
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rigid spirocyclic diol from fructose-based 5-hydroxymethylfurfural: synthesis, life-cycle assessment, and polymerization for renewable polyesters and poly(urethane-urea)s†
Niklas
Warlin
 a,
Maria Nelly
Garcia Gonzalez
b,
Smita
Mankar
a,
Nitin G.
Valsange
a,
Mahmoud
Sayed
c,
Sang-Hyun
Pyo
c,
Nicola
Rehnberg
ad,
Stefan
Lundmark
e,
Rajni
Hatti-Kaul
*c,
Patric
Jannasch
*a and
Baozhong
Zhang
*a
a,
Maria Nelly
Garcia Gonzalez
b,
Smita
Mankar
a,
Nitin G.
Valsange
a,
Mahmoud
Sayed
c,
Sang-Hyun
Pyo
c,
Nicola
Rehnberg
ad,
Stefan
Lundmark
e,
Rajni
Hatti-Kaul
*c,
Patric
Jannasch
*a and
Baozhong
Zhang
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: patric.jannasch@chem.lu.se; baozhong.zhang@chem.lu.se
bEnvironmental and Energy Systems Studies, Lund University, Lund, Sweden
cBiotechnology, Department of Chemistry, Lund University, P.O. Box 124, SE-22100, Lund, Sweden. E-mail: rajni.hatti-kaul@biotek.lu.se
dBona Sweden AB, Box 210 74, 200 21 Malmö, Sweden
ePerstorp AB, Innovation, Perstorp Industrial Park, 284 80 Perstorp, Sweden
First published on 13th November 2019
Abstract
There is currently an intensive development of sugar-based building blocks toward the production of renewable high-performance plastics. In this context, we report on the synthesis of a rigid diol with a spirocyclic structure via a one-step acid-catalyzed acetalation of fructose-sourced 5-hydroxymethylfurfural and pentaerythritol. Preliminary life cycle assessment (LCA) indicated that the spiro-diol produced 46% less CO2 emission than bio-based 1,3-propanediol. Polymerizations of the spiro-diol together with another sugar-based flexible 1,6-hexanediol for the production of polyesters and poly(urethane-urea)s were investigated, and reasonably high molecular weights were achieved when up to 20 and 60 mol% spiro-diol was used for polyesters and poly(urethane-urea)s, respectively. The glass transition temperatures (Tgs) of the polyesters and poly(urethane-urea)s significantly increased upon the incorporation of the rigid spirocyclic structure. On the other hand, it was observed that the spiro-diol was heat-sensitive, which could cause coloration and partial crosslinking when >10% (with respect to dicarboxylate) was used for the polyester synthesis at high temperatures. The results indicated that the polymerization conditions have to be carefully controlled under these conditions. However, when the spiro-diol was used for the synthesis of polyurethanes at lower temperature, the side reactions were insignificant. This suggests that the new spiro-diol can be potentially suitable toward the production of sustainable rigid polyurethane materials like coatings or foams, as well as renewable polyesters after further optimization of the polymerization conditions.
Introduction
Sugar-based chemicals play an important role in the recent advance of renewable green materials, because of the abundance of natural polysaccharide resources like cellulose, hemicellulose, or starch.1 Chemicals produced from various sugar resources have been intensively investigated toward various applications such as chemicals, plastics, additives, surfactants and cosmetics.2–5 Particularly, sugar-based monomers and polymers have received enormous attention, due to their great potential to resolve the sustainability and environmental challenges caused by conventional fossil-based plastics.6,7 For example, furan dicarboxylic acid (FDCA) is the most actively explored sugar-based monomer today,8,9 which can be used to replace fossil-based terephthalic acid in PET production, yielding a 100% bio-based poly(ethylene 2,5-furandicarboxylate) or PEF. To a large extent, PEF can mimic PET regarding the material properties, so it is expected to become the material for the next generation bio-based beverage bottles.10–16Recently, the development of sugar-based (or in a broad sense any bio-based) monomers and polymers has been greatly focused on a so-called “bioadvantage” strategy,17 which means to develop high value-added monomers and high-performance plastics without the competition from the low cost fossil-based counter parts. In this direction, rigid monomers have attracted increasing attention because they can usually improve the thermal and mechanical performance of the resulting plastics. In fossil-based plastic industry, the use of rigid monomers to yield high performance polymers has been under rapid development recently. For example, fossil-based (or potentially bio-based) 1,4-cyclohexanedimethanol (CHDM)18–20 has been used as a comonomer to produce a PET-like polyester called PETG, which has higher Tg and alkaline resistance compared with PET.21 A fossil-based cyclic rigid diol, cis/trans-2,2,4,4-tetramethyl-1,3-cyclobutanediol (CBDO), has also been used to prepare polyesters with high impact resistance and superior optical clarity.22 Furthermore, the combination of both CHDM and CBDO in a polyester structure results in a high performance polyester Tritan™, commercialized by Eastman Chemical Co. Recently, a partially bio-based rigid diol with a spirocylic acetal structure has been introduced by Perstorp AB to produce a high performance polyester, Akestra™, which has Tg ≈90 to 110 °C (depending on the content of the rigid diol), and can thus be potentially used in hot-filling applications.23,24 Inspired by the industrial advances, many bio-based rigid monomers toward high-performance polymers (e.g. polyesters, polyurethanes and polycarbonates) have been reported based on a variety of biomass resources.25–31,107
Sugar-based rigid building blocks have also been intensively investigated. For example, isosorbide (a bicyclic sugar-based diol) was reported to produce diversified polymer structures with increased Tg.32–38 Sugar-based polyols like sorbitol and mannitol have been converted into rigid monomers (e.g. diols, diamines, dicarboxylates) with bicyclic structures and used in polymerizations of polyesters, polyurethanes and polyureas.39–43 Aldaric acids (sugar acids)-derived dicarboxylate monomers were prepared and used for polyester synthesis.44,45 Alditol (sugar-alcohol) was converted into cyclic acetalized tartrate monomers for the production of copolyesters with enhanced performance.46–48 Quinic acid (a sugar-based molecule in coffee beans) was reported for the production of cyclic rigid diol toward high-performance polycarbonates.49 However, none of these examples has yet reached close to industrial production and commercialization, which is probably due to various reasons like raw material scarcity, high production cost, or complicated synthesis and purification. Therefore, there is still a strong driving force to develop more suitable sugar-based rigid building blocks toward affordable high-performance biopolymers, using a simple and eco-friendly synthesis protocol.
Among sugar-based chemicals, 5-hydroxymethylfurfural (HMF) has recently been recognized as an important platform molecule,50,108 which can be conveniently produced by the dehydration of fructose,51 an inexpensive abundant natural resource.52–54 HMF contains an aldehyde and an alcohol in its structure, so it can be conveniently converted into many bis-functional monomers. The most notable example is the oxidation of HMF to FDCA toward PEF production.8,9 Many other HMF-derived monomers have also been reported, such as 2,5-bis(hydroxymethyl)furan, 2,5-bis(aminomethyl)furan, 5-hydroxymethyl-2-vinylfuran and 2,5-bis[(2-oxiranylmethoxy)-methyl]-furan, which can be used for the production of polyesters, polyamides, polyurethanes, vinyl polymers, and epoxy polymers.55–58 However, not many rigid diols have been prepared from HMF and investigated for polymer production. 2,5-Bis(hydroxymethyl)furan could be conveniently prepared by the reduction of HMF, but polyesters derived from this monomer usually suffer from low molecular weight and poor physical properties.59–61 Aldol condensation of HMF with acetone produced an unsaturated diol, but this was only investigated for the production of bio-fuels after hydrogenation.62 A patent issued by Stepan Company reports the reaction of HMF with glycerol to produce a rigid diol with cyclic acetal structures, which could potentially be used for the production of polyesters and polyurethanes.63 However, the preparation and physical properties of these biopolymer materials have not yet been reported. As such, the potential of HMF-based rigid diols toward high-performance bioplastics remains largely unexplored. Another important but largely ignored issue is whether or not newly developed bio-based chemicals (and the resulting biopolymers) are truly environmentally friendly. Most often, it is just taken for granted that bio-based monomers and polymers are environmentally benign without any proper assessment on their environmental impact such as greenhouse gas emissions.
Herein, we present a simple, high-yielding and eco-friendly synthesis of a novel bio-based spirocyclic diol (denoted as Monomer S) using fructose-derived HMF and bio-based pentaerythritol from Perstorp AB. The environmental impact of Monomer S has been evaluated by a cradle-to-grave life cycle assessment (LCA) of its CO2 emissions. The results show that Monomer S has a lower CO2 emission profile compared with bio-based 1,3-propanediol, which is commonly used for industrial polymer synthesis (e.g. to produce Sorona™). Step-growth polymerizations of Monomer S for the preparation of polyesters and poly(urethane-urea)s were investigated. The thermal and physical properties of the obtained copolymers showed a clear dependence on the incorporated content of the spirocylic structures. Particularly, the incorporation of Monomer S significantly increased the Tg of the resulting polymers, which showed the potential of the spiro-diol S as a green monomer for the production of high performance polyurethane or polyester materials. With the current conditions we use, polyesters containing up to 5% spiro-diol can be prepared without significant side reactions. When >10% spiro-diol was used, polyesters that were prepared under higher temperatures showed coloration and partial crosslinking, which indicated that the conditions have to be further optimized. Polyurethanes with up to 60% spirocyclic structure were synthesized at relatively low temperatures, which showed insignificant side reaction or coloration.
Experimental
Materials
p-Toluenesulfonic acid monohydrate (p-TsOH, 98.5%), oxalic acid (99%), dimethyl terephthalate (99%), 1,6-hexanediol (HD, 97%), dibutyltin oxide (DBTO, 98%), sulfuric acid (95–97%), 2-propanol (2-PrOH, 99.8%), dimethyl sulfoxide (DMSO, 99.9%), 2-butanone (>99%), DMSO-d6 (99.9% atom D) and chloroform-d (99.8% atom D) were purchased from Sigma-Aldrich. Methanol, chloroform (99.1%, stabilized with 0.6% ethanol), HPLC-grade chloroform (stabilized with 0.002% amylene), 1-propanol, tert-butanol (t-BuOH), sodium hydrogen carbonate (NaHCO3, 99.7%), sodium hydroxide (NaOH, 99.1%), acetone, dichloromethane (DCM), ethyl acetate, dimethyl formamide (DMF), n-heptane, and acetonitrile were purchased from VWR Chemicals. HMF (98%) was purchased from Nanjing Confidence Chemical Co., Ltd. Bio-based pentaerythritol (Voxtar™, 99%) was obtained by a courtesy of Perstorp AB. Ethanol (99.7%) was purchased from Solveco. HCl solution (37%) was purchased from Scharlau. Citric acid monohydrate (99.5%) was purchased from Duchefa biochemie. Formic acid (95%) and tetrahydrofuran (THF) were purchased from Honeywell. Acetic acid (99.8%) was purchased from Acros Organics. Isophorone diisocyanate (IPDI, 99.9%) was purchased from Evonik. Ethylene diamine (100%) was purchased from ECEM. Dibutyltin laurate (Metatin katalysator 712 ES) was purchased from Dow Chemicals. All solvents were of analytical grade or higher, and all reagents and chemicals were used without further purification.Analytical methods
1H and 13C NMR measurements were performed on a Bruker DR X400 spectrometer at 400.13 MHz and 100.61 MHz, respectively. Chemical shifts were reported as δ values (ppm). HRMS was taken on a Micromass QTOF mass spectrometer (ESI). Size exclusion chromatography (SEC) measurements for the polyesters were carried out using Malvern Viscotek TDAmax instrument with a 2 × PL-Gel Mix-B LS column set (2 × 30 cm) equipped with OmniSEC triple detectors (refractive index, viscosity and light scattering) and chloroform as eluent at 35 °C at a flow rate of 1 mL min−1. Calibration was performed with a narrow polystyrene standard Mp = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, Đ = 1.03 (Polymer Laboratories Ltd, Agilent Technologies and Water Associates). The intrinsic viscosity [η], Mark–Houwink–Sakurada parameters (K and a), and the hydrodynamic radii (Rh) of polymers were measured by SEC. Size exclusion chromatography (SEC) measurements for the poly(urethane-urea)s were carried out using Agilent 1100/1200 Infinity HPLC System equipped with three columns (GPC column PSS GRAM 3000 Å, 10 μm; GPC column PSS GRAM 1000 Å, 10 μm; GPC column PSS GRAM 30 Å, 10 μm) connected in sequence at 40 °C in DMAc with LiBr (5 g L−1) at a flow rate of 1 mL min−1. Calibration was carried out with ReadyCal-Kit poly(methyl methacrylate) standards Mp = 202–2200000 Da. Fourier transform infrared (FTIR) spectra were measured with an attenuated total reflection (ATR) setup using a Bruker Alpha FT-IR spectrometer. The polyesters were dissolved in DCM and successively placed on the cell. The poly(urethane-urea)s were taken directly from the reaction solution and placed on the cell. The measurement was performed after evaporation of the solvent. For all samples 64 successive scans over the range of 400–4000 cm−1 was recorded. TGA measurements were carried out on a TA instrument mode TGA Q500. The samples were first heated to 120 °C for 20 minutes to remove any trace of water, and the measurement was performed by heating from 50–550 °C with a heating rate of 10 °C min−1. DSC measurements were performed on a DSC Q2000 analyzer from TA instruments. Data was recorded from −50 to 220 °C, and Tg was determined from the second heating cycle. The synthesized polyesters (PHT, PHST-3, PHST-10, PHST-16 and PHST-19) and commercial sample (Akestra90) were made into films of dimensions (17.5 mm × 6 mm × 1 mm) for dynamic mechanical analysis (DMA). The polymers were placed into a PTFE mold, and then melted under vacuum before hot pressing to remove any air trapped in the solid polymer. PHT and PHST films were made by hot pressing at 150 °C under 1000 kg, and were then cooled to 100 °C under maintained pressure before being removed from the hot press. Akestra90 was hot pressed at 180 °C, cooled to 100 °C with a rate of 5 °C per min and then cooled rapidly to room temperature. DMA measurements were performed in a stretching mode using TA instruments Q800 analyzer. The measured samples were heated from −50 °C to 115 °C at a heating rate of 3 °C min−1 and frequency of 1 Hz with strain of 0.1%. The purified poly(urethane-urea) samples were dissolved in THF (50 mg mL−1) and cast on glass slides (76 × 26 mm) by spreading 0.3 mL of the polymer solution over one end of the slide. The polymer solution was spread evenly using the needle of the syringe, and was then placed under a funnel overnight to have THF evaporated slowly. The glass slides were subsequently placed under vacuum for 24 h to ensure complete removal of the solvent. Water contact angle (θ) was measured in a picture (taken with a Nikon Bellows PB-6 camera) of a single water droplet placed onto the dry polymer film.
