
Sample pre-concentration on a digital microfluidic platform for rapid AMR detection in urine†
Sumit
Kalsi
ab,
Martha
Valiadi
ab,
Carrie
Turner
c,
Mark
Sutton
c and
Hywel
Morgan
*ab
aElectronics and Computer Science, University of Southampton, Southampton SO17 1BJ, UK. E-mail: hm@ecs.soton.ac.uk; Tel: +44 (0)2380 593330
bInstitute for Life Sciences, University of Southampton, Southampton SO17 1BJ, UK
cNational Infections Service, Public Health England, Porton Down, Salisbury SP4 0JG, UK
First published on 23rd November 2018
There is a growing need for rapid diagnostic methods to support stewardship of antibiotics. We describe an analytical platform for sample concentration to detect antimicrobial resistance (AMR) genes directly from human urine for the diagnosis of urinary tract infections (UTIs) that are resistant to antibiotics. A sample-processing unit concentrates plasmid DNA directly from urine using magnetic beads, followed by isothermal amplification of target genes. The sample pre-concentration unit interfaces with a digital microfluidic platform (DMF) and scales the sample volume by 500-fold, pre-concentrating DNA from 1 mL into a 2 μL droplet for downstream processing. Tests with a clinical strain of Klebsiella pneumoniae (NCTC 13443), spiked into human urine demonstrated a limit of detection of 104 cfu mL−1 and a “sample to answer” detection in approximately 30 minutes.Introduction
Antimicrobial resistance (AMR) is a growing worldwide public health problem, and is expected to become the primary cause of death and claim 10 million lives per year by 2050.1 The latest guidance recommends that by 2020, no antibiotics should be prescribed to patients unless resistance to that antibiotic has been excluded by a diagnostic test.1 Currently, routine testing is not performed and the tests that are in use to detect antibiotic resistant bacteria, rely on cell culture and/or molecular detection that require dedicated laboratories, expert users and days to complete. One of the most common bacterial infections is urinary tract infections (UTIs) which account for substantial health costs and morbidity.2 UTIs are the second most common cause of bacteremia in hospitalized patients and are responsible for as many as 40% of nosocomial infections.3 Organisms including Escherichia coli and Klebsiella pneumoniae that produce extended spectrum β-lactamase (ESBL) and carbapenemase enzymes have emerged as a major healthcare issue over the last 10–20 years.4 Amongst the wide range of ESBL enzymes, the CTX-M enzyme family is the most prevalent, conferring resistance to key β-lactam antibiotics, including 3rd generation cephalosporins5–7 in at least 26 bacterial species, residing in both nosocomial and community environments.7 Therefore, a gene encoding the widespread CTX-M-15 isoform, blaCTX-M-15, which encodes a protein with enhanced catalytic activity7 is an ideal target candidate for rapid point-of-care (PoC) detection of antibiotic resistant pathogens.Numerous nucleic acid-based assays have been developed and implemented for pathogen detection on microdevices, particularly since the advent of isothermal amplification techniques.8–12 One attractive method is recombinase polymerase amplification (RPA) which is growing in popularity owing to its low incubation temperature, ease of primer design, and sensitivity and high tolerance to impurities eliminating the need for elaborated DNA purification protocols.13,14 Previously, we have demonstrated a programmable digital microfluidic (DMF) platform to perform RPA for rapid detection of AMR encoding genes with an LoD of approximately 10 copies within 10 minutes.12,15 The device enables automated and simultaneous manipulation of many nanolitre (nL) droplets of liquid on an array of thousands of individually addressable microelectrodes.16 Our previous publications used DNA samples extracted and purified using commercial kits with sample preparation separate from the analysis chip. In many diagnostic platforms, sample preparation often remains a rate-limiting step, and in the case of UTIs, clinical samples of several mL must also be scaled down by at least 2–3 orders of magnitude to the μL (or nL) volumes required for PoC assays. This requires technologies for pre-concentration of target cells or the genetic material from large volumes of the clinical sample.
A number of different approaches have been developed to pre-concentrate samples, for example, capturing bacterial cells from solution using aptamer or antibody coated magnetic microparticles17–20 or microfluidic chip surfaces21 or ion exchange beads,22–24 but all of these approaches are either restricted to a limited range or have variable capture efficiency depending on the species and on the composition of the sample matrix. Cell lysis to release nucleic acids into solution is the most direct way of performing molecular analysis of a whole bacterial community within a clinical sample. Nucleic acid extraction on microdevices is usually based on a miniaturized version of bench-top techniques that include cell lysis, DNA capture onto a solid surface (beads or columns), followed by washing and finally elution for subsequent analysis. Cell lysis often requires pre-concentration by centrifugation off-chip.25,26 Subsequent steps are then incorporated into microfluidic devices. Microfluidic sample preparation has been demonstrated extensively for blood samples. For example, extraction of RNA27 or DNA28 from human whole blood has been implemented on a DMF platform, with a subsequent downstream analysis off device. However, bacterial cells are generally hard to lyse and samples must be subjected to more aggressive procedures. These may include a combination of lysis by heat, enzymes or mechanical forces. Heat lysis is very simple, and has been successfully used for complete extraction and detection of DNA of Staphylococcus and Streptococcus from saliva samples.29 However, when target cells are very dilute, heat lysis requires a pre-concentration step to achieve clinically relevant detection limits. Mechanical forces generated by microstructures have been used to improve the enzymatic lysis of bacteria in human urine, before DNA recovery on a solid phase extraction (SPE) column for purification with off-chip analyses.30 van Heirstraeten et al. (2014) scaled down a complete standard DNA extraction protocol utilizing an SPE column, extracting Gram-positive and negative bacteria, and virus DNA from swab samples.31 A centrifugal Lab-on-a-Disk analysis system demonstrated a complete “sample in – answer out” assay to detect bacterial pathogens.32 This included DNA extraction after cell lysis, DNA capture on magnetic beads, washing and release for multiplex PCR. The authors demonstrated a LoD between 2 and 5 cfu in 200 μL serum samples with a “sample-to-result” time of under 4 hours. Choi et al. (2016) demonstrated an RPA assay using a centrifugal platform for the identification of three different organisms in contaminated milk with a LoD of 4 cells per 3.2 μL of milk (1250 cells per ml) within 30 min.33 Mosley et al. (2016) described a simple method for lysing and extracting DNA from the Gram-negative bacteria Helicobacter pylori in stool samples by the addition of guanidine hydrochloride.34 Magnetic microparticles were used to capture the DNA from the crude lysate where the DNA bead sample was cleaned by pulling the beads through an immiscible mineral oil, through an adaptation of the IFAST (immiscible filtration assisted by surface tension) technique developed by the Beebe group.35 Recently, the Wheeler group demonstrated a pre-concentration technique (liquid intake by paper) that allows interfacing of larger volume to the digital microfluidics platform. They concentrated protein biomarkers using magnetic beads.36,37
In this publication, we describe a simple sample pre-concentration unit compatible with a DMF platform, and analyse processed urine samples for identification of AMR. Bacterial plasmids are pivotal in the transfer and acquisition of resistance genes,38,39 therefore we ensured that the extraction and concentration method would be suitable for both plasmid and genomic DNA. Viable bacteria suspended in a urine sample (typically 1 mL) are heat lysed (on a Thermomixer) to release genetic material in the presence of a chaotropic salt and plasmid binding beads. The sample with DNA immobilised on the magnetic beads is processed with the pre-concentration unit that interfaces directly with the DMF platform. The DNA loaded beads are pulled from the main sample through an immiscible oil/aqueous interface directly onto the DMF platform, reducing the sample volume from 1 mL to 2 μL in a single step. No wash steps are performed and the entire assay takes less than 30 minutes with a LoD of 104 bacteria cfu mL−1, which is compatible with the requirements for direct detection of bacteria causing UTIs and in line with clinical recommendations in the UK.2 The utility of the assay was demonstrated by spiking a clinical isolate of Klebsiella pneumoniae into human urine followed by detection of the blaCTX-M-15 gene. This provides evidence that the technology could be applied to enable rapid detection of this problematic multidrug resistant (MDR) bacteria, as a model for detecting commonly occurring UTI pathogens direct from urine samples.
