DOI:
10.1039/C8MD00395E
(Research Article)
Med. Chem. Commun., 2019,
10, 72-79
Design and synthesis of DNA-intercalative naphthalimide-benzothiazole/cinnamide derivatives: cytotoxicity evaluation and topoisomerase-IIα inhibition†
Received
13th August 2018
, Accepted 1st November 2018
First published on 8th November 2018
Abstract
A new series of different naphthalimide-benzothiazole/cinnamide derivatives were designed, synthesized and tested for their in vitro cytotoxicity on selected human cancer cell lines. Among them, derivatives 4a and 4b with the 6-aminobenzothiazole ring and 5g with the cinnamide ring displayed potent cytotoxic activity against colon (IC50: 3.715 and 3.467 μM) and lung cancer (IC50: 4.074 and 3.890 μM) cell lines when compared to amonafide (IC50: 5.459 and 7.762 μM). Later, the DNA binding studies for these selected derivatives (by CD, UV/vis, fluorescence spectroscopy, DNA viscosity, and molecular docking) suggested that these new derivatives significantly intercalate between two strands of DNA. In addition, the most potent derivatives 4a and 4b were also found to inhibit DNA topoisomerase-II.
Introduction
Nowadays, cancer is considered to be one of the most common diseases which causes death and has always been an unsolved problem worldwide. The development of selective, efficient and safer drugs for cancer chemotherapy remains a high priority and an unmet medical need. Therefore, discovery of new anticancer agents with improved pharmacological profiles including selectivity remains a fascinating challenge in the field of cancer chemotherapy. DNA plays an important role in the process of cell growth and division in a controlled manner, and consequently this macromolecule continues to be one of the main molecular targets in the design of antineoplastic agents.1 Moreover, DNA topoisomerase (topo) is recognized as an important cellular target in anticancer drug discovery.2–6
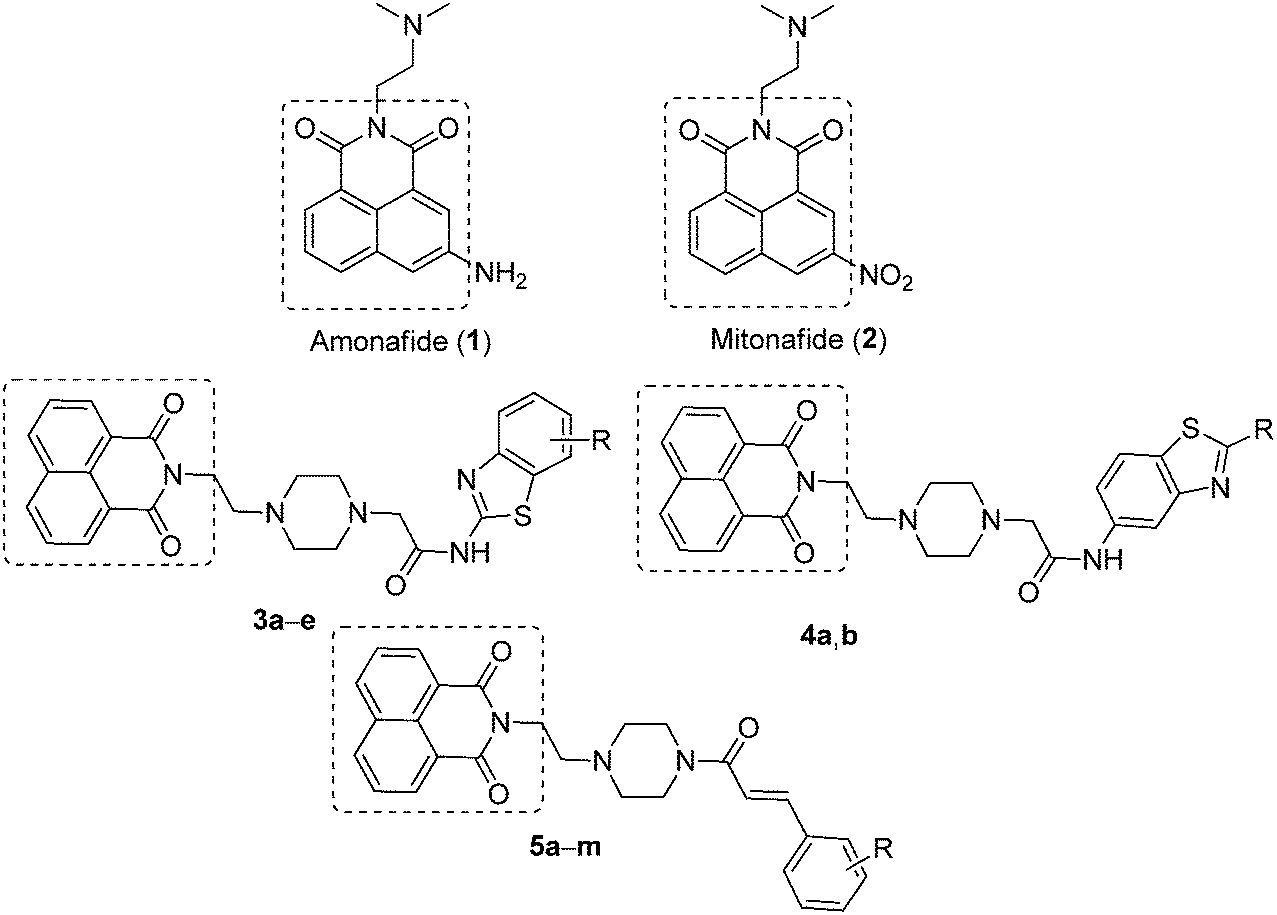
A naphthalimide (NI) core is the most well-known unit that exhibits DNA-intercalating properties and topoisomerase II inhibition,7 and considered as one of the most versatile fluorophore units owing to its unique photophysical properties.8 These intercalators form and stabilize a ternary drug–DNA–topoisomerase complex and it has been proposed that these kinds of derivatives bind at the protein/DNA interface in such a way that the planar ring system intercalates into DNA, while the side chains would interact with the enzyme.9 The well-known DNA-interactive agents such as amonafide (1) and mitonafide (2, Fig. 1) were the most potent naphthalimide-based topo-II inhibitors.10–12 In particular, amonafide showed potent antitumor activity against advanced breast cancer, but apparently, its clinical development was regrettably terminated due to its poor therapeutic index or dose-limiting bone marrow toxicity.13,14 Accordingly, extensive efforts including the modification of the side chain, the aromatic ring system, and the substituents on the ring have been attempted to search for more selective naphthalimide derivatives to improve the potency and reduce the adverse effects.15,16 Braña et al. have designed and synthesized a series of heterocyclic fused naphthalimide derivatives as better cytotoxic agents than amonafide.17 Our research group has also made significant attempts at synthesizing naphthalimide derivatives to enhance cytotoxicity.18 As a continuation of our efforts to develop effective anticancer agents, we designed and synthesized a new series of naphthalimide-based derivatives containing 2-aminobenzothiazole, 6-aminobenzothiazole and cinnamides (Fig. 1), linked through a piperazine spacer for establishing a better structure–activity relationship (SAR). Most of these derivatives from this series exhibited superior anticancer activities in comparison with amonafide against human cancer cell lines and also acted as topoisomerase-II inhibitors.
 |
| | Fig. 1 Lead naphthalimides (1 and 2) and newly designed naphthalimide derivatives 3a–e, 4a,b and 5a–m. | |
Results and discussion
Chemistry
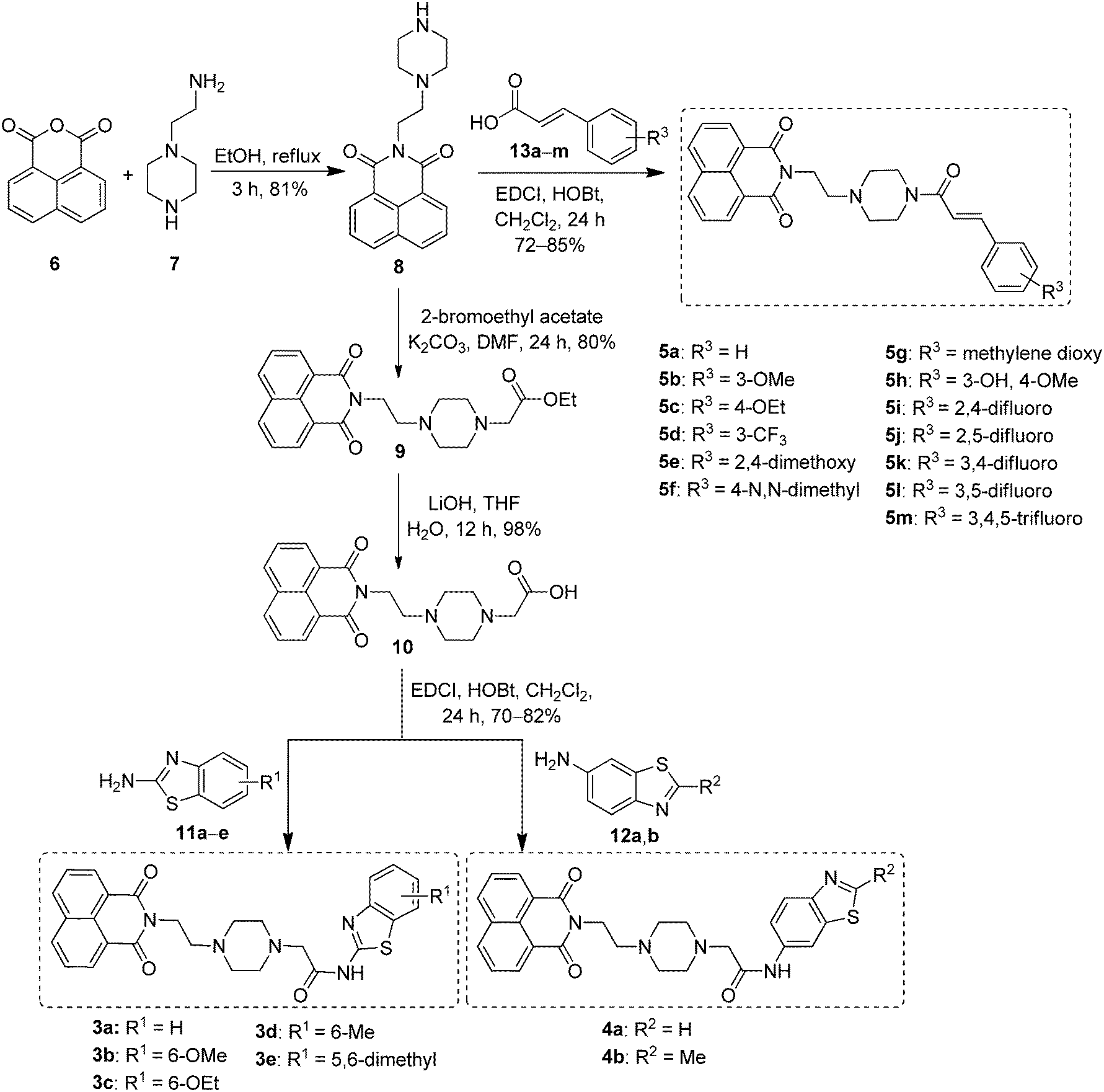
The synthesis of newly designed naphthalimide-amidobenzothiazole derivatives 3a–e, 4a–b and 5a–m is depicted in Scheme 1. Naphthalimide ethylpiperazine (8) was prepared in good yield by the reaction of 1,8-naphthalic anhydride (6) with amino ethyl piperazine (7) in ethanol under reflux. Then naphthalimide ethylpiperazine (8) was esterified using K2CO3 in dry DMF with α-bromoethyl acetate to furnish naphthalimide ester (9). Hydrolysis of this ester with LiOH–H2O afforded naphthalimide acid (10). Derivatives 3a–e were successfully synthesized by the coupling of naphthalimide acid (10) with different 2-aminobenzothiazoles 11a–e in the presence of EDCI/HOBt and obtained in good yields. Similarly, other derivatives 4a,b were synthesized using 6-aminobenzothiazoles 12a,b, as shown in Scheme 1. Later, another series of new naphthalimide-cinnamide derivatives 5a–m was also synthesized, as shown in Scheme 1. These amide derivatives were obtained by the acid amine coupling reaction of naphthalimide ethylpiperazine (8) with different trans-cinnamic acids 13a–m by using EDCI/HOBt and obtained in good yields.
 |
| | Scheme 1 Synthesis of naphthalimide-amidobenzothiazole derivatives 3a–e, 4a,b and naphthalimide-cinnamide derivatives 5a–m. | |
Biological studies
In vitro cytotoxicity evaluation.
In an effort to define a structure–activity relationship (SAR) for 1 and to identify more potent forms, several derivatives of the compound were designed and synthesized. The newly synthesized derivatives 3a–e, 4a,b and 5a–m were evaluated for their in vitro cytotoxic activity against a panel of three human cancer cell lines, namely HT-29 (colon), A549 (lung), and MCF-7 (breast), by employing the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay.19 These cell lines were procured from the National Centre for Cell Sciences (NCCS, Pune, India (ESI)†). The assay was performed thrice, and the average of the cytotoxicity data, expressed as IC50 values, is summarized in Table 1 and compared with that of amonafide as the positive control. The screening results demonstrated that these derivatives displayed promising cytotoxic activity with IC50 values ranging from 3.46 to 34.15 μM against different cancer cell lines compared with the parent compound 1. The most active derivatives 4a and 4b showed IC50 values ranging from 3.46–5.08 and 3.71–7.90 μM, respectively, on the tested cancer cell lines. The antiproliferative activity of the hybrid derivatives can be further characterized in terms of their substitution patterns. In particular, compounds 3b and 3e with electron-donating groups (6-OMe and 5,6-dimethyl) on the phenyl ring of benzothiazole show significant cytotoxic activity compared to amonafide. The cytotoxicity data (Table 1) clearly indicate that 5g,j are more potent analogs that exhibit significant antiproliferative activity with IC50 values ranging from 3.80–4.78 and 4.17–6.39 μM against both the HT-29 and A549 cancer cell lines. It is observed that compound 5f with the 4-N,N-dimethyl cinnamic acid moiety is more active against the HT-29 cancer cell line, whereas 5l with the 3,5-difluoro cinnamic acid moiety is more active against the A549 cancer cell line. Overall, these results demonstrated that the amide derivatives achieved with 6-aminobenzothiazoles showed better activity compared to 2-aminobenzothiazoles. All the synthesized derivatives were selectively more potent in the HT-29 and A549 cell lines compared to MCF-7.
Table 1
In vitro cytotoxicity (IC50 values in μM) data of naphthalimide derivatives 3a–e, 4a,b and 5a–m against a panel of human cancer cellsa
| Compound |
HT-29b |
A549c |
MCF-7d |
|
50% growth inhibitory concentration and the values are the average of three individual experiments after 48 h of drug treatment.
Colon cancer – HT-29.
Lung cancer – A549.
Breast cancer – MCF-7.
|
|
3a
|
10.85 ± 0.1 |
14.79 ± 0.3 |
26.47 ± 0.5 |
|
3b
|
04.22 ± 0.2 |
04.76 ± 0 |
07.94 ± 0.6 |
|
3c
|
11.10 ± 0.2 |
16.55 ± 0.3 |
17.36 ± 0.3 |
|
3d
|
04.07 ± 0.1 |
08.07 ± 0.3 |
15.04 ± 0.2 |
|
3e
|
04.98 ± 0.2 |
06.20 ± 0.3 |
12.64 ± 0.4 |
|
4a
|
03.72 ± 0.3 |
4.074 ± 0.3 |
07.91 ± 0.4 |
|
4b
|
03.47 ± 0.2 |
03.89 ± 0.3 |
05.08 ± 0.3 |
|
5a
|
16.04 ± 0.3 |
21.92 ± 0.3 |
22.95 ± 0.4 |
|
5b
|
15.60 ± 0.2 |
21.72 ± 0.3 |
34.15 ± 0.4 |
|
5c
|
12.00 ± 0.2 |
19.44 ± 0.2 |
28.85 ± 0.3 |
|
5d
|
16.96 ± 0.1 |
19.74 ± 0.3 |
26.36 ± 0.2 |
|
5e
|
23.94 ± 0.2 |
27.20 ± 0.4 |
28.65 ± 0.3 |
|
5f
|
04.01 ± 0.2 |
08.26 ± 0.3 |
13.12 ± 0.4 |
|
5g
|
03.80 ± 0.1 |
04.79 ± 0.3 |
18.00 ± 0.3 |
|
5h
|
10.29 ± 0.2 |
14.15 ± 0.3 |
13.69 ± 0.2 |
|
5i
|
17.41 ± 0.1 |
16.17 ± 0.3 |
23.48 ± 0.4 |
|
5j
|
04.18 ± 0.2 |
06.39 ± 0.2 |
11.24 ± 0.3 |
|
5k
|
13.42 ± 0.1 |
18.40 ± 0.3 |
28.29 ± 0.4 |
|
5l
|
08.39 ± 0.2 |
06.41 ± 0.3 |
13.66 ± 0.5 |
|
5m
|
13.81 ± 0.1 |
14.80 ± 0.3 |
19.50 ± 0.4 |
|
Std
|
05.46 ± 0.1 |
07.76 ± 0.2 |
4.05 ± 0.3 |
Further, the most potent derivatives 4a and 4b from this series were selected for the NCI-60 cell line anticancer screening program by the National Cancer Institute (NCI), USA. After preliminary screening on the tumor cell lines, these derivatives were tested for five dose concentrations on a panel of 59 human tumor cell lines derived from 9 different cancer types: leukemia, non-small lung, colon, CNS, melanoma, ovarian, renal, prostate and breast. The results expressed as GI50 values for the test compounds are illustrated in the ESI as Table S1.† The five dose assay20 results revealed that these two compounds possess significant anticancer activity against different cancer cell lines with GI50 values in the range of 1.19–43.3 μM. The most active compounds 4a and 4b in the MTT assay retained their activity in the NCI screening data. These compounds exhibited promising growth inhibition with GI50 values ranging from 1.38–43.3 and 1.19–14.1 μM, respectively.
DNA binding affinity studies
Circular dichroism (CD).
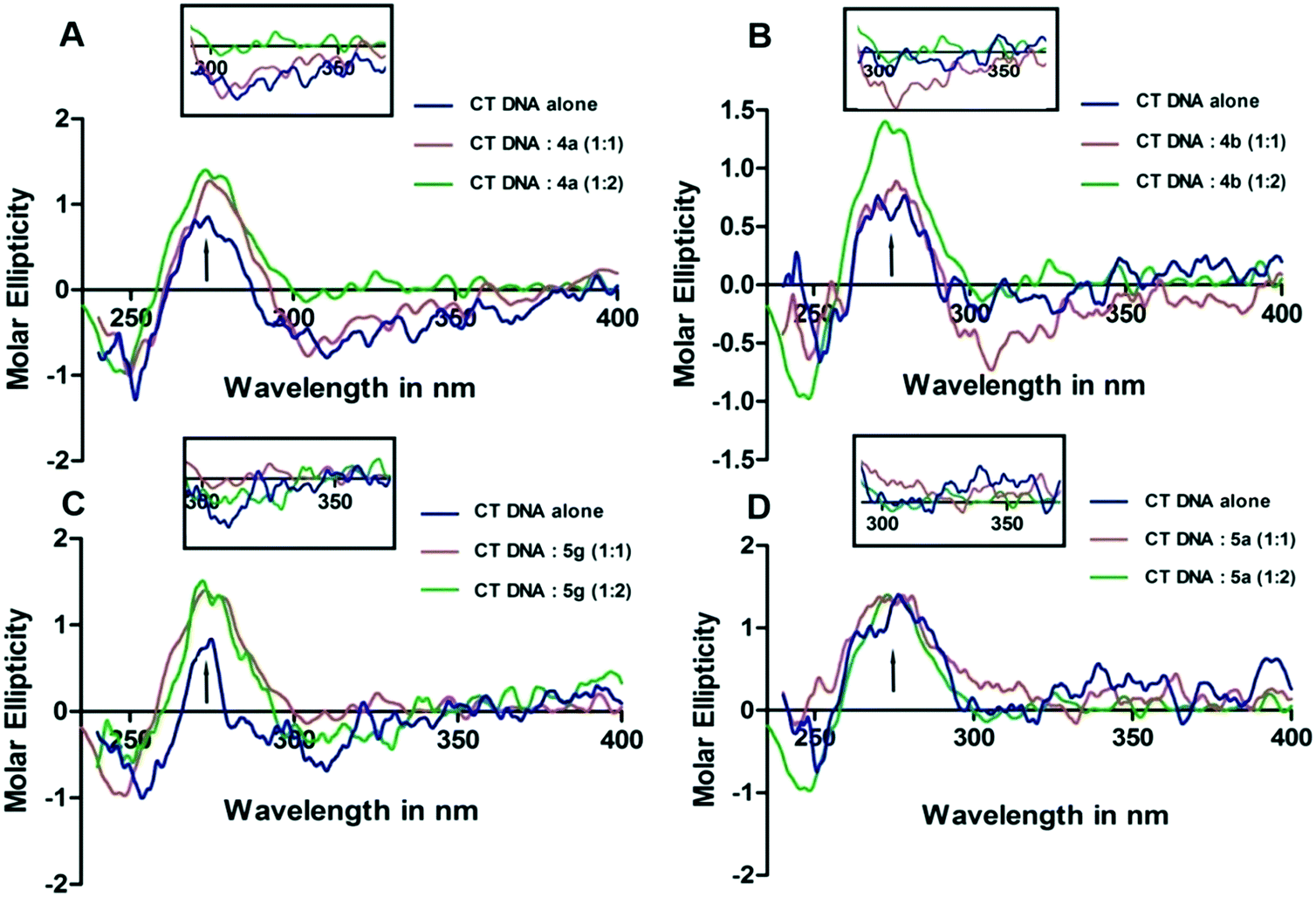
Circular dichroism studies were performed in order to analyze the interaction of DNA with small molecules which indicated changes that are likely to take place with respect to the DNA conformation. DNA is a biological macromolecule, which is responsible for cell proliferation and several other biological functions. Hence, the interaction and changes in the DNA topology will provide sufficient information on the nature of DNA–complex interactions and conformational changes in the derivatives binding to DNA. The CD spectrum was recorded while keeping the CT-DNA concentration constant (15 μM) and increasing the compound concentration from 15 μM up to 30 μM. The wavelength scan was done from 240 nm to 400 nm. The CD spectrum of CT-DNA exhibits a positive band at 275 nm and a negative band at 245 nm. These indicate π–π base stacking and right-hand helicity, the characteristic profile of B-form DNA. In the present study, addition of 4a,b and 5a,b derivatives to CT-DNA results in positive CD, indicating continuous hyperchromicity even at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 stoichiometry of DNA:compound. This suggests the stabilization of the DNA structure on compound interaction.21 The intensities of the negative band did not alter much; this indicates less effect on DNA helicity during interaction. 4a,b and 5g exhibited lower intensity with negative ICD in the region starting from 300 nm to 400 nm. The ICD exhibited by 4a,b and 5g derivatives on interaction with CT-DNA in the 300 nm to 350 nm region was shown separately in the inset. This indicates that the 4a,b and 5g molecules possibly interact with and stabilize CT-DNA. Having relatively less effect on the derivatives, the intensity of the positive CD band was noticed when 5a was added to CT-DNA, whereas 5g has brought a moderate change in the positive CD band. The appearance of negative ICD in 4a,b and 5g indicates that the derivatives may be intercalating between the two strands of DNA. The CD spectra of 4a,b, 5a and 5g on interaction with CT DNA are shown in Fig. 2.
:1 and 1:2 stoichiometry of DNA:compound. This suggests the stabilization of the DNA structure on compound interaction.21 The intensities of the negative band did not alter much; this indicates less effect on DNA helicity during interaction. 4a,b and 5g exhibited lower intensity with negative ICD in the region starting from 300 nm to 400 nm. The ICD exhibited by 4a,b and 5g derivatives on interaction with CT-DNA in the 300 nm to 350 nm region was shown separately in the inset. This indicates that the 4a,b and 5g molecules possibly interact with and stabilize CT-DNA. Having relatively less effect on the derivatives, the intensity of the positive CD band was noticed when 5a was added to CT-DNA, whereas 5g has brought a moderate change in the positive CD band. The appearance of negative ICD in 4a,b and 5g indicates that the derivatives may be intercalating between the two strands of DNA. The CD spectra of 4a,b, 5a and 5g on interaction with CT DNA are shown in Fig. 2.
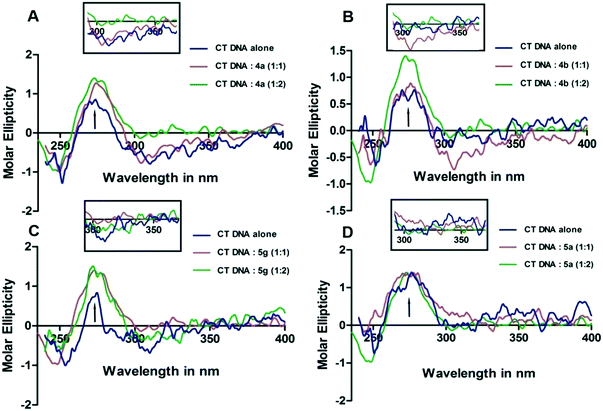 |
| | Fig. 2 The CD spectra of CT-DNA (240 to 400 nm) in 100 mM TE (pH 7.0, 25 °C). 15 μM and 30 μM 4a (A), 4b (B), 5g (C) and 5a (D) were added to 15 μM CT-DNA prepared in 100 mM TE (pH 7.0). The arrows indicate the direction in which the CD peaks move after the interaction of 4a,b, 5a and 5g with CT-DNA. The ICD observed in the wavelength range starting from 300 nm to 350 nm is shown separately in the inset. | |
UV-visible spectral study
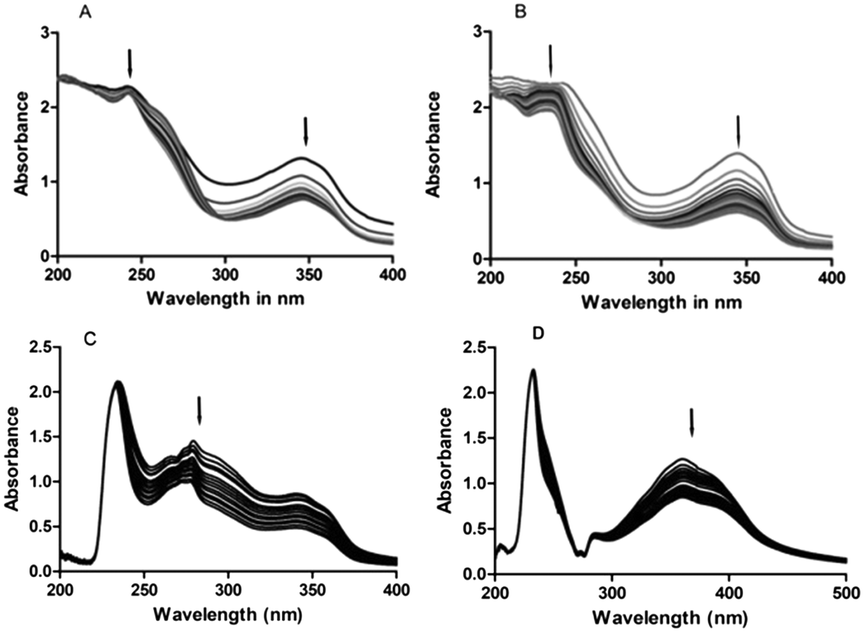
These studies indicate the possible changes that occur in the CT-DNA topology during the course of interaction of the compounds with CT-DNA. The UV-vis spectral studies conducted with CT-DNA and 4a exhibited prominent absorption bands at 244 nm and 347 nm. Addition of 4a to 25 μM CT-DNA in an equal amount showed a continuous decrease in the intensity of the absorption bands. Of 244 nm and 347 nm absorption peaks, the absorption band at 244 nm exhibited a slight bathochromic shift from 244 nm to 246 nm (about 2 nm red shift). Further, an isosbestic point was noticed at 248 nm and 299 nm when DNA interacted with 4a. On the other hand, 4b exhibited absorption bands at 235 nm and 345 nm. Similarly, the absorption band of 4b exhibited hypochromicity with the addition of DNA. The hypochromicity of the complex's absorption band is more evident at 345 nm than 235 nm. 5g exhibited absorption bands at 231 nm and 356 nm. The UV-vis absorption band at 231 nm was more evident compared to those of 4a and 4b. Continuous hypochromicity without much bathochromic shift of the absorption bands in the case of 4b and 5g and a slight bathochromic shift in the case of 4a on the addition of DNA and the CD results indicate that these derivatives probably bind to DNA and they may intercalate with CT-DNA.22 A effect is noticed in the UV-vis spectra of CT-DNA-5a. The Kd values obtained when 4a,b, 5g and 5a complexes interact with CT-DNA are 132 μM, 121 μM, 136 μM and 144 μM, respectively. The UV-vis titration data were fitted to eqn (1) to obtain the dissociation constant (Kd) taking into account the total amount of the 1:1 ratio of the derivative:CT-DNA complex formed, independent of the complex binding site(s). The Kd values were calculated using the formula Kd = [CT-DNA][derivative]/[CT-DNA derivative].
The results obtained from the UV-vis spectral data reveal that 4a,b and 5g interact with DNA possibly through intercalation between the two strands of CT-DNA. The UV-vis spectra obtained for 4a,b, 5a and 5g are shown in Fig. 3.
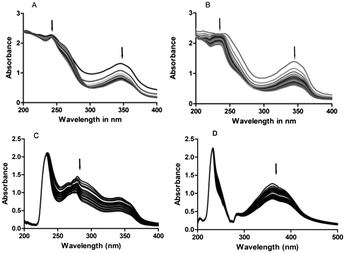 |
| | Fig. 3 UV-vis absorption spectra of compounds 4a,b, 5a and 5g (25 μM) in 100 mM TE (pH 7.0, 25 °C). (A) 4a, (B) 4b, (C) 5a and (D) 5g. 25 μM 4a,b, 5a and 5g solution and 25 μM CT DNA were prepared in 100 mM TE (pH 7.0). The arrows indicate the direction in which the absorption bands are moving. | |
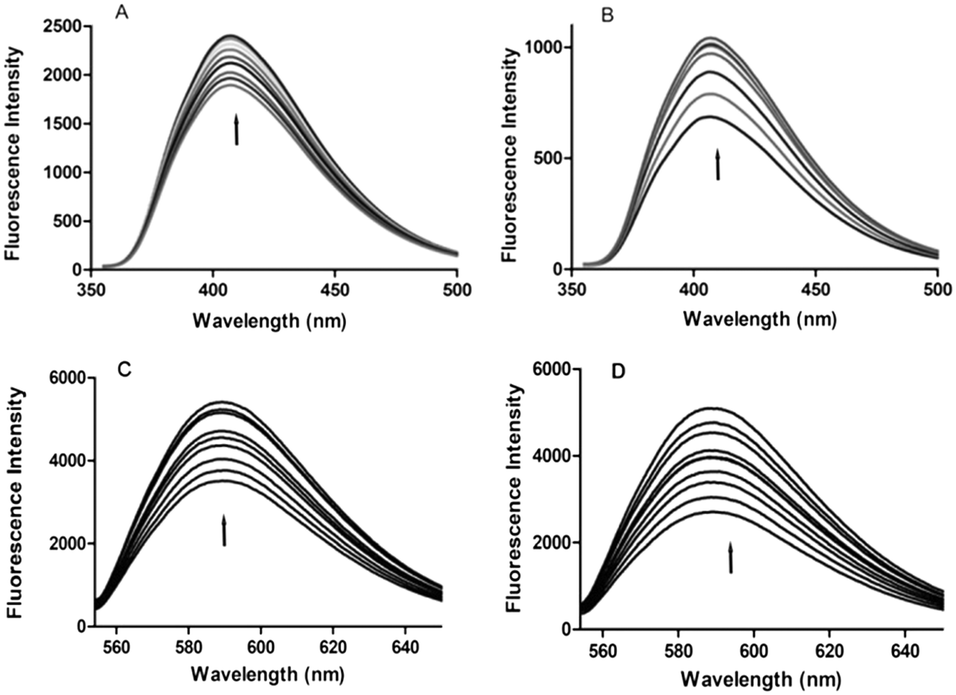
Fluorescence spectroscopy
Fluorescence titration is widely used to understand the type of interaction between DNA and small fluorescent molecules. This method is also useful for studying the electronic environment around the DNA–complex.23 Since 4a,b, 5a and 5g are fluorescent substances, their interaction with DNA can be monitored even at lower concentrations. The emission spectra of 4a and 4b show a prominent peak at 407 nm, whereas 5a and 5g exhibit emission maxima at 586 nm. On continuous addition of CT-DNA to 4a,b, 5a and 5g, the emission peak intensity increased gradually. It was noticed that upon addition of CT-DNA, the fluorescence emission intensities of 5a and 5g exhibited a higher increase when compared to those of 4a and 4b. From these results, we speculate that all the derivatives may come close to the DNA bases possibly through an intercalative mode of interaction.24 The increase in emission intensity of a peak can be attributed to the non-radioactive energy transfer between the excited fluorescent derivatives and DNA bases, wherein these derivatives are the donors and the DNA bases are the acceptors. Their spectra are shown in Fig. 4.
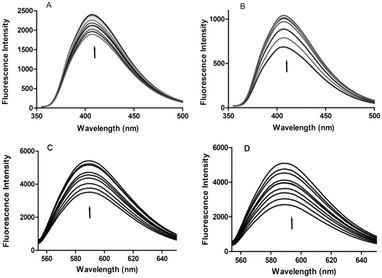 |
| | Fig. 4 Fluorescence emission spectra of compounds 4a,b, 5a and 5g (10 μM) titrated with increasing concentrations of CT-DNA (in multiples of 0.5 μM). (A) 4a, (B) 4b, (C) 5a and (D) 5g. 10 μM 4a,b, 5a and 5g derivatives were taken in the fluorescence cuvette and titrated with increasing concentrations of CT-DNA (in multiples of 0.5 μM). The arrows indicate the direction in which the absorption bands are moving. | |
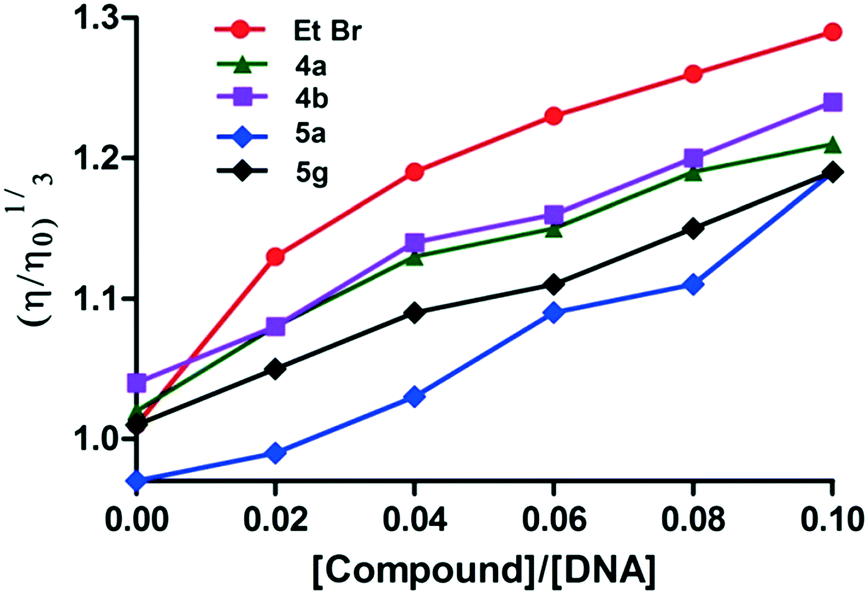
Viscosity studies
The relative viscosity of DNA generally increases when a molecule intercalates with DNA and this enhanced viscosity would be due to the increase in the axial length of the DNA helix on the molecule's intercalation. Hence, intercalation of a molecule results in an increase of the viscosity of DNA solutions.25 Meanwhile, reduction in the relative viscosity is typically observed with molecules binding covalently to DNA.26 Moreover, the viscosity does not change or show a slight change when molecules bind to the surface of DNA.27 According to the spectroscopic studies, the active derivatives 4a,b and 5g interact with CT-DNA; viscosity studies were also carried out to confirm the intercalating nature of these derivatives with CT DNA. These studies demonstrated that the viscosity of the DNA solution increased upon the addition of these derivatives, wherein the change in viscosity was larger in the case of 4b compared to 4a. Similarly, between 5a and 5g, derivative 5a has a weaker interaction with CT-DNA compared to 5g. Therefore, this study also indicates that among the derivatives, 4a and 4b have stronger interaction with CT-DNA which is closely followed by 5g. Among the derivatives studied, 5a has the weakest interaction with CT-DNA. The viscosity data indicate that among the derivatives studied, 4a,b and 5g derivatives may intercalate with CT-DNA. In the present study, the interaction between CT-DNA and ethidium bromide (EtBr), a well-known intercalator, was considered as the control. The viscosity of DNA increased continuously with the addition of ethidium bromide (EtBr), due to the intercalation of EtBr with CT-DNA. The graph plotted between (η/ηo)1/3 and compound/CT-DNA is shown in Fig. 5.
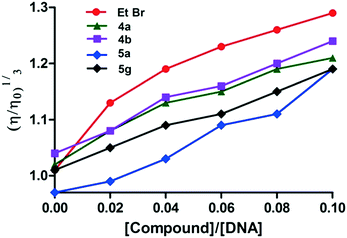 |
| | Fig. 5 Change in the CT-DNA (150 μM) viscosity before and after the addition of 4a,b, 5a and 5g (15 μM). Variation in the viscosity of CT-DNA (150 μM) on addition of ethidium bromide (15 μM) which was considered as the control. | |
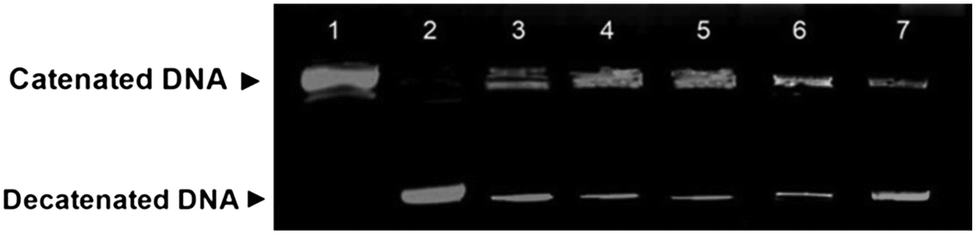
Topoisomerase II inhibition assay
Topoisomerases control the topology of DNA and these enzymes play a significant role in DNA replication, transcription, segregation and recombination.28 Type I topoisomerases (topo I) make a single nick in the DNA strand, while type II topoisomerases (topo II) make similar nicks on both the strands of DNA. Topo II is a nuclear enzyme that influences the DNA topology by catalyzing the DNA cleavage and by relegating the phosphodiester bonds; as a result, topo II initiates multiple recombination/repair pathways that trigger cell death.29 Topo II inhibition studies were carried out using a topo II drug screening kit (TG 1009, Topogen, USA). Catenated DNA and topo II enzyme (5 units) were incubated in the presence of 10 μM amonafide for 30 min (lane 3 in Fig. 6), which is considered as a positive control in the present experiment. The experimental results revealed that decatenation did not take place (by forming minicircles) and the ternary complex (kDNA + topo II and amonafide complex) remained close to the origin. The studies, when repeated with catenated kDNA (200–300 ng) in the presence of 10 μM 4b,a, 5a and 5g (lanes 4, 5, 6 and 7, respectively, in Fig. 6), revealed less formation of decatenated DNA and most of the DNA in the presence of 4a,b and 5g derivatives did not enter the gel. But with 5a, the formation of the ternary complex (kDNA + topo II and 5a) is lower compared to that of the other derivatives. Among the derivatives considered, 5a exhibited moderately higher cytotoxicity in the MTT assay. Considering this aspect, the 5a derivative was considered as the negative control throughout this study. The topo II inhibition by 5a was comparatively lower when compared to that by 4a,b and 5g. These results support the cytotoxicity data, where 4a,b and 5g showed maximum cytotoxicity compared to 5a. This suggests that a ternary complex is not formed with kDNA in the absence of any derivatives and with topo II alone as depicted in lanes 1 and 2, respectively, in Fig. 6. The formation of the ternary complex is also an indication that these derivatives intercalate with DNA. From these studies, it is evident that all the derivatives (mainly 4a,b and 5g) intercalate with DNA and interfere with the catalytic activity of the topo II enzyme-like amonafide and thus they function as catalytic inhibitory compounds.
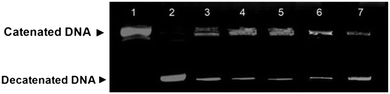 |
| | Fig. 6 Topo II inhibition assay. To about 200–300 ng of kDNA, about 5 units of Topo II was added and incubated at 37 °C for 30 minutes. Lane 1- kDNA alone; lane 2- kDNA and topo II; lane 3- kDNA, topo II and amonafide; lane 4- kDNA, topo II and 4b; lane 5- kDNA, topo II and 4a; lane 6- kDNA, topo II and 5g; lane 7- kDNA, topo II and 5a. | |
Molecular modelling studies
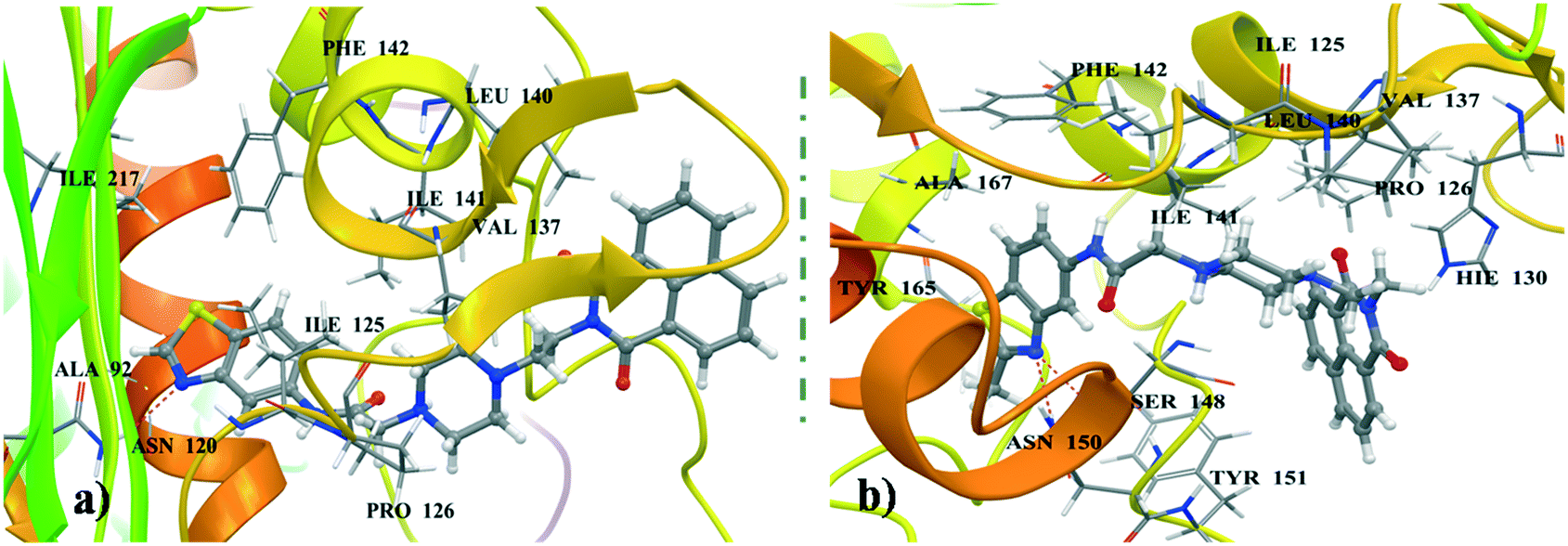
Most of the naphthalimide-containing derivatives show their anticancer activity by intercalation into DNA30 and through inhibiting topoisomerase enzymes. In order to understand their binding poses and interaction patterns, the most active molecules 4a and 4b in the present study were selected for molecular docking studies. All the coordinates of the protein crystal structures were obtained from the RCSB Protein Data Bank. Energy minimizations and necessary corrections of the proteins and DNA were performed by using the protein preparation wizard of Maestro version 11.1 (Schrödinger 2017-1).31 These docking studies were performed using Glide 7.4 (Schrödinger 2017-1).
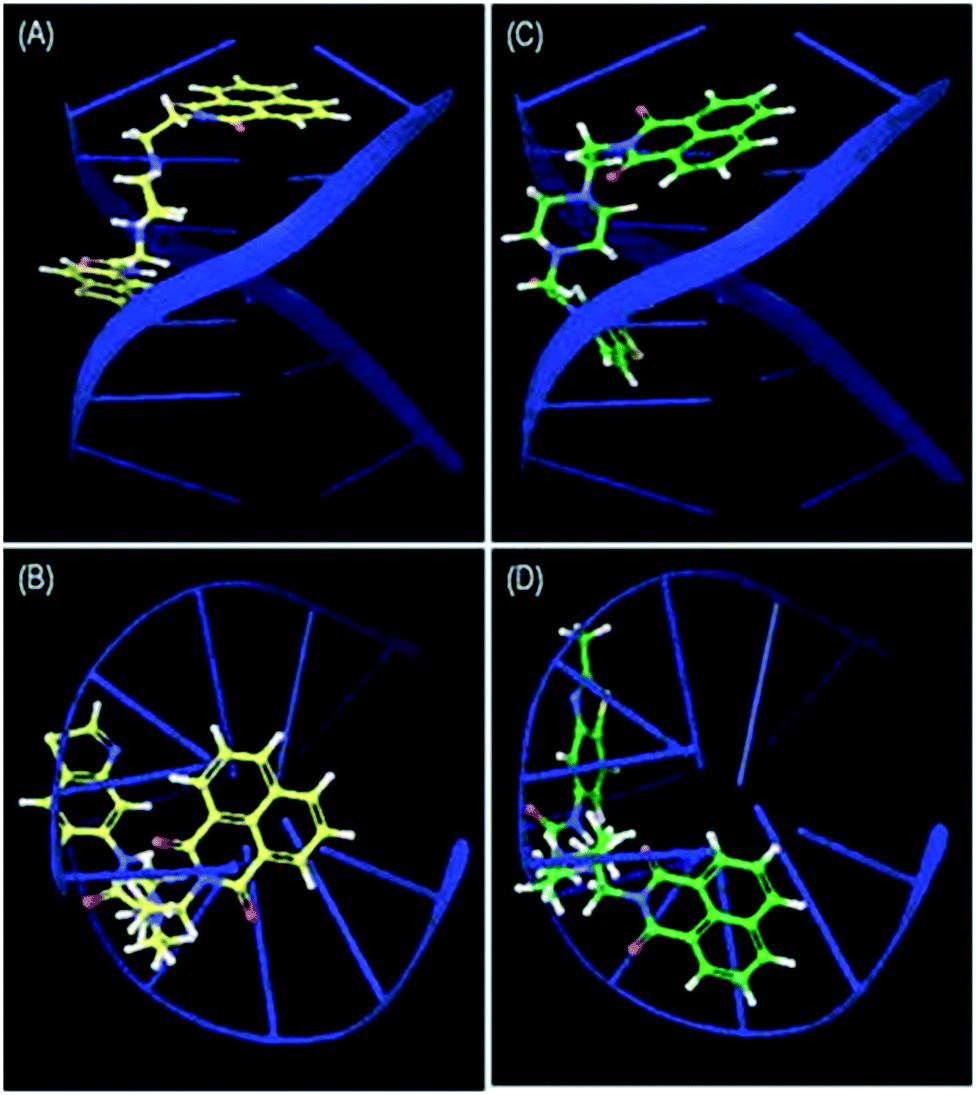
Docking studies were performed for the representative derivatives 4a and 4b at the ATP binding site of topoisomerase-IIα (PDB ID: 1ZXN).32 The results revealed that these derivatives were well accommodated in the binding site of topoisomerase-IIα. Further, for ligands 4a and 4b some common interactions similar to those of the co-crystallized ligand were observed (Fig. 7). In naphthalimide derivative 4a, the nitrogen of benzothiazole shows hydrogen-bonding interaction with Asn120, and hydrophobic interactions are observed with Ala92, Ile125, Pro126, Val137, Leu140, Ile141, Phe142, and Ile217. Meanwhile, in 4b, the naphthalimide ring forms π–π stacking interactions with Hie130, hydrogen-bonding interactions with Ser148 and Asn150 and hydrophobic contacts with Ile125, Pro126, Val137, Leu140, Ile141, Phe142, Tyr151, Tyr165, and Ala167. DNA docking was also performed in the DNA duplex d(GAAGCTTC)2 (PDB ID: 1NAB)33 using XP Glide 7.4 (Schrödinger 2017-1). The docked pose for the top scored derivatives 4a and 4b showed intercalation of the naphthalimide ring between the G–C base pairs of DNA, whereas the piperazine linker and benzothiazole were extended out to the minor groove (Fig. 8). These molecules were well stabilized in the active site of DNA by hydrogen-bonding and π–π stacking interactions between piperazine and benzothiazole motifs with DNA base pairs, respectively.
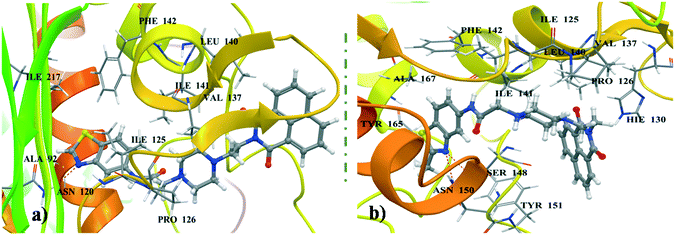 |
| | Fig. 7 (a) Binding pose of 4a in topoisomerase-IIα. (b) Binding pose of 4b in topoisomerase-IIα. | |
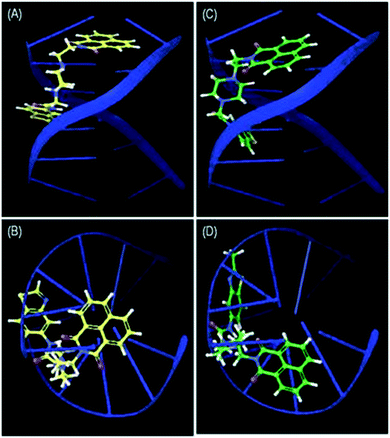 |
| | Fig. 8 Side view (A) and top view (B) of the intercalation of 4a and side view (C) and top view (D) of the intercalation of 4b between the G–C base pairs of d(GAAGCTTC)2. | |
Conclusion
In conclusion, a series of new naphthalimides were designed and synthesized and their cytotoxic activities were evaluated. The majority of these naphthalimides potentially inhibited the growth of HT-29 and A549 cancer cell lines selectively. Interestingly, hybrid derivatives 4a and 4b which have an amide bond at the 6-position displayed promising cytotoxic activity compared to the reference drug amonafide. We also investigated the most active derivatives 4a and 4b for their antitumor mechanism of action, which potentially inhibit DNA topo II. Moreover, the DNA interactions of the representative derivatives were studied by CD, UV-visible and fluorescence spectroscopy demonstrating that they intercalate with DNA.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank CSIR-New Delhi for the award of senior research. We acknowledge funding received from the project entitled “Affordable Cancer Therapeutics” (CSC0301) under the XIIth five year plan. Dr N. Shankaraiah gratefully acknowledges SERB, DST, Govt. of India for the research grant (YSS-2015-001709).
