
Ammonia-salt solvent promotes cellulosic biomass deconstruction under ambient pretreatment conditions to enable rapid soluble sugar production at ultra-low enzyme loadings†
Shishir P. S.
Chundawat
 *a,
Leonardo da Costa
Sousa
b,
Shyamal
Roy‡
a,
Zhi
Yang
c,
Shashwat
Gupta
a,
Ramendra
Pal
d,
Chao
Zhao§
a,
Shih-Hsien
Liu
e,
Loukas
Petridis
e,
Hugh
O'Neill
c and
Sai Venkatesh
Pingali
c
*a,
Leonardo da Costa
Sousa
b,
Shyamal
Roy‡
a,
Zhi
Yang
c,
Shashwat
Gupta
a,
Ramendra
Pal
d,
Chao
Zhao§
a,
Shih-Hsien
Liu
e,
Loukas
Petridis
e,
Hugh
O'Neill
c and
Sai Venkatesh
Pingali
c
aDepartment of Chemical & Biochemical Engineering, Rutgers The State University of New Jersey, Piscataway, NJ 08854, USA. E-mail: shishir.chundawat@rutgers.edu
bDepartment of Chemical Engineering & Materials Science, Michigan State University, East Lansing, MI 48824, USA
cCenter for Structural Molecular Biology and Neutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
dDepartment of Mechanical & Aerospace Engineering, Rutgers The State University of New Jersey, Piscataway, NJ 08854, USA
eUT/ORNL Center for Molecular Biophysics, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
First published on 29th November 2019
Abstract
Here, we report a novel ammonia![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ammonium salt solvent based pretreatment process that can rapidly dissolve crystalline cellulose into solution and eventually produce highly amorphous cellulose under near-ambient conditions. Pre-activating the cellulose I allomorph to its ammonia–cellulose swollen complex (or cellulose III allomorph) at ambient temperatures facilitated rapid dissolution of the pre-activated cellulose in the ammonia-salt solvent (i.e., ammonium thiocyanate salt dissolved in liquid ammonia) at ambient pressures. For the first time in reported literature, we used time-resolved in situ neutron scattering methods to characterize the cellulose polymorphs structural modification and understand the mechanism of crystalline cellulose dissolution into a ‘molecular’ solution in real-time using ammonia-salt solvents. We also used molecular dynamics simulations to provide insight into solvent interactions that non-covalently disrupted the cellulose hydrogen-bonding network and understand how such solvents are able to rapidly and fully dissolve pre-activated cellulose III. Importantly, the regenerated amorphous cellulose recovered after pretreatment was shown to require nearly ∼50-fold lesser cellulolytic enzyme usage compared to native crystalline cellulose I allomorph for achieving near-complete hydrolytic conversion into soluble sugars. Lastly, we provide proof-of-concept results to further showcase how such ammonia-salt solvents can pretreat and fractionate lignocellulosic biomass like corn stover under ambient processing conditions, while selectively co-extracting ∼80–85% of total lignin, to produce a highly digestible polysaccharide-enriched feedstock for biorefinery applications. Unlike conventional ammonia-based pretreatment processes (e.g., Ammonia Fiber Expansion or Extractive Ammonia pretreatments), the proposed ammonia-salt process can operate at near-ambient conditions to greatly reduce the pressure/temperature severity necessary for conducting effective ammonia-based pretreatments on lignocellulose.
:ammonium salt solvent based pretreatment process that can rapidly dissolve crystalline cellulose into solution and eventually produce highly amorphous cellulose under near-ambient conditions. Pre-activating the cellulose I allomorph to its ammonia–cellulose swollen complex (or cellulose III allomorph) at ambient temperatures facilitated rapid dissolution of the pre-activated cellulose in the ammonia-salt solvent (i.e., ammonium thiocyanate salt dissolved in liquid ammonia) at ambient pressures. For the first time in reported literature, we used time-resolved in situ neutron scattering methods to characterize the cellulose polymorphs structural modification and understand the mechanism of crystalline cellulose dissolution into a ‘molecular’ solution in real-time using ammonia-salt solvents. We also used molecular dynamics simulations to provide insight into solvent interactions that non-covalently disrupted the cellulose hydrogen-bonding network and understand how such solvents are able to rapidly and fully dissolve pre-activated cellulose III. Importantly, the regenerated amorphous cellulose recovered after pretreatment was shown to require nearly ∼50-fold lesser cellulolytic enzyme usage compared to native crystalline cellulose I allomorph for achieving near-complete hydrolytic conversion into soluble sugars. Lastly, we provide proof-of-concept results to further showcase how such ammonia-salt solvents can pretreat and fractionate lignocellulosic biomass like corn stover under ambient processing conditions, while selectively co-extracting ∼80–85% of total lignin, to produce a highly digestible polysaccharide-enriched feedstock for biorefinery applications. Unlike conventional ammonia-based pretreatment processes (e.g., Ammonia Fiber Expansion or Extractive Ammonia pretreatments), the proposed ammonia-salt process can operate at near-ambient conditions to greatly reduce the pressure/temperature severity necessary for conducting effective ammonia-based pretreatments on lignocellulose.
Introduction
Native cellulose derived from lignocellulosic biomass is a β-1,4-D-glucose polymer with extensive intra- and inter-molecular hydrogen bonding that stabilizes its ordered crystalline structure and makes it highly recalcitrant to cellulase enzyme-catalyzed degradation into soluble sugars.1–3 Disruption of cellulose crystallinity using suitable solvents or thermochemical pretreatments is thus necessary to make polysaccharides more readily accessible to cellulolytic enzymes4,5 or suitable chemicals/catalysts6,7 that facilitate cellulose hydrolysis to glucose. One major advantage of pretreatment solvents that can significantly disrupt cellulose crystallinity, while possibly selectively co-extracting lignin, is the significant reduction in enzyme usage that could make cellulosic biorefineries economically more viable.8 Most enzyme loadings reported currently for saccharification of crystalline cellulose or pretreated biomass is still at least an order of magnitude (10–15 mg enzymes per g cellulose) higher than what is likely to be commercially viable,9 similar to advanced corn grain dry-grind processes that often use <0.5–1 mg enzyme per g starch loading.10 Therefore, there is a critical need to identify pretreatments that can significantly disrupt cellulose crystallinity to reduce enzyme loadings during biomass saccharification by at-least 10-fold from current usage.Over the last 150 years several non-derivatizing solvents, like ionic liquids (IL), have been employed to readily dissolve cellulose and/or disrupt cellulose crystallinity to produce highly disordered or regenerated amorphous cellulose.11,12 Disordered or regenerated amorphous cellulose (RAC) recovered after disruption of native cellulose crystallinity has been shown to result in significantly enhanced enzymatic hydrolysis rates.13,14 However, most reported cellulose solvents are fairly expensive and difficult to recycle.5,14,15 For example, most commercially available imidazolium based IL cost >$50 kg−1,12 but there have been advances in recent years to develop lower cost IL and pretreatment-solvent recycle strategies to reduce effective IL cost to ∼$5 kg−1.5,16 Nevertheless, alternative inexpensive solvent systems with cellulose dissolution properties similar to conventional IL are still being explored.
Several research groups have studied the impact of ammonia pretreatment on cellulose crystallinity and its beneficial impact on cellulolytic enzyme activity.3,17–20 Anhydrous liquid ammonia (NH3) acts as a swelling agent of cellulose by intercalating into the crystalline structure of cellulose and disrupting the native hydrogen bonding network to form a metastable cellulose–ammonia complex.19,21–23 An unnatural crystalline cellulose allomorph, called cellulose III, is formed from these swollen cellulose–ammonia complexes upon immediate removal of ammonia. We have shown that selective ‘rewiring’ of the cellulose hydrogen bond network within cellulose III can enhance synergistic enzymatic hydrolysis rates by several fold, but hydrolysis rates for crystalline cellulose was still much lower than RAC.3 Subsequently, a novel liquid ammonia pretreatment methodology called Extractive Ammonia (EA) was developed to simultaneously convert native crystalline cellulose I into cellulose III allomorph, while also partially extracting lignin from lignocellulosic biomass like corn stover, to improve upon the traditional Ammonia Fiber Expansion (AFEX) process.24 However, although ammonia is inexpensive (<$0.5 kg−1) and readily recoverable due to its high volatility,25,26 effective biomass pretreatments with ammonia alone during EA or AFEX pretreatments require high severity operating pressures (250–1500 psi) and temperatures (90–130 °C). Furthermore, although crystalline cellulose III has improved enzymatic digestibility compared to native cellulose I, it is still more recalcitrant towards hydrolysis than truly amorphous cellulose (or RAC) produced in IL-based pretreatments.
One strategy to reduce ammonia-based pretreatment severity to ambient conditions while fully disrupting cellulose crystallinity would be to add a co-solvent/chemical that can reduce NH3 partial pressure while also facilitating rapid cellulose dissolution. We have recently shown that co-solvents like ethanol can reduce the operating pressure during EA pretreatment by ∼2-fold without hindering the formation of the cellulose–ammonia complex that leads to cellulose III.27 However, inclusion of ammonium salts can reduce the vapor pressure of NH3 by nearly 30-fold (at 25 °C),28 which can greatly reduce the thermochemical severity of a theoretical ammonia-salt treatment process compared to conventional NH3 based treatments.28,29 Furthermore, inclusion of hydrogen-bond acceptor anion (e.g., SCN− or I−) based ammonium salts with NH3 has been reported to remarkably facilitate complete dissolution of regenerated cellulose.30,31 However, native crystalline cellulose I dissolution in an ammonia–ammonium thiocyanate (NH3–NH4SCN) or A:At based ammonia-salt solvent can only take place by extensive thermal cycling of the cellulose-solvent slurry by first exposure to extreme freezing temperatures (−80 to −30 °C) followed by mechanical stirring of the slurry as the temperature is slowly raised to room temperature over 6–12 hours, followed again by multiple freeze–thaw cycles of the slurry till the cellulose is fully dissolved into solution.32,33 In the absence of multiple freeze–thaw thermal cycles, native crystalline cellulose can take several days at room temperature to be only partially dissolved in NH3–NH4SCN. Such an extreme temperature cycling requirement to achieve complete cellulose dissolution over several days makes this originally reported process not ideal for cellulosic biomass pretreatment in a biorefinery setting. Furthermore, we currently still have a poor understanding of the in situ mechanism of real-time cellulose dissolution in ammonia-salt solvents which has further led to limited advances to original process reported by Cuculo.31
One strategy to design a more efficient ammonia-salt pretreatment process would be to take advantage of recent advances in our understanding of ammonia–cellulose interactions during liquid NH3 based pretreatments. Our recent experimental3,27 and modeling34–36 studies have revealed the early mechanism of crystalline cellulose swelling that takes place nearly instantaneously in the presence of liquid NH3, which ultimately results in the formation of cellulose III only upon removal of NH3. Molecular dynamics (MD) simulations of ammonia–cellulose interactions suggested NH3 first hydrogen bonds with cellulose fibril surface exposed hydroxyl groups, which results in the rotation of the C6-hydroxymethyl groups from Trans–Gauche (TG) to Gauche–Trans (GT) conformations. This GT conformation change instantly leads to the formation of hydrophilic channels in the crystalline fibril to form an intermediate structure that resembles the cellulose–ammonia complex.34,36 Crystallographic evidence has also verified the formation of these channels that allow bulk NH3 molecules to penetrate into the fibril and cause further swelling of individual cellulose crystalline fibers.37 However, unlike true cellulose dissolving solvent systems like imidazolium acetate IL where both anionic and cationic solvent species are necessary to dissolve individual cellulose polymer chains,38 NH3 alone is unable to fully disrupt the internal cellulose crystal hydrogen bonding network to solubilize individual cellulose chains. We hypothesized that first pre-activation of native crystalline cellulose to its ammonia–cellulose swollen complex at ambient temperatures would allow improved accessibility of the ammonia-salt ions through the hydrophilic channels into the core fibril. Addition of the anionic salts like ammonium thiocyanate immediately after pre-swelling the cellulose in ammonia could allow rapid dissolution of the pre-activated cellulose under ambient pressures and temperatures. No one has so far studied the effect of pre-activation of cellulose on its dissolution kinetics in NH3–NH4SCN solvent, or studied the multi-length scale cellulose dissolution dynamics using suitable scattering techniques, or developed an ammonia-salt based pretreatment for biochemical conversion of biomass into soluble sugars.