Methods
Bacterial culture and control DNA (genomic and plasmid) extraction
Klebsiella pneumoniae NCTC 13443, isolated from a human clinical sample was obtained from the National Collection of Type Cultures (NCTC) at the Public Health England (PHE). The genome of this strain has been sequenced (EBI accession number SAMEA2742597) and is known to contain genes for the ESBL CTX-M-15 and the carbapenemase NDM-1. Routine maintenance of the bacteria on tryptone soy agar (TSA) plates, cell inoculations and cell counts of cultures in tryptone soy broth (TSB) medium was conducted as previously described.24 Overnight cultures in TSB medium were diluted 20-fold the following morning and incubated at 37 °C for a further 1 h to ensure that the cells were in the early exponential growth phase. Control DNA, containing plasmids, was obtained from an overnight culture of Klebsiella pneumoniae NCTC 13443. For the benchtop assays, DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, UK) following the manufacturer's protocol. The eluted DNA was quantified on a Qubit® fluorometer using DNA assay reagents.Test samples
Experiments were conducted using human urine spiked with Klebsiella pneumoniae NCTC 13443 to simulate clinical samples with an infection of antibiotic resistant bacteria. Urine samples from healthy volunteers were collected with ethical approval from the Southampton Research Biorepository Access Committee (Ref: 12/NW/0794). All experiments were performed in accordance with the guidelines from the University Hospital Southampton R & D policies, and approved by the ethics committee at the University Hospital Southampton. Informed consents were obtained from the human participants of this study.Approximately 50 mL of mid-stream urine was collected into sterile polypropylene tubes. Urine from four volunteers was used to evaluate the robustness of the assay for variability in sample composition, for example urine hydration.
Extraction of bacterial DNA for the benchtop assay
A benchtop protocol for extraction of DNA from the bacteria spiked into urine was designed that could be translated to the DMF platform. The protocol is shown in Fig. 1. 525 μL of unfiltered urine was spiked with 100 μL of K. pneumoniae bacteria of varying concentrations. 375 μL of GuHCl at 8 M was then added to the sample to obtain a final concentration of 3 M and a final volume of 1 mL. To specifically isolate the plasmid DNA (rather than total genomic DNA), we used MagneSil™ Red paramagnetic particles (Promega, UK). 2.5 μL of the stock solution of these beads was added to the solution and the sample incubated at 90 °C for 10 minutes with continuous mixing at 800 rpm on a thermomixer (Thermomixer comfort, Eppendorf, Germany). This bacterial lysis and plasmid capture protocol was identical for both the benchtop and DMF platform. The sample was allowed to cool to room temperature, following which the beads were concentrated on the side of the tube with a magnet as shown in Fig. 1. Approximately 950 μL of the supernatant was removed and discarded, and after re-suspending the beads, the remaining 50 μL volume was covered with a layer of dodecane (100 μL). The beads were then extracted by pulling them through the oil–urine interface with a magnet (step 6). After discarding the remaining solution, the bead pellet was re-suspended in the same tube with 5 μL of elution buffer for five minutes (Promega, UK) to elute the plasmid from the beads (step 7). Finally, the beads were concentrated on the side of the tube with a magnet and the resulting plasmid (in the elution buffer) was used in the bench-top RPA assays (step 8).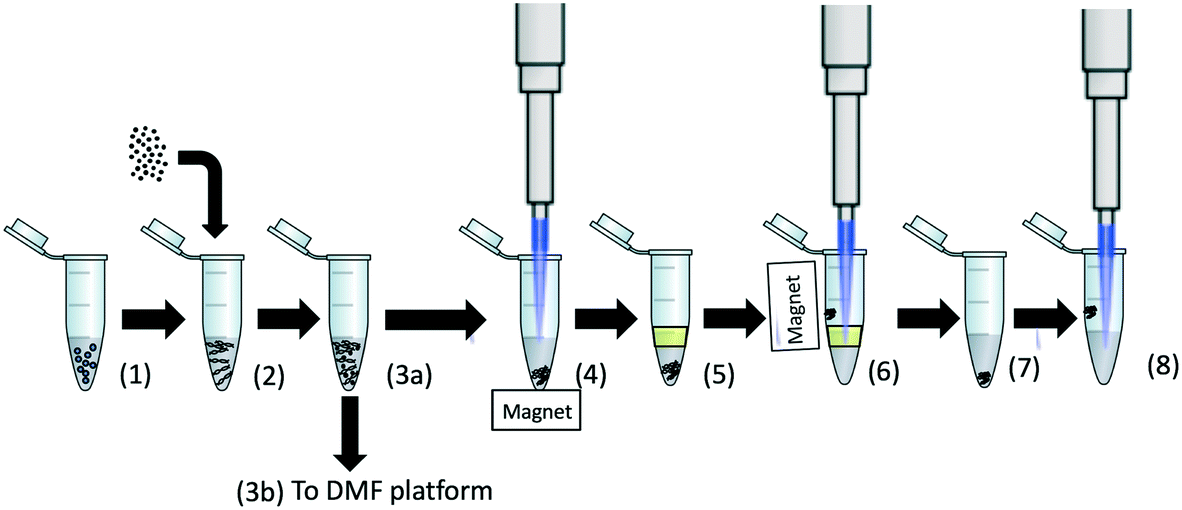 | ||
| Fig. 1 Schematic representation of the workflow for the benchtop RPA assay on urine spiked with Klebsiella pneumoniae NCTC 13443. (1) The unfiltered urine sample is spiked with bacteria, and GuHCl is added to a final concentration of 3 M in the sample (total sample volume = 1 mL). (2) Magnesil red beads (2.5 μL) are added and the sample is incubated at 90 °C for 10 minutes. (3a) The sample is cooled to room temperature. (4) The beads are concentrated into a pellet at the bottom of tube and 950 μL of the supernatant is removed. (5) 100 μL of dodecane is added. (6) The beads are pulled through an oil phase to avoid carry over of inhibitors. The remaining solution in the tube is removed and (7) the bead pellet is suspended in 5 μL of elution buffer to elute the plasmid from the beads. (8) The beads are pelleted on the sidewall and the entire volume of eluate is used in the benchtop RPA. For the DMF system, the assay is the same except that the sample is introduced to the device at point (3b). | ||
Sample pre-concentration device and DMF assembly
The assembled DMF platform is shown in Fig. 2A and B. It consists of a thin film transistor (TFT) backplane that drives the electrodes, together with a transparent glass indium tin oxide (ITO) top plate. The sample pre-concentration unit is attached to this top plate (Fig. 2C and D). The TFT backplane comprises an active matrix array of 96 × 175 electrodes, each approximately 200 μm × 200 μm in size. The TFT backplane also has 9 large volume (2.2 μL) fluid input pads at the periphery of the active matrix array. Each of these fluid input pads is made of 7 discrete electrodes [see ref. 12] with a footprint of 3 mm × 9 mm. Custom electronics were used to provide electrode actuation signals to all the different electrodes. The device is described further in (ref. 12) and an image of the DMF assembly is shown in Fig. 2B. The DMF platform was assembled by physically clamping the ITO top plate to the bottom with a 125 μm spacer. | ||
