
Recent developments in compounds acting in the DNA minor groove
Adeyemi
Rahman
,
Patrick
O'Sullivan
and
Isabel
Rozas
 *
*
School of Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160-Pearse Street, Dublin 2, Ireland. E-mail: rozasi@tcd.ie
First published on 12th December 2018
Abstract
The macromolecule that carries genetic information, DNA, is considered as an exceptional target for diseases depending on cellular division of malignant cells (i.e. cancer), microbes (i.e. bacteria) or parasites (i.e. protozoa). To aim for a comprehensive review to cover all aspects related to DNA targeting would be an impossible task and, hence, the objective of the present review is to present, from a medicinal chemistry point of view, recent developments of compounds targeting the minor groove of DNA. Accordingly, we discuss the medicinal chemistry aspects of heterocyclic small-molecules binding the DNA minor groove, as novel anticancer, antibacterial and antiparasitic agents.
1. Introduction
During many years, deoxyribonucleic acid (DNA) has been considered as an excellent target for therapies to treat diseases depending on cellular division of malignant cells (i.e. cancer), microbes (i.e. bacteria) or parasites (i.e. protozoa). It is well-known that, based on the familiar double-stranded DNA structure, the most typical modes of action of DNA targeting agents are: intercalation, alkylation, strand-cleaving and binding to the minor groove (Fig. 1).1 | ||
| Fig. 1 Key DNA–drug interactions: intercalators, alkylators, cleaving agents and minor groove binders (MGB). | ||
Intercalators are large, hydrophobic, planar molecules that insert in between DNA bases through a combination of π–π stacking and hydrophobic interactions.2 Originally, it was assumed that their mode of action was the structural disruption of DNA in such a way that important recognition proteins could not bind; it is now accepted that intercalators generally act by DNA topoisomerase II inhibition. Thus, these molecules create a ternary structure with DNA and topoisomerase II after a single strand break occurs, rendering the enzyme unable to complete the double strand breakage, and sterically blocking any other enzyme from repairing the break.3 An important example is doxorubicin (Fig. 2), which is known to bind to mitochondrial DNA and used in cancer chemotherapy (e.g. leukemias and Hodgkin's lymphoma4).
 | ||
| Fig. 2 Examples of DNA targeting agents used in clinic: intercalator (doxorubicin), cross-linking (melphalan and cisplatin), methylating (procarbazine) and cleaving (bleomycin). | ||
Methylating and cross-linking agents that form covalent bonds with DNA altering replication and transcription have been extensively studied as anticancer agents since World War II.5 Classical alkylating agents such as nitrogen mustards, (e.g. melphalan, Fig. 2) are used to treat leukemias; they form electrophilic aziridinium rings in situ establishing DNA inter-strand crosslinks.5 Cross-linking agent cisplatin (Fig. 2) and its derivatives are probably some of the chemotherapeutic drugs more widely used in clinic and signified an enormous step forward in the treatment of several cancers (i.e. the 5 years survival rate in testicular cancer has been increased from 10 to 98%).6 An example of an efficient methylating agent is procarbazine (Fig. 2), which is used in clinic to treat Hodgkin's lymphoma and brain cancers.7
DNA cleaving agents (e.g. bleomycin, Fig. 2) have also been widely used. These compounds break the DNA strands probably with the involvement of hydroxide and superoxide free radicals. Bleomycin is a natural product extracted from Streptomyces verticillus and it is used in clinic to treat a wide variety of cancers usually as a co-adjuvant.8
Finally, compounds that bind in the minor groove of DNA tend to be concave in shape, possess several aromatic rings, are cationic in nature and have the ability to form hydrogen bonds (HBs).9 Their lipophilic aromatic rings displace the structured spine of hydration along the DNA minor groove.10 Compounds acting in the DNA minor groove can disrupt protein–DNA interactions and have found application in the treatment of many diseases, including cancer, parasitic, bacterial and viral infections.10 Some of these compounds form noncovalent complexes with DNA (e.g. minor groove binders –MGBs– such as distamycin A or pentamidine, Fig. 3), while others act as alkylating MGBs (e.g. CC-1065,11Fig. 3).
 | ||
| Fig. 3 Examples of known DNA minor groove binders used in clinic: distamycin A, netropsin, CC-1065, pentamidine and furamidine. | ||
Because of their structural differences, MGBs have different pathways for cellular up-take, have specificity for definite DNA sequences and preferentially induce apoptosis in different organisms and cell types. Most notably, bis-amidines such as pentamidine, furamidine and berenil have been used clinically to treat trypanosomal infections in humans and animals. Other MGBs include polyamides such as distamycin, which lacks of anticancer effects by itself but has been the base of interesting anticancer derivatives,12 or netropsin that shows antiviral activity as well as being active against Gram-positive and Gram-negative bacteria (Fig. 3).13
Vast amount of work has been devoted to DNA targeting compounds of therapeutic interest,2,5,8–12,14 gene techniques15 or epigenetics,16 and aiming for a comprehensive review to cover all those aspects would be an impossible task. For that reason, the objective of this review is to present a medicinal chemistry overview of compounds that target the minor groove of DNA resulting in fundamental therapeutic breakthroughs, as well as the most recent MGBs designed as therapies. The review is organised according to the most relevant therapeutic applications of these compounds and, thus, novel anticancer, antibacterial and antiparasitic agents are discussed.
2. Drugs acting on the DNA minor groove as anticancer agents
The most commonly targeted macromolecule in cancer chemotherapy is DNA and those DNA-interacting anticancer agents, approved by the FDA and EMA, which shaped the field include among others nitrogen mustards, platinum-based drugs, dactinomycin, or doxorubicin.5,17 From those compounds many others are arising widening the antineoplastic arsenal and, in particular, MGBs represent a novel chemotherapeutic approach.2.1. (Di)Aryl amidine-like derivatives: amidines, guanidines and isoureas
Different aryl systems have been explored in this type of amidine-like derivatives. For example, quinoline–arylamidine hybrids have been prepared bearing anti-tumour activity.18 Thus, four series of 7-chloroquinoline and arylamidine systems joined through different linkers (1, –O– or 2, –NH–CH2–CH2–O–, Fig. 4) were synthesized, and their DNA/RNA binding properties and cytotoxic activity tested against several human cancer lines. The DNA/RNA interaction of these compounds was assessed by UV-vis and CD spectroscopy showing that binding affinity increases with the molecular length and number of groups able to form HBs. It has been recently shown that the thermodynamics of the minor groove binding is determined by the nature of the ligands.19 Thus, binding of compounds that form HBs as well as van der Waals interactions with the bases in the minor groove is driven by enthalpy, whereas highly lipophilic compounds with anionic groups bind in an entropically favourable manner due to the hydrophobic effect. Additionally, optimal binding was obtained by increasing flexibility of these hybrids which preferentially bind to DNA/RNA grooves. The antiproliferative effects were assessed on normal (MDCK1), carcinoma (HeLa and CaCo2) and leukemia cell lines (Raji and K-562) yielding GI50 values in the range of 5 to >100 μM. The most effective compounds against leukemia cell lines were flexible hybrids (carrying –NH–CH2–CH2–O– linker) with GI50 values around 5 to 35 μM and flow cytometry indicated that some of them induced cell cycle arrest at G0/G1 phase, dominantly.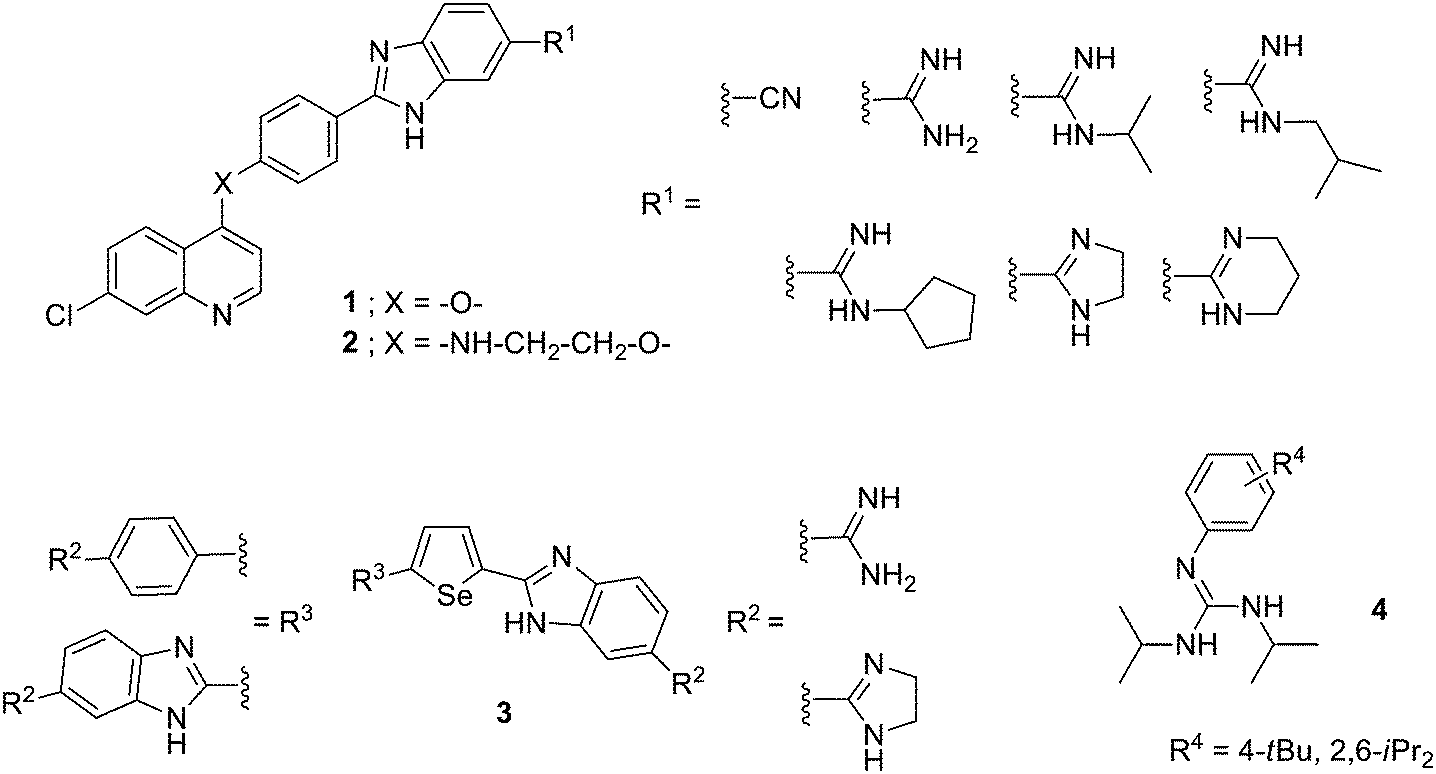 | ||
