
Molecular modelling studies of quinazolinone derivatives as MMP-13 inhibitors by QSAR, molecular docking and molecular dynamics simulations techniques†
Shanshan
Huang
,
Kairui
Feng
and
Yujie
Ren
 *
*
School of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai, China. E-mail: clab@sit.edu.cn
First published on 13th November 2018
Abstract
Matrix metalloproteinase-13 (MMP-13) is an attractive drug target for the treatment of osteoarthritis (OA). In this study, a series of quinazolinone derivatives as MMP-13 inhibitors were firstly systematically studied using QSAR, molecular docking and molecular dynamics (MD) simulation. The reliable CoMFA (q2 = 0.646, r2 = 0.992, Rpred2 = 0.829) and CoMSIA (q2 = 0.704, r2 = 0.992, Rpred2 = 0.839) models were constructed and verified by the Topomer CoMFA model. Results of contour maps indicated that the electrostatic, hydrophobic and H-bond acceptor fields primarily influenced the activity of MMP-13 inhibitors in the models. Several key residues (Ala238, Thr245, Thr247, Met253, Asn215 and Lys140) were identified as important factors to improve the activity and stability of the inhibitor through hydrogen bonding and electrostatic interaction. Based on these results, eight novel quinazolinones (D1–D8) were further designed. Additionally, all designed compounds showed good pharmacokinetic properties by ADMET predictions. Compounds D3 and D8 exhibited excellent predictive activity, and the 10 ns MD simulations analysis revealed that the hydrogen bonding interaction with residues (Ser250 and Gly248) was enhanced, and the small group in R2 and U-shaped conformation was of pivotal importance. These results provided strong guidance for the discovery and design of novel potential MMP-13 inhibitors.
Introduction
Osteoarthritis (OA), known as the “immortal cancer”, has become the leading cause of human disability.1 It is characterized by cartilage degradation and brings patients great pain and heavy financial burden.2 Matrix metalloproteinases (MMPs) are involved in many inflammatory diseases, cancers and other illnesses.3–5 MMP-13 is the most efficient collagenase in the cleavage of type II collagen.6,7 Clinical investigations reveal that patients with articular cartilage destruction have overexpressed MMP-13, suggesting that the increased MMP-13 is the cause of cartilage degradation to a large extent.8 OA is generally caused by the following process.9,10 Under the stimulation of proinflammatory cytokines IL-1β, TNF-α and bFGF, the MMP-13 decomposition is strengthened, and the metabolism of type II collagen is increased. Then the extracellular matrix is destroyed, and the cartilage degeneration is accelerated. Thus, MMP-13 plays an important role in maintaining the integrity of cartilage tissue during the development of OA. It is a compelling drug target for the treatment of OA.However, no MMP-13 inhibitor has been approved by the FDA at present.11 In recent years, several potential inhibitors of MMP-13, such as licofelone,12,13 CDDO,14 cipemastat15 and ilomastat16 have been discovered (Fig. 1). Nevertheless, they are still defective in clinical trial. For example, the interactions of licofelone and drug could increase the risk of toxicity,13 and the CDDO could cause cell mitochondrial metabolic abnormalities to cell apoptosis.17 Cipemastat and ilomastat are at risk of toxicity and dose-limiting efficacy.15,16 Therefore, MMP-13 inhibitors with high efficiency and low toxicity should be extensively explored. Recently, Nara et al.18,19 have synthesized a series of quinazolinone derivatives, and the activity tests showed that these derivatives had potent inhibitory activity against MMP-13. However, the research is currently confined to experimental synthesis and cell-level research. The theoretical study of such inhibitors has not yet been involved.
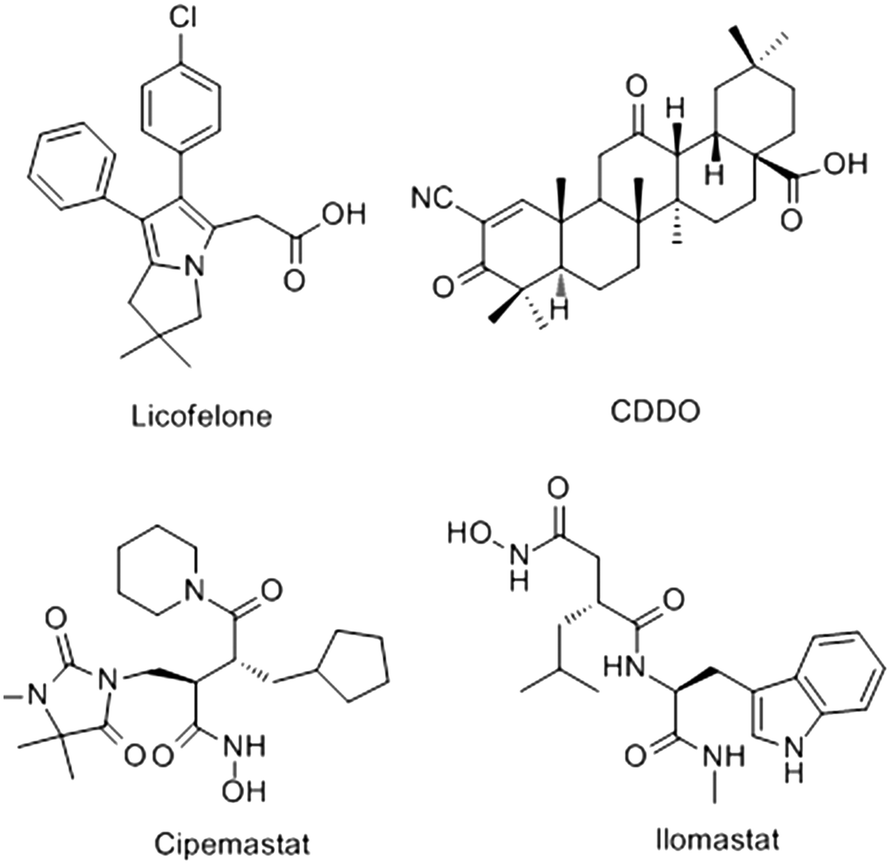 | ||
| Fig. 1 Chemical structures of the representative MMP-13 inhibitors. | ||
The quantitative structure–activity relationship (QSAR) is an accurate and effective method for drug design.20 Recently, the approach has speed up the lead optimization process by studying three dimensional features of chemicals.21 3D-QSAR analysis, including CoMFA,22 CoMSIA23 and Topomer CoMFA,24 occupies a dominant position in drug design.25 Currently, 3D-QSAR is widely used in the research and development of TRPV1 inhibitors,26 PI3K inhibitors27 and mTOR inhibitors28 for its definite and rich information. In this study, the 3D-QSAR models were constructed by a series of quinazolinone MMP-13 inhibitors, and the relationship between the structure and biological activity of the inhibitors was revealed. Then, molecular docking and MD simulation were performed to analyze the binding mode of the ligands and MMP-13 receptors. Finally, the novel quinazolinone MMP-13 inhibitors were designed, and the pharmacokinetic properties of these compounds were predicted by ADMET. This study could avoid the blindness of drug design, and provide theoretical and practical values for the design, discovery and synthesis of new potent MMP-13 inhibitors.
Materials and methods
Data sets and biological activities
The 53 quinazolinone derivatives with inhibitory activities were obtained from the Nara research group.18,19 The bioactivity values of these compounds in vitro were reported as IC50, which were converted to the corresponding pIC50 (pIC50 = −log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50). The whole compounds were randomly divided into two sets by considering both the distribution of bioactivity values and structural diversities. The training set of 45 compounds (85%) was used for the construction of the models, while the remaining 8 compounds (15%) were added to the test set to evaluate the models. The structures and pIC50 values of these compounds were displayed in Table S1.†
IC50). The whole compounds were randomly divided into two sets by considering both the distribution of bioactivity values and structural diversities. The training set of 45 compounds (85%) was used for the construction of the models, while the remaining 8 compounds (15%) were added to the test set to evaluate the models. The structures and pIC50 values of these compounds were displayed in Table S1.†
Molecular optimization and alignment
The molecular optimization and calculations were performed using the SYBYL-X 2.0 package (Tripos, Inc., USA). Energy minimization was employed by Powell method with the Tripos force field29 and Gasteiger–Huckel charge.30 The maximum iterations for minimizations were set to 10000. The energy minimization was finished when energy gradient convergence criterion was up to 0.005 kcal mol−1 Å−1.31 Other parameters were set as the default value.
Molecular alignment was regarded as the engine room of 3D-QSAR model, which directly affected the reliability and predictability of the models. In order to obtain the best possible 3D-QSAR model, three different alignment rules were adopted and the corresponding models were developed. The first alignment rule was ligand-based alignment (Alignment I). The highest activity compound 26 was chosen as the template, and the remaining compounds in training set were automatically aligned based on the common structure. The common substructure was marked in red, as shown in Fig. 2A, and the Alignment I of the training set was displayed in Fig. 3A. The second alignment rule was the docking-based alignment (Alignment II), as shown in Fig. 3B. The conformations obtained by molecular docking were considered to be the best conformations, and the CoMFA/CoMSIA models were directly modeled with it. The last rule was pharmacophore-based alignment (Alignment III) shown in Fig. 2D. The best pharmacophore model and corresponding pharmacophore features were generated using the GALAHAD32 module and the models were applied for the alignment of the compounds.
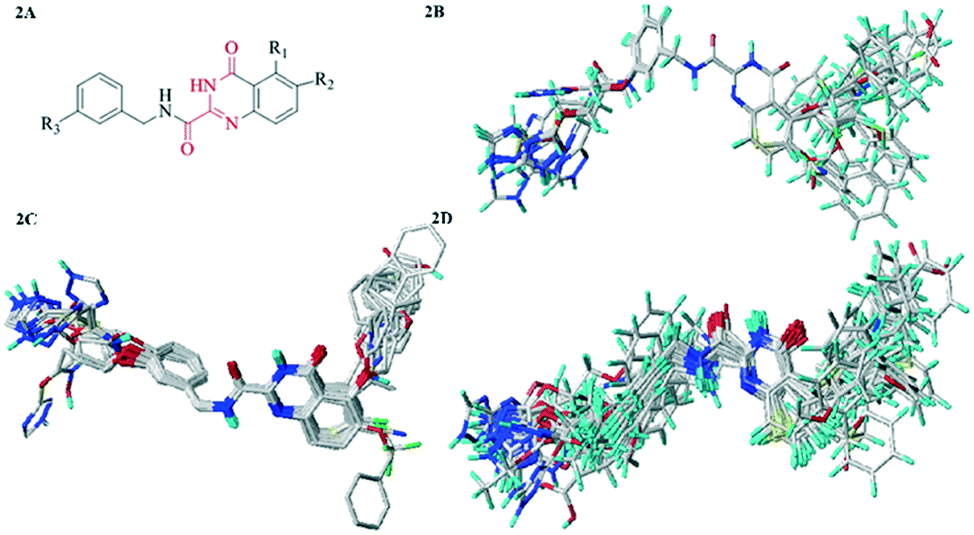 | ||
| Fig. 2 (2A) Common substructure (red). (2B) Ligand-based alignment (Alignment I). (2C) Docking-based alignment (Alignment II). (2D) Pharmacophore-based alignment (Alignment III). | ||
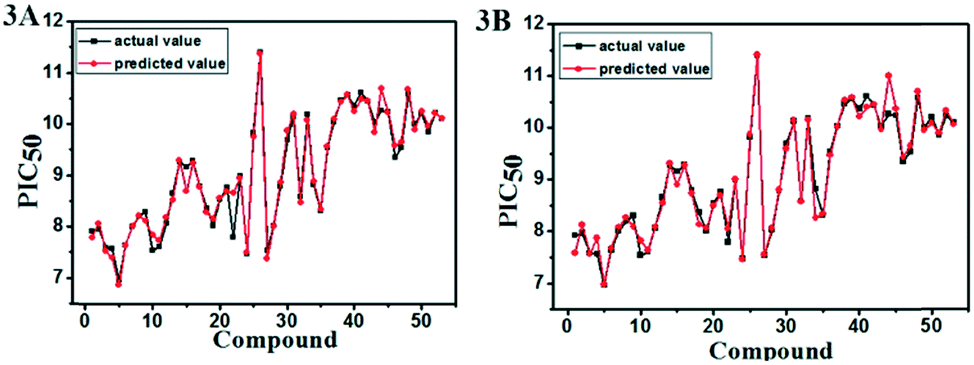 | ||
| Fig. 3 Plots of actual versus predicted pIC50 values for the total set in the CoMFA (A) and CoMSIA (B) models. | ||
A series of important statistical parameters, especially the value of the cross-validated correlation coefficient (q2), the non-cross-validated correlation coefficient (r2), the standard error of estimate (SEE) and the F-statistic values (F) are generally used for internal evaluation of a model. The statistic results of the CoMFA and CoMSIA models on the basis of three different alignment methods were summarized in Table 1. The favorable model should have low value of SEE and high values of q2, r2 and F. As can be seen from Table 1, the statistical results of the CoMFA/CoMSIA model using the Alignment I method, which leading to the lowest SEE and the highest q2, r2 and F, were superior to those of the Alignment II and Alignment III. Hence, the Alignment I method was more conducive to the further 3D-QSAR research.
| PLS statistics | Alignment methods | ||||||
|---|---|---|---|---|---|---|---|
| Alignment I | Alignment II | Alignment III | |||||
| CoMFA | CoMSIA | Topomer CoMFA | CoMFA | CoMSIA | CoMFA | CoMSIA | |
| a Field contribution: COMFA and COMSIA with different field contributions such as S (steric); E (electrostatic); H (hydrophobic); D (H-bond donor); A (H-bond acceptor). | |||||||
| q 2 | 0.646 | 0.704 | 0.592 | 0.286 | 0.288 | 0.395 | 0.553 |
| r 2 | 0.992 | 0.992 | 0.714 | 0.992 | 0.422 | 0.700 | 0.997 |
| ONC | 9 | 9 | 2 | 8 | 1 | 2 | 10 |
| SEE | 0.116 | 0.115 | — | 0.106 | 0.835 | 0.638 | 0.073 |
| F value | 464 | 471 | — | 573 | 31 | 48 | 1053 |
| Field contributiona | |||||||
| S | 0.569 | 0.149 | — | 0.378 | 0.148 | 1 | 0.127 |
| E | 0.431 | 0.293 | — | 0.622 | 0.194 | — | 0.305 |
| H | — | 0.295 | — | — | 0.216 | — | 0.296 |
| D | — | 0.108 | — | — | 0.228 | — | 0.108 |
| A | — | 0.155 | — | — | 0.214 | — | 0.165 |
CoMFA, CoMSIA and Topomer CoMFA descriptors
In SYBYL-X 2.0, the field properties of the models were calculated using a 3D cubic lattices with the spaced grid of 2.0 Å. For the CoMFA method, it was assumed that ligand–receptor binding was non-covalent. A sp3 hybridized carbon atom with +1 charge as the probe was used to detect the size and distribution of the steric and electrostatic fields.22 While in the CoMSIA model, besides the steric and electrostatic field, the hydrophobic, H-bond acceptor and donor fields were introduced.23 Topomer CoMFA was the second-generation CoMFA method. Compared with the traditional CoMFA method, Topomer CoMFA automated the generation of models,24 which enhanced the reproducibility of CoMFA and greatly improved the computational efficiency. The steric and electrostatic filed were calculated using the carbon sp3 probe and partial least-squares (PLS) regression was used for generating Topomer CoMFA model.33 In this study, each molecule was broken into three fragments, including the common skeleton (black), the region 1 (blue) and the region 2 (red), and the segmentation was shown in Fig. S1.†Validation of 3D-QSAR models
The partial least squares (PLS)34,35 was used for cross-validation analysis, which regarded the descriptors of force fields as independent variables and the pIC50 values as the dependent variables for 3D-QSAR modeling analysis. Then the leave-one-out (LOO) method was applied and a series of internal validation parameters were obtained including the q2, r2, ONC, SEE and F values. In general, for internal validation, the robust and reliable ability of the CoMFA/CoMSIA model should meet q2 > 0.5, r2 > 0.9.36 The Topomer CoMFA model satisfies q2 > 0.2 as for internal validation.However, these internal validation parameters appeared to be the necessary but not the sufficient condition for a model with high predictive power. Therefore, a set of external validation methods were adopted for evaluating the predictive ability of 3D-QSAR models. The correlation coefficient Rpred2 and MAE (mean absolute error) were expressed as the following formulas ((1) and (2)). For a high predictive ability of the model, the Rpred2 value should be higher than 0.5,37,38 the MAE value should meet MAE ≤ 0.1 × training set range.39
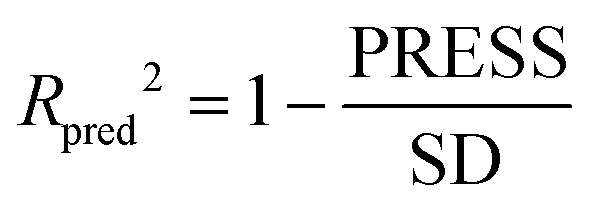 | (1) |
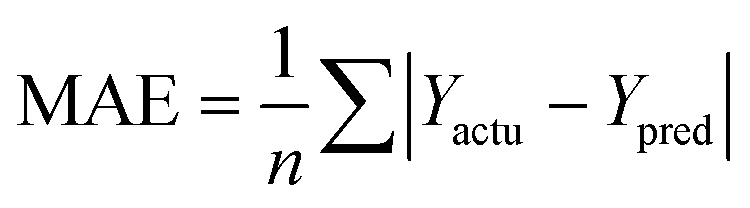 | (2) |
In eqn (1): the PRESS value was the sum of squared deviations between the actual and the predicted pIC50 of the test compounds. The SD meant the sum of squared deviation between pIC50 of test molecules and the average pIC50 of the training molecules. In eqn (2): the Ypred and Yactu were the value of predicted and actual activities.
The fitness and predictability of the models were also checked based on Golbraikh and Tropsha statistical characteristics criteria40,41 for external validation. The three criteria were evaluated the predicted pIC50 values:
| (1) R2 > 0.6; (2) 0.85 < k < 1.15; (3) [(R2 − R02)/R2] < 0.1. |
The value of correlation coefficient R, the slope k and correlation coefficient R02 for regressions were calculated as following formulas ((3)–(5)):
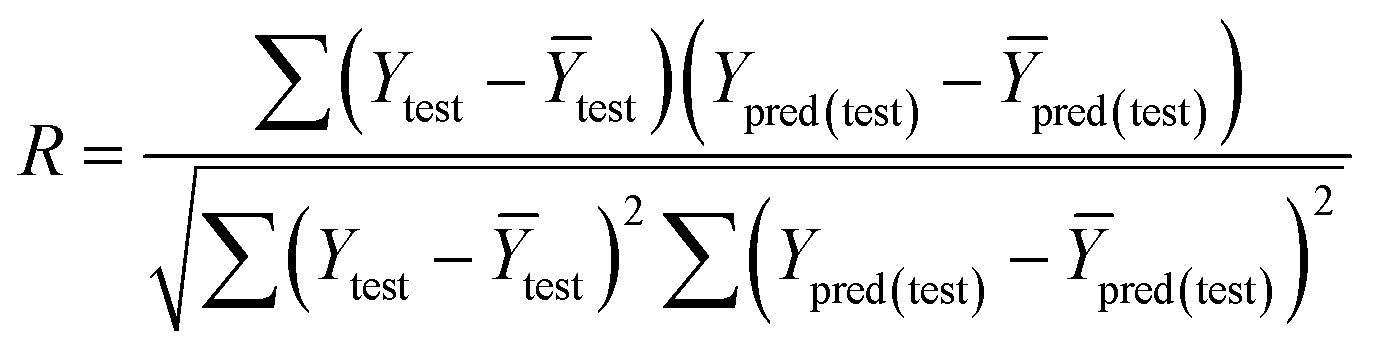 | (3) |
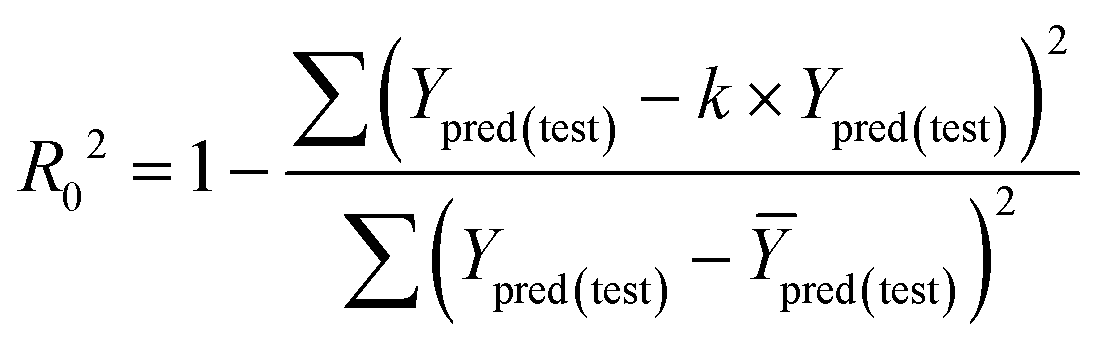 | (4) |
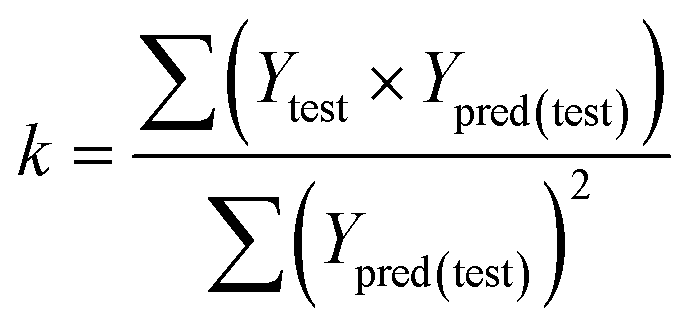 | (5) |
The predictive and reliable power of the 3D-QSAR models were further accessed by calculating additional validation parameters rm2 proposed by Roy.42 The rm2 was described as the following formula (6).
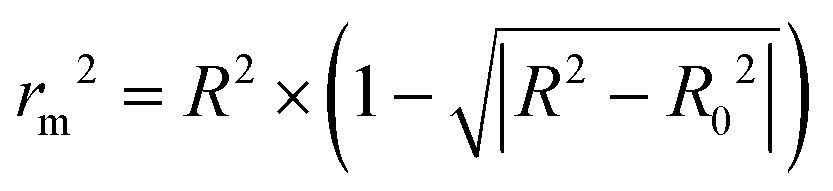 | (6) |
Preparation of the protein and molecular docking
Molecular docking, as one of the most intuitive methods of drug molecular design, has been widely used in the study of intermolecular interactions. Molecular docking was conducted using the Surflex-Dock module in the Sybyl-X 2.0. The crystal structure of the MMP-13 (PDB ID: 3wv1, resolution: 1.98 Å) was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/). Before the docking procedure, the crystal structure was pretreated including the following process: the protein was added hydrogen atoms and AMBERFF99 charges,43 then all water molecules were deleted and the original ligand was extracted. Other parameters were kept default in the settings. Finally, docking pocket was generated. All molecules were automatically docked into the active pocket of MMP-13 protein.Molecular dynamics (MD) simulations
MD simulations were running on the Amber 14.0 package. Before the simulation, the molecular mechanics method was usually adopted to optimize the biological macromolecular system. The general AMBER force field (GAFF) was used for the inhibitors, and the ff99SB force field was employed for the protein.44,45 All the protein–inhibitor complex systems were immersed in a truncated octahedron box of TIP3P water model.46 The systems were neutralized by the addition of Na+ or Cl− counterions. All the MD simulations followed the procedure of minimization, heating, density, equilibration, and production with the Sander module. Energy optimization had the flowing two steps: the first step was restrained the protein and ligand to 100 kcal mol−1 Å−2, and then the whole systems were minimized without restraint condition. The entire systems were minimized by 5000 steps of steepest descent method followed by 5000 steps of conjugated gradient method. After minimization, the systems were gradually heated in the NVT ensemble from 0 to 300 K over 20 ps and maintained at this point for the following MD simulations. The SHAKE algorithm47 was applied to all bonds involving hydrogen atoms, and the Langevin dynamics was used for temperature control. Finally, 10 ns MD simulations with the target temperature of 300 K and the pressure of 1 atm were performed. The coordinate trajectories were recorded per 2 ps throughout the MD runs. All analyses were conducted using Visual Molecular Dynamics software.48 RMSD and RMSF analysis were presented for selected compounds using the Xmgrace program.The binding free energy is an important indicator for reflecting the degree of binding compactness. The positive and negative of the binding free energies reflect the possibility of the ligand binding to the receptor. In this paper, the method of MM/PBSA (Molecular mechanics/Poisson Boltzmann Surface Area)49 and MM/GBSA (Molecular Mechanics/Generalized Born Surface Area)50 were used to calculate the binding free energy. The corresponding formulas were as follows:
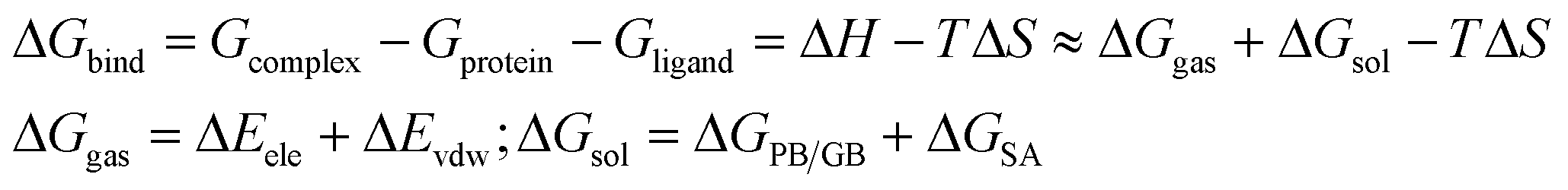
In the formulas, ΔGbind was the final binding free energies, and Gcomplex, Gprotein and Gligand were the free energies of the complex, MMP-13 protein and ligand, respectively. ΔGgas was the gas-phase interaction energy between the protein and the ligand, which consisted of ΔEele (electrostatic energy) and ΔEvdw (van der Waals energy). ΔGsol was the solvation free energy, which was the sum of electrostatic solvation energy ΔGPB/GB (polar contribution) and non-electrostatic solvation energy ΔGSA (nonpolar contribution). In the actual calculation, the conformational entropy (TΔS) was not only time-consuming, but also had little influence on the total free energy, hence, −TΔS was often ignored.51
By using the MM/PBSA module in Amber 14, the contribution of each residue energy is approximately divided into the vacuum interaction energy. The polar solvation energy calculated by GB model and the nonpolar solvation energy calculated by LCPO model, which are decomposed into the contributions of the main chain atoms and side chain atoms of the residues.
All energy components were calculated using 50 snapshots extracted from the last 2 ns trajectory in MD simulations.
ADMET predictions
The ADMET (absorption, distribution, metabolism, excretion, toxicity) predictions are the key factor in the success of drug design.52 During the past year, about 60% of the drugs have failed due to the poor nature of ADME or excessive toxicity.53 The ADMET properties were predicted using ADMET descriptors. The module used six mathematical models to quantify the prediction characteristics by a set of rules of the threshold ADMET characteristics based on the available drug information. The six properties of ADMET, including aqueous solubility, blood brain barrier penetration (BBB), cytochrome P450-2D6 (CYP2D6) enzyme inhibition, hepatotoxicity, human intestinal absorption (HIA) and plasma-protein binding (PPB) were predicted for pharmacokinetic properties. The aqueous solubility which was one of the key ingredients in medicine, had the level 1 (no, very low but possible), 2 (yes, low), 3 (yes, good) and 4 (yes, optimal). The blood brain barrier affected the entry of drug into the brain tissue. The level 0, 1, 2, 3, and 4 of BBB properties denoted very high, high, medium, low and undefined, respectively. The cytochrome CYP2D6 enzyme had an important impact on the process of drug metabolism. The level 0 (ADMET CYP2D6 probability <0.5) meant that compounds were unlikely to inhibit the CYP2D6 enzyme, and the level 1 (ADMET CYP2D6 probability >0.5) meant that compounds were likely to inhibit the enzyme. As an important organ for drug metabolism, the liver was vulnerable to damage. The hepatotoxicity predicted a wide range of potential human hepatotoxicity of structurally diverse compounds. The HIA mainly depended on intestinal enzymes and intestinal mucosal cells on the metabolism and barrier function. The level 0, 1, 2, 3 and 4 of HIA properties meant good absorption, moderate absorption, low absorption and very low absorption, respectively. The PPB rate referred to the ratio of drugs to plasma protein binding in plasma. The level 0, 1 and 2 of PPB properties represented binding < 90%, 90% < binding < 95% and binding > 95%, respectively.Although the compound 26 showed high bioactivity, it also exhibited poor oral bioavailability in all tested species. Compound 26 had a carboxyl function with relatively high acidity, which allowed the formation of salts to improve solubility. Hence, the monosodium salt of compound 26 was selected for the favorable stability, nonhygroscopic property, high degree of crystallinity and good reproducibility.18 The new designed compounds (D1–D8) were also formed the corresponding monosodium salt (D1-Na–D8-Na) for further prediction of ADMET characteristics.
Results and discussion
Analysis of CoMFA, CoMSIA and Topomer CoMFA
The statistical results for the final CoMFA, CoMSIA and Topomer CoMFA models were summarized in Table 1. In the best CoMFA model, the PLS regression analysis obtained the q2 of 0.646 and the ONC of 9. Then, the non-cross validation gave the r2 of 0.992 with the SEE of 0.116 and the F-statistic value of 464.725. The contributions of steric and electrostatic fields to the CoMFA model were 43.1% and 56.9%, respectively. In the CoMSIA model, the PLS regression analysis showed the q2 of 0.704, the ONC of 9, the r2 of 0.992, the SEE of 0.115 and the F value of 471.823. The contributions of steric, electrostatic, hydrophobic, H-bond acceptor and H-bond donor fields were 14.9%, 29.3%, 29.5%, 15.5% and 10.8%, respectively. It can be found that the electrostatic, hydrophobic and H-bond acceptor field made major contributions among the five fields. The data sets used in the Topomer CoMFA model were the same as that of CoMFA, and its statistical parameters were shown in Table 1. The Topomer CoMFA model gave the q2 of 0.592 with the ONC of 2 and the r2 of 0.714. In short, all these results were in an acceptable range, suggesting the reliability and robustness of the 3D-QSAR model.In addition, the eight compounds with different structures and large span of activities were selected as test sets to verify the predictive capability of the model. As shown in Table 2, the results of the Rpred2 value obtained by the CoMFA and CoMSIA models were 0.829 and 0.839, respectively. The values of MAEtraining (0.078 for CoMFA, 0.070 for CoMSIA) and MAEtest (0.293 for CoMFA, 0.312 for CoMSIA) were both less than the 0.1× the training set range (0.394). The correlation coefficient R2 values for the CoMFA and CoMSIA model were 0.922 and 0.933, respectively. The R02 values (0.990 for CoMFA, 0.998 for CoMSIA) were close to R2. The values of rm2 (0.531 for CoMFA, 0.557 for CoMSIA) were less than 0.5. The values of k (0.989 for CoMFA, 0.996 for CoMSIA) were close to 1. These statistical results of external validation were further verified that the CoMFA and CoMSIA models had high predictability. Moreover, the reliability and predictability of the CoMSIA model were superior to CoMFA, hence the CoMSIA model got more attention in the following.
| Parameter | CoMFA | CoMSIA |
|---|---|---|
| R pred 2 | 0.829 | 0.839 |
| PRESStest | 1.221 | 1.151 |
| PRESStraining | 0.466 | 0.473 |
| RMSEP | 0.391 | 0.379 |
| RMSEE | 0.168 | 0.164 |
| R | 0.922 | 0.932 |
| R 2 | 0.850 | 0.870 |
| k | 0.989 | 0.996 |
| R 0 2 | 0.990 | 0.999 |
| r m 2 | 0.531 | 0.558 |
| (R2 − R02)/R2 | −0.165 | −0.148 |
| MAEtest | 0.293 | 0.312 |
| MAEtraining | 0.078 | 0.070 |
Analysis of CoMFA, CoMSIA and Topomer CoMFA contour maps
The Fig. 4A was the contour map of the steric field in CoMFA model. The green contours indicated that the bulky groups were conducive to activity, whereas yellow ones meant the bulky groups would result in the decreased activity. The R1 linker chains and R2 positions of compound 26 were surrounded by a few pieces yellow contour, indicating that the bulky groups in this position had the negative effects on the biological activity. These were consistent with the experimental data. For instance, compounds 19 and 20 with small groups in R1 linker chains exhibited higher potency than compound 22. The similar phenomenon could also be observed in R2 substituent. The order of inhibitory activity was: 26 (R1 = –O(CH2)2Ph) > 16 (R1 = –O(CH2)3Ph) > 17 (R1 = –O(CH2)2OCH2Ph); 8 (R2 = CN) > 7 (R2 = Ph) > 5 (R2 = OBn). The ortho-position of benzene ring in R1 was neared by the green contour, which meant the bulky groups were beneficial to the activity here. For example, the activities of compounds 38 (R2 = –S) and 41 (R2 = –F) were higher than that of compound 40 (R2 = –H). Therefore, in the future design, small groups should be introduced to the link chain of R1 and R2 position, and large groups should bring into the ortho-position of benzene ring in R1 as far as possible.
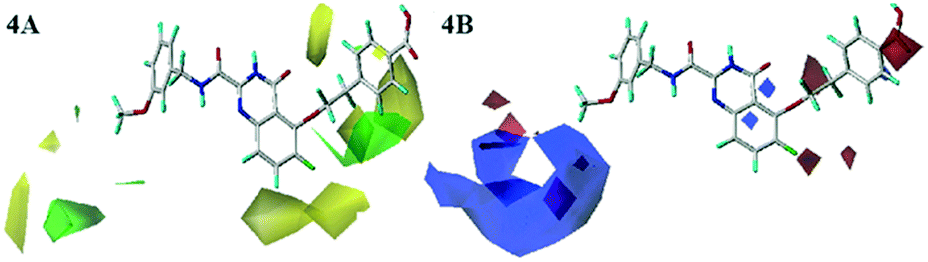 | ||
| Fig. 4 Steric (4A) and electrostatic (4B) contour maps of the CoMFA model. | ||
In the electrostatic contour map of the CoMFA model, the electrostatic favorable and unfavorable regions were represented by blue and red contours, respectively. As shown in Fig. 4B, the R3 position far from the skeleton was wrapped by a large blue contour. Therefore, the activity of compound 30 was larger than that of compounds 27 and 28. There were two small pieces of red contours shown in the link chain and the para-position of the phenyl ring, respectively. It was suggested that the positions were suitable for the electronegative groups. This observation was consistent with the results, for example, the order of inhibitory activities was as follows: 21 (–O) > 20 (–C), 26 (–COOH) > 25 (F). So, the R3 position should introduce the electropositive groups, and the link chain and benzene ring of R1 should introduce the electronegative charged groups, which would contribute to the increment of inhibitor activity.
The steric and electrostatic contour maps of CoMSIA model were shown in Fig. 5A and B, whose information were basically similar to those of CoMFA model. Hence, they were not discussed here. In the hydrophobic field of CoMSIA model, hydrophobic favorable and unfavorable areas were exhibited by yellow and white contours, respectively. As shown in Fig. 5C, the link chain and the position above the benzene ring in R1 were neared by a middle yellow contour, which indicated that the hydrophobic groups in the positions were favorable. This could be confirmed that the activities of compounds 20, 18 and 19 were gradually decreased due to the weakening of the hydrophobicity in this position. There were two white regions in the R1 and R3 groups indicated that these positions were suitable for hydrophilic groups. The activities sequence of the compounds were 29 > 28 > 27, which were mainly ascribed to the introduction of hydrophilic groups at the R3 position. Therefore, it is necessary to introduce the hydrophobic groups in the linker chain of R1 and hydrophilic groups in benzene ring of R1 and R3 position, when considering the design of new inhibitors.
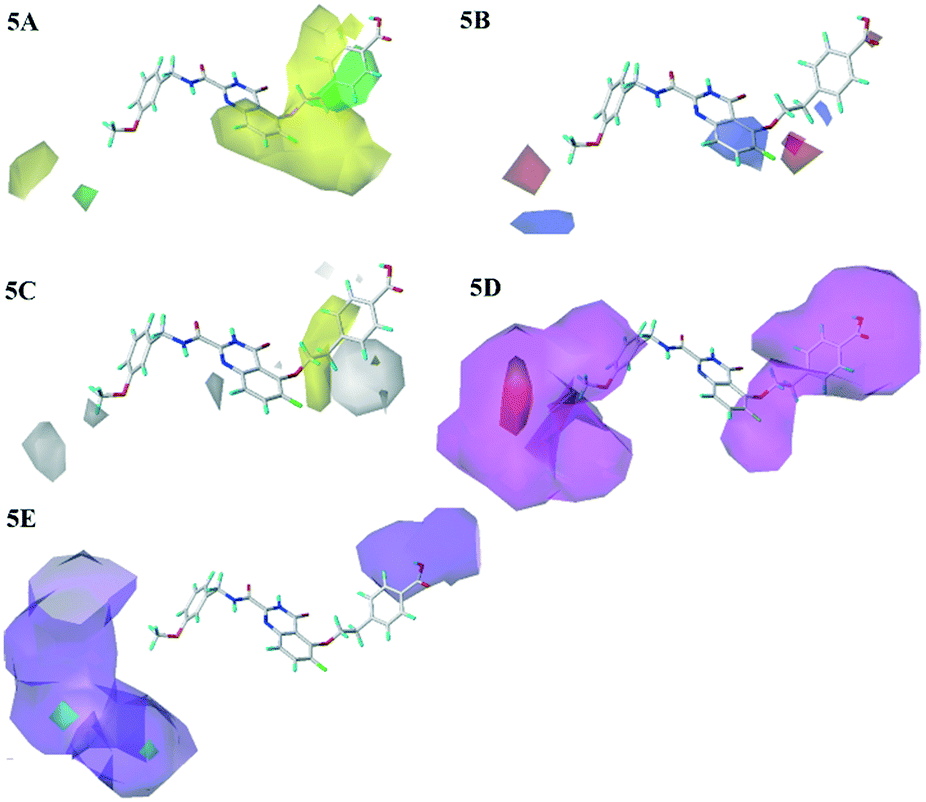 | ||
| Fig. 5 Steric (5A), electrostatic (5B), hydrophobic (5C), H-bond acceptor (5D) and H-bond donor (5E) contour maps of the CoMSIA model. | ||
The H-bond acceptor field of the CoMSIA model was described in Fig. 5D. The H-bond acceptor field was indicated by magenta and red contours. The R1, R2 and R3 regions of the 26 were wrapped in magenta contours, only a small piece of red contour appeared in the R3 position, which suggested that the H-bond acceptor groups in these regions would make for the bioactivities. The H-bond donor field of the CoMSIA model was shown in Fig. 5E, where the cyan and purple contours represented the favorable and unfavorable the H-bond donor areas, respectively. The position of R1 and R3 was covered by large purple contours, and the cyan contours were hardly observed. It showed that the H-donor groups at these positions were adverse to the activity of the compounds. Additionally, the magenta contours of the H-bond acceptor field almost coincide with the purple contours of the H-bond donor field, which indicated the consistency of the established models. These phenomena could be explained from the following examples: 14 (R1 = OPh) > 13 (R1 = Ph); 9 (R1 = F) > 1 (R1 = H), 12 (R1 = CN) > 10 (R1 = Me). Therefore, the introduction of H-bond acceptor groups should be considered in future designs.
With respect to the structurally similar compounds 26 and 25, a large magenta contour of the H-bond acceptor field appeared in the R1 region. Meanwhile, the position was closed to a red contour of the electrostatic field, which indicated that the H-bond acceptor and electronegative groups were beneficial to the biological activities. The carboxyl group of 26 and the fluorine group of 25 were H-bond acceptor, and the electronegativity of carboxyl was larger than that of fluorine. Therefore taking the influence of two fields into considering, the activity of compound 26 was higher than 25.
Molecular docking
Molecular docking determines the binding mode of ligand and receptor, thus provides theoretical basis for the design of new compounds. Before molecular docking, the docking accuracy was verified by re-docking the original ligands. As illustrated in the Fig. 6, the re-docking results showed that the re-docked ligand (blue) and the original ligand (red) almost overlapped in the U-shaped configurations, indicating that the position in the receptor cavity was correct. What's more, the quinazolinone ring was deeply embedded in the S1' pocket of the MMP-13 receptor, and thus produced hydrogen bonds between the amino acids (Ala238, Thr245 and Thr247). The fluorine group in the R2 position was hydrogen bonded to the backbone amide nitrogen of Met253, the carboxylic group in the terminal phenyl ring formed the hydrogen bond with the ε-amino group of Lys140. These results were consistent with literature provided by the Nara research group.18 Overall, the Surflex-Dock program can successfully reproduce the original docking conformation of MMP-13 protein.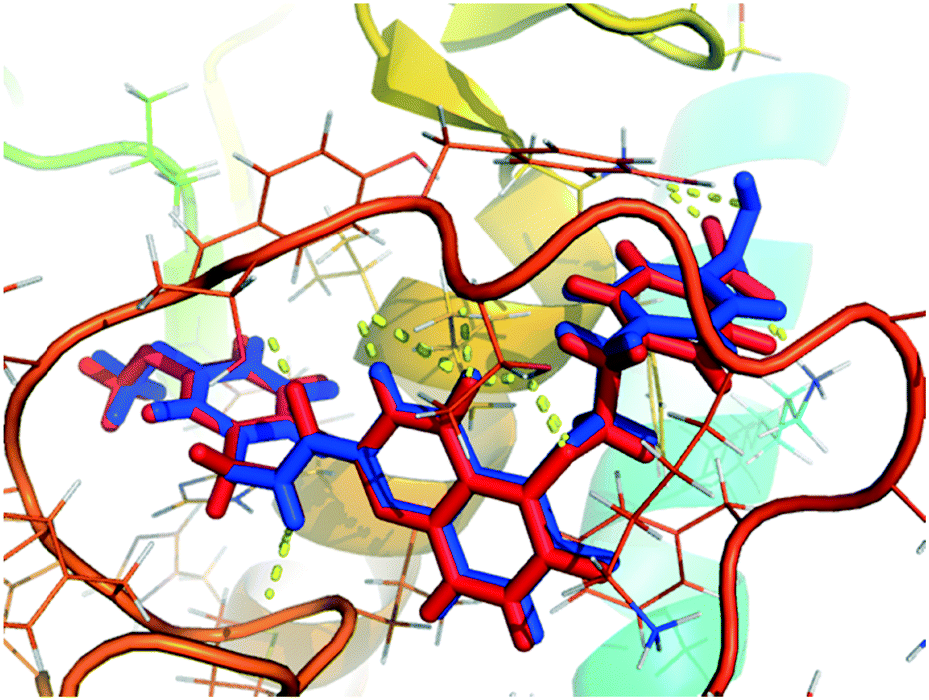 | ||
| Fig. 6 The re-docking result of the re-docked ligand (blue) overlapping with original ligand (red) in the MMP-13 protein. | ||
Subsequently, all these compounds in this study were docked into MMP-13 receptor to analyze the bonding patterns. As shown in Fig. 7, all the molecules maintained the similar U-shaped configurations in the receptor cavity. What's more, most compounds were buried deeply in the S1' pockets, and then formed hydrogen bonds with the key residues Ala238, Thr245 and Thr247. Meanwhile, the most potent compound 26 was used as the template to explore the docking interactions. As showed in Fig. 8, the compound 26 was observed to tightly bind to the active site of MMP-13 in the U-shaped conformation via nine hydrogen bonds interaction between the residues Ala238 (2.12 Å, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HN–), Thr245 (2.10 Å, –H⋯O–), Thr245 (1.92 Å, –CO⋯HN–), Thr247 (2.16 Å, –H⋯O–), Thr247 (1.92 Å, –H⋯O–), Thr247 (2.21 Å, –H⋯O–), Met253 (2.22 Å, –H⋯F–), Asn215 (2.09 Å, –H⋯O–) and Lys140 (2.07 Å, –H⋯O–). Obviously, the compound 26 functioned as the H-bond acceptor and formed hydrogen bonds with residues Thr245, Thr247, Met253, Asn215 and Lys140. It was in agreement with the large magenta contours of H-acceptor field of the CoMSIA model. In addition, the R1 and R3 positions had electrostatic effects with residues Asn215, Lys249, Lys140, Ser250, Pro242, Phe241 and Ile243, which were likely to be closely related to the red blocks of the CoMFA/CoMSIA model. The link chain in R1 position was inlaid with hydrophobic pocket consisting of residues Phe252, Phe247 and Met253, which corresponded to the yellow blocks in hydrophobic filed of the CoMSIA model. The benzene ring of the R1 substituent extended into the hydrophilic pocket composed of residues Asn215, Lys249, Lys140 and Gly248, which was consistent with the white block in the CoMSIA model. And van der Waals forces were generated at this position with residues Phe252, Phe217 and Gly248 to maintain molecular stability. These results suggested that the molecular docking was agreement with the generated CoMFA/CoMSIA model.
O⋯HN–), Thr245 (2.10 Å, –H⋯O–), Thr245 (1.92 Å, –CO⋯HN–), Thr247 (2.16 Å, –H⋯O–), Thr247 (1.92 Å, –H⋯O–), Thr247 (2.21 Å, –H⋯O–), Met253 (2.22 Å, –H⋯F–), Asn215 (2.09 Å, –H⋯O–) and Lys140 (2.07 Å, –H⋯O–). Obviously, the compound 26 functioned as the H-bond acceptor and formed hydrogen bonds with residues Thr245, Thr247, Met253, Asn215 and Lys140. It was in agreement with the large magenta contours of H-acceptor field of the CoMSIA model. In addition, the R1 and R3 positions had electrostatic effects with residues Asn215, Lys249, Lys140, Ser250, Pro242, Phe241 and Ile243, which were likely to be closely related to the red blocks of the CoMFA/CoMSIA model. The link chain in R1 position was inlaid with hydrophobic pocket consisting of residues Phe252, Phe247 and Met253, which corresponded to the yellow blocks in hydrophobic filed of the CoMSIA model. The benzene ring of the R1 substituent extended into the hydrophilic pocket composed of residues Asn215, Lys249, Lys140 and Gly248, which was consistent with the white block in the CoMSIA model. And van der Waals forces were generated at this position with residues Phe252, Phe217 and Gly248 to maintain molecular stability. These results suggested that the molecular docking was agreement with the generated CoMFA/CoMSIA model.
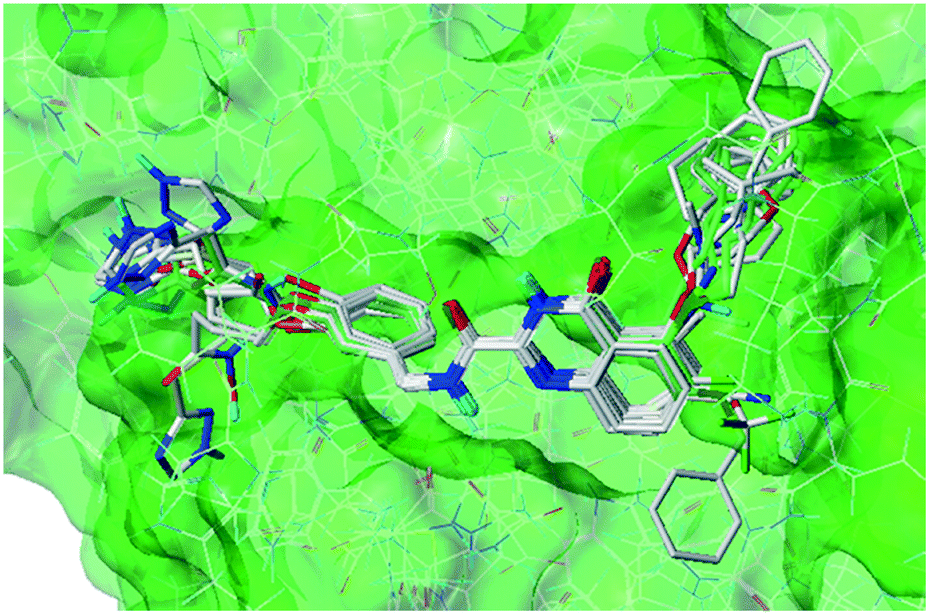 | ||
| Fig. 7 Docking results of all the compounds in MMP-13 protein. | ||
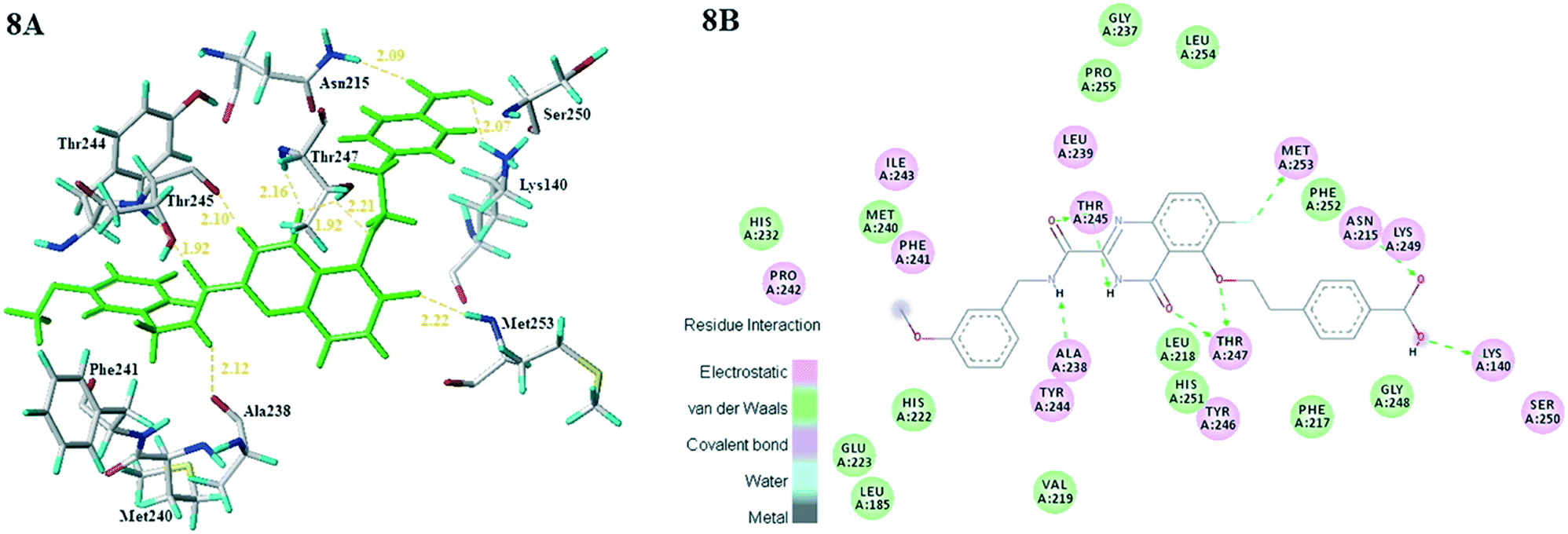 | ||
| Fig. 8 (8A) Docking result of compound 26 (green) in the active site of MMP-13. (8B) 2D interaction map of the docking results of the compound 26 in the active site of MMP-13. | ||
Further analysis of compounds 8 and 10, it was found that they had the similar U-shaped conformation. The compound 10 in the active pocket of MMP-13 moved downward due to the shifting of quinazolinone, resulting in no electrostatic interactions with the residues Met253 and Lys249. Meanwhile, the CN group of the compound 8 at R2 position formed hydrogen bonds with residue Met253 and electrostatic hydrogen bonds with residue Met253 and electrostatic interaction with Lys249. These were likely to explain the better inhibitory activity of compound 8. Therefore, the electrostatic and H-bonding interaction between compounds and residues (Met253 and Lys249) made a difference for the bioactivity.
Moreover, the compounds 26, 25 and 15 with similar structure and different activity were compared for the deep analysis. In R2 position, compounds 26 and 25 formed hydrogen bonds with residue Met253, and electrostatic effects with residues Met253 and Lys249. The bioactivities of 26 and 25 were greater than 15, which further explained the importance of residues Met253 and Lys249. Compared with compounds 26 and 25, compound 15 had no hydrogen bonding or electrostatic interactions with residues Lys140 and Asn215. In combination with the contour maps, the residues Lys140 and Asn215 were adjacent to the red contours of the electrostatic field. The electronegativity of the carboxyl group in 26 was greater than that of F in 25, hence the activity of 26 was greater than 25. It was suggested that the residues Lys140 and Asn215 had an important impact on the bioactivities. At the same time, it was also verified that the result of molecular docking was consistent with that of 3D-QSAR.
Finally, the highest activity of 26 and the lowest activity of 5 were compared. It was interesting to find that they were similar in the substituents of R2. However the positions of their substituents were different. Although the two molecular skeletons were superimposed into the active site, compound 26 with U-shaped conformation was perfectly docked into the active pocket and 5 with the different S-shaped conformation gone beyond the pocket. In combination with the contour maps, the R2 position of compound 5 was wrapped in a large yellow contour of the steric field, which suggested that the bulk groups were unfavorable to bioactivity. So, large groups should not be introduced at this location. The quinazolinone skeletons of both compounds 26 and 5 could produce H-bonding and electrostatic interactions with the residues Ala238, Thr245 and Thr247, which played an important role in the stability of the compounds in the receptor. Besides, the lowest activity of 5 had neither electrostatic or H-bonds interactions with the residues of Asn215 and Lys140.
Therefore, in the future molecular design, it could be concluded that enhancing H-bond interaction with Ala238, Thr245 and Thr247, and electrostatic interaction with Met253, Asn215 and Lys140 were significant to increase the bioactivity and stability of the inhibitor. Additionally, it is necessary to ensure that the compounds are inserted into MMP-13 protein in U-shaped conformation. And the group should be small in R2, and it should be guaranteed to form electrostatic interaction with the residues Met253, Lys249. At the same time, the electronegativity group should be introduced into the R1 benzene ring and electrostatic interactions are formed with the residue Asn215 and Lys140. In short, these results were mutually validated with 3D-QSAR results.
Design of new MMP-13 inhibitors
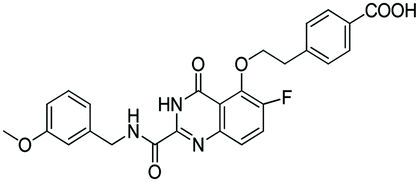
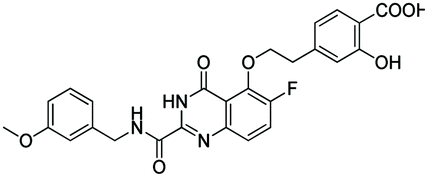
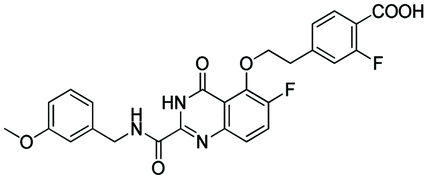
According to the 3D-QSAR model and molecular docking results, some meaningful SAR information about quinazolinone scaffolds as potent MMP-13 inhibitors was obtained, as shown in Fig. 9. In general, the site of the compound was affected by several fields rather than a single field, so the field effects should be taken into account when designing the compound. The inhibitor 26 exhibited the highest activity and was taken as the template to design new compounds. In the benzene ring of R1 position, there were the white block of hydrophilic field and green blocks of the steric field, suggesting hydrophilic and large substituents favored the biological activity. Hence the OH was introduced to this position and thus the designed compounds D1 were designed. In addition, this position was also wrapped in large magenta contours of H-bond acceptor field, indicating that the H-bond receptor groups in this position were beneficial to the activity. Therefore, the F was introduced, thus compounds D2, D3 and D4 were designed. Meanwhile, the benzene ring was replaced by pyridine ring to weaken the electron density and enhance the H-bond interaction with MMP-13 receptor. Then the compounds D5–D7 were designed. In the link chain of R1 position, there was wrapped by a red contour of electrostatic field and large magenta contours of H-bond acceptor field, indicating that the electronegative and H-bond receptor groups were favored the biological activity, so the electronegative O atom was introduced to this position. And the CH3 and Cl groups were introduced to the benzene ring as a result of green block of the steric field. Then the compound D8 was designed. The activities of the new eight compounds were predicted by CoMFA and CoMSIA models, and the structure and prediction of designed compounds were shown in Table 3. The results showed that the novel designed compounds had higher activity than the template 26, which further verified the reliable and predictive capability of the 3D-QASR model.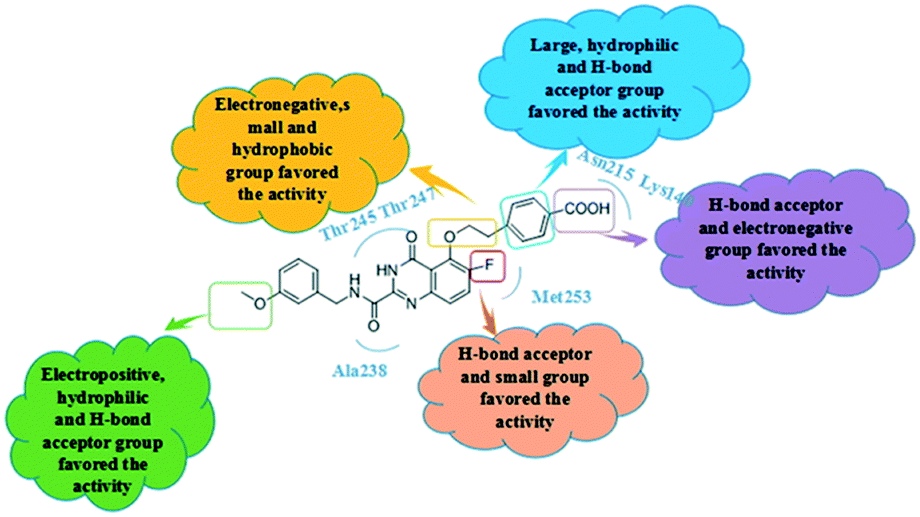 | ||
| Fig. 9 Summary of structure–activity relationship derived from 3D-QSAR studies. | ||
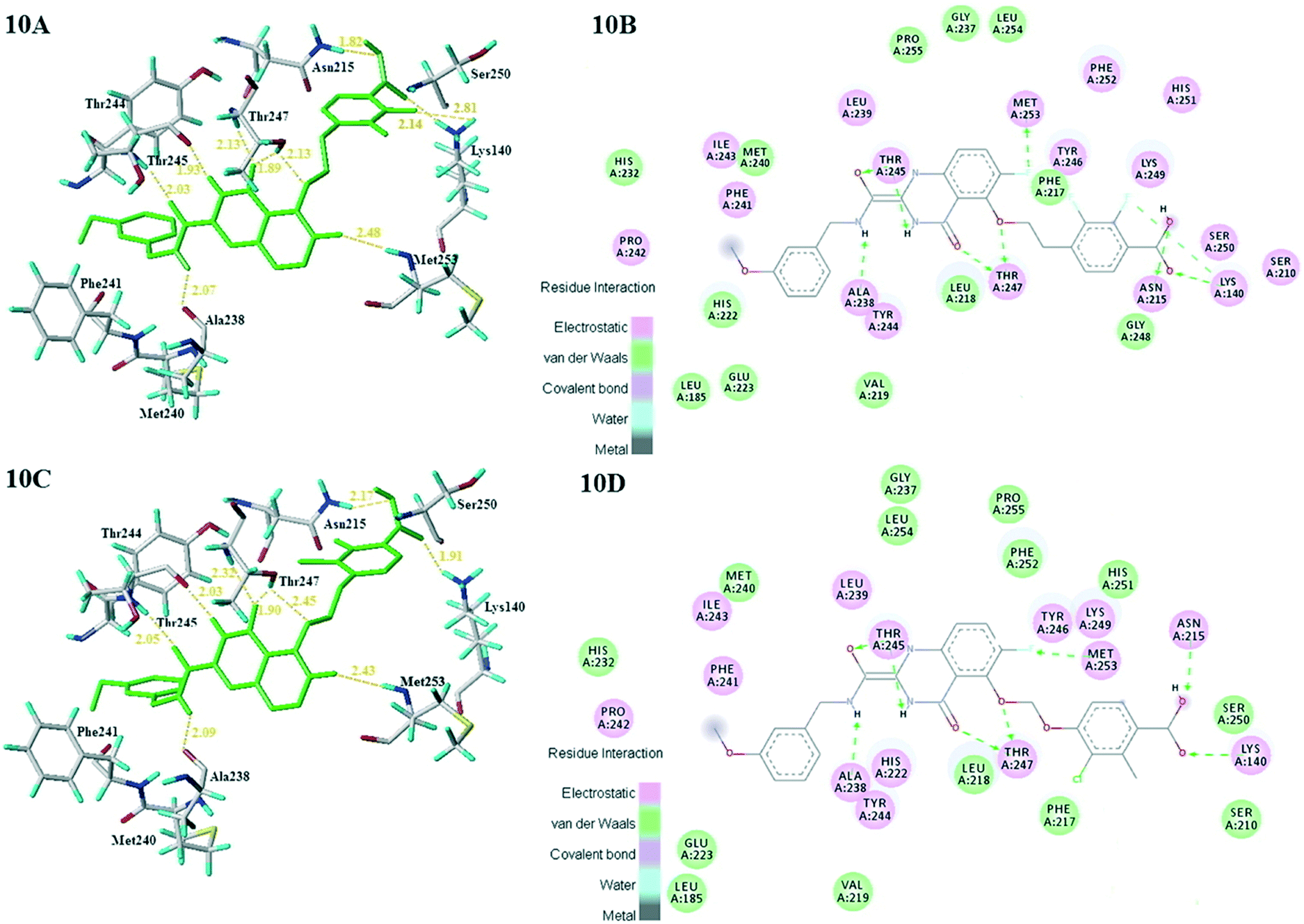 | ||
| Fig. 10 Docking results of newly designed compound D3 (10A) and D8 (10C) in the active site of MMP-13. 2D interaction map of the docking results of newly designed compound D3 (10B) and D8 (10D) in the active site of MMP-13. | ||
| No. | Structures | Predicted activities (pIC50) | |
|---|---|---|---|
| CoMFA | CoMSIA | ||
| 26 |
|
11.377 | 11.408 |
| D1 |
|
11.414 | 11.510 |
| D2 |
|
11.423 | 11.511 |
| D3 |
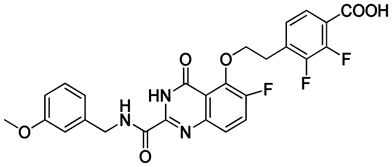
|
11.410 | 11.682 |
| D4 |
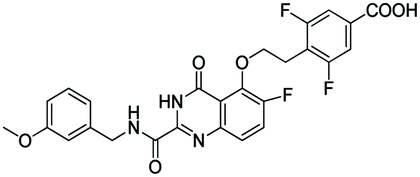
|
11.426 | 11.611 |
| D5 |
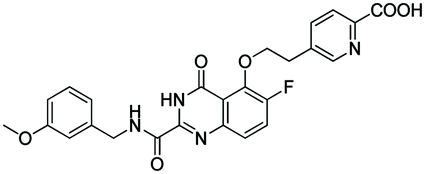
|
11.509 | 11.418 |
| D6 |
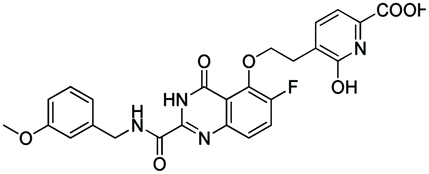
|
11.588 | 11.527 |
| D7 |
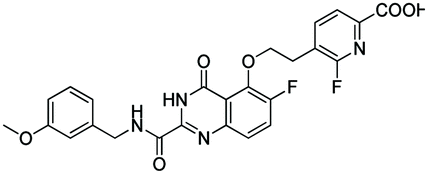
|
11.671 | 11.650 |
| D8 |
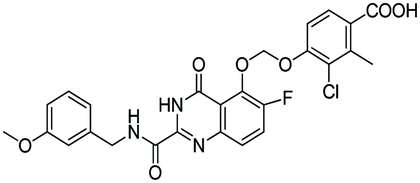
|
11.502 | 11.778 |
What's more, compounds D1–D8 were docked into the active site of MMP-13 receptor to explore their binding modes. The 3D binding patterns of compounds D1–D8 were showed in ESI.† The docking results indicated that their binding modes were similar to that of the co-crystallized ligands. The most potent compounds D3 and D8 that predicted by CoMSIA model were choose for the in-depth analysis. The compound D3 formed ten hydrogen bonds with the key residues, and their H-bond distances were observed to be 2.07 Å (–HN⋯CO–, Ala238), 2.03 Å (–H⋯O–, Thr245), 1.93 Å (–CO⋯HN–, Thr245), 2.13 Å (–H⋯O–, Thr247), 1.89 Å (–H⋯O–, Thr247), 2.13 Å (–H⋯O–, Thr247), 2.48 Å (–H⋯F–, Met253), 1.82 Å (–H⋯O–, Asn215), 2.14 Å (–H⋯O–, Lys140) and 2.81 Å (–F⋯H–, Lys140), respectively. The additional H-bond interaction of the newly introduced fluorine group with the residue Lys140 was responsible for the high affinity for MMP-13. The relative short distance of H-bond interactions between compound D3 and the residues Ala238, Thr245, Thr247 and Asn215, had important effects on stability of compounds in the active pocket. The quinazolinone ring produced van der Waals interactions with the residues Phe217. The R1 and the R3 positions formed electrostatic interactions with the residues Ser210 and His251, and the hydrophobic interaction was observed between benzene ring of R1 with the residue Phe217. These interactions further enhanced the stability of the ligand in the receptor. The compound D8 was immobilized in the pocket via nine hydrogen bonds with the key residues, and their H-bond distances were 2.09 Å (–HN⋯CO–, Ala238), 2.05 Å (–H⋯O–, Thr245), 2.03 Å (–CO⋯HN–, Thr245), 2.32 Å (–H⋯O–, Thr247), 1.90 Å (–H⋯O–, Thr247), 2.45 Å (–H⋯O–, Thr247), 2.43 Å (–H⋯F–, Met253), 2.17 Å (–H⋯O–, Asn215) and 1.91 Å (–H⋯O–, Lys140), respectively. The H-bond distances between compound D8 and residues (Ala238, Thr245 and Lys140) were shortened as compared to the template compound 26. The interaction of compound D8 with the surrounding residues was almost identical with that of D3. Simultaneously, the docking results could also prove the predictive power of the 3D-QSAR model. For example, the compound D3 with the introduced fluorine group formed a new hydrogen bond with residue Lys140, which was consistent with the magenta patches in the H-bond acceptor field of the CoMSIA model. In addition, the introduced fluorine group rendered the compound D3 form hydrophilic interaction with the residues Tyr246 and Ser250, which corresponded to the hydrophilic white patches of the CoMSIA model. The R3 position produced electrostatic interactions with the residues Pro242, Phe241 and Ile243, which was likely related to the blue patches of the CoMFA model in this position. Together, these results demonstrated the consistency of the molecular docking and 3D-QSAR models, and also proved the rationality of the designed compounds, indicating that the 3D-QSAR model could be used to guide the rational design of new compounds (Fig. 10).
Analysis of MD simulation
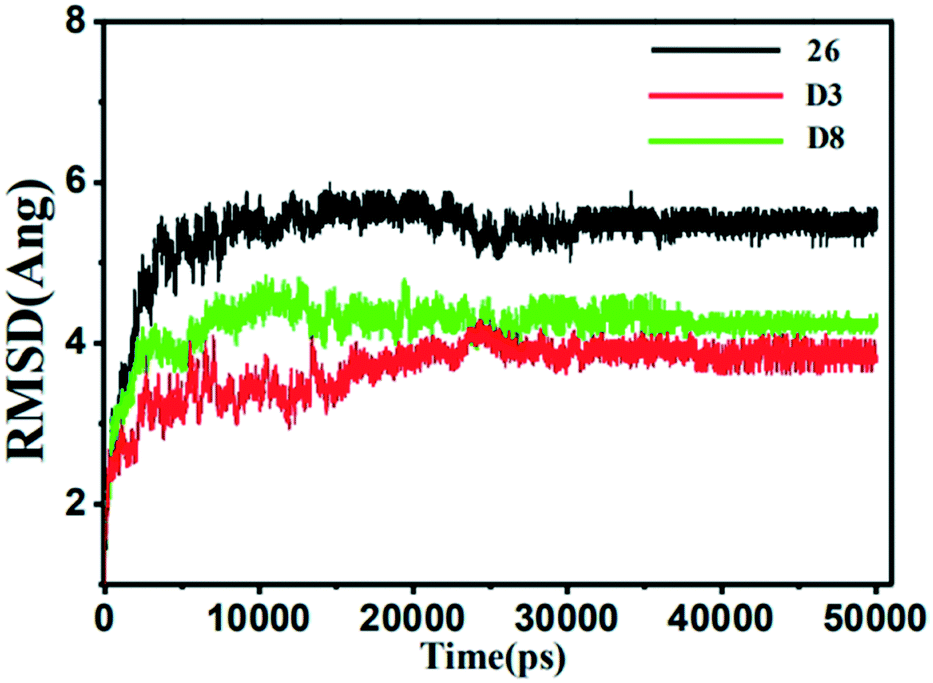 | ||
| Fig. 11 The root-mean-square deviation (RMSD) of the compound 26 (black), D3 (red) and D8 (green) in all three MMP-13 complexes obtained from 10 ns MD simulation. | ||
The superposition of the final structure of complex 26-MMP-13 extracted from MD simulation (blue) and the original docked structure (red) was showed in Fig. 12. The final simulation structure and the initial docked structure were in the same binding pocket excepting for a slight drift. Moreover, it can be found that hydrogen bonds with residue Ala238, Thr245, Thr247, Gly248 and Ser250, electrostatic interaction with residue Met253, Lys140, Ile243, Ser250, Asn215, Lys249, Ala238, Phe241, had not changed in the binding sites. The complex 26-MMP-13 preserved the similar initial docking conformation, which illustrated the reliability of docking pocket. However, compared with the initial docking structure, the two hydrogen bonds between compound 26 and Thr247 were decreased, and two new hydrogen bonds were formed between 26 and residues Gly248 and Ser250. The molecules 26 showed slight up-shifting motion after the MD simulation, resulting in the distance between 26 and residues Met253 and Thr247 increased. In addition, the electrostatic field force and van der Waals interaction between the molecules and the residues (such as His261, His260, Ser211 and Leu218) were increased, which were beneficial to the stability of the molecule in the MMP-13 receptor.
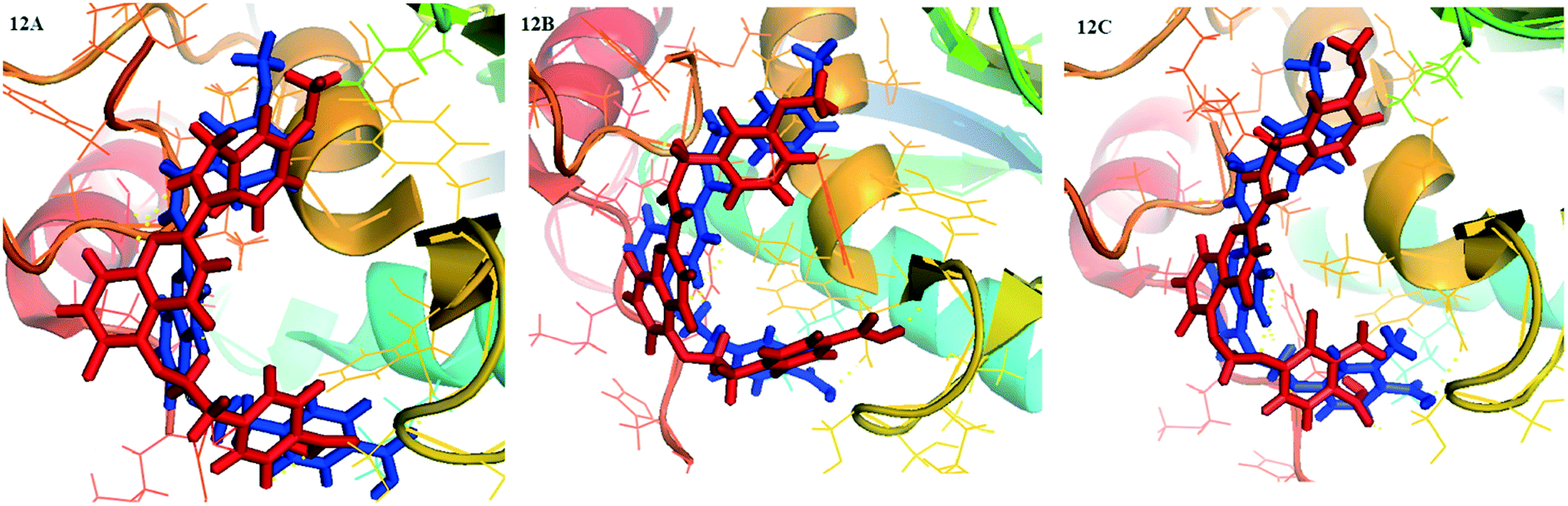 | ||
| Fig. 12 Structural comparison between initial (red) and representative snapshots from MD simulation (blue) of 26-MMP-13 (12A), D3-MMP-13 (12B) and D8-MMP-13 (12C). | ||
The MD-simulated 26, D3 and D8 still preserved H-bonding interactions with the residues Ala238, Thr245 and Thr247, and maintained the docking U-shaped conformation in the process of MD simulation. As shown in Fig. 13, the quinazolinone ring of three compounds produced electrostatic interactions with the residues Gly248, Tyr245, Tyr246 and Tyr247 in order to keep the stability of compounds in the MMP-13 receptor. Due to the up-shifting of the molecules in the MD simulations, the R2 and R1 benzene rings of compounds D3 and D8 produced additional hydrogen bonds with the residues Ser250 and Gly248. These results may be the reason for the higher predicted activity and binding affinity of compounds D3 and D8 than 26. The R3 position went deep into the hydrophobic pocket consisting of the residues Phe241 and His222, which facilitated the stability of compounds in MMP-13 receptors. The carboxyl group on R1 could act as the H-bond receptor and form hydrogen bonds with the surrounding the residues Ser250 and Lys140, which played an essential role in bonding affinity and activity of the inhibitor. Therefore, the carboxyl group on the benzene ring should be reserved in the future design of the quinazolinone derivatives.
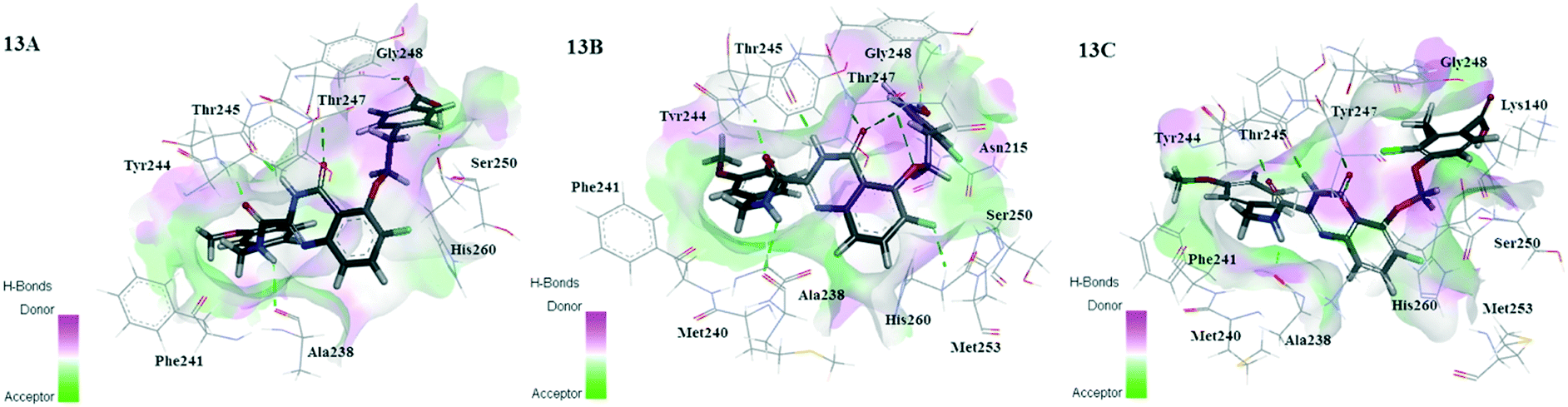 | ||
| Fig. 13 The binding mode of 26-MMP-13 (13A), D3-MMP-13 (13B) and D8-MMP-13 (13C) after MD simulation. | ||
The RMSF (root-mean-square fluctuation) is calculated for the motion change of each residue, so as to determine the flexibility of a certain region of the protein during the MD simulation process. The RMSF value versus the residue number of the three inhibitor–protein complexes was depicted in Fig. 14. The three complexes had similar dynamic fluctuation trends and RMSF distributions, indicating that these inhibitors had similar binding mode with MMP-13.
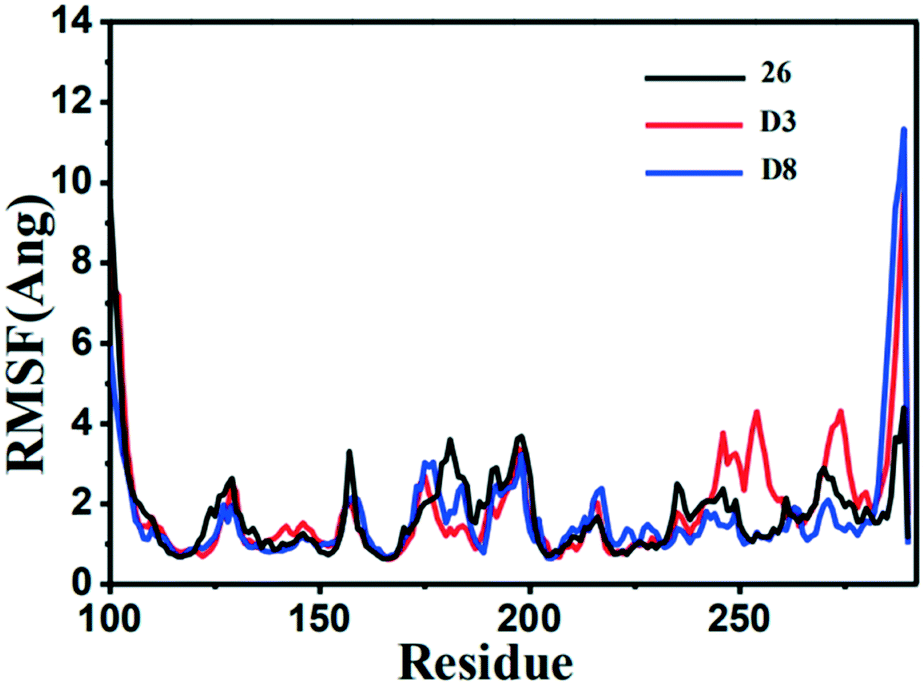 | ||
| Fig. 14 The root-mean-square fluctuation (RMSF) of the compound 26 (black), D3 (red) and D8 (green) in all three MMP-13 complexes obtained from 10 ns MD simulation. | ||
It was also shown that the RMSF value of the majority of protein residues in each complex was lower than 4 Å. The residues (Leu184, Phe201, and Asp202) with higher fluctuation values were around the flexible loop regions. The key residues, including Thr245, Thr247, Ala238, Met253, Asn215 and Lys140, showed rigid behavior with lower RMSF values, indicating that the compounds had stable nature with the complex. All these results supported the reliability of MD simulation. Furthermore, the RMSF fluctuations in the complexes D3-MMP-13 and D8-MMP-13 were observed to be the lower than the complex 26-MMP-13, implying they had the relatively lower structural mobility than the 26-MMP-13. These results suggested that D3 and D8 exhibited the relatively favorable binding affinity with MMP-13.
Based on the MD simulation, binding free energies were calculated using the MM-PBSA method to assess the energetic aspects of the three representative complexes. The 50 snapshots were extracted from last 2 ns trajectory and binding free energies terms of complexes were listed in Table 4. The results showed that the ΔGbind of compounds 26, D3 and D8 by MM/PBSA were −42.6219, −43.2322 and −44.3403 kcal mol−1, respectively, by MM/GBSA were −51.9742, −52.4444, −57.2545 kcal mol−1, respectively, which were in good agreement with the predicted activity of the CoMSIA model. Furthermore, the ΔGbind of MMP-13 with D3 and D8 were more negative than that of 26, which indicated that D3 and D8 bonded with MMP-13 more tightly than 26. As listed in Table 4, it could be observed that the contribution of the van der Waals energy ΔEvdw was the leading factor for total binding energy. The van der Waals energy ΔEvdw for 26, D3 and D8 were −66.7878, −68.1115 and −61.9011 kcal mol−1, respectively. Electrostatic energy ΔEele was the second largest contributor to total energy, and the electrostatic energies of compounds 26, D3 and D8 were −50.2873, −63.3876 and −47.3668 kcal mol−1, respectively. In addition, the non-polar solvation energy ΔGSA was beneficial to the binding free energy, whereas the polar solvation energy was not conducive to the binding free energy. The MM/PBSA and MM/GBSA prediction showed that van der Waals and electrostatic interactions were the main factor in the recognition of ligand to MMP-13, which was confirmed the results of 3D-QSAR.
| Contribution | 26-MMP-13 | D3-MMP-13 | D8-MMP-13 | 5-MMP-13 | ||||
|---|---|---|---|---|---|---|---|---|
| Mean (kcal mol−1) | Std. | Mean (kcal mol−1) | Std. | Mean (kcal mol−1) | Std. | Mean (kcal mol−1) | Std. | |
| ΔEvdw | −66.7878 | 3.2783 | −68.1115 | 2.1566 | −61.9011 | 3.2584 | −51.9732 | 1.5279 |
| ΔEele | −50.2837 | 8.4969 | −63.3876 | 6.2982 | −47.3668 | 6.6622 | −29.8305 | 4.0362 |
| ΔGgas | −117.0689 | 8.465 | −131.5015 | 5.7853 | −109.2639 | 7.7436 | −81.7967 | 4.6909 |
| ΔGGB | 73.1047 | 5.3665 | 86.7853 | 8.2685 | 60.1228 | 4.4232 | 50.0586 | 3.1046 |
| ΔGSA | −8.01 | 0.1124 | −7.7282 | 0.311 | −8.1134 | 0.2149 | −5.8757 | 0.2979 |
| ΔGsol(GB) | 65.0947 | 7.7436 | 79.0571 | 8.1296 | 52.0094 | 4.3908 | 44.1829 | 2.8213 |
| ΔGbind(GB) | −51.9742 | 4.0108 | −52.4444 | 3.4678 | −57.2545 | 2.25 | −37.6138 | 2.1384 |
| ΔGPB | 79.8582 | 8.2863 | 93.852 | 5.2411 | 70.5474 | 5.286 | 62.8713 | 5.2331 |
| ΔGSA | −5.4113 | 0.1244 | −5.5826 | 0.0794 | −5.6238 | 0.1138 | −4.849 | 0.1725 |
| ΔGsol(PB) | 74.447 | 8.2279 | 88.2694 | 5.7853 | 64.9236 | 5.2976 | 58.0223 | 5.0933 |
| ΔGbind(PB) | −42.6219 | 4.5729 | −43.2322 | 1.8197 | −44.3403 | 4.9159 | −23.7744 | 2.5999 |
In conclusion, the key residues Ala238, Thr245, Thr247, Met253, Asn215 and Lys140 with regard to hydrogen bonding and electrostatic interactions were significant for the activity and stability of the inhibitor. The MD simulation confirmed the docking results, and further verified the accuracy and stability of the 3D-QSAR model.
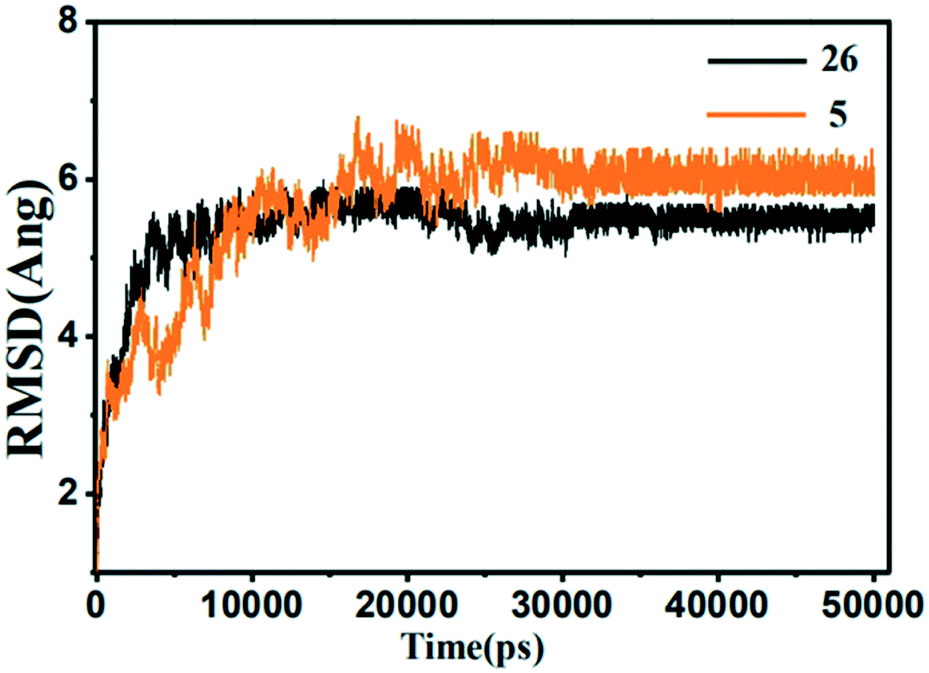 | ||
| Fig. 15 The root-mean-square deviation (RMSD) of the compound 26 (black) and 5 (orange) in the MMP-13 complexes obtained from 10 ns MD simulation. | ||
From the energy point of view, the ΔGbind(PB) −23.7744 kcal mol−1 and ΔGbind(GB) −37.6138 kcal mol−1 of 5 was higher than that of 26, which was in accordance with the activity measured by the experiment, that is, the activity of 5 was lower than that of 26. And the van der Waals energy and electrostatic energy of 5 and 26 were the main contribution for total binding energy. From the structural point of view, the quinazolinones ring of the 5 generated electrostatic interactions with the residues Tyr244, His260, Thr247, His261 and Ile243. And 5 only form a hydrogen bond with the residue His260. Compared to the template 26, the key residues (Ala238, Thr245, Asn215, Met253, and Lys140) disappeared. The lowest activity of 5 indicated that these key residues had important effect on the activity and stability of the compounds. In conclusion, the U-shaped conformation of the compound, the hydrogen bonding and electrostatic interactions with the key residues Ala238, Thr245, Thr247, Asn215, Met253, Lys140, were critical to the stability and activity of the compound.
Analysis of residual decomposition
In order to identify the key residues in favor of inhibitor activity and, to understand the interaction between the inhibitors and the residues around the active pocket of MMP-13, the energy contributions of each residue in MMP-13 protein were calculated by MM/PBSA.py program. As can be seen from Fig. 16A–D, the interaction between residues and inhibitors fluctuated between 1.82 and −3.16 kcal mol−1. The results show that the residues which contribute more to binding energy are mainly distributed in residues His134, and Asn215–His260. As can be seen from Fig. 16, the key residues Asn215, Thr247, Met253 and His260 have large contribution to the binding free energy of MMP-13 complexes. The hydrogen bonds between the residues Thr247, Met253 and ligands results in higher energy values. The van der Waals interaction of Asn215, His260, His134 on benzene ring makes them have higher binding free energy values. The energy decomposition of the residues are shown in the ESI† Tables S2–S5. According to the results of energy decomposition, the contributions of residues Thr247 and Met253 are mainly hydrophobic and van der Waals. The energy decomposition well validated the binding mode of receptors and ligands. These results provide a useful clue for rationalizing drug design.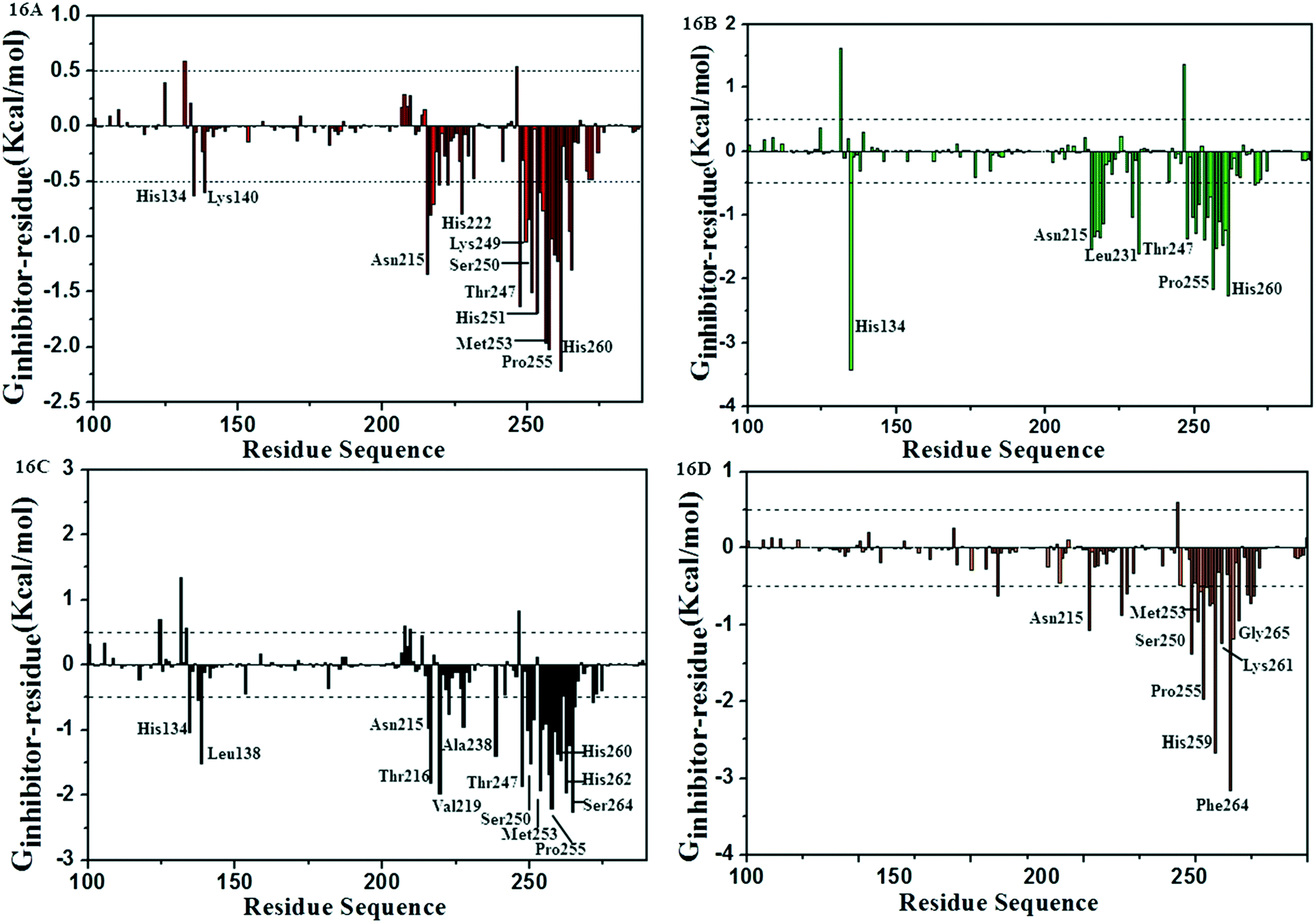 | ||
| Fig. 16 Comparison of per-residue energy decomposition of D3-MMP-13 (16A), D8-MMP-13 (16B), 26-MMP-13 (16C) and 5-MMP-13 (16D). | ||
ADMET evaluations
The prediction results for representative monosodium salt compounds (26-Na, D1-Na–D8-Na) were listed in ESI.† The aqueous solubility levels of D5-Na were 3 and the other compounds with the level 2, indicating that D5-Na had the better aqueous solubility. All the compounds had undefined BBB penetration properties (level 4). For the cytochrome P450-2D6 inhibition, all the compound showed the satisfactory effect (level 0), indicating that the molecules could readily undergo oxidation and hydroxylation in the phase-I metabolism. As for the hepatotoxicity, all the compounds had the level 1 of ADMET hepatotoxicity, suggesting that these compounds were likely to cause dose-dependent liver injuries. However, with the exception of compound D4-Na, all other molecules had low hepatotoxicity than 26-Na. The absorption levels of D2-Na, D6-Na and D8-Na were 2, suggesting that they had low absorption. The absorption levels of the remaining compounds were 1, which had moderate absorption. Then, the predicted result of ADMET PPB Level showed that compound D8-Na had a binding 90–95% (level 1) and the other compounds had binding <90% (level 0). Finally, all the compounds had the druggability potential with the value of 0.4 < AlogP98 < 5.6 and 7 < PSA-2D < 200.54,55 The results of this study showed that compound D8 had the highest predictive activity and the lowest hepatotoxicity probability, but the solubility was not good. All designed compounds except for D4, showed lower hepatotoxicity probability than the template molecule 26. In conclusion, all results showed that all the designed compounds were within the acceptable scale of ADMET, and provided the reference for further research.Conclusion
In this study, fifty-three quinazolinone derivatives as MMP-13 inhibitors were studied based on the mean of the computer-aided drug design, including 3D-QSAR, molecular docking, and MD simulations. 3D-QSAR models with high reliability and predictive abilities were developed to discover the key structural factors that influenced the MMP-13 inhibitory activity. Molecular docking and MD simulations analysis indicated that the U-conformation of the compound and the small group in R2 were crucial to the activity and stability of the inhibitors. And the hydrogen bonding and electrostatic interactions with several key residues, such as Ala238, Thr245, Thr247, Met253, Asn215 and Lys140 in the R2 and the R1 sites played a key role in ligands binding with the receptor. The results showed that the 3D-QSAR, molecular docking and MD simulations were consistent with each other, and Topomer CoMFA further verified the results of the 3D-QSAR model. Based on the above results, eight novel quinazolinone candidate compounds (D1–D8) were designed and predicted, and the ADMET results showed that these compounds had good pharmacokinetic properties. Compounds D3 and D8 with high predictive activity and small ΔGbind values could be used as good potential MMP-13 inhibitors in the field of OA.Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Onuora, Nat. Rev. Rheumatol., 2017, 13, 130 CrossRef PubMed.
- N. E. Lane and M. Corr, Nat. Rev. Rheumatol., 2017, 13, 76–78 CrossRef PubMed.
- C. Amalinei, I. D. Caruntu, S. E. Giusca and R. A. Balan, Rom. J. Morphol. Embryol., 2010, 51, 215–228 Search PubMed.
- R. W. Thompson and B. T. Baxter, Ann. N. Y. Acad. Sci., 1999, 878, 159–178 CrossRef CAS PubMed.
- L. J. McCawley and L. M. Matrisian, Mol. Med. Today, 2000, 6, 149–156 CrossRef CAS PubMed.
- P. G. Mitchell, H. A. Magna, L. M. Reeves, L. L. Lopresti-Morrow, S. A. Yocum, P. J. Rosner, K. F. Geoghegan and J. E. Hambor, J. Clin. Invest., 1996, 97, 761–768 CrossRef CAS PubMed.
- T. Klein and R. Bischoff, Amino Acids, 2011, 41, 271–290 CrossRef CAS PubMed.
- M. Wang, E. R. Sampson, H. Jin, J. Li, Q. H. Ke, H. J. Im and D. Chen, Arthritis Res. Ther., 2013, 15, R5 CrossRef CAS PubMed.
- M. B. Goldring, Curr. Rheumatol. Rep., 2000, 2, 459–465 CrossRef CAS PubMed.
- C. G. Fan, Q. Li, Y. L. Zhang, X. M. Liu, M. H. Luo, D. Abbott, W. H. Zhou and J. F. Engelhardt, J. Clin. Invest., 2004, 113, 746–755 CrossRef CAS PubMed.
- J. Y. Choi, R. Fuerst, A. M. Knapinska, A. B. Taylor, L. Smith, X. H. Cao, P. J. Hart, G. B. Fields and W. R. Roush, J. Med. Chem., 2017, 60, 5816–5825 CrossRef CAS PubMed.
- S. Laufer, Inflammopharmacology, 2001, 9, 101–112 CrossRef CAS.
- S. Tries and S. Laufer, Inflammopharmacology, 2001, 9, 113–124 CrossRef CAS.
- E. Sarah, H. Ezra, M. Michael, S. Michael and V. Matthew, Arthritis Res. Ther., 2003, 5, R285 CrossRef PubMed.
- F. J. Hemmings, M. Farhan, J. Rowland, L. Banken and R. Jain, Rheumatology, 2001, 40, 537–543 CrossRef CAS.
- R. P. Beckett, Expert Opin. Ther. Pat., 1996, 6, 1305–1315 CrossRef CAS.
- I. Samudio, S. Kurinna, P. Ruvolo, B. Korchin, H. Kantarjian, M. Beran, K. Dunner, S. Kondo, M. Andreeff and M. Konopleva, Mol. Cancer Ther., 2008, 7, 1130–1139 CrossRef CAS PubMed.
- H. Nara, K. Sato, T. Naito, H. Mototani, H. Oki, Y. Yamamoto, H. Kuno, T. Santou, N. Kanzaki, J. Terauchi, O. Uchikawa and M. Kori, J. Med. Chem., 2014, 57, 8886–8902 CrossRef CAS.
- H. Nara, A. Kaieda, K. Sato, T. Naito, H. Mototani, H. Oki, Y. Yamamoto, H. Kuno, T. Santou, N. Kanzaki, J. Terauchi, O. Uchikawa and M. Kori, J. Med. Chem., 2017, 60, 608–626 CrossRef CAS PubMed.
- H. J. Huang, H. W. Yu, C. Y. Chen, C. H. Hsu, H. Y. Chen, K. J. Lee, F. J. Tsai and C. Y. C. Chen, J. Taiwan Inst. Chem. Eng., 2010, 41, 623–635 CrossRef CAS.
- F. P. Guengerich and J. S. MacDonald, Chem. Res. Toxicol., 2007, 20, 344–369 Search PubMed.
- R. D. Cramer, D. E. Patterson and J. D. Bunce, J. Am. Chem. Soc., 1988, 110, 5959–5967 CrossRef CAS PubMed.
- G. Klebe, U. Abraham and T. Mietzner, J. Med. Chem., 1994, 37, 4130–4146 CrossRef CAS PubMed.
- R. D. Cramer, J. Med. Chem., 2003, 46, 374–388 CrossRef CAS PubMed.
- L. C. Dearden, Int. J. Quant. Struct.-Prop. Relat., 2016, 1, 1–44 Search PubMed.
- R. Kristam, V. Parmar and V. N. Viswanadhan, J. Mol. Graphics Modell., 2013, 45, 157–172 CrossRef CAS.
- J. Oluić, K. Nikolic, J. Vucicevic, Z. Gagic, S. Filipica and D. Agbabaaet, Comb. Chem. High Throughput Screening, 2017, 20, 292–303 CrossRef PubMed.
- F. Wu, X. Y. Hou, H. Luo, M. Zhou, W. J. Zhang, Z. Y. Ding and R. Li, Med. Chem. Commun., 2013, 4, 1482–1496 RSC.
- M. Clark, D. Cramer and O. N. Van, J. Comput. Chem., 1989, 10, 982–1012 CrossRef CAS.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- Z. Ul-Ha, N. Khan, S. K. Zafar and S. T. Moin, Eur. J. Pharm. Sci., 2016, 88, 26–36 CrossRef PubMed.
- N. Richmond, C. A. Abrams, P. R. N. Wolohan, E. Abrahamian, P. Willett and R. D. Clark, J. Comput.-Aided Mol. Des., 2006, 20, 567–587 CrossRef CAS PubMed.
- S. Yu, J. T. Yuan, J. H. Shi, X. J. Ruan, T. Zhang, Y. L. Wang and Y. Du, Chemom. Intell. Lab. Syst., 2015, 146, 34–41 CrossRef CAS.
- S. Wold, A. Ruhe, H. Wold and W. J. D. Lii, SIAM J. Sci. Comput., 1984, 5, 735–743 CrossRef.
- L. Ståhle and S. Wold, J. Chemom., 1987, 1, 185–196 CrossRef.
- P. P. Roy, J. T. Leonard and K. Roy, Chemom. Intell. Lab. Syst., 2008, 90, 31–42 CrossRef CAS.
- M. H. Dong, Y. J. Ren and X. D. Gao, Bioorg. Med. Chem. Lett., 2015, 25, 4118–4126 CrossRef CAS PubMed.
- P. K. Ojha and K. Roy, Chemom. Intell. Lab. Syst., 2011, 109, 146–161 CrossRef CAS.
- K. Roy, R. N. Das, P. Ambure and R. B. Aher, Chemom. Intell. Lab. Syst., 2016, 152, 18–33 CrossRef CAS.
- A. Golbraikh and A. Tropsha, J. Mol. Graphics Modell., 2002, 20, 269–276 CrossRef CAS PubMed.
- A. Tropsha, P. Gramatica and V. K. Gombar, Mol. Inf., 2003, 22, 69–77 CAS.
- P. P. Roy and K. Roy, Mol. Inf., 2010, 27, 302–313 Search PubMed.
- A. N. Jain, J. Med. Chem., 2003, 46, 499 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, G. W. F. Van, A. DiNola and J. R. Haak, J. Chem. Phys., 1998, 81, 3684–3690 CrossRef.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1998, 79, 926–935 CrossRef.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2001, 32, 889–897 Search PubMed.
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS.
- I. Grossman, Expert Opin Drug Metab Toxicol, 2009, 5, 449–462 CrossRef CAS PubMed.
- I. Khanna, Drug Discovery Today, 2012, 17, 1088 CrossRef.
- C. A. Lipinski, Drug Discovery Today, 2003, 8, 12–16 CrossRef.
- W. J. Egan and G. Lauri, Adv. Drug Delivery Rev., 2002, 54, 273–289 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00375k |
| This journal is © The Royal Society of Chemistry 2019 |