
Base-promoted aerobic oxidation of N-alkyl iminium salts derived from isoquinolines and related heterocycles†
Li-Gang
Bai‡§
,
Yue
Zhou‡
,
Xin
Zhuang
,
Liang
Zhang
,
Jian
Xue
,
Xiao-Long
Lin
,
Tian
Cai
 * and
Qun-Li
Luo
*
* and
Qun-Li
Luo
*
Key Laboratory of Applied Chemistry of Chongqing Municipality, College of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, China. E-mail: qlluo@swu.edu.cn; caitian@swu.edu.cn
First published on 28th November 2019
Abstract
Potassium tert-butoxide-promoted aerobic oxidation of N-alkyl iminium salts is reported. The reaction is atom-economical and environmentally friendly. Iminium salts derived from isoquinoline, quinoline, phenanthridine, phenanthroline, and phthalazine were successfully transformed into their corresponding unsaturated lactams with up to 95% yield under mild conditions in the absence of photocatalysts and metallic or organic catalysts. Owing to the general substrate scope, low cost, feasibility of scale up, wide availability of reagents, and green reaction conditions, this method shows great potential for preparing isoquinolones and related compounds. The method was applied for atom- and step-economical total synthesis of natural products such as norketoyobyrine.
Introduction
Oxidation is a fundamental transformation in organic chemistry and is commonly used to convert a variety of organic compounds. Oxygen (O2), also known as dioxygen or molecular oxygen, is an abundant, inexpensive, clean, and atom-economical oxidant.1 It constitutes approximately one fifth of the Earth's atmosphere, and reacts exothermically with most elements. However, oxygen has a triplet ground state, which poses a high barrier to reactions with molecules that are usually in the singlet state.2 Two activating strategies for oxidation with oxygen are commonly used. One is the activation of oxygen by converting oxygen molecules from triplet (3O2) to singlet oxygen (1O2) or other reactive oxygen species (ROS).3 The other is the activation of the substrate or catalyst by forming radicals through a single electron transfer process between the substrate and catalyst.4 These strategies often require expensive photocatalysts and/or metal complexes as promoters.5 Therefore, the development of environmentally friendly, mild, and low-cost oxidation approaches based on oxygen as the terminal oxidant would be desirable in the chemical community and is a key focus of green chemistry.6Bases are also typically electron donors.7 Oxygen molecules tend to be more reactive under an electron-rich alkaline environment than under neutral conditions. Numerous studies have reported that some organic molecules are prone to oxidation by oxygen (air) in a simple manner under alkaline conditions, whereas they can coexist in air for a long time under neutral conditions.8 We envisaged that certain photocatalytic or metal-mediated oxidations may be promoted by the use of certain bases.
Isoquinolones and quinolones are present in various natural products and bioactive molecules.9 Hence, synthetic methodologies for these structural scaffolds have drawn considerable interest.10 Oxidation of N-alkyl iminium salts derived from isoquinolines and related heterocycles is a particularly straightforward strategy for the synthesis of isoquinolones and their analogues. Traditional approaches to this oxidation rely on the use of an excess of potassium ferricyanide as the oxidant, which unavoidably generates a large amount of harmful metal waste.10e,f In contrast, oxidation with oxygen as a green oxidant is an environmentally friendly method (Scheme 1).10e–h Using limited examples, Gngnon-Dubois et al. showed an access to isoquinolones and quinolones via an ultrasonic irradiation-accelerated nucleophilic addition of quinolinium and isoquinolinium salts with potassium tert-butoxide (t-BuOK) followed by silica gel-catalyzed aerobic oxidation.10h In 2017, Fu et al. realized photocatalytic oxidation of N-alkyl iminium salts with air.10e In 2018, Huang and Fu achieved the carbene-catalyzed aerobic oxidation of N-alkyl isoquinolinium salts.10f Herein, we describe t-BuOK-promoted aerobic oxidation of N-alkyl iminium salts. Compared to the previous methods, the main advantages of our process include lower reagent costs, less organic waste generation and energy consumption, and no requirement of a special reaction setup. Through the use of this method, the total synthesis of the natural alkaloid norketoyobyrine was accomplished using a concise route starting from isoquinoline and other inexpensive commercial chemicals.
 | ||
| Scheme 1 Selected literature precedents on aerobic oxidation of azine-derived N-alkyl iminium salts (a), idea (b) and advantages (c) of this work. | ||
Results and discussion
We began our investigation by selecting 2-benzylisoquinolin-2-ium bromide 1a as a model substrate (Table 1). In a dimethyl sulfoxide (DMSO) solution of potassium tert-butoxide, electron transfer between donors and electron-deficient π systems (electron acceptors) often occurs readily.11N-Alkyl isoquinolinium salts are electron-deficient π systems. We envisaged that electron transfer from a base or other electron donor species to the salts should also readily occur in DMSO. Thus, we conducted the oxidation of 1a in air at room temperature (RT) with the use of DMSO as the solvent and t-BuOK as a base. Rewardingly, the desired isoquinolinone 2a was obtained in good yield (entry 1). Solvent screening experiments showed that tert-butanol also realized the transformation with moderate yield. Other tested solvents gave disappointing results (entries 3–8). Base screening experiments (entries 9–14) indicated that t-BuOK was superior to KOH (entry 11 vs. 1) and Cs2CO3 (entry 14 vs. 13). A decrease in the amount of t-BuOK to 2 equivalents gave the best result (entry 12).12,13|
|
|||
|---|---|---|---|
| Entry | Base (equiv.) | Solvent | Yield (%) |
| a Reaction conditions: 1a (0.3 mmol), base, solvent (3 mL) at room temperature (RT) for 16 h (entries 1–10), or solvent (1.5 mL) at RT for 24 h (entries 11–15). b Hydrogen peroxide–urea (UHP, 1.5 equiv.) was employed. | |||
| 1 | t-BuOK (3) | DMSO | 82 |
| 2 | t-BuOK (3) | t-BuOH | 52 |
| 3 | t-BuOK (3) | DMF | Trace |
| 4 | t-BuOK (3) | Dioxane | Trace |
| 5 | t-BuOK (3) | DCM | Trace |
| 6 | t-BuOK (3) | Toluene | Trace |
| 7 | t-BuOK (3) | MeCN | Messy |
| 8 | t-BuOK (3) | THF | Messy |
| 9 | t-BuOK (1.5) | MeCN | 31 |
| 10 | t-BuOK (1) | THF | 17 |
| 11 | KOH (3) | DMSO | 76 |
| 12 | t-BuOK (2) | DMSO | 95 |
| 13 | t-BuOK (1.5) | DMSO | 80 |
| 14 | Cs2CO3 (1.5) | DMSO | 33 |
| 15 | Cs 2 CO 3 (1.5) | DMSO | 86 |

Analysis of the composition of the final reaction mixture by NMR spectroscopy showed that 15%–30% of dimethyl sulfone was formed, but no dimethyl sulfide was detected.13 We inferred that ROS were produced in situ in the aerobic oxidation process, because ROS are capable of converting sulfoxides to sulfones.14 Cs2CO3 is a weaker base than t-BuOK but is not as efficient as the latter for oxidation (entry 14). Hydrogen peroxide was explored as an additional oxidant. A combination of hydrogen peroxide–urea (UHP) and Cs2CO3 also realized the transformation with high yield (entry 15), and provides a green alternative for use with strong base-sensitive substrates.
