
Benzoflavone derivatives as potent antihyperuricemic agents†
Jatinder V.
Singh
a,
Gurbachan
Mal
a,
Gurleen
Kaur
a,
Manish K.
Gupta
b,
Amritpal
Singh
a,
Kunal
Nepali
a,
Harbinder
Singh
 *a,
Sahil
Sharma
*a and
Preet Mohinder
S. Bedi
*a
*a,
Sahil
Sharma
*a and
Preet Mohinder
S. Bedi
*a
aDepartment of Pharmaceutical Sciences, Guru Nanak Dev University, Amritsar, Punjab 143005, India. E-mail: singh.harbinder40@gmail.com; ss.gq2009@gmail.com; bedi_preet@yahoo.com
bTERI-Deakin Nanobiotechnology Centre, The Energy and Resources Institute, TERI Gram, Gual Pahari, Gurugram, Haryana 122001, India
First published on 6th December 2018
Abstract
Two series of benzoflavone derivatives were rationally designed, synthesized and evaluated for their xanthine oxidase (XO) inhibitory potential. Among both series, eight compounds (NF-2, NF-4, NF-9, NF-12, NF-16, NF-25, NF-28, and NF-32) were found to exert significant XO inhibition with IC50 values lower than 10 μM. Enzyme kinetic studies revealed that the most potent benzoflavone derivatives (NF-4 and NF-28) are mixed type inhibitors of the XO enzyme. Molecular modeling studies were also performed to investigate the binding interactions of these molecules (NF-4 and NF-28) with the amino acid residues present in the active site of the enzyme. Docking results confirmed that their favorable binding conformations in the active site of XO can completely block the catalytic activity of the enzyme. Benzoflavone derivatives exhibiting potent XO enzyme inhibition also showed promising results in a hyperuricemic mice model when tested in vivo.
1. Introduction
The key role of the versatile molybdoflavoprotein xanthine oxidase (XO) has been longer familiar, today. There is a well-known relationship between xanthine oxidase and hyperuricemia. The metabolism of purines, the oxidative hydroxylation of hypoxanthine and xanthine to generate uric acid, and the production of reactive oxygen species within the human body are the primary functions of the XO enzyme.1 The potential increase in serum uric acid level (hyperuricemia) leads to deposition of sodium urate crystals in joints which results in inflammation and pain in joints. In this regard, xanthine oxidase is an important and selective target for sustaining broad-spectrum chemotherapy in hyperuricemic patients.1,2In the recent past, due to the success of allopurinol (a purine based xanthine oxidase inhibitor) in the treatment of hyperuricemia, several purine analogs like pterin and 6-formylpterin,3 2-alkylhypoxanthines,3,4 and 2-substituted 7H-pyrazolo-[4,3-e]-1,2,4-triazolo-[1,5-c]-pyrimidines5–8 were developed as potent xanthine oxidase inhibitors. However, the side effects associated with these purine-based analogs and their significant effect on the activities of purine and pyrimidine metabolizing enzymes have diverted researchers to develop non-purine based xanthine oxidase inhibitors (Fig. 1).9–11
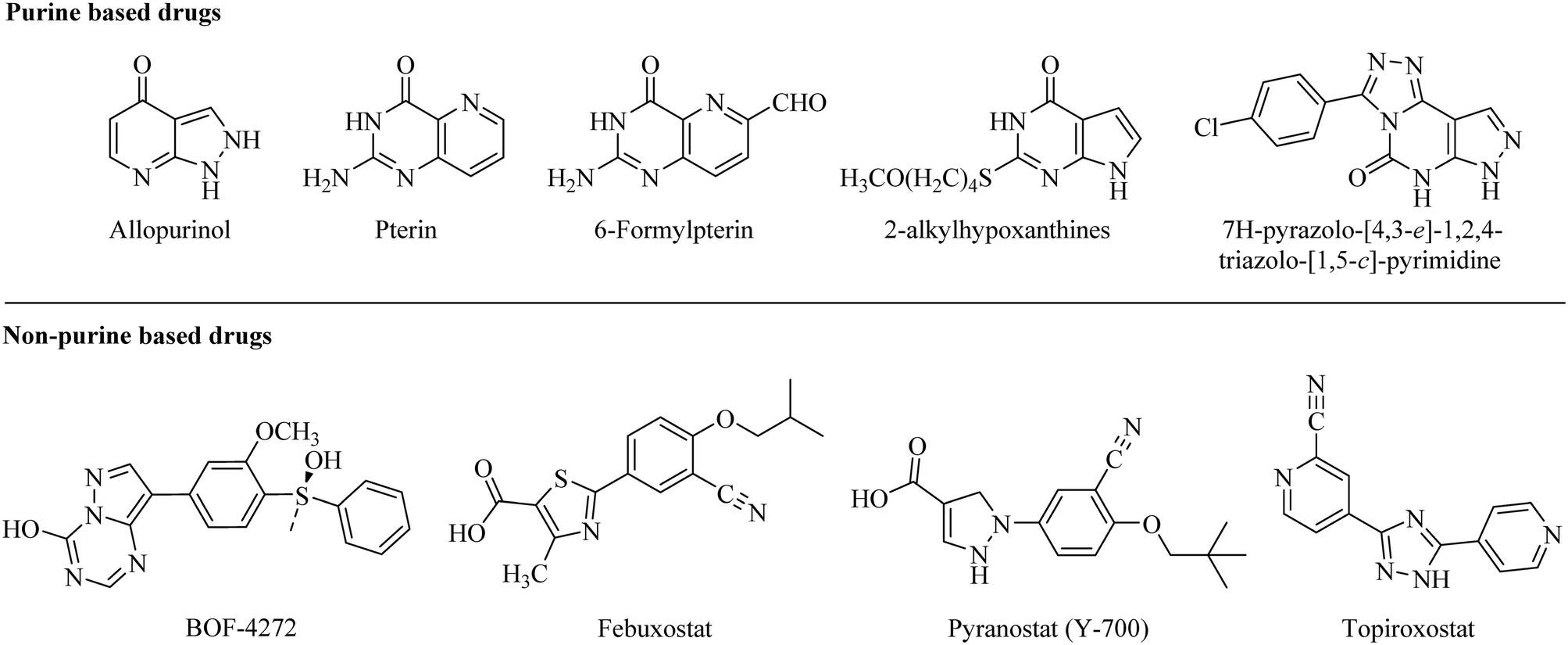 | ||
| Fig. 1 Drugs used clinically for the treatment of hyperuricemia by inhibiting the xanthine oxidase enzyme. | ||
Several non-purine based XO inhibitors have been developed so far. Among them, BOF-4272 is one of the potent XO inhibitors but the variation in its efficacy from person to person due to the difference in hepatic metabolism limits its clinical use (Fig. 1).12 In 2009, febuxostat got approved by the USFDA as the first non-purine based XO inhibitor for the treatment of hyperuricemia with prominent efficacy and longer lasting hypouricemic effect than allopurinol.13–15 But it has certain adverse effects like liver function abnormalities, headache, diarrhea, dizziness, and nausea similar to allopurinol.16–19 Furthermore, a safety clinical trial carried out by the FDA on febuxostat demonstrated that its use can increase the risk of heart-related complications.20
Working on similar lines, another notable compound, pyranostat, was developed with potent in vivo efficacy but has poor pharmacokinetic profile.21 Topiroxostat, another non-purine based XO inhibitor, was approved for use in Japan in 2013. A Japanese Phase III study revealed that the reduction of serum uric acid by topiroxostat 120 mg per day was comparable to that of 200 mg per day of allopurinol but has adverse effects similar to allopurinol (Fig. 1).22
These adverse events associated with both purine and non-purine based xanthine oxidase inhibitors prompted researchers working in this field to develop alternate scaffolds as potent XO inhibitors that can be used for the treatment of hyperuricemia.
Among the various natural products, flavones have attracted much attention due to their significant range of pharmacological activities such as antioxidant, antimutagenic, antibacterial, antiangiogenic, anti-inflammatory, antiallergic, modulators of enzymatic activities and anti-cancer activity.23–26 There are numerous reports too on the xanthine oxidase inhibitory potential of flavones. Various reported flavones exerting considerable inhibitory potential against the xanthine oxidase enzyme are depicted in Fig. 2.27–33
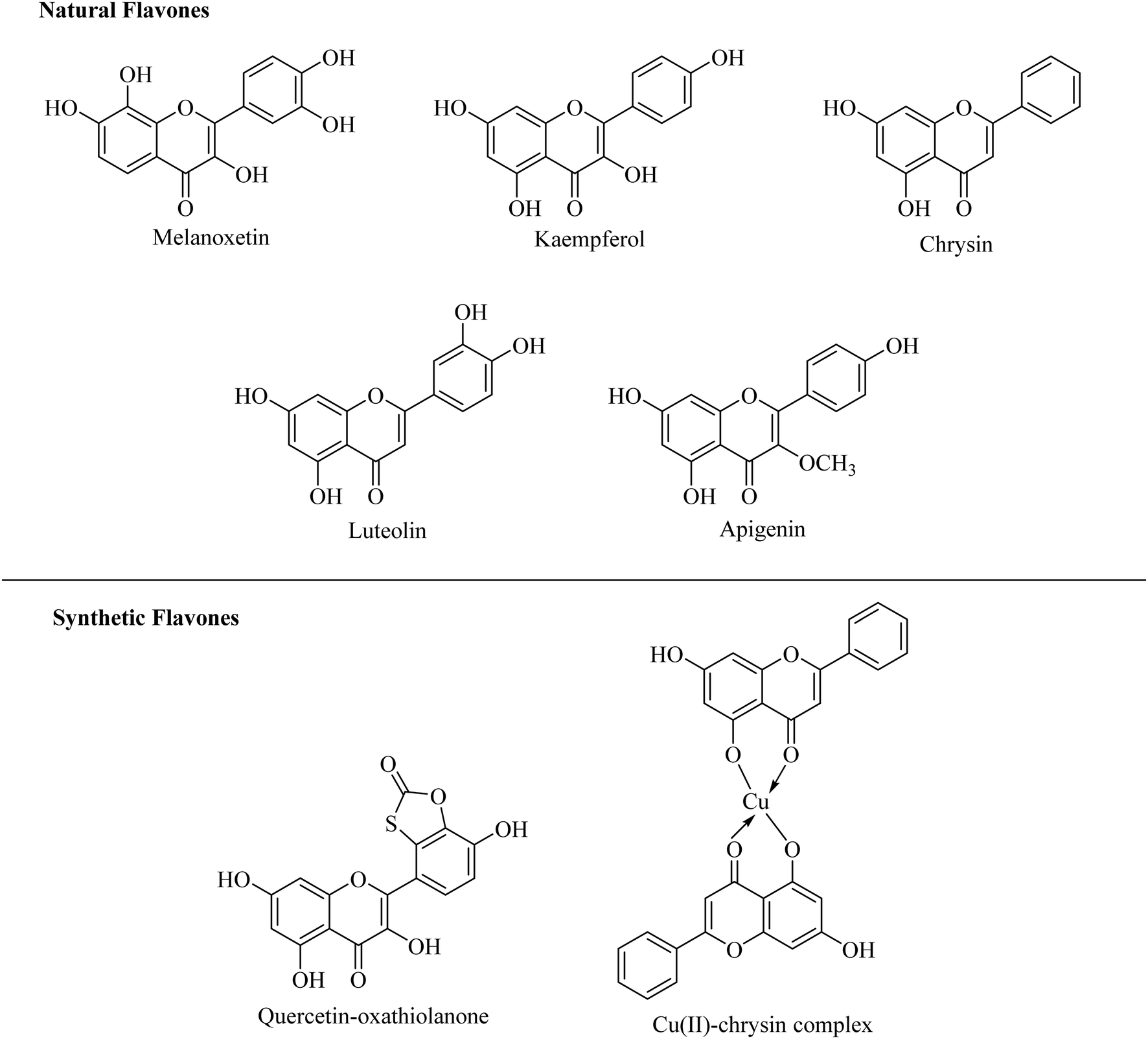 | ||
| Fig. 2 Various flavonoids as potent xanthine oxidase inhibitors. | ||
For the last five years, we have been actively involved in the development of non-purine based xanthine oxidase inhibitors.34–40 By keeping in view the significant XO inhibitory potential of various flavonoids, we have previously reported a series of 7,8-benzoflavone derivatives endowed with significant XO inhibitory potential. The design was based upon natural flavones exhibiting potent XO inhibition taking apigenin as the lead molecule. The established structure–activity relationship for flavones reveals that their XO inhibitory potential is attributed to the presence of hydroxyl groups on the benzopyran nucleus. We explored the role of the naphthyl ring as a surrogate for the hydroxyl groups present on the benzopyran moiety which actually enhanced the arene–arene and hydrophobic interactions within the active site of the xanthine oxidase enzyme.41 Among all the derivatives reported by our research group, this series was found to be a prominent class exhibiting IC50 values ranging from 0.62–41.2 μM against the XO enzyme, which inspired us to design a new series of compounds (i.e. 5,6-benzoflavones) by changing the orientation of ring-A from trans- to cis-configuration with respect to the carbonyl group of ring-C (Fig. 3). Molecular modeling studies provided a strong rationale behind the design of this new series of molecules as 5,6-benzoflavones found to be occupied well by the amino acid residues present in the active site of the XO enzyme with closer proximity as compared to the molecules of reported 7,8-benzoflavone series. Therefore, it can be suggested that the cis-orientation of ring-A with respect to the carbonyl group of ring-C is well tolerable for XO inhibitory potential.
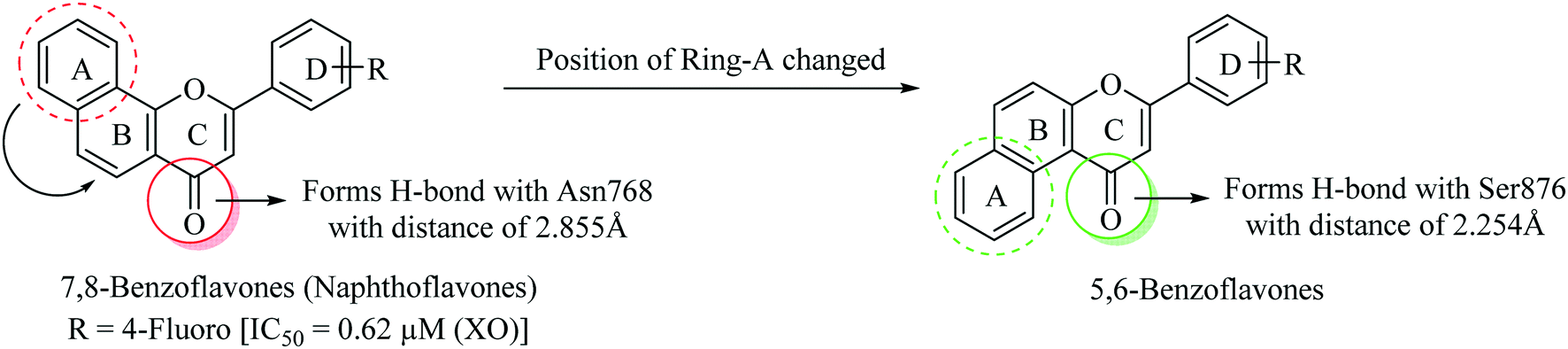 | ||
| Fig. 3 Design strategy for 5,6-benzoflavone derivatives. | ||
In the present study, all the designed compounds were synthesized and evaluated in vitro against the xanthine oxidase enzyme by using spectrophotometric assay.42,43 The mode of inhibition of the most potent compounds and their binding interactions with the amino acid residues present in the active site of the XO enzyme were also analyzed. Free radical scavenging assay was performed to examine the antioxidant potential for both the series of compounds (7,8- and 5,6-benzoflavones). Furthermore, the serum uric acid lowering ability of synthesized benzoflavones exhibiting potent XO enzyme inhibition was also investigated by using a potassium oxonate (PO) induced hyperuricemic mice model.
2. Results and discussion
2.1. Synthesis
Benzoflavone derivatives were synthesized viaScheme 1. α- and β-naphthol were subjected to Fries rearrangement. Fries rearranged products (1a and 1b) were then benzoylated using variously substituted benzoyl chlorides to obtain benzoylated products (2a and 2b) which were then subjected to Baker Venkataraman rearrangement. Baker Venkataraman rearranged products (3a & 3b) existing in the enol form (confirmed by the appearance of a singlet for two D2O exchangeable protons above 11 ppm along with the vinylic proton to carbonyl which appeared as a merged signal in a multiplet) were then cyclized by treatment with sulphuric acid to yield the desired benzoflavones. All the reactions proceeded smoothly with diverse benzoyl chlorides and products were obtained in good yields. No retro-Diels fragmentation was observed for derivatives in the mass spectrum. The structures of all the synthesized compounds were elucidated by 1H NMR, 13C NMR, MASS and elemental analysis. Spectral data were in accordance with the assumed structures.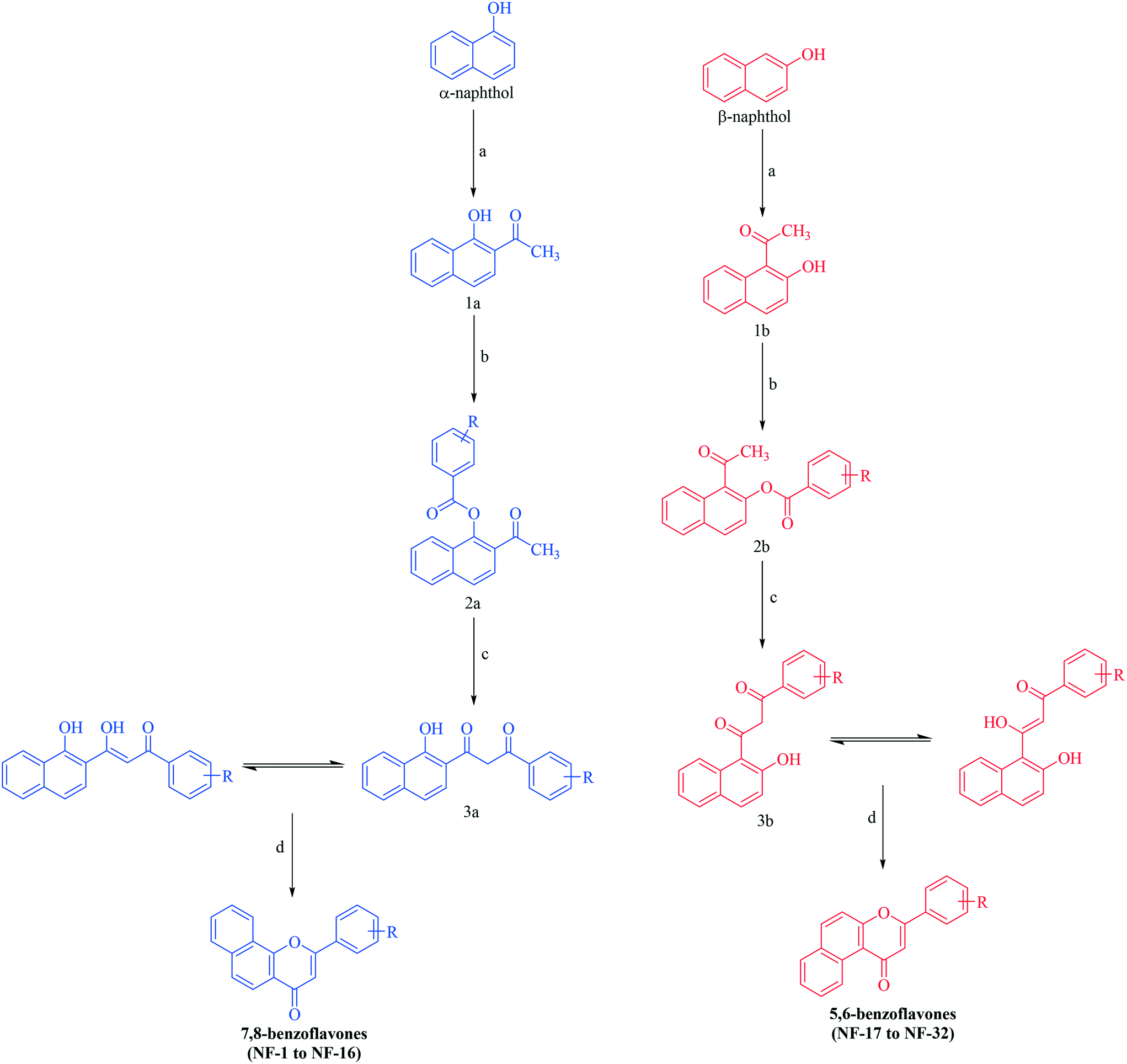 | ||
| Scheme 1 Synthesis of benzoflavone derivatives. Reagents and conditions: (a) MW, ZnCl2, CH3COOH, 20 min; (b) various substituted benzoyl chlorides, pyridine, stirring, rt, 1 h; (c) KOH, pyridine, warm, 15 min; (d) a drop of conc. H2SO4, CH3COOH, reflux, 30 min. | ||
2.2. In vitro xanthine oxidase assay
The xanthine oxidase inhibitory potential of all the synthetics was elucidated (Table 1) by using spectrophotometric assay as described in the literature. Allopurinol, febuxostat, and apigenin were used as reference compounds.42,43 Among the previously reported 7,8-benzoflavones, compounds NF-2, NF-4, and NF-12 exhibited IC50 values below 5 μM and NF-4 was found to be the most potent XO inhibitor with an IC50 value of 0.62 μM.41 The established structure–activity relationship for 7,8-benzoflavones revealed that the phenyl ring (ring-D) is very sensitive to nature as well as the positioning of the substituent present. The substituted compounds were more active than the unsubstituted ones. The overall preference order for the substituent was found to be halo and nitro > methoxy > methyl > hydrogen. Simultaneously, ortho and para positioning of the substituents on ring-D is favorable for XO inhibitory activity as compared to meta substitution. Among the series of newly designed compounds (5,6-benzoflavones), NF-28 exhibited the most potent XO inhibitory activity with an IC50 value of 5.21 μM. Compounds with ortho and para substituted phenyl rings were more potent than meta-substituted compounds (see the IC50 values of NF-19, NF-21, NF-24, and NF-27). It has been observed that the stronger inductive effect at the para position favors the xanthine oxidase inhibitory activity (see the IC50 values of NF-25, NF-28, and NF-32). The similar effect had also been seen in the previously reported results for 7,8-benzoflavones, whereas, the overall potency of 5,6-benzoflavones was lower than that of 7,8-benzoflavones, which remains unjustified. The overall results showed eight compounds (NF-2, NF-4, NF-9, NF-12, NF-16, NF-25, NF-28, and NF-32) with IC50 values comparable to that of allopurinol (IC50 < 10 μM) which were further selected to perform in vivo experiments to check their serum uric acid lowering ability by using a hyperuricemic mice model. Moreover, the type of inhibition for the most potent compounds has been analyzed by using enzyme kinetic studies.| Code | Structure | Xanthine oxidase IC50 (μM + SD) | Antioxidant IC50 (μM + SD) |
|---|---|---|---|
| a Literature value.41 b Literature value.52 | |||
| NF-1 |
|
>20 | 15.55 ± 1.78 |
| NF-2 |
|
4.94 ± 0.45 | 4.73 ± 1.23 |
| NF-3 |
|
>20 | 10.13 ± 1.98 |
| NF-4 |
|
0.62 ± 0.10 | 0.83 ± 0.14 |
| NF-5 |
|
>20 | 7.75 ± 1.55 |
| NF-6 |
|
>20 | 10.90 ± 1.89 |
| NF-7 |
|
16.80 ± 1.78 | 9.90 ± 1.67 |
| NF-8 |
|
>20 | 5.98 ± 1.45 |
| NF-9 |
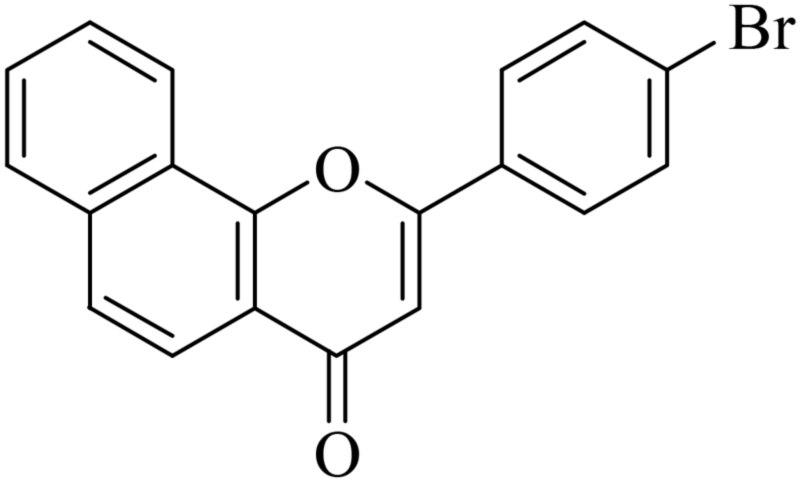
|
8.93 ± 0.67 | 0.35 ± 0.09 |
| NF-10 |
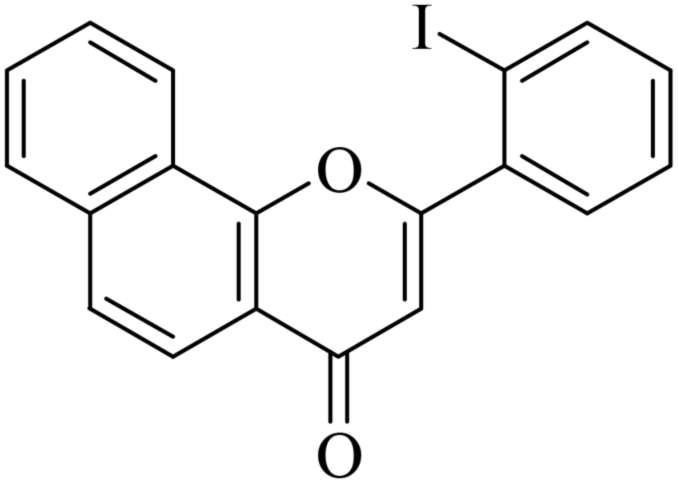
|
19.40 ± 1.89 | 7.33 ± 1.54 |
| NF-11 |
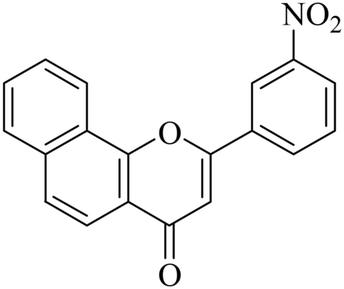
|
>20 | 8.24 ± 1.56 |
| NF-12 |
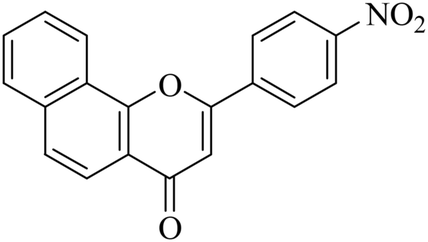
|
1.95 ± 0.18 | 0.62 ± 0.11 |
| NF-13 |
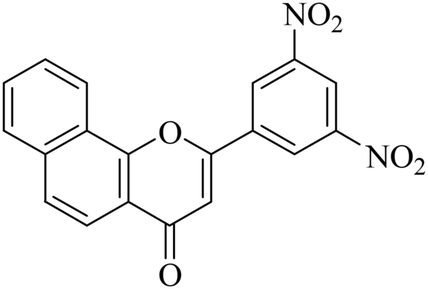
|
>20 | 5.55 ± 1.55 |
| NF-14 |
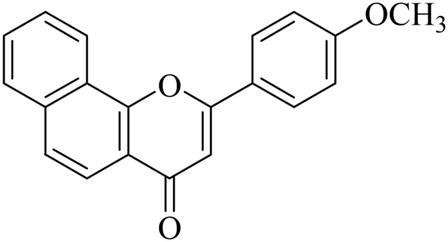
|
18.90 ± 1.81 | 4.27 ± 1.26 |
| NF-15 |
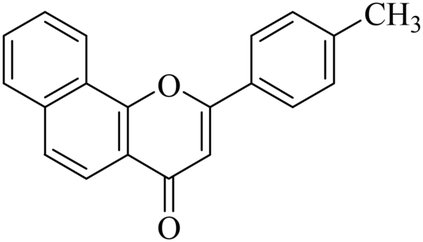
|
>20 | 6.50 ± 1.34 |
| NF-16 |
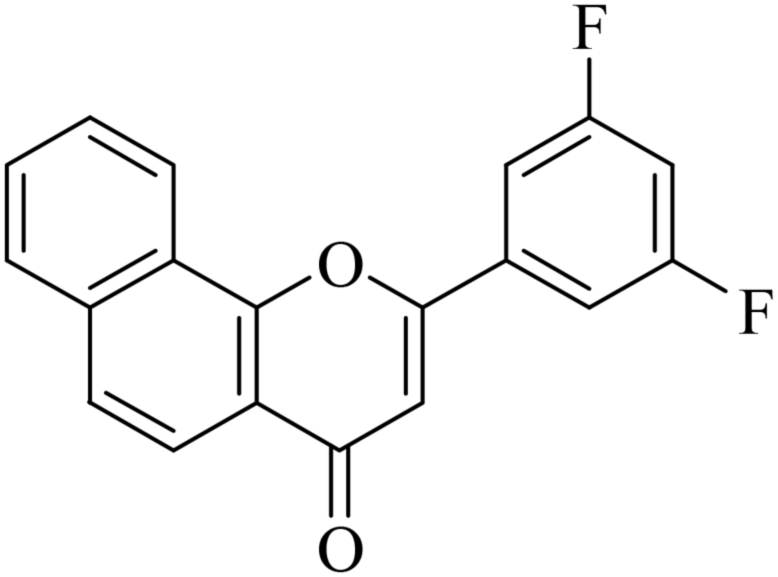
|
6.95 ± 0.62 | 3.14 ± 1.22 |
| NF-17 |
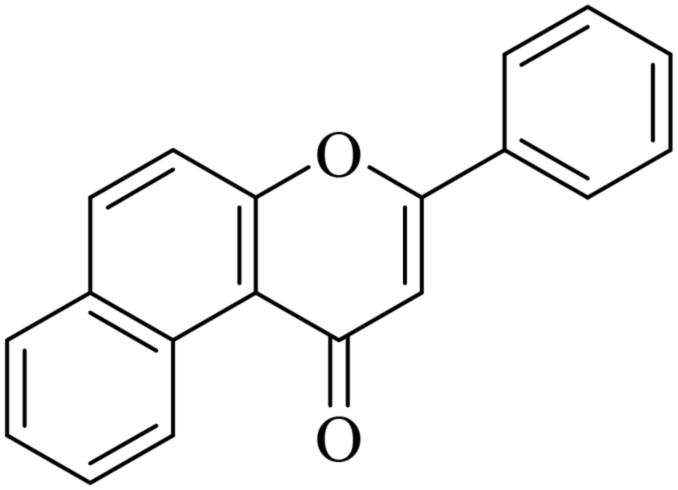
|
>20 | >20 |
| NF-18 |
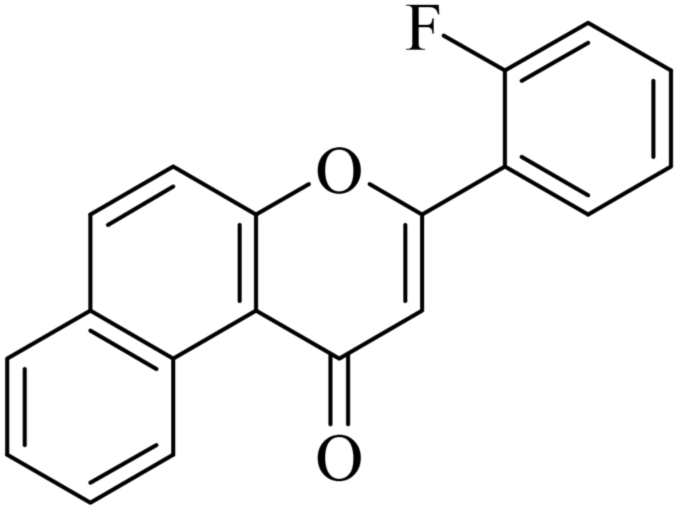
|
14.67 ± 1.58 | >20 |
| NF-19 |
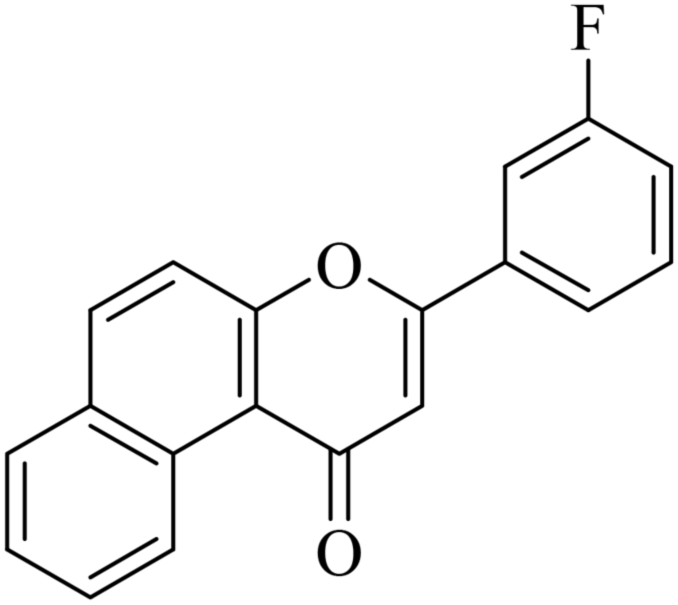
|
>20 | >20 |
| NF-20 |
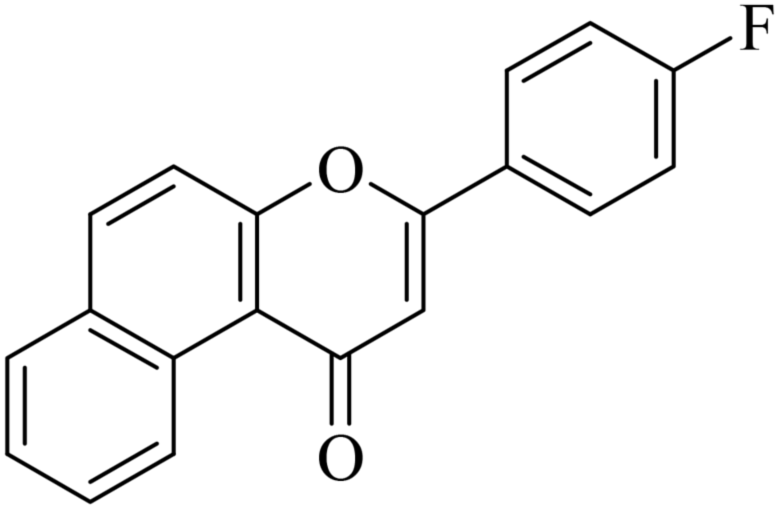
|
12.44 ± 1.34 | >20 |
| NF-21 |
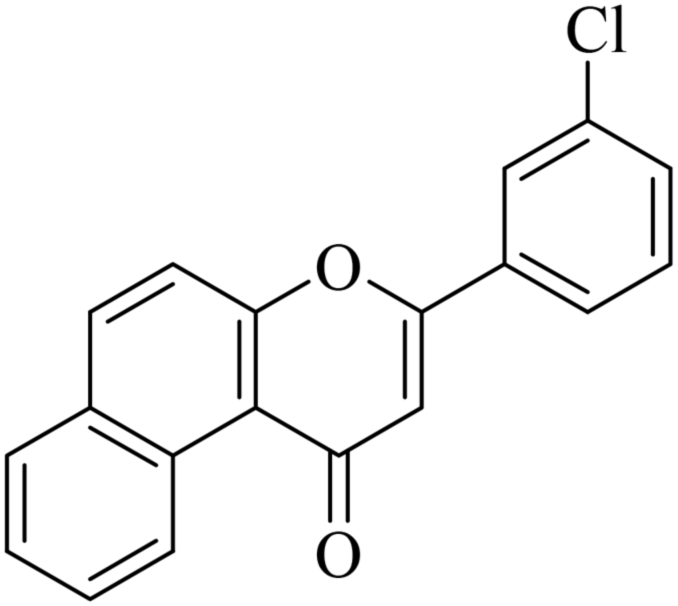
|
>20 | >20 |
| NF-22 |
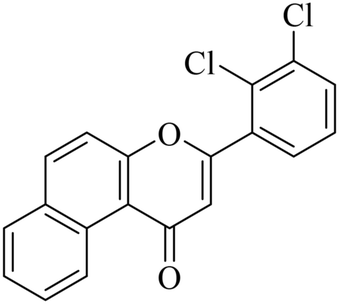
|
18.58 ± 1.23 | >20 |
| NF-23 |
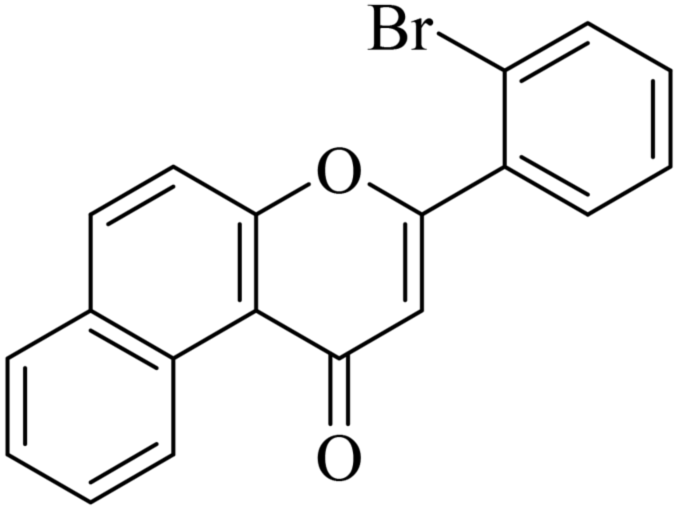
|
17.07 ± 1.63 | >20 |
| NF-24 |
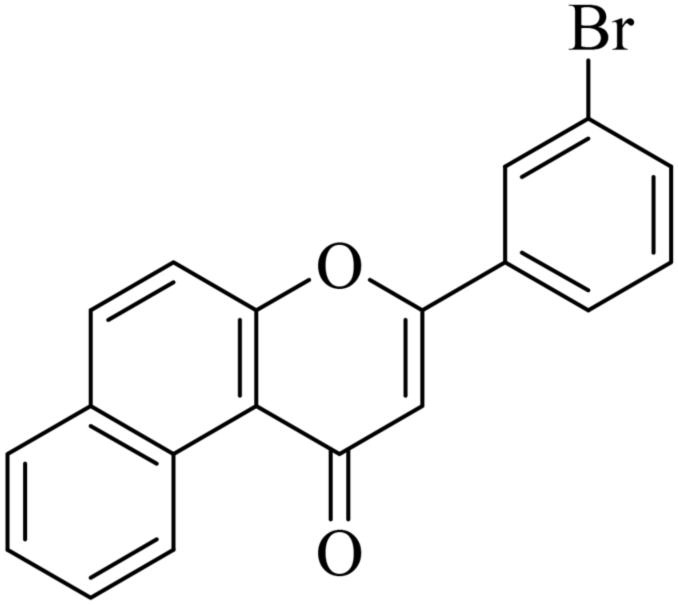
|
>20 | >20 |
| NF-25 |
|
8.90 ± 0.56 | >20 |
| NF-26 |
|
>20 | >20 |
| NF-27 |
|
>20 | >20 |
| NF-28 |
|
5.21 ± 0.65 | >20 |
| NF-29 |
|
>20 | >20 |
| NF-30 |
|
>20 | >20 |
| NF-31 |
|
>20 | >20 |
| NF-32 |
|
7.67 ± 0.79 | >20 |
| Allopurinol | 8.69 | — | |
| Febuxostat | 0.03 | — | |
| Apigenin | 1.11a | — | |
| Quercetin | — | 28.00b | |
2.3. Enzyme kinetics
Compound NF-28 was further investigated for the type of inhibition by performing enzyme kinetic studies. The Lineweaver–Burk plot (Fig. 4) observed for NF-28 was very similar to that for NF-4.41 The pattern of the graph shows that it is a form of mixed inhibition scenario where Km, Vmax, and the slope are all affected by the inhibitor. The inhibitor has increased the Km and slope (Km/Vmax) while decreasing the Vmax. Moreover, on careful observation, it was found that intersecting lines on the graph converge to the left of the y-axis and above the x-axis which indicates that the value of α (a constant that defines the degree to which inhibitor binding affects the affinity of the enzyme for the substrate) is greater than 1.44 This confirms that the inhibitor preferentially binds to the free enzyme and not the enzyme–substrate complex. Therefore, the mode of inhibition of NF-28 is mixed-type but it seems that it has a strong competitive component.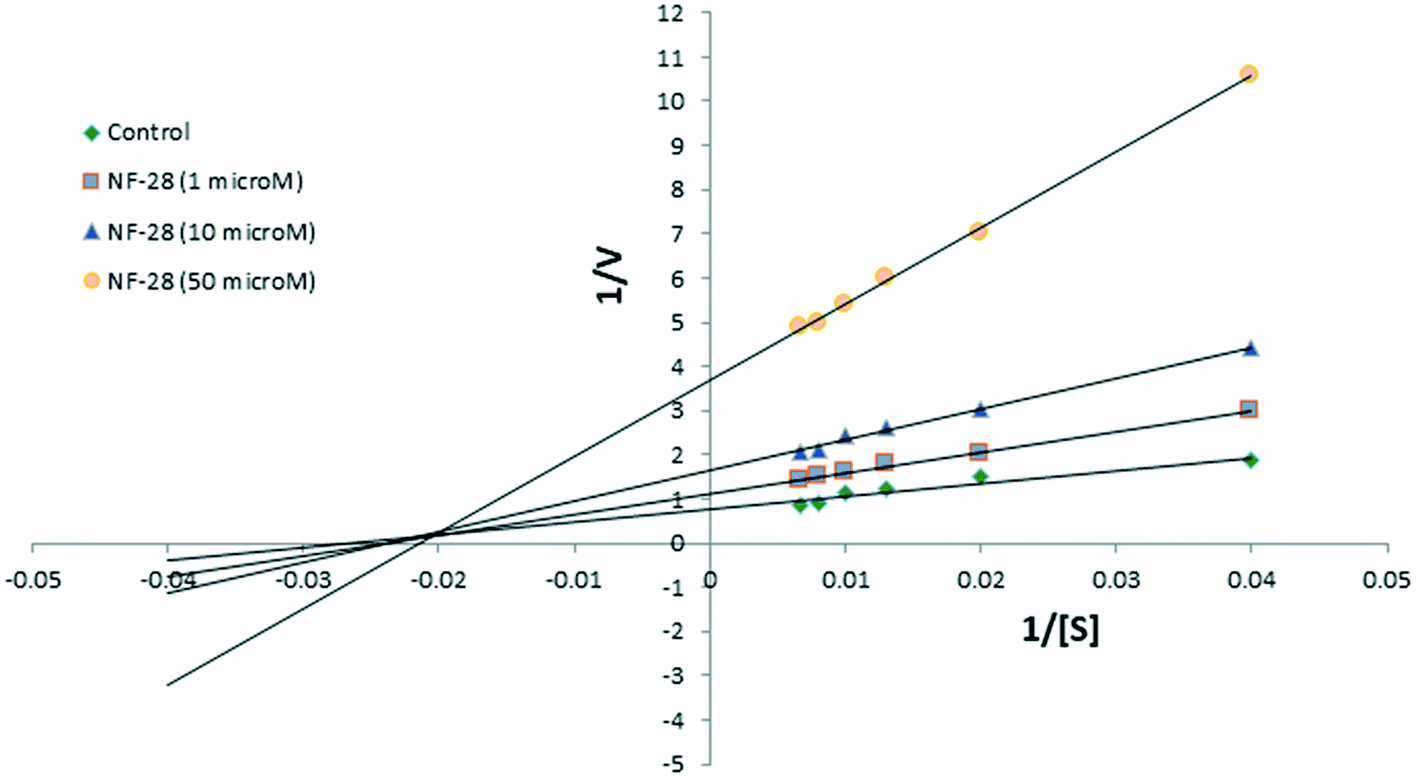 | ||
| Fig. 4 Lineweaver–Burk plot of NF-28. | ||
2.4. Docking study
To understand the binding conformation of inhibitors, the potent XO inhibitors NF-4 and NF-28 were docked into the febuxostat binding site. The docking procedure was validated by reproducing a co-crystallized conformation of febuxostat. The software used can reproduce the co-crystallized conformation of febuxostat with an RMSD value of 0.792 Å (Fig. 5), which ensures the acceptability of the selected docking protocol. Several docked conformations of NF-28 were generated and ranked according to their in silico binding affinity. The conformation with the highest binding affinity (−11.1 kcal mol−1) was selected for discussion (Fig. 6).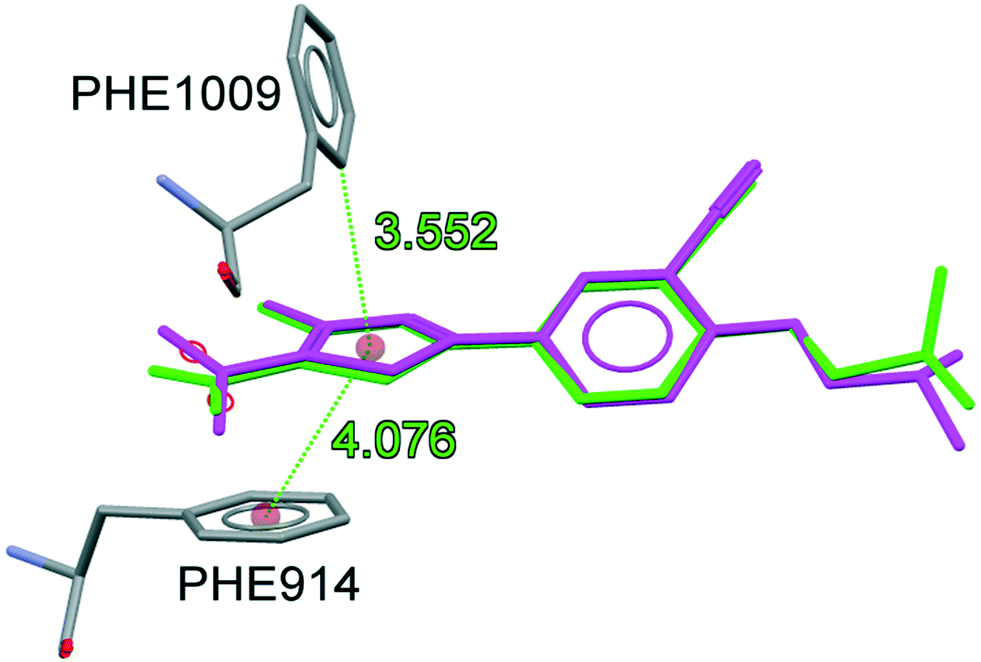 | ||
| Fig. 5 Superimposition of co-crystalized (green) and docked conformations (magenta) of febuxostat (RMSD: 0.792 Å). | ||
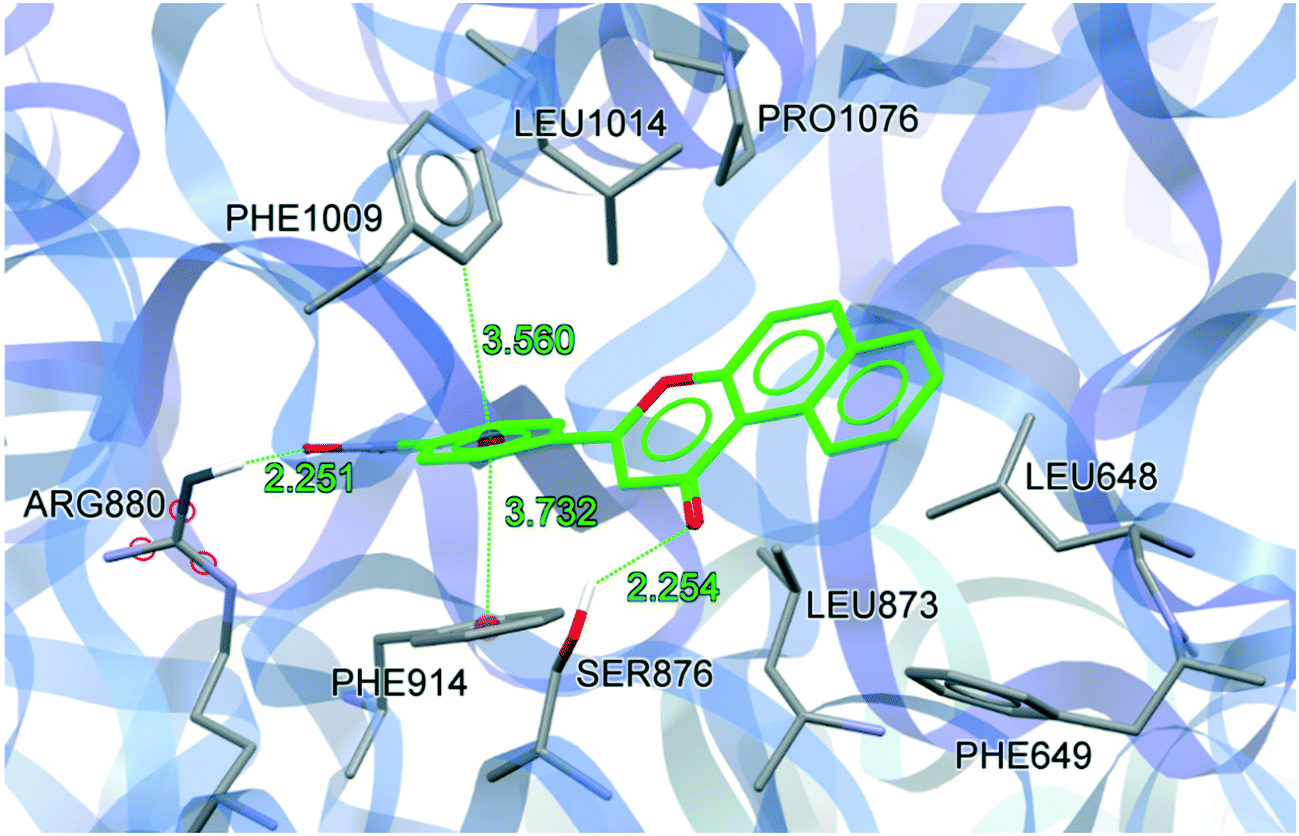 | ||
| Fig. 6 Docking conformation of NF-28 at the febuxostat binding site (hydrogen atoms which are involved in H-bond interactions are shown). | ||
The binding site residues and overall binding mode of NF-28 are like those observed with febuxostat,45 salicylic acid46 and curcumin.47NF-28 gets stabilized at the binding cavity by several types of electrostatic interactions. The major interactions between NF-28 and XO include π–π stacking with Phe914 and Phe1009 and two hydrogen bonds with Ser876 and Arg880 (Fig. 6). Rings A and B get positioned in a cavity formed by hydrophobic residues Leu648, Phe649, Leu873, Leu1014, and Pro1076. The carbonyl function at ring C has shown H-bond interaction with the hydroxyl group of Ser876 (H-bond donor; d = 2.254 Å). Another H-bond was observed between the N+–O− function at ring D and terminal –NH2 of Arg880 (H-bond donor; d = 2.251 Å). The phenyl rings of Phe914 and Phe1009 are involved in face-to-face and edge-to-face π–π stacking interactions, respectively, with ring D (d = 3.372 Å and 3.560 Å). This arrangement of energetically favorable π–π stacking interactions was also seen in the co-crystal structure of XO with febuxostat and salicylate.45,46 Its conservation argues for an important role in stabilizing the binding positions of aromatic substrates and it might well represent one of the key features of substrate recognition.
We also compared the docked conformations of the previously reported most potent compounds NF-4 and NF-28 in the febuxostat binding site using AutoDock Vina for a clear and deep perception behind the potency against XO (Fig. 7). The binding affinity of NF-4 for XO was found to be −9.5 kcal mol−1, which is approximately 14% lower than that of NF-28. The docked conformation of both inhibitors (NF-4 and NF-28) showed that, in both cases, ring-A and ring-B get positioned in a cavity formed by hydrophobic residues Leu648, Phe649, Leu873, Leu1014, and Pro1076. The major differences in the binding modes of NF-4 and NF-28 include the opposite orientation of ring-C and the higher distances between NF-4 and important binding site residues of XO (Table 2, Fig. 7). The carbonyl function at ring-C adopted an opposite orientation to that of NF-28 and is involved in H-bonding interaction with the terminal –NH2 group of Asn768 (d = 2.855 Å; 0.601 Å higher than that of NF-28 where the carbonyl function makes a H-bond with Ser876). Also, the distances between ring-D and Phe914 (face-to-face π–π stacking interactions) and the 4-fluoro group at ring-D and Arg880 were also observed to be higher than those of NF-28 (Table 2). Therefore, according to computational studies, this can suggest that ring-A in cis orientation with the carbonyl function at ring-C is more favorable for binding than its trans orientation and thus a series of 5,6-benzoflavones could be more active than the reported 7,8-benzoflavone series which was the rationale behind the synthesis of the current series of molecules. But according to in vitro results, the molecules of the previous series were found to be more active than the molecules of the new series.
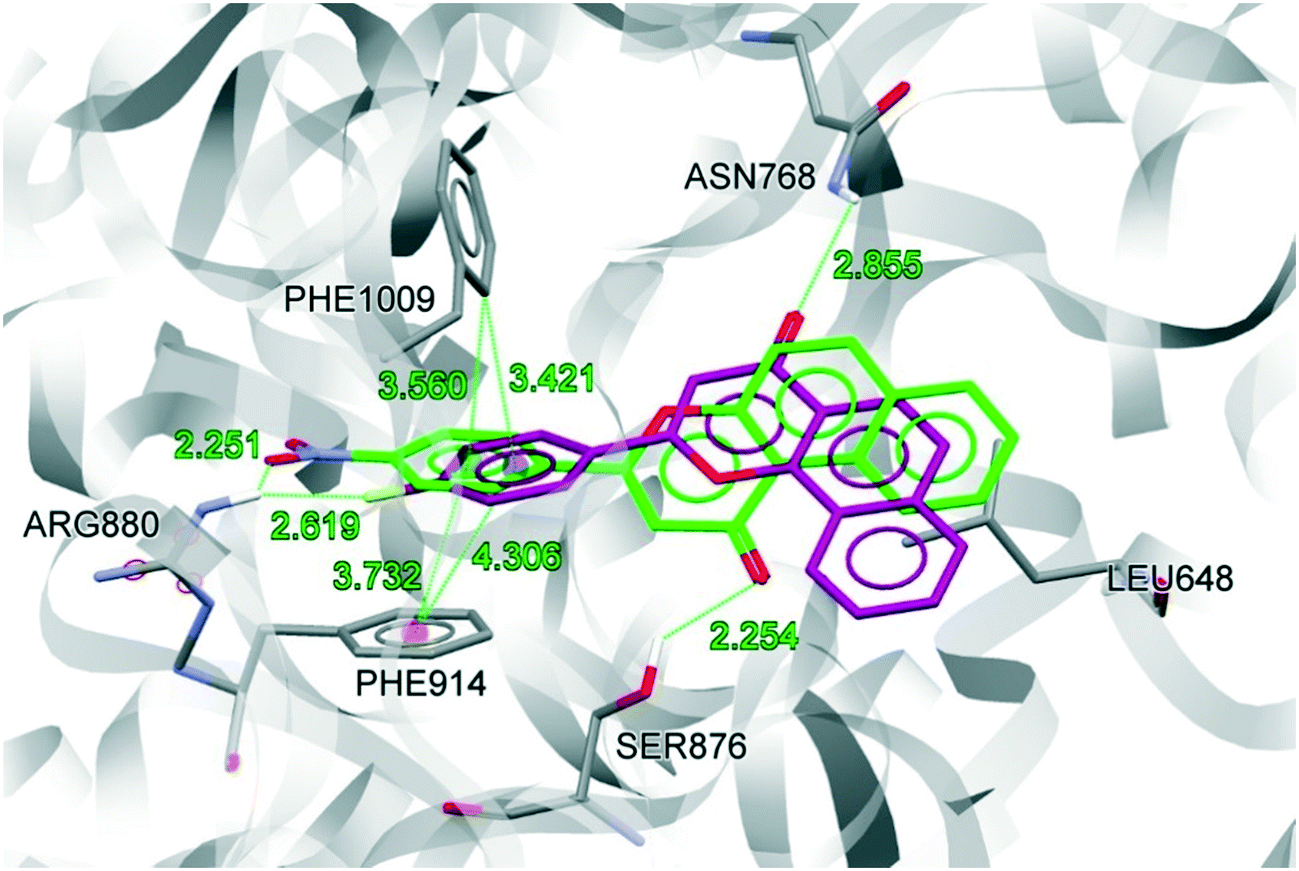 | ||
| Fig. 7 Superimposition of the docked conformations of NF-4 (magenta) and NF-28 (green). | ||
| S. no. | Residues | Distancea Å | Differenceb | |
|---|---|---|---|---|
| NF-4 | NF-28 | |||
| a Refer to Fig. 7 for bond details. b Difference between NF-4 and NF-28. c Difference in H-bond distance involving Asn768 and Ser876. | ||||
| 1 | Asn768 | 2.855 | — | 0.601c |
| 2 | Ser876 | — | 2.254 | |
| 3 | Arg880 | 2.619 | 2.251 | 0.368 |
| 4 | Phe914 | 4.306 | 3.732 | 0.574 |
| 5 | Phe1009 | 3.421 | 3.560 | −0.139 |
2.5. Antioxidant activity (DPPH assay)
During the oxidative hydroxylation of hypoxanthine and xanthine to produce uric acid, the reduction of oxygen at the flavin center simultaneously generated a superoxide anion radical or hydrogen peroxide.1 Therefore, it would be beneficial if the compounds with XO inhibitory potential can possess antioxidant potential as well. 1,1-Diphenyl-2-picrylhydrazyl (DPPH) radical scavenging assay was used to determine the free radical scavenging ability of all the synthesized compounds. The results of DPPH assay (Table 1) revealed that some of the compounds have significant antioxidant potential. Among the active compounds, NF-9 was found to be the most potent antioxidant with an IC50 value of 0.35 μM, followed by NF-4 and NF-12 with IC50 values of 0.83 μM and 0.62 μM, respectively. Surprisingly, 5,6-benzoflavones were found to be less potent as compared to 7,8-benzoflavones with IC50 values greater than 20 μM for all the compounds. Compounds with para substitution on the phenyl ring (ring D) are more active than the ortho and meta-substituted compounds whereas the unsubstituted compound (NF-1) showed moderate antioxidant activity.2.6. In silico study
Furthermore, in silico physicochemical properties like absorption, distribution, metabolism, and excretion (ADME) of the synthesized compounds were determined by http://www.chemaxon.com/) and PreADMET (http://preadmet.bmdrc.org/). Particularly, Caco-2 cell, MDCK cell, blood brain barrier (BBB) & skin permeabilities, human intestinal absorption and plasma protein binding affinities were predicted by the above-mentioned applications and are summarized in Table 3. The results indicate that the compounds are predicted to have lower blood–brain barrier permeation which is less likely to cause neurotoxicity. In other words, we can say that the synthesized compounds cannot alter the normal activity of neuronal cells. The basicity and lipophilicity of all the synthesized compounds were determined with ChemAxon software MarvinSketch and the results are shown in Table 4 which defines the compliance of all the synthesized compounds with the Lipinski rule of five. Tabular values indicate that a) all the molecules have a molecular weight in the range of 286–408 which lies in the limit of 180–500; b) compounds have no H-bond donating property; c) compounds follow the H-bond acceptor criteria (<10); d) molar refractivities are consistent and in the range of 83.64–98.07 which lies well in the permitted range of 40–130 and e) log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of all the compounds is lower than 5.6 which indicates that the compounds are not very lipophilic. These results suggest that all the compounds follow the Lipinski rule of five and ADME properties which makes them pharmacologically efficient for use in the animal model. Therefore, eight potent xanthine oxidase inhibitors were chosen for in vivo experiments to evaluate their serum uric acid lowering capacity.
P of all the compounds is lower than 5.6 which indicates that the compounds are not very lipophilic. These results suggest that all the compounds follow the Lipinski rule of five and ADME properties which makes them pharmacologically efficient for use in the animal model. Therefore, eight potent xanthine oxidase inhibitors were chosen for in vivo experiments to evaluate their serum uric acid lowering capacity.
| Compound | Absorption | Distribution | ||||
|---|---|---|---|---|---|---|
| Human intestinal absorption (HIA)% | In vitro Caco-2 cell permeability (nm s−1) | In vitro MDCK cell permeability (nm s−1) |
In vitro skin permeability (logKp) cm h−1 |
In vitro plasma protein binding (%) | In vivo blood brain barrier penetration (C. brain/C. blood) | |
| NF-1 | 100 | 56.74 | 35.60 | −2.64 | 94.25 | 2.89 |
| NF-2 | 100 | 54.20 | 21.03 | −2.95 | 95.32 | 2.63 |
| NF-3 | 100 | 54.21 | 29.51 | −2.97 | 98.77 | 2.05 |
| NF-4 | 100 | 55.39 | 11.79 | −2.93 | 98.84 | 0.23 |
| NF-5 | 100 | 47.71 | 26.74 | −2.71 | 95.46 | 0.96 |
| NF-6 | 100 | 49.57 | 29.92 | −2.62 | 99.82 | 0.98 |
| NF-7 | 100 | 46.32 | 0.19 | −2.56 | 100 | 1.89 |
| NF-8 | 100 | 46.46 | 0.14 | −2.58 | 99.96 | 0.85 |
| NF-9 | 100 | 46.45 | 0.08 | −2.58 | 100 | 0.45 |
| NF-10 | 100 | 46.34 | 0.26 | −2.62 | 100 | 2.07 |
| NF-11 | 91.92 | 18.77 | 1.74 | −3.67 | 99.48 | 3.33 |
| NF-12 | 91.92 | 6.12 | 0.20 | −3.67 | 99.80 | 2.10 |
| NF-13 | 75.68 | 17.99 | 0.09 | −3.72 | 100 | 2.17 |
| NF-14 | 98.80 | 57.27 | 9.21 | −2.79 | 93.78 | 0.07 |
| NF-15 | 100 | 56.23 | 15.97 | −2.55 | 93.85 | 0.60 |
| NF-16 | 100 | 52.39 | 19.45 | −3.11 | 100 | 1.11 |
| NF-17 | 100 | 56.73 | 50.02 | −2.65 | 95.97 | 2.75 |
| NF-18 | 100 | 54.71 | 20.11 | −2.91 | 98.40 | 2.17 |
| NF-19 | 100 | 54.75 | 27.06 | −2.94 | 100 | 0.28 |
| NF-20 | 100 | 55.39 | 3.48 | −2.94 | 100 | 0.24 |
| NF-21 | 100 | 47.91 | 26.61 | −2.70 | 96.47 | 0.42 |
| NF-22 | 100 | 50.14 | 20.56 | −2.62 | 100 | 0.80 |
| NF-23 | 100 | 46.44 | 0.16 | −2.57 | 100 | 1.27 |
| NF-24 | 100 | 46.57 | 0.12 | −2.58 | 100 | 0.45 |
| NF-25 | 100 | 46.46 | 0.01 | −2.58 | 100 | 0.46 |
| NF-26 | 100 | 46.45 | 0.23 | −2.62 | 100 | 1.07 |
| NF-27 | 98.49 | 21.43 | 0.42 | −2.86 | 95.25 | 0.01 |
| NF-28 | 98.49 | 15.17 | 0.33 | −2.86 | 96.00 | 0.01 |
| NF-29 | 96.03 | 20.38 | 0.06 | −2.83 | 94.55 | 0.04 |
| NF-30 | 98.80 | 57.28 | 2.31 | −2.80 | 95.48 | 0.08 |
| NF-31 | 100 | 56.22 | 5.38 | −2.56 | 95.75 | 0.36 |
| NF-32 | 100 | 73.77 | 10.53 | −3.12 | 100 | 0.30 |
| Compound | Molecular weight | No. of H-bond donors | No. of H-bond acceptors | Molar refractivity | logP |
No. of Lipinski violation |
|---|---|---|---|---|---|---|
| NF-1 | 272 | 0 | 2 | 83.42 | 3.96 | 0 |
| NF-2 | 290 | 0 | 2 | 83.64 | 4.10 | 0 |
| NF-3 | 290 | 0 | 2 | 83.64 | 4.10 | 0 |
| NF-4 | 290 | 0 | 2 | 83.64 | 4.10 | 0 |
| NF-5 | 306 | 0 | 2 | 88.23 | 4.56 | 0 |
| NF-6 | 341 | 0 | 2 | 93.03 | 5.16 | 0 |
| NF-7 | 351 | 0 | 2 | 91.04 | 4.73 | 0 |
| NF-8 | 351 | 0 | 2 | 91.04 | 4.73 | 0 |
| NF-9 | 351 | 0 | 2 | 91.04 | 4.73 | 0 |
| NF-10 | 398 | 0 | 2 | 96.78 | 4.89 | 0 |
| NF-11 | 317 | 0 | 4 | 90.75 | 3.90 | 0 |
| NF-12 | 317 | 0 | 4 | 90.75 | 3.90 | 0 |
| NF-13 | 362 | 0 | 6 | 98.07 | 3.84 | 0 |
| NF-14 | 302 | 0 | 3 | 89.88 | 3.80 | 0 |
| NF-15 | 286 | 0 | 2 | 88.46 | 4.47 | 0 |
| NF-16 | 308 | 0 | 2 | 83.85 | 4.24 | 0 |
| NF-17 | 272 | 0 | 2 | 83.42 | 3.90 | 0 |
| NF-18 | 290 | 0 | 2 | 83.64 | 4.10 | 0 |
| NF-19 | 290 | 0 | 2 | 83.64 | 4.10 | 0 |
| NF-20 | 290 | 0 | 2 | 83.64 | 4.10 | 0 |
| NF-21 | 306 | 0 | 2 | 88.23 | 4.56 | 0 |
| NF-22 | 341 | 0 | 2 | 93.03 | 5.16 | 0 |
| NF-23 | 351 | 0 | 2 | 91.04 | 4.73 | 0 |
| NF-24 | 351 | 0 | 2 | 91.04 | 4.73 | 0 |
| NF-25 | 351 | 0 | 2 | 91.04 | 4.73 | 0 |
| NF-26 | 398 | 0 | 2 | 96.78 | 4.89 | 0 |
| NF-27 | 317 | 0 | 4 | 90.75 | 3.90 | 0 |
| NF-28 | 317 | 0 | 4 | 90.75 | 3.90 | 0 |
| NF-29 | 362 | 0 | 6 | 98.07 | 3.84 | 0 |
| NF-30 | 302 | 0 | 3 | 89.88 | 3.80 | 0 |
| NF-31 | 286 | 0 | 2 | 88.46 | 4.47 | 0 |
| NF-32 | 308 | 0 | 2 | 83.85 | 4.24 | 0 |
2.7. In vivo antihyperuricemic activity
Owing to their XO inhibitory potential, eight compounds were subjected for the evaluation of antihyperuricemic effects by using a potassium oxonate-induced hyperuricemic mice model. The mean serum uric acid level of normal mice was found to be 1.8 ± 0.2 mg dL−1. A significant enhancement was observed in the serum uric acid level (5.9 ± 0.42 mg dL−1) with the administration of potassium oxonate (PO) and xanthine (Xn). Allopurinol and febuxostat were used as standard drugs for comparison, which reduced the serum uric acid level of potassium oxonate-induced hyperuricemic mice to 1.8 and 1.5 mg dL−1, respectively. The serum uric acid levels of potassium oxonate-induced hyperuricemic mice treated with the synthesized compounds exhibited a dose-dependent hypouricemic effect. All the compounds showed a significant reduction in serum uric acid when tested at three dose levels (1 mg kg−1, 5 mg kg−1 & 10 mg kg−1). Compounds NF-2, NF-9, NF-16, NF-25 and NF-32 decreased the uric acid level ranging from 1.8–2.8 mg dL−1 even at 1 mg kg−1 which is comparable to standard drugs. These five compounds were further evaluated at a dose level of 0.5 mg kg−1 but a significant decrease in their hypouricemic effect was observed at this concentration. Two compounds NF-25 and NF-32 were found to exhibit a hypouricemic effect comparable to febuxostat at a dose level of 5 mg kg−1. Compound NF-28 was found to be less potent in vivo and showed a significant hypouricemic effect only at a dose level of 10 mg kg−1 whereas compounds NF-4 and NF-12 were not able to reduce serum uric acid considerably even at a dose level of 10 mg kg−1. The overall study revealed that six compounds (NF-2, NF-9, NF-16, NF-25, NF-28, and NF-32 except for NF-4 & NF-12) can reduce the serum uric acid to its normal level. Careful examination of Fig. 8 and 9 shows that there is no significant difference between the hypouricemic effect of standard drugs used and compounds NF-2, NF-9, NF-16, NF-25 and NF-32 at 5 and 10 mg kg−1. Thus, the overall potency order of the compounds in comparison with febuxostat (at 5 mg kg−1) is as follows: NF-32 > NF-25 > NF-2 > NF-9 > NF-16 > NF-28 > NF-12 > NF-4. Therefore, it is expected that these potent compounds can act as clinical candidates for the treatment of hyperuricemia.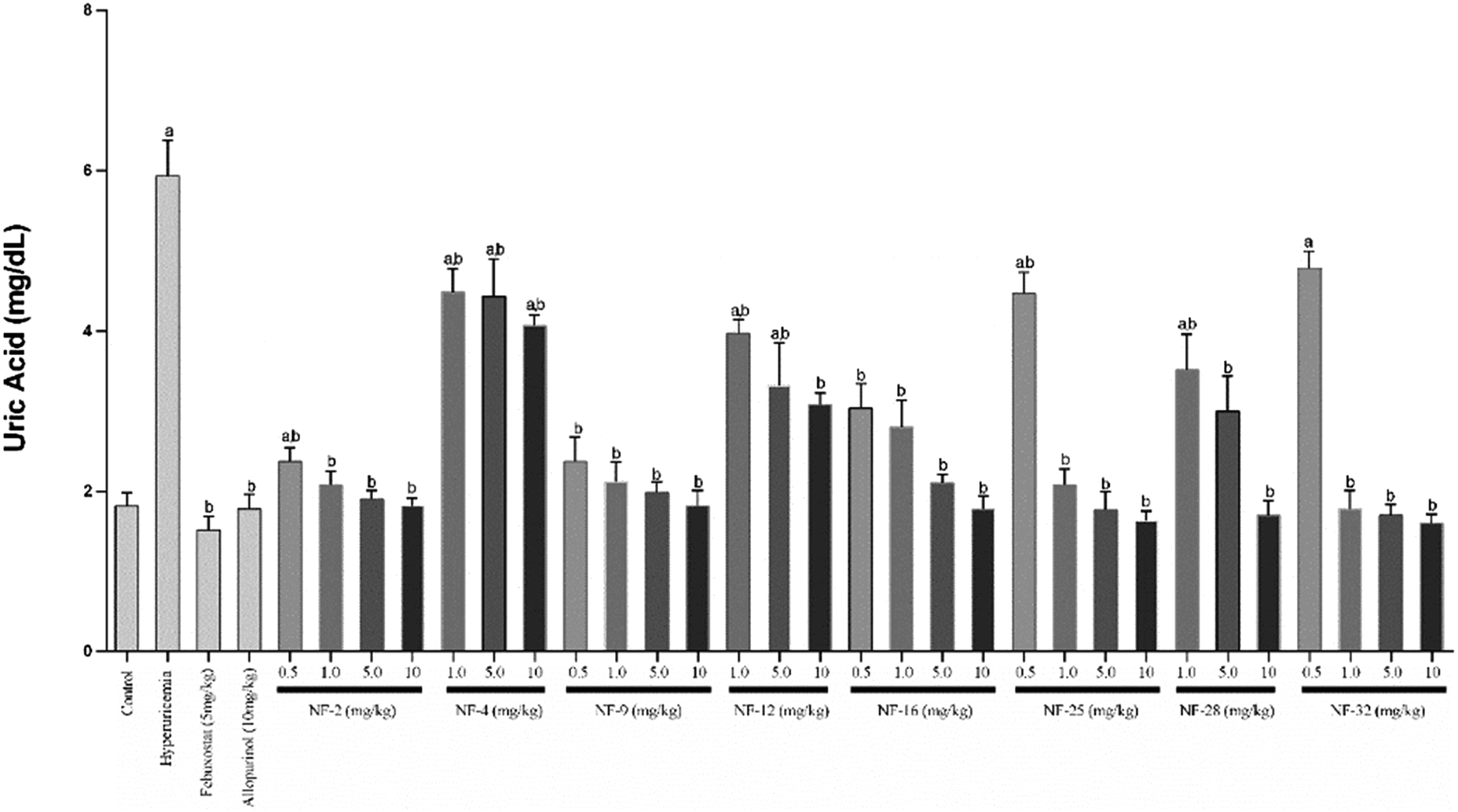 | ||
| Fig. 8 Effect of compounds on the serum uric acid levels in a potassium oxonate-induced hyperuricemic mice model. The values are shown as the mean ± S.E.M (n = 6). ap < 0.05 compared with the control (vehicle), bp < 0.05 compared with the hyperuricemic control. | ||
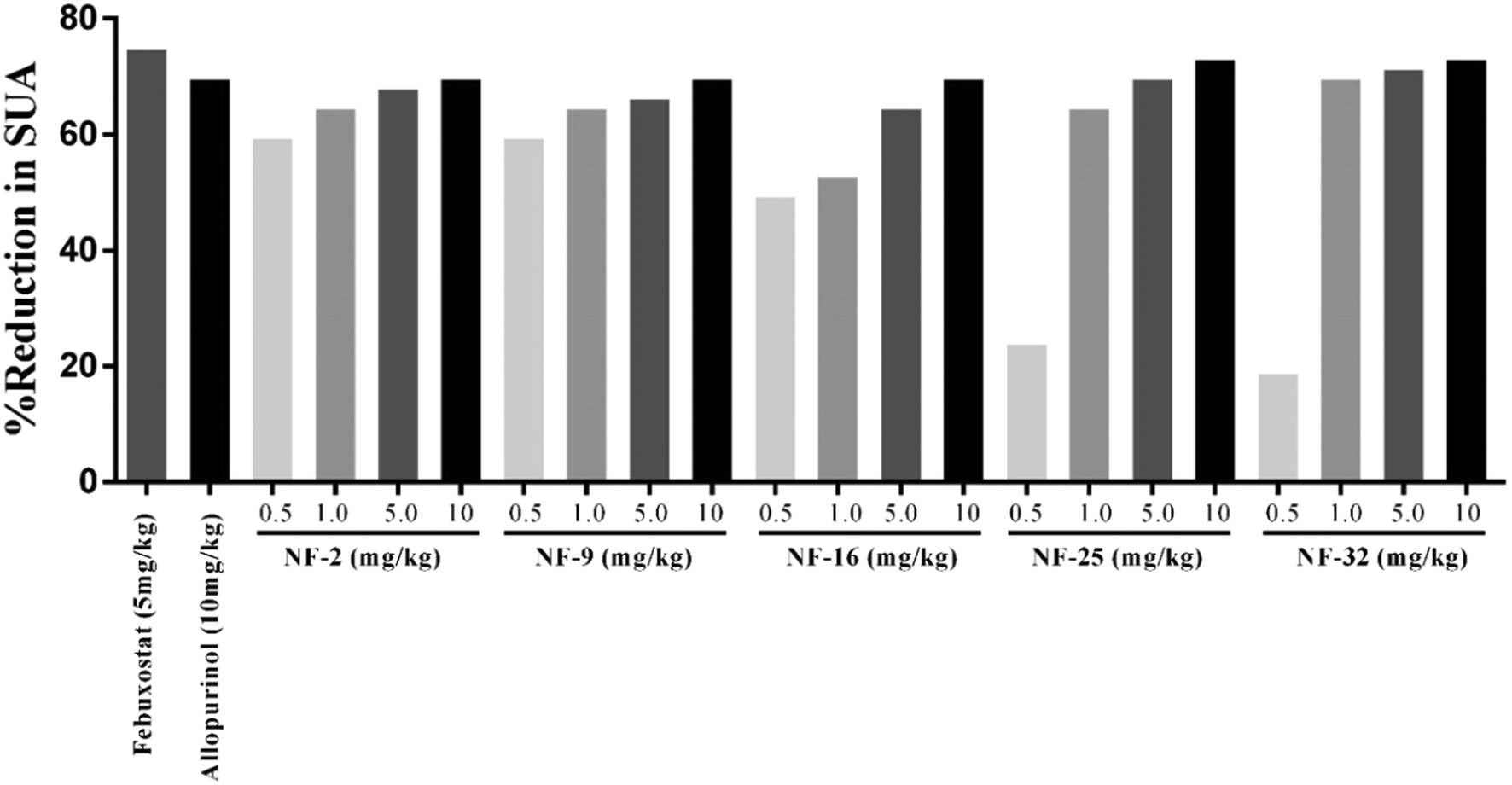 | ||
| Fig. 9 Percent reduction in serum uric acid (SUA) level in a hyperuricemic mice model [the SUA level in the control (1.8 mg dL−1) is 70% lower than the SUA level in hyperuricemic mice (5.9 mg dL−1)]. | ||
2.8. Acute toxicity study
Acute toxicity studies were performed for the two highly potent compounds (NF-2 and NF-32), according to OECD guidelines 2001.48 No gross behavioral abnormality was found in any of the three groups of animals within 24 h after the administration of test compounds.3. Conclusion
The present study is a follow-up to a previous report on xanthine oxidase inhibitors, 7,8-benzoflavones (naphthoflavones), in which 5,6-benzoflavone derivatives were rationally designed and synthesized. All the synthetics were evaluated in vitro to check their inhibitory potential against xanthine oxidase by using spectrophotometric assay. Many compounds exhibited significant XO inhibition among which eight compounds exhibited remarkable XO inhibitory potential. The biological assay elucidated some interesting aspects about the structure–activity relationship which stated that the substituents on the phenyl ring (ring-D) remarkably influence the XO inhibitory activity. The para position on ring-D is the most reliable one for the inhibitory activity of the compounds. The mode of inhibition was also analyzed by using a Lineweaver–Burk plot which showed a mixed type inhibition pattern for the most potent compounds against XO enzyme. The docking study suggests that the compounds NF-4 and NF-28 fit well in the active site of the XO enzyme and completely block its catalytic assembly. The compounds were also found to exhibit antioxidant potential when evaluated by DPPH assay. Moreover, in silico parameters revealed that compounds with improved XO inhibitory potential can be used for in vivo analysis. Eight potent compounds were tested for their hypouricemic effect in a potassium oxonate-induced hyperuricemic mice model. The results of the in vivo experiment showed that the compounds can considerably reduce the serum uric acid comparable to allopurinol and febuxostat. It was found noteworthy that no sign of toxicity was observed when the mice were administered with a 2000 mg kg−1 dose of benzoflavones. Thus, the overall study distinguished three compounds NF-2, NF-25 and NF-32 with a profound pharmacological profile in both in vitro and in vivo screening which can act as clinical candidates for the treatment of hyperuricemia.4. Future perspective
In the recent past, the search for purine and non-purine based xanthine oxidase inhibitors for the treatment of hyperuricemia has been successfully carried out. But various side effects associated with both purine and non-purine based scaffolds limit their clinical use. The development of potential xanthine oxidase inhibitors based on natural moieties like flavonoids is the future perspective in this area of research. As observed, the modification of the flavone moiety to benzoflavone was found to result in an improved and significant xanthine oxidase inhibitory activity. Their potent antioxidant potential as well as their ability to reduce the high serum uric acid level in mice makes them a more effective and resourceful medication for hyperuricemia. By further optimization of these promising hit molecules by pharmacokinetic and chronic toxicity studies, we can conclude whether the benzoflavones are fit for clinical use or not.5. Experimental
5.1. Materials and measurements
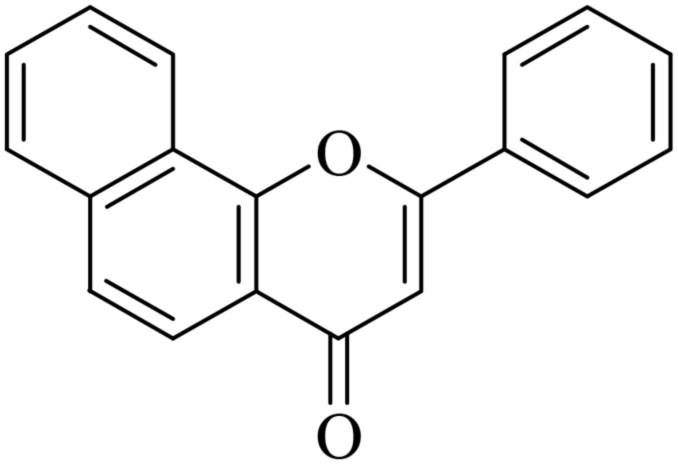
The reagents were procured from Sigma Aldrich, CDH and Loba, India and used without further purification. All yields refer to isolated products after purification. Products were characterized by comparison with authentic samples and by spectroscopic methods (1H, 13C NMR and MASS). 1H NMR and 13C NMR spectra were recorded on a JEOL AL 300 NMR spectrometer. The spectra were measured in CDCl3 relative to TMS (0.00 ppm). In 1H NMR chemical shifts were reported in δ values using tetramethylsilane as an internal standard with the number of protons, multiplicities (s – singlet, bs – broad singlet, d – doublet, m – multiplet) and coupling constants (J) in Hz (Hertz) in the solvent indicated. HRMS was recorded on a micrOTOF-QII Bruker Daltonik LC-MS/MS high-resolution mass spectrometer. Melting points were determined in open capillaries and were uncorrected.5.1.7.1. 2-Phenyl-4H-benzo[h]chromen-4-one (NF-1). 41 Yield: 72%; mp: 167–169 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.56 (1H, bs, ArH), 8.13 (1H, d, J = 8.4 Hz, ArH), 7.91–7.99 (3H, m, ArH), 7.69–7.76 (6H, m, ArH), 6.98 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 108.6, 120.1, 120.6, 122.3, 124.0, 125.4, 126.2, 127.2, 128.2, 129.2, 129.3, 131.6, 131.8, 136.0, 153.6, 162.8, 178.3. MS: m/z: 273 (M+ + 1); anal. calcd. for C19H12O2: C, 83.81; H, 4.44; found: C, 83.53; H, 4.68.
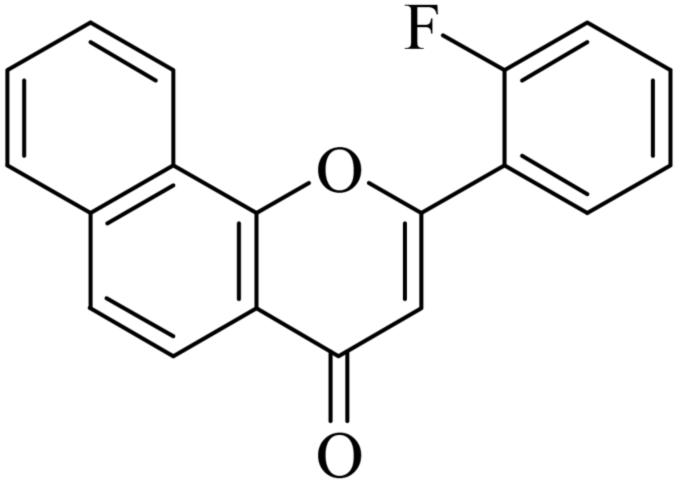
5.1.7.2. 2-(2-Fluorophenyl)-4H-benzo[h]chromen-4-one (NF-2). 41 Yield: 71%; mp: 118–120 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.55 (1H, m, ArH), 8.17 (1H, d, J = 8.7 Hz, ArH), 7.92–8.05 (2H, m, ArH), 7.69–7.79 (3H, m, ArH), 7.55 (1H, m, ArH), 7.24–7.41 (2H, m, ArH), 6.91 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 113.2, 113.4, 117.0, 117.3, 120.1, 120.4, 120.7, 122.4, 124.0, 124.4, 124.8, 125.4, 127.2, 128.2, 129.0, 129.3, 132.8, 133.0, 136.0, 153.7, 158.3, 158.8, 162.2, 178.2. MS: m/z: 291 (M+ + 1); anal. calcd. for C19H11FO2: C, 78.61; H, 3.82; F, 6.54; found: C, 78.73; H, 3.54.
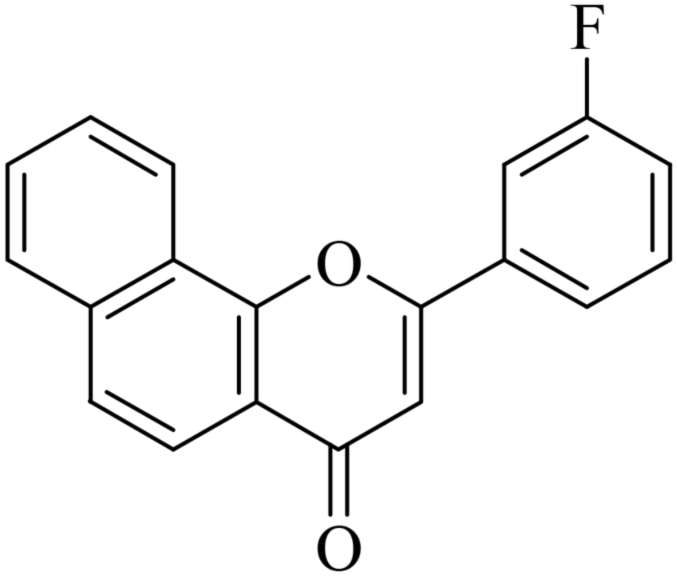
5.1.7.3. 2-(3-Fluorophenyl)-4H-benzo[h]chromen-4-one (NF-3). 41 Yield: 71%; mp: 143–145 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.57 (1H, bs, ArH), 8.15 (1H, d, J = 8.7 Hz, ArH), 7.95 (1H, m, ArH), 7.78 (2H, d, J = 8.4 Hz, ArH), 7.71–7.74 (3H, m, ArH), 7.55 (1H, m, ArH), 7.55 (1H, m, ArH), 7.28 (1H, d, J = 8.4 Hz, ArH), 6.95 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 108.3, 112.1, 112.4, 117.3, 117.6, 119.2, 119.6, 120.9, 121.2, 123.0, 124.6, 126.3, 127.3, 128.4, 129.8, 129.9, 133.0, 133.1, 135.0, 152.5, 160.2, 160.4, 163.7, 177.104. MS: m/z: 291 (M+ + 1); anal. calcd. for C19H11FO2: C, 78.61; H, 3.82; F, 6.54; found: C, 78.83; H, 3.64.
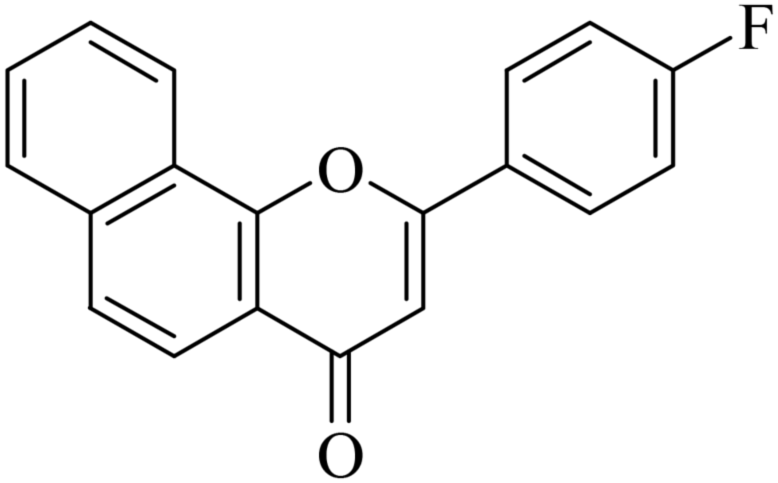
5.1.7.4. 2-(4-Fluorophenyl)-4H-benzo[h]chromen-4-one (NF-4). 41 Yield: 78%; mp: 154–156 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.55 (1H, bs, ArH), 8.14 (1H, d, J = 8.7 Hz, ArH), 7.92–8.07 (3H, m, ArH), 7.77 (1H, d, J = 8.7 Hz, ArH), 7.64–7.74 (2H, m, ArH), 7.23–7.29 (2H, m, ArH), 6.91 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 108.5, 116.3, 116.6, 120.0, 120.6, 122.2, 124.0, 125.5, 127.2, 128.0, 128.3, 128.4, 128.5, 129.3, 136.0, 153.5, 161.8, 178.1. MS: m/z: 291 (M+ + 1); anal. calcd. for C19H11FO2: C, 78.61; H, 3.82; F, 6.54; found: C, 79.00; H, 3.53.
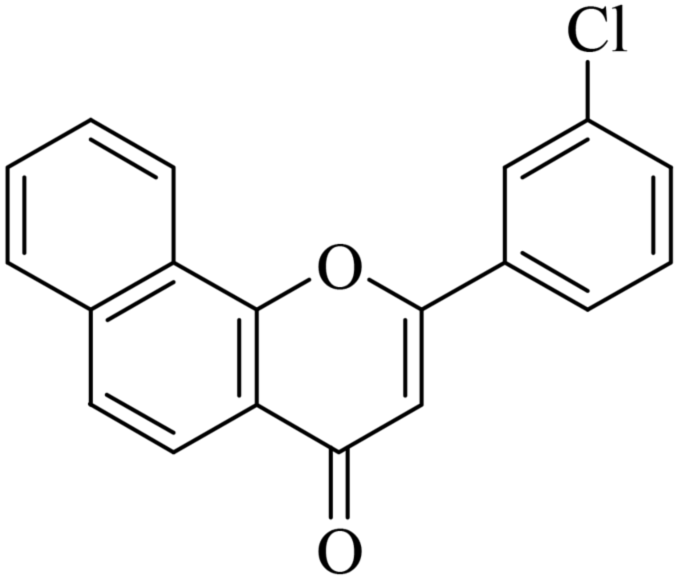
5.1.7.5. 2-(3-Chlorophenyl)-4H-benzo[h]chromen-4-one (NF-5). 41 Yield: 76%; mp: 140–142 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.57 (1H, bs, ArH), 8.15 (1H, d, J = 8.7 Hz, ArH), 7.99 (1H, s, ArH), 7.93 (1H, m, ArH), 7.87 (1H, d, J = 6.9 Hz, ArH), 7.71–7.80 (3H, m, ArH), 7.48–7.56 (2H, m, ArH), 6.94 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 109.4, 120.2, 120.6, 122.3, 124.0, 124.3, 125.6, 126.3, 127.3, 128.3, 129.4, 130.5, 131.5, 133.7, 135.4, 136.1, 153.5, 161.1, 178.0. MS: m/z: 307 (M+ + 1); anal. calcd. for C19H11ClO2: C, 74.40; H, 3.61; Cl, 11.56; found: C, 74.45; H, 3.31.
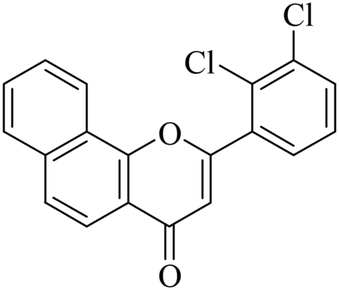
5.1.7.6. 2-(2,3-Dichlorophenyl)-4H-benzo[h]chromen-4-one (NF-6). 41 Yield: 83%; mp: 65–72 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.31 (1H, d, J = 8.1 Hz, ArH), 8.18 (1H, d, J = 8.4 Hz, ArH), 7.97 (1H, d, J = 8.1 Hz, ArH), 7.82 (1H, d, J = 8.7 Hz, ArH), 7.64–7.75 (3H, m, ArH), 7.59 (1H, d, J = 7.8 Hz, ArH), 7.41 (1H, m, ArH), 6.79 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 114.1, 120.5, 122.5, 125.7, 127.3, 127.8, 128.1, 129.2, 129.6, 132.7, 161.8. MS: m/z: 342 (M+ + 1); anal. calcd. for C19H10Cl2O2: C, 66.89; H, 2.95; Cl, 20.78; found: C, 67.10; H, 2.77; Cl, 20.98.
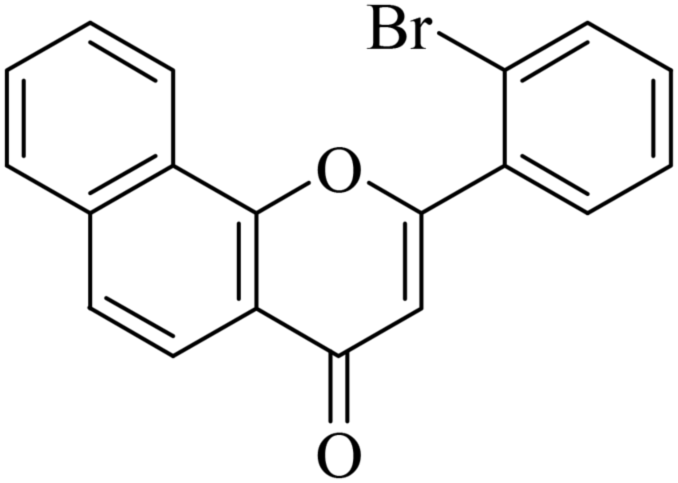
5.1.7.7. 2-(2-Bromophenyl)-4H-benzo[h]chromen-4-one (NF-7). 41 Yield: 67%; mp: 143–145 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.57 (1H, d, J = 8.1 Hz, ArH), 8.19 (1H, d, J = 8.1 Hz), 7.94 (1H, d, J = 7.8 Hz, ArH), 7.65–7.82 (5H, m, ArH), 7.40–7.53 (2H, m, ArH), 6.76 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 113.9, 120.1, 120.6, 121.8, 122.6, 124.1, 125.5, 127.2, 127.8, 128.1, 129.4, 131.2, 132.0, 134.1, 134.1, 136.0, 154.0, 163.3, 178.0. MS: m/z: 352 (M+ + 1); anal. calcd. for C19H11BrO2: C, 64.98; H, 3.16; Br, 22.75; found: C, 65.13; H, 3.36.
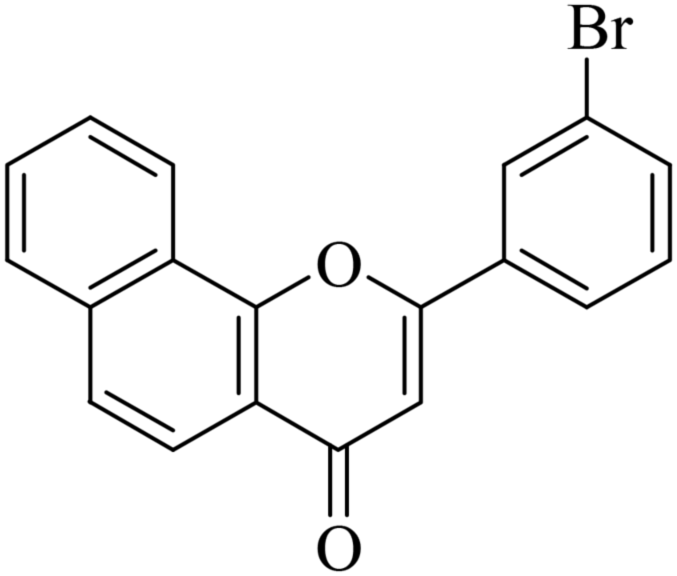
5.1.7.8. 2-(3-Bromophenyl)-4H-benzo[h]chromen-4-one (NF-8). 41 Yield: 74%; mp: 130–132 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.48 (1H, m, ArH), 8.09 (2H, m, ArH), 7.69–7.89 (6H, m, ArH), 7.41 (1H, m, ArH), 6.93 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 109.2, 120.0, 120.5, 122.2, 123.3, 123.8, 124.8, 125.6, 127.3, 128.2, 129.1, 129.4, 130.0, 130.7, 133.7, 134.4, 136.0, 153.5, 161.0, 178.0. MS: m/z: 352 (M+ + 1); anal. calcd. for C19H11BrO2: C, 64.98; H, 3.16; Br, 22.75; found: C, 65.24; H, 2.98.
5.1.7.9. 2-(4-Bromophenyl)-4H-benzo[h]chromen-4-one (NF-9). 41 Yield: 77% mp: 150–152 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.57 (1H, m, ArH), 8.18 (1H, d, J = 8.4 Hz, ArH), 7.96 (1H, m, ArH), 7.89 (2H, d, J = 8.4 Hz, ArH), 7.79 (1H, d, J = 8.7 Hz, ArH), 7.70–7.73 (4H, m, ArH), 6.96 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 108.9, 120.2, 120.7, 122.2, 125.6, 126.3, 127.3, 127.6, 128.3, 129.4, 130.8, 132.5, 136.1, 161.7, 178.2. MS: m/z: 350 (M+); anal. calcd. for C19H11BrO2: C, 64.98; H, 3.16; Br, 22.75; found: C, 64.67; H, 2.96.
5.1.7.10. 2-(2-Iodophenyl)-4H-benzo[h]chromen-4-one (NF-10). 41 Yield: 69%; mp: 140–141 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.61 (1H, m, ArH), 8.18 (1H, m, ArH), 8.06 (1H, m, ArH), 7.93 (1H, m, ArH), 7.78–7.92 (1H, m, ArH), 7.55–7.70, (4H, m, ArH), 7.24 (1H, m, ArH), 6.68 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 95.3, 113.6, 120.2, 120.6, 122.8, 124.1, 125.6, 127.2, 128.1, 128.5, 129.4, 130.8, 131.9, 136.0, 138.0, 140.5, 154.0, 165.2, 178.2. MS: m/z: 399 (M+ + 1); anal. calcd. for C19H11IO2: C, 57.31; H, 2.78; I, 31.87; found: C, 57.62; H, 2.94.
5.1.7.11. 2-(3-Nitrophenyl)-4H-benzo[h]chromen-4-one (NF-11). 41 Yield: 79%; mp: 139–141 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.89 (1H, s), 8.59 (1H, d, J = 8.4 Hz, ArH), 8.43 (1H, d, J = 7.6 Hz, ArH), 8.30 (1H, d, J = 7.8 Hz, ArH), 8.16 (1H, d, J = 8.7 Hz, ArH), 7.95 (1H, m, ArH), 7.71–7.83 (4H, m, ArH), 7.04 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 110.1, 120.3, 120.5, 121.1, 122.2, 123.9, 125.8, 125.9, 127.6, 128.4, 129.6, 130.4, 131.7, 133.7, 136.2, 148.8, 153.5, 159.9, 177.8. MS: m/z: 318 (M+ + 1); anal. calcd. for C19H11NO4: C, 71.92; H, 3.49; N, 4.41; found: C, 72.13; H, 3.10; N, 4.24.
5.1.7.12. 2-(4-Nitrophenyl)-4H-benzo[h]chromen-4-one (NF-12). 41 Yield: 76%; mp: 73–75 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.60 (1H, m, ArH), 8.45 (2H, d, J = 9.00 Hz, ArH), 8.18 (3H, m, ArH), 8.01 (1H, m, ArH), 7.67–7.86 (3H, m, ArH), 7.08 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 110.8, 120.3, 120.5, 122.2, 124.4, 126.0, 127.1, 127.5, 128.4, 129.7, 137.7, 160.0, 177.9. MS: m/z: 318 (M+ + 1); anal. calcd. for C19H11NO4: C, 71.92; H, 3.49; N, 4.41; found: C, 71.65; H, 3.15; N, 4.75.
5.1.7.13. 2-(3,5-Dinitrophenyl)-4H-benzo[h]chromen-4-one (NF-13). 41 Yield: 81%; mp: 65–67 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 9.27 (1H, s, ArH), 9.20 (2H, s, ArH), 8.51 (1H, m, ArH), 8.10 (1H, m, ArH), 7.97 (1H, d, J = 8.7 Hz, ArH), 7.84 (1H, d, J = 8.1 Hz, ArH), 7.74–7.77 (2H, m, ArH), 6.98 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 26.8, 111.2, 118.3, 120.4, 122.0, 124.4, 124.9, 125.8, 127.3, 127.9, 128.5, 130.0. MS: m/z: 363 (M+ + 1); anal. calcd. for C19H10N2O6: C, 62.76; H, 2.93; N, 7.35; found: C, 62.59; H, 3.13; N, 7.29.
5.1.7.14. 2-(4-Methoxyphenyl)-4H-benzo[h]chromen-4-one (NF-14). 41 Yield: 73%; mp: 57–58 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.62 (1H, m, ArH), 8.15 (2H, d, J = 8.4 Hz, ArH), 8.07 (1H, m, ArH), 7.97 (1H, d, J = 8.7 Hz, ArH), 7.86 (1H, d, J = 8.1 Hz, ArH), 7.74–7.77 (2H, m, ArH), 7.13 (2H, d, J = 8.7 Hz, ArH), 6.98 (1H, s, –CH–), 3.95 (3H, s, OCH3); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 55.9, 107.0, 115.1, 119.9, 120.4, 122.6, 123.7, 123.9, 125.7, 128.1, 128.6, 129.9, 135.8, 153.0, 162.5, 177.2. MS: m/z: 303 (M+ + 1); anal. calcd. for C20H14O3: C, 79.46; H, 4.67; found: C, 79.34; H, 4.97.
5.1.7.15. 2-p-Tolyl-4H-benzo[h]chromen-4-one (NF-15). 41 Yield: 72%; mp: 68–70 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.63 (1H, d, J = 9.3 Hz, ArH), 8.18 (1H, d, J = 8.4 Hz, ArH), 7.81–7.95 (3H, m, ArH), 7.71–7.81 (3H, m, ArH), 7.39 (2H, d, J = 8.4 Hz, ArH), 6.97 (1H, s, –CH–), 2.40 (3H, s, CH3); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 21.5, 107.9, 120.0, 120.6, 122.3, 124.0, 125.3, 126.1, 127.1, 128.2, 128.9, 129.2, 129.9, 135.9, 142.3, 153.5, 163.0, 178.3. MS: m/z: 287 (M+ + 1); anal. calcd. for C20H14O2: C, 83.90; H, 4.93; found: C, 84.13; H, 4.73.
5.1.7.16. 2-(3,5-Difluorophenyl)-4H-benzo[h]chromen-4-one (NF-16). Yield: 76%; mp: 124–139 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 8.55–8.58 (1H, m, ArH), 8.15 (1H, d, J = 8.7 Hz, ArH), 7.95–7.98 (1H, m, ArH), 7.79–7.82 (1H, m, ArH), 7.73–7.77 (2H, m, ArH), 7.52–7.55 (2H, m, ArH), 7.01–7.06 (1H, m, ArH), 6.92 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 113.2, 115.4, 115.4, 115.8, 116.1, 126.8, 128.5, 130.1, 132.1, 133.7, 134.6, 135.8, 142.4, 159.8, 171.5, 184.2. MS: m/z: 309 (M+ + 1); anal. calcd. for C19H10F2O2: C, 74.03; H, 3.27; F, 12.33; found: C, 73.93; H, 3.17; F, 12.64.
5.1.7.17. 3-Phenyl-1H-benzo[f]chromen-1-one (NF-17). Yield: 91%; mp: 157–162 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 9.78 (1H, d, J = 9.0 Hz, ArH), 7.77–7.79 (1H, m, ArH), 7.73–7.74 (1H, m, ArH), 7.56–7.61 (3H, m, ArH), 7.36–7.47 (5H, m, ArH), 6.93 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 108.6, 119.1, 120.4, 122.5, 124.4, 125.3, 126.4, 127.5, 128.3, 129.3, 129.2, 131,6, 131.34, 136.6, 153.3, 162.2, 178.3. MS: m/z: 273 (M+ + 1); anal. calcd for C19H12O2: C, 83.81; H, 4.44; found: C, 83.59; H, 4.54.
5.1.7.18. 3-(2-Fluorophenyl)-1H-benzo[f]chromen-1-one (NF-18). Yield: 76%; mp: 105–109 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.06 (1H, d, J = 8.4 Hz, ArH), 8.13 (1H, d, J = 9.0 Hz, ArH), 7.91–7.99 (2H, m, ArH), 7.75–7.81 (1H, m, ArH), 7.35–7.66 (5H, m, ArH), 7.13 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 115.1, 115.2, 116.9, 117.0, 117.5, 123.7, 124.3, 124.6, 126.7, 127.2, 128.1, 128.9, 129.3, 129.5, 130.4, 130.6, 132.7, 132.7, 135.6, 137.4, 156.4, 157.5, 180.2. MS: m/z: 291 (M+ + 1); anal. calcd. for C19H11FO2: C, 78.61; H, 3.82; F, 6.54; found: C, 78.55; H, 3.91; F, 6.44.
5.1.7.19. 3-(3-Fluorophenyl)-1H-benzo[f]chromen-1-one (NF-19). Yield: 78%; mp: 123–128 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.03 (1H, d, J = 8.6 Hz, ArH), 8.12–8.14 (1H, m, ArH), 7.92–7.95 (2H, m, ArH), 7.75–7.79 (1H, m, ArH), 7.62–7.66 (3H, m, ArH), 7.34–7.36 (2H, m, ArH), 7.14 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 114.1, 115.4, 116.9, 117.5, 117.5, 123.7, 124.3, 124.6, 126.7, 127.2, 128.1, 128.9, 129.3, 129.8, 130.4, 130.3, 132.7, 132.4, 135.6, 137.4, 156.4, 157.5, 180.4. MS: m/z: 291 (M+ + 1); anal. calcd. for C19H11FO2: C, 78.61; H, 3.82; F, 6.54; found: C, 78.74; H, 3.75; F, 6.55.
5.1.7.20. 3-(4-Fluorophenyl)-1H-benzo[f]chromen-1-one (NF-20). Yield: 77%; mp: 130–136 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.04 (1H, d, J = 8.5 Hz, ArH), 8.13–8.15 (1H, m, ArH), 7.91–7.94 (2H, m, ArH), 7.75–7.79 (2H, m, ArH), 7.62–7.66 (2H, m, ArH), 7.22 (2H, d, J = 8.9 Hz, ArH), 7.13 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 114.1, 115.4, 116.9, 117.5, 117.5, 123.7, 124.3, 124.6, 126.7, 127.2, 128.1, 128.9, 129.3, 129.8, 130.4, 130.3, 132.7, 132.4, 135.6, 137.4, 156.4, 157.5, 180.4. MS: m/z: 291 (M+ + 1); anal. calcd. for C19H11FO2: C, 78.61; H, 3.82; F, 6.54; found: C, 78.72; H, 3.64; F, 6.59.
5.1.7.21. 3-(3-Chlorophenyl)-1H-benzo[f]chromen-1-one (NF-21). Yield: 78%; mp: 160–166 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.07 (1H, m, ArH), 8.11–8.17 (3H, m, ArH), 8.00–8.04 (2H, m, ArH), 7.66 (1H, s, ArH), 7.23–7.30 (3H, m, ArH), 6.96 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 115.4, 117.4, 117.6, 126.2, 127.6, 128.3, 129.0, 130.3, 131.6, 132.3, 133.3, 134.3, 135.4, 136.5, 153.5, 161.1, 180.0. MS: m/z: 307 (M+ + 1); anal. calcd. for C19H11ClO2: C, 74.40; H, 3.61; Cl, 11.56; found: C, 74.50; H, 3.59; Cl, 11.60.
5.1.7.22. 3-(2,3-Dichlorophenyl)-1H-benzo[f]chromen-1-one (NF-22). Yield: 84%; mp: 160–165 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.08 (1H, m, ArH), 8.15–8.19 (3H, m, ArH), 7.98–8.03 (2H, m, ArH), 7.32–7.36 (3H, m, ArH), 6.81 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 115.9, 117.2, 117.5, 126.8, 127.1, 127.6, 128.2, 129.0, 129.4, 130.4, 130.6, 131.6, 132.5, 133.7, 134.6, 135.8, 157.7, 159.7, 179.7. MS: m/z: 341 (M+ + 1); anal. calcd. for C19H10Cl2O2: C, 66.89; H, 2.95; Cl, 20.78; found: C, 66.99; H, 2.85; Cl, 20.91.
5.1.7.23. 3-(2-Bromophenyl)-1H-benzo[f]chromen-1-one (NF-23). Yield: 75%; mp: 153–159 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.08 (1H, d, J = 8.7 Hz, ArH), 8.13 (1H, d, J = 9.0 Hz, ArH), 7.94 (1H, d, J = 7.5 Hz, ArH), 7.40–7.80 (7H, m, ArH), 6.78 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 115.1, 116.8, 117.0, 117.1, 117.5, 119.9, 124.6, 124.6, 126.7, 127.1, 128.1, 128.9, 129.3, 130.6, 132.7, 132.7, 135.6, 157.5, 180.3. MS: m/z: 352 (M+ + 1); anal. calcd. for C19H11BrO2: C, 64.98; H, 3.16; Br, 22.75; found: C, 65.05; H, 3.03; Br, 22.88.
5.1.7.24. 3-(3-Bromophenyl)-1H-benzo[f]chromen-1-one (NF-24). Yield: 78%; mp: 168–171 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.04 (1H, d, ArH), 8.15 (1H, d, J = 9.0 Hz, ArH), 7.95 (1H, m, ArH), 7.77–7.83 (3H, m, ArH), 7.63 (1H, s, ArH), 7.23–7.33 (3H, m, ArH), 6.98 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 110.5, 117.2, 117.4, 126.0, 126.7, 127.1, 127.4, 128.1, 129.3, 130.3, 130.4, 130.6, 132.3, 135.6, 157.3, 159.7, 179.9. MS: m/z: 350 (M+ + 1); anal. calcd. for C19H11BrO2: C, 64.98; H, 3.16; Br, 22.75; found: C, 65.02; H, 3.09; Br, 22.79.
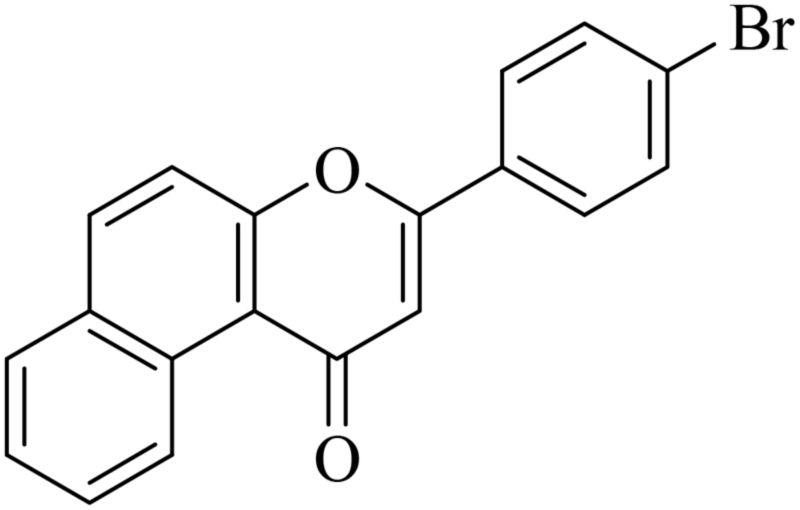
5.1.7.25. 3-(4-Bromophenyl)-1H-benzo[f]chromen-1-one (NF-25). Yield: 80%; mp: 220–225 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.06 (1H, d, J = 8.7 Hz, ArH), 8.14 (1H, d, J = 9.0 Hz, ArH), 7.93 (1H, d, J = 8.1 Hz, ArH), 7.78–7.85 (3H, m, ArH), 7.61–7.70 (4H, m, ArH), 6.97 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 110.5, 117.2, 117.4, 126.0, 126.7, 127.1, 127.4, 128.1, 129.3, 130.3, 130.4, 130.6, 132.3, 135.6, 157.3, 159.7, 180.0. MS: m/z: 351 (M+); anal. calcd. for C19H11BrO2: C, 64.98; H, 3.16; Br, 22.75; found: C, 64.78; H, 3.00; Br, 22.65.
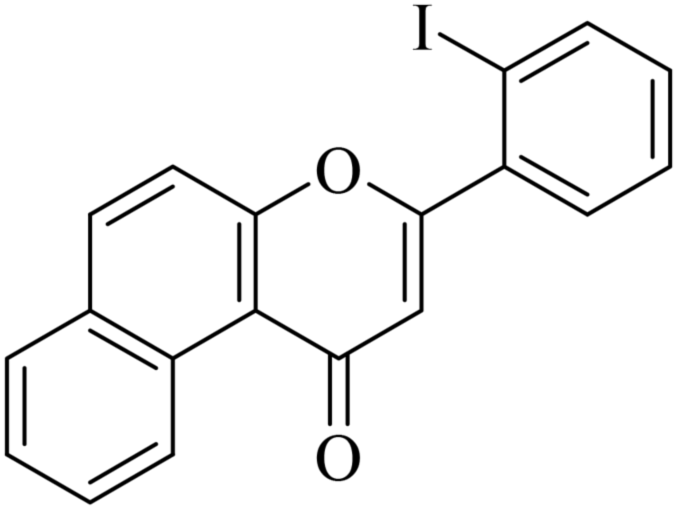
5.1.7.26. 3-(2-Iodophenyl)-1H-benzo[f]chromen-1-one (NF-26). Yield: 69%; mp: 141–146 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.05 (1H, m, ArH), 8.19–8.25 (3H, m, ArH), 8.11–8.13 (2H, m, ArH), 7.82–7.84 (1H, m, ArH), 7.31–7.36 (3H, m, ArH), 6.96 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 110.3, 113.6, 119.2, 120.6, 122.8, 124.1, 125.6, 127.2, 128.1, 128.5, 129.4, 130.8, 131.9, 136.0, 138.0, 140.5, 154.0, 165.2, 180.1. MS: m/z: 398 (M+ + 1); anal. calcd. for C19H11IO2: C, 57.31; H, 2.78; I, 31.87; found: C, 57.42; H, 2.88; I, 31.93.
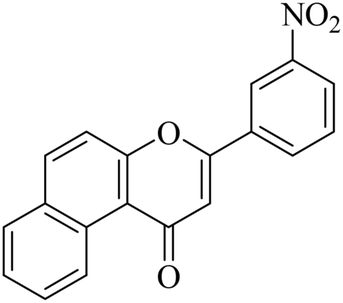
5.1.7.27. 3-(3-Nitrophenyl)-1H-benzo[f]chromen-1-one (NF-27). Yield: 79%; mp: 139–141 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.15 (1H, m, ArH), 8.42 (1H, m, ArH), 8.11–8.14 (3H, m, ArH), 7.78–7.85 (2H, m, ArH), 7.62–7.65 (2H, m, ArH), 7.07 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 112.4, 115.9, 117.3, 124.2, 124.3, 126.7, 126.9, 127.1, 128.2, 128.3, 129.3, 129.6, 136.1, 136.1, 137.4, 157.4, 158.1, 179.7. MS: m/z: 318 (M+ + 1); anal. calcd. for C19H11NO4: C, 71.92; H, 3.49; N, 4.41; found: C, 72.00; H, 3.33; N, 4.34.
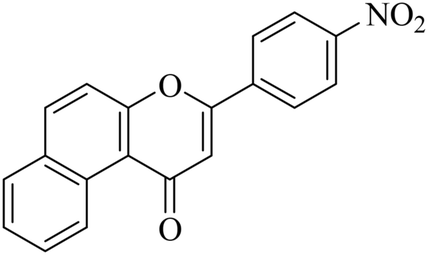
5.1.7.28. 3-(4-Nitrophenyl)-1H-benzo[f]chromen-1-one (NF-28). Yield: 76%; mp: 102–106 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.14 (1H, d, J = 8.4 Hz, ArH), 8.41 (1H, d, J = 8.7 Hz, ArH), 8.14–8.19 (3H, m, ArH), 7.95 (1H, d, J = 8.4 Hz, ArH), 7.78–7.80 (1H, m, ArH), 7.65–7.68 (2H, m, ArH), 7.08 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 112.4, 115.9, 117.3, 124.2, 124.3, 126.7, 126.9, 127.1, 128.2, 128.3, 129.3, 129.6, 136.1, 136.1, 137.4, 157.4, 158.1, 179.7. MS: m/z: 318 (M+ + 1); anal. calcd. for C19H11NO4: C, 71.92; H, 3.49; N, 4.41; found: C, 71.85; H, 3.35; N, 4.65.
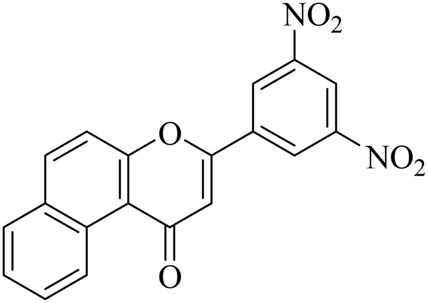
5.1.7.29. 3-(3,5-Dinitrophenyl)-1H-benzo[f]chromen-1-one (NF-29). Yield: 81%; mp: 101–106 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.15 (1H, s, ArH), 9.08 (1H, s, ArH), 8.65–8.70 (2H, m, ArH), 8.11 (1H, m, ArH), 7.97–7.99 (1H, m, ArH), 7.77–7.84 (3H, m, ArH), 7.07 (1H, s, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 112.4, 115.9, 117.3, 124.2, 124.3, 126.7, 126.9, 127.1, 128.2, 128.3, 129.3, 129.6, 136.1, 136.1, 137.4, 157.4, 158.1, 180.7. MS: m/z: 363 (M+ + 1); anal. calcd. for C19H10N2O6: C, 62.76; H, 2.93; N, 7.35; found: C, 62.66; H, 3.03; N, 7.30.
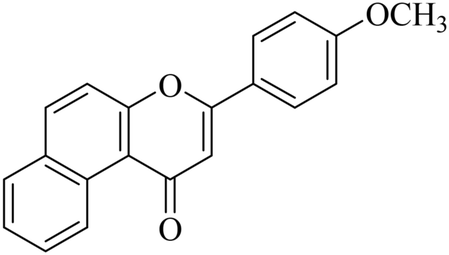
5.1.7.30. 3-(4-Methoxyphenyl)-1H-benzo[f]chromen-1-one (NF-30). Yield: 88%; mp: 72–78 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.11 (1H, m, ArH), 8.13–8.17 (3H, m, ArH), 8.07 (1H, m, ArH), 7.97 (1H, m, ArH), 7.41 (2H, d, J = 8.5 Hz, ArH), 7.01–7.03 (2H, m, ArH), 6.95 (1H, s, –CH–), 3.96 (3H, s, OCH3); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 56.9, 108.2, 114.1, 118.4, 119.5, 121.6, 123.4, 123.9, 126.7, 127.1, 128.3, 130.1, 130.6, 135.7, 153.3, 162.5, 177.7. MS: m/z: 303 (M+ + 1); anal. calcd for C20H14O3: C, 79.46; H, 4.67; found: C, 79.57; H, 4.42.
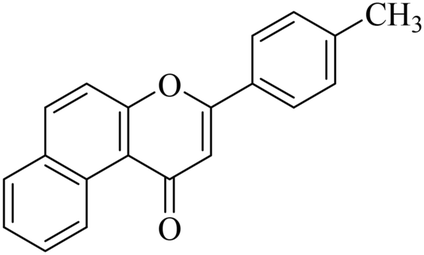
5.1.7.31. 3-p-Tolyl-1H-benzo[f]chromen-1-one (NF-31). Yield: 85%; mp: 96–102 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 10.04 (1H, m, ArH), 8.12–8.18 (3H, m, ArH), 8.01–8.06 (2H, m, ArH), 7.33 (2H, d, J = 8.4 Hz, ArH), 7.11–7.15 (2H, m, ArH), 7.01 (1H, s, –CH–), 2.45 (3H, s, CH3); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 26.4, 110.2, 119.9, 122.6, 123.7, 123.9, 126.3, 127.4, 128.1, 128.6, 129.0, 129.9, 130.4, 135.3, 138.5, 153.0, 162.2, 180.2. MS: m/z: 287 (M+ + 1); anal. calcd. for C20H14O2: C, 83.90; H, 4.93; found: C, 83.80; H, 4.98.
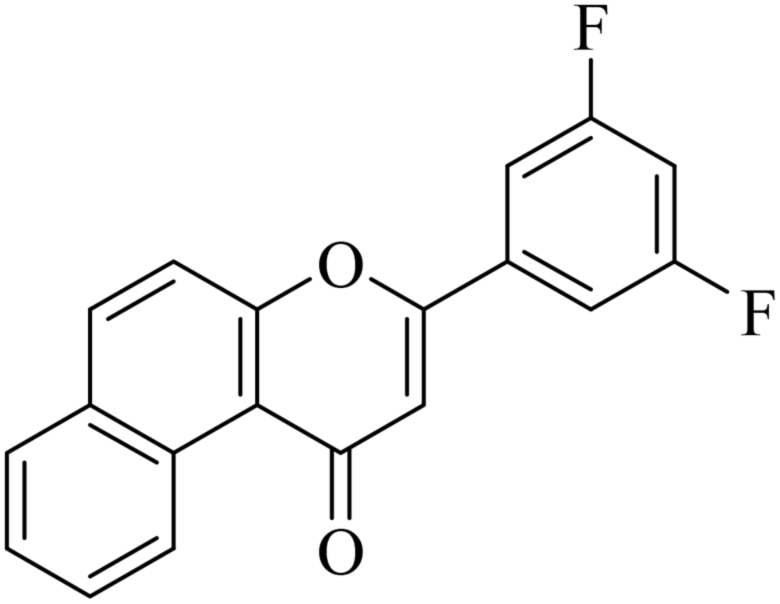
5.1.7.32. 3-(3,5-Difluorophenyl)-1H-benzo[f]chromen-1-one (NF-32). Yield: 78%; mp: 124–139 °C; 1H NMR (CDCl3, 300 MHz, δ, TMS = 0): 9.80 (1H, d, J = 8.4 Hz, ArH), 7.97 (1H, d, J = 9.0 Hz, ArH), 7.75 (1H, d, J = 8.1 Hz, ArH), 7.42–7.58 (4H, m, ArH), 7.29–7.33 (2H, m, ArH), 6.76–6.85 (1H, m, –CH–); 13C NMR (CDCl3, 75 MHz, δ, TMS = 0): 114.5, 115.4, 116.9, 117.5, 117.5, 123.7, 128.1, 128.6, 129.9, 130.4, 135.3, 153.0, 158.4, 162.2, 179.9. MS: m/z: 309 (M+ + 1); anal. calcd. for C19H10F2O2: C, 74.03; H, 3.27; F, 12.33; found: C, 73.99; H, 3.34; F, 12.15.
5.2. In vitro xanthine oxidase assay
All the synthesized compounds were evaluated against the xanthine oxidase enzyme. Bovine milk xanthine oxidase (grade 1, ammonium sulfate suspension, Sigma-Aldrich) activity was assayed spectrophotometrically by measuring the uric acid formation at 293 nm using a Hitachi U-3010 UV-visible spectrophotometer at 25 °C. The reaction mixture contains 50 mM potassium phosphate buffer (pH 7.6), 75 μM xanthine and 0.08 units of xanthine oxidase. Inhibition of xanthine oxidase activity of the synthetics at different concentrations (1, 5, 10, 25, 50 μM) was measured by monitoring the decrease in uric acid formation at 293 nm at 25 °C. The enzyme was pre-incubated for 5 min with a test compound and dissolved in DMSO (1% v/v), and the reaction was started by the addition of xanthine. The final concentration of DMSO (1% v/v) did not interfere with the enzyme activity. All the experiments were performed in triplicate and values were expressed as the mean of three experiments.42,435.3. Enzyme kinetics study
Potent XO enzyme inhibitors were further investigated for the type of inhibition and an enzyme kinetics study was carried out. The Lineweaver–Burk plot was established from which we could calculate the Km, Vmax of the slope of the inhibitor and the value of α (a constant that defines the degree to which inhibitor binding affects the affinity of the enzyme for the substrate).445.4. Docking study
The three-dimensional structure of bovine milk xanthine dehydrogenase (XDH) complexed with febuxostat (a potent XO inhibitor) was retrieved from the RCSB protein data bank (PDB code 1N5X).45 For docking simulations, hydrogen atoms were added to the protein structure using MGL Tools – 1.5.6. The 2D structure of the ligands was drawn in ChemDraw and subjected to energy minimization using the MM2 force field as implemented in Chem 3D Ultra software (ChemDraw Ultra 6.0 and Chem3D Ultra). Docking was performed using the AutoDock Vina software.49 For the docking performance, a grid box was generated by fixing the number of points in the x, y and z directions to 40, each. The spacing was adjusted to 1.00 Å. The center of the grid box was fixed to the point of 96, 52 and 38.5.5. Antioxidant activity (DPPH assay)
All the synthesized compounds were tested for their antioxidant potential by using 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay.50 Different concentrations of test compounds and 0.004% w/v DPPH solution were prepared in methanol. 1.5 mL DPPH solution and 1 mL methanol was taken as a control while methanol alone was taken as a blank. The reaction mixture contained 1.5 mL of 0.004% w/v DPPH solution and 1 mL of test compounds in a test tube. The reaction mixture was shaken at 37 °C for 1 min and placed in the dark for 30 min. Absorption measurements were then recorded using a spectrophotometer (SYSTRONICS-Serial no. 2231 at 517 nm). The ability to scavenge the DPPH radical (% inhibition) was calculated using the following equation: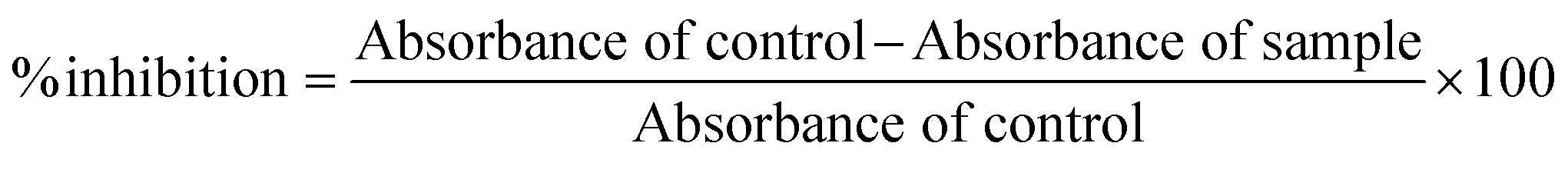
The concentration of antioxidant substance causing a 50% (IC50) decrease in the concentration of free radicals was also determined. All experiments were performed in triplicate and values were then expressed as the mean.
5.6. In vivo antihyperuricemic activity
000 rpm for 5 minutes at −2 °C to obtain the serum. The fresh serum was used for further uric acid detection by using an ERBA diagnostic kit.
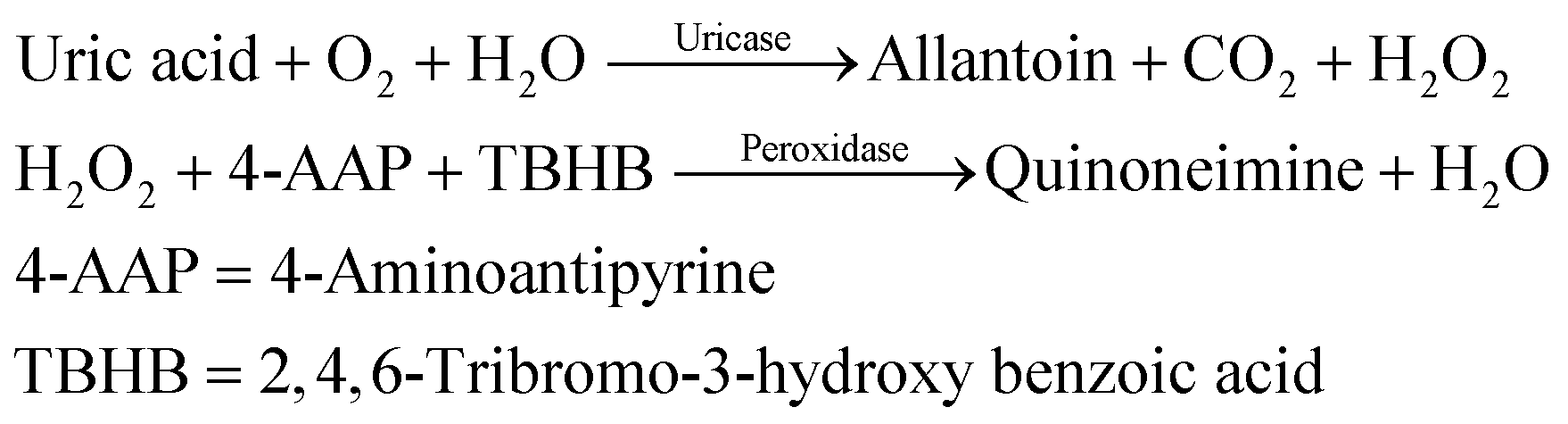
The reaction mixture contained 30 μL of serum (sample) and 1.5 mL of enzymatic reagent. This reaction mixture was mixed and incubated for 5 minutes at 37 °C. The absorbance was recorded at 505 nm by using a colorimeter and further used to calculate the concentration of uric acid by following the given formula:
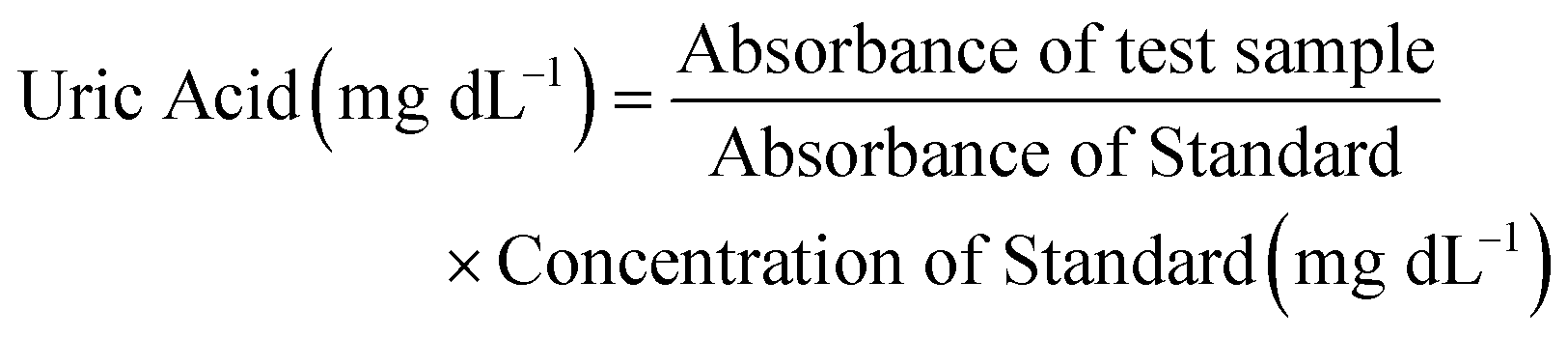
5.7. Acute toxicity study
Three groups of animals each consisting of 3 mice were employed for acute toxicity studies for potent compounds. The first group was treated with the vehicle and served as the vehicle control. The remaining groups were treated with a single higher dose (2000 mg kg−1) of compounds. All the treatments were administered immediately after 4 h of fasting. The animals were observed continuously for any signs and symptoms of toxicity for 24 h after treatment.48Conflicts of interest
The authors confirm that they have no conflicts of interest.Acknowledgements
The authors are grateful to the University Grants Commission for providing funds under the Rajiv Gandhi National Fellowship (RGNF) and they are thankful to Guru Nanak Dev University, Amritsar for providing various facilities to carry out the research work.References
- R. Ojha, J. Singh, A. Ojha, H. Singh, S. Sharma and K. Nepali, Expert Opin. Ther. Pat., 2010, 27, 311–345 CrossRef PubMed.
- R. Kumar, Darpan, S. Sharma and R. Singh, Expert Opin. Ther. Pat., 2011, 21, 1071–1108 CrossRef CAS PubMed.
- K. Oettl and G. Reibneggar, Biochim. Biophys. Acta, Gen. Subj., 1999, 1430, 387–395 CrossRef CAS.
- (a) G. Biagi, I. Giorgi, F. Pacchini, O. Livi and V. Scartoni, Farmaco, 2001, 56, 809–813 CrossRef CAS PubMed; (b) D. E. Brien, R. H. Springer, T. N. A. Albert, K. Senga, J. P. Miller and D. G. Streeter, J. Heterocycl. Chem., 1985, 22, 601–634 CrossRef.
- T. Nagamatsu, T. Fujita and K. Endo, J. Chem. Soc. Perkin. 1, 2000, 1, 33–38 RSC.
- T. Nagamatsu, M. Ukai, F. Yoneda and D. J. Brown, Chem. Pharm. Bull., 1985, 33, 3113–3121 CrossRef CAS.
- T. Nagamatsu and H. Yamasaki, J. Chem. Soc., Chem. Commun., 1995, 2041–2043 RSC.
- H. I. Ali, T. Fujita, E. Akaho and T. Nagamatsu, J. Comput.-Aided Mol. Des., 2010, 24, 57–75 CrossRef CAS PubMed.
- P. Pacher, A. Nivorozhkin and C. Szabo, Pharmacol. Rev., 2006, 58, 87–114 CrossRef CAS PubMed.
- R. Hille, Eur. J. Inorg. Chem., 2006, 10, 1913–1926 CrossRef.
- F. Borges, E. Fernandes and F. Roleira, Curr. Med. Chem., 2002, 9, 195–217 CrossRef CAS PubMed.
- S. Seiji, T. Kunihiko and N. Takeshi, A novel xanthine dehydrogenase inhibitor (BOF-4272), in Purine and Pyrimidine Metabolism in Man VII, Volume 309A of the series Advances in Experimental Medicine and Biology, ed. R.A. Harkness, G. B. Elion and N. Zollner, Springer, New York (US), 1991, pp. 135–138 Search PubMed.
- Y. Osada, M. Tsuchimoto, H. Fukushima, K. Takahashi, S. Kondo, M. Hasegawa and K. Komoriya, Eur. J. Pharmacol., 1993, 241, 183–188 CrossRef CAS PubMed.
- K. Komoriya, Y. Osada, M. Hasegawa, H. Horiuchi, S. Kondo, R. C. Couch and T. B. Griffin, Eur. J. Pharmacol., 1993, 250, 455–460 CrossRef CAS PubMed.
- M. A. Becker, J. Kisicki, R. Khosravan, J. Wu, D. Mulford, B. Hunt, P. MacDonald and N. Joseph-Ridge, Nucleosides, Nucleotides Nucleic Acids, 2004, 23, 35–40 Search PubMed.
- B. L. Love, R. Barrons, A. Veverka and K. M. Snider, Pharmacotherapy, 2010, 30, 594–608 CrossRef CAS PubMed.
- M. A. Becker, H. R. J. Schumacher, R. L. Wortmann, P. A. MacDonald, D. Eustace, W. A. Palo, J. Streit and N. Joseph-Ridge, N. Engl. J. Med., 2005, 353, 2450–2461 CrossRef CAS PubMed.
- H. R. J. Schumacher, M. A. Becker, R. L. Wortmann, P. A. Macdonald, B. Hunt, J. Streit, C. Lademacher and N. Joseph-Ridge, Arthritis Rheum., 2008, 59, 1540–1548 CrossRef CAS PubMed.
- M. Becker, H. Schumacher and P. A. MacDonald, Arthritis Rheum., 2007, 56(9 Suppl), S322 Search PubMed.
- https://www.fda.gov/Drugs/DrugSafety/ucm584702.htm retrieved on 2-july-2018 .
- (a) S. Ishibuchi, H. Morimoto, T. Oe, T. Ikebe, H. Inoue, A. Fukunari, M. Kamezawa, I. Yamada and Y. Naka, Bioorg. Med. Chem. Lett., 2001, 11, 879–882 CrossRef CAS PubMed; (b) I. Yamada, A. Fukunari, T. Osajima, M. Kamezawa, H. Mori and J. Iwane, Nucleosides, Nucleotides Nucleic Acids, 2004, 23, 1123–1125 CrossRef CAS PubMed.
- S. E. Sattui and A. L. Gaffo, Ther. Adv. Musculoskeletal Dis., 2016, 8, 145–159 CrossRef CAS PubMed.
- T. P. T. Cushnie and A. J. Lamb, Int. J. Antimicrob. Agents, 2011, 38, 99–107 CrossRef CAS PubMed.
- M. Kawai, T. Hirano, S. Higa, J. Arimitsu, M. Maruta, Y. Kuwahara, T. Ohkawara, K. Hagihara, T. Yamadori, Y. Shima, A. Ogata, I. Kawase and T. Tanaka, Allergol. Int., 2007, 56, 113–123 CrossRef CAS PubMed.
- A. Garcia-Lafuente, E. Guillamon, A. Villares, M. A. Rostagno and J. A. Martınez, Inflammation Res., 2009, 58, 537–552 CrossRef CAS PubMed.
- D. Ravishankar, A. K. Rajora, F. Greco and H. M. I. Osborn, Int. J. Biochem. Cell Biol., 2018, 45, 2821–2831 CrossRef PubMed.
- Y. T. Tung, C. A. Hsu, C. S. Chen, S. C. Yang, C. C. Huang and S. T. Chang, J. Agric. Food Chem., 2010, 58, 9936–9941 CrossRef CAS PubMed.
- Y. Wang, G. Zhang, J. Pan and D. Gong, J. Agric. Food Chem., 2015, 63, 526–534 CrossRef CAS PubMed.
- S. Lina, G. Z. Y. b. Liao and J. Pan, Int. J. Biol. Macromol., 2015, 81, 274–282 CrossRef PubMed.
- S. Lin, G. Zhang, Y. Liao, J. Pan and D. Gong, J. Agric. Food Chem., 2015, 63, 7784–7794 CrossRef CAS PubMed.
- F. Borges, E. Fernandes and F. Roleira, Curr. Med. Chem., 2002, 9, 195–217 CrossRef CAS PubMed.
- U. Takahama, Y. Koga, S. Hirota and R. Yamauchi, Food Chem., 2011, 126, 1808–1811 CrossRef CAS PubMed.
- S. Lin, L. Zeng, G. Zhang, Y. Liao and D. Gong, Spectrochim. Acta, Part A, 2017, 178, 71–78 CrossRef CAS PubMed.
- R. Dhiman, S. Sharma, G. Singh, K. Nepali and P. M. S. Bedi, Arch. Pharm. Chem. Life Sci., 2012, 346, 7–16 CrossRef PubMed.
- S. Shukla, D. Kumar, R. Ojha, M. K. Gupta, K. Nepali and P. M. S. Bedi, Arch. Pharm. Chem. Life Sci., 2014, 347, 1–10 CrossRef PubMed.
- S. Sharma, K. Sharma, R. Ojha, D. Kumar, G. Singh, K. Nepali and P. M. S. Bedi, Bioorg. Med. Chem. Lett., 2014, 24, 495–500 CrossRef CAS PubMed.
- H. S. Virdi, S. Sharma, S. Mehndiratta, P. M. S. Bedi and K. Nepali, J. Enzyme Inhib. Med. Chem., 2014, 30, 1–7 Search PubMed.
- R. Kaur, F. Naaz, S. Sharma, S. Mehndiratta, M. K. Gupta, P. M. S. Bedi and K. Nepali, Med. Chem. Res., 2015, 24, 3334–3349 CrossRef CAS.
- C. Kaur, S. Dhiman, H. Singh, M. Kaur, S. Bhagat, M. Gupta, S. Sharma and P. M. S. Bedi, J. Chem. Pharm. Res., 2015, 7, 127–136 CAS.
- M. Kaur, A. Kaur, S. Mankotia, H. Singh, A. Singh, J. V. Singh, M. K. Gupta, S. Sharma, K. Nepali and P. M. S. Bedi, Eur. J. Med. Chem., 2017, 131, 14–28 CrossRef CAS PubMed.
- H. Singh, S. Sharma, R. Ojha, M. K. Gupta, K. Nepali and P. M. S. Bedi, Bioorg. Med. Chem. Lett., 2014, 24, 4192–4197 CrossRef CAS PubMed.
- J. Escribano, F. Gracia-Canovas and F. Garcia-Carmona, Biochem. J., 1988, 254, 829–833 CrossRef CAS PubMed.
- Y. Takano, K. Hase-Aoki, H. Horiuchi, L. Zhao, Y. Kasahara, S. Kondo and M. A. Becker, Life Sci., 2005, 76, 1835–1847 CrossRef CAS PubMed.
- R. A. Copeland, Evaluation of Enzyme Inhibitors in Drug Discovery, Wiley, Hoboken, 2005 Search PubMed.
- K. Okamoto, B. T. Eger, T. Nishino, S. Kondo, E. F. Pai and T. Nishino, J. Biol. Chem., 2003, 278, 1848–1855 CrossRef CAS PubMed.
- C. Enroth, B. T. Eger, K. Okamoto, T. Nishino, T. Nishino and E. F. Pai, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10723–10728 CrossRef CAS.
- L. Shen and H. F. Ji, Bioorg. Med. Chem. Lett., 2009, 19, 5990–5993 CrossRef CAS PubMed.
- OECD Guidelines for the Testing of Chemicals, ed. OECD, OECD Publishing, Paris, France, 2001, pp. 1–14 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- H. J. Tang, X. W. Zhang, L. Yang, W. Li, J. H. Li, J. X. Wang and J. Chen, Eur. J. Med. Chem., 2016, 124, 637–648 CrossRef CAS PubMed.
- Z. Qin, S. Wang, Y. Lin, Y. Zhao, S. Yang, J. Song, T. Xie, J. Tian, S. Wu and G. Du, Acta Pharm. Sin. B, 2018, 8, 306–315 CrossRef PubMed.
- I. L. Martins, C. Charneira, V. Gandin, J. L. Ferreira da Silva, G. C. Justino, J. P. Telo, A. J. S. C. Vieira, C. Marzano and A. M. M. Antunes, J. Med. Chem., 2015, 58, 4250–4265 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00512e |
| This journal is © The Royal Society of Chemistry 2019 |