
Preparation of NIR absorbing axial substituted tin(IV) porphyrins and their photocytotoxic properties†
Balaji
Babu
a,
Edith
Amuhaya
b,
David
Oluwole
a,
Earl
Prinsloo
 c,
John
Mack
*a and
Tebello
Nyokong
a
c,
John
Mack
*a and
Tebello
Nyokong
a
aCentre for Nanotechnology Innovation, Department of Chemistry, Rhodes University, Makhanda 6140, South Africa. E-mail: j.mack@ru.ac.za
bSchool of Pharmacy and Healthy Sciences, United States International University - Africa, Nairobi, Kenya
cBiotechnology Innovation Centre, Rhodes University, Makhanda 6140, South Africa
First published on 19th October 2018
Abstract
Sn(IV) porphyrins ([Sn(IV)TTP(3PyO)2] (5) and [Sn(IV)TPP(3PyO)2] (6) [tetrathienylporphyrin (TTP), tetraphenylporphyrin (TPP), and pyridyloxy (PyO)]) were prepared and characterized and their photocytotoxicity upon irradiation with 625 nm light has been studied. The presence of the 3PyO axial ligands was found to limit the aggregation and enhance the solubility of 5 and 6 in DMF/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The photophysical properties and photodynamic therapy (PDT) activity of the meso-2-thienyl and meso-phenyl-substituted Sn(IV) porphyrins are compared. 5 and 6 were found to be photocytotoxic in MCF-7 cancer cells when irradiated with a Thorlabs M625L3 LED at 625 nm but remained nontoxic in the dark. The PDT activity of Sn(IV) meso-tetra-2-thienylporphyrin 5 was found to be significantly enhanced relative to its analogous tetraphenylporphyrin 6. There is a marked red-shift of the Q00 band of 5 into the therapeutic window due to the meso-2-thienyl rings, and 5 has an unusually high singlet oxygen quantum yield value of 0.83 in DMF. The results demonstrate that readily synthesized axially ligated Sn(IV) meso-arylporphyrins are potentially suitable for use as singlet oxygen photosensitizers in biomedical applications and merit further in depth investigation in this context.
:1). The photophysical properties and photodynamic therapy (PDT) activity of the meso-2-thienyl and meso-phenyl-substituted Sn(IV) porphyrins are compared. 5 and 6 were found to be photocytotoxic in MCF-7 cancer cells when irradiated with a Thorlabs M625L3 LED at 625 nm but remained nontoxic in the dark. The PDT activity of Sn(IV) meso-tetra-2-thienylporphyrin 5 was found to be significantly enhanced relative to its analogous tetraphenylporphyrin 6. There is a marked red-shift of the Q00 band of 5 into the therapeutic window due to the meso-2-thienyl rings, and 5 has an unusually high singlet oxygen quantum yield value of 0.83 in DMF. The results demonstrate that readily synthesized axially ligated Sn(IV) meso-arylporphyrins are potentially suitable for use as singlet oxygen photosensitizers in biomedical applications and merit further in depth investigation in this context.
Introduction
Porphyrins are a class of tetrapyrrolic compounds with 18 delocalised π-electrons, which are found to have a wide range of applications, due to their intense absorption and emission bands in the visible region. Different strategies have been studied to functionally modify the porphyrin core to make it more suitable for use in applications such as catalysis, solar photovoltaic cells, chemical sensors, and electrochemical devices.1–4 In recent years, there has been growing interest in the use of a wide range of different porphyrin derivatives in photodynamic therapy (PDT).5,6 Photofrin®, the first commercially available PDT drug, is an oligomeric mixture of porphyrin derivatives which absorbs relatively weakly in the therapeutic window (620–850 nm) where there is maximum light penetration in tissue and its use can leave the skin photosensitized for prolonged periods.7–9 Efforts were subsequently made to develop a second generation of photosensitizer dyes that absorb more strongly in the therapeutic window.10 A significant drawback of using highly planar monomeric porphyrinoid ligands and their metal complexes in this context has been their tendency to form H-aggregates.11,12 Porphyrins and other porphyrinoids that have been considered for use in PDT, such as phthalocyanines12e–h and naphthalocyanines,12i,j can form supramolecular assemblies through π–π stacking and this can limit their solubility in aqueous solvents and complicate their intravenous administration. Different approaches have been developed to prevent the aggregation-induced deactivation of porphyrin excited states. The axial substitution of metal porphyrins with bulky groups helps to block porphyrin ligands from approaching one another thus preventing aggregation.12d,13–17 Heavy diamagnetic metal ions, e.g. Zn2+, Sn4+, Pd2+ and In3+, are preferred in this context since they promote intersystem crossing (ISC) and hence the energy transfer from the triplet manifold to dioxygen to form singlet oxygen.18,19The meso-substituents of tetraphenylporphyrins (TPPs) greatly influence their physicochemical properties. The substituents which can extend the delocalization of the π-electrons in the porphyrin core tend to absorb at longer wavelengths since there is a narrowing of the HOMO–LUMO gap. The main Q and B (or Soret) bands of meso-thienyl-substituted porphyrins have been reported to lie at longer wavelengths than those in the spectra of conventional tetraphenylporphyrins.20,21 Replacing a six-membered phenyl ring with a smaller five-membered 2-thienyl group enhances the free rotation of the meso-aryl substituent, and this results in a stronger interaction with the π-system of the porphyrin macrocycle and makes aggregation in solution more likely. Since an effective PDT agent should absorb strongly in the therapeutic window, it is important to enhance the absorption of the Q bands of the porphyrin ring in this spectral region.22,23 With this goal in mind, the aim of this study was to prepare and characterize the Sn(IV) complexes of meso-2-thienyl-substituted porphyrins (TTPs) with 3-pyridyloxy (3PyO) axial ligands [Sn(IV)(TTP)(3PyO)2] (Scheme 1) and to study their photophysical properties and PDT efficiency. To further explore the effect of introducing 2-thienyl rings, the analogous phenyl-substituted Sn(IV) porphyrin [Sn(IV)(TPP)(3PyO)2] was prepared to provide a control compound.
 | ||
| Scheme 1 Synthetic route for Sn(Por)(3PyO)2 complexes. | ||
Results and discussion
Synthesis and structural characterization
Free base TTP (1) and TPP (2) were prepared from the corresponding aldehydes.20a Sn(IV) tetraarylporphyrins with trans-dihydroxy axial ligands [(Sn(IV)(Por)(OH)2)] (3 and 4) were prepared by treating the free base porphyrins with SnCl2·2H2O in pyridine followed by hydrolysis with excess aqueous ammonia (Scheme 1).243 and 4 were then heated at reflux in CHCl3 for 4 h with two equivalents of 3-hydroxypyridine to yield axially ligated target compounds 5 and 6 in quantitative yield. The target compounds were isolated and purified by neutral alumina column chromatography and characterized by MALDI-TOF MS, 1H NMR, UV-visible absorption and emission spectroscopy. The dyes were found to be highly soluble in polar solvents such as dimethylformamide (DMF) and dimethylsulfoxide (DMSO), however. Attempts were made to quaternize the pyridine nitrogens of the axial ligands, but these proved unsuccessful.The aromatic protons of the 3PyO axial ligands resonate at a higher field than those of the porphyrin ligand in the 1H NMR spectra of 5 (Fig. S1, see the ESI†), since the axial ligand experiences strong shielding from the ring current of the porphyrin macrocyle.25,26 Integration of the pyridine ring proton signals confirmed that the stoichiometry of the axial ligation is 2:1. The MALDI-TOF mass spectrum of 5 (Fig. S2, see the ESI†) contains a major molecular ion peak corresponding to [Sn-3PyO]+ with a minor [Sn-(3PyO)2]+ peak corresponding to the loss of one and two axial ligands, respectively.25,26
Photophysical properties
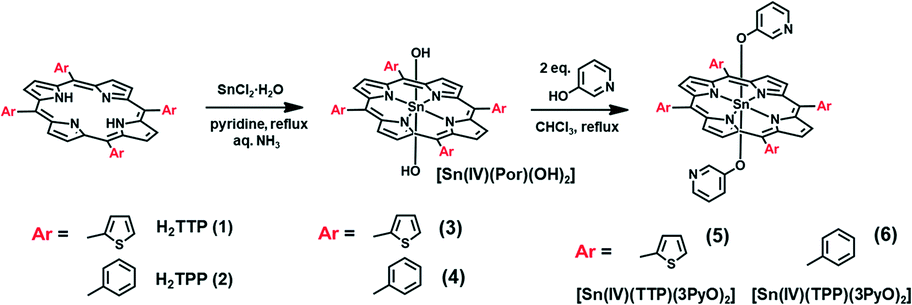
The UV-visible absorption spectra of free base porphyrins 1 and 2 and Sn(IV) porphyrins 5 and 6 are shown in Fig. 1, and their photophysical data are summarized in Table 1. The free base porphyrin spectra contain an intense B band and four weak Q bands. The Q and B bands of the TTP compounds are red-shifted compared to those of TPP (Fig. 1), due in part to the easier free rotation of the meso-aryl rings. As is usually the case upon metallation, only two Q bands are observed in the spectra of the Sn(IV) porphyrins due to the four-fold symmetry (Fig. 1). The absorption maximum of 5 is red-shifted to 613 nm, which falls on the edge of the therapeutic window. The absorption spectra of Sn(IV) porphyrins 3 and 4 that have the hydroxyl rather than the 3PyO axial ligands exhibited similar absorption maxima (Fig. S3, see the ESI†) to those of 5 and 6, respectively. This demonstrates that the axial substitution does not have a significant influence on the optical properties and electronic structures of the Sn(IV) porphyrins.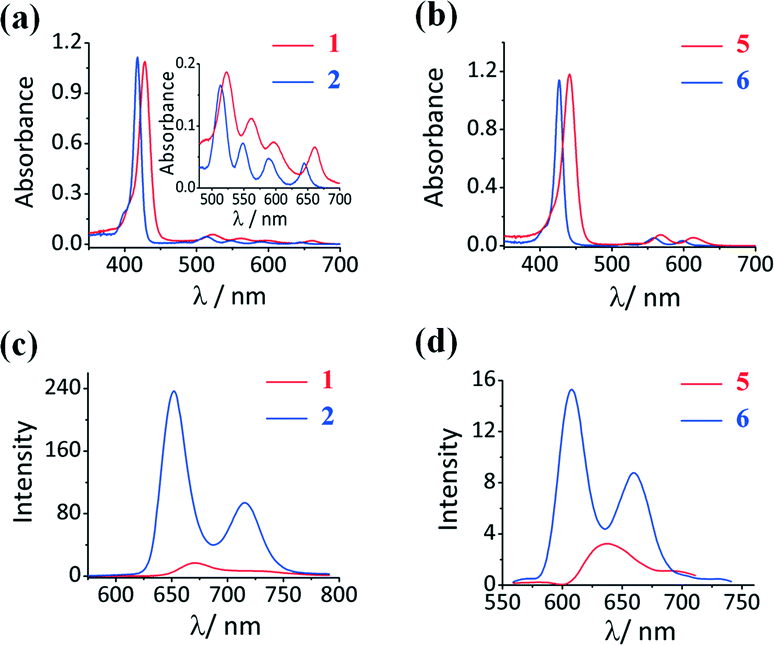 | ||
| Fig. 1 UV-vis spectra of (a) free base porphyrins 1 and 2, and (b) Sn(IV) porphyrins 5 and 6 in DMF (TOP). Emission spectra of (c) free base porphyrins 1 and 2, and (d) Sn(IV) porphyrins 5 and 6 in DMF with λex set at the B band maximum (BOTTOM). | ||
| λ max (nm) | λ em (nm) | Φ F | τ F (ns) | τ T (μs) | Φ Δ | |||
|---|---|---|---|---|---|---|---|---|
| B band | Q bands | |||||||
| 1 | H2TTP | 428 | 523, 562, 597, 660 | 670, 728(sh) | 0.02 | 1.26 | 9.0 | 0.55 |
| 2 | H2TPP | 418 | 513, 548, 589, 644 | 652, 715 | 0.13 | 11.48 | 9.6 | 0.42 |
| 5 | [Sn(IV)TTP(3PyO)2] | 441 | 568, 613 | 637, 694(sh) | <0.01 | 1.76 | 7.7 | 0.83 |
| 6 | [Sn(IV)TPP(3PyO)2] | 426 | 559, 598 | 607, 660 | 0.04 | 5.42 | 5.1 | 0.60 |
The free base and Sn(IV) porphyrins fluoresce when excited at their B band maximum. The fluorescence quantum yield (ΦF) values in Table 1 show that the meso-thienyl free base and Sn(IV) porphyrins are only weakly emissive with emission bands that are red-shifted compared to their meso-phenyl porphyrin analogues. The decrease in the ΦF and fluorescence lifetime (τF) values for 2-thienyl-substituted porphyrins can be attributed primarily to the heavy atom effect arising from the presence of four sulfur atoms on the periphery of the porphyrin ring. The fluorescence spectra of free base meso-thienyl porphyrin and 5 contain broad emission bands (Fig. 1), whereas those of the phenyl analogues contain two well-resolved emission bands. Interestingly, metallation with a central Sn(IV) ion decreases the ΦF value drastically compared to the free porphyrin analogues, due to the heavy atom effect. The transient absorption curve of 5 was recorded in DMF (Fig. S4, see the ESI†) and the triplet state lifetime (τT) was determined to be 7.7 μs. The stability of the Sn(IV) porphyrins was studied in DMF by using the UV-visible absorption spectroscopic technique. There was no change in the spectral features of the compounds over 4 h.
Aggregation studies
Aggregation due to π–π stacking is a commonly encountered issue with macrocyclic complexes such as porphyrins and metal porphyrins, and this is problematic since it facilitates internal conversion to the ground state, which suppresses the rate of ISC and in turn decreases the singlet oxygen generation ability. Various methodologies have been adopted to suppress aggregation. The incorporation of axial ligands through metal bonding prevents the metal porphyrins from approaching each other thereby suppressing the aggregation. The Sn(IV) ion of 5 and 6 has a high affinity towards phenolate ligands, which bind in a trans-axial mode. This enables the preparation of 5 and 6 with the 3PyO axial ligands.The presence of two trans-axial ligands prevents the aggregation of 5 and 6. The UV-visible absorption spectra of 5 and 6 were measured at different concentrations in dimethylformamide (DMF) to prove this hypothesis. As shown in Fig. 2, the spectra of 5 and 6 are typical of those observed for non-aggregated metal porphyrins, with narrow and intense B bands at 426 and 441 nm, respectively, and Q00 bands at 598 and 613 nm. The spectra were found to strictly obey the Beer–Lambert law, suggesting that these complexes exist only as monomers in DMF solution. In contrast, the analogous free base porphyrin structures exhibit marked deviations from linearity at higher concentrations due to aggregation (Fig. S5, see the ESI†). Aggregation studies were also carried out in 1:1 DMF/H2O (v/v) for both the free base and Sn(IV) porphyrins (Fig. 3). Broadened and less intense B bands were observed in the free base porphyrin spectra as would usually be anticipated in this context due to aggregation. In contrast, no changes were observed in the spectra of the axially substituted Sn(IV) porphyrins in 1:1 DMF/H2O (v/v), so it is reasonable to conclude that metalation with tin and axial ligation above and below the porphyrin ring play an important role in preventing aggregation even in the presence of smaller 2-thienyl meso-aryl rings.
 | ||
| Fig. 2 UV-visible absorption spectra of (a) 5 and (b) 6 at different concentrations in DMF. The inset plots show the B-band absorbance vs. concentration of the metal porphyrin. | ||
 | ||
| Fig. 3 UV-visible absorption spectra of (a) 5 and (b) 6 in DMF (blue) and 1:1 DMF/H2O (v/v) (red). | ||
Theoretical calculations
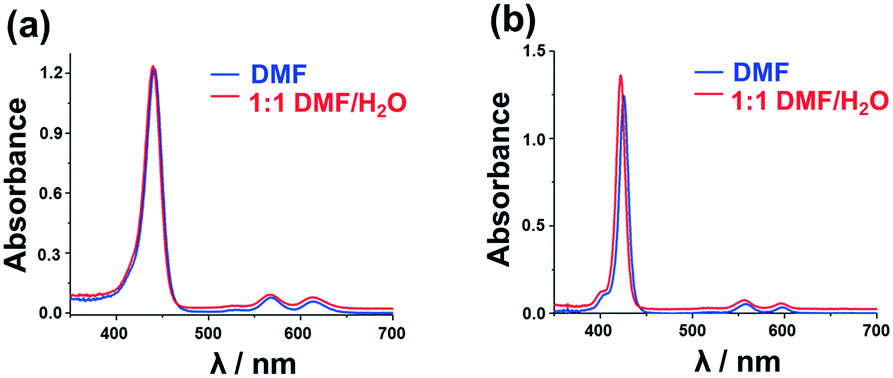
A series of DFT and TD-DFT calculations were carried out using the Gaussian 09 software package.27 The electronic structures of the porphyrins can be understood in the context of the Gouterman's 4-orbital model28,29 which describes the optical properties in terms of a 16 atom 18 π-electron system on the inner ligand perimeter arranged in an ML = 0, ±1, ±2, ±3, ±4, ±5, ±6, ±7 and 8 sequence in ascending energy terms. The HOMO and LUMO of the C16H162− parent hydrocarbon perimeter have ML = ±4 and ±5 properties, respectively, and the ΔML = ±1 and ±9 transitions between these orbitals give rise to allowed B and forbidden Q bands, respectively.In the context of the Michl's perimeter model,30 the four frontier π-MOs derived from the HOMO and LUMO of the parent hydrocarbon perimeter are referred to as the a, s, −a and −s MOs (Fig. 4), respectively, depending on whether there is a nodal plane (a or −a MOs) or large MO coefficients (s or −s MOs) aligned with the y-axis. This nomenclature aids the comparison of the electronic structures of porphyrin-related molecules with differing symmetries. When the predicted MO energies of 1, 2, 5 and 6 are compared (Fig. 4), it becomes clear that there is a narrowing of the average HOMO–LUMO gaps of 1 and 5 relative to those of 2 and 6 when the four MOs derived from the HOMO and LUMO of the parent hydrocarbon perimeter are taken into consideration, due to the incorporation of the meso-thienyl rings. There is a destabilization of the a MO relative to the s, −a and −s MOs, since it lacks large MO coefficients on the meso-carbons and this results in a narrowing of the HOMO–LUMO gap and a significant red shift of the Q and B bands (Fig. 5), making the dye more suitable for use as a photosensitizer in the therapeutic window.
 | ||
| Fig. 4 The angular nodal patterns and energies of the a, s, −a and −s MOs of 5 (TOP). MO energies for 1, 2, 5 and 6 at the CAM-B3LYP/SDD level of theory (BOTTOM). Occupied MOs are highlighted with small back squares and large red diamonds are used to denote the HOMO–LUMO band gaps and are plotted against a secondary axis. Black squares are used to highlight the s MOs. | ||
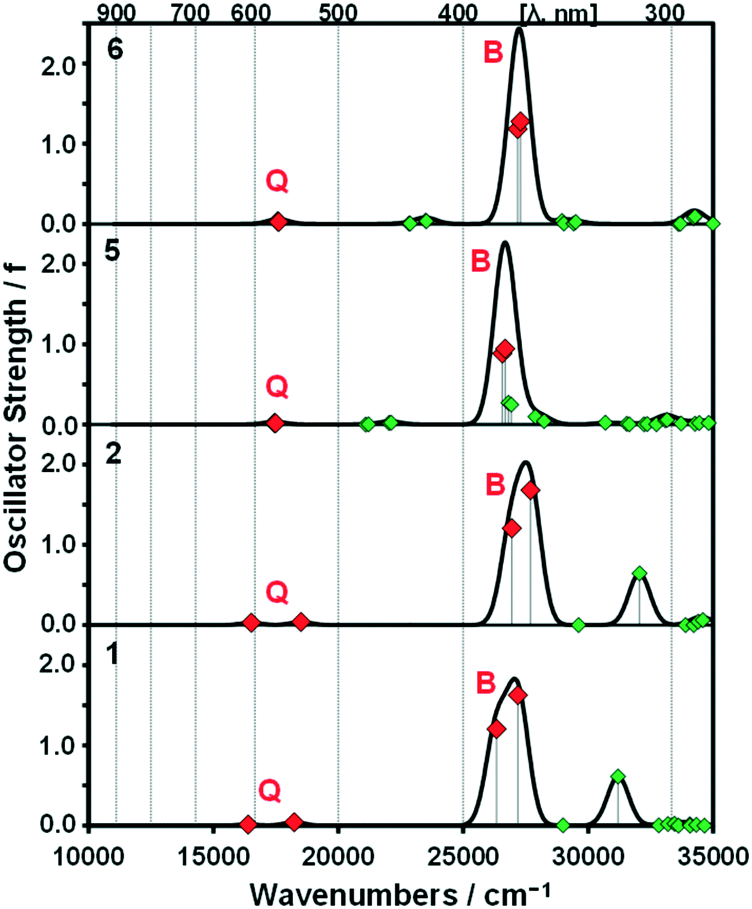 | ||
| Fig. 5 Calculated TD-DFT spectra for B3LYP-optimized geometries of 1, 2, 5 and 6 at the CAM-B3LYP/SDD level of theory. Red diamonds are used to highlight the Q and B bands of Gouterman's 4-orbital model.28 Simulated spectra were generated using the Chemcraft program with a fixed bandwidth of 2000 cm−1.31 Details of the calculations are provided in Table S1, see the ESI.† | ||
Singlet oxygen generation
Singlet oxygen is one of the major cytotoxic agents that is produced by irradiation of photosensitizer dyes during PDT.5a,23a The singlet oxygen generation efficiencies of the free base and Sn(IV) porphyrins were evaluated in DMF by using 1,3-diphenylisobenzofuran (DPBF) as a singlet oxygen scavenger. Zinc tetraphenylporphyrin was used as the reference compound, Table 1. The singlet oxygen quantum yield value of the free base meso-thienyl-substituted porphyrin (ΦΔ = 0.55) is higher than that of their meso-phenyl substituted analogues (ΦΔ = 0.42). The presence of four heavy sulfur atoms enhances the rate of ISC and increases the population of the T1 state. The ΦΔ value of Sn(IV) porphyrin 5 (ΦΔ = 0.83) is substantially higher than the corresponding free base porphyrin, due to the heavy atom effect associated with the central metal ion. A similar trend is observed for 6 and the free base tetraphenylporphyrin. It is clear from Table 1 that 5 is a more efficient photosensitizer than its phenyl analog 6. The incorporation of 2-thienyl groups at the meso-position results not only in a significant red shift of the Q bands but also in a significant increase in the ΦΔ value, thus making the dye more suitable for use in PDT.PDT studies
The in vitro photocytotoxic potential of Sn(IV) porphyrins 5 and 6 was determined by using the WST-1 assay (Fig. 6) on human breast adenocarcinoma (MCF-7) cells based on a protocol reported in the literature.32 The concentration range studied was from 3.1 to 50 μM. The Sn(IV) porphyrins were dissolved in media containing <0.5% DMSO and were initially treated and incubated for 24 h in the dark at 37 °C. A Modulight® ML7200 series illumination source set-up fitted with a Thorlabs M625L3 LED for 10 and 20 min (625 nm, 144 and 288 J cm−2) was used for the in vitro PDT treatment. Appropriate dark controls were used to determine whether there was any dark toxicity. Interestingly, both 5 and 6 exhibited minimal dark toxicity over the entire concentration range that was studied.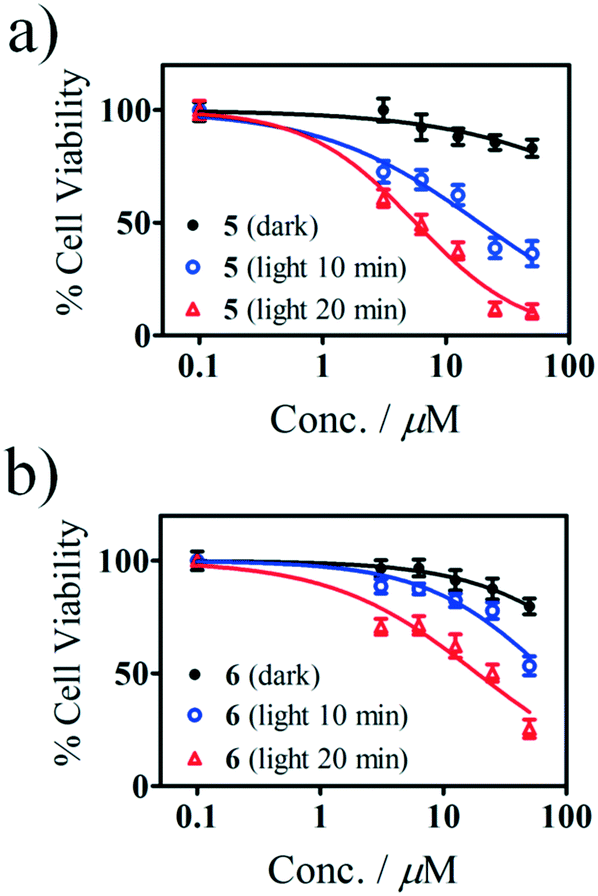 | ||
| Fig. 6 Cytotoxicity of a) 5 and b) 6 in MCF-7 cells after 24 h of incubation in the dark followed by photo-irradiation at 625 nm with a Thorlabs M625L3 LED as determined with the WST-1 assay. The results for the dark-treated cells are depicted as black circles, while those for the photoexposed cells are given as blue (10 min) and red triangles (20 min). | ||
The IC50 values are given in Table 2 along with those of the Photofrin and cisplatin drugs. It was found that 20 min of irradiation resulted in better photocytotoxicity than 10 min for both 5 and 6. As anticipated, the meso-thienyl-substituted compound 5 exhibited a significantly greater PDT activity than its tetraphenyl-substituted analogue 6 with IC50 values of 5.6 and 18.7 μM, respectively, for 20 min of irradiation time. This demonstrates that incorporating 2-thienyl rings has a highly significant impact on the photocytotoxicity towards the MCF-7 cells as was anticipated given its higher ΦΔ value, low aggregation in solution and enhanced absorbance of the Q band in the therapeutic window relative to 6. Since low dark toxicity and high photocytotoxicity are two of the main criteria for PDT agents, the results, when combined with the aggregation studies, demonstrate that structurally modified Sn(IV) tetraarylporphyrin complexes similar to 5 merit further consideration for use in PDT.
:1 DMF/H2O, and comparable values * for Photofrin® and cisplatin33
| IC50 (μM) darka | IC50 (μM) 10 minb | IC50 (μM) 20 minb | PA (m2 s W−1) | |
|---|---|---|---|---|
| a The IC50 values for 5 and 6 after 24 h of incubation in the dark without any photoexposure. b The IC50 values of 5 and 6 after 24 h of incubation in the dark followed by photoexposure to a 625 nm Thorlabs M625L3 LED (240 mW cm−2) for 10 and 20 min. *Photofrin® has an IC50 value of 1.0 (±0.1) μM (2 h of exposure; white bulb; fluence rate: 5.5 × 10−2 mW cm−2) in MCF-7 cells.33a *Cisplatin has an IC50 value of 2.0 (±0.3) μM for 96 h of treatment in MCF-7 cells.33b | ||||
| 5 | >50 | 18.6 (±1.2) | 5.60 (±1.1) | 0.7 |
| 6 | >50 | >50 | 18.7 (±1.1) | 0.4 |
Determination of the photodynamic activity (PA) by using uric acid (UA) as a chemical dosimeter
The photodynamic activity (PA) of photosentizers, PS (1, 2, 5, and 6), were studied by the uric acid method.34 Uric acid is known to react with singlet oxygen to form triuret, sodium oxalate, allantoxaidin and CO2. Uric acid absorbs in the UV region (λmax = 288 nm in 1:1 DMF/H2O), and has no absorption band where free base porphyrins (1, 2) and tin porphyrins (5, 6) absorb, which assures the photosensitizers as the unique light absorber species. The 1O2 produced by the PS degrades uric acid when the mixture of UA and PS is irradiated by 625 nm light from the Thorlabs M625L3 LED. This is monitored by the decrease in the absorption band of UA at 288 nm.
The decay curves of UA in the solution containing a mixture of 5 and UA are characterized by the decrease in the absorption at 288 nm with irradiation time (Fig. 7), and a similar behavior was observed for 6. The absorption bands of 5 and 6 remain unchanged after irradiation which demonstrates that these dyes are photostable under these irradiation conditions. The decrease in uric acid absorption at 288 nm is greater for 5 compared to 6 (Fig. 7c) which shows that 5 is more efficient in producing singlet oxygen due to the difference in absorbance values at 625 nm. Although the free base porphyrins (TTP and TPP) exhibit similar behavior (Fig. S6, see the ESI†), there is clear evidence of aggregation in the spectra recorded for 1 in 1:1 (DMF/H2O), due to the absence of a central metal ion with axial ligands, which would become even more problematic as the fraction of water is increased in the context of cell studies. The photodynamic activity (PA) values were determined by Fischer's expression from the decay curve and are given in Table 2.34a The PA value of 5 is still higher than that of 6 when the difference in absorbance is factored in. The results are in agreement with those derived from the DPBF method in DMF.
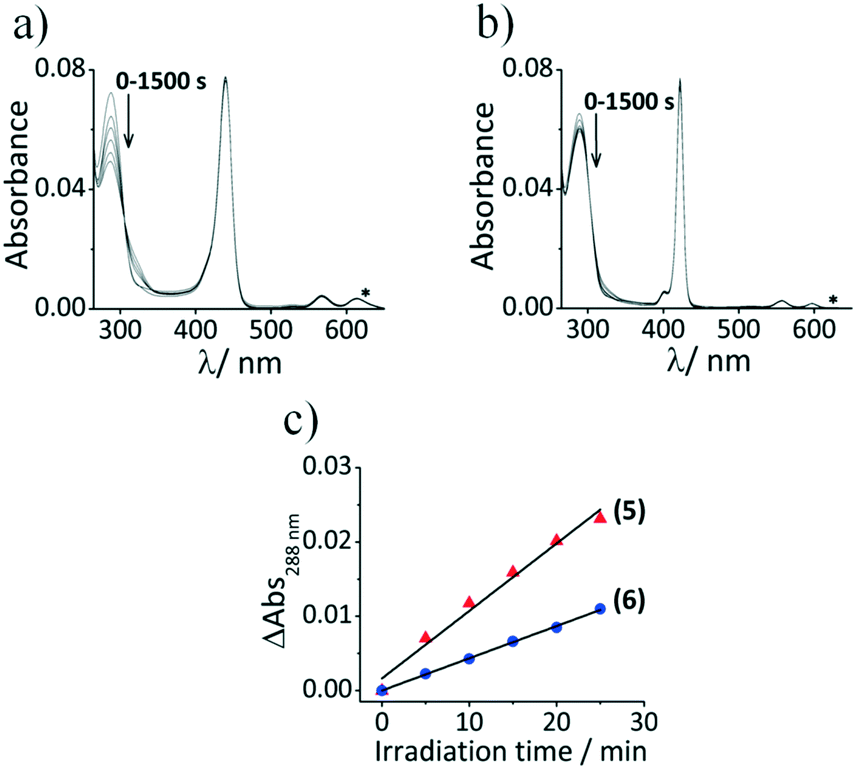 | ||
| Fig. 7 Absorption spectra of (a) UA + 5 and (b) UA + 6 in DMF/H2O (1:1) after different irradiation times with 625 nm light. (c) Decay curves of the UA absorption band at 288 nm as a function of the irradiation time for UA + 5 (solid red triangles) and UA + 6 (solid blue circles). The wavelength of irradiation obtained using the Thorlabs M625L3 LED (625 nm) is marked by an asterisk. | ||
Experimental
Materials
Ultra-pure water was obtained from a Milli-Q water system (Millipore Corp, Bedford, MA, USA). DMF and trypan blue were purchased from Sigma Aldrich. Cultures of the MCF-7 cell were obtained from Cellonex®. 10% (v/v) heat-inactivated fetal calf serum (FCS), and 100 unit per mL penicillin–100 μg per mL streptomycin–amphotericin B were obtained from Biowest®. Dulbecco's phosphate-buffered saline (DPBS) and Dulbecco's modified Eagle's medium (DMEM) were purchased from Lonza®.Equipment
UV-visible absorption and fluorescence emission spectra were measured on a Shimadzu UV-2550 spectrophotometer and a Varian Eclipse spectrofluorimeter using a 360–1100 nm filter. The fluorescence lifetimes were measured using a time-correlated single photon counting set-up (FluoTime 300, Picoquant GmbH) with a diode laser (LDH-P-670, Picoquant_GmbH, 20 MHz repetition rate, 44 ps pulse width). The triplet state lifetimes and transient absorption curves were recorded in DMF at 480 or 500 nm using an Edinburgh Instruments LP980 spectrometer and an Ekspla NT-342B laser to provide an excitation wavelength of 425 nm. The MS data were collected on a Bruker® AutoFLEX III Smart-beam TOF/TOF mass spectrometer using α-cyano-4-hydrocinnamic acid as the matrix in the positive ion mode.Cell studies – dark toxicity
The cultures of the MCF-7 breast cancer cell lines were grown in DMEM containing L-glutamine and phenol red, supplemented with 10% heat-inactivated FCS and 100 unit per ml penicillin–100 μg per ml streptomycin–amphotericin B. The cells were grown in a T75 flask and incubated at 37 °C and 5% CO2, until a cell confluence of 80–100% was achieved. A fluorescence LED inverted microscope was used to view the cells under phase contrast (Zeiss-AxioVert). Following which the cells were rinsed with DPBS and lifted using trypsin. The cells were then counted using a hemocytometer upon the addition of 50 μl of DPBS and 100 μl trypan blue and cultured (from a known volume). The cells were seeded and allowed to attach in 100 μl of supplemented DMEM containing phenol red in a 96-well tissue culture plates at a density of around 10000 cells per well, and the cells were then incubated at 37 °C and 5% CO2 for 24 h. After this time, the attached cells were rinsed with 100 μl DPBS, followed by the addition of 100 μl cultured medium containing gradient concentrations of the respective compounds. The control cells were given fresh supplemented (or with 0.1–0.8% DPBS or DMSO in) placebo medium. The plates were re-incubated at 37 °C and 5% CO2 in the dark for 24 h. Following this, the wells were then rinsed with 100 μl DPBS; the medium was replaced with a fresh one and the cells were incubated overnight.
Photodynamic therapy test
The PDT effects were determined by incubating the attached cells seeded as above with gradient concentrations of 5 and 6 in a 96-well plate at a density of 10000 cells per well in the culture medium (100 μl). The plate was incubated at 37 °C and 5% CO2 in the dark for 24 h, and the plate was then washed with 100 μl DPBS and the medium was replaced with supplemented DMEM without phenol red. The 96-well plate was irradiated using the illumination kit of a Modulight® 7710-680 medical laser fitted with a Thorlab M625L3 light emitting diode that was found to provide an irradiance of 240 mW cm−2 (measured with a Coherent FieldmaxII TOP energy/power meter fitted with a Powermax PM10 sensor). The irradiation time was varied between 10 and 20 min. After irradiation, the medium was replaced with a fresh one containing phenol red. The surviving cells were quantified 24 h after treatment using the WST-1 assay (from Roche®), which is known to be a sensitive assay for evaluating toxicity and cell proliferation in monolayer cultures. The absorbance of the cells was determined by using a Synergy 2 multi-mode microplate reader (BioTek®) at an excitation wavelength of 450 nm. The cytotoxicity of the test compounds was measured as the percentage ratio of the absorbance of the treated cells to the untreated controls. The IC50 values were determined by nonlinear regression analysis by using GraphPad Prism 5 software.
Uric acid – photodynamic activity (PA)
In order to verify the photodynamic activity of compounds, the uric acid test was carried out.34 An equal volume of UA (4.3 μg mL−1) and sensitizer (0.15 μg mL−1) was mixed and illuminated (Thorlabs M625L3 LED, 625 nm, I0 = 240 mW cm−2, t = 1500 s). The absorbance of the mixture is measured at regular intervals of time after irradiation. The photodynamic activity of the photosensitizer was determined in units of m2 s W−1 following the decrease of UA absorbance at 288 nm (ΔAUA) as a function of irradiation time (t) by using eqn (1):33| PA = (ΔAUA × 105)/(I0·t·APS) | (1) |
Synthesis
Free base tetrathienylporphyrin and tetraphenylporphyrin and the [(Sn(IV)(Por)(OH)2)] complexes were prepared by using a previously reported procedure.20a,24Synthesis of 5 and 6: 1 equiv. of [(Sn(IV)(Por)(OH)2)] was dissolved in a minimal volume of dry CHCl3 and an excess of 3-hydroxypyridine was added. The mixture was refluxed for 4 h and the completion of the reaction was monitored by UV-visible absorption spectroscopy by following the red shift of the B band. The solvent was evaporated, and the resulting residue was purified on a neutral alumina column by using 9:1 CHCl3/MeOH (v/v) as the eluent to give the target compounds 5 and 6 as purple solids.
[Sn(IV)(TTP)(3PyO)2] (5): 1H NMR (CDCl3, 600 MHz): δ 2.20 (2H, d), 3.25 (2H, d), 5.62 (2H, m), 7.06 (2H, d), 7.56 (4H, m), 7.95 (8H, d), 9.31 (8H, s). MS (MALDI-TOF): m/z calcd for [Sn-3PyO]+ 849.99; found 850.27. [Sn-(3PyO)2]2+ 755.96; found 756.19.
[Sn(IV)(TPP)(3PyO)2] (6): 1H NMR (CDCl3, 600 MHz): δ 2.20 (2H, d), 3.24 (2H, d), 5.61 (2H, m), 7.03 (2H, d), 7.82 (12H, m), 8.21 (8H, d), 9.16 (8H, s). MS (MALDI-TOF): m/z calcd for [M]+ 920.19; found 920.72.
Conclusions
As has been reported previously, the incorporation of meso-thienyl rings results in a significant red shift of the Q and B bands of the porphyrin ligand. A Sn(IV) complex with bulky 3-pyridyloxy axial ligands was found to have an unusually high ΦΔ value of 0.83 and does not exhibit aggregation properties in polar solvents such as DMF and 1:1 DMF/water. When its photodynamic therapy activity was tested in media containing <0.5% DMSO by using the WST-1 assay on MCF-7 cells, there was clear evidence of enhanced photocytotoxicity relative to the analogous tin tetraphenylporphyrin complex. Further studies with other easily synthesized axially ligated Sn(IV) tetraarylporphyrin complexes with unusually high ΦΔ values are underway to identify dyes of this type that can provide superior absorption properties in the therapeutic window through a further red shift of the Q00 band. This should enable the use of lower light doses comparable to those that are required with the existing photosensitizer dyes that have been approved for clinical use. The axial ligands can also be modified to incorporate suitable targeting groups for selective accumulation in neoplastic tissues.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Department of Science and Technology (DST) and the National Research Foundation (NRF) of South Africa through the DST/NRF South African Research Chairs Initiative for Professor of Medicinal Chemistry and Nanotechnology (uid: 62620), a South Africa-Kenya CSUR grant from the NRF of South Africa to JM (uid: 105809). Photophysical measurements were made possible by the Laser Rental Pool Programme of the Council for Scientific and Industrial Research (CSIR) of South Africa. Theoretical calculations were carried out at the Centre for High Performance Computing in Cape Town.References
- (a) M. Kielmann, C. Prior and M. O. Senge, New J. Chem., 2018, 42, 7529 RSC; (b) M. Van de Boom, A. Gulino, S. Giuffrida, P. Mineo, M. Purrazzo, E. Scamporrino, G. Ventimiglia and I. Fragalà, J. Phys. Chem. B, 2006, 110, 16781 CrossRef PubMed; (c) A. Gulino, P. Mineo, S. Bazzano, D. Vitalini and I. Fragala, Chem. Mater., 2005, 17, 4043 CrossRef.
- (a) A. Gulino, P. Mineo, E. Scamporrino, D. Vitalini and I. Fragalà, Chem. Mater., 2004, 16, 1838 CrossRef; (b) P. Mineo, E. Scamporrino, E. Spina and D. Vitalini, Sens. Actuators, B, 2013, 188, 1284 CrossRef; (c) Y. Aoyama, A. Yamagishi, M. Asagawa, H. Toi and H. Ogoshi, J. Am. Chem. Soc., 1988, 110, 4076 CrossRef; (d) T. Mizutani, K. Wada and S. Kitagawa, J. Am. Chem. Soc., 1999, 121, 11425 CrossRef.
- (a) E. Mikros, F. Gaudemer and A. Gaudemer, Inorg. Chem., 1991, 30, 1806 CrossRef; (b) C. Verchere-Beaur, E. Mikros, M. Perre-Fauvet and A. Gaudemer, J. Inorg. Biochem., 1990, 40, 127 CrossRef; (c) V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, J. Am. Chem. Soc., 2001, 123, 2979 CrossRef; (d) E. Scamporrino, P. Mineo, S. Dattilo, D. Vitalini and E. Spina, Macromol. Rapid Commun., 2007, 28, 1546 CrossRef.
- (a) N. Angelini, N. Micali, P. Mineo, E. Scamporrino, V. Villari and D. Vitalini, J. Phys. Chem. B, 2005, 109, 18645 CrossRef; (b) P. Mineo, N. Micali, V. Villari, M. G. Donato and E. Scamporrino, Chem. – Eur. J., 2012, 18, 12452 CrossRef; (c) H. Lee, K. I. Hong and W. D. Jang, Coord. Chem. Rev., 2018, 354, 46 CrossRef; (d) A. Thomas, F. Kuttassery, S. Mathew, S. N. Remello, Y. Ohsaki, D. Yamamoto, Y. Nabetani, H. Tachibana and H. Inoue, J. Photochem. Photobiol., A, 2018, 358, 402 CrossRef; (e) K. Kurimoto, T. Yamazaki, Y. Suzuri, Y. Nabetani, S. Onuki, S. Takagi, T. Shimada, H. Tachibana and H. Inoue, Photochem. Photobiol. Sci., 2014, 13, 154 RSC.
- (a) M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340 RSC; (b) A. Naik, R. Rubbiani, G. Gasser and P.-D. B. Spingler, Angew. Chem., 2014, 126, 7058 CrossRef; (c) N. J. Patel, Y. Chen, P. Joshi, P. Pera, H. Baumann, J. R. Missert, K. Ohkubo, S. Fukuzumi, R. R. Nani, M. J. Schnermann, P. Chen, J. Zhu, K. M. Kadish and R. K. Pandey, Bioconjugate Chem., 2016, 27, 667 CrossRef.
- (a) A. Sour, S. Jenni, A. O. Suárez, J. Schmitt, V. Heitz, F. Bolze, P. L. de Sousa, C. Po, C. S. Bonnet, A. Pallier, É. Tóth and B. Ventura, Inorg. Chem., 2016, 55, 4545 CrossRef; (b) A. A. Srivatsan, J. R. Missert, S. K. Upadhyay and R. K. Pandey, J. Porphyrins Phthalocyanines, 2015, 19, 109 CrossRef; (c) P. M. Antoni, A. Naik, I. Albert, R. Rubbiani, S. Gupta, P. Ruiz-Sanchez, P. Munikorn, J. M. Mateos, V. Luginbuehl, P. Thamyongkit, U. Ziegler, G. Gasser, G. Jeschke and B. Spingler, Chem. – Eur. J., 2015, 21, 1179 CrossRef.
- D. Kessel, Photodiagn. Photodyn. Ther., 2004, 1, 3 CrossRef.
- S. Yano, S. Hirohara, M. Obata, Y. Hagiya, S. I. Ogura, A. Ikeda, H. Kataoka, M. Tanaka and T. Joh, J. Photochem. Photobiol., C, 2011, 12, 46 CrossRef.
- Y. You, S. L. Gibson, R. Hilf, S. R. Davies, A. R. Oseroff, I. R. Roy, T. Y. Ohulchanskyy, E. J. Bergey and M. R. Detty, J. Med. Chem., 2003, 46, 3734 CrossRef CAS.
- (a) R. R. Allison, G. H. Downie, R. Cuenca, X.-H. Hu, C. J. H. Childs and C. H. Sibata, Photodiagn. Photodyn. Ther., 2004, 1, 27 CrossRef CAS; (b) L. B. Josefsen and R. W. Boyle, Met.-Based Drugs, 2008, 276109 Search PubMed; (c) A. B. Ormond and H. S. Freeman, Materials, 2013, 6, 817 CrossRef CAS.
- (a) C. A. Hunter and J. K. M. Sanders, Photochem. Photobiol., 2013, 88, 1011 Search PubMed; (b) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525 CrossRef CAS; (c) N. Micali, V. Villari, M. A. Castriciano, A. Romeo and L. M. Scolaro, J. Phys. Chem. B, 2006, 110, 8289 CrossRef CAS.
- (a) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef; (b) O. Ohno, Y. Kaizu and H. Kobayashi, J. Chem. Phys., 1993, 99, 418 CrossRef; (c) D. L. Akins, H. R. Zhu and C. Guo, J. Phys. Chem., 1996, 100, 5420 CrossRef CAS; (d) Y. Mikata, T. Sawaguchi, T. Kakuchi, M. Gottschaldt, U. S. Schubert, H. Ohi and S. Yano, Eur. J. Org. Chem., 2010, 663 CrossRef CAS; (e) S. Makhseed, M. Machacek, W. Alfadly, A. Tuhl, M. Vinodh, T. Simunek, V. Novakova, P. Kubat, E. Rudolf and P. Zimcik, Chem. Commun., 2013, 49, 11149 RSC; (f) A. Wang, X. Chen, L. Zhang, G. Zhang, L. Zhou, S. Lu, J. Zhou and S. Wei, J. Photochem. Photobiol., A, 2014, 288, 1 CrossRef CAS; (g) Z. Jiang, J. Shao, T. Yang, J. Wang and L. Jia, J. Pharm. Biomed. Anal., 2014, 87, 98 CrossRef CAS; (h) C. Jing, R. Wang, H. Ou, A. Li, Y. An, S. Guo and L. Shi, Chem. Commun., 2018, 54, 3985 RSC; (i) M. M. Zuk, B. D. Rihter, M. E. Kenney, M. A. J. Rodgers and M. Kreimer Birnbaum, Photochem. Photobiol., 1996, 63, 132 CrossRef CAS; (j) L. Luan, L. Ding, W. Zhang, J. Shi, X. Yu and W. Liu, Biorg. Med. Chem. Lett., 2013, 23, 3775 CrossRef CAS.
- T. Lazarides, S. Kuhri, G. Charalambidis, M. K. Panda, D. M. Guldi and A. G. Coutsolelos, Inorg. Chem., 2012, 51, 4193 CrossRef CAS.
- V. S. Shetti and M. Ravikanth, Inorg. Chem., 2010, 49, 2692 CrossRef CAS.
- Z. Biyiklioglu and H. Alp, Dalton Trans., 2015, 44, 18993 RSC.
- L. M. Gomis, E. M. Barea, F. F. Lázaro, J. Bisquert and Á. S. Santos, J. Porphyrins Phthalocyanines, 2011, 15, 1004 CrossRef.
- A. J. Pearson, T. Plint, S. T. E. Jones, B. H. Lessard, D. Credgington, T. P. Bender and N. C. Greenham, J. Mater. Chem. C, 2017, 5, 12688 RSC.
- N. J. Patel, Y. Chen, P. Joshi, P. Pera, H. Baumann, J. R. Missert, K. Ohkubo, S. Fukuzumi, R. R. Nani, M. J. Schnermann, P. Chen, J. Zhu, K. M. Kadish and R. K. Pandey, Bioconjugate Chem., 2016, 27, 667 CrossRef CAS.
- (a) M. P. Donzello, E. Viola, M. Giustini, C. Ercolani and F. Monacelli, Dalton Trans., 2012, 41, 6112 RSC; (b) I. Baldea, R.-M. Ion, D. E. Olteanu, I. Nenu, D. Tudor and A. G. Filip, Clujul Med., 2015, 88, 175 CrossRef.
- (a) C. Brückner, P. C. D. Foss, J. O. Sullivan, R. Pelto, M. Zeller, R. R. Birge and G. Crundwell, Phys. Chem. Chem. Phys., 2006, 8, 2402 RSC; (b) I. Gupta and M. Ravikanth, J. Photochem. Photobiol., A, 2006, 177, 156 CrossRef CAS; (c) X. Sun, J. Zhang and B. He, J. Photochem. Photobiol., A, 2005, 172, 283 CrossRef CAS.
- (a) J. Rochford, S. Botchway, J. J. McGarvey, A. D. Rooney and M. T. Pryce, J. Phys. Chem. A, 2008, 112, 11611 CrossRef CAS; (b) A. Ghosh, S. M. Mobin, R. Frohlich, R. J. Butcher, D. K. Maity and M. Ravikanth, Inorg. Chem., 2010, 49, 8287 CrossRef CAS PubMed; (c) N. M. Boyle, J. Rochford and M. T. Pryce, Coord. Chem. Rev., 2010, 254, 77 CrossRef CAS.
- (a) D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 380 CrossRef CAS PubMed; (b) K. Szacilowski, W. Macyk, A. Drzewiecka-Matuszek, M. Brindell and G. Stochel, Chem. Rev., 2005, 36, 2647 CrossRef.
- (a) M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS; (b) E. Ruggiero, S. A. de Castro, A. Habtemariam and L. Salassa, Dalton Trans., 2016, 45, 13012 RSC.
- D. P. Arnold, J. Chem. Educ., 1988, 65, 1111 CrossRef CAS.
- (a) I. A. Khodov, G. A. Alper, G. M. Mamardashvili and N. Z. Mamardashvili, J. Mol. Struct., 2015, 1099, 174 CrossRef CAS; (b) S. V. Bhosale, C. Chong, C. Forsyth, S. J. Langford and C. P. Woodward, Tetrahedron, 2008, 64, 8394 CrossRef CAS.
- (a) Y. Tong, D. G. Hamilton, J.-C. Meillon and J. K. M. Sanders, Org. Lett., 1999, 1, 1343 CrossRef CAS; (b) G. D. Fallon, S. J. Langford, M. A.-P. Lee and E. Lygris, Inorg. Chem. Commun., 2002, 5, 715 CrossRef CAS; (c) M. J. Crossley, P. Thordarson and R. A.-S. Wu, J. Chem. Soc., Perkin Trans. 1, 2001, 1, 2294 RSC.
- M. J. Frisch, et al., Gaussian 09, see the ESI for full reference and a detailed description of the computational methods Search PubMed.
- M. Gouterman, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 3, pp. 1–165 Search PubMed.
- J. Mack, Chem. Rev., 2017, 117, 3444 CrossRef CAS PubMed.
- (a) J. Michl, J. Am. Chem. Soc., 1978, 100, 6801 CrossRef CAS; (b) J. Michl, Tetrahedron, 1984, 40, 3845 CrossRef CAS.
- Chemcraft - graphical software for visualization of quantum chemistry computations, https://www.chemcraftprog.com Search PubMed.
- (a) M. Managa, J. Britton, E. K. Amuhaya and T. Nyokong, J. Lumin., 2017, 185, 34 CrossRef CAS; (b) D. O. Oluwole, C. M. Tilbury, E. Prinsloo, J. Limson and T. Nyokong, Polyhedron, 2016, 106, 92 CrossRef CAS; (c) R. O. Ogbodu and T. Nyokong, Spectrochim. Acta, Part A, 2015, 151, 174 CrossRef CAS PubMed.
- (a) F. Ménard, V. Sol, C. Ringot, R. Granet, S. Alves, C. L. Morvan, Y. Queneau, N. Ono and P. Krausz, Bioorg. Med. Chem., 2009, 17, 7647 CrossRef PubMed; (b) I. Ott, K. Schmidt, B. Kircher, P. Schumacher, T. Wiglenda and R. Gust, J. Med. Chem., 2005, 48, 622 CrossRef CAS PubMed.
- (a) F. Fischer, G. Graschew, H. J. Sinn, W. Maier-Borst, W. J. Lorenz and P. M. Schlag, Clin. Chim. Acta, 1998, 274, 89 CrossRef CAS PubMed; (b) M. N. O. Moritz, J. L. S. Gonçalves, I. A. P. Linares, J. R. Perussi and K. T. de Oliveira, Photodiagn. Photodyn. Ther., 2017, 17, 39 CrossRef CAS PubMed; (c) R. S. Cavalcante, H. Imasato, V. S. Bagnato and J. R. Perussi, Laser Phys. Lett., 2009, 6, 64 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, additional photophysical data and details of calculations. See DOI: 10.1039/c8md00373d |
| This journal is © The Royal Society of Chemistry 2019 |