With the optimized conditions in hand (Table 1, entry 12), we selected a series of isoquinolinium salts 1 to evaluate the reaction scope (Table 2). The reactivity of isoquinolinium salts containing various N-alkyl groups (1a–1r) was first investigated. Substrates bearing an N-benzyl or a linear N-alkyl group gave the corresponding isoquinolones in very good to excellent yields (2a–2e). Substrates bearing a cyclopropylmethyl, 2-methoxyethyl or a branched alkyl group also led to the oxidation products in good yields (2f–2h). In contrast, N-diphenylmethyl, N-allyl, and N-cinnamyl isoquinolinium salts failed to produce the intended isoquinolones 2i–2k under the standard conditions, presumably because of the relatively strong acidity of these N-alkyl groups. Through the use of UHP and a weak base, 2i–2k were obtained successfully. Substituents on the N-benzyl group of isoquinolinium salts had little effect on the oxidation, except for the nitro groups (2qvs.21–2p). The nitro substituents are strongly electron withdrawing and capable of increasing the acidity of the benzyl hydrogen atom, which might obstruct the oxidation of 1q in a strongly alkaline solution.1,8c,11c Through the use of UHP and Cs2CO3, 1q was converted into 2q with acceptable yield. Bis(isoquinolinium) salt 1r was oxidized to 2r in 73% yield under standard conditions. Substituted isoquinolinium salts 1s–1w were then evaluated. The reactivity of the 6-position substituted substrates was similar to that of their unsubstituted counterparts (2s–2tvs.2a–2e). However, 3-bromo and 5-bromo isoquinolinium salts generated a certain amount of the debrominative oxygenation product 2a, accompanied by the desired isoquinolones in moderate yields (2u–2v). Furthermore, 5-nitro-isoquinolone 2w was obtained in poor yield, which further suggests that the nitro group prevented oxidation under standard conditions.

We next extended our method to other N-alkyl iminium salts. All reactions proceeded smoothly and led to the formation of the desired unsaturated lactams 4 in mostly good yields under optimal conditions (Table 3). Debrominative oxygenation of 5-bromo quinolinium 3g was also observed (13% of 4b plus 51% of 4g). UHP-mediated oxidation overcame the debromination and produced 4g in good yield. Phenanthridinium salts showed lower reactivity than the other tested iminium salts and required a prolonged reaction time of 36 h for full conversion (4l–4n).

Analysis of the unexpected outcomes of the oxidation helped us probe the reaction mechanism. Under standard conditions, the oxidation of N-diphenylmethyl substrate 1i generated 76% of benzophenone and 64% of isoquinolin-1(2H)-one 2i′, whereas the oxidation of N-(1-phenyl)ethyl substrate 1h gave 76% of the intended oxidation product 2h (eqn (1) and (2)).
 | (1) |
 | (2) |
t-BuOK-mediated oxidation of 1j mainly yielded the double bond-shifted product 2j′,15 whereas the UHP-mediated method gave the “normal” oxidation product 2j (eqn (3) and (4)). Under standard conditions, 2j quantitatively converted to (E)-2j′, whereas (E)-2j′ and (Z)-2j′ did not isomerize into each other (eqn (5) and (6)). Meanwhile, 1k was oxidized into cyclic 2k′, rather than the “normal” oxidation product 2k (eqn (7)).
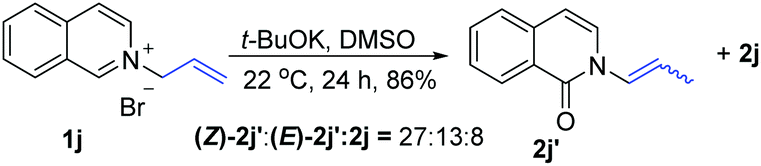 | (3) |
 | (4) |
 | (5) |
 | (6) |
 | (7) |
Mechanistic studies (Scheme 2a) indicated that the yield was hardly affected when the reaction was performed in the dark but sharply decreased under an inert atmosphere. The reaction was greatly limited by the radical inhibitor butylated hydroxytoluene (BHT, 2 equivalents),16 but accelerated by the free radical 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO, 2 equivalents).
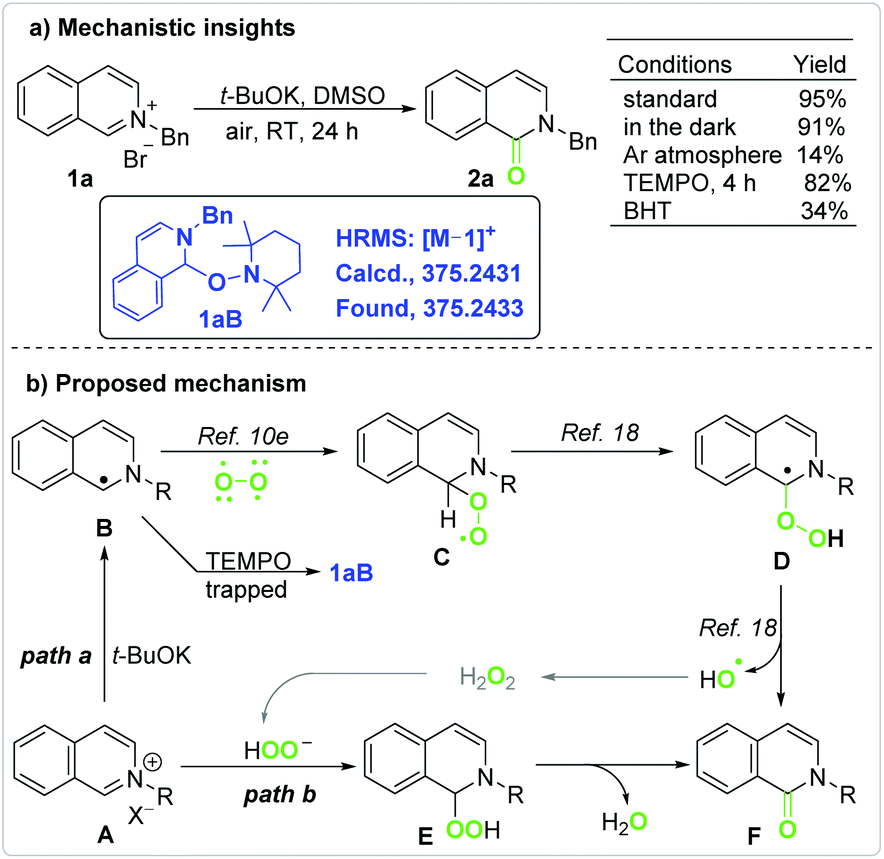 | ||
| Scheme 2 Mechanistic insights (a) and the proposed mechanism (b). | ||
The reaction mechanism was further studied by high resolution mass spectroscopy (HRMS) with an ESI source (Fig. S6–S11†).13 Under standard conditions, mass peaks at m/z = 220.11 (M − 79 peak of 1a) and 274.06 (M + 39 peak of 2a) were detected; however, that at m/z = 375.24 was not detected in the reaction mixture. On addition of TEMPO to the reaction mixture, a new mass peak at m/z = 375.24 (M − 1 peak of 1aB, Scheme 2a) was detected, which resulted from a radical intermediate being trapped by TEMPO. In the absence of t-BuOK, the addition of TEMPO did not lead to the formation of either 2a or 1aB. These observations indicate that the oxidation of 1a was actually initiated by t-BuOK.
According to previous work and the above results, a possible mechanism for the t-BuOK-promoted aerobic oxidation is proposed in Scheme 2b. The reaction was initiated along path a. A single electron transfer (SET) from the base to iminium cation A provides radical B.11,17 The cross coupling of carbon radical B with diradical molecular oxygen leads to alkylperoxyl radical C.10e Alkylperoxyl radical C is converted to α-hydroperoxy-α-carbon radical Dvia hydrogen atom transfer (HAT).18 Radical D gives the final product F and a hydroxyl radical.18 A hydroxyl radical evolves into hydrogen peroxide or other ROS that further oxidize A to yield Fvia pathways such as path b.3,19
The utility of our protocol is exemplified in Scheme 3. Gram-scale preparation by this oxidation was feasible. The reaction efficiency was slightly affected when 1a was used on a 6 mmol scale for both t-BuOK-promoted aerobic oxidation and UHP-mediated oxidation. The bromination of oxidation product 2a with N-bromosuccinimide (NBS) under mild conditions led to 4-bromo isoquinolinone 5 in excellent yield (Scheme 3a).
 | ||
| Scheme 3 Gram-scale synthesis and bromination of 2a (a), total syntheses of norketoyobyrine and its related compounds (b), and formal synthesis of topoisomerase I inhibitor 10 (c). Conditions: (i) t-BuOK (2 equiv.); (ii) UHP (1.5 equiv.), Cs2CO3 (1.5 equiv.); (iii) BF3·Et2O, MeCN, 85 °C; and (iv) CeCl3·7H2O (0.1 equiv.), air, i-PrOH, 50 °C. | ||
Our oxidative method was successfully applied to the atom- and step-economical total synthesis of natural products (Scheme 3b). Consequently, norketoyobyrine (9) was synthesized in 61% total yield via the three-step sequence of oxidation, cyclization and dehydrogenative oxidation from isoquinolinium salt 6. Reduction of intermediate 8 (dihydronorketoyobyrine) delivered demethoxycarbonyldihydrogambirtannine (8a) via a known process.20 Gao et al. reported an elegant strategy for constructing natural products such as 9.9a Our synthetic precursors for norketoyobyrine were more easily prepared compared to those using Gao et al.'s method. Our method used no protective groups and no unwanted carbon atoms were removed. Special and/or expensive reagents were avoided in each step.
Our oxidative method was also applicable to the formal synthesis of topoisomerase I inhibitor 10 (Scheme 3c).9c Compared with other green methods for the synthesis of 10 reported in the most recent literature,10f our protocol required neither the NHC catalyst nor cooling of the reaction system to far below ambient temperature. These features are beneficial in terms of reducing organic waste generation and energy consumption.
Conclusions
In conclusion, we report here a mild and environmentally benign aerobic oxidative method for the simple synthesis of a wide variety of isoquinolinones and related heterocyclic derivatives. In the absence of commonly used promoters, such as metallic reagents, photocatalysts, and other additives, a series of N-heterocycle-derived N-alkyl iminium salts were successfully oxidized into their corresponding unsaturated lactams with the sole use of a base, a solvent, and ambient atmosphere at room temperature. The reaction was scaled up to produce grams of N-alkyl isoquinolin-1(2H)-one under mild conditions. Mechanistic studies indicated that the oxidation was initiated by t-BuOK, and the experimental observations were preliminarily in agreement with a radical mechanism. Through the use of the method, a highly efficient total synthesis of the natural alkaloid norketoyobyrine was accomplished in a concise manner starting from isoquinoline and other inexpensive commercial chemicals. The method combines several advantages, such as broad generality, low cost, wide availability of reagents, limited production of harmful waste, and ease of use. Overall, the present method ranks among the most economical and greenest methods reported for the oxidation of N-alkyl iminium salts.Experimental
General procedures for the oxidation of N-alkyl iminium salts 1 & 3
The isolated products were confirmed by 1H NMR and 13C NMR. The characterization data are described in the ESI.† The exact time for each reaction is shown in Tables 2 and 3.Gram-scale preparation of 2a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to provide N-benzyl isoquinolin-1(2H)-one 2a as a yellow solid (1.32 g, 93% yield).
:1) to provide N-benzylisoquinolin-1(2H)-one 2a as a yellow solid (1.22 g, 86%).
:1) to provide N-benzyl isoquinolin-1(2H)-one 2a as a yellow solid (1.32 g, 93% yield).
:1) to provide N-benzylisoquinolin-1(2H)-one 2a as a yellow solid (1.22 g, 86%).
Bromination of 2a with N-bromosuccinimide
A 10 mL reaction flask was charged with a solution of N-benzylisoquinolin-1(2H)-one 2a (71 mg, 0.3 mmol) and NBS (133 mg, 0.75 mmol) in DMSO (1.5 mL). The reaction mixture was continuously stirred at 40 °C until 2a was consumed as indicated by TLC (ca. 18 h). Then it was diluted with ethyl acetate (5 mL) and water (5 mL) and extracted with EtOAc (3 × 5 mL). The combined organic phase was washed with water and saturated brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 5:1) to provide N-benzyl-4-bromoisoquinolin-1(2H)-one 5 as a white solid (87 mg, 93%).10c1H NMR (600 MHz, CDCl3) δ 8.40 (d, J = 8.0 Hz, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.69–7.60 (m, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.29–7.26 (m, 3H), 7.25–7.20 (m, 3H), 5.12 (s, 2H). 13C NMR (151 MHz, CDCl3) δ 161.4, 136.3, 135.47, 133.0, 131.8, 129.0, 128.5, 128.15, 128.10, 127.9, 126.6, 125.9, 100.2, 51.8.
Total synthesis of norketoyobyrine (9)
:1) to provide 2-(2-(1H-indol-3-yl)ethyl) isoquinolin-1(2H)-one 7 as a yellow solid (519 mg, 90% yield), m. p. 120–130 °C. 1H NMR (600 MHz, CDCl3) δ 8.49 (d, J = 8.0 Hz, 1H), 8.04 (s, 1H), 7.70 (d, J = 7.9 Hz, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.49 (d, J = 7.4 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.43–7.33 (m, 1H), 7.21 (t, J = 7.4 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 6.94 (s, 1H), 6.78 (d, J = 7.3 Hz, 1H), 6.32 (d, J = 7.3 Hz, 1H), 4.30 (t, J = 7.3 Hz, 2H), 3.42–3.14 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 162.1, 137.2, 136.4, 132.1, 132.0, 127.8, 127.3, 126.6, 126.3, 125.8, 122.5, 122.2, 119.6, 118.7, 112.4, 111.2, 105.4, 50.3, 25.0. HRMS (ESI-TOF) calcd for C19H16N2NaO [M + Na]+: 311.1155; found 311.1153.
:10) to provide 24 mg of a yellow solid. The 1H NMR spectrum indicated that it consisted of dihydronorketoyobyrine 8 and norketoyobyrine 9 in a ratio of 4 to 1. Thus, the total yield of the two compounds was 85%. Dihydronorketoyobyrine 8 (major).20a1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 8.00 (d, J = 7.6 Hz, 1H), 7.55 (d, J = 6.9 Hz, 1H), 7.47 (t, J = 6.3 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.40–7.33 (m, 2H), 7.13–7.06 (m, 1H), 7.02 (t, J = 7.4 Hz, 1H), 5.15–4.94 (m, 2H), 3.60 (dd, J = 15.8, 3.8 Hz, 1H), 2.96 (dd, J = 12.4, 3.1 Hz, 2H), 2.89 (d, J = 11.7 Hz, 1H), 2.82–2.69 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 164.3, 137.4, 136.9, 134.19, 132.49, 129.3, 128.4, 127.7, 127.6, 126.7, 121.7, 119.2, 118.4, 111.6, 107.8, 52.2, 39.6, 34.8, 21.1. Norketoyobyrine 9 (minor).9a1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 8.25 (d, J = 8.0 Hz, 1H), 7.71 (t, J = 7.5 Hz, 1H), 7.62 (d, J = 5.6 Hz, 1H), 7.58 (d, J = 5.3 Hz, 1H), 7.41 (br.s, 2H), 7.22 (t, J = 7.6 Hz, 1H), 7.12–7.06 (m, 2H), 4.41 (t, J = 6.5 Hz, 2H), 3.10 (t, J = 6.6 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 161.2, 138.0, 136.1, 132.6, 132.3, 128.1, 127.5, 126.07, 125.9, 125.5, 124.5, 123.5, 119.4, 119.1, 112.5, 111.6, 99.0, 40.4, 19.3.
:1, 0.1 mmol) in i-PrOH (4 mL). The flask was placed in an oil-bath at 50 °C and stirred until the solid was completely dissolved. Then CeCl3·7H2O (0.01 mmol, 4 mg) was added. The reaction mixture was stirred until 8 was consumed as indicated by TLC (ca. 2.5 h). It was cooled to RT, neutralized to pH 8–9 with saturated NaHCO3, and completely extracted with EtOAc (8 × 3 mL). The combined organic layers were washed with saturated brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford the crude product, which was purified by silica gel chromatography (PE/CH2Cl2 = 1:5) to give norketoyobyrine 9 as a yellow solid (23 mg, 80%).9a1H NMR (400 MHz, DMSO-d6) δ 11.70 (s, 1H), 8.24 (d, J = 8.5 Hz, 1H), 7.75–7.67 (m, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.59 (d, J = 7.9 Hz, 1H), 7.47 (d, J = 8.1 Hz, 0H), 7.43 (d, J = 8.4 Hz, 1H), 7.22 (t, J = 7.6 Hz, 1H), 7.11–7.03 (m, 2H), 4.40 (t, J = 6.6 Hz, 2H), 3.09 (t, J = 6.6 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 161.7, 138.5, 136.6, 133.1, 132.8, 128.6, 128.0, 126.6, 126.4, 126.0, 125.0, 124.0, 120.0, 119.6, 113.0, 112.1, 99.5, 40.9, 19.8.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from the NSFC of China (20971105), the Natural Science Foundation of Chongqing (cstc2017jcyjAX0423), and the Fundamental Research Funds for the Central Universities (XDJK2019AA003).Notes and references
- K. Fang, G. Li and Y. She, J. Org. Chem., 2018, 83, 8092 CrossRef CAS.
- W. T. Borden, R. Hoffmann, T. Stuyver and B. Chen, J. Am. Chem. Soc., 2017, 139, 9010 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302 CrossRef CAS.
- Y. Li, S. A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499 CrossRef CAS.
- W. Fan and P. Li, Angew. Chem., Int. Ed., 2014, 53, 12201 CrossRef CAS.
- M. Liu, H. Wang, H. Zeng and C.-J. Li, Sci. Adv., 2015, 1, e1500020 CrossRef.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS.
- For selected examples, see: (a) W. E. Doering, G. Cortes and L. H. Knox, J. Am. Chem. Soc., 1947, 69, 1700 CrossRef CAS; (b) W. E. Doering and R. W. Haines, J. Am. Chem. Soc., 1954, 76, 482 CrossRef CAS; (c) G. E. Lewis, J. Org. Chem., 1965, 30, 2433 CrossRef CAS; (d) T. Siu, D. Qin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4713 CrossRef CAS; (e) Y.-F. Liang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 548 CrossRef CAS.
- (a) K. Li, J. Ou and S. Gao, Angew. Chem., Int. Ed., 2016, 55, 14778 CrossRef CAS; (b) K. H. Raitio, J. R. Savinainen, J. Vepsäläinen, J. T. Laitinen, A. Poso, T. Jävinen and T. Nevalainen, J. Med. Chem., 2006, 49, 2022 CrossRef CAS; (c) D. B. Khadka and W.-J. Cho, Bioorg. Med. Chem., 2011, 19, 724 CrossRef CAS PubMed.
- For selected examples, see: (a) C. S. Yeung, T. H. H. Hsieh and V. M. Dong, Chem. Sci., 2011, 2, 544 RSC; (b) D. Wang, R. Zhang, R. Deng, S. Lin, S. Guo and Z. Yan, J. Org. Chem., 2016, 81, 11162 CrossRef CAS PubMed; (c) W.-K. Luo, X. Shi, W. Zhou and L. Yang, Org. Lett., 2016, 18, 2036 CrossRef CAS PubMed; (d) T.-H. Wang, W.-C. Lee and T.-G. Ong, Adv. Synth. Catal., 2016, 358, 2751 CrossRef CAS; (e) Y. Jin, L. Ou, H. Yang and H. Fu, J. Am. Chem. Soc., 2017, 139, 14237 CrossRef CAS; (f) G. Wang, W. Hu, Z. Hu, Y. Zhang, W. Yao, L. Li, Z. Fu and W. Huang, Green Chem., 2018, 20, 3302 RSC; (g) A. Motaleb, A. Bera and P. Maity, Org. Biomol. Chem., 2018, 16, 5081 RSC; (h) M. Gngnon-Dubois and A. Meola, Synth. Commun., 1995, 25, 2999 CrossRef.
- (a) G. A. Russell, E. G. Jansen, H.-D. Becker and F. Smentowski, J. Am. Chem. Soc., 1962, 84, 2652 CrossRef CAS; (b) G. A. Russell, A. J. Moye and K. Nagpal, J. Am. Chem. Soc., 1962, 84, 4154 CrossRef CAS; (c) G. A. Russell and E. G. Janzen, J. Am. Chem. Soc., 1962, 84, 4153 CrossRef CAS; (d) G. A. Russell, E. G. Janzen and E. T. Strom, J. Am. Chem. Soc., 1962, 84, 4155 CrossRef CAS.
- The NMR spectra of the reaction mixture showed that the substrate (1a) was completely converted to the intended compound (2a). Considering the environmental friendliness and excellent yield obtained by the use of t-BuOK, we did not further try organic bases.
- For details, see the ESI.†.
- For an example of oxidation of sulfoxides to sulfones with peroxides, see: S.-K. Wang, X. You, D.-Y. Zhao, N.-J. Mou and Q.-L. Luo, Chem. – Eur. J., 2017, 23, 11757 CrossRef CAS.
- The isolated major isomer of 2j′ was the Z-configuration that is less stable than the E-configuration. The ratio of Z/E isomers is approximately 2:1 at 22 °C. When the reaction temperature was increased to 33 °C, the ratio of Z/E isomers decreased to 1:1 (Fig. S5†). For an explanation of the unexpected stereoselectivity, see the ESI.†.
- For an example of a free radical mechanism being affected by TEMPO and BHT, see ref. 14.
- (a) SET between ground-state t-BuOK and neutral molecule benzophenone occurred at 70 °C. See: G. Nocera, A. Young, F. Palumbo, K. J. Emery, G. Coulthard, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2018, 140, 9751 CrossRef CAS. Accordingly, SET between ground-state t-BuOK and cation acceptor A can occur at RT since cations are more susceptible to electrons than neutral molecules. (b) For an example of t-BuOK-initiated free radical reactions at RT, see: W.-T. Li, W.-H. Nan and Q.-L. Luo, RSC Adv., 2014, 4, 34774 RSC; (c) t-BuOK may induce certain species to yield other electron donor(s) in DMSO. It cannot be ruled out that other in situ formed electron donors initiate the oxidation. For examples of t-BuOK-promoted free radical reactions, see ref. 11.
- I. Hermans, J. Peeters and P. A. Jacobs, J. Org. Chem., 2007, 72, 3057 CrossRef CAS.
- The oxidation with UHP-Cs2CO3 supports path b. Only one pathway for reactive oxygen species-mediated oxidation is shown; others may be operative.
- (a) G. D. Pandey and K. P. Tiwari, Synth. Commun., 1980, 10, 523 CrossRef CAS; (b) I. Ninomiya, T. Naito and H. Takasugi, J. Chem. Soc., Perkin Trans. 1, 1976, 1865 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data of the products; 1H NMR, 13C NMR and MS spectra for new compounds. See DOI: 10.1039/c9gc03629f |
| ‡ These authors contributed equally. |
| § New address: College of Chemistry and Molecular Sciences, Wuhan University, 430072 Wuhan, China. |
| This journal is © The Royal Society of Chemistry 2020 |