| Fig. 2 (A) Schematic representation and (B) an image of the assembled DMF platform. The assembly consists of the driver (TFT) electronics backplane, a conducting ITO top electrode plate and the sample pre-concentration unit. Custom electronics is used to provide the DMF actuation signals. (C) Plan view of the ITO top electrode plate of the DMF device with the sample pre-concentration unit. This is fabricated from PMMA using a laser cutter and bonded to the top glass using double sided adhesive tape which provides the fluidic channel. (D) Side view of the same. The bead sample is introduced into the chamber and slowly fills the channel defined by the spacer until it hits the capillary stop. A magnet is placed beneath the TFT substrate and translated to guide the beads through the aperture in the ITO top plate into the elution buffer reservoir, see Fig. 3. Drawing not to scale. | ||
To analyse urine samples directly on the DMF platform, a sample pre-concentration unit was designed to directly interface with the top plate of the device. A schematic diagram of this unit is shown in Fig. 2C and D. It is designed to enable the same work-flow as that of the bench-top assay (Fig. 1) delivering magnetic particles (with bound plasmids) to fluid input pads of the DMF device (see below).12 The pre-concentration unit comprises a 1.2 mL volume chamber made from a 10 mm thick PMMA sheet (Techsoft, UK) by laser micromachining (Epilog Laser, UK). A thin spacer (275 μm) forms a microfluidic channel beneath the chamber (Fig. 2D). The channel is 7.2 mm long and terminates with a capillary stop (diameter 1 mm) (see Fig. 2D). The fluid channel crosses a 1 mm opening in the ITO top plate through which the bead–plasmid mixture is extracted onto the fluid input pads. The channel was cut from a 175 μm clear PMMA sheet (Goodfellow, UK) and stuck to both surfaces with a 50 μm double-side adhesive (467MP, 3M, USA), total depth = 275 μm.
Sample pre-concentration protocol on DMF
Before pre-concentration of the sample and its subsequent amplification on DMF, RPA reagent drops were pipetted on the TFT backplane. ∼2 μL droplets were dispensed onto the fluid input reservoirs as follows: 2.2 μL each of the RPA mastermix, magnesium acetate, no template control (NTC, nuclease free water), positive control (DNA 100 pg uL−1) and 2 μL of elution buffer (Promega, UK). The elution buffer droplet was placed on the fluid input reservoir pad 4 (see Fig. 2A). These pad electrodes were electrically actuated to pin the droplets to the substrate whilst the device was loaded with dodecane. The dodecane fills the gap between the top plate and TFT backplane and stops at the hole in the top plate. Fig. 3 and S1† shows the assay flow for plasmid capture and pre-concentration on the DMF platform. To begin an assay, 1 mL of the urine sample containing lysed bacteria and magnetic beads with plasmids was pipetted into the chamber of the sample concentration unit. The shallow channel at the base of the unit slowly fills with urine by capillary action until it stops at the capillary stop as shown in Fig. 3(1) and S1(1).† The dodecane in the hole in the DMF top plate forms an immiscible interface with the urine. The beads in the urine were pulled to the bottom of the chamber with a magnet (Fig. 3(2) and S1(2)†). The bead pellet was then transported along the microfluidic channel by manually moving the magnet to bring the bead pellet over the hole in the top plate (Fig. 3(3) and S1(3)†) and onto an empty fluid input reservoir pad (Fig. 3(4) and S1(4)†). The plasmids were eluted off the beads by pulling the bead pellet through the oil in the DMF device onto a neighbouring fluid reservoir pad that contained elution buffer (Fig. S3(5) and S1(5)†). The beads were dispersed by moving the magnet and incubated for 5 minutes in the elution buffer. The “waste” beads were pulled off the pad into the oil leaving the bacterial-plasmids in the reservoir droplet (Fig. 3(6)). Fig. S2† shows photographs of this sequence. To initiate the DNA amplification assay, different daughter droplets were dispensed from the respective reservoirs (including the sample) onto the DMF chip for amplification by RPA (Fig. 3(6–8)). | ||
| Fig. 3 Protocol for sample pre-concentration and DNA amplification on the DMF platform. Reservoir electrodes are pre-loaded with reagents, represented by different colors: red magnesium acetate, yellow RPA mastermix, green NTC, blue elution buffer and purple positive DNA control; (1) urine sample spiked with bacteria (lysed) with magnetic beads as prepared in Fig. 1 pipetted into the sample chamber. (2) The urine sample slowly fills the channel between the sample pre-concentration unit and the ITO glass plate, stopping at the capillary stop. (3) Using a permanent magnet, the beads are pelleted and pulled along this channel and then through the aperture in the glass top plate onto the TFT back plane (4). (5) The beads are dragged through the oil in the DMF device onto the reservoir pad with elution buffer, and incubated for five minutes to release the plasmids from the beads. (6) Droplets are dispensed from the reagent reservoirs. (7) The plasmids, NTC and positive control droplets are merged and mixed with the RPA mastermix. (8) Magnesium acetate droplets are dispensed and mixed. Black dotted arrows represent the direction of droplet movement for mixing. Figure not to scale. | ||
The efficiency of the bead extraction process was evaluated using optical absorbance measurements. A 1 mL urine sample with beads was loaded into the pre-concentration unit. The beads were pulled onto the DMF platform and then moved to the edge of the top plate (with the magnet) through the oil for recovery into an Eppendorf tube. The beads were re-suspended into 1 mL of the same urine sample and absorbance measurements used to estimate bead recovery. In a separate experiment, the residual sample volume carried over by the beads through the oil phase was estimated using fluorescein dye. The beads were suspended in 1 μM fluorescein in PBS (1 mL), and this solution was loaded into the sample chamber of the pre-concentration unit. The beads were pulled onto the TFT backplane and through the oil for recovery. The beads were then re-suspended to their original volume in PBS, and fluorescence measurements were used to quantify the amount of fluorescein carried over to the TFT backplane in the interstitial space of the packed beads.
Detection of the blaCTX-M-15 gene
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 for the RPA reaction mix, sample and magnesium acetate, respectively. The RPA reaction mix (enzymes, rehydration solution, primers and probe) was prepared so that the final assay component concentrations in the reaction droplet were identical to those in the 50 μL benchtop assay.
:1:1 for the RPA reaction mix, sample and magnesium acetate, respectively. The RPA reaction mix (enzymes, rehydration solution, primers and probe) was prepared so that the final assay component concentrations in the reaction droplet were identical to those in the 50 μL benchtop assay.
The protocol for performing the assay on the DMF chip is shown in Fig. 3(steps 6 to 8). Daughter droplets were aliquoted from the reservoirs; DNA, NTC and positive control samples were mixed with the RPA mastermix by moving droplets back and forth along the device as described previously.15 Magnesium acetate was then mixed and the DNA amplification was initiated by heating the device to 39 °C. In order to image all droplets at the same time, a custom wide-field imaging system was developed,15 comprising a high sensitivity SLR camera (Canon 5D, Mark III) with a macro lens (55 mm focal length) and an LED illuminator (Nathaniel Group, USA). Two different filters were used, an excitation (590–650 nm, Semrock, UK) and an emission filter on the camera (670–740 nm, Semrock, UK).
For a typical assay, six (45 nL) droplets were used: three sample, two NTC and a positive control. These droplets were mixed with the RPA mastermix (180 nL) for 30 seconds and then with magnesium acetate (45 nL) for 20 seconds. The entire sequence of droplet manipulation was programmed into the DMF software. During the assay, sample droplets were mixed continuously for 20 s and then the fluorescence signal was acquired for 2.5 seconds, repeated at 30 second intervals. Image processing and analysis was performed using a custom MATLAB script.15
Results and discussion
Protocol optimisation: extraction and pre-concentration on the benchtop platform
The procedure for the pre-concentration of target DNA, particularly plasmids, onto magnetic beads was adapted from ref. 35 and based on filtration through immiscible phase. The method provides a single step purification and pre-concentration. Although this DNA isolation protocol is simple and does not involve any wash steps, the beads do carry over a finite volume of both urine and guanidine in the process. Urine is known to contain several PCR inhibitors,40 so the robustness of the protocol was evaluated by measuring any possible inhibition of the RPA. Unfiltered urine was added to the RPA mix (1 μl in a 50 μL reaction mixture) and the time to positivity (TTP) was measured for an equivalent of 90000 copies of purified DNA (genomic and plasmids). The change in the TTP was compared against a reference value, obtained with the same amount of DNA suspended in 1 μL of nuclease free water. A slight increase in the TTP was observed as shown in Fig. 4A (urine from four different volunteers). The conductivity of the four samples was 0.6, 0.8, 1.3 and 1.0 mS cm−1. As reported previously,40 samples containing small amounts of urine can be amplified successfully. Our results show that the reaction becomes slower (evident from the increase in TTP) and does not appear to have any correlation with the amount of salt in the urine.
 | ||
| Fig. 4 (A) Effect of the addition of unfiltered urine to the RPA reaction mixture (1:50 dilution) plotted as a change in the TTP compared with the mean TTP of the positive control (purified K. pneumoniae plasmid DNA, 90000 copies). The DNA was added to the RPA reaction mix prepared as per manufacturer's recommendation, except that the 1 μL of nuclease free water is replaced by urine. Data from duplicates. (B) Capture efficiency of plasmid DNA onto the beads at different concentrations of GuHCl in PBS and urine, quantified as the change in TTP compared with that of the positive control. Purified plasmid DNA (90000 copies) was added to either phosphate buffer saline (PBS) or urine containing different concentrations of GuHCl and captured on 2.5 μL of magnetic beads. Plasmid was eluted into 5 μL of elution buffer. Experiments were performed using the protocol shown in Fig. 1. Data from duplicates. | ||
It is also known that denaturing reagents such as guanidine can inhibit enzyme activity during DNA amplification.41 To assess the effect of guanidine on RPA, and also to determine the optimal concentration of guanidine for plasmid DNA capture, purified control DNA (genomic and plasmid) was spiked into either PBS or urine with varying concentrations of GuHCl. Plasmid DNA was captured on magnetic beads (as in Fig. 1) and eluted into 5 μL elution buffer. The change in TTP compared with that of the positive control (no guanidine and no PBS/urine) was measured. The data is shown in Fig. 4B. For PBS, there is a clear correlation between increasing GuHCl and increasing TTP, indicating a reduction in the capture efficiency and/or inhibition of the RPA enzymes by the carried over guanidine. A similar trend is seen for the urine samples, except that the increase in the TTP is significantly less than that for PBS. The DNA spiked in urine sample without any GuHCl was also captured to some extent and successfully amplified, implying that the DNA was captured on beads in the absence of any chaotropic agent. However the TTP was significantly increased (by ∼4 min). From this data, the optimal concentration of GuHCl in the final protocol was fixed at 3 M to enable fast diagnosis. For 3 M GuHCl, the change in TTP of ∼1 min is within the 3 sigma of the effect produced by the addition of urine (Fig. 4A), suggesting excellent capture efficiency and downstream amplification of target genes.
Full bench-top assay: plasmid DNA extraction and amplification
The effect of heat on the DNA extraction efficiency was also evaluated (for two different volunteer urine samples), with the addition of GuHCl at 3 M or 5 M. Samples were either left to incubate at room temperature or heated (90 °C for 10 min) and the DNA was processed as described previously. Table 1 shows the average TTP (min) for replicate experiments. For a given sample, the optimal conditions are 3 M GuHCl with heat. Although DNA amplification was observed for samples that were not heated, they had a much longer TTP suggesting poor DNA capture and/or sample lysis.| Assay variable | TTP (min) no heat | TTP (min) heat |
|---|---|---|
| 5 M GuHCl, urine, 7.5 mS cm−1, 1.3 × 107 cfu mL−1Klebsiella pneumoniae | 22.7* | 18.3* |
| 5 M GuHCl, urine, 7.8 mS cm−1, 1.9 × 106 cfu mL−1Klebsiella pneumoniae | ** | 23.8 ± 3.1 |
| 3 M GuHCl, urine, 7.5 mS cm−1, 1.3 × 107 cfu mL−1Klebsiella pneumoniae | 20.5 ± 3.4 | 16.7 ± 1.3 |
| 3 M GuHCl, urine, 7.8 mS cm−1, 1.9 × 106 cfu mL−1Klebsiella pneumoniae | 18.9* | 16 ± 1.1 |
Fig. 5A shows data from a typical bench-top experiment performed using the protocol shown in Fig. 1. Consistent amplification of DNA extracted from Klebsiella pneumoniae was observed with a LoD of 103 cfu mL−1 (TTP ∼27 min). Fig. 5B shows the TTP vs. the log10 concentration of bacteria in urine. This data is representative for a sample, but the TTP values vary from volunteer to volunteer and also depend on the time of day the urine was taken. The plot shows a linear trend from 106 cfu mL−1 to 104 cfu mL−1 and an exponential increase in the TTP from 104 cfu mL−1 to 103 cfu mL−1 (with TTP doubling). Assuming that 1 cfu corresponds to 1 blaCTX-M-15 gene on 1 plasmid and comparing the TTP in Fig. 5B to calibration plots for purified DNA (Fig. S3†), the plasmid capture efficiency from lysed bacteria in urine is estimated to be >80% for a bacterial load of >104 cfu mL−1 and <5% for a bacterial load of <104 cfu mL−1.
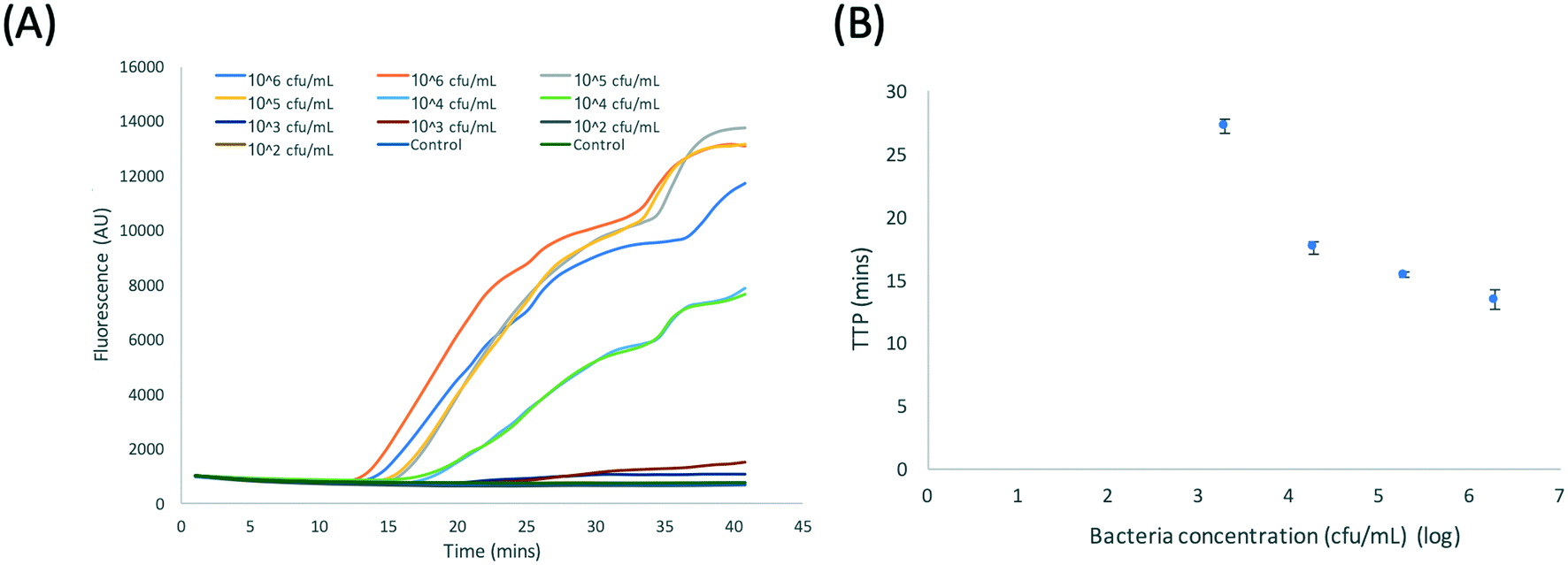 | ||
| Fig. 5 (A) RPA amplification curves for DNA extracted from Klebsiella pneumoniae NCTC 13443 using the benchtop protocol shown in Fig. 1. (B) Plot of the time to positivity (TTP) vs. log10 bacteria concentration. Data from duplicates. | ||
Plasmid DNA extraction on the sample pre-concentration unit
The pre-concentration protocol was designed to streamline the plasmid purification and pre-concentration process into a single step, significantly reducing assay time. The unit interfaces the macro (mL) sample volume to micro-fluidic requirements (μL volume) and could be used on any microfluidic device. The extraction of beads through the oil will carry over a finite volume of residual guanidine and urine. For the device, this residual volume was experimentally determined (using fluorescein dye) to be ∼300 nL for a 2 μL reservoir droplet volume. Based on this measurement, the guanidine in the sample should be further diluted by approximately 7×, when the beads are re-suspended in the elution buffer reservoir.The area of the hole in the top substrate is ∼13 mm2, and is filled with oil which prevents the urine sample flowing into the DMF device. The efficiency with which magnetic beads were pulled through the sample pre-concentration unit was experimentally calculated to be ∼80% (n = 3). Some of the beads were lost during the procedure, for example, adhesion to the top glass substrate or at the circumference of the hole (see image in Fig. S2†).
The plasmid capture efficiency for the pre-concentration unit was evaluated as follows. Purified whole DNA (plasmid and genomic) was extracted using the DNeasy Blood and Tissue Kit and spiked into PBS containing 3 M GuHCl (1 mL) with 2.5 μL magnetic beads (Magnesil Red). The beads were processed on the DMF device (Fig. 3, steps 1 to 4), pulled to the edge of the device and re-suspended in 5 μL of elution buffer. Fig. S4† shows the relationship between the amount of plasmids retrieved against the amount of total DNA added, demonstrating an extraction efficiency of ∼13% (for purified sample, no cell debris). The spiked DNA contained both genomic and plasmid DNA. The Magnesil red beads preferentially bind plasmid DNA and thus should preferentially concentrate the plasmid into the elution volume. Since the approximate amount of plasmid present in total genetic content of bacterium can be around 10–20% of the total genome content in some cases,42 a capture efficiency of 13% demonstrates efficient plasmid extraction.
Full DMF assay: plasmid DNA extraction and amplification
Before transferring the protocol to the DMF platform, the RPA assay was first implemented on the DMF device with purified DNA. Fig. 6A shows time-lapse images of DNA amplification in droplets. Note the presence of background fluorescence due to a polymer coating on the TFT backplane. In this image, the 6 (270 nL) droplets consisted of 3 reaction droplets, each with 800 DNA copies plus 3 NTC droplets. The three positive droplets give high fluorescence signals as seen in the image. Fig. 6B shows a plot of the fluorescence as a function of time with a TTP for 800 copies in approximately 3.2 min. Fig. 6C shows amplification curves for a titration down to 7 copies per droplet; the TTP vs. log10(concentration) is plotted in Fig. 6D. This shows a typical LoD of 10 copies, similar to our previous reported work where fluorescence was measured with a confocal laser imaging system. In the present case, the TTP was also improved due to optimisation of the forward primer sequence (sequence information available on request).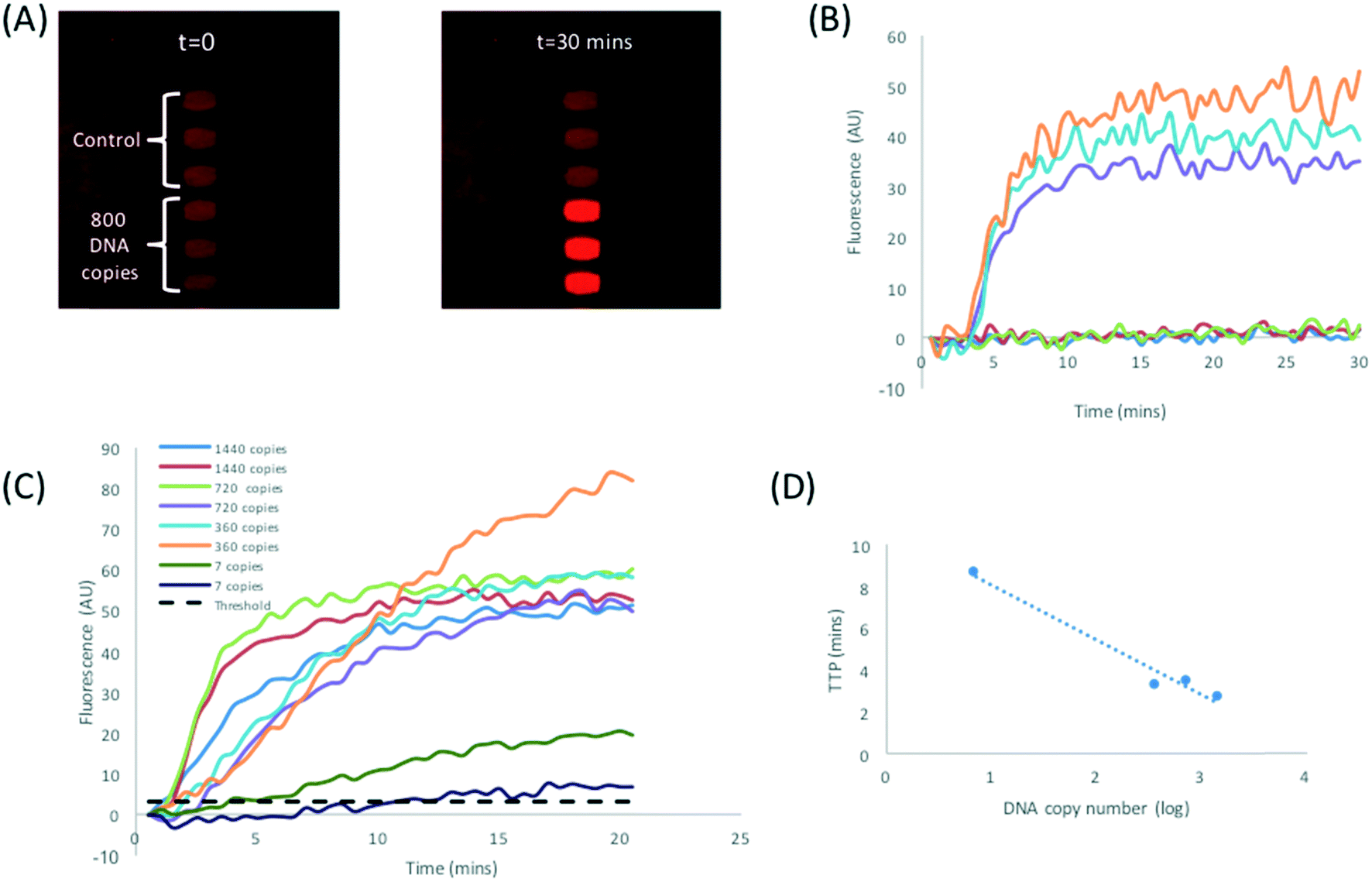 | ||
| Fig. 6 (A) Time lapse images showing RPA amplification for 270 nL volume droplets processed on the DMF platform. Purified plasmid DNA was loaded directly onto reservoir electrodes, dispensed and mixed with RPA reagents using a custom programmed sequence. (B) RPA amplification curve for the image shown in (A). Each reaction droplet contained 800 copies of DNA (C) RPA amplification curves for purified DNA loaded directly on reservoir electrodes. (D) TTP with respect to DNA concentration. Data is the average of duplicates shown in Fig. 6C. | ||
Finally, the functionality of the DNA extraction and pre-concentration unit integrated with the DMF device was tested using bacteria (Klebsiella pneumoniae NCTC 13443) spiked into urine from healthy volunteers. The sample was processed as shown in Fig. 1(1–3b) and 3. The images in Fig. 7B are example time lapse images of the amplification of the target plasmid, pre-concentrated and eluted on the DMF platform from urine containing 1.7 × 105 cfu mL−1 (Fig. 7C). The TTP for this particular reaction is less than 5 minutes. Fig. 7D shows similar data for a higher concentration (6 × 104 cfu mL−1), with a TTP of approximately 5 minutes. Note that these two experiments were performed with different volunteer samples, which explains the difference in the TTP. Nonetheless, the data demonstrates that the assay is sufficiently sensitive to detect clinically relevant levels of bacteria in urine for an uncomplicated UTI, which is in the region of 105 cfu mL−1.3 The combined assay is also reasonably fast, with a total test time of approximately 30 minutes, comprising 10 minutes of sample incubation, 5 minutes of sample processing and droplet dispensing and 10 to 15 minutes for the RPA. The LoD for the assay on the bench is 103 cfu mL−1; the same assay performed on the DMF platform has an LoD of approximately 10 times worse at 104 cfu ml−1 although the positive controls (purified DNA) could be detected down to 10 copies. This may be attributed to the low volume used for the assay (45 nL droplets from a 2 μL reservoir). With a drop in the plasmid extraction efficiency for concentrations lower than 104 cfu mL−1 to <5% (Fig. 5B and S3†), the number of plasmids extracted for 103 cfu mL−1 will be approximately 30 copies (assuming 1 plasmid per bacteria). With a bead recovery efficiency of ∼80%, this decreases to 24 copies in an elution volume of 2 μL. The 45 nL daughter droplet then has less than one copy per droplet. We anticipate that an increase in sample volume (10 mL) should be able to enhance the limit of detection on the platform.
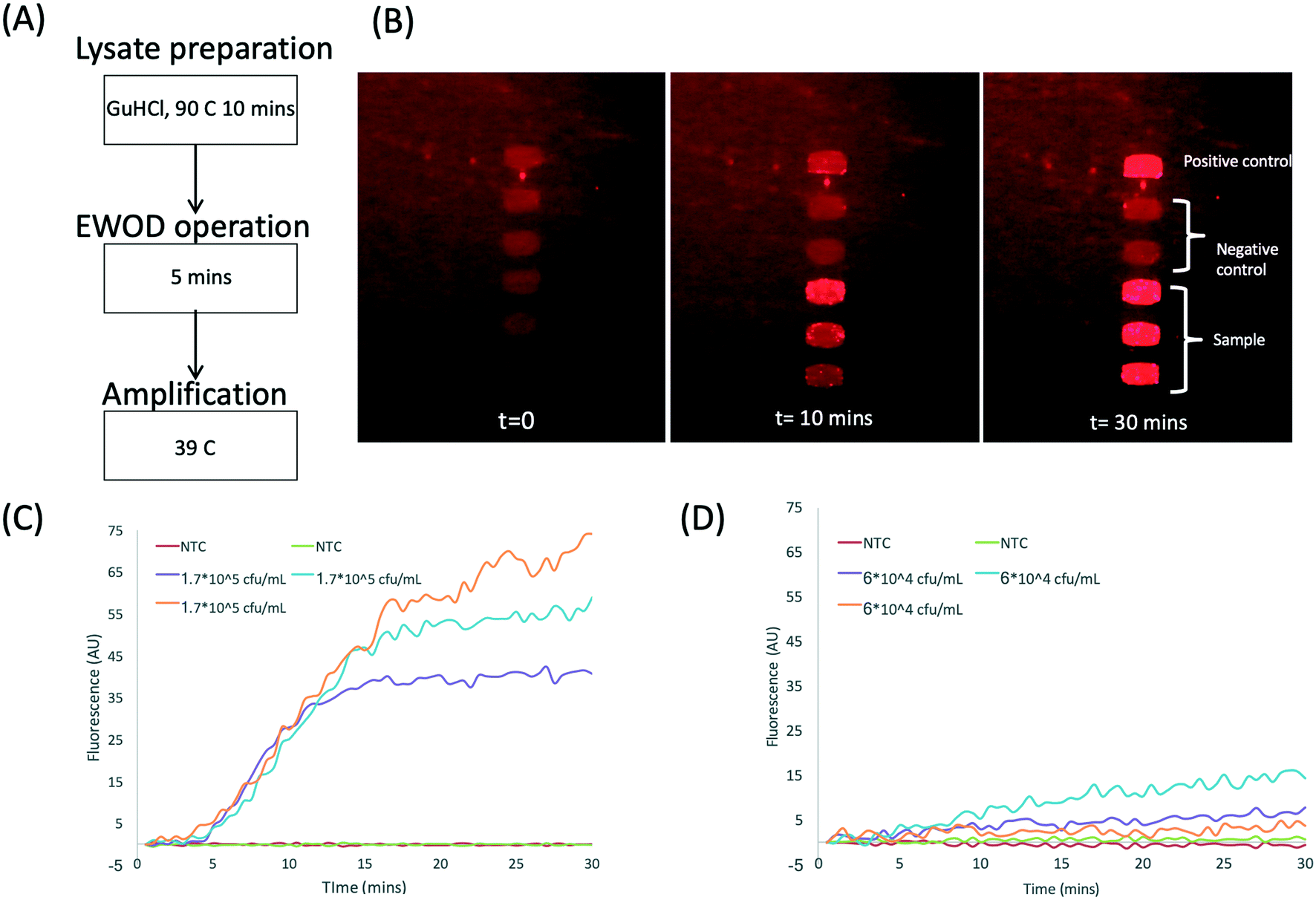 | ||
| Fig. 7 (A) The procedure used to determine antibiotic resistance in bacteria on the DMF platform. (B) Time lapse images showing RPA amplification in 270 nL droplets. 1.7 × 105 cfu mL−1 is spiked into urine and processed as shown in Fig. 2. (C and D) RPA amplification curve for two different bacteria concentrations in urine from two healthy volunteers. | ||
Conclusions
We have developed a simple pre-concentration device to convert mL sample volumes to μL volume requirements of the DMF platform. The device concentrates a sample using plasmid specific magnetic beads into μL volumes for processing on a digital microfluidic platform. The system measures the presence of a target gene that confers antibiotic resistance to bacteria present in urine. The platform benefits from advantages of single step sample purification and pre-concentration at an immiscible interface, together with automated fluidic operations on the DMF platform. Using a clinical strain of Klebsiella as a target organism, we demonstrate a nearly complete “sample to answer” assay time of approximately 30 minutes, and sufficient sensitivity to measure resistance in uncomplicated UTIs. The RPA chemistry is robust and can tolerate a small amount of contamination with urine and GuHCl, so that only a single bead wash step in the elution buffer is required. The plasmids were extracted by lysing the Gram negative bacteria using heat in a fast and simple process. Although this was done off-chip, it could be integrated following a re-design of the architecture to include a heater or IR laser. The combination of a simple sample processing cartridge with a fast programmable DMF provides a new platform technology which could have many different applications in diagnostics of microorganisms in a range of sample matrices.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Ben Hadwen, Chris J. Brown, and Jonathan Buse of Sharp Laboratories Europe for many useful discussions and the development of a measurement jig. This work has been supported by the National Institute for Health Research (NIHR) i4i Programme grant II-ES-0511-21002 “Rapid detection of infectious agents at point of triage (PoT)”. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research, Public Health England or the Department of Health. All data supporting this study are openly available from the University of Southampton repository at “https://doi.org/10.5258/SOTON/D0731”.References
- J. O’Neill, Tackling drug-resistant infections globally: Final report and recommendations. The review on Antimicrobial resistance, 2016 Search PubMed.
- L. Abbo and T. Hooton, Antibiotics, 2014, 3, 174–192 CrossRef PubMed.
- M. Grabe, R. Bartoletti, T. E. Bjerklund Johansen, T. Cai, M. Cek, B. Koves, K. G. Naber, R. S. Pickard, P. Tenke and F. Wagenlehner, Guidelines on Urological Infections, 2015 Search PubMed.
- J. D. D. Pitout, P. Nordmann and L. Poirel, Antimicrob. Agents Chemother., 2015, 59, 5873–5884 CrossRef CAS PubMed.
- R. Bonnet, Antimicrob. Agents Chemother., 2004, 48, 1–14 CrossRef CAS PubMed.
- M. M. D'Andrea, F. Arena, L. Pallecchi and G. M. Rossolini, Int. J. Med. Microbiol., 2013, 303, 305–317 CrossRef PubMed.
- W.-H. Zhao and Z.-Q. Hu, Crit. Rev. Microbiol., 2013, 39, 79–101 CrossRef CAS PubMed.
- X. Fang, Y. Liu, J. Kong and X. Jiang, Anal. Chem., 2010, 82, 3002–3006 CrossRef CAS PubMed.
- C.-H. Wang, K.-Y. Lien, J.-J. Wu and G.-B. Lee, Lab Chip, 2011, 11, 1521 RSC.
- D. M. Tourlousse, F. Ahmad, R. D. Stedtfeld, G. Seyrig, J. M. Tiedje and S. A. Hashsham, Biomed. Microdevices, 2012, 14, 769–778 CrossRef CAS PubMed.
- T.-H. Kim, J. Park, C.-J. Kim and Y.-K. Cho, Anal. Chem., 2014, 86, 3841–3848 CrossRef CAS PubMed.
- S. Kalsi, M. Valiadi, M.-N. Tsaloglou, L. Parry-Jones, A. Jacobs, R. Watson, C. Turner, R. Amos, B. Hadwen, J. Buse, C. Brown, M. Sutton and H. Morgan, Lab Chip, 2015, 15, 3065–3075 RSC.
- R. K. Daher, G. Stewart, M. Boissinot and M. G. Bergeron, Clin. Chem., 2016, 62, 947–958 CrossRef CAS PubMed.
- L. Lillis, J. Siverson, A. Lee, J. Cantera, M. Parker, O. Piepenburg, D. A. Lehman and D. S. Boyle, Mol. Cell. Probes, 2016, 30, 74–78 CrossRef CAS PubMed.
- S. Kalsi, S. Sellars, C. Turner, J. Sutton and H. Morgan, Micromachines, 2017, 8, 111 CrossRef.
- B. Hadwen, G. R. Broder, D. Morganti, A. Jacobs, C. Brown, J. R. Hector, Y. Kubota and H. Morgan, Lab Chip, 2012, 12, 3305 RSC.
- N. Beyor, L. Yi, T. S. Seo and R. A. Mathies, Anal. Chem., 2009, 81, 3523–3528 CrossRef CAS PubMed.
- D. Dai, D. Holder, L. Raskin and C. Xi, BMC Microbiol., 2011, 11, 59 CrossRef CAS PubMed.
- S. H. Suh, H. P. Dwivedi and L.-A. Jaykus, LWT--Food Sci. Technol., 2014, 56, 256–260 CrossRef CAS.
- C.-H. Wang, C.-J. Chang, J.-J. Wu and G.-B. Lee, Nanomedicine, 2014, 10, 809–818 CrossRef CAS PubMed.
- K. Tsougeni, G. Papadakis, M. Gianneli, A. Grammoustianou, V. Constantoudis, B. Dupuy, P. S. Petrou, S. E. Kakabakos, A. Tserepi, E. Gizeli and E. Gogolides, Lab Chip, 2016, 16, 120–131 RSC.
- Z. Guo, Y. Liu, S. Li and Z. Yang, Rapid Commun. Mass Spectrom., 2009, 23, 3983–3993 CrossRef CAS PubMed.
- K. Yang, D. M. Jenkins and W. W. Su, J. Microbiol. Methods, 2011, 86, 69–77 CrossRef CAS PubMed.
- M. Valiadi, S. Kalsi, I. G. F. Jones, C. Turner, J. M. Sutton and H. Morgan, Biomed. Microdevices, 2016, 18, 1–10 CrossRef CAS PubMed.
- Y.-K. Cho, J.-G. Lee, J.-M. Park, B.-S. Lee, Y. Lee and C. Ko, Lab Chip, 2007, 7, 565 RSC.
- L. X. Kong, A. Perebikovsky, J. Moebius, L. Kulinsky and M. Madou, J. Lab. Autom., 2016, 21, 323–355 CrossRef PubMed.
- M. J. Jebrail, A. Sinha, S. Vellucci, R. F. Renzi, C. Ambriz, C. Gondhalekar, J. S. Schoeniger, K. D. Patel and S. S. Branda, Anal. Chem., 2014, 86, 3856–3862 CrossRef CAS PubMed.
- P.-Y. Hung, P.-S. Jiang, E.-F. Lee, S.-K. Fan and Y.-W. Lu, Microsyst. Technol., 2017, 23, 313–320 CrossRef CAS.
- E. A. Oblath, W. H. Henley, J. P. Alarie and J. M. Ramsey, Lab Chip, 2013, 13, 1325 RSC.
- M. D. Kulinski, M. Mahalanabis, S. Gillers, J. Y. Zhang, S. Singh and C. M. Klapperich, Biomed. Microdevices, 2009, 11, 671–678 CrossRef CAS PubMed.
- L. Van Heirstraeten, P. Spang, C. Schwind, K. S. Drese, M. Ritzi-Lehnert, B. Nieto, M. Camps, B. Landgraf, F. Guasch, A. H. Corbera, J. Samitier, H. Goossens, S. Malhotra-Kumar and T. Roeser, Lab Chip, 2014, 14, 1519–1526 RSC.
- G. Czilwik, T. Messinger, O. Strohmeier, S. Wadle, F. von Stetten, N. Paust, G. Roth, R. Zengerle, P. Saarinen, J. Niittymäki, K. McAllister, O. Sheils, J. O'Leary and D. Mark, Lab Chip, 2015, 15, 3749–3759 RSC.
- G. Choi, J. H. Jung, B. H. Park, S. J. Oh, J. H. Seo, J. S. Choi, D. H. Kim and T. S. Seo, Lab Chip, 2016, 16, 2309–2316 RSC.
- O. Mosley, L. Melling, M. D. Tarn, C. Kemp, M. M. N. Esfahani, N. Pamme and K. J. Shaw, Lab Chip, 2016, 16, 2108–2115 RSC.
- S. M. Berry, E. T. Alarid and D. J. Beebe, Lab Chip, 2011, 11, 1747 RSC.
- B. Seale, C. Lam, D. G. Rackus, M. D. Chamberlain, C. Liu and A. R. Wheeler, Anal. Chem., 2016, 88, 10223–10230 CrossRef CAS PubMed.
- D. G. Rackus, R. P. S. de Campos, C. Chan, M. M. Karcz, B. Seale, T. Narahari, C. Dixon, M. D. Chamberlain and A. R. Wheeler, Lab Chip, 2017, 17, 2272–2280 RSC.
- P. M. Bennett, Br. J. Pharmacol., 2008, 153(Suppl 1), S347–S357 CAS.
- M. Rozwandowicz, M. S. M. Brouwer, J. Fischer, J. A. Wagenaar, B. Gonzalez-Zorn, B. Guerra, D. J. Mevius and J. Hordijk, J. Antimicrob. Chemother., 2018, 73, 1121–1137 CrossRef CAS PubMed.
- K. Krõlov, J. Frolova, O. Tudoran, J. Suhorutsenko, T. Lehto, H. Sibul, I. Mäger, M. Laanpere, I. Tulp and Ü. Langel, J. Mol. Diagn., 2014, 16, 127–135 CrossRef PubMed.
- P. Rod, Dealing with Amplification Inhibitors: Reagent choice matters, https://www.promega.com/-/media/files/promega-worldwide/north-america/promega-us/webinars-and-events/2014/dealing-with-amplification-inhibitors-webinar-feb-2014.pdf?la=en, (accessed 10 January 2017).
- C. Zhong, D. Peng, W. Ye, L. Chai, J. Qi, Z. Yu, L. Ruan and M. Sun, PLoS One, 2011, 6, e16025 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc01249k |
| This journal is © The Royal Society of Chemistry 2019 |