
Design, synthesis, and in vitro biological evaluation of novel benzimidazole tethered allylidenehydrazinylmethylthiazole derivatives as potent inhibitors of Mycobacterium tuberculosis†
Goverdhan
Surineni‡
 ab,
Yamin
Gao‡
ab,
Muzammal
Hussain
ab,
Zhiyong
Liu
a,
Zhili
Lu
a,
Chiranjibi
Chhotaray
ab,
Md Mahmudul
Islam
ab,
H. M. Adnan
Hameed
ab and
Tianyu
Zhang
*ab
ab,
Yamin
Gao‡
ab,
Muzammal
Hussain
ab,
Zhiyong
Liu
a,
Zhili
Lu
a,
Chiranjibi
Chhotaray
ab,
Md Mahmudul
Islam
ab,
H. M. Adnan
Hameed
ab and
Tianyu
Zhang
*ab
aState Key Laboratory of Respiratory Disease, Guangzhou Institutes of Biomedicine and Health (GIBH), Chinese Academy of Sciences (CAS), Guangzhou-510530, China. E-mail: zhang_tianyu@gibh.ac.cn; Fax: (+86)20 3201 5270; Tel: (+86)20 3201 5270
bUniversity of Chinese Academy of Sciences (UCAS), Beijing, China
First published on 25th October 2018
Abstract
Tuberculosis (TB) has become one of the most significant public health problems in recent years. Antibiotic therapy remains the mainstay of TB control strategies, but the increasing resistance of mycobacterial species has heightened alarm, requiring the development of novel drugs in order to improve treatment outcomes. Here, as an effort to identify novel and effective antitubercular agents, we designed and synthesized a series of novel substituted benzimidazolallylidenehydrazinylmethylthiazole derivatives via a multi-component molecular hybridization approach with single molecular architecture. Our design strategy involved assembling the antitubercular pharmacophoric fragments benzimidazole, 2-aminothiazole and substituted α,β-unsaturated ketones via condensation reactions. All the newly synthesized compounds were fully characterized via NMR and mass spectral data and evaluated for in vitro biological activity against the H37Ra strain of Mycobacterium tuberculosis. From the biological evaluation data, we identified some effective compounds, of which 8g and 7e were the most active ones (both having MIC values of 2.5 μg mL−1). In addition, compound 8g exhibited a lower cytotoxicity profile. We conceive that compound 8g may serve as a chemical probe of interest for further lead optimization studies with the general aim of developing novel and effective antitubercular agents.
1. Introduction
Tuberculosis (TB) is one of the serious infectious diseases caused by Mycobacterium tuberculosis (Mtb) and becomes lethal when co-infected with HIV.1–3 According to recent reports by the World Health Organization (WHO), worldwide, 10.4 million new tuberculosis patients were identified in 2016, from which 1.67 (16.1%) million deaths were recorded.4 The majority of TB infected people can be cured with early diagnosis and proper treatment. Antibiotic therapy remains the mainstay of TB control strategies. However, the long duration of treatment (at least 6 months), toxicity with low efficacy, and health burden are the main barriers regarding treating TB patients using drugs.5 In addition, the increasing resistance of Mycobacterium species to existing drugs has heightened alarm about TB in the international health community.6 In particular, the emergence of multi-drug resistant strains (MDR-TB), which are resistant to the first-line drugs isoniazid and rifampicin, and more recently the emergence of extensively drug-resistant strains (XDR-TB), which are MDR-TB strains also resistant to any fluoroquinolone drug and to at least one of the three injectable drugs used for TB treatment (amikacin, capreomycin and kanamycin), have complicated treatment outcomes.7 In 2016, around 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new TB patients were diagnosed as having resistance to rifampicin (RRTB); among these, 490000 patients had MDR-TB while 8000 patients had XDR-TB. The lack of new drugs as well as druggable targets, which could be utilized for developing more efficient anti-TB agents, are major hurdles to tackling this resistance.6,8–11 Clearly, there is a pressing need to identify and develop novel drug-based antimycobacterial therapies in order to improve treatment outcomes.8 However, the drug development pipeline in this regard remains woefully thin, with little chemical diversity.12–15
000 new TB patients were diagnosed as having resistance to rifampicin (RRTB); among these, 490000 patients had MDR-TB while 8000 patients had XDR-TB. The lack of new drugs as well as druggable targets, which could be utilized for developing more efficient anti-TB agents, are major hurdles to tackling this resistance.6,8–11 Clearly, there is a pressing need to identify and develop novel drug-based antimycobacterial therapies in order to improve treatment outcomes.8 However, the drug development pipeline in this regard remains woefully thin, with little chemical diversity.12–15
In recent years, synthetic heterocyclic hybrid molecules have exhibited therapeutic potential against many diseases.16–19 Particularly, heterocyclic compounds bearing benzimidazole and aminothiazole moieties have shown a broad range of medicinal advantages, having demonstrated antitubercular, antihelminthic, antifungal, anticancer, and antiulcer activities.20 Among the benzimidazole-derived heterocyclics, trisubstituted SB-P3G2 (I) and SB-P8B2 (II) have shown good activity against drug-resistant and susceptible strains of Mtb, and have also exhibited efficacy in a murine Mtb infection model.21,22 Similarly, aminobenzimidazole 1 (AB-1) (III) has been demonstrated as a potent inhibitor of GyrB, while exhibiting a minimum inhibitory concentration (MIC) of 1 μg L−1 against the H37Ra Mtb strain.23 There are also some benzimidazole-derived hybrid molecules that have been developed as potent drugs and are being effectively used for the treatment of different diseases. These include: albendazole (IV), having antimicrobial activity;24,25 omeprazole (V), as an antiulcer drug;26,27 and bendamustine (VI), regarded as one of the effective anticancer drugs (Fig. 1).28,29
 | ||
| Fig. 1 Synthetic bioactive analogs of benzimidazole. | ||
Similarly to benzimidazole hybrids, 2-aminothiazoles are also a familiar group of heterocyclic compounds that exhibit a broad range of biological activities, and their advantages as medicines have been well established.30,31 For example, the thiazole nucleus is an integral component (main core) of all available penicillins, which have transmuted the therapy of bacterial diseases.32 In fact, aminothiazole derivatives have attracted the medicinal community with regards to the synthesis of combinatorial heterocyclic compound series possessing various biological or therapeutic activities, particularly against Mtb.16,33 The synthetic dibenzofuran-, carbazole- and dibenzothiophene-derived thiazolamine derivatives VII–IX have been shown to possess potent anti-tubercular activity against the H37Ra Mtb strain, with an MIC range of 0.78–1.56 μg mL−1, respectively.33,34 Similarly, the naturally occurring antibiotic thiolactomycin (TLM) analogs X and XI, which primarily act by inhibiting the FAS-II β-ketoacyl-ATP synthase condensing enzyme, have been demonstrated to halt mycolic acid biosynthesis and then subsequently induce Mtb cell death.35–38 Moreover, nitazoxanide (NTZ, XII) and its active metabolite tizoxanide (TIZ, XIII) are other examples of aminothiazole-based drugs that have been manifested to inhibit replicative and non-replicative strains of Mtb.39,40 The good remedial properties of these related drugs have inspired medicinal chemists to synthesize a number of novel clinical agents to cure various types of diseases.16,18,41
Indeed, the molecular hybridization approach has emerged as a useful structural modification approach for the design of ligands with new molecular architectures.42 In this work, in an effort to identify new antitubercular agents, we report the synthesis and biological evaluation of a novel series of benzimidazolallylidenehydrazinylmethylthiazole derivatives that we designed through the hybridization of antitubercular pharmacophores like benzimidazole, aminothiazole and unsaturated aromatic substitution units. The designed scaffold (Fig. 3) originated from the structures of the antitubercular agents VII, VIII and IX (Fig. 2). The newly designed hybridized molecule comprises three parts: (1) a 2-aminothiazole ring, as an antitubercular active core unit; (2) benzimidazole, as an antitubercular pharmacophoric backbone unit; and (3) substituted aromatic unsaturated groups appended to the benzimidazole and aminothiazole units, for enhancing the pharmacophoric impact and to induce a lipophilic nature. The presence of an allyl group may cause a decrease in lipophilicity and, as a result, diminish the compound's ability to penetrate the thick lipophilic cell wall of Mtb.43 Therefore, variations in the designed hybrid molecule were accomplished with the choice of substituted aromatic unsaturated groups, aiming for greater lipophilicity, which may assist in potentiating the anti-TB activity of the designed series. The target compounds 8a–o were synthesized through a multi-step synthetic sequence utilizing simple condensation reactions. The in vitro antimycobacterial screening of the designed library led to the identification of an important hit compound, which may be pursued for future lead optimization studies.
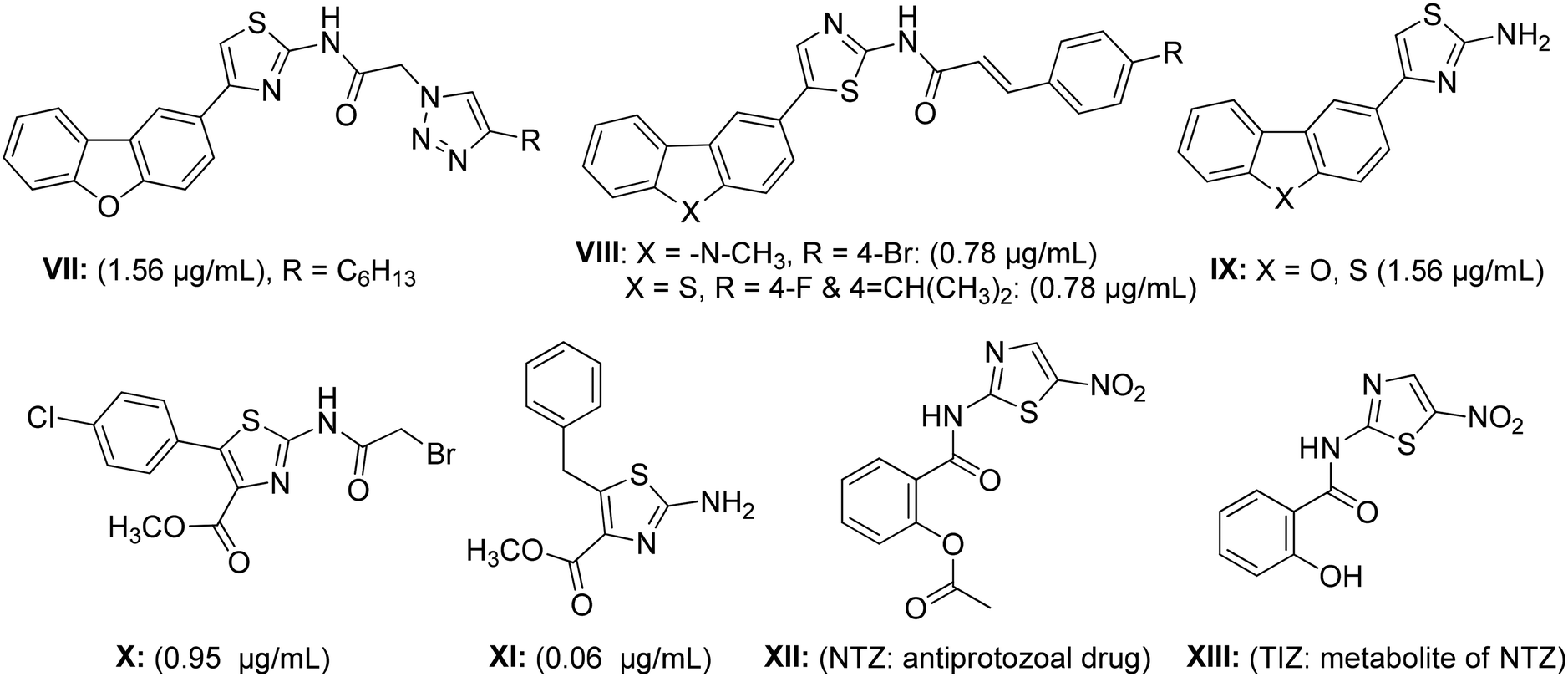 | ||
| Fig. 2 Synthetic bioactive analogs of aminothiazole. | ||
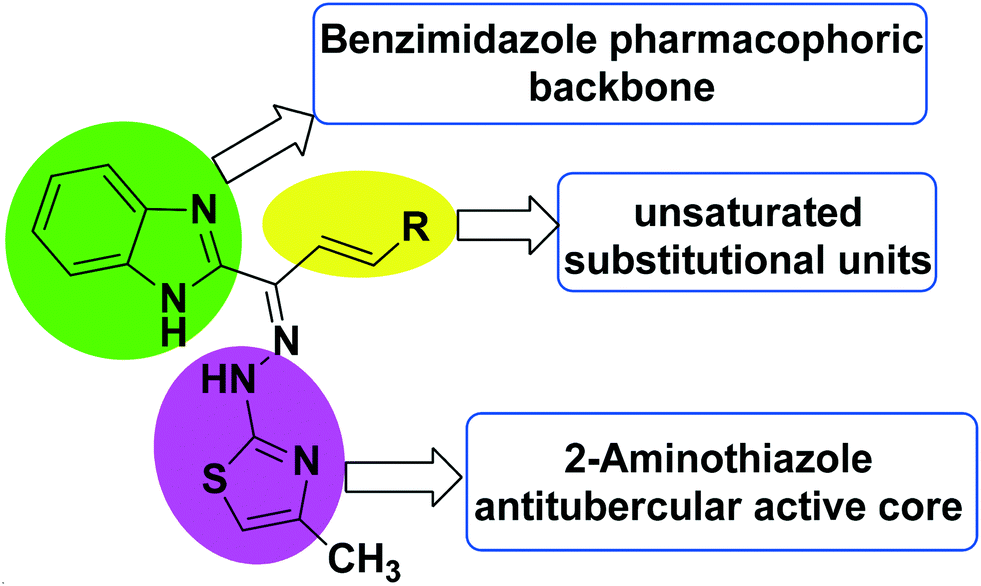 | ||
| Fig. 3 The design strategy for the synthesis of benzimidazolallylidenehydrazinylmethylthiazole derivatives. | ||
2. Results and discussion
2.1. Chemistry
Initially, as depicted in Scheme 1, we started with the synthesis of 1-(1H-benzo[d]imidazol-2-yl)ethanone (4). For this purpose, we conducted the cyclisation of benzene-1,2-diamine (1) in the presence of lactic acid (2) and 4N hydrochloric acid to form 1-(1H-benzo[d]imidazol-2-yl)ethanol (3), and then oxidized compound 3 with chromium trioxide in the presence of acetic acid to form the required 1-(1H-benzo[d]imidazol-2-yl)ethanone (4) in 62% yield. All compounds were fully characterized via NMR and ESI-mass spectral analysis.44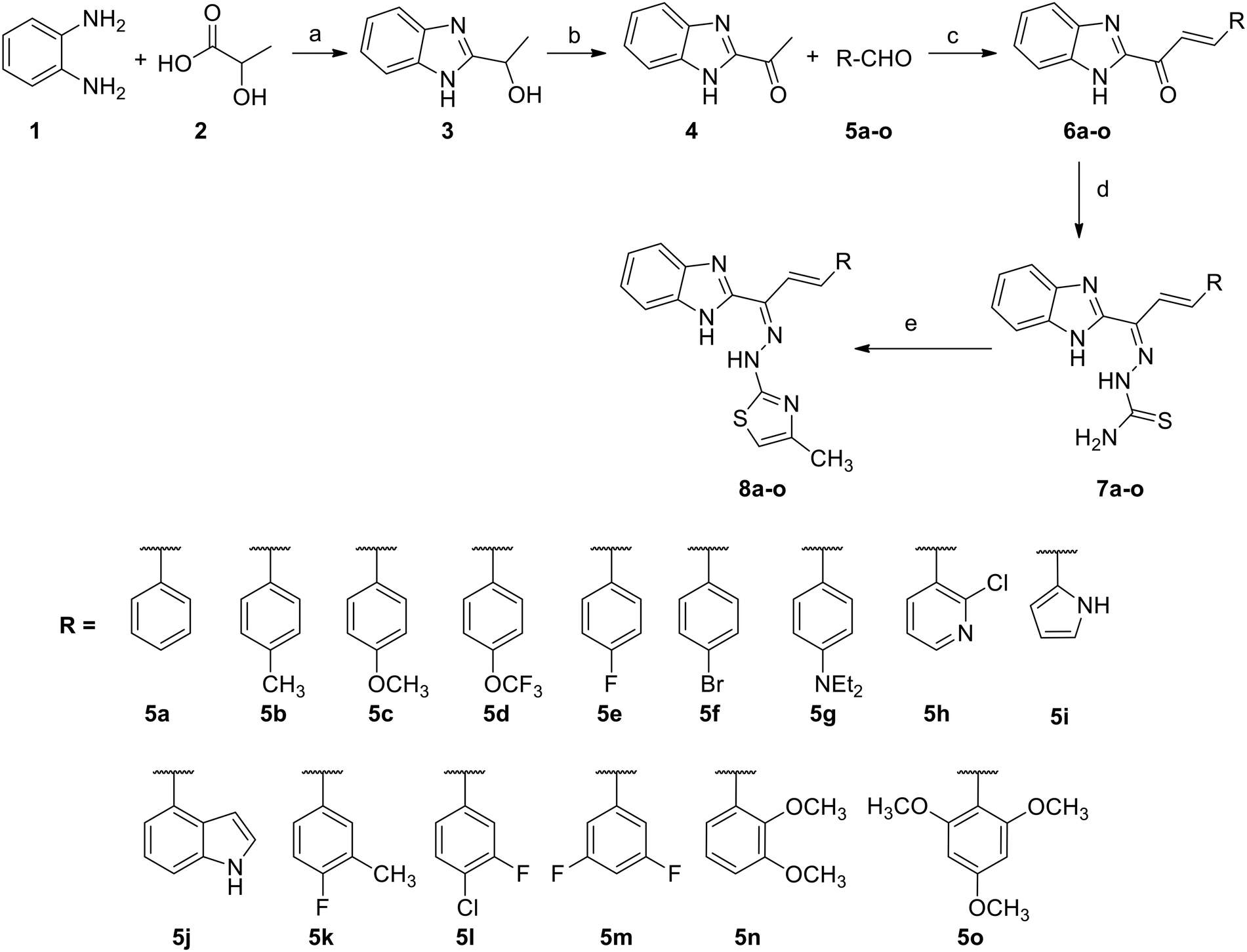 | ||
| Scheme 1 The synthesis of novel benzimidazolylallylidenehydrazinecarbothioamide (7a–o) and benzimidazolallylidenehydrazinylmethylthiazole (8a–o) derivatives. Reagents and conditions: (a) 4N HCl, reflux, 10 h; (b) CrO3, AcOH, 90–105 °C, 30 min; (c) NaOH, EtOH, 0 °C – RT, 8 h; (d) NH2SNHNH2, conc. HCl, EtOH, reflux, 5 h; and (e) ClCH2COCH3, MeOH, reflux, 3 h. | ||
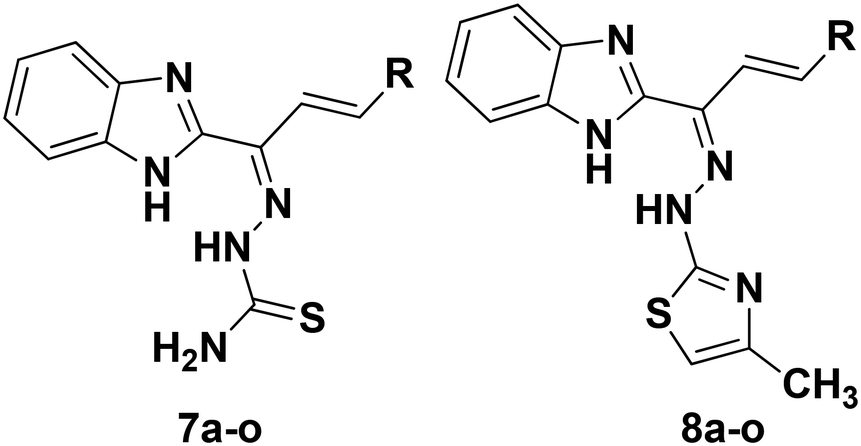
Compound 4 was treated with the various substituted aromatic aldehydes 5a–o and sodium hydroxide under aldol condensation conditions to form the substituted (E)-1-(1H-benzo[d]imidazol-2-yl)-3-phenylprop-2-en-1-one derivatives 6a–o. Compounds 6a–o were then condensed with thiosemicarbazide in the presence of concentrated hydrochloric acid and ethanol under reflux to form the benzimidazolylallylidenehydrazinecarbothioamide derivatives 7a–o in good yields. Finally, the substituted benzimidazolylallylidenehydrazinecarbothioamides 7a–o were treated with chloroacetone in methanol under reflux to form the new desired benzimidazolallylidenehydrazinylmethylthiazole derivatives 8a–o in good yields (Table 1). All synthesized compounds were fully characterized using 1H and 13C NMR, IR and mass (ESI-MS & HR-MS) spectral data.44,45 The logP and clogP values required to assess the lipophilic character of the new analogs were calculated using the Chembiodraw 12.0 program (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Producta | Yieldb (%) | Logpc |
clogPc |
MIC90 (μg mL−1) |
| a Benzimidazolyl substituted arylpropenone (1.0 mmol), thiosemicarbazide (1.1 mmol) and benzimidazolylallylidenehydrazinecarbothioamide (1.0 mmol), and chloroacetone (1.1 mmol). b Isolated yield. c Calculated using Chembiodraw 12.0. | ||||||
| 1. | 5a | 7a | 65 | 2.93 | 4.93 | 5 |
| 8a | 60 | 5.07 | 4.99 | 4 | ||
| 2. | 5b | 7b | 62 | 3.42 | 5.43 | 5 |
| 8b | 60 | 5.56 | 5.49 | 10 | ||
| 3. | 5c | 7c | 55 | 2.80 | 4.85 | 10 |
| 8c | 53 | 4.95 | 4.91 | 10 | ||
| 4. | 5d | 7d | 56 | 4.46 | 5.96 | 5 |
| 8d | 54 | 6.60 | 6.02 | 10 | ||
| 5. | 5e | 7e | 61 | 3.09 | 5.08 | 2.5 |
| 8e | 58 | 5.23 | 5.14 | 5 | ||
| 6. | 5f | 7f | 54 | 3.76 | 5.80 | 4 |
| 8f | 55 | 5.90 | 5.86 | 10 | ||
| 7. | 5g | 7g | 50 | 3.89 | 6.16 | 10 |
| 8g | 52 | 6.03 | 6.22 | 2.5 | ||
| 8. | 5h | 7h | 58 | 2.49 | 4.23 | 10 |
| 8h | 55 | 4.64 | 4.29 | 10 | ||
| 9. | 5i | 7i | 60 | 1.47 | 3.54 | 100 |
| 8i | 52 | 3.62 | 3.60 | 100 | ||
| 10. | 5j | 7j | 53 | 2.47 | 4.92 | 5 |
| 8j | 61 | 4.61 | 4.98 | 10 | ||
| 11. | 5k | 7k | 63 | 3.58 | 5.58 | 4 |
| 8k | 65 | 5.72 | 5.64 | 10 | ||
| 12. | 5l | 7l | 52 | 3.65 | 5.79 | 5 |
| 8l | 60 | 5.79 | 5.85 | 10 | ||
| 13. | 5m | 7m | 55 | 3.25 | 5.22 | 5 |
| 8m | 58 | 5.39 | 5.28 | 10 | ||
| 14. | 5n | 7n | 62 | 2.68 | 4.59 | 5 |
| 8n | 61 | 4.82 | 4.65 | 10 | ||
| 15. | 5o | 7o | 58 | 2.55 | 4.93 | >50 |
| 8o | 55 | 4.69 | 4.99 | >50 | ||
| 16. | Rifampicin | — | — | — | — | 0.25 |
2.2. Pharmacology
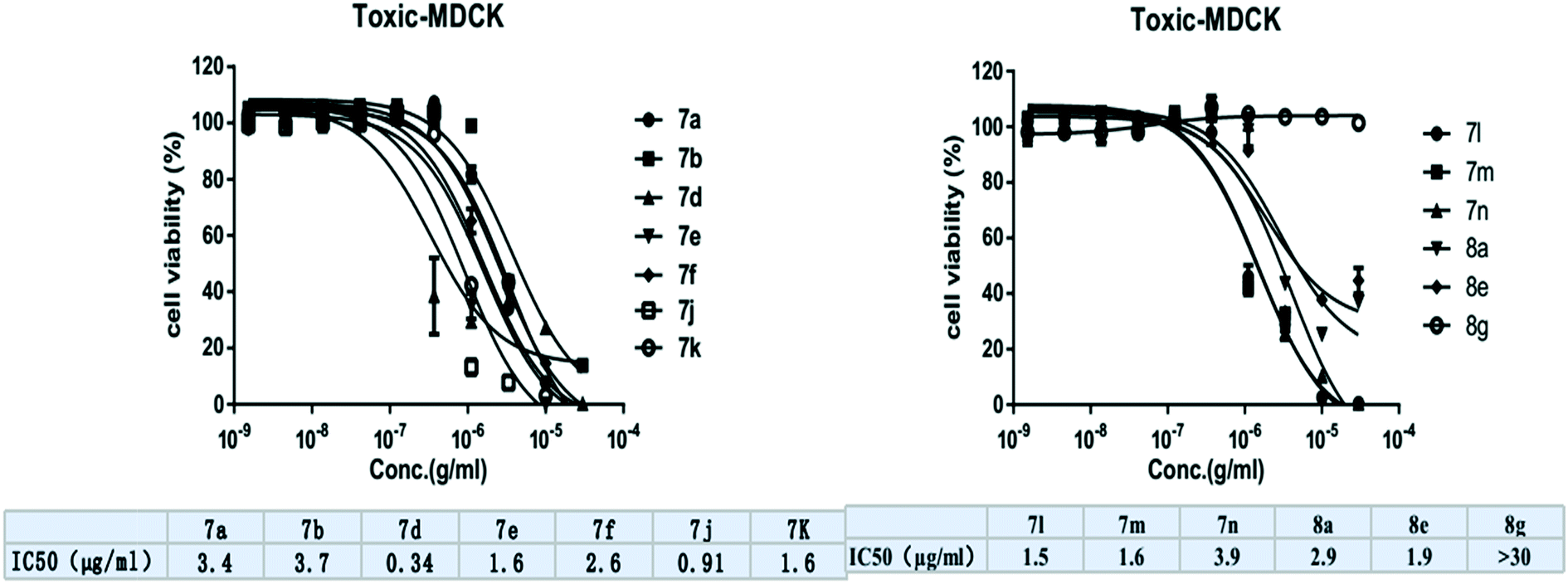 | ||
| Fig. 4 IC50 values of the novel benzimidazolylallylidenehydrazinecarbothioamides 7a–o and benzimidazolallylidenehydrazinylmethylthiazoles 8a–o against MDCK cells. | ||
2.3. Structure–activity relationship (SAR)
To begin the SAR studies, we first investigated the antitubercular potentials of the benzimidazolylallylidenehydrazinecarbothioamide derivatives (7a–o) through modifications to the olefinic system. We found that p-bromo phenyl (MIC: 4 μg mL−1; 7f) and p-fluoro phenyl (MIC: 2.5 μg mL−1; 7e) substitutions were a little more potent compared to simple phenyl (7a; MIC: 5 μg mL−1) and p-tollyl (7b; MIC: 5 μg mL−1) substitutions. We also probed the Mtb inhibition activities by introducing di-substituted phenyl systems into the benzimidazolylallylidenehydrazinecarbothioamide. In this case, di-halo substituted phenyl rings with 4-chloro-3-fluoro (7l) and 3,5-di-fluoro phenyl (7m) substitutions (both having MIC values of 5 μg mL−1) were slightly less potent than the phenyl ring with 4-fluoro-3-methyl substitution (7k; MIC: 4 μg mL−1). This indicated that mono- or di-halo substituted phenyl derivatives exhibit more potent Mtb inhibition with 4-fluoro phenyl substitutions.Next, we modified the phenyl ring with electron donating groups, for example with methoxy substitution at different positions. In this case, 2,3-dimethoxy phenyl (7n; MIC: 5 μg mL−1) demonstrated more potent activity than p-methoxy phenyl (7c; MIC: 10 μg mL−1) substitution; whereas, 2,4,6-trimethoxy phenyl substitution was found to be worse in activity (7o; MIC: > 50 μg mL−1). From the perspective of electron withdrawing substitutions on the phenyl ring, we found that p-trifluoromethoxy substitution (7d; MIC: 5 μg mL−1) resulted in better activity compared to p-methoxy substitution on the phenyl ring (7c; MIC: 10 μg mL−1). We further altered the p-trifluoromethoxy substitution with an electron donating group, for example with p-N,N-diethyl substitution on the phenyl ring (7g). However, the potency was slightly decreased (MIC: 10 μg mL−1). Based on these findings, we concluded that strong electronegative groups at the p-position of the phenyl ring exhibit a good activity profile (i.e. potency) compared to p-electropositive groups.
We also investigated the activity profiles of the newly synthesized compounds by altering the phenyl substitutions with heterocyclic groups on the olefinic chain. We observed that the potency of indole substitution (7j; MIC: 5 μg mL−1) was better in comparison to 2-chloropyridine (7h; MIC: 10 μg mL−1) substitution and was far better than pyrrole (7i; MIC: 100 μg mL−1) substitution.
Finally, we studied the activity profiles of aminothiazole derivatives (8a–o) derived from benzimidazolylallylidenehydrazinecarbothioamides (7a–o), following the same SAR pattern as for 7a–o. Here, the Mtb activity decreased from all kinds of substitutions, except the electron donating group (p-N,N-diethyl phenyl) on 7g, which resulted in a 4-fold increase in potency when it was converted to its corresponding aminothiazole 8g (MIC: 2.5 μg mL−1). Moreover, no change in activity was observed when 7c, 7i, and 7o were converted to their corresponding 8c (MIC: 10 μg mL−1), 8i (MIC: 100 μg mL−1), and 8o (MIC > 50 μg mL−1) aminothiazole derivatives.
3. Conclusions
We have designed and synthesized a series of novel benzimidazolylallylidenehydrazinecarbothioamide derivatives 7a–o and benzimidazolallylidenehydrazinylmethylthiazole derivatives 8a–ovia multi-step condensation reactions. All new derivatives were fully analyzed via NMR and mass spectral analysis. We also explored the SAR of the synthesized compounds in relation to antitubercular activity against the H37Ra Mtb strain. From in vitro antimycobacterial screening assays, we have identified compounds 7e and 8g (MIC: 2.5 μg mL−1) as the most active antitubercular agents among the synthesized series. Moreover, compound 8g exhibited a lower cytotoxicity profile. We conceive that compound 8g may be regarded as a drug-like hit compound and may serve as a chemical probe of interest for further SAR and lead optimization studies aimed at developing novel and effective antitubercular agents.4. Experimental section
4.1. Chemistry
4.1.4.1. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-phenylprop-2-en-1-one (6a). Yield: 65%; 1H NMR (400 MHz, CDCl3): δ 10.83 (br s, 1H), 8.18–8.07 (m, 2H), 7.96 (d, J = 14.2 Hz, 1H), 7.76 (t, J = 5.6 Hz, 2H), 7.59 (d, J = 14.1 Hz, 1H), 7.46–7.36 (m, 5H); MS (ESI): m/z 271 (M + Na)+.
4.1.4.2. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(p-tolyl)prop-2-en-1-one (6b). Yield: 68%; 1H NMR (500 MHz, CDCl3): δ 10.38 (br s, 1H), 8.08 (s, 3H), 7.96 (d, J = 13.9 Hz, 1H), 7.66 (d, J = 8.1 Hz, 2H), 7.56 (d, J = 14.3 Hz, 1H), 7.44 (t, J = 8.1 Hz, 1H), 7.38 (t, J = 8.5 Hz, 1H), 7.24 (s, 1H), 2.41 (s, 3H); MS (ESI): m/z 285 (M + Na)+.
4.1.4.3. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-methoxyphenyl)prop-2-en-1-one (6c). Yield: 67%; 1H NMR (400 MHz, CDCl3): δ 10.52 (br s, 1H), 8.04 (d, J = 8.1 Hz, 2H), 7.95 (d, J = 13.8 Hz, 1H), 7.73 (d, J = 8.5 Hz, 2H), 7.58 (d, J = 14.0 Hz, 1H), 7.46–7.35 (m, 2H), 6.96 (d, J = 8.5 Hz, 2H), 3.87 (s, 3H); MS (ESI): m/z 301 (M + Na)+.
4.1.4.4. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-(trifluoromethoxy)phenyl)prop-2-en-1-one (6d). Yield: 60%; 1H NMR (400 MHz, CDCl3): δ 10.57 (br s, 1H), 8.08 (q, J = 8.1 Hz, 7.8 Hz, 2H), 7.96 (d, J = 13.5 Hz, 1H), 7.78 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 13.8 Hz, 1H), 7.45 (t, J = 7.0 Hz, 1H), 7.39 (t, J = 7.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H); MS (ESI): m/z 333 (M + H).
4.1.4.5. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)prop-2-en-1-one (6e). Yield: 58%; 1H NMR (400 MHz, CDCl3): δ 10.55 (br s, 1H), 8.05 (s, 2H), 7.95 (d, J = 13.9 Hz, 1H), 7.76 (q, J = 5.5 Hz, 2H), 7.58 (d, J = 13.9 Hz, 1H), 7.48–7.36 (m, 2H), 7.13 (t, J = 8.5 Hz, 1H); MS (ESI): m/z 289 (M + Na)+.
4.1.4.6. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-bromophenyl)prop-2-en-1-one (6f). Yield: 62%; 1H NMR (400 MHz, DMSO-d6): δ 13.19 (s, 1H), 8.14 (d, J = 14.1 Hz, 1H), 7.93 (d, J = 15.2 Hz, 1H), 7.71 (s, 1H), 7.68–7.56 (m, 5H), 7.35 (s, 2H); MS (ESI): m/z 327 (M + H)+.
4.1.4.7. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-(diethylamino)phenyl)prop-2-en-1-one (6g). Yield: 65%; 1H NMR (400 MHz, DMSO-d6): δ 13.21 (br s, 1H), 7.96–7.76 (m, 3H), 7.63 (d, J = 14.2 Hz, 2H), 7.57 (d, J = 14.8 Hz, 1H), 7.41–7.23 (m, 2H), 6.72 (d, J = 8.5 Hz, 2H), 3.46 (q, J = 7.07 Hz, 6.80 Hz, 4H), 1.19 (t, J = 6.8 Hz, 6H); MS (ESI): m/z 320 (M + H)+.
4.1.4.8. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2-chloropyridin-3-yl)prop-2-en-1-one (6h). Yield: 55%; 1H NMR (400 MHz, DMSO-d6): δ 13.64 (br s, 1H), 8.63 (d, J = 8.5 Hz, 1H), 8.55 (d, J = 14.2 Hz, 1H), 8.22 (q, J = 6.5 Hz, 7.1 Hz, 2H), 7.83 (s, 2H), 7.60 (q, J = 15.5 Hz, 1H), 7.43 (s, 2H); MS (ESI): m/z 282 (M − H)+.
4.1.4.9. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(1H-pyrrol-2-yl)prop-2-en-1-one (6i). Yield: 58%; 1H NMR (400 MHz, DMSO-d6): δ 13.09 (br s, 1H), 11.82 (s, 1H), 7.83 (s, 2H), 7.77 (d, J = 13.8 Hz, 1H), 7.58 (d, J = 14.2 Hz, 1H), 7.39–7.25 (m, 2H), 7.05 (s, 1H), 6.70 (s, 1H), 6.25 (s, 1H); MS (ESI): m/z 260 (M + Na).
4.1.4.10. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(1H-indol-4-yl)prop-2-en-1-one (6j). Yield: 60%; 1H NMR (400 MHz, DMSO-d6): δ 13.47 (br s, 1H), 11.55 (br s, 1H), 8.29 (s, 2H), 7.91 (d, J = 15.2 Hz, 1H), 7.65–7.56 (m, 4H), 7.44–7.33 (m, 2H), 7.23 (d, J = 14.8 Hz, 1H), 6.91 (s, 1H); MS (ESI): m/z 288 (M + H).
4.1.4.11. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-fluoro-3-methyl phenyl)prop-2-en-1-one (6k). Yield: 62%; 1H NMR (500 MHz, DMSO-d6): δ 8.12 (d, J = 15.4 Hz, 1H), 7.92 (t, J = 8.5 Hz, 2H), 7.75 (s, 3H), 7.38 (s, 2H), 7.28 (d, J = 14.5 Hz, 1H), 2.34 (s, 2H); MS (ESI): m/z 281 (M + H)+.
4.1.4.12. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-chloro-3-fluorophenyl)prop-2-en-1-one (6l). Yield: 59%; 1H NMR (400 MHz, DMSO-d6): δ 13.55 (s, 1H), 8.19 (d, J = 16.1 Hz, 1H), 8.05–7.95 (m, 1H), 7.90 (d, J = 15.6 Hz, 1 Hz), 7.80 (t, J = 4.0 Hz, 1H), 7.76 (d, J = 15.2 Hz, 1H), 7.71 (d, J = 8.2 Hz, 1H), 7.61 (d, J = 4.1 Hz, 1H), 7.44 (d, J = 8.1 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H); MS (ESI): m/z 301 (M + H)+.
4.1.4.13. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(3,5-difluorophenyl)prop-2-en-1-one (6m). Yield: 57%; 1H NMR (400 MHz, DMSO-d6): δ 12.79 (br s, 1H), 9.01 (d, J = 15.9 Hz, 1H), 8.65 (d, J = 7.8 Hz, 1H), 7.83 (d, J = 15.9 Hz, 1H), 7.76 (d, J = 15.6 Hz, 1H), 7.55 (d, J = 7.5 Hz, 1H), 7.49 (t, J = 8.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.27 (m, 2H); MS (ESI): m/z 285 (M + H)+.
4.1.4.14. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2,3-dimethoxyphenyl)prop-2-en-1-one (6n). Yield: 62%; 1H NMR (400 MHz, DMSO-d6): δ 13.48 (s, 1H), 8.15 (q, J = 8.4 Hz, 8.1 Hz, 2H), 7.87 (s, 1H), 7.60 (s, 1H), 7.50 (d, J = 14.5 Hz, 1H), 7.38 (t, J = 14.8 Hz, 2H), 7.24–7.18 (m, 2H), 3.85 (s, 6H); MS (ESI): m/z 309 (M + H)+.
4.1.4.15. (E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2,4,6-trimethoxyphenyl)prop-2-en-1-one (6o). Yield: 65%; 1H NMR (400 MHz, DMSO-d6): δ 13.22 (br s, 1H), 8.27 (s, 2H), 7.84 (d, J = 14.2 Hz, 1H), 7.56 (d, J = 14.8 Hz, 1H), 7.42–7.25 (m, 2H), 6.35 (s, 2H), 3.95 (s, 6H), 3.88 (s, 3H); MS (ESI): m/z 339 (M + H)+.
4.1.5.1. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-phenylallylidene)hydrazinecarbothioamide (7a). Yield: 65%; m.p: 143–144 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.76 (br s, 1H), 11.53 (br s, 1H), 8.67 (d, J = 8.2 Hz, 2H), 8.49 (d, J = 15.1 Hz, 1H), 7.87–7.79 (m, 3H), 7.75 (t, J = 7.8 Hz, 1H), 7.53 (d, J = 15.5 Hz, 1H), 7.49 (m, 3H), 7.30 (t, J = 7.5 Hz, 1H), 7.21 (t, J = 7.5 Hz, 1H); 13C NMR (125 MHz, DMSO-d6): δ 180.0, 150.2, 142.0, 137.4, 130.1129.5, 128.9, 128.1, 124.4, 122.5, 116.3, 112.0; IR (KBr): 3250, 2925, 1572, 1452, 1352, 1150, 952, 757 cm−1; MS (ESI): m/z 322 (M + H)+.
4.1.5.2. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(p-tolyl)allylidene)hydrazinecarbothioamide (7b). Yield: 62%; m.p: 132–134 °C; 1H NMR (500 MHz, CDCl3 + DMSO-d6): δ 12.68 (s, 1H), 11.44 (s, 1H), 8.61 (br s, 2H), 8.49 (d, J = 15.5 Hz, 1H), 7.76–7.67 (m, 2H), 7.66–7.63 (m, 1H), 7.50 (d, J = 7.1 Hz, 1H), 7.35 (d, J = 14.8 Hz, 1H), 7.34–7.21 (m, 3H), 6.88 (d, J = 7.5 Hz, 1H), 2.37 (s, 3H); 13C NMR (125 MHz, CDCl3 + DMSO-d6): δ 179.0, 149.2, 141.1, 139.1, 133.7, 129.3, 129.2, 128.3, 128.0, 127.27, 127.24, 114.3, 21.0; IR (KBr): 3252, 2923, 1575, 1435, 1345, 1085, 735 cm−1; MS (ESI): m/z 336 (M + H)+.
4.1.5.3. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-methoxyphenyl)allylidene)hydrazinecarbothioamide (7c). Yield: 55%; m.p: 116–118 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.66 (s, 1H), 11.39 (s, 1H), 8.59 (br s, 2H), 8.48 (d, J = 8.5 Hz, 2H), 7.79 (d, J = 14.2 Hz, 1H), 7.69–7.61 (m, 2H), 7.30–7.20 (m, 3H), 6.98 (d, J = 14.6 Hz, 1H), 6.93 (d, J = 8.5 Hz, 1H), 6.62 (d, J = 8.5 Hz, 1H), 3.83 (s, 3H); IR (KBr): 3252, 2856, 1575, 1345, 1183, 955, 735 cm−1; MS (ESI): m/z 352 (M + H)+.
4.1.5.4. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-trifluoromethoxyphenyl)allylidene)hydrazinecarbothioamide (7d). Yield: 56%; m.p: 128–130 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.77 (br s, 1H), 11.56 (br s, 1H), 8.69 (d, J = 21.7 Hz, 2H), 8.50 (d, J = 14.9 Hz, 1H), 7.96 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 15.2 Hz, 1H), 7.44 (d, J = 8.0 Hz, 2H), 7.34–7.17 (m, 2H); 13C NMR (125 MHz, DMSO-d6): δ 180.1, 150.1, 149.6, 140.1, 136.8, 136.7, 136.3, 130.7, 129.8, 122.2, 121.9, 117.5; IR (KBr): 3250, 2920, 1565, 1475, 1355, 1152, 955 cm−1; MS (ESI): m/z 406 (M + H)+.
4.1.5.5. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)allylidene)hydrazinecarbothioamide (7e). 1H NMR (500 MHz, CDCl3 + DMSO-d6): Yield: 61%; HPLC purity: 95%; m.p: 135–137 °C; δ 11.78 (s, 1H), 8.87 (d, J = 15.1 Hz, 2H), 7.88 (q, J = 8.5 Hz, 7.8 Hz, 2H), 7.82 (q, J = 7.2 Hz, 6.9 Hz, 2H), 7.70 (d, J = 15.9 Hz, 1H), 7.58–7.50 (m, 3H), 7.26 (t, J = 8.5 Hz, 2H); 13C NMR (75 MHz, CDCl3 + DMSO-d6): δ 179.3, 146.6, 143.8, 139.0, 138.4, 130.2, 130.1, 129.47, 129.40, 127.5, 125.5, 115.7, 115.5, 115.3, 115.1, 114.6, 114.0; IR (KBr): 3320, 2865, 1575, 1455, 1350, 1175, 952, 735 cm−1; MS (ESI): m/z 340 (M + H)+.
4.1.5.6. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-bromophenyl)allylidene)hydrazinecarbothioamide (7f). 1H NMR (400 MHz, DMSO-d6): yield: 54%; m.p: 155–157 °C; δ 12.77 (s, 1H), 11.57 (s, 1H), 8.68 (d, J = 8.3 Hz, 2H), 8.47 (d, J = 15.9 Hz, 2H), 7.84–7.71 (m, 3H), 7.65 (d, J = 15.3 Hz, 1H), 7.53 (d, J = 14.9 Hz, 1H), 7.36–7.16 (m, 3H); 13C NMR (125 MHz, DMSO-d6): δ 179.0, 149.1, 139.5, 135.7, 135.3, 131.5, 129.8, 122.3, 116.1; IR (KBr): 3265, 2920, 1565, 1435, 1352, 935, 752 cm−1; MS (ESI): m/z 402 (M + 2)+.
4.1.5.7. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-(diethylamino)phenyl)allylidene)hydrazinecarbothioamide (7g). Yield: 50%; m.p: 152–154 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.90 (br s, 1H), 8.25 (br s, 1H), 7.69 (d, J = 15.6 Hz, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.31 (t, J = 7.8 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 6.96 (d, J = 8.3 Hz, 2H), 6.57 (d, J = 14.2 Hz, 2H), 5.82 (dd, J = 8.3 Hz & 7.5 Hz, 1H), 3.28 (q, J = 7.0 Hz, 4H), 1.05 (t, J = 6.8 Hz, 6H); 13C NMR (125 MHz, DMSO-d6): δ 176.5, 147.2, 146.4, 144.8, 134.3, 128.3, 126.5, 124.1, 122.0, 119.4, 111.5, 111.1, 43.4, 12.2; IR (KBr): 3256, 1626, 1435, 1352, 1225, 1055, 785 cm−1; MS (ESI): m/z 393 (M + H)+.
4.1.5.8. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2-chloropyridin-3-yl)allylidene)hydrazinecarbothioamide (7h). Yield: 58%; m.p: 82–84 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.84 (s, 1H), 11.66 (s, 1H), 8.94 (d, J = 15.6 Hz, 1H), 8.77 (d, J = 17.8 Hz, 2H), 8.68 (d, J = 7.8 Hz, 1H), 8.46 (s, 1H), 7.88 (d, J = 15.6 Hz, 1H), 7.75 (d, J = 8.0 Hz, 1H), 759 (q, J = 7.04 Hz, 2H), 7.34 (t, J = 7.8 Hz, 1H), 7.25 (t, J = 7.8 Hz, 1H); IR (KBr): 3255, 1565, 1455, 1235, 1055, 852, 785 cm−1; MS (ESI): m/z 357 (M + H)+.
4.1.5.9. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(1H-pyrrol-2-yl)allylidene)hydrazinecarbothioamide (7i). Yield: 60%; m.p: 110–112 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.86 (br s, 1H), 11.78 (br s, 1H), 7.72 (d, J = 14.5 Hz, 1H), 7.58 (d, J = 14.2 Hz, 2H), 7.33 (t, J = 8.0 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H), 6.77–6.57 (m, 2H), 6.02 (dd, J = 8.0 Hz, 7.5 Hz, 1H), 5.94–5.86 (m, 3H); 13C NMR (125 MHz, DMSO-d6): δ 176.5, 147.9, 144.7, 134.3, 130.7, 124.1, 122.0, 119.4, 117.6, 111.5, 106.8, 105.3; IR (KBr): 3275, 2865, 1652, 1535, 1425, 1250, 1055, 783 cm−1; MS (ESI): m/z 309 (M − H)+.
4.1.5.10. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(1H-indol-4-yl)allylidene)hydrazinecarbothioamide (7j). Yield: 53%; m.p: 147–149 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.78 (br s, 1H), 11.50 (br s, 1H), 11.30 (br s, 1H), 9.02 (d, J = 8.1 Hz, 1H), 8.64 (d, J = 8.2 Hz, 2H), 7.83 (d, J = 15.9 Hz, 1H), 7.77 (d, J = 15.6 Hz, 1H), 7.57–7.44 (m, 2H), 7.35–7.15 (m, 3H), 6.74 (s, 1H); 13C NMR (125 MHz, DMSO-d6): δ 180.0, 150.5, 140.7, 137.8, 137.2, 128.7, 128.4, 127.2, 124.4, 122.4, 121.9, 120.5, 118.4, 115.5, 113.5, 100.4; IR (KBr): 3257, 1621, 1456, 1413, 1378, 1256, 1145, 817, 729, 661 cm−1; MS (ESI): m/z 361 (M + H)+.
4.1.5.11. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-fluoro-3-methylphenyl)allylidene)hydrazinecarbothioamide (7k). Yield: 63%; m.p: 140–142 °C; 1H NMR (500 MHz, DMSO-d6): δ 12.79 (br s, 1H), 11.48 (s, 1H), 8.68 (d, J = 8.0 Hz, 1H), 8.45 (d, J = 15.6 Hz, 1H), 7.73 (t, J = 7.8 Hz, 3H), 7.53 (d, J = 15.2 Hz, 1H), 7.33 (m, 3H), 2.30 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 180.0, 163.1, 161.1, 150.1, 140.9, 133.6, 132.2, 128.27, 128.21, 125.5, 125.3, 116.0, 115.9, 14.9; IR (KBr): 3252, 2853, 1565, 1455, 1055, 745 cm−1; MS (ESI): m/z 354 (M + H)+.
4.1.5.12. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-chloro-3-fluorophenyl)allylidene)hydrazinecarbothioamide (7l). Yield: 52%; m.p: 122–124 °C;1H NMR (400 MHz, DMSO-d6): δ 12.78 (br s, 1H), 11.54 (s, 1H), 8.02 (d, J = 8.3 Hz, 1H), 7.83 (t, J = 8.0 Hz, 1H), 7.78 (d, J = 14.5 Hz, 1H), 7.73 (d, J = 7.5 Hz, 1H), 7.69 (d, J = 15.2 Hz, 1H), 7.63 (t, J = 8.8 Hz, 2H), 7.29 (t, J = 7.3 Hz, 1H), 7.22 (t, J = 7.4 Hz, 1H); IR (KBr): 3270, 2851, 1592, 1488, 1456, 1370, 1357, 1172, 964, 766 cm−1; MS (ESI): m/z 374 (M + H)+.
4.1.5.13. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(3,5-difluorophenyl)allylidene)hydrazinecarbothioamide (7m). Yield: 55%; m.p: 142–144 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.81 (br s, 1H), 11.58 (br s, 1H), 8.80 (s, 1H), 7.72 (s, 1H), 8.46 (d, J = 8.2 Hz, 1H), 7.86 (s, 1H), 7.75 (d, J = 14.3 Hz, 1H), 7.63 (t, J = 8.3 Hz, 2H), 7.55 (d, J = 7.8 Hz, 1H), 7.43 (d, J = 14.8 Hz, 1H), 7.34–7.23 (m, 2H); IR (KBr): 3255, 1552, 1452, 1232, 1153, 785 cm−1; MS (ESI): m/z 358 (M + H)+.
4.1.5.14. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2,3-dimethoxyphenyl)allylidene)hydrazinecarbothioamide (7n). Yield: 62%; m.p: 132–134 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.76 (s, 1H), 11.50 (s, 1H), 8.82 (d, J = 14.2 Hz, 1H), 8.66 (d, J = 8.3 Hz, 2H), 7.78–7.63 (m, 3H), 7.53 (d, J = 14.5 Hz, 1H), 7.30–7.20 (m, 1H), 7.18–7.03 (m, 1H), 3.83 (s, 6H); 13C NMR (125 MHz, DMSO-d6): δ 179.0, 152.6, 149.2, 147.5, 143.3, 136.2, 135.4, 133.5, 130.3, 123.8, 123.4, 121.4, 119.4, 118.2, 115.7, 113.5, 110.9, 60.9, 55.6; IR (KBr): 3312, 2825, 1452, 1365, 1175, 958, 765 cm−1; MS (ESI): m/z 382 (M + H)+.
4.1.5.15. (Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2,4,6-trimethoxyphenyl)allylidene)hydrazinecarbothioamide (7o). Yield: 58%; m.p: 136–138 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.89 (br s, 1H), 7.99 (br s, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.43 (s, 1H), 7.34 (t, J = 14.5 Hz, 1H), 7.26 (t, J = 7.5 Hz, 1H), 6.23 (d, J = 14.1 Hz, 1H), 3.80 (s, 6H), 3.54 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 176.7, 161.1, 147.3, 146.4, 144.6, 135.3, 124.8, 122.8, 120.3, 112.3, 111.9, 56.0, 55.2; IR (KBr): 3273, 1685, 1465, 1225, 1065, 798, 745 cm−1; MS (ESI): m/z 412 (M + H)+.
4.1.6.1. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-phenylallylidene)hydrazinyl)-4-methylthiazole (8a). 1H NMR (500 MHz, CDCl3): Yield: 60%; m.p: 97–99 °C; δ 7.54 (br s, 1H), 7.34 (d, J = 14.1 Hz, 1H), 7.29–7.23 (m, 7H), 7.22 (d, J = 14.2 Hz, 2H), 6.29 (s, 1H), 2.34 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 147.1, 137.3, 132.2, 129.5, 129.0, 128.9, 128.3, 128.1, 127.8, 127.3, 124.4, 123.2, 122.8, 17.7; IR (KBr): 3321, 2971, 1620, 1535, 1486, 1252, 732 cm−1; MS (ESI): m/z 360 (M + H)+; HRMS (ESI): calcd for C20H18N5S (M + H)+: 360.1312, found: 360.1311.
4.1.6.2. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(p-tolyl)allylidene) hydrazinyl)-4-methylthiazole (8b). Yield: 60%; m.p: 92–94 °C; 1H NMR (500 MHz, DMSO-d6): δ 8.01 (s, 1H), 7.60 (d, J = 7.5 Hz, 2H), 7.56 (d, J = 14.5 Hz, 1H), 7.48 (t, J = 7.5 Hz 3H), 7.42 (d, J = 7.8 Hz, 2H), 7.35 (d, J = 14.8 Hz, 2H), 7.18 (t, J = 8.5 Hz, 2H), 6.27 (s, 1H), 2.33 (s, 3H), 2.28 (s, 3H); 13C NMR (125 MHz, CDCl3 + DMSO-d6): δ 169.2, 147.4, 138.8, 138.4, 138.0, 133.7, 131.9, 129.37, 129.34, 127.4, 127.1, 125.6, 120.5, 113.9, 20.9, 14.2; IR (KBr): 3317, 2919, 1717, 1562, 1483, 1431, 1376, 1238, 1081, 822 cm−1; MS (ESI): m/z 374 (M + H)+; HRMS (ESI): calcd for C21H20N5S (M + H)+: 374.1482, found: 374.1484.
4.1.6.3. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-methoxyphenyl)allylidene)hydrazinyl)-4-methylthiazole (8c). Yield: 53%; m.p: 97–99 °C; 1H NMR (500 MHz, DMSO-d6): δ 7.85-7.83 (m, 3H), 7.70 (d, J = 8.5 Hz, 2H), 7.60 (t, J = 7.5 Hz, 2H), 7.46 (d, J = 15.2 Hz, 1H), 7.07 (d, J = 14.8 Hz, 2H), 6.56 (s, 1H), 3.85 (s, 3H), 2.26 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 161.2, 140.1, 135.2, 132.5, 132.4, 130.3, 130.1, 129.8, 127.0, 126.8, 115.2, 115.0, 114.9, 114.3, 56.2, 15.2; IR (KBr): 3315, 2932, 1612, 1542, 1483, 1368, 1250, 823 cm−1; MS (ESI): m/z 390 (M + H); HRMS (ESI): calcd for C21H20N5OS (M + H)+: 390.1404, found: 390.1409.
4.1.6.4. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-(trifluoromethoxy)phenyl)allylidene)hydrazinyl)-4-methylthiazole (8d). Yield: 54%; m.p: 119–121 °C; 1H NMR (400 MHz, CDCl3): δ 7.68–7.43 (m, 4H), 7.23 (s, 1H), 7.10 (d, J = 14.6 Hz, 1H), 7.03 (d, J = 14.8 Hz, 3H), 6.88 (t, J = 7.8 Hz, 1H), 6.24 (s, 1H), 2.32 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 167.6, 147.9, 145.7, 135.3, 139.4, 128.3, 127.9, 127.3, 122.6, 120.7, 120.4, 120.2, 118.7, 117.0, 103.6, 16.6; IR (KBr): 3327, 2950, 1617, 1530, 1482, 1365, 1250, 830 cm−1; MS (ESI): m/z 444 (M + H)+.
4.1.6.5. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)allylidene)hydrazinyl)-4-methylthiazole (8e). Yield: 58%; m.p: 104–106 °C; 1H NMR (400 MHz, CDCl3): δ 7.63–7.52 (m, 2H), 7.35–7.25 (m, 4H), 7.18 (d, J = 15.1 Hz, 1H), 7.13–7.01 (m, 1H), 6.96 (t, J = 14.8 Hz, 1H), 6.91–6.78 (m, 1H), 6.28 (s, 1H), 2.35 (s, 3H); IR (KBr): 2952, 1540, 1432, 1285, 1150, 982, 735 cm−1; MS (ESI): m/z 378 (M + H)+; HRMS (ESI): calcd for C20H17FN5S (M + H)+: 378.1216, found: 378.1216.
4.1.6.6. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-bromophenyl)allylidene)hydrazinyl)-4-methylthiazole (8f). Yield: 55%; m.p: 130–132 °C; 1H NMR (400 MHz, DMSO-d6): δ 7.63 (s, 1H), 7.57–7.40 (m, 6H), 7.34–7.20 (m, 4H), 6.33 (s, 1H), 2.25 (s, 3H); 13C NMR (125 MHz, CDCl3 + DMSO-d6): 166.1, 151.9, 145.9, 135.5, 131.35, 131.32, 131.2, 129.7, 128.7, 128.4, 121.0, 112.1, 21.8; IR (KBr): 2968, 1543, 1524, 1485, 1292, 1133, 972, 721 cm−1; MS (ESI): m/z 440 (M + 2)+; HRMS (ESI): calcd for C20H18BrN5S (M + 2)+: 440.0407, found: 440.0398.
4.1.6.7. 4-((1E,3Z)-3-(1H-Benzo[d]imidazol-2-yl)-3-(2-(4-methylthiazol-2-yl)hydrazono)prop-1-en-1-yl)-N,N-diethylaniline (8g). Yield: 52%; HPLC purity: 99.5%; m.p: 128–130 °C; 1H NMR (400 MHz, CDCl3): δ 10.70 (br s, 1H), 7.75 (s, 1H), 7.44 (s, 1H), 7.27 (s, 3H), 7.13 (d, J = 14.8 Hz, 2H), 6.55 (d, J = 8.0 Hz, 2H), 6.15 (s, 1H), 6.48 (q, J = 15.0 Hz, 1H), 3.29 (q, J = 7.0 Hz, 4H), 1.11 (t, J = 7.2 Hz, 6H); 13C NMR (125 MHz, CDCl3 + DMSO-d6): 163.4, 148.5, 146.6, 144.6, 143.9, 127.0, 126.5, 122.4, 111.7, 110.8, 103.2, 43.5, 16.9, 11.9; IR (KBr): 2967, 1613, 1518, 1265, 1191, 1096, 1077, 709 cm−1; MS (ESI): m/z 431 (M + H)+; HRMS (ESI): Calcd for C24H27N6S (M + H)+: 431.2057, found: 431.2048.
4.1.6.8. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2-chloropyridin-3-yl)allylidene)hydrazinyl)-4-methylthiazole (8h). Yield: 55%; m.p: 113–115 °C; 1H NMR (400 MHz, DMSO-d6): δ 8.24 (d, J = 14.3 Hz, 1H), 8.13 (s, 1H), 7.75–7.67 (m, 2H), 7.59 (d, J = 14.8 Hz, 1H), 7.55–7.47 (m, 3H), 7.22 (m, 1H), 6.25 (s, 1H), 2.31 (s, 3H); IR (KBr): 3252, 2920, 1720, 1550, 1435, 1357, 1227, 728 cm−1; MS (ESI): m/z 395 (M + H)+.
4.1.6.9. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(1H-pyrrol-2-yl)allylidene)hydrazinyl)-4-methylthiazole (8i). Yield: 52%; m.p: 135–137 °C;1H NMR (400 MHz, DMSO-d6): δ 12.87 (br s, 1H), 11.81 (br s, 1H), 7.60 (d, J = 14.2 Hz, 2H), 7.24 (s, 3H), 6.73 (s, 1H), 6.40 (s, 1H), 6.10 (d, J = 14.5 Hz, 1H), 5.98 (s, 1H), 5.78–5.64 (m, 1H), 2.30 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 164.6, 148.2, 145.2, 144.6, 129.4, 118.0, 106.9, 105.8, 104.2, 17.0; IR (KBr): 3285, 2920, 1542, 1482, 1350, 1230, 975, 832 cm−1; MS (ESI): m/z 349 (M + H)+; HRMS (ESI): Calcd for C18H17N6S (M + H): 349.1281, found: 349.1281.
4.1.6.10. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(1H-indol-4-yl)allylidene)hydrazinyl)-4-methyl thiazole (8j). Yield: 61%; m.p: 118–120 °C1; H NMR (400 MHz, CDCl3): δ 7.56 (s, 2H), 7.35–7.24 (m, 5H), 7.17 (d, J = 14.1 Hz, 1H), 7.02 (d, J = 14.9 Hz, 1H), 6.96 (t, J = 8.5 Hz, 2H), 6.81 (t, J = 8.5 Hz, 1H), 6.27 (s, 1H), 2.34 (s, 3H); IR (KBr): 3520, 2930, 1735, 1552, 1435, 1232, 975, 732 cm−1; MS (ESI): m/z 399 (M + H)+.
4.1.6.11. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-fluoro-3-methylphenyl)allylidene)hydrazinyl)-4-methylthiazole (8k). Yield: 65%; m.p: 137–139 °C; 1H NMR (500 MHz, CDCl3): δ 7.51 (s, 2H), 7.31–7.21 (m, 3H), 7.12 (d, J = 14.7 Hz, 1H), 7.01 (d, J = 15.3 Hz, 1H), 6.93–6.67 (m, 2H), 6.26 (s, 1H), 2.33 (s, 3H), 2.15 (s, 3H); IR (KBr): 3252, 2975, 1735, 1535, 1235, 1085, 785 cm−1; MS (ESI): m/z 392 (M + H)+.
4.1.6.12. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(4-chloro-3-fluorophenyl)allylidene)hydrazinyl)-4-methylthiazole (8l). Yield: 60%; m.p: 108–110 °C; 1H NMR (400 MHz, CDCl3): δ 7.56 (m, 1H), 7.40–7.19 (m, 5H), 7.16–6.98 (m, 3H), 6.28 (d, J = 14.3 Hz, 1H), 2.35 (s, 3H); IR (KBr): 2850, 1635, 1535, 1185, 952, 735 cm−1; MS (ESI): m/z 412 (M + H); HRMS (ESI): Calcd for C20H16ClFN5S (M + H)+: 412.0813, found: 412.0820.
4.1.6.13. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(3,5-difluorophenyl)allylidene)hydrazinyl)-4-methylthiazole (8m). Yield: 58%; m.p: 102–104 °C; 1H NMR (400 MHz, CDCl3): δ 7.61–7.59 (m, 2H), 7.32–7.30 (m, 2H), 7.18 (s, 2H), 6.92 (d, J = 14.2 Hz, 2H), 6.70 (t, J = 15.2 Hz, 1H), 6.31 (s, 1H), 2.36 (s, 3H); 13C NMR (125 MHz, CDCl3 + DMSO-d6): δ 164.3, 162.3, 146.7, 140.9, 129.0, 124.8, 110.0, 109.8, 104.8, 103.4, 103.2, 103.0, 17.6; IR (KBr): 3252, 2925, 1735, 1485, 1352, 1250, 835 cm−1; MS (ESI): m/z 396 (M + H)+; HRMS (ESI): calcd for C20H16F2N5S (M + H)+: 396.1124, found: 396.1125.
4.1.6.14. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2,3-dimethoxyphenyl)allylidene)hydrazinyl)-4-methylthiazole (8n). Yield: 61%; m.p: 95–97 °C; 1H NMR (400 MHz, DMSO-d6): δ 7.90 (s, 1H), 7.74–7.59 (m, 2H), 7.43 (d, J = 14.5 Hz, 1H), 7.32 (s, 2H), 7.21 (s, 1H), 7.10 (t, J = 7.8 Hz, 1H), 6.93 (d, J = 14.2 Hz, 1H), 6.37 (s, 1H), 3.87 (s, 6H), 2.31 (s, 3H); IR (KBr): 3225, 2923, 1432, 1235, 1132, 982, 725 cm−1; MS (ESI): m/z 420 (M + H)+; HRMS (ESI): calcd for C27H23O2N2S (M + H)+: 420.1526, found: 420.1527.
4.1.6.15. 2-((Z)-2-((E)-1-(1H-Benzo[d]imidazol-2-yl)-3-(2,4,6-trimethoxyphenyl)allylidene)hydrazinyl)-4-methylthiazole (8o). Yield: 55%; m.p: 99–101 °C; 1H NMR (400 MHz, DMSO-d6): δ 12.94 (br s, 1H), 7.76–7.60 (m, 2H), 7.26 (s, 2H), 6.41 (d, J = 14.1 Hz, 1H), 6.36–6.27 (m, 3H), 6.12–6.07 (m, 1H), 3.79 (s, 9H), 2.09 (s, 3H); 13C NMR (125 MHz, CDCl3 + DMSO-d6): δ171.3, 162.8, 160.7, 159.1, 148.3, 145.3, 143.8, 122.6, 108.6, 103.1, 91.2, 54.9, 54.7, 17.1; IR (KBr): 3325, 2950, 1732, 1535, 1132, 958, 832 cm−1; MS (ESI): m/z 450 (M + H)+; HRMS (ESI): calcd for C27H23O2N2S (M + H)+: 450.1624, found: 450.1626.
4.2. Antimycobacterial evaluation assays
The unmarked autoluminescent M. tuberculosis H37Ra (UalRa) was homogenized with sterile glass beads in a 50 mL tube containing 5 ml of Middlebook 7H9 medium plus 0.05% Tween 80, 10% v/v oleic acid albumin dextrose catalase (OADC) supplement (7H9-OADC-Tween80). When the OD600 value reached 0.3–0.5, the relative light unit (RLU) count was determined. by putting a 1.5 mL transparent tube containing 200 μL of culture on the detection hole of the luminometer. When the RLU reached 2 million, the effective concentrations of compounds 7a–o and 8a–o were assessed over a 3-fold range increasing from 0.000001 μg mL−1 to 10 μg mL−1, prepared in 25 μl of UAlRa broth cuture (RLU diluted to 2000–4000) grown in 7H9 broth without Tween80. In the treatment group, DMSO was used as a negative control and rifampicin (RIF; 10 μg mL−1, 1 μg mL−1 and 0.1 μg mL−1) was used as a positive control. RLU counts were determined daily, for 5 days. In the analysis of the data, the MIClux value is the lowest drug concentration that can achieve an RLUdrug/RLUDMSO ration of less than 10% after treatment.4.3. MDCK cellular toxicity assays
MDCK cells were maintained in DMEM high glucose (4.5 g l−1) powder medium with 10% FBS and 1% PS. Cells in the log phase were used. 500 cells per well were seeded in 384-PP plates with a 50 μl volume and the media was replaced with 25 μl after 24 h. Compounds were dissolved to 30 μg ml−1 with DMSO, and a 3-fold serial dilution of the compounds from 30 μg ml−1 to 0.00152 μg ml−1 was performed. 75 nL of compound solution was correspondingly added using Echo520 to the 384-PP plate, and 75 nL of DMSO instead of compound solution was used as the 0% inhibitor control. After co-incubation for 72 h, 3 μl of CCK8 (5 mg ml−1) was added. 2 h later at 37 °C, the plates were read in the Envision Multilabel Reader at 450 nm. The cell survival rate (activity%) was calculated with the formula: activity% = (OD experiment − OD blank)/(OD control − OD blank) × 100. The resulting data was then analyzed with the corresponding compound concentrations using Graph Pad Prism 5 Demo. The IC50 values were fitted using a nonlinear regression model with a sigmoidal dose response. The MDCK cells were bought from Zhongqiaoxinzhou Company, Shanghai, China.Conflicts of interest
The authors declare there are no conflicts of interest.Acknowledgements
This work was supported by the National Mega-project of China for Innovative Drugs (2018ZX09721001-003-003), and the Chinese Academy of Sciences (154144KYSB20150045). GS is especially thankful to CAS-PIFI for a Postdoctoral Fellowship and the Guangzhou Development Zone for a Postdoctoral Fellowship. T. Z. received funding from the Science and Technology Innovation Leader of Guangdong Province (2016TX03R095). M. H, M. M. I. and C. C. are supported by the CAS-TWAS President's PhD Fellowship Program for International Students. H. M. A. H. is supported by the UCAS PhD Fellowship Program.References
- S. A. Ali, T. R. Mavundla, R. Fantu and T. Awoke, Outcomes of TB treatment in HIV co-infected TB patients in Ethiopia: a cross-sectional analytic study, BMC Infect. Dis., 2016, 16(1), 640 CrossRef PubMed.
- Q. Meng, I. Sayin, D. H. Canaday, H. Mayanja-Kizza, J. Baseke and Z. Toossi, Immune Activation at Sites of HIV/TB Co-Infection Contributes to the Pathogenesis of HIV-1 Disease, PLoS One, 2016, 11(11), e0166954 CrossRef PubMed.
- G. Surineni, P. Yogeeswari, D. Sriram and S. Kantevari, Design and synthesis of novel carbazole tethered pyrrole derivatives as potent inhibitors of Mycobacterium tuberculosis, Bioorg. Med. Chem. Lett., 2015, 25(3), 485–491 CrossRef PubMed.
- WHO, Global Tuberculosis Report 2017, http://wwwwhoint/tb/publications/global_report/en/, 2017 Search PubMed.
- S. Tiberi, M. Munoz-Torrico, R. Duarte, M. Dalcolmo, L. D'Ambrosio and G. B. Migliori, New drugs and perspectives for new anti-tuberculosis regimens, Pulmonology, 2018, 24(2), 86–98 CrossRef PubMed.
- H. M. A. Hameed, M. M. Islam, C. Chhotaray, C. Wang, Y. Liu and Y. Tan, et al. Molecular Targets Related Drug Resistance Mechanisms in MDR-, XDR-, and TDR-Mycobacterium tuberculosis Strains, Front. Cell. Infect. Microbiol., 2018, 8, 114 CrossRef PubMed.
- A. Zumla, R. Hafner, C. Lienhardt, M. Hoelscher and A. Nunn, Advancing the development of tuberculosis therapy, Nat. Rev. Drug Discovery, 2012, 11(3), 171–172 CrossRef PubMed.
- M. M. Islam, H. M. A. Hameed, J. Mugweru, C. Chhotaray, C. Wang and Y. Tan, et al. Drug resistance mechanisms and novel drug targets for tuberculosis therapy, J. Genet. Genomics, 2017, 44(1), 21–37 CrossRef PubMed.
- T. M. Walker, M. Merker, A. M. Knoblauch, P. Helbling, O. D. Schoch and M. J. van der Werf, et al. A cluster of multidrug-resistant Mycobacterium tuberculosis among patients arriving in Europe from the Horn of Africa: a molecular epidemiological study, Lancet Infect. Dis., 2018, 18(4), 431–440 CrossRef PubMed.
- L. Fanzani, F. Porta, F. Meneghetti, S. Villa, A. Gelain and A. P. Lucarelli, et al. Mycobacterium tuberculosis Low Molecular Weight Phosphatases (MPtpA and MPtpB): From Biological Insight to Inhibitors, Curr. Med. Chem., 2015, 22(27), 3110–3132 CrossRef.
- F. Meneghetti, S. Villa, A. Gelain, D. Barlocco, L. R. Chiarelli and M. R. Pasca, et al. Iron Acquisition Pathways as Targets for Antitubercular Drugs, Curr. Med. Chem., 2016, 23(35), 4009–4026 CrossRef.
- G. Fadda, Tuberculosis: novel approaches to an old disease, J. Infect. Dev. Countries, 2012, 6(1), 4–5 Search PubMed.
- U. H. Manjunatha and P. W. Smith, Perspective: Challenges and opportunities in TB drug discovery from phenotypic screening, Bioorg. Med. Chem., 2015, 23(16), 5087–5097 CrossRef.