Here, we have tested the cellulose pre-activation hypothesis to explore the possibility of developing an ammonia-salt solvent-based pretreatment method that operates under ambient conditions to rapidly and completely solubilize cellulose. We have used in situ neutron scattering to follow the real-time dissolution of cellulose in the ammonia-salt solvent, and provide fundamental insights into the pretreatment mechanism. Molecular dynamics simulations were also used to support our hypothesis and to fully understand the mechanism of model cellulose polymers dissolution in A:At based solvent systems. We also conducted detailed enzymatic saccharification assays at varying enzyme loadings for the cellulose recovered after treatment with the ammonia-salt solvent to compare the impact of pretreatment on cellulase activity. Finally, we provide proof-of-concept results showing how ammonia-salt solvents can effectively pretreat and selectively fractionate complex lignocellulosic biomass like corn stover into its constituent biopolymers under ambient pretreatment conditions.
Materials and methods
Microcrystalline cellulose & biomass feedstocks
High purity (>98% cellulose content, dry weight mass basis or dwb) microcrystalline cellulose, purchased from Sigma-Aldrich (Avicel PH-101, Lot No. BCBD6923V), was used here. This original sample, called Avicel cellulose I or CI, also served as the native cellulose I allomorph standard. Pre-activated crystalline cellulose III (or CIII) was produced from CI using anhydrous liquid ammonia as reported previously.3,27 Briefly, CIII was prepared in a high-pressure stirred batch reactor at desired temperature (e.g., 25 or 90 °C) for 30 min residence time using a 6:1 ammonia-to-cellulose loading ratio (on dry cellulose weight basis or dwb). The reactor pressure was maintained constant using nitrogen gas during pretreatment, and ammonia was slowly evaporated from the reactor through a venting valve after 30 min. During this evaporation process, the temperature of the reactor was kept stabilized at 25 °C. The treated cellulose sample was then removed from the reactor and placed overnight in the fume hood to evaporate any residual ammonia. No additional washing steps were employed other than using nitrogen gas and overnight fume hood drying to remove all traces of ammonia from the treated cellulose samples. All treated cellulose samples were stored at 4 °C in a zip sealed bag prior to usage. The CIII sample had a total moisture content less than 6–7% (on total wet cellulose weight basis or twb) and was used directly for all further ammonia-salt pretreatment studies without any further drying. Lastly, pioneer 36H56 corn hybrid derived corn stover was generously provided by the Great Lakes Bioenergy Research Center and processed for handling as described earlier.24 Details regarding corn stover composition analysis method and results is provided in the ESI appendix.†
Ammonia–ammonium thiocyanate salt based pretreatments
Unless noted otherwise, commercial grade anhydrous ammonia (>99.99% NH3 purity from Airgas, Lawrenceville, GA) and ammonium thiocyanate (>99% NH4SCN purity from Sigma-Aldrich, St Louis, MO) were used here as is without any drying for all ex situ pretreatment studies. Deuterated ammonia-d3 (99% atom isotopic purity ND3, Sigma-Aldrich, St Louis, MO), deuterated ammonium-d4 thiocyanate (99% atom isotopic purity ND4SCN, Sigma-Aldrich, St Louis, MO), and deuterated water-d2 (99.9% atom isotopic purity D2O, Sigma-Aldrich, St Louis, MO) were used for all in situ neutron scattering pretreatment studies. Analytical grade Acetone and 200 Proof Ethanol solvents were procured from Sigma-Aldrich and VWR Scientific, respectively.To prepare the ammonia-salt solution for cellulose pretreatment, the required dry weight of ammonium salt was transferred into a high-pressure sealed vessel (Parr) followed by slowly loading ammonia into the vessel while weighing the reactor intermittently to achieve targeted ammonia loading. After a stable ammonia-salt solution is formed, transfer the solution into an amber stained Schott glass bottle with screw cap for storage under ambient conditions in fume hood. This colourless solution was stable at room temperature for several months and was used directly for biomass pretreatment.
Native (CI) or pre-activated microcrystalline (CIII) cellulose was pretreated using a 27:73% (weight basis; w/w) ammonia–ammonium thiocyanate solution as described below. This solvent concentration has been reported previously to be ideal for cellulose dissolution.31,39 For all ex situ ammonia-salt pretreatments, 5 g of either cellulose I or cellulose III (on twb) was added along with 95 g of ammonia-salt solution (i.e., 95 g of solution was composed of 69.4 g ammonium thiocyanate salt dissolved in 25.6 g ammonia) in a 500 mL stoppered glass bottle. The slurry was mixed using a magnetic stirrer at 500 rpm for desired pretreatment time (0.5–72 h) at ambient pressure and temperature. At the desired sampling time, 100 mL of ethanol (100%) was added as anti-solvent to precipitate cellulose out of solution. The precipitate was washed with excess ethanol (50:1, v/w), followed by five washes with 1:1 ethanol–acetone (20:1, v/w) to remove any trace residual ammonia-salt. Treated cellulose samples were stored in a never-dried state, soaked in 100% ethanol, at 4 °C prior to usage. Details regarding corn stover pretreatment and additional cellulose ultrastructure characterization using X-Ray Diffraction (XRD) and Scanning Electron Microscopy (SEM) based methods is provided in the ESI appendix (ESI M1–M4†).
Small angle neutron scattering (SANS)
SANS measurements were performed with the CG-3 Bio-SANS instrument at the High Flux Isotope Reactor (HFIR) facility of Oak Ridge National Laboratory.40 Samples for SANS studies were placed in either a 1 mm pathlength Hellma cell (Hellma Model #106-QS 1MM) or 1.0 mm pathlength titanium cells with detachable cell walls. The choice of the cell was based on sample state; viscous samples were placed in titanium cells with detachable cell walls to allow for eliminating bubbles from the illuminated region of the sample. A single instrument configuration was employed to cover the range, 0.003 < Q(Å−1) < 0.8, of scattering vectors. The scattering vector Q is related to λ, neutron wavelength and 2θ, the scattering angle as Q = 4π sin θ/λ. Using 6 Å neutron wavelength, main detector at 15.5 m from sample and wing detector at 1.4° rotation from direct beam, a sufficient overlap in scattering vectors to stitch data from the two detectors was achieved. The instrument resolution was defined placing source and sample circular apertures at 17430 mm apart and with diameters 40 mm and 14 mm, respectively. The relative wavelength spread Δλ/λ was set to 0.15. The scattering intensity profiles I(Q) versus Q, were obtained by azimuthally averaging the processed 2D images, which were normalized to incident beam monitor counts, and corrected for detector dark current, pixel sensitivity and scattering from the quartz cell walls and solvent scattering backgrounds. SANS data was analyzed using the multilevel unified equation implemented in Irena Package to elucidate the multiple levels of structural organization.41 Irena is an Igor Pro software package consisting of various structural models to analyze small-angle scattering data. For each individual level, i, the scattering signal is the sum of Guinier's exponential form and the structurally limited power-law as,42–44where i = 1, …, n. To model the cellulose SANS data, we used a total of three levels with i = 1 and i = 3 referring to the smallest and largest size structural levels, respectively. Here, Gi = ciViΔρi2 is the exponential prefactor, where ci is the concentration of the ith kind of particle; Vi is the volume of the particle and Δρi is the contrast of the ith kind of particle with respect to the solvent; Rgi is the radius of gyration describing the average size of the ith level structural unit; Bi is a Q-independent prefactor specific to the type of power-law scattering with power-law exponent Pi and Ibkg is the flat background intensity due to incoherent scattering.42
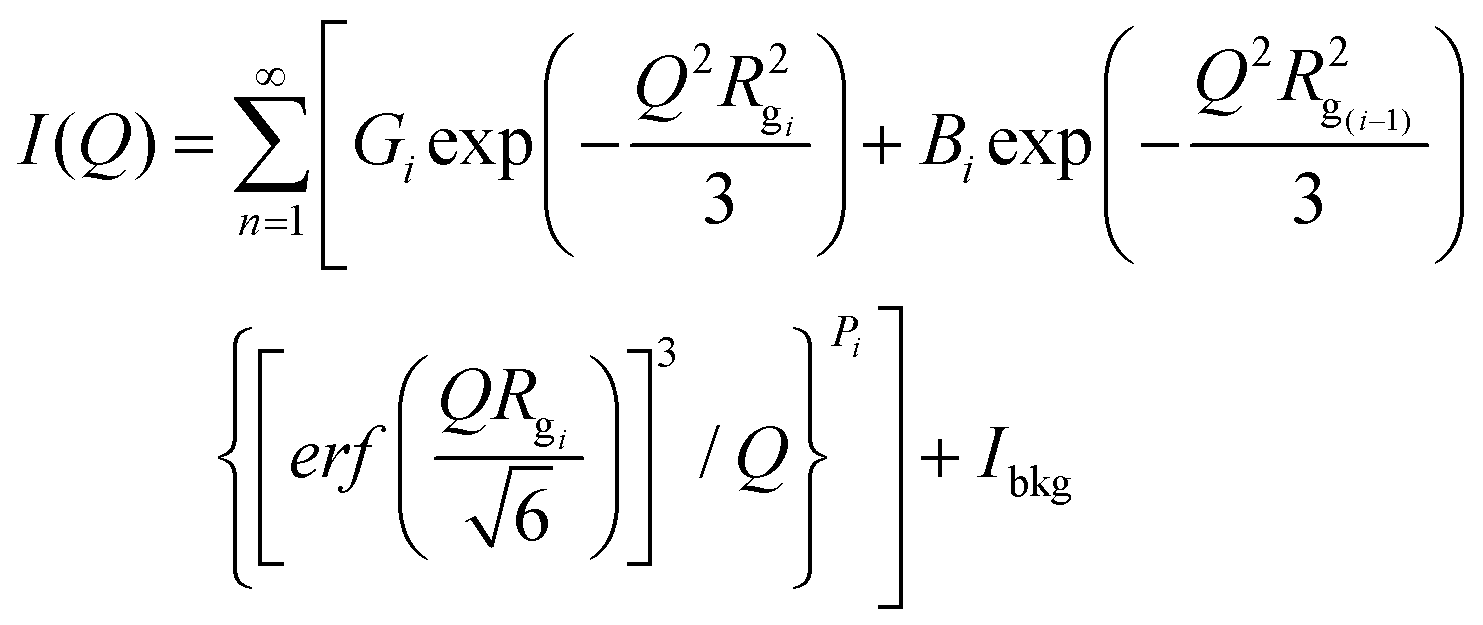
Ex situ neutron scattering experiments were performed on regenerated cellulose recovered directly after ammonia-salt treatment of cellulose samples for varying conditions. For SANS experiments, the cellulose samples were centrifuged and then resuspended in 100% D2O solvent at a 1:10 ratio to exchange the labile hydroxyl hydrogens in cellulose prior to SANS characterization of cellulose samples suspended in 100% D2O solvent. This exchange process was repeated three times. The final concentration of all ex situ pretreated and D2O solvent exchanged cellulose was 50 g L−1. In situ neutron scattering experiments were performed on native cellulose I or pre-activated cellulose III in real-time to study cellulose dissolution dynamics in deuterated and protiated ammonia-salt solvent at room temperature/pressure for varying dissolution time periods (0.5–72 h). The concentration of all in situ cellulose samples suspended in the ammonia-salt solvent was ∼2% (w/v). The deuterated or protiated ammonia-salt solutions composition was identical to the ex situ experiments (i.e., 95 g of 27:73% w/w ammonia-salt solution was composed of 69.4 g ammonium thiocyanate salt dissolved in 25.6 g ammonia).
Cellulosic biomass enzymatic hydrolysis
All pretreated cellulose samples were subjected to enzymatic hydrolysis using a commercial cellulase cocktail at 0.4% glucan loading in a 0.2 ml reaction volume using microplates based on the original high-throughput assay,45 with some minor modifications as described below. Assays for varying cellulase loading and saccharification time course were conducted in 200 μL total volume (using 0.5 ml Greiner microplates sealed with Micronic TPE capclusters) that comprised of 80 μL of 10 g L−1 cellulosic substrate slurry added along with 100 μL of 50 mM pH 4.8 sodium acetate buffer, and 20 μL of suitably diluted enzyme stock to desired protein loadings (i.e., mg total enzyme loading per gram of added cellulose per well). Stock cellulosic substrate slurry was prepared in deionized water with sodium azide (0.2% w/v) added to prevent microbial growth. The total protein loadings used during enzymatic hydrolysis ranged between 0.025 to 5 mg enzyme per g glucan of Cellic C.Tec2 enzymes (Novozymes, CA). The protein concentration (40 mg ml−1) for the enzyme stock solutions was determined using the Bradford method.46 The microplates were incubated at 50 °C with 5 rpm end-over-end mixing oven for desired saccharification time (0–120 h). All pretreated corn stover samples were subjected to enzymatic hydrolysis at 2.5% glucan loading in 10 ml reaction volume using 15 ml glass vials as described before,3 based on the original NREL protocols.47 In all assays, final reaction volume pH 4.8 was achieved using 50 mM sodium citrate buffer and sodium azide was added to prevent any microbial growth (0.1% w/v final concentration). Vials were incubated at 50 °C in an orbital shaking incubator set at 150 RPM (New Brunswick, Innova 44, Enfield, CT) for the desired saccharification time.The hydrolysate supernatants were analyzed for total reducing sugar concentrations using the standard dinitrosalicylic acid (DNS) colorimetric assay or glucose/xylose based enzymatic assay kits as reported earlier.48 Briefly, 30 μl of the cellulose hydrolysate supernatant was incubated with 60 μl of DNS stock reagent in PCR plates at 95 °C for 5 min in an Eppendorf thermal cycler. After the PCR plates cooled down to room temperature, transfer and dilute the DNS reaction mixture in DI water using a clear, flat-bottom microplate for measuring solution absorbance at 540 nm. Suitable reducing sugar standards (e.g., glucose standards ranging from 0.1–5 g l−1) were included for the DNS assay. Glucose concentrations in corn stover hydrolyzates was determined using suitable enzymatic assay kit from Megazyme (Bray, Ireland).48 All hydrolysis experiments were carried out in at least triplicates with typically less than ±10% standard deviation from reported mean values. Glucan conversion to glucose was quantified as described in the NREL protocol.47 Specific activity of the cellulase cocktail on various cellulose substrates was determined from the varying ultra-low enzyme loading assay dataset after 24 h incubation period.46 One unit of specific cellulase activity was defined as 1 μmol reducing sugars (as glucose equivalents) released per milligram enzyme per minute. Error bars reported in this work represent one standard deviation (±1σ) from mean values for replicate assays.
Molecular dynamics (MD) simulations of cellulose-A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :At solvent
:At solvent
MD simulation methods (ESI M5 and Tables S2–S4†) and detailed supporting results are provided in the ESI appendix.†
Results and discussion
Pre-activation of cellulose facilitates rapid solvent dissolution
We tested our cellulose pre-activation hypothesis by dissolving two distinct crystalline allomorphs of cellulose using an A:At solvent at room temperature for varying durations ranging from 1–24 h (Fig. 1A). To our surprise, pre-activated cellulose III started to dissolve nearly instantly upon contact with the A:At solvent (Fig. 1B), unlike native cellulose I which took over several hours to show any visible change in the cellulose slurry transparency/viscosity. In order to characterize the degree of decrystallization as a function of pretreatment duration, cellulose regenerated from the A:At solution using ethanol as anti-solvent was lyophilized and characterized by XRD and SEM. XRD clearly shows a significant drop in the equatorial crystallite peak reflections for (101), (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ), and (002) crystallographic planes for cellulose III within 1 h of A:At treatment with close to baseline signals (Fig. 1D). SEM analysis also confirmed that lyophilized A:At treated cellulose III had completely lost all fibril like features seen in native microcrystalline cellulose (ESI Fig. S1†). However, cellulose I shows a rather slow, but progressive, decrease in the equatorial reflections as a function of A:At treatment time (Fig. 1C). Even after 24 h cellulose I was only partial dissolved in A:At as indicated by the relatively strong XRD equatorial reflections for the (002) crystallographic plane in particular for recovered and regenerated samples. Details regarding analysis of cellulose crystallinity index for all lyophilized samples using XRD peak deconvolution method is provided in the ESI appendix (ESI Fig. S2 & Table S1†).
), and (002) crystallographic planes for cellulose III within 1 h of A:At treatment with close to baseline signals (Fig. 1D). SEM analysis also confirmed that lyophilized A:At treated cellulose III had completely lost all fibril like features seen in native microcrystalline cellulose (ESI Fig. S1†). However, cellulose I shows a rather slow, but progressive, decrease in the equatorial reflections as a function of A:At treatment time (Fig. 1C). Even after 24 h cellulose I was only partial dissolved in A:At as indicated by the relatively strong XRD equatorial reflections for the (002) crystallographic plane in particular for recovered and regenerated samples. Details regarding analysis of cellulose crystallinity index for all lyophilized samples using XRD peak deconvolution method is provided in the ESI appendix (ESI Fig. S2 & Table S1†).
 | ||
| Fig. 1 Pre-activation of crystalline cellulose with ammonia facilitates rapid dissolution in ammonia:ammonium thiocyanate (A:At) based ammonia-salt solvent system. (A) Schematic outlining cross-sectional view of model cellulose I fibril and how it can be pre-activated to a swollen cellulose–ammonia complex, or ultimately cellulose III allomorph, to facilitate salt penetration into crystalline matrix to fully solubilize individual cellulose chains into an A:At solution. The dissolved cellulose can be then regenerated as highly amorphous cellulose using a suitable antisolvent like ethanol. The dotted lines represent inter-molecular hydrogen-bonding between cellulose chains. (B) Picture showing how A:At solution can nearly instantly solubilize cellulose III within a few minutes of contact to give rise to a clear solution. X-ray diffraction (XRD) was carried out on lyophilized amorphous cellulose regenerated from A:At solution treated cellulose I (C) and cellulose III (D) for increasing durations of treatment time (1–24 h) at room temperature and pressure. XRD spectra reveals pre-activated cellulose III is nearly completely decrystallized within a few hours of A:At treatment while cellulose I is still mostly crystalline and intact even after 24 hours. Here, control cellulose I and cellulose III XRD spectra are shown in dark magenta; while 1, 3, 6, and 24 hours treated samples are shown in purple, cyan, green, and olive brown, respectively. Cellulose crystallite sizes for all A:At treated cellulose samples, based on the XRD peak deconvolution method, is outlined in the ESI section.† | ||
Ammonia-salt treated cellulose is easily hydrolyzed by cellulases
Next, we systematically characterized the hydrolysis activity of a commercial cellulase enzyme cocktail (Cellic C.Tec2 from Novozymes) at varying total enzyme loadings and durations of saccharification time for all A:At treated cellulose samples (Fig. 2). Firstly, we found that all A:At treated cellulose samples always resulted in a pretreated substrate that was significantly less recalcitrant towards cellulase activity. The degree of cellulose crystallinity disruption, quantified as recovered lyophilized cellulose crystallite size, was seen to be inversely correlated to the enzymatic hydrolysis yield of the never-dried recovered cellulose samples (Fig. 2A). It should be noted that lyophilization of amorphous cellulose from wet slurries can cause significant cellulose recrystallization and lower cellulase accessibility. This makes it challenging to draw clear correlations between XRD for dried samples versus enzymatic hydrolysis data for never-dried samples.49 We have also conducted enzymatic hydrolysis on all lyophilized samples to show a clear drop in overall hydrolysis yields (ESI Fig. S3†). Nevertheless, here again we see that A:At treated cellulose III was significantly more digestible than cellulose I or III controls.
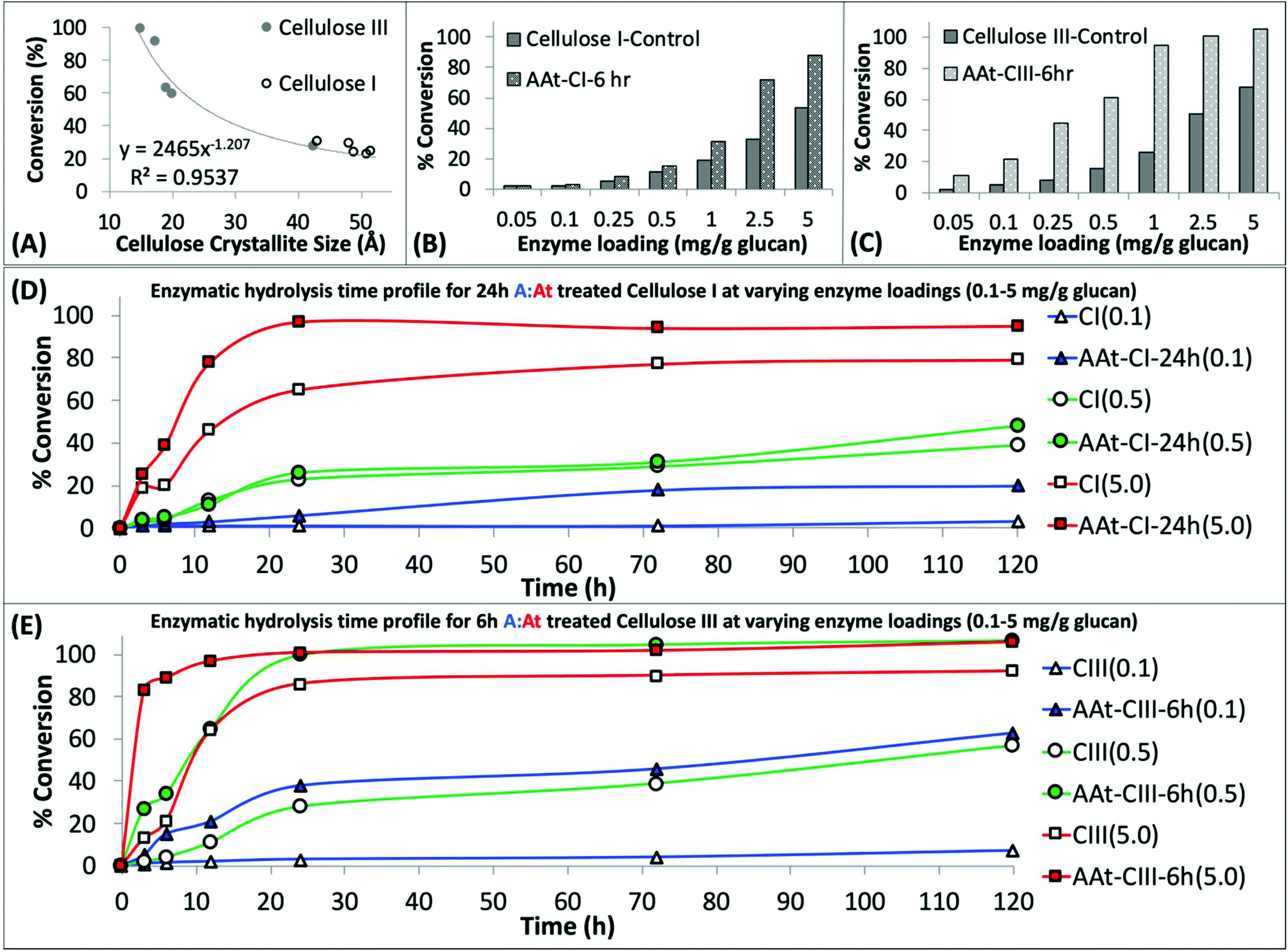 | ||
| Fig. 2 Regenerated amorphous cellulose recovered after ammonia:ammonium thiocyanate (A:At) pretreatment of crystalline cellulose can be readily hydrolyzed by cellulases. (A) A:At solvent pretreatment of cellulose results in reduction of cellulose crystallite size, estimated by deconvoluted XRD peaks analysis by Scherrer method for all cellulose I and cellulose III samples treated for varying durations. The crystallite size was inversely proportional to the cellulase catalyzed cellulose-to-glucose saccharification yield. See ESI† appendix for details on crystallite size analysis details. Cellulase enzyme dosage response studies revealed that A:At treated pre-activated cellulose III (C) gives higher hydrolysis yields versus both native and A:At pretreated cellulose I (C) after 24 hours of saccharification. Here, cellulose I and cellulose III were both treated by A:At for 6 h for sake of comparison. Lastly, detailed cellulase hydrolysis kinetics time course profile over 0–120 h is shown for native/A:At treated cellulose I (D) and cellulose III (E) at three different cellulase enzyme loadings (i.e., 0.1, 0.5, and 5 mg enzyme per g glucan). Here, 24 h A:At treated cellulose I and 6 h A:At treated cellulose III were compared for their relative hydrolysis time profiles. | ||
Furthermore, we studied the effect of total cellulase loading (i.e., mg enzyme per g cellulose) for all never-dried A:At pretreated cellulose samples. Representative 24 h saccharification results for cellulose allomorph controls and their respective 6 h A:At treated samples are shown here (Fig. 2B and C). These results clearly demonstrate that A:At treated cellulose III required much lower enzyme loadings to achieve near-theoretical conversion. Relative initial specific activity of the cellulase cocktail was determined to be close to an order of magnitude higher for A:At treated cellulose III versus native cellulose I (∼9.1 vs. 1 μmol glucose released per mg enzyme per min, respectively). Specific cellulase activity on control amorphous cellulose (i.e., PASC) under identical assay conditions was ∼6.5 μmol mg−1 min−1. Interestingly, relative specific activity of the cellulase cocktail was comparable for A:At treated cellulose I and cellulose III control (∼1.4 μmol glucose released per mg enzyme per min). Similar enzyme loading effects on cellulose conversion trends were seen for other pretreatment conditions as well, therefore, detailed data is not reported here. See ESI† for representative saccharification data reported for other A:At pretreated samples.
However, since neither native cellulose I or cellulose III gave maximum theoretical yields after 24 h, we also conducted kinetic time course saccharification assays over 5-days to determine the maximum conversion possible at ultra-low (i.e., 0.1, 0.5 mg g−1 glucan loading) and low (i.e., 5 mg g−1 glucan loading) enzyme loadings. The saccharification time course assay revealed that both cellulose allomorphs never achieved complete hydrolysis even at the highest 5 mg g−1 loading after 5 days (Fig. 2D). This is not surprising since slow but steady deactivation of cellulase enzymes at the air–liquid interface has been shown to be main factor responsible for incomplete cellulose digestion at low enzyme loadings of 5 mg g−1 glucan.50 However, A:At pretreated cellulose samples were able to achieve near-theoretical conversion even at the 0.5 mg g−1 glucan loading within 24 h (Fig. 2E). Improved substrate accessibility and reduced recalcitrance of the A:At pretreated cellulose III sample to hydrolytic cellulases is clearly beneficial to achieve rapid saccharification at ultra-low enzyme loadings that overcomes the challenges of enzyme deactivation during prolonged multi-day durations. Even with a much lower enzyme loading of 0.1 mg g−1 glucan, A:At pretreated cellulose III was able to digest nearly 2/3rds of the substrate within 5 days while native cellulose I and control cellulose III allomorphs both showed only 5–7% conversion after 5 days. In fact, close to 2/3rds hydrolysis of native cellulose I was only seen at ∼50-fold higher enzyme loading unlike A:At pretreated cellulose III.
SANS analysis of never-dried A:At treated cellulose ultra-structure
The never-dried cellulose recovered after A:At pretreatment could not be used to glean detailed structural information using XRD, and therefore SANS was used to characterize the never-dried cellulose samples (Fig. 3 and ESI Fig. S4†). Here, all samples were ex situ pretreated using protiated A:At solvent for 24 h and the regenerated amorphous cellulose recovered was directly analyzed without lyophilization, after exchange into D2O. All Avicel based cellulose samples were analyzed with three structural levels of the Unified Fit model: high-Q (>0.06 Å−1), mid-Q (0.008 < Q < 0.06 Å−1), and low-Q regions (<0.008 Å−1), as illustrated in Fig. 3C and the model fit values of the structural parameters are listed in Table 1.
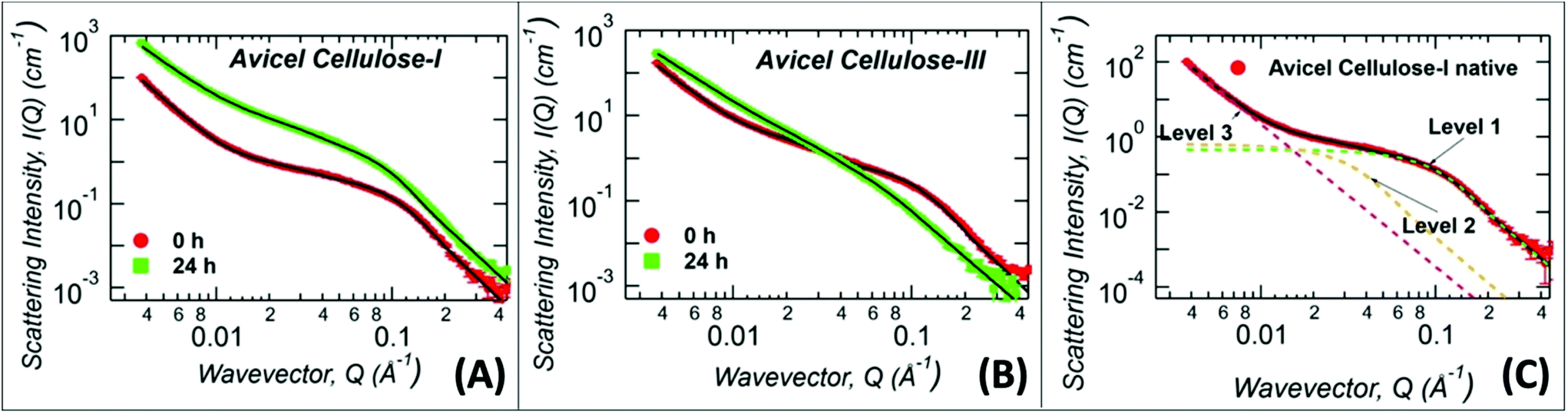 | ||
| Fig. 3
Ex situ ammonia:ammonium thiocyanate (A:At) pretreatment derived regenerated amorphous cellulose (RAC) were characterized by Small Angle Neutron Scattering (SANS). (A) Protiated A:At pretreated cellulose I (for 24 hours treatment time at room temperature/pressure prior to recovery of RAC) SANS scattering profile is shown here in green squares, while control untreated cellulose I (0 h) is shown in red dots. (B) Protiated A:At pretreated cellulose III (for 24 hours treatment time at room temperature/pressure prior to recovery of RAC) SANS scattering profile is shown here in green squares, while control cellulose III (0 h) is shown in red dots. (C) A representative unified model fit curve is shown as a solid black line fitted to cellulose I data (in red), along with individual levels shown as green (level 1), gold (level 2), magenta (level 3) dashed lines. The solid black lines in (A) and (B) are the unified fits to the experimental SANS data with results shown in Table 1. Note that all samples were exchanged in to D2O solvent prior to analysis and were never dried. | ||
:At treated (for 24 h) samples are shown here. Here, ‘#’ denotes parameters that were fixed during the model fitting process
| Sample type | High-Q Rg (Å) | High-Q exponent, P | Mid-Q Rg (Å) | Mid-Q exponent, P | Low-Q exponent, P |
|---|---|---|---|---|---|
| CI control | 20 ± 1 | 4# | 60 ± 10 | 4 # | 4.0 ± 0.1 |
| AAt-CI 24 h-treated | 23 ± 2 | 4# | 64 ± 6 | 4 # | 3.0 ± 0.1 |
| CIII control | 18.6 ± 0.6 | 4# | 68 ± 5 | 4 # | 3.3 ± 0.1 |
| AAt-CIII 24 h-treated | — | — | 61 ± 4 | 3.6 ± 0.2 | 2.67 ± 0.02 |
High-Q Rg represents the crystalline cellulose microfibril cross-sectional dimension,44,51 while the mid-Q Rg represents the size of the cellulose microfibril bundles or aggregates which are not necessarily crystalline in arrangement. Note that, Rg, is a shape independent parameter and should be multiplied by a factor of 2.3 to convert the high-Q Rg and mid-Q Rg values to true cellulose microfibril and bundle cross-sectional diameter, respectively. Both the cellulose microfibril size (high-Q Rg) and the cellulose bundle size (mid-Q Rg) show marginal change upon treatment of cellulose I with A:At (Fig. 3A). This indicates minimal penetration and disruption of A:At into the crystalline core of cellulose even after extensive 24 hours of dissolution time. However, in the low-Q region, which is sensitive to length scales up to 200 nm, the SANS profile follows a power-law decay. A power-law exponent of 4 represents a smooth surface while 3 represents a highly rough surface. The power-law exponent decreased from 4.0 ± 0.1 to 3.0 ± 0.1 after A:At treatment of cellulose I indicating that the surface morphology of the larger aggregates became rough upon treatment. This implies that A:At only disrupts larger aggregates of cellulose-I by slowly disrupting its surfaces while smaller crystalline core structures are still largely intact.
In contrast, the high-Q cellulose microfibril size feature disappeared completely after A:At pretreatment of ammonia pre-activated cellulose III, indicating a complete loss of the crystalline arrangement of the cellulose microfibril structure (Fig. 3B). This observation implies that after 24 h of dissolution, A:At solvent had fully penetrated into the cellulose microfibrils and disrupted its core crystalline structure. Interestingly, the mid-Q Rg (60–68 Å) showed a marginal difference after A:At treatment. Unlike the features responsible for SANS signal in the high-Q region, the mid-Q feature represents larger cellulose structures. These larger entangled structures mostly arise upon precipitation of highly solvated cellulose chains from an A:At solution into an aqueous environment. Note that the larger cellulose structures detected in this Q-region do not need to be highly crystalline. However, in the high-Q region, the largest contrast is seen between cellulose structures with minimal solvent penetration and the solvent. Here, crystalline cellulose has the most contrast with respect to the solvent. In addition, the low-Q power-law exponent decreased from 3.3 ± 0.1 to 2.67 ± 0.02 after treatment of cellulose III by A:At solution. This change in the power-law exponent suggests that the amorphous cellulose recovered after A:At treatment has significantly higher solvent penetration and sufficiently disrupted microfibril structure.51 This smaller power-law exponent qualitatively explains why A:At treated cellulose III results in a substrate with significantly higher rates of enzymatic hydrolysis unlike A:At treated cellulose I.
Mechanism of cellulose dissolution in A:At solvent system
Since A:At treatment duration had a critical impact on the recalcitrance of the recovered regenerated cellulose after pretreatment, it was necessary to carefully study the subtle structural changes in cellulose in situ during the A:At treatment process using SANS (Fig. 4). Ex situ analysis revealed that the native allomorphs microfibril size (Rg) is 20 ± 1 Å and 18.6 ± 0.6 Å for Avicel based cellulose I and cellulose III, respectively (Table 1). These sizes are consistent with literature reported size of Rg = 18.5 ± 0.5 Å for Avicel PH-105 microcrystalline cellulose.44,51 In addition, the cellulose microfibril bundles in the mid-Q region are also of similar sizes, Rg ∼ 60 ± 10 and 68 ± 5 Å, for cellulose I and cellulose III, respectively. The only clear distinction between cellulose I and cellulose III structures is the power-law exponent in the tertiary region, 4.0 ± 0.1 and 3.3 ± 0.1, respectively. Cellulose III exhibits a high degree of surface roughness of the large structures unlike the smoother surfaces of cellulose I. However, current set of ex situ SANS analysis of recovered cellulose after A:At pretreatment provide limited mechanistic information on how subtle differences in cellulose structure impacts real-time A:At penetration and subsequent dissolution of pre-activated cellulose III. Therefore, we used SANS to characterize the ultrastructure of cellulose during real-time, in situ dissolution using deuterated A:At solvent over a duration of 0–72 h at room temperature (Fig. 4).
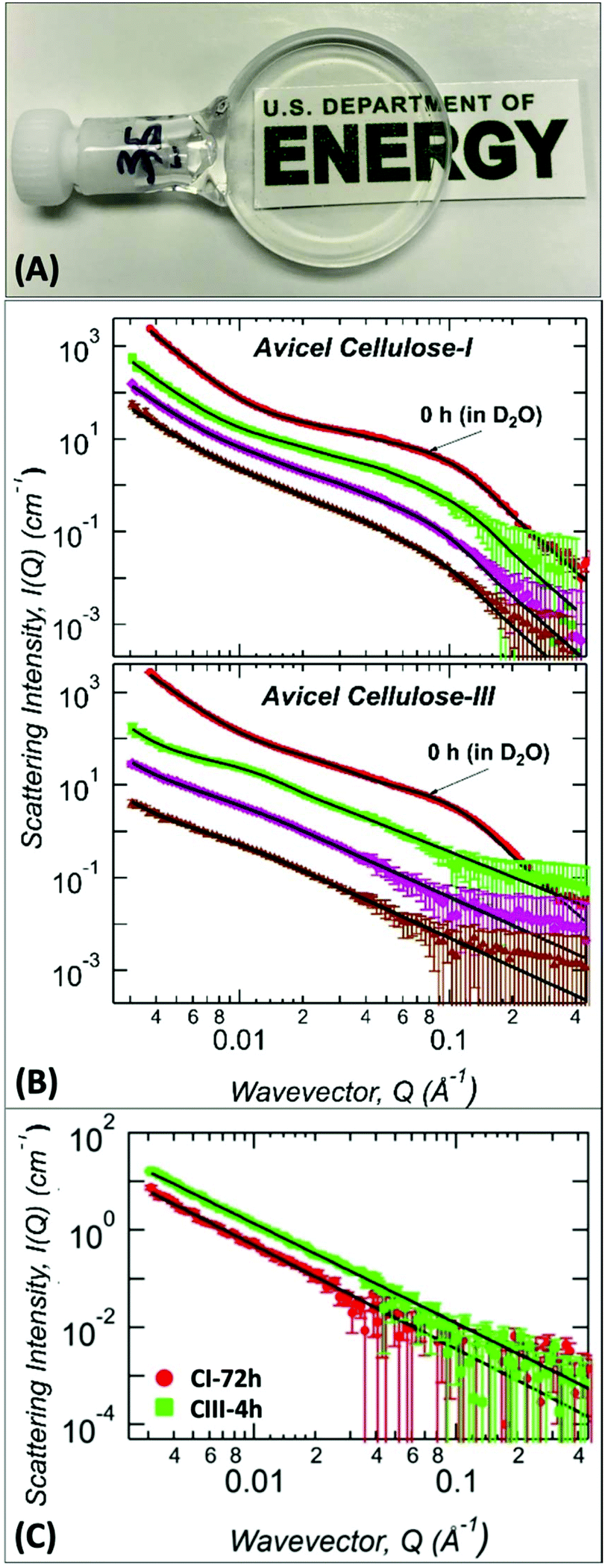 | ||
| Fig. 4
In situ ammonia:ammonium thiocyanate (A:At) dissolution of cellulose and characterization by Small Angle Neutron Scattering (SANS). (A) Visual example SANS sample holder cell containing a clear solution of 2% (w/v) cellulose dissolved in deuterated A:At solution prepared using pre-activated cellulose III. (B) SANS profiles snapshot of untreated and deuterated A:At solvent treated (top) cellulose-I and (bottom) cellulose-III samples dissolved real-time for varying time periods. The curves are untreated controls are suspended in 100% D2O (red dot); deuterated A:At solvent treated for 0.5 h (green square), 4 h (magenta diamond), and 24 h (brown up triangle) are shown here. The scattering curves are vertically shifted for clarity. (C) SANS profiles snapshot of protiated A:At solvent treated (green square) cellulose-I after 72 h and (red dot) cellulose-III samples after 4 h dissolution times, respectively. In all plots, solid black lines are the Unified fits to the experimental SANS data with results shown in Table 2. | ||
Structural parameters of the in situ A:At treated cellulose was obtained from unified fit models of SANS data as before (Table 2). For cellulose I, the size of cellulose microfibrils marginally increased from 20 to 23–25 Å within 24 h of treatment (high-Q region). However, most of this change occurred after the first few hours of treatment. Cellulose bundles (or aggregates) increased mostly within the first 4 h of treatment (mid-Q region) and bundle associated Rg value remained constant up to 24 h. The surface morphology of the larger structures showed a gradual surface roughening during the treatment as indicated by the steady decrease in the low-Q power-law exponent. However, after 24 h of treatment, A:At had only penetrated the large structures of native cellulose I, which was sufficient to distinguish the highly entangled larger network from the intact crystalline core.
:At solvents for varying durations of treatment time are shown here. Here, ‘#’ denotes parameters that were fixed during the model fitting process
| Sample type | High-Q Rg (Å) | High-Q exponent, P | Mid-Q Rg (Å) | Mid-Q exponent, P | Low-Q exponent, P |
|---|---|---|---|---|---|
| Avicel cellulose-I | |||||
| In 100% D2O | 20 ± 1 | 4# | 60 ± 10 | 4 # | 4.0 ± 0.1 |
| 0.5 h in ND3:ND4SCN |
20 ± 3 | 4# | 80 ± 16 | 4 # | 3.4 ± 0.2 |
| 4 h in ND3:ND4SCN |
25 ± 3 | 4# | 110 ± 16 | 4 # | 3.2 ± 0.1 |
| 24 h in ND3:ND4SCN |
23 ± 2 | 4# | 94 ± 8 | 4 # | 2.8 ± 0.1 |
| 72 h in NH3:NH4SCN* |
— | — | — | 2.05 ± 0.03 | — |
| Avicel cellulose-III | |||||
| In 100% D2O | 18.6 ± 0.6 | 4# | 68 ± 5 | 4 # | 3.3 ± 0.1 |
| 0.5 h in ND3:ND4SCN |
— | — | 153 ± 6 | 1.8 ± 0.1 | — |
| 4 h in ND3:ND4SCN |
— | — | 280 ± 32 | 2.0 ± 0.1 | — |
| 24 h in ND3:ND4SCN |
— | — | 300 ± 41 | 2.1 ± 0.1 | — |
| 4 h in NH3:NH4SCN* |
— | — | — | 2.14 ± 0.07 | — |
Cellulose III, unlike cellulose I, undergoes rapid deconstruction upon contact with A:At solvent. The cellulose microfibril feature in the high-Q region is not present within 30 min of treatment suggesting that A:At solvent readily penetrates and rapidly swells the cellulose crystalline core microfibrils. As a consequence, the microfibril features are not at all visible. Only changes in the cellulose bundle size (based on observed mid-Q Rg values) is observed likely due to rearrangement of the cellulose microfibrils within the bundles. Cellulose III bundle size increased by 100% from 0.5 h to 24 h of A:At treatment. A similar change in mid-Q Rg values is observed as seen earlier for cellulose-I. However, most of this change occurs within the first 4 h of treatment (i.e., 153 to 300 Å) and the bundles are much larger in size than those observed with cellulose I (i.e., 94 to 110 Å). In agreement with the trend seen for the radius of gyration, the bulk morphology obtained from the mid-Q exponent P suggests that the cellulose microfibril conformation transitions from a slightly stiff to flexible conformation at 0.5 h to 24 h, i.e., 1.8 to 2.1.42 At the beginning of the treatment (∼0.5 h), the thiocyanate anions are likely associated with the surface of closely-packed individual cellulose strands, thus inducing self-repulsion between cellulose-ion clusters to give rise to a stiffer polymer conformation. As the treatment proceeds, increasing solvation and A:At solvent penetration are expected to occur along with slow diffusion of the solvated cellulose chains into the bulk solvent, thereby making the cellulose chains dispersed in the solvent to be progressively more random oriented and thus have increased conformational flexibility.
The cellulose C6-hydroxymethyl group conformation likely plays a critical role on facilitating the rapid cellulose III microfibrils dissolution and ultimately aiding the solvation of individual cellulose polymer chains in A:At based solvent (Fig. 5). We further hypothesize that individual solvated cellulose chains likely adopt a predominantly Gauche–Gauche (GG) and GT conformations due to strong hydrogen-bonding with cation–anion clusters that facilitate extensive cellulose solvation and prevent recrystallization of fully solvated cellulose polymer chains. Molecular dynamics simulations were carried out to test this hypothesis, characterize the hydrogen bonding interactions between cellulose–A:At, and gain understanding of the molecular mechanism of model cellulose polymer chain (i.e., cellohexaose) solvation in an A:At solvent. We found individually solvated cellohexaose chains adopt both Gauche–Trans (GT) and Gauche–Gauche (GG) hydroxymethyl conformations in A:At solvent due to strong interactions of cellulose with the ammonium cations that completely disrupt intrachain cellulose O2–O6 hydrogen bonding (ESI Table S5†). The simulations also reveal the formation of ammonium cation/ammonia cationic clusters in the solvent that facilitated dissolution of solvated cellulose fibrils. Raman spectroscopic analysis for concentrated A:At solutions also revealed the presence of strong interactions of thiocyanate–ammonia and thiocyanate–ammonium ions,52 which further validate our MD simulation findings. The strong interactions of cellulose with the A:At solvent facilitate extensive cellulose solvation and likely prevent recrystallization of fully solvated cellulose polymer chains. See ESI appendix for details (ESI M5, Fig. S5–S12 and Tables S5†). Finally, the MD simulation results support earlier hypothesis that dissolution of cellulose in ammonia-salt solutions would depend on the physical properties of the associated salt (e.g., anion size, solvation sphere, strength of interaction between ammonia and salt ions). Therefore, this suggests that the alternative ammonia-salt conditions with appropriate physical properties (e.g., ammonium iodide) might be also able to rapidly dissolve cellulose if the cellulose has been pre-activated by ammonia.
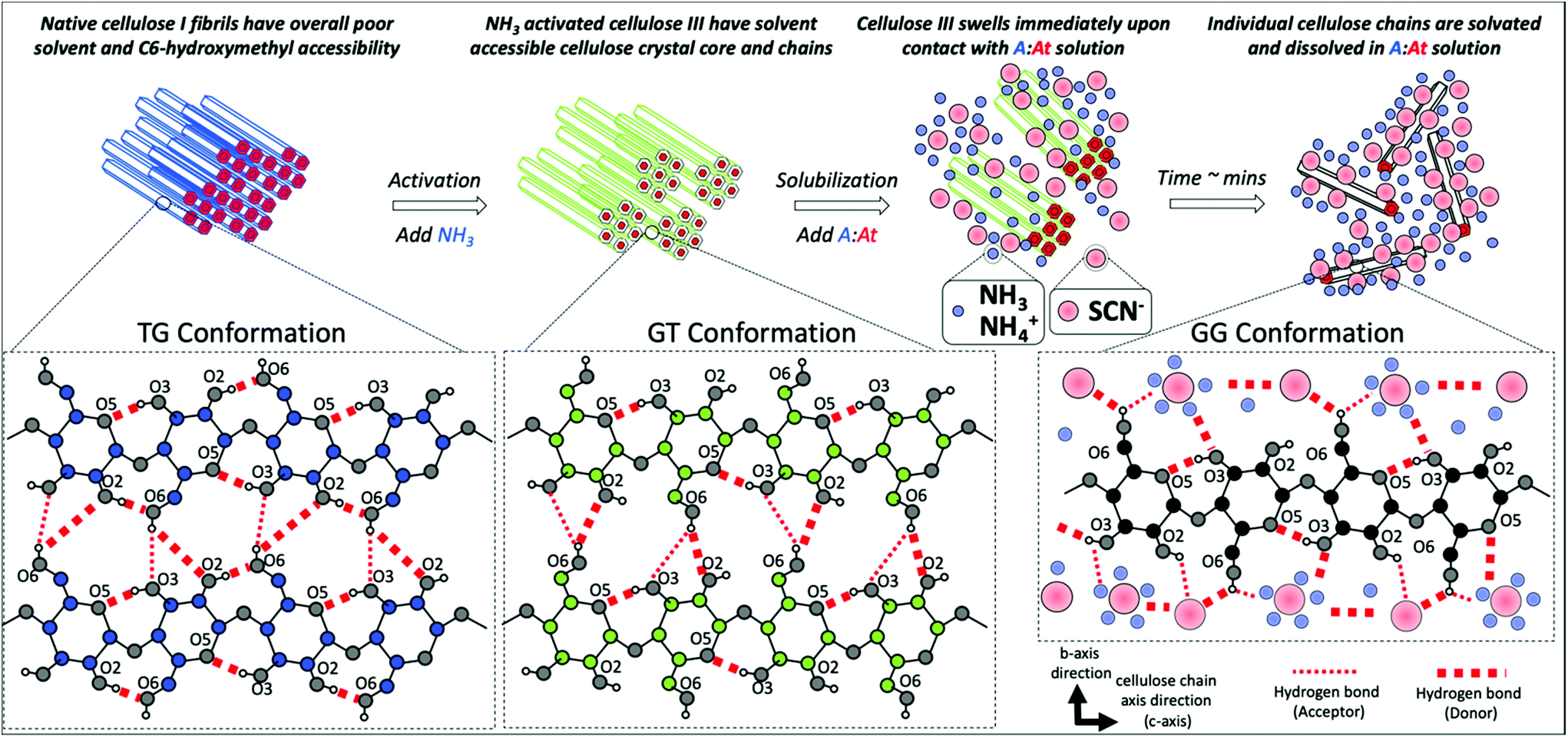 | ||
| Fig. 5 Hypothesized role of cellulose C6-hydroxymethyl group conformation on rapid microfibrils dissolution and ultimately solvation of individual cellulose polymer chains in an ammonia:ammonium thiocyanate (A:At) based solvent. Native cellulose I fibrillar structure is resistant to rapid dissolution in A:At largely due to a Trans–Gauche (TG) conformation of the C6-hydroxymethly group. However, ammonia pre-activation results in the formation of a slightly less tightly packed crystalline allomorph (i.e., cellulose III), with a predominantly Gauche–Trans (GT) conformation of the C6-hydroxymethyl group, and reduced intra-sheet hydrogen bonding. The GT-conformation and the altered accessible crystal morphology facilitates A:At solvent penetration into the core crystalline matrix and causes rapid swelling of the microfibrils. Finally, we hypothesize that individual solvated cellulose chains adopt a predominantly Gauche–Gauche (GG) and GT conformations due to strong hydrogen-bonding with ammonia/ammonium cation solvated thiocyanate ionic clusters that facilitates extensive solvation and prevents recrystallization of fully solvated cellulose polymer chains. Here, key cellulose intrasheet based interchain and intrachain hydrogen bonding (shown as dotted red lines for likely donor and/or acceptor bonds formed) for native cellulose Iβ, cellulose III, or soluble single cellulose chains is depicted in the dotted inset boxes with carbon atoms depicted in blue, green, or black filled circles, respectively. Oxygen and select hydrogen atoms are depicted as gray and white filled circles, respectively. Oxygen atoms are numbered according to the attached carbon atom. Ammonia/ammonium cations and thiocyanate anions are depicted by light blue and light red filled circles, respectively. Please note that all elements depicted in the schematic are not representative of actual relative scale. | ||
Lastly, we have analyzed the in situ SANS profiles for cellulose I dissolved in protiated A:At solvent after 3 days. Interestingly, the high-Q cellulose microfibril features and cellulose bundle features were not observed for both 4 h and 72 h A:At treated cellulose III and cellulose I, respectively (Fig. 4C). However, the use of protiated solvents makes it difficult to resolve smaller structures. The reason is that the signal of cellulose microfibril and bundles/aggregates is reduced due to lower contrast. Also, incoherent background scattering from the hydrogen atoms in the system is significantly increased which makes the overall signal-to-noise rather poor. Nevertheless, the mid-Q exponent values of 2.05 observed suggests that significant disruption takes place in the cellulose I structure only after extended duration of incubation with the A:At solvent unlike pre-activated cellulose III.
One of the other limitations of the previous experimental approach to validate the polymorph interconversion process to explain the mechanism of cellulose I dissolution in A:At solvent was the regenerated cellulose samples analysis recovered after each A:At thermal cycle treatment step using XRD or NMR.32,33 However, no in situ studies were conducted to study real-time cellulose dissolution dynamics in A:At solutions. Although, Groot and co-workers used static light scattering (SLS) to characterize the structure of dissolved cellulose to determine the polymer persistence length and characterize the cellulose structure upon being fully solvated in A:At solutions.39,53 But one of the major challenges associated with light scattering methods is the weak scattering observed due to the very low contrast in the refractive index of A:At and cellulose that ultimately resulted in radius of gyration (Rg) measurements with poor accuracy (±30–50% error) and low reliability since the Rg values estimated were close to the lower detection limit of classical SLS methods (∼15 nm). Nevertheless, the radius of gyration predicted using SLS was predicted to be about ∼21 ± 11 nm for model cellulose fully dissolved in A:At solution. SANS is an effective alternative technique for dynamic multilength scale in situ or static multilength scale ex situ characterization of complex ultrastructures within highly heterogenous samples like cellulose or even whole plant cell walls.54,55 We have now used deuterated A:At based solutions to overcome limitations associated with the poor contrast between the solvent and cellulose for maximizing the signal collected during neutron scattering. Real-time SANS characterization of dissolution of pre-activated cellulose III has revealed that the cellulose microfibril features are lost within a few minutes and the final radius of gyration (mid-Q range Rg) obtained within a few hours was stabilized at about ∼29 ± 5 nm. These values are consistent with observations made for molecular solutions of cellulose for other solvents like IL.55–57
Lignocellulosic biomass pretreatment under ambient conditions
Finally, we were interested in generating proof-of-concept data to show that the A:At solvent system could be used to directly pretreat and fractionate lignocellulosic biomass like corn stover into its constituent components similar to other reported IL. Interestingly, addition of untreated corn stover to A:At solvent almost instantly gave rise to a dark brown colored and slightly viscous gel-like solution (Fig. 6A). Corn stover was incubated at 4% (w/v) solids loading in the A:At solvent under ambient conditions for 24 h prior to addition of ethanol as anti-solvent to precipitate any solubilized polysaccharides to perform a material balance analysis. The supernatant was then removed after centrifugation and the solid biomass pellet was washed to remove any residual A:At. We found about ∼15–20% of the starting material by weight was extracted from corn stover by A:At solvent (Fig. 6B). Compositional analysis revealed that A:At solvent was able to readily extract close to ∼80–85% of original available total lignin, while leaving behind most of the polysaccharides (i.e., glucans and xylans) largely intact with significantly less than ∼5% total sugar loss in the extractives stream. Overall lignin extraction yields for the A:At solvent based biomass pretreatment process were nearly double of our previous reported value for the EA process (∼40%),24 but with similar polysaccharide extraction yields. However, unlike the current proposed A:At pretreatment process performed at 25 °C and ∼15 psi pressure, the EA process was operating at a much higher thermochemical severity (e.g., ∼100–130 °C at ∼1200–1500 psi operational pressures). Furthermore, the A:At solvent pretreated residual corn stover polysaccharides were shown to be readily hydrolyzed using a low enzyme loading (∼2 mg g−1 glucan) into soluble sugars like glucose (Fig. 6C). There is obviously room for further process improvement as no optimization has been currently performed on either the solvent system composition or exact pretreatment conditions that maximize treated cellulosic biomass conversion yields.
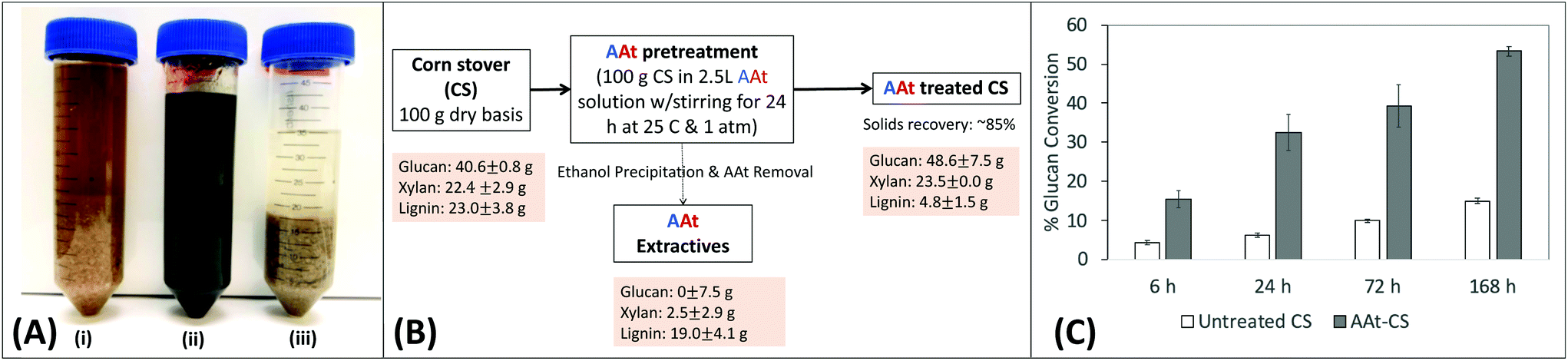 | ||
| Fig. 6 Lignocellulosic biomass ambient conditions pretreatment and selective lignin fractionation using an ammonia:ammonium thiocyanate (A:At) based solvent system. (A) Representative images of corn stover (CS) suspended in water (i), A:At solvent during treatment (ii), and after ethanol precipitation for dissolved solids recovery (suspended here in 100% ethanol) (iii). (B) Mass balance and composition analysis (i.e., glucan, xylan, and total lignin) for corn stover before and after A:At pretreatment are shown here. Here, all mass balance and compositional analysis experiments were done in two replicate batches conducted on two separate days each (see ESI M4† for details) and the relevant mean values and corresponding standard deviations are reported here. The A:At CS extractives composition was indirectly estimated based on the untreated CS and A:At CS substrate composition as well as based on the ∼85% solids mass recovery values. (C) Cellulase activity assay studies reveal that A:At treated corn stover gives higher cellulose hydrolysis yields versus untreated corn stover during all saccharification time points. | ||
Conclusions
Saturated solutions of ammonia–ammonium thiocyanate (A:At) were first reported as a possible non-derivatizing solvent system to fully dissolve regenerated amorphous cellulose, like viscose cellulose, by Scherer in 1931.30 However, mixed results were reported for the dissolution ability of several ammonia-salt solutions on more crystalline forms of cellulose like cotton. It was only half a century later in the 1980s that Cuculo and co-workers at North Carolina State University developed a multi-stage thermal cycling based treatment process using extremely low freezing temperatures to more effectively dissolve crystalline cellulose like cotton linters into A:At type solvents.31 This process involved exposing the cellulose to A:At solvent first to a low freezing temperature (e.g., −80 to −20 °C) for several hours to days followed by slowly bringing the temperature up to 20–40 °C with constant mechanical stirring. The low temperature cycle process was then repeated again multiple times to facilitate complete dissolution. Cuculo and co-workers hypothesized about the mechanism of native crystalline cellulose I dissolution in A:At solvent, due to extreme thermal cycling steps, took place via intermediate polymorph transformations similar in structures to cellulose II and III.32,33 They invoked the interplay of entropic vs. enthalpic thermodynamic effects driving ammonia–thiocyanate vs. ammonia–cellulose interactions at the extreme ranges of the thermal cycling temperatures to rationalize the polymorph transformation mechanism. However, the critical role of the cellulose ultrastructure modification to either cellulose-ammonia complex, or cellulose III allomorph, on the real-time dissolution kinetics of cellulose in A:At solvent was not explored. We have now shown that pre-activation of cellulose by ammonia to form a metastable cellulose–ammonia complex, or cellulose III allomorph, facilitates near-instantaneous swelling and penetration of A:At solution to rapidly dissolve cellulose within a few mins. Furthermore, this pre-activation step allows cellulose dissolution at near ambient conditions with no requirement for extreme thermal cycling temperatures or extensive mechanical mixing of the cellulose solution. Molecular dynamics simulations provided detailed insight into the highly dynamic hydrogen bonding interactions between cellulose and ammonium cation/ammonia cationic clusters that disrupted intramolecular cellulose hydrogen bonds. Our MD analysis also provides further support to the polymorph interconversion hypothesis that explains why pre-activated cellulose III like allomorphs are rapidly solvated and fully dissolved in A:At type solvents unlike cellulose I at room temperature without any necessity for extensive thermal cycling.
We have also conducted detailed enzymatic assays to show that amorphous cellulose regenerated from a molecular solution of A:At prepared using pre-activated cellulose III can be rapidly digested by cellulases at ultra-low enzyme loadings (0.1–1 mg g−1 glucan) to soluble sugars like glucose, unlike native crystalline cellulose I and cellulose III as well. Also, we have confirmed A:At based solvents can selectively extract lignin from complex lignocellulosic biomass like corn stover while significantly reducing recalcitrance of residual polysaccharides to enzymatic hydrolysis even at low enzyme loadings. The proposed A:At pretreatment process can be theoretically conducted at ambient conditions unlike the traditional EA or other anhydrous liquid ammonia based pretreatments. Compared to ionic liquids like 1-ethyl-3-methylimidazolium acetate, the dynamic viscosity of the A:At solution alone is nearly 3 orders of magnitude lower under comparable process conditions (see ESI M6† for detailed analysis of A:At solvent and A:At/cellulose solution viscosity). This clearly highlights that A:At solvent is an ideal low viscosity solvent for cellulose dissolution at room temperature unlike conventional ILs for cellulose pretreatment. Furthermore, since efficient cellulose deconstruction is likely dependent on the exact A:At processing conditions (e.g., cellulose loading, dissolution temperature, mixing/stirring/shear rate), we believe there is ample opportunity to further reduce total processing time (i.e., under 1 h) and solvent loading (i.e., by increasing cellulose loading) by further optimizing pretreatment conditions as well as optimizing enzymatic saccharification conditions to achieve near-theoretical hydrolysis yields similar to currently mature IL-based processes. Our promising lignocellulosic biomass fractionation-type pretreatment results inspire confidence about the possibility of selectively optimizing and upgrading the carbohydrate and aromatics enriched product streams into diverse bioproducts using either standalone or hybrid catalytic/biocatalytic processing routes in the near future. Cuculo and co-workers have already reported that A:At type solvents are non-derivatizing and do not cause any cellulose degradation upon long-term incubation with cellulose at temperatures close to 25 °C.31,58 However, ammonia-based solvents/pretreatments (e.g., AFEX/EA) are reported to form minor glucosamine derivatives due to the reaction of ammonia with reducing sugar aldehydes as well as amides due to ammonolytic reactions of lignin-carbohydrate ester linkages.24,59,60 It is currently unclear if A:At could further alter the core lignin structure during similar ammonolytic-type reactions (similar to AFEX61 or EA62 pretreatment) at room temperature during pretreatment that facilitates lignocellulose fractionation and how exactly such lignin structural modifications would impact either downstream fermentation process for sugar-upgrading or catalytic conversion processes for lignin valorization.
Regarding safety, health, and environmental or SH&E factors impacting overall ‘sustainability’, toxicity is indeed an important consideration for commercialization/scale-up of any biorefinery process employing chemicals. From a SH&E sustainability point of view, classical solvents (e.g., ethanol) have clear SH&E guidelines to allow ranking of such solvents as first reported in a 2016 Green Chemistry study,63 but it is challenging to determine similar solvent ranking scores for poorly studied ionic liquids or chemically complex mixtures (e.g., A:At) based solvent systems. Nevertheless, based on the available Global Harmonized System (GHS) ratings, we can readily estimate the combined solvent/chemical SH&E ranking score for ammonium thiocyanate alone to be 8 (i.e., classified as hazardous based on the 2016 report for ranking solvent ‘greenness’ using a combined SH&E criteria score).63 This poor ranking score is mostly based of an environmental criteria score of 5 due to the high aquatic environmental toxicity rating of ammonium thiocyanate (i.e., H412 GHS rating). Thus, an ammonia-salt solvent system employing ammonium thiocyanate as salt would likely be also classified as hazardous using similar SH&E criteria. However, we believe our current study provides a clear mechanistic basis for developing similar one-pot biomass fractionation by ammonia-salt solvent systems using alternative salts that have more favorable SH&E ratings (e.g., ammonium chloride or iodide). Ammonium iodide was originally also reported by Scherer in 1931,30 along with ammonium thiocyanate, to also dissolve regenerated viscose cellulose in ammonia-salt solutions but ammonium iodide based solvents were reported to only have partial solubility for crystalline cotton cellulose. However, it is very likely that pre-activation of cellulose by ammonia alone would also facilitate dissolution of pre-activated crystalline cellulose III in ammonia–ammonium iodide solutions. While Cuculo and co-workers revisited the ammonia–ammonium thiocyanate solvent system for cellulose dissolution using an improved cellulose-solvent slurry freeze/thaw method in the 1980s,31 no additional work has been reported in the open literature on the use of alternative ammonium salts for cellulose dissolution. Similarly, while ammonium chloride is known to also dissolve in liquid ammonia to form stable ammonia-salt solutions at room temperature,64 no data is currently available on the suitability of an equivalent ammonia–ammonium chloride solvent system for enabling rapid cellulose dissolution. Detailed future studies would be necessary to explore alternative ammonium salts (e.g., ammonium iodides or chlorides) that are potentially less toxic and have better SH&E sustainability ratings than ammonium thiocyanate to be used in similar one-pot ammonia pre-activation based rapid cellulose dissolution process. For example, the current safety and health criteria score for ammonium chloride is 1 (due to its desired low flammability) and 2 (mostly due to its undesired acute toxicity upon oral ingestion), respectively. The GHS ratings for ammonium iodide (or chloride) suggest these alternative chemicals are less hazardous than ammonium thiocyanate and likely have a combined SH&E solvent rating criterion similar to other ammonium or quaternary ammonium-based salts currently used in agricultural systems.65 Furthermore, due to the world-wide prevalent use of ammonia and ammoniacal salts as fertilizers in our current agricultural systems, an appropriate choice of a suitable ammonia-salt system with ideal SH&E ratings would facilitate systematic integration of such chemicals into our current agro-industrial economy for large-scale implementation of ammonia/ammonia-salt based biorefineries.
Regarding technoeconomic factors impacting ‘sustainability’, our currently proposed approach from a process intensification point-of-view combines elements of a traditional AFEX or EA based pretreatment process along with Scherrer/Cuculo's reported ammonia–ammonium thiocyanate solvent system to achieve a new one-pot process with likely signification reduction in overall capital costs and/or energy usage for cost-effective conversion of cellulosic biomass to fermentable sugars in a biorefinery. We believe our proposed process simplification and pretreatment mechanism-based process intensification would lead to a more economic and energetically sustainable process. Reducing the overall thermal and pressure severity employed by use of ammonia-based solvents could promote development of more sustainable and safer processing strategies for biomass refining. Furthermore, ammonia-salt based solvents have a significantly lower cost than traditional ionic liquids which could also significantly lower the economic barrier for implementing such pretreatment processes in biorefineries. Recent work has focused on the development of low-cost ionic liquids to address this very issue for cellulosic biomass pretreatment. For example, quaternary ammonium salts66 and lignin-derived bionic liquids67 have been recently reported as alternative low-cost ionic liquids for biomass pretreatment. However, most previous work that have used such ionic liquids (e.g., triethylammonium hydrogen sulfate) often require elevated temperatures (i.e., 120–130 °C) for effective cellulosic biomass pretreatment due to the high viscosity of such solvent systems at lower temperatures. But we have shown it is possible to pretreat/fractionate lignocellulosic biomass at room temperature/pressure using our low-viscosity model ammonia-salt solvent system,58 which could reduce safety related hazards associated with handling any solvents/chemicals at elevated temperatures. Nevertheless, a detailed technoeconomic and life cycle analysis of similar processes (including solvent recycle) will need to be pursued in the near future to properly evaluate the feasibility of ammonia-salt based solvents for commercial applications.
Author contributions
SPSC conceived the project. SPSC, LS, and RP prepared the ammonia-salt solutions. SPSC, LS, SVP, and HO carried out the SANS experiments; while ZY and SVP performed all SANS data analysis. SR, SG, and CZ performed all post-treatment sample characterization, enzymatic processing, and data analysis. SHL and LP performed the MD simulations and relevant data analysis. SPSC wrote the manuscript. All authors contributed to reviewing and editing the final paper.Conflicts of interest
SPSC has filed for patent applications on processes to produce cellulose-III enriched cellulosic biomass for biofuels production (US20130244293A1 and WO2011133571A2). Other authors have no conflicts to declare.Acknowledgements
SPSC acknowledges support from the ORAU 2016 Ralph E. Powe Award, ORNL Neutron Sciences User Facility, US National Science Foundation CBET awards (1604421 and 1846797), Rutgers Global Grant, Rutgers Division of Continuing Studies, Rutgers School of Engineering, and the Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494). MD and SANS studies on Bio-SANS were supported by the Office of Biological and Environmental Research (OBER) funded Center for Structural Molecular Biology (CSMB) under contract FWP ERKP291 and the Bio-Fuels Science Focus Area under contract FWP ERKP752, using the High Flux Isotope Reactor supported by the Office of Basic Energy Sciences (BES), U. S. Department of Energy (DOE). SR and CZ acknowledge support from University Grants Commission of India (Raman Fellowship Program) and China Scholarship Council, respectively, to support their one-year postdoctoral research work at SPSC's lab at Rutgers University. Lastly, we are very grateful to Benjamin Esposito and Jia-Mei Hong from SPSC's Rutgers research group for their critical experimental contributions to the project.This manuscript has been co-authored by UT-Battelle, LLC, under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
References
- G. T. Beckham, J. F. Matthews, B. Peters, Y. J. Bomble, M. E. Himmel and M. F. Crowley, J. Phys. Chem. B, 2011, 115, 4118–4127 CrossRef CAS.
- P. J. Weimer, A. D. French and T. A. Calamari Jr., Appl. Environ. Microbiol., 1991, 57, 3101–3106 CAS.
- S. P. S. Chundawat, G. Bellesia, N. Uppugundla, L. Sousa, D. Gao, A. Cheh, U. Agarwal, C. Bianchetti, G. Phillips, P. Langan, V. Balan, S. Gnanakaran and B. E. Dale, J. Am. Chem. Soc., 2011, 133, 11163–11174 CrossRef CAS.
- Y.-H. P. Zhang, S.-Y. Ding, J. R. Mielenz, J.-B. Cui, R. T. Elander, M. Laser, M. E. Himmel, J. R. McMillan and L. R. Lynd, Biotechnol. Bioeng., 2007, 97, 214–223 CrossRef CAS.
- F. Xu, J. Sun, N. V. S. N. M. Konda, J. Shi, T. Dutta, C. D. Scown, B. A. Simmons and S. Singh, Energy Environ. Sci., 2016, 9, 1042–1049 RSC.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Science, 2014, 343, 277–280 CrossRef CAS.
- M. Tyufekchiev, P. Duan, K. Schmidt-Rohr, S. Granados Focil, M. T. Timko and M. H. Emmert, ACS Catal., 2018, 8, 1464–1468 CrossRef CAS.
- T. Y. Nguyen, C. M. Cai, R. Kumar and C. E. Wyman, ChemSusChem, 2015, 8, 1716–1725 CrossRef CAS.
- E. Johnson, Biofuels, Bioprod. Biorefin., 2016, 10, 164–174 CrossRef CAS.
- D. Kumar and V. Singh, Biotechnol. Biofuels, 2016, 9, 228 CrossRef.
- T. Liebert, in Cellulose Solvents: For Analysis, Shaping and Chemical Modification, American Chemical Society, 2010, vol. 1033, pp. 3–54 Search PubMed.
- S. Singh and B. A. Simmons, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, John Wiley & Sons, Ltd, 2013, pp. 223–238 Search PubMed.
- S. P. S. Chundawat, G. T. Beckham, M. Himmel and B. E. Dale, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 121–145 CrossRef CAS.
- A. Pinkert, K. N. Marsh and S. Pang, Ind. Eng. Chem. Res., 2010, 49, 11121–11130 CrossRef CAS.
- D. Klein-Marcuschamer, B. A. Simmons and H. W. Blanch, Biofuels, Bioprod. Biorefin., 2011, 5, 562–569 CrossRef CAS.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098 RSC.
- H. Schleicher, C. Daniels and B. Philipp, J. Polym. Sci., Part C: Polym. Symp., 1974, 251–260 CAS.
- A. Mittal, R. Katahira, M. Himmel and D. Johnson, Biotechnol. Biofuels, 2011, 4, 41 CrossRef CAS.
- A. J. Barry, F. C. Peterson and A. J. King, J. Am. Chem. Soc., 1936, 58, 333–337 CrossRef CAS.
- K. Igarashi, M. Wada and M. Samejima, FEBS J., 2007, 274, 1785–1792 CrossRef CAS.
- M. Lewin and L. G. Roldan, J. Polym. Sci., Part C: Polym. Symp., 1971, 213–229 CAS.
- W. E. Davis, A. J. Barry, F. C. Peterson and A. J. King, J. Am. Chem. Soc., 1943, 65, 1294–1299 CrossRef CAS.
- G. L. Clark and E. A. Parker, J. Phys. Chem., 1937, 41, 777–786 CrossRef CAS.
- L. da Costa Sousa, M. Jin, S. P. S. Chundawat, V. Bokade, X. Tang, A. Azarpira, F. Lu, U. Avci, J. Humpula, N. Uppugundla, C. Gunawan, S. Pattathil, A. M. Cheh, N. Kothari, R. Kumar, J. Ralph, M. G. Hahn, C. E. Wyman, S. Singh, B. A. Simmons, B. E. Dale and V. Balan, Energy Environ. Sci., 2016, 9, 1215–1223 RSC.
- E. Sendich, M. Laser, S. Kim, H. Alizadeh, L. Laureano-Perez, B. Dale and L. Lynd, Bioresour. Technol., 2008, 99, 8429–8435 CrossRef CAS PubMed.
- L. Tao, A. Aden, R. T. Elander, V. R. Pallapolu, Y. Y. Lee, R. J. Garlock, V. Balan, B. E. Dale, Y. Kim, N. S. Mosier, M. R. Ladisch, M. Falls, M. T. Holtzapple, R. Sierra, J. Shi, M. A. Ebrik, T. Redmond, B. Yang, C. E. Wyman, B. Hames, S. Thomas and R. E. Warner, Bioresour. Technol., 2011, 102, 11105–11114 CrossRef CAS PubMed.
- L. da C. Sousa, J. Humpula, V. Balan, B. E. Dale and S. P. S. Chundawat, ACS Sustainable Chem. Eng., 2019, 7, 14411–14424 CrossRef CAS.
- H. W. Foote and M. A. Hunter, J. Am. Chem. Soc., 1920, 42, 69–78 CrossRef CAS.
- S. P. S. Chundawat, B. Bals, T. Campbell, L. Sousa, D. Gao, M. Jin, P. Eranki, R. Garlock, F. Teymouri, V. Balan and B. E. Dale, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, John Wiley & Sons, Ltd, 2013, pp. 169–200 Search PubMed.
- P. C. Scherer, J. Am. Chem. Soc., 1931, 53, 4009–4013 CrossRef CAS.
- S. M. Hudson and J. A. Cuculo, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 3469–3481 CrossRef CAS.
- J. A. Cuculo, C. B. Smith, U. Sangwatanaroj, E. O. Stejskal and S. S. Sankar, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 241–247 CrossRef CAS.
- J. A. Cuculo, C. B. Smith, U. Sangwatanaroj, E. O. Stejskal and S. S. Sankar, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 229–239 CrossRef CAS.
- G. Bellesia, S. P. S. Chundawat, P. Langan, B. E. Dale and S. Gnanakaran, J. Phys. Chem. B, 2011, 115, 9782–9788 CrossRef CAS PubMed.
- R. Parthasarathi, G. Bellesia, S. P. S. Chundawat, B. E. Dale, P. Langan and S. Gnanakaran, J. Phys. Chem. A, 2011, 115, 14191–14202 CrossRef CAS.
- G. Bellesia, S. P. S. Chundawat, P. Langan, A. Redondo, B. E. Dale and S. Gnanakaran, J. Phys. Chem. B, 2012, 116, 8031–8037 CrossRef CAS.
- D. Sawada, L. Hanson, M. Wada, Y. Nishiyama and P. Langan, Cellulose, 2014, 21, 1117–1126 CrossRef CAS.
- Y. Li, J. Wang, X. Liu and S. Zhang, Chem. Sci., 2018, 9, 4027–4043 RSC.
- A. W. De Groot, D. E. Guinnup, M. H. Thiel and J. A. Cuculo, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 557–563 CrossRef CAS.
- G. W. Lynn, W. Heller, V. Urban, G. D. Wignall, K. Weiss and D. A. A. Myles, Physica B: Condens. Matter, 2006, 385–386, 880–882 CrossRef CAS.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1996, 29, 134–146 CrossRef CAS.
- G. Beaucage, J. Appl. Crystallogr., 1995, 28, 717–728 CrossRef CAS.
- S. V. Pingali, V. S. Urban, W. T. Heller, J. McGaughey, H. M. O'Neill, M. Foston, D. A. Myles, A. J. Ragauskas and B. R. Evans, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 1189–1193 CrossRef CAS PubMed.
- S. P. S. Chundawat, V. Balan and B. E. Dale, Biotechnol. Bioeng., 2008, 99, 1281–1294 CrossRef CAS PubMed.
- S. P. S. Chundawat, M. S. Lipton, S. O. Purvine, N. Uppugundla, D. Gao, V. Balan and B. E. Dale, J. Proteome Res., 2011, 10, 4365–4372 CrossRef CAS PubMed.
- M. G. Resch, J. O. Baker and S. R. Decker, Tech. Rep. NREL/TP-5100–63351, Lab. Anal. Proced, 2015, pp. 1–9 Search PubMed.
- D. Gao, S. P. S. Chundawat, C. Krishnan, V. Balan and B. E. Dale, Bioresour. Technol., 2010, 101, 2770–2781 CrossRef CAS PubMed.
- N. Sathitsuksanoh, Z. Zhu, S. Wi and Y. H. Percival Zhang, Biotechnol. Bioeng., 2011, 108, 521–529 CrossRef CAS PubMed.
- S. Bhagia, R. Dhir, R. Kumar and C. E. Wyman, Sci. Rep., 2018, 8, 1350 CrossRef PubMed.
- S. V. Pingali, V. S. Urban, W. T. Heller, J. McGaughey, H. O'Neill, M. Foston, D. A. Myles, A. Ragauskas and B. R. Evans, Biomacromolecules, 2010, 11, 2329–2335 CrossRef CAS PubMed.
- A. T. Lemley and J. J. Lagowski, J. Phys. Chem., 1974, 78, 708–713 CrossRef CAS.
- A. W. De Groot, D. E. Guinnup, M. H. Theil and J. A. Cuculo, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 547–556 CrossRef CAS.
- G. Cheng, X. Zhang, B. Simmons and S. Singh, Energy Environ. Sci., 2015, 8, 436–455 RSC.
- V. S. Raghuwanshi, Y. Cohen, G. Garnier, C. J. Garvey, R. A. Russell, T. Darwish and G. Garnier, Macromolecules, 2018, 51, 7649–7655 CrossRef CAS.
- D. M. Rein, R. Khalfin, N. Szekely and Y. Cohen, Carbohydr. Polym., 2014, 112, 125–133 CrossRef CAS.
- M. Yang, W. Zhao, S. Wang, C. Yu, S. Singh, B. Simmons and G. Cheng, Cellulose, 2019, 26, 2243–2253 CrossRef CAS.
- S. M. Hudson, J. A. Cuculo and L. C. Wadsworth, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 651–670 CrossRef CAS.
- A. W. Degroot, F. I. Carroll and J. A. Cuculo, J. Polym. Sci., Part A: Polym. Chem., 1986, 24, 673–680 CrossRef CAS.
- S. P. S. Chundawat, R. Vismeh, L. Sharma, J. Humpula, L. Sousa, C. K. Chambliss, A. D. Jones, V. Balan and B. E. Dale, Bioresour. Technol., 2010, 101, 8429–8438 CrossRef CAS PubMed.
- S. P. S. Chundawat, B. S. Donohoe, L. Sousa, T. Elder, U. P. Agarwal, F. Lu, J. Ralph, M. E. Himmel, V. Balan and B. E. Dale, Energy Environ. Sci., 2011, 4, 973–984 RSC.
- L. da Costa Sousa, M. Foston, V. Bokade, A. Azarpira, F. Lu, A. J. Ragauskas, J. Ralph, B. Dale and V. Balan, Green Chem., 2016, 18, 4205–4215 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- H. Hunt and L. Boncyk, J. Am. Chem. Soc., 1933, 55, 3528–3530 CrossRef CAS.
- EPA-SAB, Reactive Nitrogen in the United States: An Analysis of Inputs, Flows, Consequences, and Management Options, A Report of the EPA Science Advisory Board, Washington, DC, 2011 Search PubMed.
- A. George, A. Brandt, K. Tran, S. M. S. N. S. Zahari, D. Klein-Marcuschamer, N. Sun, N. Sathitsuksanoh, J. Shi, V. Stavila, R. Parthasarathi, S. Singh, B. M. Holmes, T. Welton, B. A. Simmons and J. P. Hallett, Green Chem., 2015, 17, 1728–1734 RSC.
- A. M. Socha, R. Parthasarathi, J. Shi, S. Pattathil, D. Whyte, M. Bergeron, A. George, K. Tran, V. Stavila, S. Venkatachalam, M. G. Hahn, B. A. Simmons and S. Singh, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3587–E3595 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03524a |
| ‡ Current address: Department of Chemical Engineering, Jadavpur University, Jadavpur, Kolkata, West Bengal 700032, India. |
| § Current address: School of Engineering, Zhejiang A&F University, Linan, Zhejiang 311300, People's Republic of China. |
| This journal is © The Royal Society of Chemistry 2020 |