References
-
(a) M. F. Brana and A. Ramos, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 237 CrossRef CAS PubMed;
(b) S. Nekkanti, R. Tokala and N. Shankaraiah, Curr. Med. Chem., 2017, 24, 2887 CrossRef CAS PubMed;
(c) N. Shankaraiah, S. Nekkanti, O. Ommi and P. S. S. Lakshmi, Curr. Med. Chem., 2018 DOI:10.2174/0929867325666180410110729.
-
DNA Topoisomerases in Cancer Therapy: Present and Future, ed. T. Andoh, Kluwer Academic/Plenum, New York, 2003 Search PubMed.
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 338 CrossRef CAS.
- H. K. Wang, S. L. Morris-Natschke and K. H. Lee, Med. Res. Rev., 1997, 17, 367 CrossRef CAS PubMed.
- K. J. Haglof, E. Popa and H. S. Hochster, Update Cancer Ther., 2006, 1, 117 CrossRef.
- Y. Pommier, Chem. Rev., 2009, 109, 2894 CrossRef CAS PubMed.
- D. Kaung and X. Qian, J. Med. Chem., 2010, 53, 2589–2600 CrossRef PubMed.
- P. Gopikrishna, N. Meher and P. K. Iyer, ACS Appl. Mater. Interfaces, 2018, 10, 12081–12111 CrossRef CAS PubMed.
- G. Capranico and M. Binaschi, Biochim. Biophys. Acta, 1998, 1400, 185 CrossRef CAS.
- M. F. Brana, J. M. Castellano, M. Moran, M. J. Perez de Vega, C. R. Romerdahl, X.-D. Qian, P. Bousquet, F. Emling, E. Schlick and G. Keilhauer, Anti-Cancer Drug Des., 1993, 8, 257 CAS.
- M. F. Brana, J. M. Castellano, M. Moran, M. J. Perez de Vega, D. Perron, D. Conlon, P. F. Bousquet, C. A. Romerdahl and S. P. Robinson, Anti-Cancer Drug Des., 1996, 11, 297 CAS.
- M. F. Brana, M. Cacho, A. Gradillas, B. de Pascual-Teresa and A. Ramos, Curr. Pharm. Des., 2001, 7, 1745 CrossRef CAS PubMed.
- A. Kamal, N. R. Bolla, P. S. Srikanth and A. K. Srivastava, Expert Opin. Ther. Pat., 2013, 23, 299–317 CrossRef CAS PubMed.
- G. Gellerman, Lett. Drug Des. Discovery, 2016, 13, 47 CrossRef CAS.
-
(a) K. Kokosza, G. Andrei, D. Schols, R. Snoeck and D. G. Piotrowska, Bioorg. Med. Chem., 2015, 23, 3135 CrossRef CAS;
(b) M. Lv and H. Xu, Curr. Med. Chem., 2009, 16, 4797 CrossRef CAS PubMed.
- L. Ingrassia, F. Lefranc and R. Kiss, Curr. Med. Chem., 2009, 16, 1192 CrossRef CAS.
- M. F. Braña, M. Cacho, M. A. Garcia, B. de Pascual-Teresa, A. Ramos, M. T. Dominguez, J. M. Pozuelo, C. Abradelo, M. F. Rey-Stolle and M. Yuste, J. Med. Chem., 2004, 47, 1391 CrossRef PubMed.
-
(a) A. Kamal, G. Ramakrishna, V. L. Nayak, P. Raju, A. V. Subba Rao, G. Viswanath, M. V. P. S. Vishnuvardhan, S. Ramakrishna and G. Srinivas, Bioorg. Med. Chem., 2012, 20, 789 CrossRef CAS PubMed;
(b)
A. Kamal, Novel napthalimide-benzimidazole hybrids as potential antitumour agents and process for the preparation thereof, WO Patent WO/2009/110000, 2009 Search PubMed;
(c) A. Kamal, B. Reddy and G. Reddy, Bioorg. Med. Chem. Lett., 2002, 12, 1933 CrossRef CAS PubMed;
(d) N. Shankaraiah, N. P. Kumar, R. Tokala, B. S. Gayatri, V. Talla and L. S. Santos, J. Braz. Chem. Soc. DOI:10.21577/0103-5053.20180111;
(e) N. S. Rao, V. L. Nayak, A. V. S. Rao, S. M. Ali Hussaini, S. Satish, A. Alarifi and A. Kamal, Arabian J. Chem. DOI:10.1016/j.arabjc.2016.07.014.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112 CrossRef CAS PubMed.
-
http://dtp.nci.nih.gov/branches/btb/ivclsp.html
.
- L. Wang, Y. Wen, J. Liu, J. Zhou and C. Li, Org. Biomol. Chem., 2011, 9, 2648 RSC.
- J. K. Barton, J. J. Dennenberg and J. B. Chaires, Biochemistry, 1993, 32, 2573 CrossRef.
- J. Wu, M. Zhao, K. Qian, K.-H. Lee, S. Morris–Natschke and S. Peng, Eur. J. Med. Chem., 2009, 44, 4153 CrossRef CAS PubMed.
- D. Suh and J. B. Chaires, Bioorg. Med. Chem., 1995, 3, 723 CrossRef CAS PubMed.
- N. Shahabadi, S. Kashanian and M. Purfoulad, Spectrochim. Acta, Part A, 2009, 72, 757 CrossRef PubMed.
- J. M. Kelly, A. B. Tossi, D. J. Mcconnell and C. Ohui-gin, Nucleic Acids Res., 1985, 13, 6017–6034 CrossRef CAS PubMed.
- C. Metcalfe, C. Rajput and J. A. Thomas, J. Inorg. Biochem., 2006, 100, 1314 CrossRef CAS PubMed.
- J. L. Nitiss and W. T. Beck, Eur. J. Cancer, 1996, 32A(6), 958 CrossRef CAS.
- Y. Pommier, E. Leo and H. L. Zhang, Chem. Biol., 2010, 17, 421 CrossRef CAS PubMed.
- R. Tandon, V. Luxami, H. Kaur, N. Tandon and K. Paul, Chem. Rec., 2017, 17, 1 CrossRef.
-
Schrödinger suite 2017-1, Schrödinger, LLC, New York, 2017 Search PubMed.
- H. Wei, A. J. Ruthenburg, S. K. Bechis and G. L. Verdine, J. Biol. Chem., 2005, 280, 37041 CrossRef CAS.
- C. Tempeini, L. Messori, P. Orioli, C. Di Bugno, F. Animati and G. Ughetto, Nucleic Acids Res., 2003, 31, 1464 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00395e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *c,
V.
Lakshma Nayak
a,
Satish
Sunkari
ab,
Ramya
Tokala
*c,
V.
Lakshma Nayak
a,
Satish
Sunkari
ab,
Ramya
Tokala