- M. Schito, M. Maeurer, P. Kim, D. Hanna and A. Zumla, Translating the Tuberculosis Research Agenda: Much Accomplished, but Much More to Be Done, Clin. Infect. Dis., 2015, 61(Suppl 3), S95–101 CrossRef.
- E. S. Zuniga, J. Early and T. Parish, The future for early-stage tuberculosis drug discovery, Future Microbiol., 2015, 10(2), 217–229 CrossRef.
- D. Das, P. Sikdar and M. Bairagi, Recent developments of 2-aminothiazoles in medicinal chemistry, Eur. J. Med. Chem., 2016, 109, 89–98 CrossRef PubMed.
- S. Kaur Manjal, R. Kaur, R. Bhatia, K. Kumar, V. Singh and R. Shankar, et al. Synthetic and medicinal perspective of thiazolidinones: A review, Bioorg. Chem., 2017, 75, 406–423 CrossRef.
- R. S. Keri, C. K. Rajappa, S. A. Patil and B. M. Nagaraja, Benzimidazole-core as an antimycobacterial agent, Pharmacol. Rep., 2016, 68(6), 1254–1265 CrossRef.
- N. Shrivastava, M. J. Naim, M. J. Alam, F. Nawaz, S. Ahmed and O. Alam, Benzimidazole Scaffold as Anticancer Agent: Synthetic Approaches and Structure-Activity Relationship, Arch. Pharm., 2017, 350(6), 28544162 CrossRef.
- P. Bhattacharya, J. T. Leonard and K. Roy, Exploring QSAR of thiazole and thiadiazole derivatives as potent and selective human adenosine A3 receptor antagonists using FA and GFA techniques, Bioorg. Med. Chem., 2005, 13(4), 1159–1165 CrossRef.
- S. E. Knudson, D. Awasthi, K. Kumar, A. Carreau, L. Goullieux and S. Lagrange, et al. Cell division inhibitors with efficacy equivalent to isoniazid in the acute murine Mycobacterium tuberculosis infection model, J. Antimicrob. Chemother., 2015, 70(11), 3070–3073 CrossRef.
- S. E. Knudson, K. Kumar, D. Awasthi, I. Ojima and R. A. Slayden, In vitro-in vivo activity relationship of substituted benzimidazole cell division inhibitors with activity against Mycobacterium tuberculosis, Tuberculosis, 2014, 94(3), 271–276 CrossRef.
- S. Chopra, K. Matsuyama, T. Tran, J. P. Malerich, B. Wan and S. G. Franzblau, et al. Evaluation of gyrase B as a drug target in Mycobacterium tuberculosis, J. Antimicrob. Chemother., 2012, 67(2), 415–421 CrossRef.
- E. Abate, D. Elias, A. Getachew, S. Alemu, E. Diro and S. Britton, et al. Effects of albendazole on the clinical outcome and immunological responses in helminth co-infected tuberculosis patients: a double blind randomised clinical trial, Int. J. Parasitol., 2015, 45(2-3), 133–140 CrossRef.
- E. Karagoz, M. B. Selek, E. Aydin, M. Hatipoglu, V. Turhan and A. Acar, et al. Recurrent toxocariasis due to chronic urticaria and successful treatment with prolonged albendazole therapy, Turkiye Parazitol Derg., 2015, 39(1), 83–85 CrossRef.
- L. Mitrov-Winkelmolen, M. W. van Buul-Gast, D. J. Swank, H. Overdiek, R. H. N. van Schaik and D. J. Touw, The Effect of Roux-en-Y Gastric Bypass Surgery in Morbidly Obese Patients on Pharmacokinetics of (Acetyl)Salicylic Acid and Omeprazole: the ERY-PAO Study, Obes. Surg., 2016, 26(9), 2051–2058 CrossRef.
- G. S. Noland, S. Blount and M. Gonzalez, Post-Mass Drug Administration Transmission Assessment Survey for Elimination of Lymphatic Filariasis in La Cienaga, Dominican Republic, Am. J. Trop. Med. Hyg., 2015, 93(6), 1292–1294 CrossRef.
- P. Dubey, B. Gidwani, R. Pandey and S. S. Shukla, In vitro and in vivo evaluation of PEGylated nanoparticles of bendamustine for treatment of lung cancer, Artif. Cells, Nanomed., Biotechnol., 2016, 44(6), 1491–1497 Search PubMed.
- T. Robak, A. Hellmann, J. Kloczko, J. Loscertales, E. Lech-Maranda and J. M. Pagel, et al. Randomized phase 2 study of otlertuzumab and bendamustine versus bendamustine in patients with relapsed chronic lymphocytic leukaemia, Br. J. Haematol., 2017, 176(4), 618–628 CrossRef.
- S. M. Gomha, M. M. Edrees and F. M. Altalbawy, Synthesis and Characterization of Some New Bis-Pyrazolyl-Thiazoles Incorporating the Thiophene Moiety as Potent Anti-Tumor Agents, Int. J. Mol. Sci., 2016, 17(9), 27618013 Search PubMed.
- M. Sun, Q. Xu, J. Xu, Y. Wu, Y. Wang and D. Zuo, et al. Synthesis and bioevaluation of N,4-diaryl-1,3-thiazole-2-amines as tubulin inhibitors with potent antiproliferative activity, PLoS One, 2017, 12(3), e0174006 CrossRef.
- M. T. Chhabria, S. Patel, P. Modi and P. S. Brahmkshatriya, Thiazole: A Review on Chemistry, Synthesis and Therapeutic Importance of its Derivatives, Curr. Top. Med. Chem., 2016, 16(26), 2841–2862 CrossRef CAS.
- G. Surineni, S. K. Marvadi, P. Yogeeswari, D. Sriram and S. Kantevari, Dibenzofuran, dibenzothiophene and N-methyl carbazole tethered 2-aminothiazoles and their cinnamamides as potent inhibitors of Mycobacterium tuberculosis, Bioorg. Med. Chem. Lett., 2018, 28(9), 1610–1614 CrossRef CAS.
- G. Surineni, P. Yogeeswari, D. Sriram and S. Kantevari, Click-based synthesis and antitubercular evaluation of dibenzofuran tethered thiazolyl-1,2,3-triazolyl acetamides, Bioorg. Med. Chem. Lett., 2016, 26(15), 3684–3689 CrossRef CAS.
- G. R. Bommineni, K. Kapilashrami, J. E. Cummings, Y. Lu, S. E. Knudson and C. Gu, et al. Thiolactomycin-Based Inhibitors of Bacterial beta-Ketoacyl-ACP Synthases with in Vivo Activity, J. Med. Chem., 2016, 59(11), 5377–5390 CrossRef CAS.
- D. R. Durairaj and P. Shanmughavel, In Silico Drug Design of Thiolactomycin Derivatives Against Mtb-KasA Enzyme to Inhibit Multidrug Resistance of Mycobacterium tuberculosis, Interdiscip. Sci.: Comput. Life Sci., 2017, 28856604 Search PubMed.
- G. Sabbagh and N. Berakdar, Docking studies of flavonoid compounds as inhibitors of beta-ketoacyl acyl carrier protein synthase I (Kas I) of Escherichia coli, J. Mol. Graphics Modell., 2015, 61, 214–223 CrossRef CAS.
- X. Tang, J. Li and B. S. Moore, Minimization of the Thiolactomycin Biosynthetic Pathway Reveals that the Cytochrome P450 Enzyme TlmF Is Required for Five-Membered Thiolactone Ring Formation, ChemBioChem, 2017, 18(12), 1072–1076 CrossRef CAS.
- A. A. Abdel-Magied, M. M. Hammouda, A. Mosbah and A. A. El-Henawy, In vitro activity of nitazoxanide against some metronidazole-resistant and susceptible Trichomonas vaginalis isolates, J. Infect. Chemother., 2017, 23(4), 230–233 CrossRef CAS.
- E. P. Harausz, K. A. Chervenak, C. E. Good, M. R. Jacobs, R. S. Wallis and M. Sanchez-Felix, et al. Activity of nitazoxanide and tizoxanide against Mycobacterium tuberculosis in vitro and in whole blood culture, Tuberculosis, 2016, 98, 92–96 CrossRef CAS.
- S. Jain, S. Pattnaik, K. Pathak, S. Kumar, D. Pathak and S. Jain, et al. Anticancer Potential of Thiazole Derivatives: A Retrospective Review, Mini-Rev. Med. Chem., 2018, 18(8), 640–655 CrossRef CAS.
- C. Viegas-Junior, A. Danuello, Bolzani. V. da Silva, E. J. Barreiro and C. A. Fraga, Molecular hybridization: a useful tool in the design of new drug prototypes, Curr. Med. Chem., 2007, 14(17), 1829–1852 CrossRef CAS.
- M. W. Majewski, S. Cho, P. A. Miller, S. G. Franzblau and M. J. Miller, Syntheses and evaluation of substituted aromatic hydroxamates and hydroxamic acids that target Mycobacterium tuberculosis, Bioorg. Med. Chem. Lett., 2015, 25(21), 4933–4936 CrossRef CAS.
- L. T. Wu, Z. Jiang, J. J. Shen, H. Yi, Y. C. Zhan and M. Q. Sha, et al. Design, synthesis and biological evaluation of novel benzimidazole-2-substituted phenyl or pyridine propyl ketene derivatives as antitumour agents, Eur. J. Med. Chem., 2016, 114, 328–336 CrossRef CAS.
- G. B. de Oliveira Filho, M. V. O. Cardoso, J. W. P. Espindola, E. S. D. A. Oliveira, R. S. Ferreira and P. L. Coelho, et al. Structural design, synthesis and pharmacological evaluation of thiazoles against Trypanosoma cruzi, Eur. J. Med. Chem., 2017, 141, 346–361 CrossRef CAS.
- J. Tang, B. Wang, T. Wu, J. Wan, Z. Tu and M. Njire, et al. Design, Synthesis, and Biological Evaluation of Pyrazolo[1,5-a]pyridine-3-carboxamides as Novel Antitubercular Agents, ACS Med. Chem. Lett., 2015, 6(7), 814–818 CrossRef CAS.
- F. Yang, M. M. Njire, J. Liu, T. Wu, B. Wang and T. Liu, et al. Engineering more stable, selectable marker-free autoluminescent mycobacteria by one step, PLoS One, 2015, 10(3), e0119341 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary data associated with this article can be found in the online version. Copies of 1H, 13C NMR and mass (ESI-MS and HRMS) spectra of all new compounds are incorporated. See DOI: 10.1039/c8md00389k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |