
Design and synthesis of heteroaromatic-based benzenesulfonamide derivatives as potent inhibitors of H5N1 influenza A virus†
Yongshi
Yu‡
 ab,
Tazeem‡
bc,
Zhichao
Xu
b,
Liaoqi
Du
a,
Mengyu
Jin
b,
Chune
Dong
b,
Hai-Bing
Zhou
*b and
Shuwen
Wu
*a
ab,
Tazeem‡
bc,
Zhichao
Xu
b,
Liaoqi
Du
a,
Mengyu
Jin
b,
Chune
Dong
b,
Hai-Bing
Zhou
*b and
Shuwen
Wu
*a
aState Key Laboratory of Virology, College of Life Sciences, Wuhan University, Wuhan 430072, China. E-mail: shuwenwu@hotmail.com
bHubei Province Key Laboratory of Allergy and Immunology, Hubei Province Engineering and Technology Research Center for Fluorinated Pharmaceuticals, Wuhan University School of Pharmaceutical Sciences, Wuhan 430071, China. E-mail: zhouhb@whu.edu.cn
cDepartment of Chemistry, Shia P. G. College (University of Lucknow), Lucknow, Uttar Pradesh 226020, India
First published on 23rd November 2018
Abstract
Influenza A virus is an enveloped negative single-stranded RNA virus that causes febrile respiratory infection and represents a clinically challenging threat to human health and even lives worldwide. Even more alarming is the emergence of highly pathogenic avian influenza (HPAI) strains such as H5N1, which possess much higher mortality rate (60%) than seasonal influenza strains in human infection. In this study, a novel series of heteroaromatic-based benzenesulfonamide derivatives were identified as M2 proton channel inhibitors. A systematic investigation of the structure–activity relationships and a molecular docking study demonstrated that the sulfonamide moiety and 2,5-dimethyl-substituted thiophene as the core structure played significant roles in the anti-influenza activity. Among the derivatives, compound 11k exhibited excellent antiviral activity against H5N1 virus with an EC50 value of 0.47 μM and selectivity index of 119.9, which are comparable to those of the reference drug amantadine.
Influenza is a common and serious infectious respiratory disease caused by influenza viruses belonging to the Orthomyxoviridae family.1 Despite decades of surveillance and interventions, seasonal influenza viruses continue to cause epidemics around the world each year. According to the World Health Organization, seasonal influenza epidemics result in 3–5 million flu patients and 290
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 650000 deaths worldwide each year.2 Due to its higher antigenic mutation, influenza A (flu A) virus is a greater threat to human health compared to flu B and C strains. Five flu A pandemics have occurred in the past 100 years: 1918 H1N1 (Spanish flu), 1957 H2N2 (Asian flu), 1968 H3N2 (Hong Kong flu), 2005 H5N1 (Bird flu) and 2009 H1N1 (Swine flu).3–8 Among them, the 1918 pandemic was the most severe, leading to a catastrophic 50 million deaths globally.3,9–11 Worrisome, highly pathogenic avian influenza (HPAI) viruses have recently emerged, including H5N1 (ref. 12–15) and H7N9.16,17 These viruses possess higher mortality compared to seasonal influenza viruses.18 HPAI H5N1 flu strains have been shown to be transmissible among humans after acquiring only one or a few mutations, generating considerable concern among researchers.14,19,20 Although vaccines and antiviral agents are essential measures for influenza treatment, anti-influenza agents seem to be more effective at preventing highly contagious influenza infections due to the frequent variation of influenza virus through antigenic drift and antigenic shift.8 Currently, two classes of anti-influenza drugs are applied for the prophylaxis and treatment of influenza infection (Fig. 1): M2 proton channel inhibitors (amantadine and rimantadine) and neuraminidase inhibitors (oseltamivir, zanamivir and peramivir). However, the drug resistance of H5N1 viral strains limits the usable clinical range of these drugs, making the identification of novel anti-influenza agents imperative.21–24
000 to 650000 deaths worldwide each year.2 Due to its higher antigenic mutation, influenza A (flu A) virus is a greater threat to human health compared to flu B and C strains. Five flu A pandemics have occurred in the past 100 years: 1918 H1N1 (Spanish flu), 1957 H2N2 (Asian flu), 1968 H3N2 (Hong Kong flu), 2005 H5N1 (Bird flu) and 2009 H1N1 (Swine flu).3–8 Among them, the 1918 pandemic was the most severe, leading to a catastrophic 50 million deaths globally.3,9–11 Worrisome, highly pathogenic avian influenza (HPAI) viruses have recently emerged, including H5N1 (ref. 12–15) and H7N9.16,17 These viruses possess higher mortality compared to seasonal influenza viruses.18 HPAI H5N1 flu strains have been shown to be transmissible among humans after acquiring only one or a few mutations, generating considerable concern among researchers.14,19,20 Although vaccines and antiviral agents are essential measures for influenza treatment, anti-influenza agents seem to be more effective at preventing highly contagious influenza infections due to the frequent variation of influenza virus through antigenic drift and antigenic shift.8 Currently, two classes of anti-influenza drugs are applied for the prophylaxis and treatment of influenza infection (Fig. 1): M2 proton channel inhibitors (amantadine and rimantadine) and neuraminidase inhibitors (oseltamivir, zanamivir and peramivir). However, the drug resistance of H5N1 viral strains limits the usable clinical range of these drugs, making the identification of novel anti-influenza agents imperative.21–24
 | ||
| Fig. 1 Approved anti-influenza drugs for influenza treatment. | ||
As a consequence, some small molecules that inhibit influenza A H5N1 virus have been created through the structural modification of approved anti-influenza drugs or using the hit-and-lead discovery strategy (Fig. 2).
 | ||
| Fig. 2 Structures of known small-molecule inhibitors of H5N1 influenza A virus. | ||
In 2010, based on high-throughput screening, Severson and coworkers reported quinazolinone analogues as novel inhibitors; compound 1 exhibited an excellent EC50 value of 0.032 μM for both H5N1 and H1N1 viruses.25 In 2011, Liu and co-workers found that 3-fluoro-N-(2-(piperidin-1-yl)ethyl)-5-(trifluoromethyl)benzamide hydrochloride (2, Fig. 2) could inhibit HPAI H5N1 virus infection by blocking viral entry with an IC50 value of 27.03 μM.26 More recently, polyphenol (3, Fig. 2) was identified as entry inhibitor against H5N1 virus with an EC50 value of 1.78 μM, superior to that of the reference drug amantadine.27 In 2017, Liu's group discovered a series of oligo-thiophene compounds as HA inhibitors that act by inhibiting the membrane fusion between the H5N1 virus and the endosome of the host cell. The most potent inhibitor (4, Fig. 2) displayed good anti-H5N1 activity with an IC50 value of 0.029 μM.28 In the same year, our group developed furan–carboxamide derivatives as novel inhibitors of H5N1 influenza A virus. The most potent compound (5, Fig. 2) showed low-micromolar activity against H5N1 virus with an EC50 value of 1.25 μM, which was comparable with reference drug amantadine.29 The systematic investigation of the structure–activity relationships (SARs) of furan–carboxamide analogues indicated that heteroaromatic groups (e.g., furan and thiophene moieties) and sulfur atoms played significant roles in the inhibition of H5N1 virus. Moreover, furan–carboxamide derivatives containing para-substituents on the phenyl rings showed more potent anti-H5N1 activity than those with meta- or ortho-substituents on the phenyl rings. A molecular docking investigation of this series of furan–carboxamide analogues indicated that they cannot efficiently interact with the M2 protein cavity due to the long linker. Our long-term aim is the development of antivirals with better inhibitory potency and fewer side effects than currently used drugs;29–33 hence, to develop novel heteroaromatic-based benzenesulfonamide compounds as anti-influenza A/H5N1 agents, an attempt was made to modify the structural features of furan–carboxamide derivatives by shortening the linker length and transforming the heteroaromatic core structure (Scheme 1).
 | ||
| Scheme 1 Strategy for designing heteroaromatic-based benzenesulfonamide derivatives as novel inhibitors of H5N1 influenza A virus. | ||
Results and discussion
Chemistry
The synthesis of the designed heteroaromatic-based benzenesulfon-amide derivatives is shown in Scheme 2. All designed compounds were synthesized via the sulfonylation reaction of substituted benzenesulfonyl chloride analogues with diversified amines. Most amines were formed starting from corresponding carboxylic acid 6, as shown in Scheme 2. First, heteroaromatic formic acid 6 reacted with thionyl chloride to produce aromatic formyl chloride (7). Formamide 8 was then formed via the amination reaction of 7 and ammonium hydroxide in the presence of dichloromethane (DCM). Next, the key amine 9 was synthesized through reduction reaction in the presence of lithium aluminum hydride followed by the sulfonylation reaction to generate the final products (11–12) in 20%–60% yields.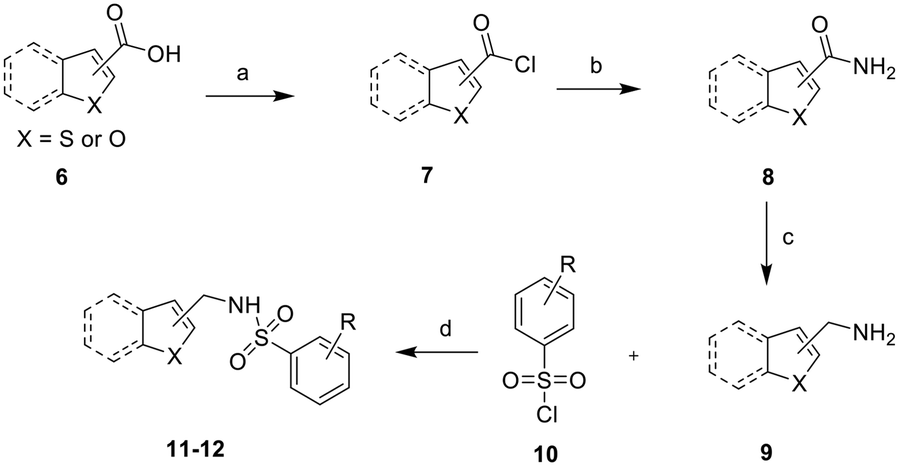 | ||
| Scheme 2 Synthetic route for heteroaromatic-based benzenesulfon-amide derivatives. Reagents and conditions: (a) SOCl2, toluene, reflux; (b) NH4OH, DCM, rt; (c) LiAlH4, THF, reflux; (d) Et3N, DCM, rt. | ||
Structure–activity relationship (SAR) studies
All synthesized novel heteroaromatic-based arylsulfonamide compounds were tested for anti-influenza activity against both amantadine-sensitive H5N1 virus and amantadine-resistant H5N1 virus using plaque reduction assays in MDCK cells. The reference anti-influenza drugs amantadine (AMD) and oseltamivir phosphate were used for comparison. The results of this extensive biological evaluation are described in Tables 1 and 2. As a global observation, the heteroaromatics as core structures played significant roles in the activity against amantadine-sensitive H5N1 virus. Originally, the designed heteroaromatic-based benzenesulfonamide compound 11a containing furan as a core structure and 4-NO2 as the R substituent showed good potency against H5N1 virus (EC50 = 1.06 μM; Table 1, entry 1). To explore the effects of R substituents on the anti-influenza activity, compounds 11b–e were synthesized by introducing electron-withdrawing groups (4-CN, 4-F, 4-Cl and 4-Br) (Table 1, entries 2–5). Unfortunately, analogues 11b–e all displayed decreased inhibitory potency. Considering that sulfur and oxygen atoms have similar chemical properties, bioisostere analogues 11g–k were synthesized (Table 1, entries 7–11). Surprisingly, we found that all compounds displayed superior anti-H5N1 activity with the exception of compound 11i (containing 4-F), which showed no inhibition potency. Among 11g–k, the most potent analogue 11k exhibited an EC50 value of 0.47 μM, comparable with those of the reference drugs amantadine and oseltamivir phosphate (Table 1, entries 11 vs. 15 and 16). We also explored the influence of other heteroaromatic core structures on the anti-influenza activity in the presence of 4-NO2 as the R substituent. We found that analogue 11f showed dramatically decreased inhibition potency when a furan moiety was the core structure (Table 1, entries 1 vs. 6). Moreover, introducing a thiophene or 2-chloro-thiophene moiety as the core structure also resulted in slightly decreased anti-influenza activity (Table 1, entries 1 and 12–14). Meanwhile, we tested the antiviral activity against amantadine-resistant H5N1 virus in MDCK cells. Unfortunately, these benzenesulfonamide analogues showed no inhibition potency (Table 1, entries 1–14).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cmpd | Core structure | R | EC50 (μM)b AMD-sensitive H5N1 | EC50 (μM)c AMD-resistant H5N1 | CC50 (μM)d | SIe |
| a All data were obtained from at least three independent experiments. b EC50, concentration that effectively inhibited amantadine-sensitive H5N1 virus plaque formation by 50%. c EC50, concentration that effectively inhibited amantadine-resistant H5N1 virus plaque formation by 50%. d CC50, concentration that inhibited cell growth by 50% compared with control cultures. e Selectivity index (SI) was determined for effective compounds by dividing CC50 by EC50 (amantadine-sensitive H5N1 virus). f NA: no activity (EC50 > 100 μM). g —: no detection. h —: no selectivity index. | |||||||
| 1 | 11a |
|
4-NO2 | 1.06 ± 0.26 | NA | >152.10 ± 40.60 | >143.5 |
| 2 | 11b | 4-CN | 4.03 ± 1.62 | NA | 105.39 ± 22.04 | 26.1 | |
| 3 | 11c | 4-F | 8.08 ± 1.84 | NA | 138.01 ± 36.00 | 17.1 | |
| 4 | 11d | 4-Cl | 1.37 ± 0.27 | NA | 55.71 ± 16.34 | 40.7 | |
| 5 | 11e | 4-Br | 1.80 ± 0.52 | NA | 29.63 ± 10.75 | 16.5 | |
| 6 | 11f |
|
4-NO2 | 47.19 ± 17.25 | NA | >236.30 | >5.0 |
| 7 | 11g |
|
4-NO2 | 1.29 ± 0.43 | NA | >204.36 | >158.4 |
| 8 | 11h | 4-CN | 0.62 ± 0.098 | NA | >217.69 | >351.1 | |
| 9 | 11i | 4-F | NAf | NA | —g | —h | |
| 10 | 11j | 4-Cl | 0.70 ± 0.16 | NA | 52.88 ± 14.56 | 75.5 | |
| 11 | 11k | 4-Br | 0.47 ± 0.11 | NA | 56.35 ± 14.43 | 119.9 | |
| 12 | 11l |
|
4-NO2 | 1.47 ± 0.77 | NA | 138.77 ± 28.83 | 94.4 |
| 13 | 11m |
|
4-NO2 | 1.91 ± 0.94 | NA | 133.41 ± 27.82 | 69.8 |
| 14 | 11n |
|
4-NO2 | 70.05 ± 25.87 | NA | 127.72 ± 28.85 | 1.8 |
| 15 | AMD | 0.43 ± 0.13 | NA | >100.00 | >235.3 | ||
| 16 | Oseltamivir phosphate | 0.39 ± 0.10 | 0.34 ± 0.02 | >163.26 | >418.6 | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cmpd | Core structure | R | EC50b (μM) AMD-sensitive H5N1 | EC50c (μM) AMD-resistant H5N1 | CC50d (μM) | SIe |
| a All data were obtained from at least three independent experiments. b EC50, concentration that effectively inhibited amantadine-sensitive H5N1 virus plaque formation by 50%. c EC50, concentration that effectively inhibited amantadine-resistant H5N1 virus plaque formation by 50%. d CC50, concentration that inhibited cell growth by 50% compared with control cultures. e Selectivity index (SI) was determined for the effective compounds by dividing CC50 by EC50 (amantadine-sensitive H5N1 virus). f NA: no activity (EC50 > 100 μM). g —: no detection. h —: no selectivity index. | |||||||
| 1 | 12a |
|
4-NO2 | 1.38 ± 0.29 | NA | >191.45 | >138.7 |
| 2 | 12b | 4-CN | 20.28 ± 8.34 | NA | 122.41 ± 32.59 | 6.0 | |
| 3 | 12c | 4-F | 3.42 ± 0.81 | NA | 98.64 ± 29.25 | 28.8 | |
| 4 | 12d | 4-Cl | 1.98 ± 0.68 | NA | 69.56 ± 15.39 | 35.1 | |
| 5 | 12e | 4-Br | 7.22 ± 1.60 | NA | 87.11 ± 21.71 | 12.1 | |
| 6 | 12f | 4-CF3 | 38.56 ± 14.73 | NA | 94.78 ± 14.27 | 2.5 | |
| 7 | 12g | 4-Me | 10.49 ± 3.78 | NA | >210.13 | >20.0 | |
| 8 | 12h | 2-F | NAf | NA | —g | —h | |
| 9 | 12i | 3-F | NA | NA | — | — | |
| 10 | 12j | 4-COCH3 | 12.04 ± 3.85 | NA | >193.09 | >16.0 | |
| 11 | 12k |
|
4-NO2 | 49.97 ± 14.87 | NA | >191.45 | >3.8 |
| 12 | 12l | 4-CN | NA | NA | — | — | |
| 13 | 12m | 4-F | NA | NA | — | — | |
| 14 | 12n | 4-Cl | NA | NA | — | — | |
| 15 | 12o | 4-Br | 40.70 ± 12.11 | NA | >174.47 | >4.3 | |
| 16 | 12p | 4-CF3 | 44.83 ± 16.51 | NA | >179.60 | >4.0 | |
| 17 | 12q | 4-Me | 41.96 ± 13.74 | NA | >210.13 | >5.0 | |
| 18 | 12r | 2-F | NA | NA | — | — | |
| 19 | 12s | 3-F | NA | NA | — | — | |
| 20 | AMD | 0.43 ± 0.13 | NA | >100.00 | >235.3 | ||
| 21 | Oseltamivir phosphate | 0.39 ± 0.10 | 0.34 ± 0.02 | >163.26 | >418.6 | ||
Next, we further investigated the SAR when using a benzothiophene moiety as the core structure. Initially, we synthesized compound 12a with a C3-substituted benzothiophene moiety as the core structure and 4-NO2 as the R substituent. Interestingly, 12a maintained potency against the amantadine-sensitive H5N1 virus when the benzothiophene moiety was the core structure, indicating that our hypothesis was rational (Table 1, entries 7, 12–13vs.Table 2, entry 1).
To further explore the influence of the R substituent on the antiviral activity, we designed and synthesized compounds 12b–g and 12j containing electron-withdrawing groups (4-CN, 4-F, 4-Cl, 4-Br, 4-CF3 and 4-COCH3) and an electron-donating group (4-Me), respectively (Table 2, entries 2–7 and 10). All compounds displayed inferior activity against amantadine-sensitive H5N1 virus compared to compound 12a; in particular, the potency of analogue 12f was approximately 28 times lower than that of 12a, likely due to the stronger electron-withdrawing property of the 4-CF3 group (Table 2, entries 1 vs. 6). Meanwhile, analogues 12k–q were synthesized to study the effect of the C2-substituted benzothiophene moiety as a core structure on the biological activity. These analogues all exhibited over 30-fold decreased potency compared to the corresponding analogues 12a–g containing C3-substituted benzothiophene moiety as the core structure (Table 2, entries 1–7 and 11–17). We also synthesized analogues 12h–i and 12r–s to study the anti-H5N1 activity of analogues containing ortho- and meta-substituents as R groups. The biological results indicated no inhibitory activity against amantadine-sensitive H5N1 virus, showing that a para-substituent as the R group is important for the anti-influenza activity (Table 2, entry 3 vs. 8–9 and entry 13 vs. 18–19), which is consistent with our prediction. Among benzothiophene-based benzenesulfonamide derivatives, the C3-substituted compound 12a displayed potent antiviral activity against amantadine-sensitive H5N1 virus with an EC50 of 1.38 μM (Table 2, entry 1). Next, we evaluated the activity against amantadine-resistant H5N1 virus in MDCK cells. Unfortunately, these benzothiophene-based benzene–sulfonamide derivatives displayed no potency (Table 2, entries 1–19).
Fig. 3 intuitively shows that the 2,5-dimethyl-substituted thiophene moiety was a more potent core structure than the 2,5-dimethyl-substituted furan moiety or benzo-thiophene moiety in terms of anti-influenza activity against amantadine-sensitive H5N1 virus (Fig. 3A and B). Furthermore, the para-substituent was superior to the meta- or ortho-substituent as an R group in terms of activity.
 | ||
| Fig. 3 Graphical representation of the effects of the core structure and R substituent of heteroaromatic-based benzenesulfonamide compounds on activity against amantadine-sensitive H5N1 virus in MDCK cells. A) The effects of 2,5-dimethylfuran or 2,5-dimethylthiophene moiety as core structure and R substituent of compounds 11a–e and 11g–k on antiviral activity; B) the effects of benzothiophene as core structure and R substituent of compounds 12a–g and 12k–q on antiviral activity. | ||
The anti-proliferative activities of the synthesized compounds against MDCK cells were also evaluated to monitor potential cytotoxic effects. As expected, none of the heteroaromatic-based benzenesulfonamide derivatives obviously inhibited the growth of MDCK cells at a concentration of 50 μM, with the exception of compound 11e, consolidating the safety of this series of compounds (Tables 1 and 2).
Preliminary mechanistic studies based on a time-course experiment with compound 11k and reference drugs amantadine and favipiravir (T-705) demonstrated that the novel heteroaromatic-based benzenesulfonamide derivatives prevented early-stage virus replication (Fig. 4). Similar to the M2 proton channel inhibitor amantadine, compound 11k likely blocked channel function to inhibit the uncoating of the viral RNPs.34
 | ||
| Fig. 4 Results of the time-course experiment. Compound 11k (c = 0.68 μg mL−1), AMD (amantadine, c = 0.26 μg mL−1) and T-705 (favipiravir, c = 0.63 μg mL−1) were added to MDCK cells that had been infected with H5N1 virus at an MOI of 2 at the indicated time. | ||
To further explore the possible drug target of this series of novel heteroaromatic-based benzenesulfonamide derivatives, we performed a yeast growth restoration assay.35–38 The M2 proton channel inhibitor amantadine and neuraminidase (NA) inhibitor oseltamivir were included as reference drugs. The yeast toxicities of the heteroaromatic-based benzenesulfonamide derivatives were also evaluated to eliminate the contribution of toxic effects. As shown in Table 3, this series of compounds exhibited good potency against WT M2 and displayed little yeast toxicity in the yeast growth restoration assay, which was almost consistent with the anti-H5N1 activity results in MDCK cells (Table 3vs.Tables 1 and 2). Moreover, compounds 11a, 11h, 11j and 11k showed excellent activity against WT M2 in comparison to amantadine (Table 3, entries 1, 8, 10–11 and 34). In contrast, the NA inhibitor oseltamivir showed no inhibitory potency against WT M2 (Table 3, entry 35). Thus, the heteroaromatic-based benzenesulfonamide analogues inhibited WT M2 expression in yeast similarly to the M2 inhibitor amantadine, demonstrating that M2 protein was the drug target of this series of compounds. Unfortunately, these novel heteroaromatic-based benzenesulfonamide derivatives displayed no potency against mutant S31N/V27A M2 (Table 3).
| Entry | Cmpd | EC50a (μM) WT M2 | EC50 (μM) V27A/S31N M2 | Yeast toxicityc |
|---|---|---|---|---|
| a EC50: concentration that effectively inhibited the expression of M2 protein by 50%. b NA: denotes inactive at the highest concentration tested. c Yeast toxicity: concentration that inhibited the proliferation of the control yeast strain by 50%. | ||||
| 1 | 11a | 1.71 ± 0.64 | NAb | >215.9 |
| 2 | 11b | 12.74 ± 5.51 | NA | 168.8 ± 34.4 |
| 3 | 11c | 24.00 ± 7.06 | NA | >236.5 |
| 4 | 11d | 3.07 ± 1.00 | NA | 123.43 ± 16.68 |
| 5 | 11e | 4.65 ± 0.87 | NA | 72.6 ± 11.6 |
| 6 | 11f | 81.48 ± 21.25 | NA | >237.4 |
| 7 | 11g | 2.21 ± 0.92 | NA | >205.3 |
| 8 | 11h | 1.40 ± 0.33 | NA | >218.7 |
| 9 | 11i | NA | NA | 120.2 ± 26.7 |
| 10 | 11j | 1.08 ± 0.32 | NA | 114.1 ± 22.2 |
| 11 | 11k | 1.58 ± 0.56 | NA | 133.2 ± 19.4 |
| 12 | 11l | 2.38 ± 0.67 | NA | >224.6 |
| 13 | 11m | 2.01 ± 0.67 | NA | 157.5 ± 20.1 |
| 14 | 11n | NA | NA | >201.3 |
| 15 | 12a | 3.44 ± 1.15 | NA | >192.3 |
| 16 | 12b | 45.67 ± 15.22 | NA | >204.0 |
| 17 | 12c | 6.22 ± 2.18 | NA | 133.8 ± 24.9 |
| 18 | 12d | 5.33 ± 1.77 | NA | 121.4 ± 20.7 |
| 19 | 12e | 15.25 ± 5.26 | NA | 134.1 ± 23.7 |
| 20 | 12f | NA | NA | >180.4 |
| 21 | 12g | 22.68 ± 6.30 | NA | >211.08 |
| 22 | 12h | NA | NA | 59.1 ± 12.4 |
| 23 | 12i | NA | NA | >208.5 |
| 24 | 12j | 27.79 ± 11.58 | NA | >194.0 |
| 25 | 12k | NA | NA | >192.3 |
| 26 | 12l | NA | NA | >204.0 |
| 27 | 12m | NA | NA | >208.5 |
| 28 | 12n | NA | NA | >198.3 |
| 29 | 12o | NA | NA | >176.2 |
| 30 | 12p | NA | NA | >180.4 |
| 31 | 12q | NA | NA | >211.08 |
| 32 | 12r | NA | NA | >180.5 |
| 33 | 12s | NA | NA | >180.5 |
| 34 | Amantadine | 0.66 ± 0.20 | NA | >443.0 |
| 35 | Oseltamivir phosphate | NA | NA | >214.5 |
To further investigate the structure–binding relationships of these heteroaromatic-based benzenesulfonamide derivatives, we conducted a docking study to explore the binding mode of 11k with the M2 protein of the H5N1 virus. Docking experiments were carried out based on the published M2 protein (PDB: 2KQT)39 with amantadine. AutoDockTool 4.2 (ref. 40) was used to study the binding modes of 11k, and the results indicated a good binding score (−5.87 kcal mol−1). As shown in Fig. 5A–C, the 2,5-dimethyl-thiophene moiety of compound 11k enters into the M2 protein cavity formed by residues Ala-30, Ser-31, Ile-33, Gly-34 and Ile-35, demonstrating that the 2,5-dimethyl-thiophene moiety played a significant role in the anti-influenza activity. Moreover, one oxygen atom of the sulfonamide moiety formed a hydrogen bond with the backbone residue His-37, and the nitrogen atom of the sulfonamide moiety formed another hydrogen bond with residue Gly-34, verifying that the sulfonamide moiety is necessary in this series of compounds (Fig. 5B–D).
 | ||
| Fig. 5 (A) The solid surface type between 11k with M2 protein (oxygen atoms are shown in red, sulfur atoms are shown in yellowish orange, and nitrogen atoms are shown in blue). (B) The transparent surface type between 11k with M2 protein (same as above). (C) The hydrophobic interactions and hydrogen bond interactions formed between 11k with the backbone residues of M2 protein. (D) Computer modeling of the complex structures of M2 protein with 11k based on the published structure (PDB: 2KQT) of M2 protein with amantadine. | ||
Conclusions
In summary, we have designed and synthesized a novel series of heteroaromatic-based benzenesulfonamide derivatives as M2 proton channel inhibitors. Several of the synthesized compounds show sub-micromolar activity against amantadine-sensitive H5N1 virus and low cytotoxicity in MDCK cells. The SAR investigation and molecular docking results indicated that the 2,5-dimethylthiophene core structure and sulfonamide moiety played key roles in the inhibition activity. Among the synthesized compounds, 11k is the most effective derivative with an EC50 value of 0.47 μM and selectivity index of 119.98, which are comparable with those of reference drugs amantadine and oseltamivir phosphate. A further mechanistic investigation of these potent M2 inhibitors is underway and will be reported in due course.Experimental section
Materials and methods
Reagents and materials were obtained from commercial suppliers unless otherwise noted. Reactions were monitored by thin layer chromatography. Column chromatography purification was performed using 230–400-mesh silica gel. NMR spectra were measured on Bruker DRX and DMX spectrometers at 400 MHz for 1H spectra and 100 MHz for 13C spectra. Spectra were calibrated using the residual solvent signal. All final products were characterized by 1H NMR, 13C NMR and MS analyses.Representative procedure for the synthesis of 2,5-dimethylfuran-3-carboxamide 8a
Thionyl chloride (0.14 mL, 1.872 mmol) was added to a solution of 2,5-dimethylfuran-3-carboxylic acid 6a (174.8 mg, 1.248 mmol) in toluene (5 mL), and the mixture was heated under reflux for 2 h. After the reaction was completed, the solution was concentrated. Without further purification, the crude product 2,5-dimethylfuran-3-carbonyl chloride (7a) was immediately used in the next step. Ammonium hydroxide (25% aqueous solution, 1 mL) was then added dropwise to a solution of intermediate 7a in anhydrous dichloromethane (5 mL) at 0 °C. The mixture was stirred at room temperature for 1 h. The mixture was then extracted with ethyl acetate (3 × 30 mL) and brine, dried over anhydrous sodium sulfate, and concentrated by vacuum pump. The residue was purified by flash column chromatography and eluted with petroleum ether/ethyl acetate (2:1) to produce the 2,5-dimethylfuran-3-carboxamide (8a) as a white solid.
Representative procedure for the synthesis of N-((2,5-dimethylfuran-3-yl)methyl)-4-nitrobenzenesulfonamide 11a
Lithium aluminum hydride (128.4 mg, 3.384 mmol) was added to a solution of 8a (157.0 mg, 1.128 mmol) in anhydrous THF (5 mL) at 0 °C. The solution was warmed to ambient temperature and heated under reflux for 4 h. After completion, the reaction was quenched by the sequential addition of 0.13 mL H2O, 0.13 mL1 5% NaOH solution, and 0.39 mL H2O. Ethyl acetate was then added to the above mixture followed by stirring at room temperature for 30 min. Next, the filtrate was dried over anhydrous sodium sulfate and concentrated by vacuum pump. Without further purification, the crude (2,5-dimethylfuran-3-yl)methanamine 9a was directly used in the next step. Triethylamine (0.19 mL, 1.354 mmol) was added to a solution of the key amine intermediate 9a (141.2 mg, 1.128 mmol) in anhydrous dichloromethane at 0 °C followed by the addition of 4-nitrobenzene-sulfonyl chloride 10a (275.0 mg, 1.241 mmol). The mixture was stirred at room temperature for 5 h. After concentration under vacuum pump, the residue was purified by flash column chromatography and eluted with petroleum ether/ethyl acetate (10:1) to produce N-((2,5-dimethylfuran-3-yl)methyl)-4-nitrobenzenesulfonamide 11a in 40% yield as a yellow solid: mp 127–129 °C, 1H NMR (400 MHz, CDCl3) δ 8.35–8.30 (m, 2H), 8.01–7.96 (m, 2H), 5.61 (s, 1H), 4.95 (t, J = 5.4 Hz, 1H), 3.95 (d, J = 5.6 Hz, 2H), 2.10 (s, 6H). 13C NMR (100 MHz, CDCl3) δ150.48, 149.92, 147.64, 146.18, 128.29, 124.15, 114.42, 106.18, 38.59, 13.23, 11.34. HRMS (ESI) calcd for C13H14N2O5S [M + Na]+ 333.0521, found 333.0520.
Plaque reduction assay
The sequence of the M2 protein of amantadine-sensitive H5N1 virus [A/chicken/Hubei/327/2004 (H5N1)] is MSLLTEVETPTRNEWEC RCSDSSDPLVVAASIIGILHLILWILDRLFFKCIYRRLKYGLKRGPSTAGVPESMREEYRQEQQSAVDVDDGHFVNIELE. Amantadine-resistant H5N1 virus was obtained by screening amantadine-resistant H5N1 flu strains based on amantadine-sensitive H5N1 virus [A/chicken/Hubei/327/2004 (H5N1)]. We confirmed that the mutation site of the M2 protein of amantadine-resistant H5N1 virus was M2-S31 N by sequencing M2 protein. M2-S31N is the most common amantadine-resistant mutation site among isolated influenza A viruses (H5N1, H1N1, etc.).MDCK (Madin–Darby canine kidney) cells were purchased from the American Type Culture Collection. A confluent monolayer of MDCK cells was prepared in six-well plates. The cells were infected with amantadine-sensitive H5N1 virus [A/chicken/Hubei/327/2004 (H5N1)] and amantadine-resistant H5N1 virus at 100 PFU per well in DMEM supplemented with 0.001% DEAE-dextran, 2 mg mL−1 TPCK-treated trypsin, and 0.5% agarose in the presence of various concentrations of test compounds for 40 min at 37 °C. Medium containing 0.001% DEAE-dextran, 2 mg mL−1 TPCK-treated trypsin, 0.5% agarose and test compounds at the same concentrations was then added. After 2–3 days of incubation at 37 °C in a CO2 incubator, cell monolayers were fixed with 3% formaldehyde and stained with 0.5% crystal violet, and the EC50 values were determined. Amantadine was included in all experiments as a reference compound.
Cytotoxicity assay
The cytotoxicities of compounds in MDCK cells were tested using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Briefly, cells were seeded at a density of 2 × 104 per well into 96-well plates. The next day, the cell medium was removed, and fresh media containing DMEM plus test compounds at various concentrations were added to each well. After 24 h, 5 mg mL−1 MTT solution was added to each well, and the plates were incubated for 4 h at 37 °C in a CO2 incubator. Successively, a solubilization solution was added to lyse the cells. After 3 h of incubation at 37 °C, absorbance was measured at 575 nm using an ELISA plate reader (Tecan Sunrise).Time-of-addition assay
MDCK cells were seeded in 48-well plates and infected with amantadine-sensitive H5N1 virus at an MOI of 2. Compound 11k and AMD were added at the time of infection (0 h) and at 1, 2, 3, 4, 6 and 8 h after infection. Viral titers were determined at 12 h after infection by plaque assay as described above. Compound 11k was added at a concentration of 1.9 μM. T-705 (4.0 μM) and AMD (1.7 μM) were used as control treatments.Determination of M2 inhibitory activity by yeast growth restoration assay
The coding sequences of the wild-type, V27A/S31N-mutated M2 gene from the H5N1 strain [A/chicken/Hubei/327/2004(H5N1)] were introduced downstream of the GAL1 promoter by PCR amplification followed by recombination cloning in yeast.35 Plasmids were rescued from the yeast strains, and the coding sequences were verified. The yeast strains were propagated in synthetic complete (SC) medium (pH 6.5) lacking leucine and containing glucose to repress M2 gene expression. After harvesting the yeast by centrifugation, the pellets were washed with water to remove traces of glucose and then suspended to A600 = 0.01 in liquid SC medium (pH 6.5) lacking leucine and containing 2% galactose. Samples (100 μL) were distributed into 96-well plates. The compounds were transferred to the yeast plates at different concentrations, and the plates were incubated at 30 °C in a humidified incubator. After 46–48 h, the cells were suspended by vortexing, and A600 was measured. The percentage of growth restoration was calculated as described previously.36 Compounds leading to growth restoration were retested in triplicate at different concentrations against WT M2, V27A/S31N M2 and empty plasmid strains to confirm their activity and determine EC50.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the NSFC (81772236, 81773557 and 81573279), Major Project of Technology Innovation Program of Hubei Province (2016ACA126, 2018ACA123), NSFHP (2017CFA024), the Fundamental Research Funds for the Central Universities of China (2042017kf0288), and Open Research Fund Program of the Hubei Province Engineering and Technology Research Center for Fluorinated Pharmaceuticals for support of this research.Notes and references
- E. J. Schrauwen and R. A. Fouchier, Emerging Microbes Infect., 2014, 3, e9 CrossRef PubMed
.
- X. Wu, X. Wu, Q. Sun, C. Zhang, S. Yang, L. Li and Z. Jia, Theranostics, 2017, 7, 826–845 CrossRef CAS PubMed
- C. Paules and K. Subbarao, Lancet, 2017, 390, 697–708 CrossRef
- J. K. Taubenberger and D. M. Morens, Emerging Infect. Dis., 2006, 12, 15–22 CrossRef PubMed
- C. Scholtissek, W. Rohde, V. Von Hoyningen and R. Rott, Virology, 1978, 87, 13–20 CrossRef CAS PubMed
- G. J. D. Smith, D. Vijaykrishna, J. Bahl, S. J. Lycett, M. Worobey, O. G. Pybus, S. K. Ma, C. L. Cheung, J. Raghwani, S. Bhatt, J. S. M. Peiris, Y. Guan and A. Rambaut, Nature, 2009, 459, 1122–1125 CrossRef CAS PubMed
- V. N. Petrova and C. A. Russell, Nat. Rev. Microbiol., 2018, 16, 47–60 CrossRef CAS PubMed
- K. Das, J. Med. Chem., 2012, 55, 6263–6277 CrossRef CAS PubMed
- M. Biggerstaff, S. Cauchemez, C. Reed, M. Gambhir and L. Finelli, BMC Infect. Dis., 2014, 14, 480 CrossRef PubMed
- N. P. A. S. Johnson and J. Mueller, Bull. Hist. Med., 2002, 76, 105–115 CrossRef PubMed
- P. Palese, Nat. Med., 2004, 10, S82–S87 CrossRef CAS PubMed
- A. N. Abdel-Ghafar, T. Chotpitayasunondh, Z. Gao, F. G. Hayden, D. H. Nguyen, M. D. de Jong, A. Naghdaliyev, J. S. M. Peiris, N. Shindo, S. Soeroso and T. M. Uyeki, N. Engl. J. Med., 2008, 358, 261–273 CrossRef CAS PubMed
- L. Fiebig, J. Soyka, S. Buda, U. Buchholz, M. Dehnert and W. Haas, Euro. Surveill., 2011, 16, 19941 Search PubMed
- Y. Guan, L. L. M. Poon, C. Y. Cheung, T. M. Ellis, W. Lim, A. S. Lipatov, K. H. Chan, K. M. Sturm-Ramirez, C. L. Cheung, Y. H. C. Leung, K. Y. Yuen, R. G. Webster and J. S. M. Peiris, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8156–8161 CrossRef CAS PubMed
- J. Yin, S. Liu and Y. Zhu, Virol. Sin., 2013, 28, 3–15 CrossRef CAS PubMed
- H. Yu, B. J. Cowling, L. Feng, E. H. Y. Lau, Q. Liao, T. K. Tsang, Z. Peng, P. Wu, F. Liu, V. J. Fang, H. Zhang, M. Li, L. Zeng, Z. Xu, Z. Li, H. Luo, Q. Li, Z. Feng, B. Cao, W. Yang, J. T. Wu, Y. Wang and G. M. Leung, Lancet, 2013, 382, 138–145 CrossRef
- D. Xiang, X. Shen, Z. Pu, D. M. Irwin, M. Liao and Y. Shen, J. Infect. Dis., 2018, 217, 1699–1707 CrossRef PubMed
- M. von Itzstein, Nat. Rev. Drug Discovery, 2007, 6, 967–974 CrossRef CAS PubMed
- K. K. W. To, K. H. L. Ng, T.-L. Que, J. M. C. Chan, K.-Y. Tsang, A. K. L. Tsang, H. Chen and K.-Y. Yuen, Emerg. Microbes Infect., 2012, 1, e25 CrossRef PubMed
- G. Neumann, Future Virol., 2015, 10, 971–980 CrossRef CAS PubMed
- P. J. Collins, L. F. Haire, Y. P. Lin, J. Liu, R. J. Russell, P. A. Walker, J. J. Skehel, S. R. Martin, A. J. Hay and S. J. Gamblin, Nature, 2008, 453, 1258–1261 CrossRef CAS PubMed
- H. Leonov, P. Astrahan, M. Krugliak and I. T. Arkin, J. Am. Chem. Soc., 2011, 133, 9903–9911 CrossRef CAS PubMed
- E. A. Govorkova, T. Baranovich, P. Seiler, J. Armstrong, A. Burnham, Y. Guan, M. Peiris, R. J. Webby and R. G. Webster, Antiviral Res., 2013, 98, 297–304 CrossRef CAS PubMed
- A. Jacob, R. Sood, K. V. Chanu, S. Bhatia, R. Khandia, A. K. Pateriya, S. Nagarajan, U. Dimri and D. D. Kulkarni, Microb. Pathog., 2016, 91, 35–40 CrossRef CAS PubMed
- A. M. Joseph, C. Xi, B. J. Colleen, A. Subramaniam, H. Judith, F. S. Donald, W. N. James, N. Diana, X. Xiaolin, J. Fuli, M. Clinton, I. S. Melinda, E. L. White and E. S. William, J. Biomol. Screening, 2010, 16, 73–81 Search PubMed
- S. Liu, R. Li, R. Zhang, C. C. S. Chan, B. Xi, Z. Zhu, J. Yang, V. K. M. Poon, J. Zhou, M. Chen, J. Münch, F. Kirchhoff, S. Pleschka, T. Haarmann, U. Dietrich, C. Pan, L. Du, S. Jiang and B. Zheng, Eur. J. Pharmacol., 2011, 660, 460–467 CrossRef CAS PubMed
- J. Yang, J. X. Yang, F. Zhang, G. Chen, W. Pan, R. Yu, S. Wu and P. Tien, Bioorg. Med. Chem. Lett., 2014, 24, 2680–2684 CrossRef CAS PubMed
- Z. Zhu, Z. Yao, X. Shen, Z. Chen, X. Liu, J. R. Parquette and S. Liu, Eur. J. Med. Chem., 2017, 130, 185–194 CrossRef CAS PubMed
- Y. Yu, J. Zheng, L. Cao, S. Li, X. Li, H.-B. Zhou, X. Liu, S. Wu and C. Dong, RSC Adv., 2017, 7, 9620–9627 RSC
- X. Han, H. Wu, W. Wang, C. Dong, P. Tien, S. Wu and H.-B. Zhou, Org. Biomol. Chem., 2014, 12, 8308–8317 RSC
- X. Han, H. Wu, C. Dong, P. Tien, W. Xie, S. Wu and H.-B. Zhou, RSC Adv., 2015, 5, 10005–10013 RSC
- J. Pan, X. Han, N. Sun, H. Wu, D. Lin, P. Tien, H.-B. Zhou and S. Wu, RSC Adv., 2015, 5, 55100–55108 RSC
- X. Han, N. Sun, H. Wu, D. Guo, P. Tien, C. Dong, S. Wu and H.-B. Zhou, J. Med. Chem., 2016, 59, 2139–2150 CrossRef CAS PubMed
- L. H. Pinto and R. A. Lamb, J. Biol. Chem., 2006, 281, 8997–9000 CrossRef CAS PubMed
- S. Kurtz, G. Luo, K. M. Hahnenberger, C. Brooks, O. Gecha, K. Ingalls, K. Numata and M. Krystal, Antimicrob. Agents Chemother., 1995, 39, 2204–2209 CrossRef CAS PubMed
- A. D. Balgi and M. Roberge, Methods Mol. Biol., 2009, 486, 125–137 CrossRef CAS PubMed
- A. D. Balgi, J. Wang, D. Y. H. Cheng, C. Ma, T. A. Pfeifer, Y. Shimizu, H. J. Anderson, L. H. Pinto, R. A. Lamb, W. F. DeGrado and M. Roberge, PLoS One, 2013, 8, e55271 CrossRef CAS PubMed
- A. S. Arns, A. D. Balgi, Y. Shimizu, T. A. Pfeifer, N. Kumar, F. S. Shidmoossavee, S. Sun, S. S. H. Tai, O. Agafitei, J. B. Jaquith, E. Bourque, M. Niikura and M. Roberge, Eur. J. Med. Chem., 2016, 120, 64–73 CrossRef PubMed
- Y. Wu, B. Canturk, H. Jo, C. Ma, E. Gianti, M. L. Klein, L. H. Pinto, R. A. Lamb, G. Fiorin, J. Wang and W. F. DeGrado, J. Am. Chem. Soc., 2014, 136, 17987–17995 CrossRef CAS PubMed
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of 11–12. See DOI: 10.1039/c8md00474a |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |