
Synthesis and evaluation of novel purple acid phosphatase inhibitors†
Waleed M.
Hussein
ab,
Daniel
Feder
a,
Gerhard
Schenk
 ac,
Luke W.
Guddat
a and
Ross P.
McGeary
*a
ac,
Luke W.
Guddat
a and
Ross P.
McGeary
*a
aThe University of Queensland, School of Chemistry and Molecular Biosciences, Brisbane, QLD 4072, Australia. E-mail: r.mcgeary@uq.edu.au; Tel: +61 7 33653955
bHelwan University, Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Ein Helwan, Helwan, Egypt
cThe University of Queensland, Australian Centre for Ecogenomics, Brisbane, QLD 4072, Australia
First published on 13th November 2018
Abstract
Transgenic studies in animals have demonstrated a direct association between the level of expression of purple acid phosphatase (PAP; also known as tartrate-resistant acid phosphatase) and the progression of osteoporosis. Consequently, PAP has emerged as a promising target for the development of novel therapeutic agents to treat this debilitating disorder. PAPs are binuclear hydrolases that catalyse the hydrolysis of phosphorylated substrates under acidic to neutral conditions. A series of phenyltriazole carboxylic acids, prepared by the reactions of azide derivatives with propiolic acid through copper(I)-catalysed azide–alkyne cycloaddition click reactions, has been assessed for their inhibitory effect on the catalytic activity of pig and red kidney bean PAPs. The binding mode of most of these compounds is purely uncompetitive with Kiuc values as low as ∼23 μM for the mammalian enzyme. Molecular modelling has been used to examine the binding modes of these triazole compounds in the presence of a substrate in the active site of the enzyme in order to rationalise their activities and to design more potent and specific derivatives.
Introduction
Purple acid phosphatases (PAPs) are binuclear metalloenzymes found in animals, plants and fungi.1 The PAP active site contains two metal ions that assist in the hydrolysis of phosphate esters and anhydrides under acidic to neutral conditions.2 PAPs are the only known metallohydrolases that require a heterovalent dimetallic active site to promote hydrolysis.1 While animal PAPs generally employ a redox-active FeIII–FeII/III combination, in plants FeIII–ZnII and FeIII–MnII centres are found.1–3PAPs can dephosphorylate small substrates such as adenosine diphosphate (ADP), adenosine triphosphate (ATP) and para-nitrophenyl phosphate (pNPP),1a–d,3a as well as larger substrates such as the phosphoproteins, osteopontin and sialoprotein.4 Several biological roles for PAPs have been suggested. PAP from the allantoic fluid of pregnant sows is implicated in the transport of iron from mother to foetus during gestation.5 Consequently, this enzyme is also referred to as uteroferrin (Uf).2a,3b–d In humans, PAP is involved in the inflammatory response of antigen-presenting cells6 and in the process of bone remodelling in the bone resorptive space.4b,7
Osteoporosis is a disease that affects mainly the elderly and in particular post-menopausal women. In the developed world, mortality and morbidity rates are increasing together with life expectancy.8 In Europe and the United States, 30% women are osteoporotic; 40% of post-menopausal women and 30% of men will experience osteoporotic fractures in their lifetime.9
In humans, elevated PAP levels in the blood are directly correlated with the onset and/or progression of osteoporosis.10 In mice that overexpress PAP, a decrease in bone density is observed, consistent with the onset of osteoporosis.4a In mice where the gene encoding for PAP is selectively disrupted, there is an increase in bone mineralisation, symptomatic of osteopetrosis (the opposite phenotype of osteoporosis).10a These observations suggest that PAP inhibitors could be used to treat osteoporosis. There is also the possibility that potent PAP inhibitors will find additional clinical applications in the treatment of other conditions which are associated with elevated levels of this enzyme in the serum, e.g. AIDS encephalopathy,11 Gaucher's disease,12 hairy cell leukemia,13 Alzheimer's disease14 and bone metastases.15
Known inhibitors of PAPs include fluoride and tetrahedral oxoanions such as phosphate, vanadate, tungstate, arsenate and molybdate, but they have not been further developed into anti-osteoporotic drugs due to their lack of specificity and their potential to be cytotoxic.1a–d,2c,d,3d,4b,16 Vanadate (VO43−) is a strong inhibitor of pig and red kidney bean PAP (rkbPAP), with Ki values of 0.040 mM and 0.030 mM, respectively, which is approximately 50 times more potent than the inhibitory effect of phosphate (Ki ∼ 2 mM).1,16b This observation is consistent with similar studies that indicate that the orthovanadate anion is significantly more potent at inhibiting protein phosphatases than orthophosphate.17 This difference is likely to be due to vanadate being able to stabilise a five-coordinate trigonal bipyramidal complex at the active site, which resembles the transition state of phosphoryl transfer reactions.17
Myers and coworkers synthesised and tested the inhibitory activity against rkbPAP for a group of alkylphosphonic acids that contain metal-binding groups such as thiol, carboxylate or phosphonate.18 The results of their study did not yield strong drug leads due to the modest potencies of the inhibitors (IC50 = 80–3000 μM), however, they did demonstrate that metal-binding moieties are important for the enzyme-binding affinities of these inhibitors.18 A subsequent study by Valizadeh and colleagues described the design of a suite of modified phosphotyrosine-containing tripeptides, and showed that these compounds exhibit inhibitory activity against several mammalian and plant PAPs in the mid-micromolar range.19
Based on the work of Schwender et al.,20 our group designed and developed a series of α-alkoxy-substituted naphthylmethylphosphonic acids, some of which showed low micromolar inhibitory activity, with the best inhibitor having a Ki value of 4 μM against rkbPAP and 17 μM against pig PAP.21 We have also described the synthesis, PAP inhibition and computational modelling of a series of structurally analogous α-amino-substituted naphthylmethylphosphonic acids, which display Ki values of 5 μM against rkbPAP and 8 μM against pig PAP.22 Our group also synthesised a series of acyl derivatives of 6-aminopenicillanic acid and these showed potent inhibition against PAP, with Ki values in a low micromolar range.23
Human PAP is the clinically relevant target for the development of novel PAP inhibitors, but to date this enzyme can only be obtained in small quantities using a baculoviral expression system,2b and since large amounts of enzyme are required for kinetic testing and crystallisation studies, pig and rkbPAPs have been used as models for human PAP to test the in vitro effects of potential inhibitors. The validity of using pig and rkbPAPs as models of the human enzyme is supported by the observations that: (i) most of the amino acids in the active site that coordinate the metal ions are conserved across all PAPs,1,2c–g,24 (ii) pig and human PAPs share 88% amino acid sequence identity and the amino acid residues within the active site are highly conserved in human and pig PAPs,25 and (iii) previous studies have shown that pig, human and rkbPAPs catalyse the hydrolysis of similar substrates with similar catalytic efficiencies.1a–d,2c–g,3a,d It should be noted that in the human genome two genes encoding PAPs have been identified, one that is very similar to the pig enzyme (a monomeric enzyme of ∼35 kDa), and one that resembles rkbPAP (a putatively homodimeric form with ∼55 kDa subunits).26 The function of this larger form is currently unknown but based on the arguments above, PAP inhibitors tested against pig and rkbPAP are expected to affect both human PAP isoforms similarly.
Results and discussion
PAP has been identified as an attractive target for the development of chemotherapeutics to treat osteoporosis.1–4 Through the screening of a Maybridge™ compound library, we previously identified a promising inhibitor, p-tolyl thiazole carboxylic acid, with Ki values of 340 μM for rkbPAP and 42 μM for pig PAP.27 Here, a set of analogues of this thiazole inhibitor was synthesised in which the thiazole ring was replaced by 1,2,3-triazole ring to afford compounds 1a–f (Scheme 1).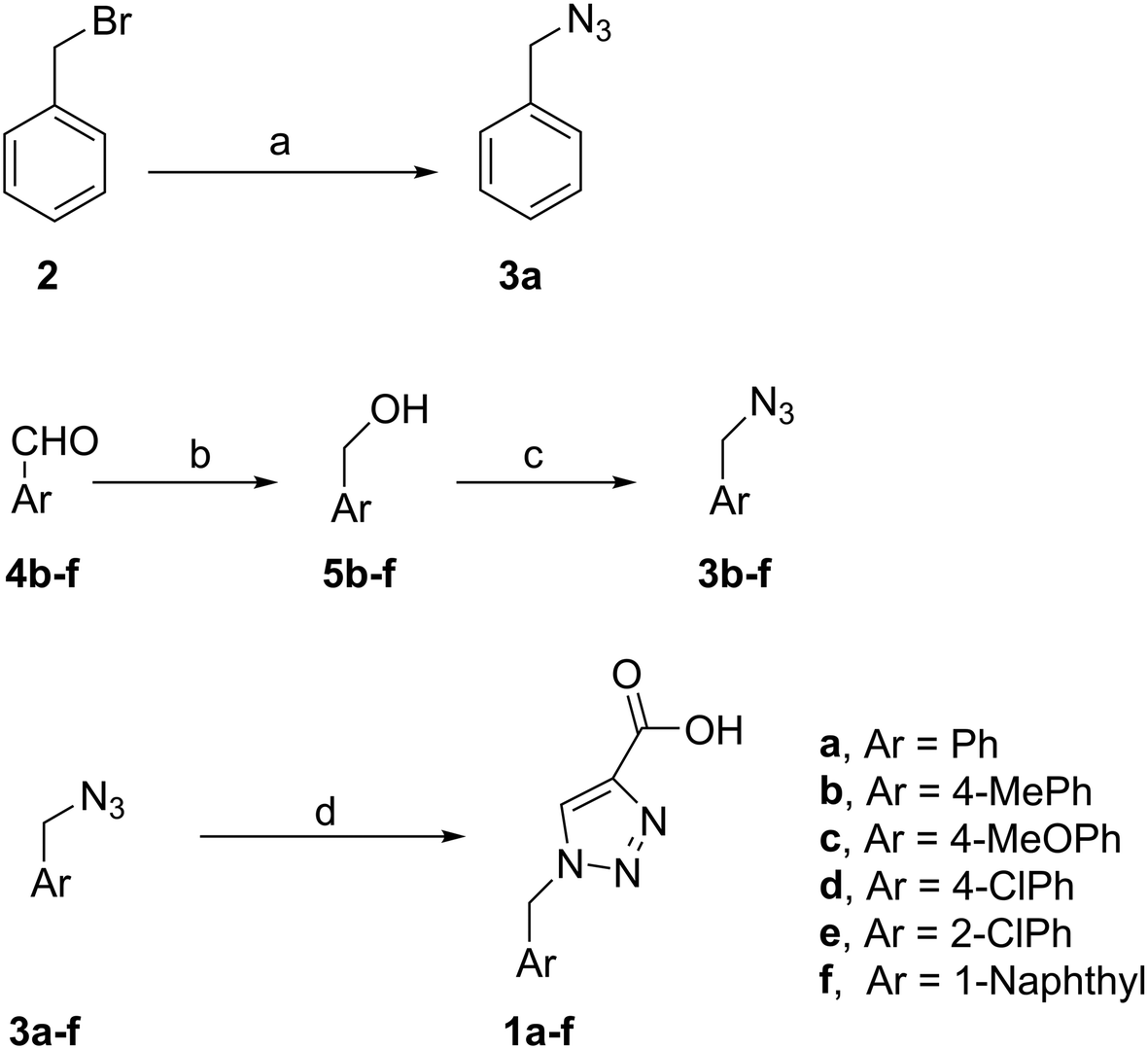 | ||
Scheme 1 Reagents and conditions: (a) NaN3, (CH3)2CO/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), r.t., 24 h. 3a (70%); (b) NaBH4, 2 M, NaOH, MeOH, r.t., 4 h. 5b (51%); 5c (44%); 5d (52%); 5e (61%); 5f (57%); (c) (PhO)2PON3, DBU, dry toluene, 0 °C, 2 h. Then r.t., 16 h. 3b (81%); 3c (89%); 3d (87%); 3e (84%); 3f (92%); (d) propiolic acid, sodium ascorbate, Cu(AcO)2, H2O/tBuOH (1:1), r.t., 14 h. 1a (90%); 1b (83%); 1c (87%); 1d (61%); 1e (75%); 1f (70%). :1), r.t., 24 h. 3a (70%); (b) NaBH4, 2 M, NaOH, MeOH, r.t., 4 h. 5b (51%); 5c (44%); 5d (52%); 5e (61%); 5f (57%); (c) (PhO)2PON3, DBU, dry toluene, 0 °C, 2 h. Then r.t., 16 h. 3b (81%); 3c (89%); 3d (87%); 3e (84%); 3f (92%); (d) propiolic acid, sodium ascorbate, Cu(AcO)2, H2O/tBuOH (1:1), r.t., 14 h. 1a (90%); 1b (83%); 1c (87%); 1d (61%); 1e (75%); 1f (70%). | ||
Chemistry
To synthesise benzyl azide (3a), benzyl bromide (2) was treated with sodium azide in acetone:water (4:1) to give 3a in 70% yield. The rest of the benzyl azide derivatives 3b–f were synthesised over two steps. First, reduction of benzaldehyde derivatives 4b–f to their corresponding benzyl alcohols 5b–f (44–61% yield) using sodium borohydride, followed by conversion the hydroxyl group to the azide moiety using diphenylphosphoryl azide in dry toluene to give 3b–f in 81–92% yield (Scheme 1). The final products 1a–f were obtained in 61–90% yield via the copper(I)-catalysed azide alkyne cycloaddition click reaction between azide derivatives 3a–f and propiolic acid in the presence of sodium ascorbate and copper(II) acetate in t-butanol:water mixture (1:1).
Initial assessment of inhibitory potential of novel compounds
Initial percentage inhibition assays were performed at compound concentrations of 100 μM to test the abilities of 1a–f to inhibit rkbPAP and fully reduced pig PAP in the presence of the chromophoric substrate para-nitrophenyl phosphate (pNPP) at 5 mM. The assay procedure was adapted from similar studies with PAPs.22,23,27Mode and magnitude of inhibitor binding
Subsequently, more detailed catalytic studies were performed to determine the magnitude of the competitive (Kic) and/or uncompetitive (Kiuc) inhibitor dissociation constants, representing the equilibrium constants for the enzyme-inhibitor and enzyme-substrate-inhibitor complexes, respectively (Table 1). Details of the data collection and numeric analyses are described in the Experimental section and follow a similar procedure we employed for other systems (e.g. the antibiotic-degrading metallo-β-lactamases).28 Compounds 1a–f showed either no or only weak competitive binding to either of the two enzymes. However, in particular, compounds 1a, 1b and 1c displayed uncompetitive inhibition against the mammalian PAP, with Kiuc values of 23, 76 and 48 μM, respectively. Of particular importance is that inhibitors 1a–f exclusively bind in an uncompetitive mode to pig PAP, whereas they bind largely in a competitive mode (albeit very weakly) to the plant enzyme. This difference in binding mode to active sites that share considerable similarity (see above) suggests that the inhibitors engage in different binding interactions. Furthermore, since there is a second human PAP isoform that is homologous to rkbPAP26 it may be possible to selectively inhibit only the PAP isoform associated with bone-related ailments.| Compound | PAP | K ic (μM) | K iuc (μM) | MolDock score/(kcal mol−1) | Ligand efficiencya |
|---|---|---|---|---|---|
| a Ligand efficiency is calculated as the binding free energy (in kcal mol−1) divided by the number of heavy atoms. | |||||
| 1a | Pig | — | 23 | −81.52 | −5.60 |
| 1a | rkb | — | 963 | ||
| 1b | Pig | — | 76 | −82.74 | −5.18 |
| 1b | rkb | 951 | — | ||
| 1c | Pig | — | 48 | −81.41 | −4.77 |
| 1c | rkb | — | — | ||
| 1d | Pig | — | 103 | −76.30 | −4.94 |
| 1d | rkb | 1073 | — | ||
| 1e | Pig | — | 276 | −68.14 | −4.72 |
| 1e | rkb | 438 | — | ||
| 1f | Pig | — | 184 | −88.70 | −4.67 |
| 1f | rkb | 2276 | — | ||
Docking studies
Computational docking studies were undertaken with Molegro Virtual Docker (MVD) to predict the binding mode of 1a–f with human PAP (Fig. 1). Firstly, the substrate p-NPP was docked into the active site of human PAP. In its optimal conformation, p-NPP is predicted to bind through its phosphate group in a μ-1,3 bidentate mode to the metal centre, with the phenyl ring and nitro group positioned in a groove formed by the metal-coordinating residue H221 and the sidechains of residues F54 and F242. Compounds 1a–f were then each docked to this hypothetical human PAP–p-NPP complex. The lowest energy poses predicted that all compounds 1a–f would bind in a positively charged pocket under the mammalian repression loop between the side-chains of R248 and H249 and the side-chains of S148 and Q149. The similarity of the predicted binding modes (Fig. 2) for these compounds is consistent with their inhibition constants (Table 1). Docking scores were within the range of −68.14 and −88.70 kcal mol−1 for all ligands and ligand efficiencies were highest for 1a–1d (Table 1). This finding is consistent with their improved inhibitory activity when compared to 1e–1f. Compound 1a showed the highest ligand efficiency and a predicted binding mode that is different from the other inhibitors in this series, likely due to its smaller size, allowing it to bind more complementarily to the enzyme (Fig. 1). | ||
| Fig. 1 Surface and stick representation of the MVD docking results for compounds 1a–f (green surface) (in respective order from left to right) to the docked hPAP–p-NPP complex (grey and yellow surfaces, respectively). The inhibitors are predicted to bind in a groove between the mammalian PAP repression loop and residues R248 and K252. Metal ions are shown in green and are marked with an asterisk. | ||
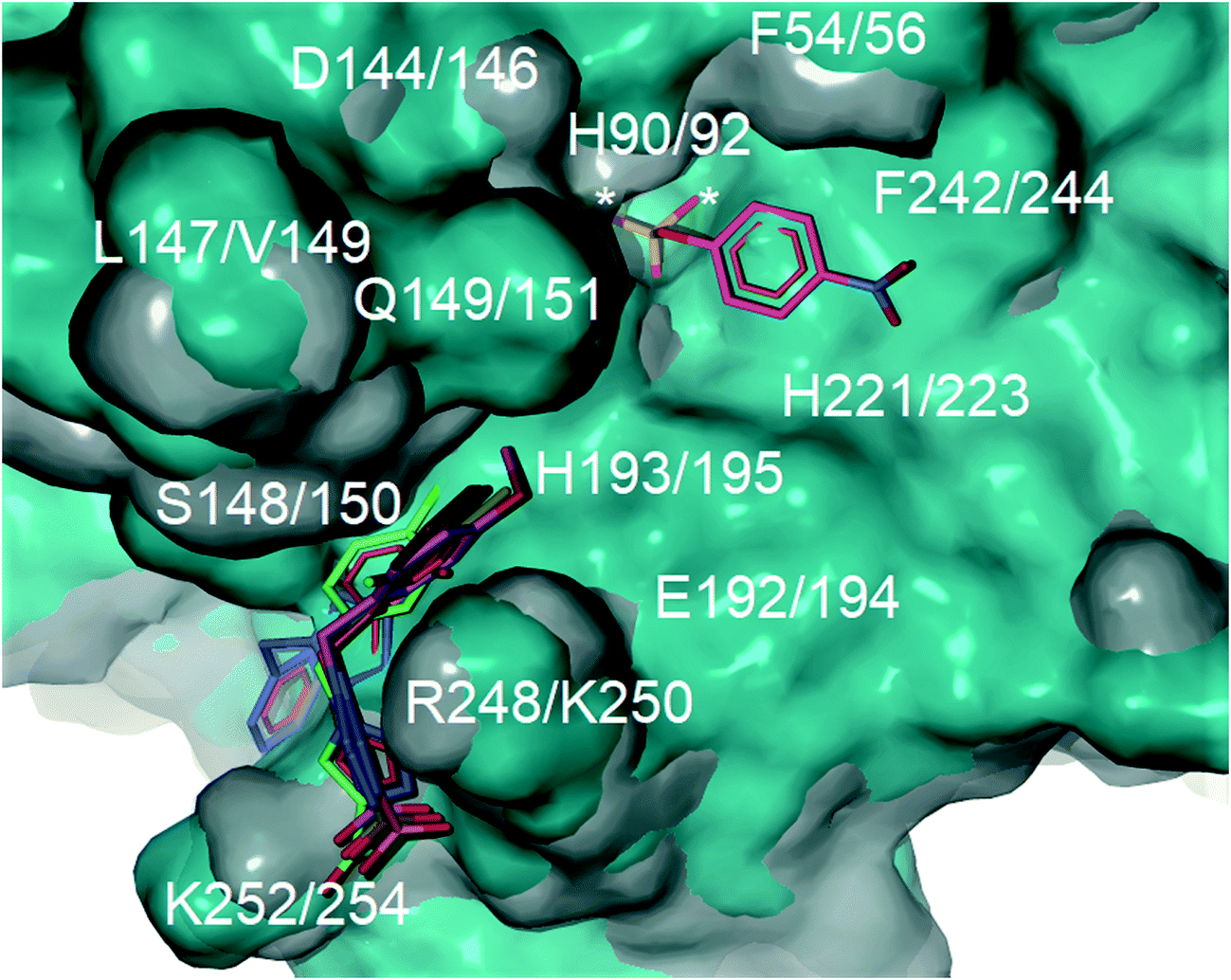 | ||
| Fig. 2 Superimposition of the crystal structure of human PAP (PDB ID: 1WAR, grey) and pig PAP (PDB ID 5UQ6, cyan) showing that the binding site for p-NPP (pink carbons) and the groove formed under the repression loop are conserved between the two enzymes. The only difference relevant to inhibitor binding being R248/K250. The docked poses for inhibitors 1a–f (carbons coloured by ID) and for p-NPP are superimposed on the crystal structures. The active site is marked with an asterisk. Labels show both human PAP (first) and pig PAP (second) numbering. | ||
Superimposition of the crystal structure of pig PAP onto the crystal structure of human PAP shows that the only notable difference in the surface predicted to bind 1a–f and p-NPP is a conservative substitution of R248 in human PAP to K250 in pig PAP (Fig. 2). Therefore, the mode and magnitude of inhibition are likely to be conserved between pig and human PAPs.
Conclusions
Six novel inhibitors have been discovered that inhibit mammalian PAP in an uncompetitive manner. Docking and kinetic studies were used to identify the most probable binding site for these inhibitors. The inhibitors are all predicted to bind in a pocket under the mammalian repression loop, a structural feature that is unique to mammalian PAPs and absent in plant PAPs. This prediction is supported by the fact that these compounds inhibit rkbPAP, by and large, competitively. These findings thus provide the basis for the design of improved human PAP inhibitors that could exploit the structural features of the mammalian repression loop as well as the immediate environment surrounding the metal centre.Experimental
Enzyme preparation and purification
PAP from red kidney bean (rkbPAP) was purified following a previously published protocol.29 Briefly, red kidney beans (Phaseolus vulgaris) were ground in a Waring blender and suspended in 0.5 M sodium chloride. The suspension was filtered through a muslin cloth, followed by ethanol fractionation and ammonium sulfate precipitation. Purification was achieved by ion-exchange chromatography using a CM-cellulose column followed by gel filtration on a Sephadex S-300 column. The resulting preparation was concentrated to 23.8 mg mL−1 using a Millipore Amicon centrifugal concentrator and stored at 4 °C in 0.5 M sodium chloride. Pig PAP was extracted from the uterine fluid of a pregnant sow and purified by ion-exchange chromatography using CM-cellulose followed by gel filtration on a Sephadex G-75.30 Purified pig PAP was concentrated to 8.1 mg mL−1 and stored at −20 °C in 100 mM acetate buffer at pH 4.9. Protein concentrations were determined by measuring the absorbance at 280 nm using extinction coefficients of 1.41 for a 1 mg mL−1 solution (28.6 μM) of pig PAP and 2.1 for a 1 mg mL−1 solution (9.1 μM) of rkbPAP. SDS-Page analysis showed that the enzymes were >95% pure.Enzyme kinetics
Inhibition assays for both pig PAP and rkbPAP were performed in 96-well 400 μL multi-titre plates using a UV/vis multiplate spectrophotometer. Prior to its use in kinetic assays pig PAP was fully reduced with 0.77 mM β-mercaptoethanol for ten minutes at 37 °C. Kinetic measurements were carried out at pH 4.9 (0.1 M sodium acetate buffer with 25% DMSO) at 25 °C using para-nitrophenyl phosphate (pNPP) as substrate at concentrations of 1, 3, 5, 7.5, 10 and 12.5 mM. The rate of product formation was measured at λ = 405 nm (ε = 343 M−1 cm−1).3a The concentration of enzyme used was 20.9 nM for pig PAP and 7.4 nM for rkbPAP, while the concentration of the inhibitors ranged from 50 μM to 300 μM. The data were analysed by non-linear regression using the general inhibition equation (eqn (1)) and the program WinCurveFit (Kevin Raner software).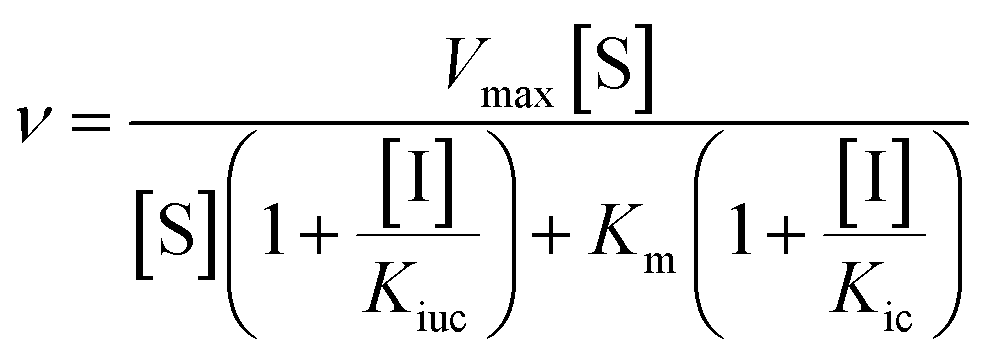 | (1) |
In this equation, Kic and Kiuc represent the equilibrium dissociation constants for competitive and uncompetitive inhibitor binding, respectively, while v, Vmax, Km, [S] and [I] represent the rate of product formation, the maximum rate of product formation, the Michaelis constant, the substrate concentration and the inhibitor concentration, respectively.
Docking studies
Docking studies were undertaken with Molegro Virtual Docker (MVD)31 using the MolDock SE algorithm. The ligand search space was confined to an 8 Å sphere originating from the metal centre of PAP. For human PAP, the coordinates used were those of recombinant human PAP in complex with phosphate (PDB code 1WAR)25a with the phosphate anion omitted from the active site. Water molecules were removed from all coordinate files prior to docking.Chemistry
For TLC staining, KMnO4 was used. KMnO4 dye ingredients: 3 g potassium permanganate; 20 g potassium carbonate; 5 mL 5% sodium hydroxide and 300 mL water.Light petroleum (LP, b.p. 40–60 °C) was distilled before use. Flash chromatography was carried out with Merck Kieselgel 60 as described by Still.32 NMR spectra were recorded on 300, 400 and 500 MHz spectrometers (Bruker, Rheinstetten, Germany). Chemical shifts are reported in parts per million (ppm) on a δ scale, relative to the solvent peak (CDCl3δH 7.24, δC 77.0; (CD3)2SO δH 2.49, δC 39.5). Coupling constants (J) are reported in hertz and peak multiplicities described as singlet (s), doublet (d), triplet (t), quartet (q), septet (sept), multiplet (m), or broad (br). High resolution electrospray ionisation accurate mass measurements were recorded in positive and negative mode on a quadrupole – time of flight instrument (Bruker) with an ESI† source. Accurate mass measurements were carried out with external calibration using sodium formate as reference calibrant and/or Agilent tune mix (mw > 500). Low- and high-resolution electron impact ionisation mass measurements were recorded using perfluorokerosene-H as reference calibrant.
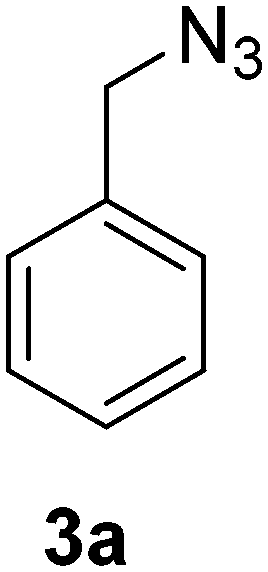
Synthesis of benzyl azide (3a)33
A solution of sodium azide (1.95 g, 30 mmol) in water (10 mL) was added to a stirred solution of benzyl bromide (2) (3.42 g, 2.38 mL, 20 mmol) in acetone (40 mL). The reaction mixture was stirred for 24 h at room temperature. Acetone was evaporated in vacuo and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layer was dried over Na2SO4, filtered and evaporated in vacuo to afford the benzyl azide (3a) as a yellow oil (1.86 g, 70%) Rf: 0.60 (2% EtOAc in LP, UV and KMnO4 dip). 1H NMR (300 MHz, CDCl3) δ 4.34 (2H, s), 7.32–7.44 (5H, m); 13C NMR (75 MHz, CDCl3) δ 54.7, 128.1, 128.2, 128.8, 135.3 in agreement with that described by Campbell-Verduyn.33
General method for preparation of benzyl alcohol derivatives (5b–f)
These compounds 5b–f were prepared using the general method described by Zartler and Shapiro.34 A solution of sodium borohydride (0.37 eq., 173 mg, 4.58 mmol) in 2 M NaOH (247 μL) diluted with water (2.22 mL) was added dropwise to a solution of aromatic aldehydes (4b–f) (12.38 mmol) in methanol (12.5 mL) at 18–25 °C. The reaction mixture was stirred for 4 h at room temperature. The reaction mixture was evaporated in vacuo; methanol (2 × 10 mL) was added and evaporated in vacuo. HCl (5%, 50 mL) was added to the residue and extracted with diethyl ether (2 × 100 mL). The combined organic layer was dried over anhydrous Na2SO4, and evaporated in vacuo to afford 5b–f.
p-Tolylmethanol (5b)
The crude product was purified by silica flash column chromatography (0–10% EtOAc in LP) to afford 5b as white solid (770 mg, 51%) Rf: 0.14 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (400 MHz, CDCl3) δ 2.40 (3H, s), 2.61 (1H, s, OH), 4.61 (2H, s), 7.20 (2H, d, J 7.6 Hz), 7.26 (2H, d, J 7.9 Hz) in agreement with that described by Shaikh.3513C NMR (100 MHz, CDCl3) δ 21.0, 64.9, 127.0, 129.1, 137.1, 137.8.
(4-Methoxyphenyl)methanol (5c)
The crude product was purified by silica flash column chromatography (0–60% EtOAc in LP) to afford 5c as yellow oil (755 mg, 44%) Rf: 0.09 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (300 MHz, CDCl3) δ 2.68 (1H, s, OH), 3.79 (3H, s), 4.56 (2H, s), 6.87 (2H, d, J 8.7 Hz), 7.26 (2H, d, J 8.8 Hz); 13C NMR (75 MHz, CDCl3) δ 55.2, 64.7, 113.8, 128.5, 133.0, 159.0 in agreement with that described by Shaikh.35
(4-Chlorophenyl)methanol (5d)
The crude product was purified by silica flash column chromatography (0–10% EtOAc in LP) to afford 5d as white solid (911 mg, 52%) Rf: 0.14 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (400 MHz, CDCl3) δ 2.45 (1H, s, OH), 4.62 (2H, s), 7.26 (2H, d, J 8.5 Hz), 7.32 (2H, d, J 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 64.3, 128.2, 128.6, 133.2, 139.2 in agreement with that described by Shaikh.35
(2-Chlorophenyl)methanol (5e)
The crude product was purified by silica flash column chromatography (0–10% EtOAc in LP) to afford 5e as white solid (1.08 g, 61%) Rf: 0.17 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (400 MHz, CDCl3) δ 2.37 (1H, s, OH), 4.78 (2H, s), 7.23–7.31 (2H, m), 7.37 (1H, dd, J 1.6 Hz, J 7.6 Hz), 7.47–7.50 (1H, m); 13C NMR (100 MHz, CDCl3) δ 62.7, 127.0, 128.6, 128.7, 129.3, 132.6, 138.1 in agreement with that described by Shaikh.35
Naphthalen-1-ylmethanol (5f)
The crude product was purified by silica flash column chromatography (0–10% EtOAc in LP) to afford 5f as yellow solid (1.12 g, 57%) Rf: 0.15 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (300 MHz, CDCl3) δ 2.05 (1H, s, OH), 5.09 (2H, s), 7.40–7.56 (4H, m), 7.80 (1H, d, J 7.9 Hz), 7.85–7.89 (1H, m), 8.07–8.10 (1H, m) in agreement with that described by Ragnarsson.3613C NMR (75 MHz, CDCl3) δ 63.5, 123.6, 125.2, 125.3, 125.8, 126.3, 128.5, 128.6, 131.2, 133.7, 136.2.
General method for preparation of azide derivatives (3b–f)
These compounds 3b–f were prepared using the general method described by Thompson.37
Diphenyl phosphoryl azide (2.16 g, 7.80 mmol, 1.21 eq.) was added to a solution of alcohol 5b–f (790 mg, 6.50 mmol of 5b, 1 eq.) in dry toluene (11 mL) at 0 °C under argon atmosphere. DBU (1.19 g, 7.80 mmol, 1.21 eq.) was added to the reaction mixture. The reaction mixture was stirred for 2 h at 0 °C then for 16 h at 20 °C. The resulting two-phase mixture was washed with water (2 × 10 mL) and 5% HCl (10 mL). The organic layer was evaporated in vacuo and purified by flash column chromatography.
1-(Azidomethyl)-4-methylbenzene (3b)
The crude product was purified by silica flash column chromatography (5% EtOAc in LP) to afford 3b as colourless oil (766 mg, 81%) Rf: 0.66 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (300 MHz, CDCl3) δ 2.41 (3H, s), 4.32 (2H, s), 7.25 (4H, s); 13C NMR (75 MHz, CDCl3) δ 21.1, 54.5, 128.2, 129.4, 132.2, 138.0 in agreement with that described by Kim.38
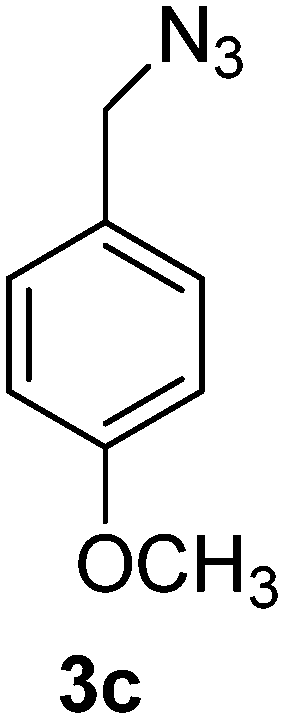
1-(Azidomethyl)-4-methoxybenzene (3c)
The crude product was purified by silica flash column chromatography (5% EtOAc in LP) to afford 3c as colourless oil (554 mg, 89%) Rf: 0.58 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (400 MHz, CDCl3) δ 3.84 (3H, s), 4.29 (2H, s), 6.94 (2H, d, J 8.7 Hz), 7.27 (2H, d, J 8.7 Hz); 13C NMR (100 MHz, CDCl3) δ 54.3, 55.2, 114.1, 127.3, 129.7, 159.6 in agreement with that described by Pietruszka.39
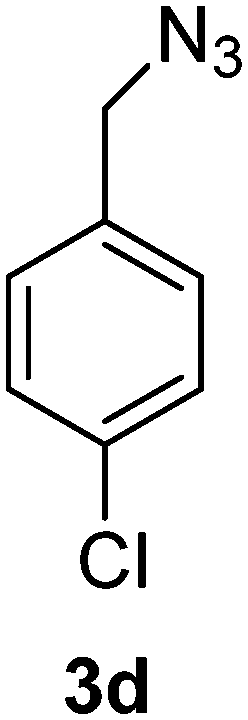
1-(Azidomethyl)-4-chlorobenzene (3d)
The crude product was purified by silica flash column chromatography (5% EtOAc in LP) to afford 3d as colourless oil (556 mg, 87%) Rf: 0.64 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (300 MHz, CDCl3) δ 4.33 (2H, s), 7.26 (2H, d, J 8.7 Hz), 7.37 (2H, d, J 8.6 Hz); 13C NMR (75 MHz, CDCl3) δ 54.0, 129.0, 129.5, 133.8, 134.2 in agreement with that described by Elson.40
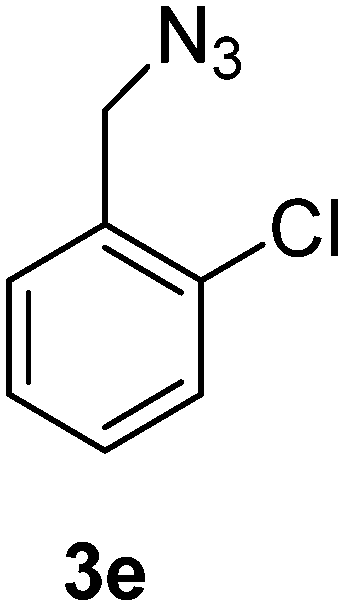
1-(Azidomethyl)-2-chlorobenzene (3e)
The crude product was purified by silica flash column chromatography (5% EtOAc in LP) to afford 3e as colourless oil (539 mg, 84%) Rf: 0.69 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (400 MHz, CDCl3) δ 4.48 (2H, s), 7.25–7.30 (2H, m), 7.36–7.43 (2H, m); 13C NMR (100 MHz, CDCl3) δ 52.2, 127.1, 129.6, 129.7, 130.0, 133.3, 133.7 in agreement with that described by Ikawa.41
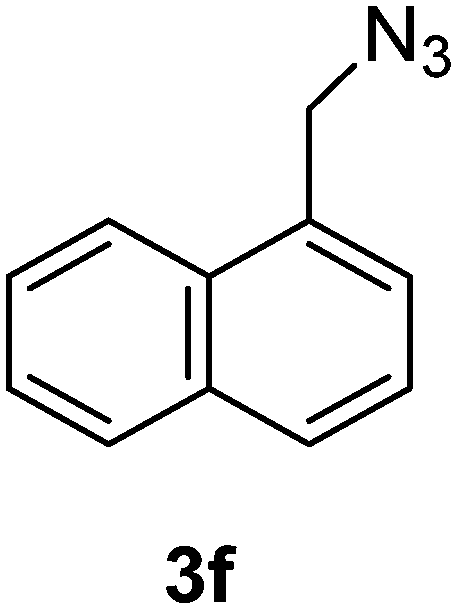
1-(Azidomethyl)naphthalene (3f)
The crude product was purified by silica flash column chromatography (5% EtOAc in LP) to afford 3f as colourless oil (645 mg, 92%) Rf: 0.72 (10% EtOAc in LP, UV and KMnO4 dip). 1H NMR (300 MHz, CDCl3) δ 4.76 (2H, s), 7.47–7.48 (2H, m), 7.52–7.67 (2H, m), 7.86–7.94 (2H, m), 8.04–8.07 (1H, m) in agreement with that described by Amegadzie.4213C NMR (75 MHz, CDCl3) δ 52.9, 123.4, 125.1, 126.1, 126.6, 127.2, 128.7, 129.3, 130.9, 131.3, 133.8.
General method for preparation of 1,2,3-triazole carboxylic acid derivatives (1a–f)
These compounds 1a–f were prepared using the general method described by Mindt.43 To a stirred solution of azides 3a–f (3.00 mmol) in t-butanol (15 mL), propiolic acid (210 mg, 185 μL, 3.00 mmol) was added, followed by a solution of sodium ascorbate (119 mg, 0.6 mmol) and copper(II) acetate monohydrate (60 mg, 0.30 mmol) in water (15 mL). The greenish blue reaction mixture was stirred for 14 h. The reaction mixture was evaporated in vacuo and the residue was filtered through a plug of silica to afford the triazole compounds 1a–f.
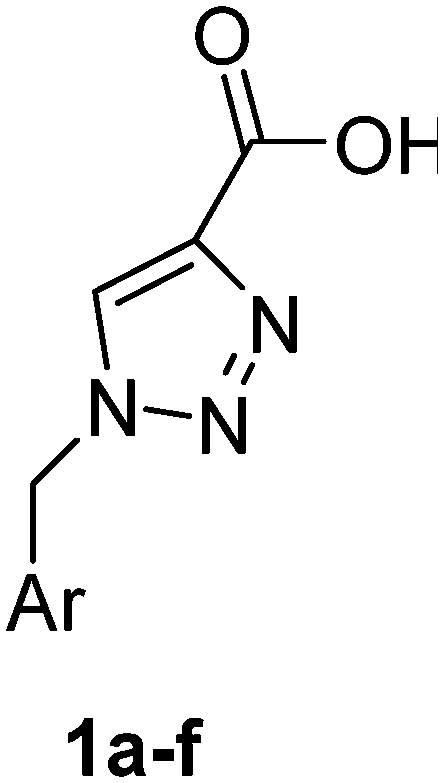
1-Benzyl-1H-1,2,3-triazole-4-carboxylic acid (1a)43
The crude product was purified with 60% EtOAc in LP followed by 50% methanol in CHCl3 to afford 1a as a green solid (548 mg, 90%). Rf: 0.15 (60% EtOAc in LP and 3 drops acetic acid, UV and KMnO4 dip). ESI-MS, m/z: 226 [M + Na]+. HRMS calculated for C10H9N3NaO2+ 226.0592, found 226.0589. 1H NMR (300 MHz, DMSO-d6) δ 5.63 (2H, s), 7.31–7.40 (5H, m), 8.74 (1H, s); 13C NMR (75 MHz, DMSO-d6) δ 53.0, 128.0, 128.3, 128.76, 128.84, 135.6, 206.6 in agreement with that reported by Mindt.43
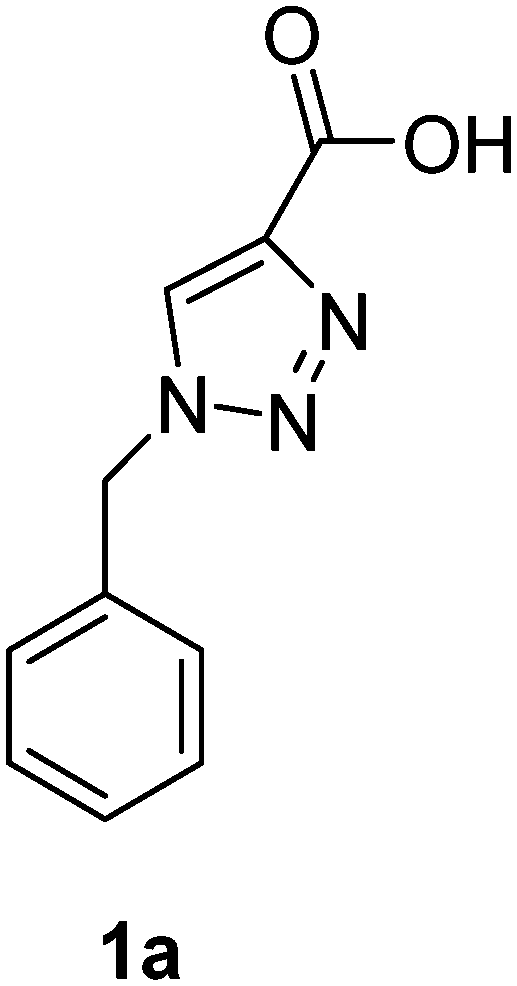
1-(4-Methylbenzyl)-1H-1,2,3-triazole-4-carboxylic acid (1b)
The crude product was purified with 60% EtOAc in LP followed by 50% methanol in CHCl3 to afford 1b as a green solid (543 mg, 83%). Rf: 0.19 (60% EtOAc in LP and 3 drops acetic acid, UV and KMnO4 dip). ESI-MS, m/z: 240 [M + Na]+ and 216 [M − H]−. HRMS calculated for C11H10N3O2− 216.0773, found 216.0788. 1H NMR (400 MHz, DMSO-d6) δ 2.26 (s, 3H), 5.60 (2H, s), 7.16 (2H, d, J 6.6 Hz), 7.21 (2H, d, J 7.5 Hz), 8.84 (1H, br s); 13C NMR (100 MHz, DMSO-d6) δ 20.7, 52.5, 127.9, 128.1, 129.3, 129.4, 132.6, 137.7.
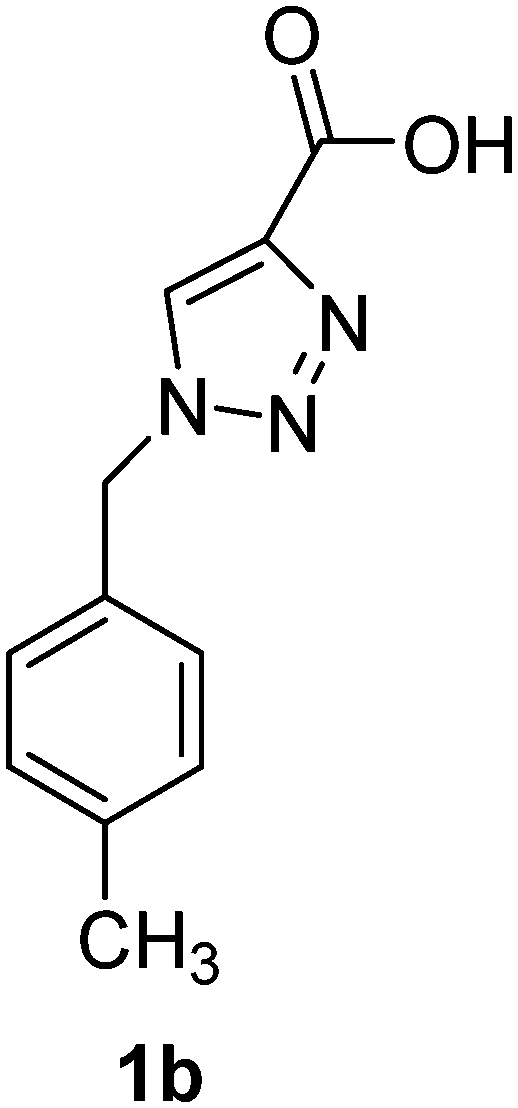
1-(4-Methoxybenzyl)-1H-1,2,3-triazole-4-carboxylic acid (1c)
The crude product was purified with 60% EtOAc in LP followed by 50% methanol in CHCl3 to afford 1c as a yellow solid (607 mg, 87%). Rf: 0.15 (60% EtOAc in LP and 3 drops acetic acid, UV and KMnO4 dip). ESI-MS, m/z: 256 [M + Na]+. HRMS calculated for C11H11N3NaO3+ 256.0698, found 256.0696. 1H NMR (300 MHz, DMSO-d6) δ 3.72 (s, 3H), 5.57 (2H, s), 6.92 (2H, d, J 7.8 Hz), 7.31 (2H, d, J 8.0 Hz), 8.74 (1H, br s); 13C NMR (75 MHz, DMSO-d6) δ 52.1, 55.1, 114.09, 114.14, 127.4, 128.1, 129.5, 129.8, 159.3.
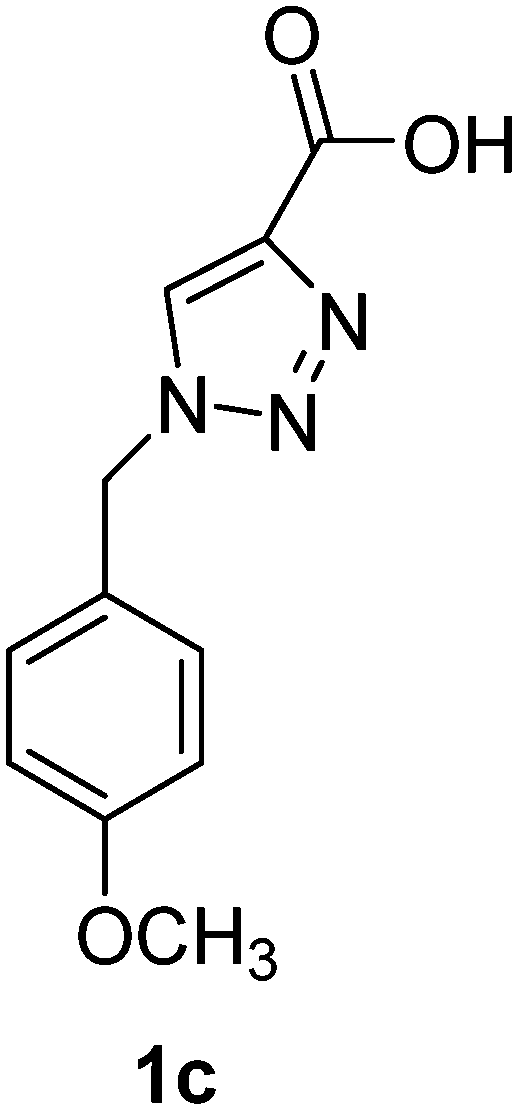
1-(4-Chlorobenzyl)-1H-1,2,3-triazole-4-carboxylic acid (1d)
The crude product was purified with 60% EtOAc in LP followed by 50% methanol in CHCl3 to afford 1d as a light brown solid (433 mg, 61%). Rf: 0.16 (60% EtOAc in LP and 3 drops acetic acid, UV and KMnO4 dip). ESI-MS, m/z: 260 [M + Na]+. HRMS calculated for C10H8ClN3NaO2+ 260.0203, found 260.0195. 1H NMR (300 MHz, DMSO-d6) δ 5.61 (2H, s), 7.34 (2H, d, J 8.5 Hz), 7.43 (2H, d, J 8.5 Hz), 8.56 (1H, s); 13C NMR (75 MHz, DMSO-d6) δ 52.1, 125.0, 128.8, 129.9, 132.9, 133.6, 134.8, 162.5.
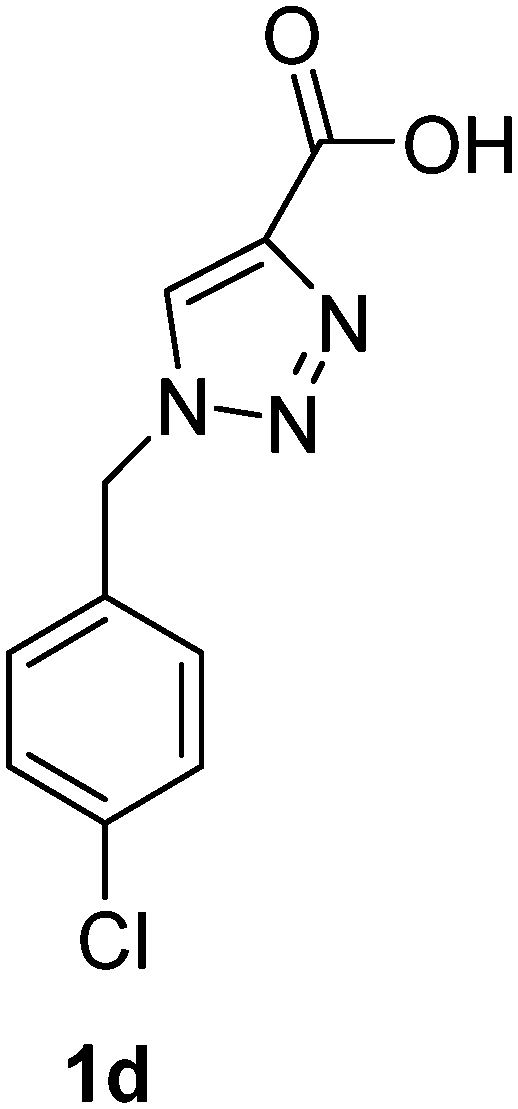
1-(2-Chlorobenzyl)-1H-1,2,3-triazole-4-carboxylic acid (1e)
The crude product was purified with 60% EtOAc in LP followed by 50% methanol in CHCl3 to afford 1e as a brown solid (530 mg, 75%). Rf: 0.16 (60% EtOAc in LP and 3 drops acetic acid, UV and KMnO4 dip). ESI-MS, m/z: 260 [M + Na]+. HRMS calculated for C10H8ClN3NaO2+ 260.0203, found 260.0199. 1H NMR (300 MHz, DMSO-d6) δ 5.72 (2H, s), 7.23 (1H, dd, J 2.1 Hz, J 6.9 Hz), 7.33–7.42 (2H, m), 7.51 (1H, dd, J 1.8 Hz, J 7.5 Hz), 8.50 (1H, s); 13C NMR (75 MHz, DMSO-d6) δ 50.7, 127.8, 128.3, 129.6, 130.3, 130.5, 132.6, 133.1, 142.7, 162.6.
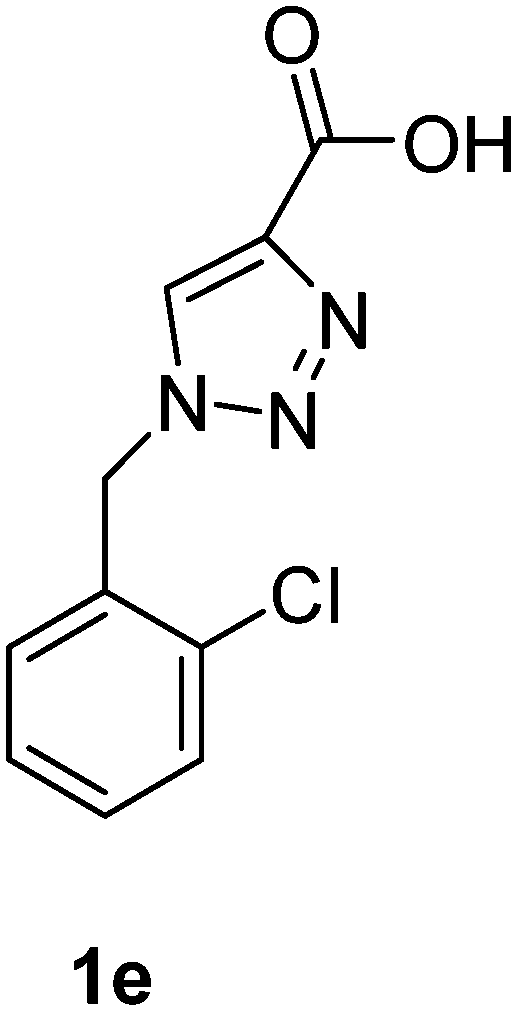
1-(Naphthalen-1-ylmethyl)-1H-1,2,3-triazole-4-carboxylic acid (1f)
The crude product was purified with 60% EtOAc in LP followed by 50% methanol in CHCl3 to afford 1f as a brown solid (529 mg, 70%). Rf: 0.16 (60% EtOAc in LP and 3 drops acetic acid, UV and KMnO4 dip). ESI-MS, m/z: 276 [M + Na]+. HRMS calculated for C14H11N3NaO2+ 276.0749, found 276.0734. 1H NMR (300 MHz, DMSO-d6) δ 6.07 (2H, s), 7.41 (1H, d, J 6.9 Hz), 7.48–7.61 (3H, m), 7.95 (2H, t, J 9.3 Hz), 8.18 (1H, d, J 7.7 Hz), 8.30 (1H, s); 13C NMR (75 MHz, DMSO-d6) δ 50.7, 123.2, 125.6, 126.2, 126.8, 127.3, 128.7, 129.0, 130.6, 131.5, 133.3, 133.5, 163.1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr. Tri Le for assistance with NMR studies and Mr. Graham MacFarlane for MS measurements. This work was funded by Australian Research Council Grants DP0986292 and DP150104358.Notes and references
- (a) N. Mitic, S. J. Smith, A. Neves, L. W. Guddat, L. R. Gahan and G. Schenk, Chem. Rev., 2006, 106, 3338–3363 CrossRef PubMed; (b) G. Schenk, N. Mitic, L. R. Gahan, D. L. Ollis, R. P. McGeary and L. W. Guddat, Acc. Chem. Res., 2012, 45, 1593–1603 CrossRef PubMed; (c) G. Schenk, N. Mitic, G. R. Hanson and P. Comba, Coord. Chem. Rev., 2013, 257, 473–482 CrossRef; (d) N. Mitic, M. Miraula, C. Selleck, K. S. Hadler, E. Uribe, M. M. Pedroso and G. Schenk, in Advances in Protein Chemistry and Structural Biology: Metal-containing Enzymes, ed. C. Z. Christov, Elsevier, Oxford, 2014, vol. 97, pp. 49–81 Search PubMed; (e) A. Reithmeier, T. Lundbaeck, M. Haraldsson, M. Frank, B. Ek-Rylander, P.-G. Nyholm, A.-L. Gustavsson and G. Andersson, Chem. Biol. Drug Des., 2018, 92, 1255–1271 CrossRef PubMed.
- (a) B. C. Antanaitis and P. Aisen, Adv. Inorg. Biochem., 1983, 5, 111–136 Search PubMed; (b) A. R. Hayman and T. M. Cox, J. Biol. Chem., 1994, 269, 1294–1300 Search PubMed; (c) S. J. Smith, A. Casellato, K. S. Hadler, N. Mitic, M. J. Riley, A. J. Bortoluzzi, B. Szpoganicz, G. Schenk, A. Neves and L. R. Gahan, J. Biol. Inorg. Chem., 2007, 12, 1207–1220 CrossRef PubMed; (d) G. Schenk, R. A. Peralta, S. C. Batista, A. J. Bortoluzzi, B. Szpoganicz, A. K. Dick, P. Herrald, G. R. Hanson, R. K. Szilagyi, M. J. Riley, L. R. Gahan and A. Neves, J. Biol. Inorg. Chem., 2008, 13, 139–155 CrossRef PubMed; (e) N. Mitic, C. J. Noble, L. R. Gahan, G. R. Hanson and G. Schenk, J. Am. Chem. Soc., 2009, 131, 8173–8179 CrossRef PubMed; (f) N. Mitic, K. S. Hadler, L. R. Gahan, A. C. Hengge and G. Schenk, J. Am. Chem. Soc., 2010, 132, 7049–7054 CrossRef PubMed; (g) G. Schenk, C. L. Boutchard, L. E. Carrington, C. J. Noble, B. Moubaraki, K. S. Murray, J. de Jersey, G. R. Hanson and S. Hamilton, J. Biol. Chem., 2001, 276, 19084–19088 CrossRef.
- (a) N. Mitic, M. Valizadeh, E. W. W. Leung, J. de Jersey, S. Hamilton, D. A. Hume, A. I. Cassady and G. Schenk, Arch. Biochem. Biophys., 2005, 439, 154–164 CrossRef; (b) B. P. Gaber, J. P. Sheridan, F. W. Bazer and R. M. Roberts, J. Biol. Chem., 1979, 254, 8340–8342 Search PubMed; (c) P. Ling and R. M. Roberts, J. Biol. Chem., 1993, 268, 6896–6902 Search PubMed; (d) M. B. Twitchett, G. Schenk, M. A. S. Aquino, D. T. Y. Yiu, T. C. Lau and A. G. Sykes, Inorg. Chem., 2002, 41, 5787–5794 CrossRef; (e) P. V. Bernhardt, G. Schenk and G. J. Wilson, Biochemistry, 2004, 43, 10387–10392 CrossRef; (f) G. Schenk, Y. B. Ge, L. E. Carrington, C. J. Wynne, I. R. Searle, B. J. Carroll, S. Hamilton and J. de Jersey, Arch. Biochem. Biophys., 1999, 370, 183–189 CrossRef.
- (a) K. Hollberg, K. Hultenby, A. R. Hayman, T. M. Cox and G. Andersson, Exp. Cell Res., 2002, 279, 227–238 CrossRef; (b) G. W. Oddie, G. Schenk, N. Z. Angel, N. Walsh, L. W. Guddat, J. De Jersey, A. I. Cassady, S. E. Hamilton and D. A. Hume, Bone, 2000, 27, 575–584 CrossRef.
- P. R. Nuttleman and R. M. Roberts, J. Biol. Chem., 1990, 265, 12192–12199 Search PubMed.
- A. J. Janckila, D. H. Neustadt, Y. R. Nakasato, J. M. Halleen, T. Hentunen and L. T. Yam, Clin. Chim. Acta, 2002, 320, 49–58 CrossRef.
- J. M. Halleen, S. Raisanen, J. J. Salo, S. V. Reddy, G. D. Roodman, T. A. Hentunen, P. P. Lehenkari, H. Kaija, P. Vihko and H. K. Vaananen, J. Biol. Chem., 1999, 274, 22907–22910 CrossRef.
- F. Guillemin, L. Martinez, M. Calvert, C. Cooper, T. Ganiats, M. Gitlin, R. Horne, A. Marciniak, J. Pfeilschifter, S. Shepherd, A. Tosteson, S. Wade, D. Macarios and N. Freemantle, Osteoporosis Int., 2013, 24, 3001–3010 CrossRef.
- (a) T. Sozen, L. Ozisik and N. C. Basaran, Eur. J. Rheumatol., 2017, 4, 46–56 CrossRef; (b) J. Y. Reginster and N. Burlet, Bone, 2006, 38, 4–9 CrossRef; (c) N. C. Wright, A. C. Looker, K. G. Saag, J. R. Curtis, E. S. Delzell, S. Randall and B. Dawson-Hughes, J. Bone Miner. Res., 2014, 29, 2520–2526 CrossRef.
- (a) J. M. Halleen, H. Kaija, J. J. Stepan, P. Vihko and H. K. Vaananen, Arch. Biochem. Biophys., 1998, 352, 97–102 CrossRef; (b) H. Yaziji, A. J. Janckila, S. C. Lear, A. W. Martin and L. T. Yam, Am. J. Clin. Pathol., 1995, 104, 397–402 CrossRef.
- J. Schindelmeiser, F. Gullotta and D. Munstermann, Pathol., Res. Pract., 1989, 185, 184–186 CrossRef CAS.
- J. Schindelmeiser, H. J. Radzun and D. Munstermann, Pathol., Res. Pract., 1991, 187, 209–213 CrossRef CAS.
- J. D. Hoyer, C. Y. Li, L. T. Yam, C. A. Hanson and P. J. Kurtin, Am. J. Clin. Pathol., 1997, 108, 308–315 CrossRef CAS.
- P. Lang, M. Schultzberg and G. Andersson, J. Histochem. Cytochem., 2001, 49, 379–396 CrossRef CAS.
- (a) Y. C. Chung, C. H. Ku, T. Y. Chao, J. C. Yu, M. M. Chen and S. H. Lee, Cancer Epidemiol., Biomarkers Prev., 2006, 15, 424–428 CrossRef CAS; (b) L. B. Tanko, M. A. Karsdal, C. Christiansen and D. J. Leeming, Cancer Metastasis Rev., 2006, 25, 659–668 CrossRef.
- (a) D. C. Crans, C. M. Simone, R. C. Holz and L. Que, Biochemistry, 1992, 31, 11731–11739 CrossRef CAS; (b) T. W. Elliott, N. Mitic, L. R. Gahan, L. W. Guddat and G. Schenk, J. Braz. Chem. Soc., 2006, 17, 1558–1565 CrossRef CAS.
- (a) V. I. Kuznetsov, A. C. Hengge and S. J. Johnson, Biochemistry, 2012, 51, 9869–9879 CrossRef CAS; (b) D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Q. Yang, Chem. Rev., 2004, 104, 849–902 CrossRef CAS.
- J. K. Myers, S. M. Antonelli and T. S. Widlanski, J. Am. Chem. Soc., 1997, 119, 3163–3164 CrossRef CAS.
- M. Valizadeh, G. Schenk, K. Nash, G. W. Oddie, L. W. Guddat, D. A. Hume, J. de Jersey, T. R. Burke and S. Hamilton, Arch. Biochem. Biophys., 2004, 424, 154–162 CrossRef CAS.
- C. F. Schwender, S. A. Beers, E. Malloy, K. Demarest, L. Minor and K. H. W. Lau, Bioorg. Med. Chem. Lett., 1995, 5, 1801–1806 CrossRef CAS.
- R. P. McGeary, P. Vella, J. Y. W. Mak, L. W. Guddat and G. Schenk, Bioorg. Med. Chem. Lett., 2009, 19, 163–166 CrossRef CAS.
- S. H. Mohd-Pahmi, W. M. Hussein, G. Schenk and R. P. McGeary, Bioorg. Med. Chem. Lett., 2011, 21, 3092–3094 CrossRef CAS.
- (a) W. Faridoon, M. Hussein, N. U. Islam, L. W. Guddat, G. Schenk and R. P. McGeary, Bioorg. Med. Chem. Lett., 2012, 22, 2555–2559 CrossRef CAS; (b) W. M. Hussein, D. Feder, G. Schenk, L. W. Guddat and R. P. McGeary, Eur. J. Med. Chem., 2018, 157, 462–479 CrossRef CAS.
- (a) G. Schenk, L. T. Guddat, Y. Ge, L. E. Carrington, D. A. Hume, S. Hamilton and J. de Jersey, Gene, 2000, 250, 117–125 CrossRef CAS; (b) T. Klabunde, N. Strater, B. Krebs and H. Witzel, FEBS Lett., 1995, 367, 56–60 CrossRef CAS; (c) T. Klabunde, B. Stahl, H. Suerbaum, S. Hahner, M. Karas, F. Hillenkamp, B. Krebs and H. Witzel, Eur. J. Biochem., 1994, 226, 369–375 CrossRef CAS.
- (a) N. Strater, B. Jasper, M. Scholte, B. Krebs, A. P. Duff, D. B. Langley, R. L. Han, B. A. Averill, H. C. Freeman and J. M. Guss, J. Mol. Biol., 2005, 351, 233–246 CrossRef; (b) C. Selleck, D. Clayton, L. R. Gahan, N. Mitic, R. P. McGeary, M. M. Pedroso, L. W. Guddat and G. Schenk, Chem. – Eur. J., 2017, 23, 4778–4781 CrossRef CAS.
- J. U. Flanagan, A. I. Cassady, G. Schenk, L. W. Guddat and D. A. Hume, Gene, 2006, 377, 12–20 CrossRef CAS.
- D. Feder, W. M. Hussein, D. J. Clayton, M. W. Kan, G. Schenk, R. P. McGeary and L. W. Guddat, Chem. Biol. Drug Des., 2012, 80, 665–674 CrossRef CAS.
- (a) O. K. Arjomandi, W. M. Hussein, P. Vella, Y. Yusof, H. E. Sidjabat, G. Schenk and R. P. McGeary, Eur. J. Med. Chem., 2016, 114, 318–327 CrossRef CAS; (b) W. M. Hussein Faridoon, P. Vella, N. Ul Islam, D. L. Ollis, G. Schenk and R. P. McGeary, Bioorg. Med. Chem. Lett., 2012, 22, 380–386 CrossRef CAS; (c) W. M. Hussein, S. S. Fatahala, Z. M. Mohamed, R. P. McGeary, G. Schenk, D. L. Ollis and M. S. Mohamed, Chem. Biol. Drug Des., 2012, 80, 500–515 CrossRef CAS; (d) R. P. McGeary, D. T. C. Tan, C. Selleck, M. M. Pedroso, H. E. Sidjabat and G. Schenk, Eur. J. Med. Chem., 2017, 137, 351–364 CrossRef CAS PubMed; (e) M. S. Mohamed, W. M. Hussein, R. P. McGeary, P. Vella, G. Schenk and R. H. Abd El-hameed, Eur. J. Med. Chem., 2011, 46, 6075–6082 CrossRef CAS PubMed; (f) Y. Yusof, D. T. C. Tan, O. K. Arjomandi, G. Schenk and R. P. McGeary, Bioorg. Med. Chem. Lett., 2016, 26, 1589–1593 CrossRef CAS PubMed.
- J. L. Beck, L. A. Mcconachie, A. C. Summors, W. N. Arnold, J. Dejersey and B. Zerner, Biochim. Biophys. Acta, 1986, 869, 61–68 CrossRef CAS.
- H. D. Campbell, D. A. Dionysius, D. T. Keough, B. E. Wilson, J. D. Jersey and B. Zerner, Biochem. Biophys. Res. Commun., 1978, 82, 615–620 CrossRef CAS.
- R. Thomsen and M. H. Christensen, J. Med. Chem., 2006, 49, 3315–3321 CrossRef CAS PubMed.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923–2925 CrossRef CAS.
- L. Campbell-Verduyn, P. H. Elsinga, L. Mirfeizi, R. A. Dierckx and B. L. Feringa, Org. Biomol. Chem., 2008, 6, 3461–3463 RSC.
- E. R. Zartler and M. J. Shapiro, Curr. Opin. Chem. Biol., 2005, 9, 366–370 CrossRef CAS.
- N. S. Shaikh, K. Junge and M. Beller, Org. Lett., 2007, 9, 5429–5432 CrossRef CAS PubMed.
- U. Ragnarsson, L. Grehn, L. S. Monteiro and H. L. S. Maia, Synlett, 2003, 2386–2388 CrossRef CAS.
- A. S. Thompson, G. R. Humphrey, A. M. Demarco, D. J. Mathre and E. J. J. Grabowski, J. Org. Chem., 1993, 58, 5886–5888 CrossRef CAS.
- B. H. Kim, S. M. Park and Y. S. Lee, Postech Found (UYPO) Univ Pushan Polytechnic (UYPU-Non-standard), US6884882-B1; JP2005194257-A; KR2005072620-A; KR540509-B1; JP4071738-B2.
- J. Pietruszka and G. Solduga, Eur. J. Org. Chem., 2009, 5998–6008 CrossRef CAS.
- K. E. Elson, I. D. Jenkins and W. A. Loughlin, Org. Biomol. Chem., 2003, 1, 2958–2965 RSC.
- T. Ikawa, H. Sajiki and K. Hirota, Tetrahedron, 2005, 61, 2217–2231 CrossRef CAS.
- A. K. Amegadzie, K. M. Gardinier, E. J. Hembre, P. A. Hipskind, L. N. Jungheim, B. S. Muehl, K. A. Savin, K. J. Thrasher, S. A. Boyd, A. Amegadzie, K. Gardinier, E. Hembre, P. Hipskind, L. Jungheim, B. Muehl, K. Savin, K. Thrasher and S. Boyd, Lilly & Co Eli (ELIL) WO2005000821-A1; EP1638944-A1; US2006160794-A1.
- T. L. Mindt and R. Schibli, J. Org. Chem., 2007, 72, 10247–10250 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Enzyme kinetic data for inhibitors 1a–f against pig PAP; 1H and 13C NMR spectra for compounds 1a–f, 3a–f and 5b–f. See DOI: 10.1039/c8md00491a |
| This journal is © The Royal Society of Chemistry 2019 |