| Fig. 4 Structure of (di)aryl amidine-like derivatives with anticancer activity. | ||
Boykin and Wilson have developed di-heterocyclic diamidines related to furamidine in which the central furan has been substituted by selenophene and one or both of the phenyl rings has been replaced with benzimidazole (3, Fig. 4). They have shown that these compounds inhibit the action of PU.1, which is a member of the ETS family of transcription factors. The PU.1 factor binds to specific DNA sequences by inserting a helix into the major groove along with contacts with the near minor groove. The reported di-heterocyclic diamidines target the minor groove near where PU.1 establishes contacts thus disrupting its binding.20 Considering that PU.1 is often impaired in patients with acute myeloid leukemia (AML), in collaboration with Poon and Steidl, they found that these di-amidines were able to inhibit PU.1 in AML cells, and could be considered for further therapeutic development.21
Several substituted phenyl guanidine derivatives have been developed for application in glioblastoma treatment. This type of brain tumour is highly malignant and current treatments (i.e., temozolomide) lead to only modest survival. Thus, Bravo et al. have synthesized a number of these derivatives (4, Fig. 4),22 assessed their DNA affinity by UV titrations and fluorescent intercalator displacement (FID) assays and their cytotoxicity was investigated in the C6 rat glioblastoma cell line. These phenyl-guanidines were identified as groove binders (even though other mechanisms of action could not be discarded) and some of them significantly reduced glioblastoma cell proliferation, with a higher potency than the standard drug.
Our group has worked on different bis-guanidine-like di-aryl systems as anticancer agents. In 2013, we reported the synthesis of novel bis-(N-hydroxyguanidinium) di-aromatic derivatives (5, Fig. 5) as potential MGBs and anti-tumour agents.23 Their DNA affinity was evaluated by UV thermal denaturation experiments and the antiproliferative activity of those strongly interacting to DNA was evaluated in human promyelocytic HL-60, breast carcinoma MCF-7, and neuroblastoma Kelly cell lines. The compounds tested, which have a tetrahedral linker (–CH2–, –O– or –S–), were active in the HL-60 cell line. In addition, 5b and 5c produced significant antiproliferative activity in the neuroblastoma cell line.
 | ||
| Fig. 5 Structure of Rozas' di-phenyl amidine-like derivatives with anticancer activity. | ||
Based on the results obtained from a computational study on the suitability of the isouronium and N-hydroxyguanidinium cations as HB donors/acceptors, the DNA binding of a series of bis- and mono-isouronium diaryl derivatives (6 and 7, Fig. 5) was assessed by DNA UV thermal denaturation and compared to related N-hydroxyguanidines.24 Due to the poor DNA binding observed, the nature of the linker between the phenyl rings was further explored. Thus, the corresponding amide-linked bis-isouronium derivative was prepared and its DNA binding studied indicating that it was a MGB. Subsequently, the inhibitory effect on cell viability was evaluated in HL-60 and Kelly cancer cell lines providing IC50 values comparable to those previously found for the bis-(N-hydroxyguanidine) family. In all series, compounds with the –S– linker proved to be considerably active in HL-60 cells and even more active in the Kelly cell line. However, no correlation was found between DNA affinity and cell growth inhibition; hence, activity may depend on different modes of action. Further studies into the apoptotic potential of these compounds indicated that some mono-isouroniums (12a and 12b, Fig. 6) considerably induce apoptosis in both cell lines. Moreover, the effects of compound 12b on cell viability and apoptosis in two non-cancerous cell lines (NIH3T3 and MCF-10A) indicate none or minimal toxicity.
 | ||
| Fig. 6 Different structural moieties identified in distamycin and structures of polyamides and distamycin derivatives: tallismustine, brostallicin, compound 8 prepared by Suckling and co-workers, compound 9 prepared by Dervan and co-workers and benzamide 10. | ||
2.2. Polyamides derivatives
The antibiotic distamycin A was isolated from Streptomyces distallicus in 1964,25 and even though shows no anti-tumour activity, it has been used as ‘lead compound’ for the development of new MGBs. In the structure of distamycin, three moieties can be identified (Fig. 6): (i) ‘head’ (formamide), (ii) ‘polyamide core’ or ‘DNA binding region’ (pyrroles connected by peptide bonds) and (iii) ‘tail’ (cationic amide). Accordingly, different modifications have been introduced to these three moieties in order to improve distamycin anti-tumour activity. Tallimustine (Fig. 6)26 is a derivative of distamycin A where the ‘head’ has been substituted by a nitrogen mustard and one of the pyrroles of the ‘polyamide core’ by a benzoyl group. Tallimustine has shown very good anti-tumour activity in preclinical tests; however, it also shows strong myelotoxicity. Following the good activity of this compound, novel cinnamic analogues have been recently reported showing improved activity and less myelotoxicity.27Also relevant has been the finding of a series of α-halogenoacrylamido derivatives of distamycin, which displayed a much improved activity profile. In this series of compounds, a guanidinium cation has been introduced in the ‘tail’ and an α-bromo-acrylamido group has been used as a ‘head’. Above all, brostallicin (Fig. 6), showed potent and broad anti-tumour activity (even on cell lines found to be resistant to alkylating agents) and strongly reduced myelotoxicity. Brostallicin was found to bind to the DNA minor groove but seem not to act as an alkylator in the biophysical experiments; in this sense, the authors hypothesise the intervention of some intracellular reactive nucleophilic species (e.g. glutathione) that could make the compound more reactive driving to the in vivo alkylation of DNA.28 The role of glutathione on the cytotoxicity of brostallicin and other derivatives was investigated in tumour cells characterised by high levels of glutathione such as melphalan resistant leukemia tumour cells (L1210:L-PAM), which have three times more glutathione than the wild type (i.e. L1210). They found a three-fold cytotoxicity increase in the L1210:L-PAM compare to L1210 cells.28 Considering its excellent cytotoxicity/myelotoxicity ratio and activity, brostallicin was selected for phase I clinical studies and then proceeded into phase II, the first trial completed in EU recently published.29
Based on the structure of distamycin, which contains three N-methylpyrrole (Py) systems, Lown and co-workers developed the pyrrole–imidazole (Py–Im) polyamides.30 Dervan's group has further advanced these Py–Im polyamides to a class of ‘programmable’ oligomers with high DNA binding sequence specificity and affinity.31 Based on their DNA-binding properties, improving their synthesis, dealing with their cell uptake, distribution and pharmacokinetic parameters as well as in vivo animal studies that lead to clinical trials, some of these Py–Im polyamides (8, Fig. 6) have been developed as potential drugs for therapy-resistant prostate cancer.32 Additionally, another sequence-specific DNA-binding Py–Im polyamide (HIF-PA) that interferes with the binding of hypoxia-inducible transcription factors (HIFs) to DNA has shown a very interesting activity against multiple myeloma (MM). This HIF-PA specifically binds to DNA with an affinity similar to that of HIFs and is able to overcome resistance to hypoxia-mediated apoptosis in MM cells in vitro and in xenograft models. These results provide a rationale to use polyamides to target the cellular hypoxic response of HIFs in future therapeutic agents against MM.
A series of 47 structurally diverse derivatives of distamycin has been prepared by Suckling and co-workers keeping part of the ‘polyamide core’ and introducing modifications both in the ‘tail’ and ‘head’ substructures.33 These MGBs have been evaluated for anti-lung cancer activity by screening against the melanoma B16-F10 cell line, which quickly metastasises to the lungs. Five of these compounds exhibited more cytotoxic activity than the standard gemcitabine and one of them (9, Fig. 6) showed 70-fold improved activity, a selectivity index >125 and metabolic stability becoming a novel lead for future lung cancer treatments.
Sugiyama and co-workers have also actively worked in the field of Py–Im polyamides.34 For example, taking advantage that these polyamides can penetrate the cellular membrane and localize in the nucleus without any delivery agents, they have added mitochondria-penetrating peptide (MPP) to these compounds to show specific localization in the mitochondria of HeLa cancer cells. In that way, these modified MPP-polyamides could repress a particular gene in the mitochondria by recognizing a specific DNA sequence.35 This group has also reported a variety of hairpin Py–Im polyamides conjugated with different structures showing diverse anticancer activities. For instance, conjugates of chlorambucil were found to be active against AML,36 those of seco-1-chloromethyl-5-hydroxy-1,2-dihydro-3H-benz[e]indole (CBI) connected by an indole showed antitumor activity against a variety of cancer cell lines,37 and other CBI conjugates were designed to target oncogenic KRAS mutants that can find application in the treatment of colon cancer.38 More applications of Sugiyama's Py–Im polyamides' applications are described in their recent review.34
Duocarmycins are a type of polyamides that have shown to be exceptionally potent anti-tumour agents acting by reversible and stereo electronically-controlled sequence selective DNA alkylation (i.e. alkylating MGBs),39 and thorough reviews on this class of compounds can be found in the literature.40,41 As mentioned before the duocarmycin CC-1065 (Fig. 3) has shown excellent in vitro anti-tumour activity (e.g. it is 100–400 times more potent than doxorubicin in murine leukemia cells). However, CC-1065 produces hepatotoxicity at therapeutic doses and even though new derivatives (e.g. adozelesin, the pro-drug carzelesin or the symmetrical dimer bizelesin) have been prepared this toxicity has yet to be overcome.
Finally, following the idea of heterocycles connected by amide bonds, a range of di- and triaryl benzamides were prepared and tested for their antiproliferative activity in a panel of 60 human cancer cell lines as well as for their DNA binding activity. Some of these compounds showed cytotoxicity and very high selectivity for K-562, CCRF-CEM, MOLT-4 and SR leukaemia lines.42 The most active compounds were triaryl benzamides and, in a 5-dose testing, compound 10 (Fig. 6) had a LC50 = 78.2 μM for CCRF-CEM. The DNA melting experiments with poly A–T DNA indicated that compound 10 strongly interact in the minor groove (ΔTm = 38.7 °C). All these results allowed establishing certain structure–activity relationships (SAR); thus, addition of a sidechain in the diaryl structures did not affect activity and changing the amide link direction (ArCONHR vs. ArNHCOR linkage) also gave very similar activity profiles.
2.3. Benzimidazoles, isoquinolines and pyrrolobenzodiazepines
One of the best studied MGBs is the bis-benzimidazole pibenzimol (or Hoechst 33258), which was reported to have moderate in vivo activity on the L1210 leukemia cell line. Related aromatic fused heterocycles have been prepared by Lown and co-workers incorporating not only benzimidazole, but also pyridoimidazole and imidazoquinine moieties (11, Fig. 7) and including p-methoxy substituents that are expected to influence the binding characteristics of the ligand.41 They found that these bis-benzimidazoles are active against human renal, CNS, colon and breast cancer as well as melanoma cell lines, with GI50 values between 0.01 and 100 mM. In particular, those compounds with a pyridoimidazole were, in general, more potent. | ||
| Fig. 7 Structure of bis-benzimidazoles 11, trabectedin and the PBD anthramycin indicating the relevant atoms. | ||
Combination of electrophilic groups and MGBs has proven fruitful in the clinic. The most successful of these combinations is trabectedin (ET-743 or Yondelis®, Fig. 7), an isoquinoline derived from Ecteinascidia turbinate (a sea squirt).43 In this compound, the electrophilic carbinolamine moiety is in equilibrium with a reactive iminium cation in vivo, with the pentacyclic core binding to the DNA minor groove.44 Trabectedin has been described to bind to specific sequences of the DNA minor groove, while part of its structure protrudes out of the double helix possibly interacting with proteins such as RNA polymerase II (Pol II).45–48 This isoquinoline is commercialised by Pharma Mar S.A. and Johnson & Johnson for the treatment of advanced soft tissue sarcoma.49 Additionally, trabectedin has shown activity in vitro and in vivo on a wide variety of solid tumour cell lines, human xenografts, ovarian (‘orphan drug status’ granted by FDA in 2005), breast, prostate and renal cancers, melanoma and NSCL. It is known that anti-tumour MGBs are substrates of the transport P-glycoprotein (P-gp) resulting in drug resistance;50 however, a moderate expression of P-gp seems not to reduce the sensitivity to trabectedin. It has been suggested that resistance to this compound is related to DNA transcription and repair; if this is confirmed, tumours resistant to trabectedin may develop hypersensitivity to other DNA damaging drugs, such as Pt complexes, justifying its sequential use with Pt drugs.
In 1963, the first pyrrolobenzodiazepine (PBD) was isolated (anthramycin, Fig. 7) and since then other PDBs such as tomaymycin, sibiromycin or DC-81 have been extracted from different strains of Streptomyces. PDBs have been found to bind to the minor groove of DNA forming a covalent bond with N2-guanine residues in DNA. These natural products are tricyclic systems containing an aromatic ring, a pyrrolidine (fully saturated or unsaturated at either the endocyclic C2–C3 bond or the exocyclic C2) and a 1-4-diazepin-5-one bearing a N10–C11 iminocarbinolamine moiety. The S configuration at the C11a present in all PDBs provides them with a right-handed twist that facilitates the fitting into the DNA minor groove previous to alkylation.51 These PBDs have shown strong anti-tumour activity against a variety of tumours. Both anthramycin and its derivative sibiromycin have been used in clinic against gastrointestinal and breast neoplasms, sarcomas, lymphomas, or Hodgkin's disease.
Despite PBDs not inducing bone marrow depression or hepatic renal or gastrointestinal toxicity, their use is limited by dose limiting cardiotoxicity.52 To avoid this, several groups have reported PBDs' analogues using heterocycles as the aromatic moiety; these compounds retain the anti-tumour activity, lower the cardiotoxicity by avoiding quinone-imine formation and modulate carbinolamine reactivity.53,54 It seems that combination of heterocyclic analogues with a side chain in position 2 of the benzodiazepine ring could drive to even more potent anti-tumour agents lacking cardiotoxicity.
3. Drugs targeting the DNA minor groove as antiviral, antibacterial and antifungal agents
Considering the emergence of multidrug-resistant bacterial and fungal strains that represent a serious problem to human health, there is a clear need for new classes of antimicrobial and antifungal agents. Accordingly, DNA binding agents have been widely explored to combat these infections. Targeting the bacterial DNA replication machinery, such as DNA polymerases or DNA gyrase, is a validated strategy for producing clinically useful antibiotics.55 Additionally, pyrrolobenzodiazepines developed by Thurston, which exert antibacterial activity by covalent binding to a guanine residue within the minor groove of DNA, are entering clinical trials as bactericidal agents against a range of Gram-positive bacteria.56 However, in this review we will focus exclusively in those compounds that directly bind to the DNA minor groove in a non-covalent manner, including some antivirals.3.1. Polyamides derivatives
Considering the novel mode of action of distamycin analogues containing N-terminal biaryl-motifs, a new family of derivatives has been prepared to deal with the alarming rise of multi drug-resistant Mycobacterium tuberculosis strains.57 Thus, compounds 12 (Fig. 8) were synthesised and evaluated for their DNA-sequence selectivity and anti-mycobacterial activity. Thiophene derivatives of polyamides 12 showed 10-fold higher inhibitory activity against M. tuberculosis than distamycin and one in particular exhibited high affinity for the 5′-ACATAT-3′ sequence. The MIC values obtained for these compounds (3.9–250 μg mL−1) indicate that the nature of the biaryl motif was influential on their inhibitory activity and eukaryotic cell toxicity. Thus, dithiophene systems positively influence the anti-mycobacterial activity, with the 2,2-dithiophene polyamide showing 10-fold stronger inhibitory activity (3.9 μg mL−1) against M. tuberculosis H37Rv strain than distamycin (31.25 μg mL−1). Additionally, both 2,2- and 3,3-dithiophene polyamides exhibit the highest anti-tubercular specific growth inhibitory activities. The authors propose that the S atoms in the thiophene systems help the polyamides to cross the highly lipophilic cell-wall of the mycobacteria. Interestingly, the strong anti-tubercular activity of this 2,2-dithiophene derivative does not fully correlate with its DNA sequence selectivity. It could happen that regardless of the high G–C content of the mycobacteria genome,58,59 these MGB polyamides inhibit the activity of mycobacterial transcription factors that bind to AT-rich DNA sequences, regulating pathways involved in antibiotic-induced gene expression in M. tuberculosis. | ||
| Fig. 8 Structure of polyamides 12, 13a, b, and 14a–d, developed as MGBs with antimicrobial and antifungal activity. | ||
The group of Suckling has worked widely in developing polyamides as antimicrobial and antifungal agents.60 Some of their most recent work deals with the evaluation of more effective drug therapies against M. tuberculosis. In general, MGBs show very good activity against various infectious agents, but they had not yet been screened against M. tuberculosis.61 Hence, the mycobactericidal activity of 96 MGBs developed by the authors was determined using a microplate assay and the hits obtained were further screened for their intracellular bactericidal efficacy against the HN878 strain. In order to perform some of these tests and assess the suitability of a drug delivery system, certain MGBs were encapsulated into non-ionic surfactant vesicles (NIVs).62 Thus, seven compounds were identified with MIC99 between 0.39–1.56 μM. Polyamides 13a and 13b (Fig. 8) exhibited intracellular mycobactericidal activity against HN878 (MIC50 = 4.09 and 4.19 μM), with no toxicity. When they were encapsulated into NIVs a 1.6-fold and 2.1-fold increment of mycobacterial activity was achieved. The authors concluded that MGBs have a very strong potential as anti-tubercular therapeutic agents and that NIVs are their best delivery system for intracellular M. tuberculosis infections.
In a different article, Suckling's group63 reported a series of structurally related MGB polyamides prepared and tested against M. tuberculosis and Cryptococcus neoformans (causes cryptococcal meningitis in immunocompromised patients). Compounds 14a and 14b (Fig. 8) have promising MIC80 values against C. neoformans (2 and 4 μg mL−1, respectively) and good selectivity indices. Also relevant are the results obtained for compounds 14c and 14d (Fig. 8), which have MIC99 values of 3.1 μM against M. tuberculosis and limited toxicity. The authors were able to deduce some SAR suggesting that the presence of the thiazole unit (marked in blue in Fig. 8) plays an important role in the anti-infective activity to these MGBs because the exchange of a pyrrole ring by a thiazole increases lipophilicity or DNA affinity.
In a recent study, Bashkin and co-workers reported two novel types of Py–Im polyamides with N-terminal guanidinium or tetra-methylguanidinium functionalities showing antiviral activity against different human papillomavirus (HPV) strains. They found that presence of N-terminal guanidinium results in improved antiviral activity.64 This group also studied the interactions of these antiviral polyamides with DNA.65 Previously, they had reported a series of large hairpin polyamides (with 10 or more rings) with low IC50 values against HPV and no detectable toxicity.66,67 All these Py–Im polyamides exert their antiviral activity by DNA damage response (DDR) mechanism and not by the traditional minor groove recognition process previously reported for polyamides.57,68,69 Bashkin and collaborators hypothesise that even though HBs will be established in the DNA minor groove, the cumulative effect of many hundreds of rigid, crescent polyamides binding to supercoiled DNA is more important for the antiviral activity observed.64
3.2. Amidine-like derivatives
As mentioned in the Introduction, berenil (Fig. 9) is a diaryl bis-amidine that acts as a DNA MGBs finding application in the treatment of infectious diseases such as those caused by Pneumocystis jiroveci (a yeast-like fungus that causes a type of pneumonia). The design of newer and more effective berenil derivatives requires extending the study of the sequence requirements for these MGBs to living cells. Thus, Eckdahl et al.70 have utilised microarray analysis to investigate the effects of berenil on yeast gene expression, enabling an examination of its in vivo sequence binding requirements. The results indicated both the sequence (A–T) and structural features (heteropolymeric character) by which berenil binds to DNA affecting the transcriptional regulation of genes. | ||
| Fig. 9 Structure of berenil, bis-indoles 15 developed by Opperman et al., amidino hydrazones 16 and 17, as well as compounds 18. | ||
The synthesis and biological study of amidinium derivatives of bis-indoles (15, Fig. 9) with potent and broad-spectrum antibacterial activity have been reported by Opperman et al.71 The results obtained show that these bis-indoles are A–T selective MGBs (in vitro and within bacterial cells), which correlates to their power to inhibit DNA and RNA synthesis in Gram-positive (Staphylococcus aureus) and Gram-negative (Escherichia coli) bacteria. It seems that these compounds prompt a DNA damage-inducible SOS response in these bacteria and they are potent inhibitors of cell wall biosynthesis. Actually, the bis-indoles prepared by Opperman et al. are very similar to pentamidine and other diamidine derivatives; however, their cytotoxicity prevents their application as a therapeutic agent in humans.
Other amidino derivatives have been prepared as antimicrobial agents by Shrestha et al.72 They studied two series of bis-(N-amidinohydrazones) and N-amidino-N′-aryl-bis-hydrazones (16 and 17, Fig. 9), against a large panel of bacteria (Gram-positive: Bacillus subtilis, Listeria monocytogenes, MRSA, VRE; and Gram-negative: Acinetobacter baumannii, Enterobacter cloacae, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Salmonella enterica), mycobacterium (M. smegmatis) and fungi (seven Candida albicans strains) and assessed their potential to develop resistance, to induce the production of reactive oxygen species and their toxicity. They found that these amidino hydrazones showed broad-spectrum antibacterial and antifungal activities against most of the tested strains with antibacterial MIC values between <0.5 to >500 μM and antifungal MIC between 1.0 to >31.3 μg mL−1. They also observed minimal resistance development in bacteria and fungi and weak to moderate hERG inhibition (IC50 values from 1.12 to 3.29 μM). Based on this data, the authors were able to deduce a number of SAR: (i) single phenyl rings as linkers (Fig. 9) in compounds 16 are detrimental for antimicrobial activity; (ii) R = CH3 (Fig. 9) in both compounds 16 and 17 is also unfavourable to antibacterial activity, but beneficial for antifungal effects; (iii) long alkyl chains between the two phenyl rings of the linker in series 17 decrease antibacterial effect, but increase antifungal activity; (iv) the nature and position of substituents on the Ph or Ph–Ph moieties (16 or 17, Fig. 9) have different effect on antibacterial or antifungal activities. Thus, the authors concluded that compounds 16 and 17 are potentially promising scaffolds for the discovery of novel antibacterial and antifungal agents.
Even though, in this article, Shrestha et al. do not study the potential interaction of these compounds to DNA, considering their similarity to furamidine (see Fig. 3) it is very likely that they are MGBs. An additional evidence to support this is the recent article of Lazić et al. where they report a series of related bis-amidinohydrazones (18, Fig. 9) exerting anti-Candida activity by means of their interaction with DNA.73 Hence, four bis-amidinohydrazone derivatives were prepared and their activity against a number of Candida strains (two C. albicans, C. glabrata, and C. parapsilosis) evaluated (MICs = 2–15.6 μg mL−1). Then, the authors assessed the interaction of these compounds with DNA; thus, the incubation of DNA from C. albicans with the compounds prepared resulted in the failure of ethidium bromide to intercalate to DNA. Additionally, CD experiments with one of these compounds (18 with a furan linker, Fig. 9) and dsDNA indicated that it binds to DNA but not as an intercalator; further docking studies indicate the suitability of this compound as a MGB. All these results illustrate the expanding potential of bis-amidinohydrazones, which after modifications may further progress to novel antifungal agents.
Even though amidine derivatives have been reported as anti-tubercular agents,74 no clear target has been identified in most of them and, hence, their activity cannot be related to DNA binding.
4. Drugs targeting DNA as antiparasitic agents
Parasitic diseases are caused by infection with organisms such as protozoa, worms or insects. These diseases include malaria (caused by Plasmodium falciparum), sleeping sickness (caused by Trypanosoma gambiense), Chagas disease (caused by Trypanosoma cruzi), leishmaniasis (caused for example by Leishmania donovani), filariasis (caused by Wuchereria bancrofti) or schistosomiasis (caused by Schistosoma mansoni) and are widely extended in Africa, southern Asia, and Central and South America. Currently available therapies are not adequate for most of these diseases; hence, appropriated drugs to treat these conditions are still an unmet medical objective.75In particular, a large body of research has been reported to develop new and efficient therapies for treat malaria, sleeping sickness or leishmaniasis as well as to identify novel targets. Among the latter, DNA has been recognised as a very useful target and many interesting compounds have been reported acting on the DNA minor groove.
4.1. Polyamides
Polyamides, which have already been discussed in this Review as antibacterial and antifungal agents, have also found application as antiparasitic therapies. Thus, Suckling's group prepared a series of diverse polyamides as MGBs and further studied their cytotoxicity against T. b. brucei.76 They found five compounds with nM activity (IC50 > 40 nM) that became new hits for optimisation towards novel treatments for HAT. The authors were able to deduce SARs pointing to the importance of the group connecting the ‘head’ moiety (see Fig. 6 for general scheme of polyamides basic structures) in the modulation of their antiparasitic activity. Additionally, they deduced that passive transport was not the only mechanism for cellular uptake and that some specific transporters recognised by the ‘head’ moiety could also be involved. Moreover, in that study the authors found that those derivatives with amidine groups as the ‘head’ moiety may yield antibacterial activity without antitrypanosomal action. Finally, they confirmed that, similar to their findings in the antibacterial studies, DNA is a possible target for their MGBs in trypanosomes.4.2. Amidine-like derivatives
For many decades, numerous diaryl bis-amidines have shown very important activity in the treatment of trypanosomiasis and leishmaniasis.10 Compounds discovered in the early 1970s such as DAPI, berenil (Fig. 9), stilbamidine or pentamidine (Fig. 3) are still being used for these parasitic infections.77Human African trypanosomiasis (HAT),78 which is caused by Trypanosoma brucei (rhodesiense and gambiense), evolves through two stages (first, parasites in the haemolymphatic system; then invasion, survival and proliferation in the central nervous system), which require different drugs. The previously mentioned pentamidine (Fig. 3) has been used in stage-1 HAT caused by T. b. gambiense, but since it does not cross the blood brain barrier (BBB) is not useful against the later stage of the infection. Moreover, pentamidine only can be administered parenterally with serious adverse effects (i.e. nephrotoxicity, cardiotoxicity, hepatotoxicity). Taking all this into account, and considering the potency of pentamidine as trypanocidal (in vitro IC50 around 1–10 nM), orally available analogues such as furamidine (Fig. 3) have been prepared. Specifically, its N-methoxy prodrug pafuramidine is converted systemically into furamidine by cytochrome P450 metabolism.79 Even though in a phase III clinical trial the oral prodrug showed similar efficiency to injected pentamidine in a 14 day dosing, phase I trial showed nephrotoxicity and pafuramidine development was stopped.
Considering the need for new antimalarial agents with novel modes of action, aromatic bis-amidines, based on the furamidine scaffold, were explored. Accordingly, Sauer et al. prepared a series of derivatives introducing modifications at the furan core as well as at the amidine groups (19, Fig. 10).80 These derivatives were tested in vitro against drug sensitive and resistant P. falciparum lines and human HEK293 cells showing high selectivity for the parasite. Then, SARs could be generated; thus, the N-substituted dimethylfuramidine derivatives (19, R = (CH3)2, Fig. 10) proved to be the most potent and selective anti-plasmodium agents with IC50 values in the 0.02–0.15 μM range. Moreover, 3,4-bis-alkoxymethylenfuramidines with unbranched alkoxy derivatives showed IC50 values in the high nM range against the Dd2 strain of the parasite but, bulky substituents reduced activity against both P. falciparum strains. Additionally, 3-acetamidefuramidines were not active against the parasite. Further, the nature of the linker was also explored by substituting the furan ring by urea and guanidine groups and while the guanidine derivatives showed loss in activity, the urea system exhibited similar nM activity and selectivity for the 3D7 strain as the furan analogues, whereas activity against the Dd2 strain was reduced.
 | ||
| Fig. 10 Structures of furamidine derivatives 19, 20, 21 and 22. | ||
Furthermore, several aza-analogues of furamidine have been reported to cure animal models of stage-2 trypanosomiasis. In particular, the pyridine derivative 20 (Fig. 10) shows an improved safety profile which is suitable to develop new drugs for stage-2 disease and thus 20 can be considered as a potential candidate for clinical advance.81
The mode of action of these antiparasitic diamidines is still being studied. In trypanosomes, the action of pentamidine is related to disintegration of the mitochondrial genome (kinetoplast).82 The kinetoplast is a network of DNA mini- and maxicircles, the minicircles are particularly rich in A–T and it is known that bis-amidines bind specifically A–T sequences in DNA. Considering that mammalian cells do not have a mitochondrial genome related to the kinetoplast, it could be thought that bis-amidines are specific against trypanosomes. However, bis-amidines are similarly efficient against Trypanosoma strains without kinetoplast (e.g. T. evansi) as with it (e.g. T. brucei). Therefore, the DNA in the kinetoplast is unlikely the only target and since bis-amidines also bind to genomic DNA, it is possible that these drugs target the kinetoplast in those strains that have it and genomic DNA in those strains that lack it.
A series of amidine-related dicationic flexible triaryl bis-guanidines (21, Fig. 10) have been prepared and studied as antiparasitic agents by Wilson and collaborators.83 Their in vitro activity against T. b. rhodesiense and P. falciparum was evaluated finding that some of these dications were more active against P. falciparum than pentamidine. Additionally, some of these compounds exerted moderate anti-trypanosomal effect. By means of thermal melting analysis it was possible to assess their ligand–DNA relative binding affinities, and docking studies of the dications with an A–T rich DNA systems helped to understand their binding mode with the minor groove. Hence, it was found that the 1,3-diphenoxyphenyl dications (21, Fig. 10) bind very poorly to DNA (probably because of their poor complementarity with the minor groove) in agreement with their poor activity against T. b. rhodesiense. Additionally, the excellent activity of the 1,4-diphenoxyphenyl guanidines and amidines (21, Fig. 10) against P. falciparum, seems to be only partially related to their minor groove binding and another mode of action may be present.
Boykin, Wilson and collaborators have prepared a family of bis-amidines with indole and benzimidazole bichalcophene cores (22, Fig. 10) as agents against sleeping sickness and malaria.84 Both indole and benzimidazole diamidines bind strongly to the DNA minor groove as was confirmed by UV thermal melting and CD experiments with A–T oligomers. The indole derivatives showed very good in vitro antimalarial activity while the benzimidazole analogues were in general less active. These bis-amidines also show high in vitro antitrypanosomal activity and again the indole derivatives were found to be more active than the benzimidazole series. In particular, one of these derivatives (with an indole–thiophene–furan core) showed and excellent in vivo activity curing all mice infected with T. b. rhodesiense (model of the acute stage of African sleeping sickness) at a low dose of 4–5 mg kg−1 i.p., better than the currently used drug pentamidine.
Dardonville has widely worked in DNA binding guanidines/2-aminoimidazolines as antiparasitic agents. For example, they studied the activity against T. b. rhodesiense of a library of alkane, diphenyl, and aza-alkane bis-guanidines and bis-(2-aminoimidazolines), which show structural similarity to trypanocidal agents such as synthalin, 4,4′-diguanidinodiphenylmethane and the polyamine N-1-(3-aminopropyl) propane-1,3-diamine.85 Most of these compounds exhibited low μM activity, with five of them displaying nM inhibition (23, Fig. 11). Those molecules with an excellent in vitro activity showed also a high selectivity for the parasite, signifying new antitrypanosomal lead compounds.
 | ||
| Fig. 11 Structures of guanidine and 2-aminoimidazoline derivatives 23, 24, 25, 26 and 27. | ||
In a more recent report, Dardonville's group prepared N-alkyl, N-alkoxy, and N-hydroxy bis-guanidines derivatives of the N-phenylbenzamide and 1,3-diphenylurea scaffolds (24, Fig. 11) and screened them in vitro against T. b. rhodesiense (STIB900) and P. falciparum (NF54) parasites.86 While N-alkoxy and N-hydroxy derivatives showed weak μM activity against both parasite lines, the N-alkyl analogues displayed sub μM and low nM inhibitory activity against P. falciparum and T. b. rhodesiense, respectively. Moreover, these compounds optimally bind to A–T oligomers as confirmed by surface plasmon resonance (SPR) experiments. Both the N,N′-diethyl- and N,N′-diisopropylguanidino phenylbenzamides (24, X = CONH, Fig. 11) showed favourable drug-like properties and in vivo efficacy (100% cures) in the STIB900 mouse model of acute HAT and accordingly, they should be considered for further studies.
In collaboration with Dardonville, our group studied an in-house library of guanidine and 2-aminoimidazolines as antiparasitic agents against T. b. rhodesiense (STIB900) and P. falciparum (K1).87 These dicationic diphenyl compounds exhibited in vitro activities in the nM range. Five of them (25 and 26, Fig. 11) cured 100% of treated mice upon intraperitoneal administration at 20 mg kg−1 in the difficult to cure T. b. rhodesiense STIB900 mouse model. However, despite their excellent in vitro antiplasmodial activity and ability to reduce the parasitaemia of mice infected with P. berghei, these compounds did not fully cure the animals in this model and hence they were not studied further.
In general, compounds bearing 2-aminoimidazoline dications showed better safety profiles than the guanidine analogues. Additionally, a correlation between A–T oligomers binding affinity and trypanocidal activity was found for most of the compounds studied supporting formation of a DNA complex as possible mechanism of action. However, no correlation was found between antiplasmodial activity and in vitro inhibition of ferriprotoporphyrin IX bio-mineralisation, suggesting that additional mechanisms of action are likely to be involved. Taking into account these promising results, we believe that bis-(2-aminoimidazoline) derivatives merit further investigation as antiprotozoal agents.
Considering the strong DNA minor groove binding observed for our previous series of diaromatic symmetric guanidinium and 2-aminoimidazolinium derivatives, we prepared new aminoalkyl derivatives (27, Fig. 11) with potential as DNA MGBs and antiprotozoal agents.88 The DNA affinity of these guanidines was evaluated by means of DNA thermal denaturation experiments showing medium to weak binding to A–T oligomers. Additionally, the antiprotozoal activity of these aminoalkyl MGBs was assessed in vitro against T. b. rhodesiense and P. falciparum. Whereas all compounds showed μM activity for T. b. rhodesiense, the O-linked derivatives (Fig. 11) showed promising nM activity against P. falciparum.
Considering the correlation between DNA binding and antiparasitic activity, we concluded that some of the derivatives may exert antimalarial activity by binding to the DNA minor groove, whereas based on some molecular modelling, other compounds could act through dihydrofolate reductase inhibition.
Based on the already mentioned presence of a kinetoplast in the mitochondrial DNA of T. brucei, which is rich in A–T base pairs, different bis-(2-aminoimidazolininium) phenylbenzamide systems (25, X = CONH, Fig. 11 and its 3-chloro derivative) have been developed by Dardonville and de Koning as potential drugs for HAT.89 These compounds exhibit in vitro cytotoxicity against T. brucei and in vivo activity in a mouse model of HAT. With the aim of identifying the cellular target of these compounds, different experiments were performed concluding that they act on the S-phase of T. brucei cell cycle, specifically damaging the kinetoplast. Furthermore, SPR experiments show that both compounds can displace proteins essential for mitochondrial DNA function from their DNA binding sites. Finally, the crystal structure of the complex of an A–T oligonucleotide with compound 25 (where X= CONH) was solved indicating that this derivative fits into the minor groove of DNA, displaces the water spine and interacts with other DNA molecules as a cross-linker. The authors conclude not only that both compounds are powerful trypanocides, but also that they act directly on the kinetoplast.
5. Outlook and conclusions
Compounds such as distamycin or pentamidine represent milestones in the development of DNA minor groove binders with therapeutic activity and they have opened new pathways in the development of more effective therapies. Even though problems such as selectivity still remain, many advances have been made and the DNA minor groove continues being a very important target for therapeutic applications. Thus, in cancer, selectivity issues over healthy cells have been partially overcome by considering the rate of division (cancer cells divide quickly), blood intake or the enhanced permeability and retention (EPR) effect; however, still more work should be devoted to deal with this issue. In the case of antimicrobial agents, DNA has to be confirmed as a useful and selective target in the different bacteria and fungus, and then, much medicinal chemistry research is required considering the compelling need of new antimicrobial drugs. Lastly, in the area of antiparasitic agents, mostly diamidine derivatives with different cores have been reported; therefore once the mode of action is completely understood (i.e. kinetoplast), new and diverse agents should be explored.In terms of selectivity there is another issue that seems not to be dealt with by research groups, why an anticancer DNA targeting drug is not usually a good antimicrobial or antiparasitic DNA-targeting drug? For those antiparasitic drugs that need to target the kinetoplast, the fact that mammalian cells do not have a mitochondrial genome related to the kinetoplast could account for their selectivity towards the parasite. In the case of bacteria (e.g. tuberculosis), the selectivity may be mostly due to the access to the target since transport across the mammalian cell membrane is very different to that in bacteria. Yet, more research should be devoted to this different behaviour of drugs targeting the DNA minor groove.
Compounds that act as MGBs are finding further applications considering new interesting targets such as guanine-quadruplexes (G4s). Nucleic acids within the cell are not only present as the classic Watson–Crick duplex, but also can form different non-canonical structures such as G4s, which are formed by G-rich DNA and RNA regions resulting from the stacking of several G-quartets stabilized by cations such as Na+ and K+.90,91 It has been established that G4s are novel therapeutic targets for cancer, parasitic infections and other conditions since ligands that stabilise these G4s (by stacking or binding to their side grooves) interfere with cellular function.92–95
In summary, considering the importance of DNA in cellular functions (i.e. protein coding, synthesis of specific/functional RNAs, control of gene expression) as well as in diseases caused by parasites and microorganisms, the specific targeting into the minor groove with small molecules offers a broad range of therapeutic possibilities and is still a very appealing and active field of research.
Abbreviations
| HB | Hydrogen bond |
| MGB | Minor groove binder |
| EMA | European Medicines Agency |
| FDA | US Food and Drug Administration |
| IND | Investigational new drug |
| HeLa | Human uterus carcinoma |
| HL-60, K-562, CCRF-CEM, MOLT-4, SR, L1210, Raji and L1210/L-PAM | Leukaemia cell lines |
| MDCK1 | Normal canine kidney cells |
| CaCo2 | Human epithelial colorectal adenocarcinoma cells |
| MCF-7 | Breast carcinoma |
| Kelly | Neuroblastoma |
| HEK293 | Human embryonic cells |
| MM | Multiple myeloma |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| VRE | Vancomycin-resistant Enterococcus |
| HAT | Human African trypanosomiasis |
| hERG | Human ether-a-go-go-related gene |
| IC50 | Drug concentration causing 50% inhibition of the desired activity |
| GI50 | Drug concentration for 50% of maximal inhibition of cell proliferation |
| MIC | Minimum inhibitory concentration |
| Py–Im | Pyrrole–imidazole (polyamides) |
| PBD | Pyrrolo benzodiazepine |
| NIVs | Non-ionic surfactant vesicles |
| ct-DNA | Calf thymus DNA |
| A–T | Adenosine–thymidine pair |
| UV-vis | Ultraviolet-visible spectroscopy |
| CD | Circular dichroism spectroscopy |
| ΔTm | Increment on an oligonucleotide melting/denaturation temperature (°C) |
| SPR | Surface plasmon resonance |
| (Q)SAR | (Quantitative) structure–activity relationships |
| EPR | Enhanced permeability and retention effect |
| G4s | Guanine quadruplexes |
Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work has been supported by the Science Foundation Ireland project SFI-CH3060 (P.O'S.) and an Irish Council Research postgraduate award (A.R.). The authors would like to thank the reviewers' constructive comments to this manuscript.References
- O. Kennard, Pure Appl. Chem., 1993, 65, 1213–1222 CAS.
- C. Avendaño and J. C. Menendez in Medicinal Chemistry of Anticancer Drugs, Elsevier B.V., Chapter 7: DNA intercalators and Topoisomerase inhibitors, 2008, pp. 199–228 Search PubMed.
- D. Sriram, P. Yogeeswari, R. Thirumurugan and T. Ratan Bal, Nat. Prod. Res., 2005, 19, 393–412 CrossRef CAS PubMed.
- N. Ashley and J. Poulton, Biochem. Biophys. Res. Commun., 2009, 378, 450–455 CrossRef CAS PubMed.
- W. O. Foye, Cancer Chemotherapeutic Agents, American Chemical Society, 1995 Search PubMed.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 0, 364–378 CrossRef CAS PubMed.
- J.-P. Armand, V. Ribrag, J.-L. Harrousseau and L. Abrey, Ther. Clin. Risk Manage., 2007, 3, 213–224 CrossRef CAS.
- J. Chen and J. Stubbe, Nat. Rev. Cancer, 2005, 5, 102–112 CrossRef CAS PubMed.
- S. Neidle, Principles of Nucleic Acid Structure, Elsevier, 1st edn., 2008.
- S. Neidle, Nat. Prod. Rep., 2001, 18, 291–309 RSC.
- D. L. Boger and D. S. Johnson, Angew. Chem., Int. Ed. Engl., 1996, 35, 1438–1474 CrossRef CAS.
- K. Gurova, Future Oncol., 2009, 5, 1685–1713 CrossRef CAS PubMed.
- C. Zimmer and U. Wähnert, Prog. Biophys. Mol. Biol., 1986, 47, 31–112 CrossRef CAS.
- W. C. Reinhold, A. Thomas and Y. Pommier, Trends. Cancer, 2017, 2–6 CrossRef PubMed.
- D. M. Thurtle-Schmidt and T. W. Lo, Biochem. Mol. Biol. Educ., 2018, 46, 195–205 CrossRef CAS PubMed.
- M. Pérez-Salvia and M. Esteller, Epigenetics, 2017, 12, 323–339 CrossRef PubMed.
- L. Strekowski and B. Wilson, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2007, 623, 3–13 CrossRef CAS PubMed.
- L. Krstulović, I. Stolić, M. Jukić, T. Opačak-Bernardi, K. Starčević, M. Bajić and L. Glavaš-Obrovac, Eur. J. Med. Chem., 2017, 137, 196–210 CrossRef PubMed.
- H. Y. Alniss, J. Med. Chem., 2018 DOI:10.1021/acs.jmedchem.8b00233.
- M. Munde, S. Wang, A. Kumar, C. E. Stephens, A. A. Farahat, D. W. Boykin and G. M. K. Poon, Nucleic Acids Res., 2014, 42, 1379–1390 CrossRef CAS PubMed.
- I. Antony-Debré, A. Paul, J. Leite, K. Mitchell, H. M. Kim, L. A. Carvajal, T. L. Todorova, K. Huang, A. Kumar, A. A. Farahat, B. Bartholdy, S. R. Narayanagari, J. Chen, A. Ambesi-Impiombato, A. A. Ferrando, I. Mantzaris, E. Gavathiotis, A. Verma, B. Will, D. W. Boykin, W. D. Wilson, G. M. Poon and U. Steidl, J. Clin. Invest., 2017, 127, 4297–4313 CrossRef PubMed.
- I. Bravo, C. Alonso-Moreno, I. Posadas, J. Albaladejo, F. Carrillo-Hermosilla, V. Ceña, A. Garzon, I. Lopez-Solera and L. Romero-Castillo, RSC Adv., 2016, 6, 8267–8276 RSC.
- A. Kahvedžić, S. M. Nathwani, D. Zisterer and I. Rozas, J. Med. Chem., 2013, 56, 451–459 CrossRef PubMed.
- A. Kahvedžić-Seljubac, S. M. Nathwani, D. M. Zisterer and I. Rozas, Eur. J. Med. Chem., 2016, 117, 269–282 CrossRef PubMed.
- F. E. Hahn, Distamycin A and Netropsin, in ed. J. W. Corcoran, F. E. Hahn, J. F. Snell and K. L. Arora, Mechanism of Action of Antimicrobial and Antitumor Agents, Antibiotics, Springer, Berlin, Heidelberg, 1975, vol. 3 Search PubMed.
- G. Pezzoni, M. Grandi, G. Biasoli, L. Capolongo, D. Ballinari, F. C. Giuliani, B. Barbieri, A. Pastori, E. Pesenti, N. Mongelli and F. Spreafico, Br. J. Cancer, 1991, 64, 1047–1050 CrossRef CAS PubMed.
- R. D'Alessio, C. Geroni, G. Biasoli, E. Pesenti, M. Grandi and N. Mongelli, Bioorg. Med. Chem. Lett., 1994, 4, 1467–1472 CrossRef.
- P. Cozzi, Farmaco, 2001, 56, 57–65 CrossRef CAS PubMed.
- M. Leahy, I. Ray-Coquard, J. Verweij, A. Le Cesne, F. Duffaud, P. C. Hogendoom, C. Fowst, C. de Balincourt, E. D. di Paola, M. van Glabbeke, I. Judson and J. Y. Blay, Eur. J. Cancer, 2007, 43, 308–315 CrossRef CAS PubMed.
- J. W. Lown, K. Krowicki, U. G. Bhat, A. Skorobogaty, B. Ward and J. C. Dabrowiak, Biochemistry, 1986, 25, 7408–7416 CrossRef CAS PubMed.
- P. B. Dervan, A. A. Kurmis and P. B. Finn, Molecular Recognition of DNA by Py–Im Polyamides: From Discovery to Oncology, in DNA-targeting Molecules as Therapeutic Agents, ed. M. J. Waring, RSC, 2018 Search PubMed; and references therein.
- A. A. Kurmis, F. Yang, T. R. Welch, N. G. Nickols and P. B. Dervan, Cancer Res., 2017, 77, 2207–2212 CrossRef CAS PubMed.
- F. J. Scott, M. Puig-Sellart, A. I. Khalaf, C. J. Henderson, G. Westrop, D. G. Watson, K. Carter, M. H. Grant and C. J. Suckling, Bioorg. Med. Chem. Lett., 2016, 26, 3478–3486 CrossRef CAS PubMed.
- Y. Kawamoto, T. Bando and H. Sugiyama, Bioorg. Med. Chem., 2018, 26, 1393–1411 CrossRef CAS PubMed.
- T. Hidaka, G. N. Pandian, J. Taniguchi, T. Nobeyama, K. Hashiya, T. Bando and H. Sugiyama, J. Am. Chem. Soc., 2017, 139, 8444–8447 CrossRef CAS PubMed.
- K. Morita, K. Suzuki, S. Maeda, A. Matsuo, Y. Mitsuda, C. Tokushige, G. Kashiwazaki, J. Taniguchi, R. Maeda, M. Noura, M. Hirata, T. Kataoka, A. Yano, Y. Yamada, H. Kiyose, M. Tokumasu, H. Matsuo, S. Tanaka, Y. Okuno, M. Muto, K. Naka, K. Ito, T. Kitamura, Y. Kaneda, P. P. Liu, T. Bando, S. Adachi, H. Sugiyama and Y. Kamikubo, J. Clin. Invest., 2017, 127, 2815–2828 CrossRef PubMed ; and references therein.
- G. Kashiwazaki, T. Bando, T. Yoshidome, S. Masui, T. Takagaki, K. Hashiya, G. N. Pandian, J. Yasuoka, K. Akiyoshi and H. Sugiyama, J. Med. Chem., 2012, 55, 2057–2066 CrossRef CAS PubMed ; and references therein.
- K. Hiraoka, T. Inoue, R. D. Taylor, T. Watanabe, N. Koshikawa, H. Yoda, K. Shinohara, A. Takatori, H. Sugimoto, Y. Maru, T. Denda, K. Fujiwara, A. Balmain, T. Ozaki, T. Bando, H. Sugiyama and H. Nagase, Nat. Commun., 2015, 6, 6706 CrossRef CAS PubMed.
- P. G. Baraldi, A. Bovero, F. Fruttarolo, D. Preti, M. A. Tabrizi, M. G. Pavani and R. Romagnoli, Med. Res. Rev., 2004, 24, 475–528 CrossRef CAS PubMed.
- W. A. Denny, Curr. Med. Chem., 2001, 8, 533–544 CrossRef CAS PubMed.
- B. S. P. Reddy, S. K. Sharma and J. W. Lown, Curr. Med. Chem., 2001, 8, 475–508 CrossRef CAS PubMed.
- G. S. Khan, L. I. Pilkington and D. Barker, Bioorg. Med. Chem. Lett., 2016, 26, 804–808 CrossRef CAS PubMed.
- M. D'Incalci, N. Badri, C. M. Galmarini and P. Allavena, Br. J. Cancer, 2014, 111, 646–650 CrossRef PubMed.
- X. Cai, P. J. Gray and D. D. Von Hoff, Cancer Treat. Rev., 2009, 35, 437–450 CrossRef CAS PubMed.
- L. H. Hurley and M. Zewail-Foote, Adv. Exp. Med. Biol., 2001, 500, 289–299 CrossRef CAS.
- F. Gago and L. H. Hurley, Devising a structural basis for the potent cytotoxic effects of ecteinascidin 743 Small Molecule DNA and RNA Binders: From Synthesis to Nuclear Acid Complexes, ed. M. Demeunynck, C. Bailly and W. D. Wilson, Wiley-VCH: Weinheim, Germany, 2002, pp. 643–675 Search PubMed.
- M. D'Incalci and C. M. Galmarini, Mol. Cancer Ther., 2010, 9, 2157–2163 CrossRef PubMed.
- S. Feuerhahn, C. Giraudon, M. Martinez-Diez, J. A. Bueren-Calabuig, C. M. Galmarini, F. Gago and J. M. Egly, Chem. Biol., 2011, 18, 988–999 CrossRef CAS PubMed.
- https://www.pharmamar.com/yondelis/ .
- B. Colmegna, S. Uboldi, E. Erba and M. D'Incalci, Drug Discovery Today: Technol., 2014, 11, 73–79 CrossRef PubMed.
- B. Gerratana, Med. Res. Rev., 2012, 32, 254–293 CrossRef CAS PubMed.
- C. Cargill, E. Bachmann and G. Zbinden, J. Natl. Cancer Inst., 1974, 53, 481–486 CrossRef CAS PubMed.
- J. Mantaj, P. J. M. Jackson, K. M. Rahman and D. E. Thurston, Angew. Chem., Int. Ed., 2017, 56, 462–488 CrossRef CAS PubMed.
- D. E. Thurston, D. S. Bose, P. W. Howard, T. C. Jenkins, A. Leoni, P. G. Baraldi, A. Guiotto, B. Cacciari, L. R. Kelland, M. P. Faloppe and S. Rault, J. Med. Chem., 1999, 42, 1951–1964 CrossRef CAS PubMed.
- Z. Yin, Y. Wang, L. R. Whittell, S. Jergic, M. Liu, E. Harry, N. E. Dixon, M. J. Kelso, J. L. Beck and A. J. Oakley, Chem. Biol., 2014, 21, 481–487 CrossRef CAS PubMed.
- K. M. Rahman, H. Rosado, J. B. Moreira, E. A. Feuerbaum, K. R. Fox, E. Stecher, P. W. Howard, S. J. Gregson, C. H. James, M. de la Fuente, D. E. Waldron, D. E. Thurston and P. W. Taylor, J. Antimicrob. Chemother., 2012, 67, 1683–1696 CrossRef CAS PubMed.
- F. Brucoli, J. D. Guzman, A. Maitra, C. H. James, K. R. Fox and S. Bhakta, Bioorg. Med. Chem., 2015, 23, 3705–3711 CrossRef CAS PubMed.
- R. Cortesi and E. Esposito, Mini-Rev. Med. Chem., 2010, 10, 217–230 CrossRef PubMed.
- P. B. Dervan, Bioorg. Med. Chem., 2001, 9, 2215–2235 CrossRef CAS PubMed.
- C. J. Suckling, Future Med. Chem., 2012, 4, 971–989 CrossRef CAS PubMed.
- L. Hlaka, M.-J. Rosslee, M. Ozturk, S. Kumar, S. P. Parihar, F. Brombacher, A. I. Khalaf, K. C. Carter, F. J. Scott, C. J. Suckling and R. Guler, J. Antimicrob. Chemother., 2017, 72, 3334–3341 CrossRef CAS PubMed.
- G. P. Kumar and P. Rajeshwarrao, Acta Pharm. Sin. B, 2011, 1, 208–219 CrossRef.
- F. J. Scott, R. J. O. Nichol, A. I. Khalaf, F. Giordani, K. Gillingwater, S. Ramu, A. Elliott, J. Zuegg, P. Duffy, M. J. Rosslee, L. Hlaka, S. Kumar, M. Ozturk, F. Brombacher, M. Barrett, R. Guler and C. J. Suckling, Eur. J. Med. Chem., 2017, 136, 561–572 CrossRef CAS PubMed.
- C. H. Castaneda, M. J. Scuderi, T. G. Edwards, G. D. Harris Jr, C. M. Dupureur, K. J. Koeller, C. Fisher and J. K. Bashkin, MedChemComm, 2016, 7, 2076–2082 RSC.
- E. Vasilieva, J. Niederschulte, Y. Song, G. D. Harris, K. J. Koeller, P. Liao, J. K. Bashkin and C. M. Dupureur, Biochimie, 2016, 127, 103–114 CrossRef CAS PubMed.
- T. G. Edwards, K. J. Koeller, U. Slomczynska, K. Fok, M. Helmus, J. K. Bashkin and C. Fisher, Antiviral Res., 2011, 91, 177–186 CrossRef CAS PubMed.
- T. G. Edwards, T. J. Vidmar, K. Koeller, J. K. Bashkin and C. Fisher, PLoS One, 2013, 8, e75406 CrossRef CAS PubMed.
- T. G. Edwards, M. J. Helmus, K. Koeller, J. K. Bashkin and C. Fisher, J. Virol., 2013, 87, 3979–3989 CrossRef CAS PubMed.
- C. Fisher, J. Clin. Med., 2015, 4, 204–230 CrossRef CAS PubMed.
- T. T. Eckdahl, A. D. Brown, S. N. Hart, K. J. Malloy, M. Shott, G. Yiu, L. L. Mays Hoopes and L. J. Heyer, BMC Genomics, 2008, 9, 32 CrossRef PubMed.
- T. J. Opperman, S. M. Kwasny, J. B. Li, M. A. Lewis, D. Aiello, J. D. Williams, N. P. Peet, D. T. Moir, T. L. Bowlin and E. C. Long, Antimicrob. Agents Chemother., 2016, 60, 7067–7076 CAS.
- S. K. Shrestha, L. M. Kril, K. D. Green, S. Kwiatkowski, V. M. Sviripa, J. R. Nickell, L. P. Dwoskin, D. S. Watt and S. Garneau-Tsodikova, Bioorg. Med. Chem., 2017, 25, 58–66 CrossRef CAS PubMed.
- J. Lazić, V. Ajdačić, S. Vojnovic, M. Zlatović, M. Pekmezovic, S. Mogavero, I. Opsenica and J. Nikodinovic-Runic, Appl. Microbiol. Biotechnol., 2018, 102, 1889–1901 CrossRef PubMed.
- D. Forge, D. Cappoen, J. Laurent, D. Stanicki, A. Mayence, T. L. Huang, L. Verschaeve, K. Huygen and J. J. Vanden Eynde, Eur. J. Med. Chem., 2012, 49, 95–101 CrossRef CAS PubMed.
- M. De Rycker, B. Baragaña, S. L. Duce and I. H. Gilbert, Nature, 2018, 559, 498–506 CrossRef CAS PubMed.
- F. J. Scott, A. I. Khalaf, F. Giordani, P. E. Wong, S. Duffy, M. Barrett, V. M. Avery and C. J. Suckling, Eur. J. Med. Chem., 2016, 116, 116–125 CrossRef CAS.
- M. Basselin, H. Denise, G. H. Coombs and M. P. Barrett, Antimicrob. Agents Chemother., 2002, 46, 3731–3738 CrossRef CAS.
- M. P. Barrett, C. G. Gemmell and C. J. Suckling, Pharmacol. Ther., 2013, 139, 12–23 CrossRef CAS PubMed.
- M. Z. Wang, J. Y. Saulter, E. Usuki, Y. L. Cheung, M. Hall, A. S. Bridges, G. Loewen, O. I. Parkinson, C. E. Stephens, J. L. Allen, C. Zeldin, D. W. Boykin, R. R. Tidwell, A. Parkinson, M. F. Paine and J. E. Hall, Drug Metab. Dispos., 2006, 34, 1985–1994 CrossRef CAS PubMed.
- B. Sauer, T. S. Skinner-Adams, A. Bouchut, M. J. Chua, C. Pierrot, F. Erdmann, D. Robaa, M. Schmidt, J. Khalife, K. T. Andrews and W. Sippl, Eur. J. Med. Chem., 2017, 127, 22–40 CrossRef CAS PubMed.
- T. Wenzler, D. W. Boykin, M. A. Ismail, J. E. Hall, R. R. Tidwell and R. Brun, Antimicrob. Agents Chemother., 2009, 53, 4185–4192 CrossRef CAS PubMed.
- A. Schnaufer, G. J. Domingo and K. Stuart, Int. J. Parasitol., 2002, 32, 1071–1084 CrossRef CAS PubMed.
- R. K. Arafa, T. Wenzler, R. Brun, Y. Chai and W. D. Wilson, Eur. J. Med. Chem., 2011, 46, 5852–5860 CrossRef CAS PubMed.
- A. A. Farahat, M. A. Ismail, A. Kumar, T. Wenzler, R. Brun, A. Paul, W. D. Wilson and D. W. Boykin, Eur. J. Med. Chem., 2017, 143, 1–7 Search PubMed.
- C. Dardonville and R. Brun, J. Med. Chem., 2004, 47, 2296–2307 CrossRef CAS PubMed.
- C. H. Ríos Martínez, L. Lagartera, M. Kaiser and C. Dardonville, Eur. J. Med. Chem., 2014, 81, 481–491 CrossRef PubMed.
- F. Rodríguez, I. Rozas, M. Kaiser, R. Brun, B. Nguyen, W. D. Wilson, R. N. Garcia and C. Dardonville, J. Med. Chem., 2008, 51, 909–923 CrossRef PubMed.
- C. McKeever, M. Kaiser and I. Rozas, J. Med. Chem., 2013, 56, 700–711 CrossRef CAS PubMed.
- C. R. Millan, F. J. Acosta-Reyes, L. Lagartera, G. U. Ebiloma, L. Lemgruber, J. J. Nué Martínez, N. Saperas, C. Dardonville, H. P. de Koning and J. L. Campos, Nucleic Acids Res., 2017, 45, 8378–8391 CrossRef CAS PubMed.
- Quadruplex Nucleic Acids, ed. S. Neidle and S. Balasubramanian, RSC publishing, Cambridge, 2006 Search PubMed; and references therein.
- G. Biffi, D. Tannahill, J. McCafferty and S. Balasubramanian, Nat. Chem., 2013, 5, 182–186 CrossRef CAS PubMed.
- L. M. Harris and C. J. Merrick, PLoS Pathog., 2015, 11, e1004562 CrossRef PubMed.
- J. D. Rohrer, A. M. Isaacs, S. Mizielinska, S. Mead, T. Lashley, S. Wray, K. Sidle, P. Fratta, R. W. Orrell, J. Hardy, J. Holton, T. Revesz, M. N. Rossor and J. D. Warren, Lancet Neurol., 2015, 14, 291–301 CrossRef CAS PubMed.
- N. Maizels and L. T. Gray, PLoS Genet., 2013, 9, e1003468 CrossRef CAS PubMed.
- T. Shalaby, G. Fiaschetti, K. Nagasawa, K. Shin-ya, M. Baumgartner and M. Grotzer, Molecules, 2013, 18, 12500–12537 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |