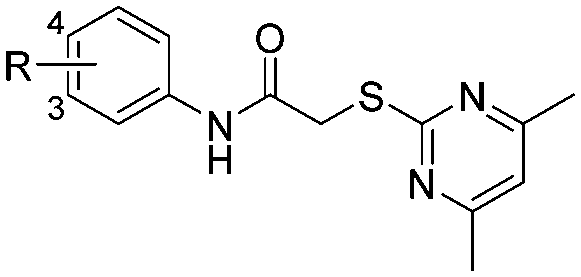
We next synthesized a series of new derivatives of 1a (8a–8b and 12a–12l, Table 1) via the synthetic routes outlined in Schemes 1 and 2 (for details, see the ESI†). Briefly, 4-iodoaniline (2a) or 3-iodoaniline (2b) reacts with ethane-1,2-dithiol in the presence of copper sulfate pentahydrate and potassium hydroxide under an argon atmosphere at 110 °C for 8 h, followed by adding iodobenzene (3) to react for another 18 h at 120 °C, resulting in the thioether intermediates (4a and 4b) in 74–78% yields (Scheme 1). Next, the key intermediates 4a–4b were directly reacted with 2-bromoacetyl bromide to give the condensation products 5a–5b in 90–92% yields. On the other hand, intermediate 4a was oxidized to sulfoxide derivative 6a (56% yield) or sulfone derivative 6b (85% yield) using 3-chlorobenzoperoxoic acid, and then reacted with 2-bromoacetyl bromide to produce intermediates 7a and 7b, respectively (Scheme 1). Finally, reactions of intermediates 5a–5b or 7a–7b with 4,6-dimethylpyrimidine-2-thiol, respectively, in the presence of potassium tert-butoxide as the acid-binding agent afford the target compounds 1a–1b (89–93% yields) or 8a–8b (91–92% yields) (Scheme 1).26
|
|
||||
|---|---|---|---|---|
| Cpd ID | R | Position | SIRT2 deacetylase Inh% @100 μM/@10 μM | SIRT2 decanoylase Inh% @100 μM/@10 μM |
| 1a |
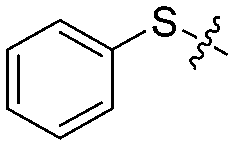
|
4 | 80 ± 5/70 ± 5 | 53 ± 6/38 ± 9 |
| 1b |
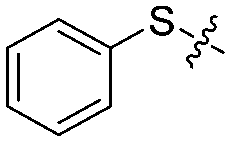
|
3 | 73 ± 5/65 ± 5 | 30 ± 10/5 ± 2 |
| 8a |
|
4 | 40 ± 5/5 ± 3 | 23 ± 5/4 ± 3 |
| 8b |
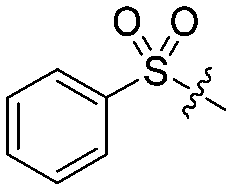
|
4 | 50 ± 8/8 ± 4 | 18 ± 6/5 ± 3 |
| 12a |
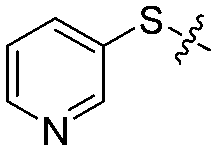
|
4 | 67 ± 6/42 ± 7 | 60 ± 6/23 ± 5 |
| 12b |
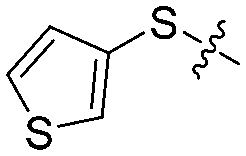
|
4 | 62 ± 5/50 ± 8 | 58 ± 7/18 ± 5 |
| 12c |
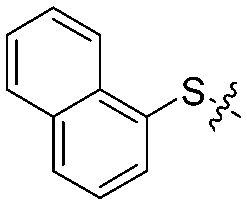
|
4 | 53 ± 4/42 ± 5 | 58 ± 3/42 ± 5 |
| 12d |
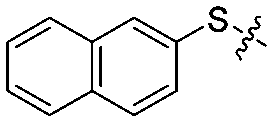
|
4 | 70 ± 5/51 ± 5 | 38 ± 6/23 ± 5 |
| 12e |
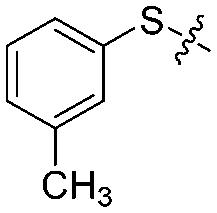
|
4 | 76 ± 3/70 ± 3 | 65 ± 8/54 ± 5 |
| 12f |
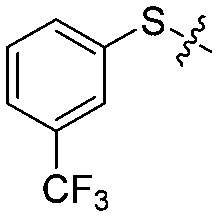
|
4 | 82 ± 5/72 ± 5 | 77 ± 8/72 ± 3 |
| 12g |
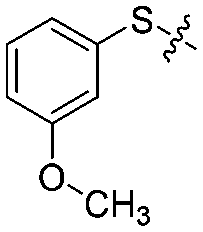
|
4 | 85 ± 3/80 ± 2 | 80 ± 5/70 ± 6 |
| 12h |
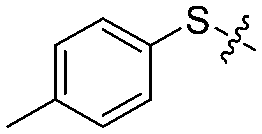
|
4 | 63 ± 6/30 ± 10 | 70 ± 5/10 ± 5 |
| 12i |
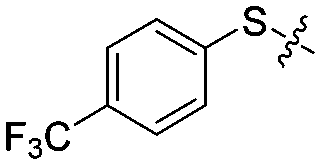
|
4 | 51 ± 7/28 ± 8 | 42 ± 5/18 ± 5 |
| 12j |
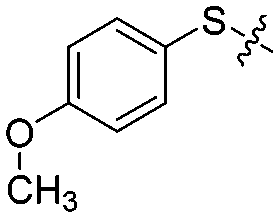
|
4 | 65 ± 8/55 ± 6 | 58 ± 5/20 ± 9 |
| 12k |
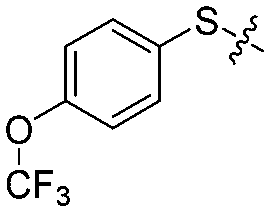
|
4 | 60 ± 8/40 ± 9 | 63 ± 6/23 ± 5 |
| 12l |
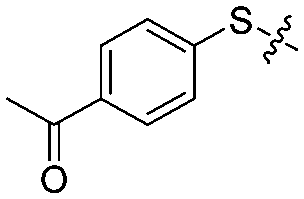
|
4 | 5 ± 3/6 ± 3 | 35 ± 5/8 ± 3 |
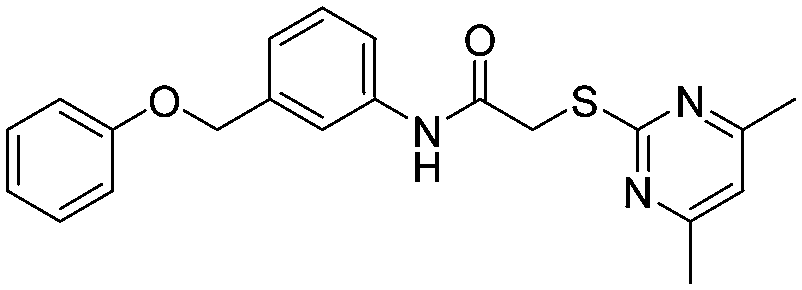 A1
A1
|
76 ± 5/53 ± 6 | 72 ± 8/32 ± 5 | ||
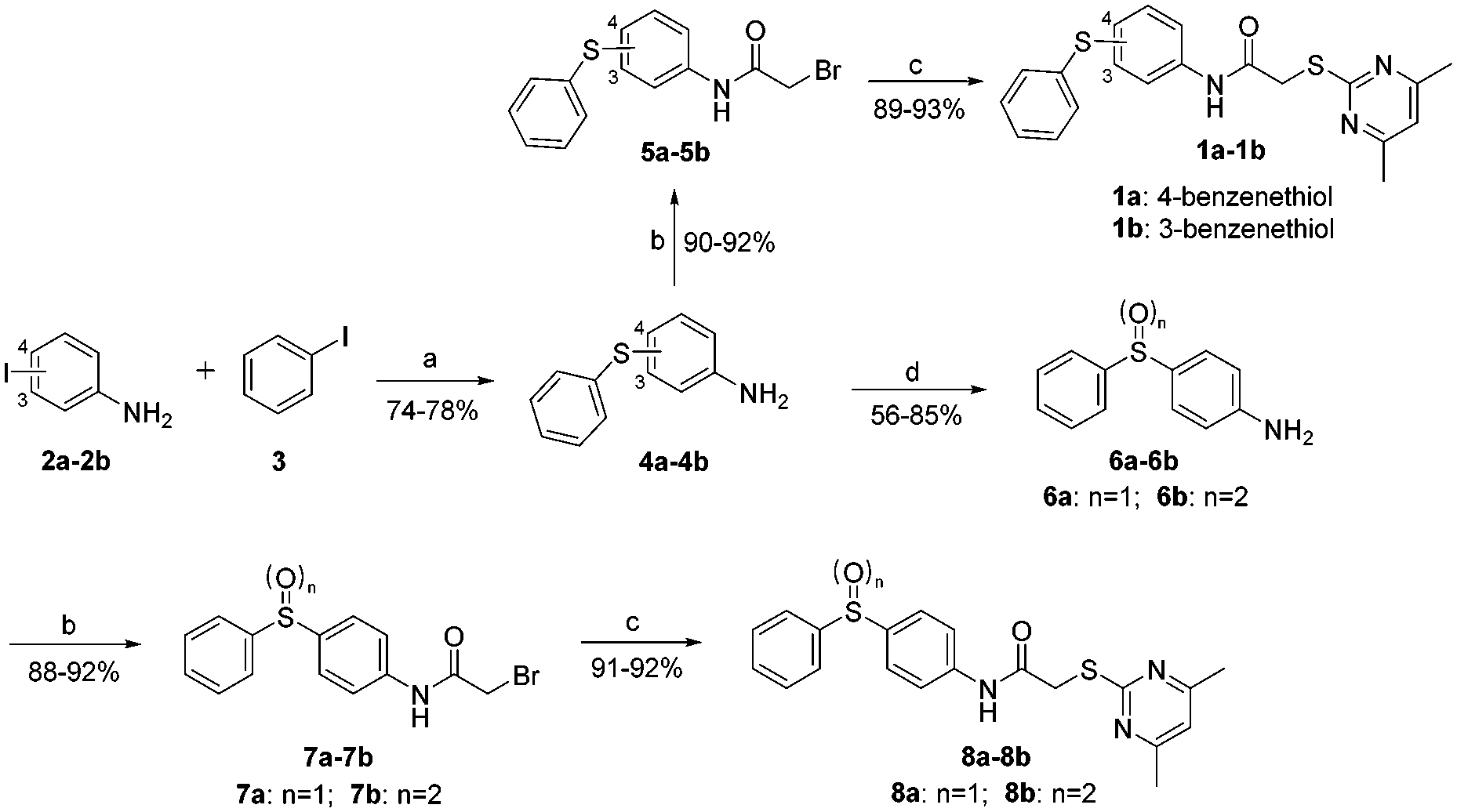 | ||
| Scheme 1 The synthesis of 1a–1b and 8a–8b. Reagents and conditions: (a) (i) ethane-1,2-dithiol, copper sulfate pentahydrate, potassium hydroxide, DMSO/H2O, 110 °C, 8 h; (ii) DMF, 120 °C, 18 h. (b) 2-Bromoacetyl bromide, TEA, DCM, 0 °C, 0.5–1 h. (c) 4,6-Dimethylpyrimidine-2-thiol, t-BuOK, DMF, r.t., 0.5 h and 12 h. (d) m-CPBA, DCM, r.t. | ||
 | ||
| Scheme 2 The synthesis of 12a–12l. Reagents and conditions: (a) (i) ethane-1,2-dithiol, copper sulfate pentahydrate, potassium hydroxide, DMSO/H2O, 110 °C, 8 h; (ii) DMF, 120 °C, 18 h. (b) 2-Bromoacetyl bromide, TEA, DCM, 0 °C, 0.5–1 h. (c) 4,6-Dimethylpyrimidine-2-thiol, t-BuOK, DMF, r.t., 0.5 h and 12 h. | ||
Synthetic access to the target compounds 12a–12l was achieved using similar methods, as described in Scheme 1. The commercially available 3-iodopyridine (9a), 3-iodothiophene (9b), 1-iodonaphthalene (9c), 2-iodonaphthalene (9d), and various substituted iodobenzenes 9e–9l were reacted with 4-iodoaniline (2a), respectively, to give the intermediates 10a–10l (55–83%), followed by a condensation reaction of acyl halide with amine (82–93%) and a t-BuOH-promoted nucleophilic substitution (79–96%) to afford the desired target compounds 12a–12l in high yields (Scheme 2).
We then performed structure–activity relationship studies using the synthesized compounds 8a–8b and 12a–12l (Table 1); the deacetylase and decanoylase activities of SIRT2 were tested using RLIK(Ac)AMC and ETDK(Ca)AMC (Fig. S10†), respectively. Compared with 1a, the oxidized derivatives 8a and 8b show lower potency to inhibit SIRT2-catalysed deacetylation on RLIK(Ac)AMC and decanoylation on ETDK(Ca)AMC. Replacement of the benzenethiol moiety of 1a with pyridine-3-thiol (12a), thiophene-3-thiol (12b), naphthalene-1-thiol (12c), and naphthalene-2-thiol (12d) also led to decreased inhibitory potency to SIRT2 catalytic activity on both RLIK(Ac)AMC and ETDK(Ca)AMC (Table 1). Compounds 12e–12g with 3-substituted benzenethiols inhibit SIRT2 deacetylase and decanoylase more potently than 1a and A1, whereas compounds 12h–12l, bearing various 4-substituted benzenethiols, show decreased inhibitory activity (Table 1). Notably, there is a clear correlation between the SIRT2 deacetylase and decanoylase inhibition of these compounds (Table 1), implying that their inhibition mode is similar to that of 1a. From the SAR results, we observe that replacement or modification of the benzenethiol motif of 1a appears prone to decreased inhibition, which may reflect the high requirements needed for specific compounds to bind with the hydrophobic pocket (Fig. 1b and c) and potently inhibit the SIRT2 catalytic activity.
For potent compounds 12f and 12g, we measured their IC50 values for inhibiting SIRT2 deacetylase and decanoylase. Compared with 1a and A1, 12f and 12g show better inhibitory activity to SIRT2 deacetylase, with IC50 values of 0.85 μM and 0.70 μM, respectively (Fig. S11†); 12f and 12g also exhibit IC50 values of 17.6 μM and 8.33 μM to SIRT2 decanoylase, respectively (Fig. S12†). Using our previously reported method, we tested 12f and 12g against other sirtuin isotypes, including SIRT1, SIRT3, SIRT5, and SIRT6, to examine the selectivity. Both compounds have no or low inhibitory activities at 100 μM to all of the tested sirtuin isotypes. These results indicate that 12f and 12g could potently and selectively inhibit SIRT2 deacetylase and decanoylase.
Conclusions
In summary, a high-quality X-ray crystal structure of SIRT2:1a was reported, which clearly reveals the mechanism of 1a inhibiting the SIRT2 deacetylase and decanoylase, i.e. via binding to the induced hydrophobic pocket and disturbing the binding of acetylated and fatty-acylated substrates. The SAR analyses of the synthesized derivatives of 1a reveal a clear correlation between their inhibition to SIRT2 deacetylase and decanoylase, and reveal the high requirements needed for compounds to bind with the hydrophobic pocket. New selective, potent inhibitors (e.g., 12f and 12g) for SIRT2 deacetylase and decanoylase were identified, which might be useful chemical tools to probe SIRT2-related molecular mechanisms. This study will aid future investigations in developing new selective inhibitors against SIRT2 deacylase to provide potential treatments for relevant diseases.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the staff of the BL19U1 beamline of the National Center for Protein Science Shanghai at Shanghai Synchrotron Radiation Facility for assistance during data collection. This work was supported by the funds from the National Natural Science Foundation of China (Grant No.: 81703355 and 81502989), the Chun-Hui Project from the Ministry of Education of China (Grant No.: 172507), and the Opening Project of Food Biotechnology (Grant No.: szjj2017-079).Notes and references
- R. H. Houtkooper, E. Pirinen and J. Auwerx, Nat. Rev. Mol. Cell Biol., 2012, 13, 225–238 CrossRef CAS PubMed.
- A. A. Sauve and D. Y. Youn, Curr. Opin. Chem. Biol., 2012, 16, 535–543 CrossRef CAS PubMed.
- H. Jing, J. Hu, B. He, Y. L. N. Abril, J. Stupinski, K. Weiser, M. Carbonaro, Y.-L. Chiang, T. Southard, P. Giannakakou, R. S. Weiss and H. Lin, Cancer Cell, 2016, 29, 297–310 CrossRef CAS PubMed.
- B. Chen, W. Zang, J. Wang, Y. Huang, Y. He, L. Yan, J. Liu and W. Zheng, Chem. Soc. Rev., 2015, 44, 5246–5264 RSC.
- L. Yang, X. Ma, Y. He, C. Yuan, Q. Chen, G. Li and X. Chen, Sci. China: Life Sci., 2017, 60, 249–256 CrossRef CAS PubMed.
- S. Liu, S. Ji, Z.-J. Yu, H.-L. Wang, X. Cheng, W.-J. Li, L. Jing, Y. Yu, Q. Chen, L.-L. Yang, G.-B. Li and Y. Wu, Chem. Biol. Drug Des., 2018, 91, 257–268 CrossRef CAS PubMed.
- A. Chalkiadaki and L. Guarente, Nat. Rev. Cancer, 2015, 15, 608–624 CrossRef CAS.
- T. M. Liu and N. Shyh-Chang, Nat. Cell Biol., 2017, 19, 412–414 CrossRef CAS PubMed.
- J. Chen, A. W. H. Chan, K.-F. To, W. Chen, Z. Zhang, J. Ren, C. Song, Y.-S. Cheung, P. B. S. Lai, S.-H. Cheng, M. H. L. Ng, A. Huang and B. C. B. Ko, Hepatology, 2013, 57, 2287–2298 CrossRef CAS PubMed.
- R. M. de Oliveira, H. Vicente Miranda, L. Francelle, R. Pinho, É. M. Szegö, R. Martinho, F. Munari, D. F. Lázaro, S. Moniot, P. Guerreiro, L. Fonseca, Z. Marijanovic, P. Antas, E. Gerhardt, F. J. Enguita, B. Fauvet, D. Penque, T. F. Pais, Q. Tong, S. Becker, S. Kügler, H. A. Lashuel, C. Steegborn, M. Zweckstetter and T. F. Outeiro, PLoS Biol., 2017, 15, e2000374 CrossRef.
- M. Gertz, F. Fischer, G. T. Nguyen, M. Lakshminarasimhan, M. Schutkowski, M. Weyand and C. Steegborn, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E2772–E2781 CrossRef CAS PubMed.
- J. S. Disch, G. Evindar, C. H. Chiu, C. A. Blum, H. Dai, L. Jin, E. Schuman, K. E. Lind, S. L. Belyanskaya, J. Deng, F. Coppo, L. Aquilani, T. L. Graybill, J. W. Cuozzo, S. Lavu, C. Mao, G. P. Vlasuk and R. B. Perni, J. Med. Chem., 2013, 56, 3666–3679 CrossRef CAS PubMed.
- H. Cui, Z. Kamal, T. Ai, Y. Xu, S. S. More, D. J. Wilson and L. Chen, J. Med. Chem., 2014, 57, 8340–8357 CrossRef CAS PubMed.
- T. Seifert, M. Malo, T. Kokkola, K. Engen, M. Fridén-Saxin, E. A. A. Wallén, M. Lahtela-Kakkonen, E. M. Jarho and K. Luthman, J. Med. Chem., 2014, 57, 9870–9888 CrossRef CAS.
- P. R. Tatum, H. Sawada, Y. Ota, Y. Itoh, P. Zhan, N. Ieda, H. Nakagawa, N. Miyata and T. Suzuki, Bioorg. Med. Chem. Lett., 2014, 24, 1871–1874 CrossRef CAS PubMed.
- M. A. Khanfar, L. Quinti, H. Wang, J. Nobles, A. G. Kazantsev and R. B. Silverman, ACS Med. Chem. Lett., 2015, 6, 607–611 CrossRef CAS PubMed.
- T. Yang, X. Chen, H.-x. Jin, G. Sethi and M.-L. Go, Eur. J. Med. Chem., 2015, 92, 145–155 CrossRef CAS PubMed.
- T. Ai, D. J. Wilson, S. S. More, J. Xie and L. Chen, J. Med. Chem., 2016, 59, 2928–2941 CrossRef CAS PubMed.
- S. Moniot, M. Forgione, A. Lucidi, G. S. Hailu, A. Nebbioso, V. Carafa, F. Baratta, L. Altucci, N. Giacché, D. Passeri, R. Pellicciari, A. Mai, C. Steegborn and D. Rotili, J. Med. Chem., 2017, 60, 2344–2360 CrossRef CAS PubMed.
- E. Therrien, G. Larouche, N. Nguyen, J. Rahil, A.-M. Lemieux, Z. Li, M. Fournel, T. P. Yan, A.-J. Landry, S. Lefebvre, J. J. Wang, K. MacBeth, C. Heise, A. Nguyen, J. M. Besterman, R. Déziel and A. Wahhab, Bioorg. Med. Chem. Lett., 2015, 25, 2514–2518 CrossRef CAS PubMed.
- M. Schiedel, T. Rumpf, B. Karaman, A. Lehotzky, J. Olah, S. Gerhardt, J. Ovadi, W. Sippl, O. Einsle and M. Jung, J. Med. Chem., 2016, 59, 1599–1612 CrossRef CAS PubMed.
- T. Rumpf, M. Schiedel, B. Karaman, C. Roessler, B. J. North, A. Lehotzky, J. Olah, K. I. Ladwein, K. Schmidtkunz, M. Gajer, M. Pannek, C. Steegborn, D. A. Sinclair, S. Gerhardt, J. Ovadi, M. Schutkowski, W. Sippl, O. Einsle and M. Jung, Nat. Commun., 2015, 6, 6263 CrossRef CAS PubMed.
- M. Schiedel, T. Rumpf, B. Karaman, A. Lehotzky, S. Gerhardt, J. Ovádi, W. Sippl, O. Einsle and M. Jung, Angew. Chem., Int. Ed., 2016, 55, 2252–2256 CrossRef CAS PubMed.
- P. Mellini, Y. Itoh, H. Tsumoto, Y. Li, M. Suzuki, N. Tokuda, T. Kakizawa, Y. Miura, J. Takeuchi, M. Lahtela-Kakkonen and T. Suzuki, Chem. Sci., 2017, 8, 6400–6408 RSC.
- S. Sundriyal, S. Moniot, Z. Mahmud, S. Yao, P. Di Fruscia, C. R. Reynolds, D. T. Dexter, M. J. E. Sternberg, E. W. F. Lam, C. Steegborn and M. J. Fuchter, J. Med. Chem., 2017, 60, 1928–1945 CrossRef CAS PubMed.
- L. Yang, X. Ma, C. Yuan, Y. He, L. Li, S. Fang, W. Xia, T. He, S. Qian, Z. Xu, G. Li and Z. Wang, Eur. J. Med. Chem., 2017, 134, 230–241 CrossRef CAS PubMed.
- L.-L. Yang, H.-L. Wang, L. Zhong, C. Yuan, S.-Y. Liu, Z.-J. Yu, S. Liu, Y.-H. Yan, C. Wu, Y. Wang, Z. Wang, Y. Yu, Q. Chen and G.-B. Li, Eur. J. Med. Chem., 2018, 155, 806–823 CrossRef CAS PubMed.
- N. Kudo, A. Ito, M. Arata, A. Nakata and M. Yoshida, Philos. Trans. R. Soc., B, 2018, 373, 20170070 CrossRef PubMed.
- W. F. Hawse and C. Wolberger, J. Biol. Chem., 2009, 284, 33654–33661 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic data collection and refinement statistics, details of chemical synthesis, and additional figures and tables. See DOI: 10.1039/c8md00462e |
| This journal is © The Royal Society of Chemistry 2019 |