000 Da, Đ = 1.03 (Polymer Laboratories Ltd, Agilent Technologies and Water Associates). The intrinsic viscosity [η], Mark–Houwink–Sakurada parameters (K and a), and the hydrodynamic radii (Rh) of polymers were measured by SEC. Size exclusion chromatography (SEC) measurements for the poly(urethane-urea)s were carried out using Agilent 1100/1200 Infinity HPLC System equipped with three columns (GPC column PSS GRAM 3000 Å, 10 μm; GPC column PSS GRAM 1000 Å, 10 μm; GPC column PSS GRAM 30 Å, 10 μm) connected in sequence at 40 °C in DMAc with LiBr (5 g L−1) at a flow rate of 1 mL min−1. Calibration was carried out with ReadyCal-Kit poly(methyl methacrylate) standards Mp = 202–2200000 Da. Fourier transform infrared (FTIR) spectra were measured with an attenuated total reflection (ATR) setup using a Bruker Alpha FT-IR spectrometer. The polyesters were dissolved in DCM and successively placed on the cell. The poly(urethane-urea)s were taken directly from the reaction solution and placed on the cell. The measurement was performed after evaporation of the solvent. For all samples 64 successive scans over the range of 400–4000 cm−1 was recorded. TGA measurements were carried out on a TA instrument mode TGA Q500. The samples were first heated to 120 °C for 20 minutes to remove any trace of water, and the measurement was performed by heating from 50–550 °C with a heating rate of 10 °C min−1. DSC measurements were performed on a DSC Q2000 analyzer from TA instruments. Data was recorded from −50 to 220 °C, and Tg was determined from the second heating cycle. The synthesized polyesters (PHT, PHST-3, PHST-10, PHST-16 and PHST-19) and commercial sample (Akestra90) were made into films of dimensions (17.5 mm × 6 mm × 1 mm) for dynamic mechanical analysis (DMA). The polymers were placed into a PTFE mold, and then melted under vacuum before hot pressing to remove any air trapped in the solid polymer. PHT and PHST films were made by hot pressing at 150 °C under 1000 kg, and were then cooled to 100 °C under maintained pressure before being removed from the hot press. Akestra90 was hot pressed at 180 °C, cooled to 100 °C with a rate of 5 °C per min and then cooled rapidly to room temperature. DMA measurements were performed in a stretching mode using TA instruments Q800 analyzer. The measured samples were heated from −50 °C to 115 °C at a heating rate of 3 °C min−1 and frequency of 1 Hz with strain of 0.1%. The purified poly(urethane-urea) samples were dissolved in THF (50 mg mL−1) and cast on glass slides (76 × 26 mm) by spreading 0.3 mL of the polymer solution over one end of the slide. The polymer solution was spread evenly using the needle of the syringe, and was then placed under a funnel overnight to have THF evaporated slowly. The glass slides were subsequently placed under vacuum for 24 h to ensure complete removal of the solvent. Water contact angle (θ) was measured in a picture (taken with a Nikon Bellows PB-6 camera) of a single water droplet placed onto the dry polymer film.
Stability evaluation of HMF
To a magnetically stirred glass vial containing either pure HMF (180 mg, 1.43 mmol) or a solution of HMF (180 mg, 1.43 mmol) in a solvent (1.0 mL) was added p-TsOH (3.0 mg, 1.7%). The mixture was stirred at a defined temperature for 5 h before visual inspection. The solvents with boiling points lower than the defined temperature were excluded from the evaluation.Screening conditions for the synthesis of Monomer S
To a well-stirred partially dissolved HMF (300 mg, 2.38 mmol) and pentaerythritol (162 mg, 1.19 mmol) in a solvent (1.5 mL) was added p-TsOH (3.0 mg, 16 μmol) at a defined temperature (25, 40, or 70 °C). The reaction mixture was then stirred at the same temperature overnight. Afterward, a drop of the reaction mixture was taken out with glass pipette, and quickly dissolved in DMSO-d6 for 1H NMR spectroscopy analysis.Fructose-based HMF synthesis
HMF was prepared by a modified method from a previous report.64 One liter of 30% (w/w) fructose solution in DMSO was placed in a 2 L flask, followed by addition of 60 g ion exchange resin (0.2 w/w equivalent to fructose), and shaking in an oil bath at 110 °C for 3 h. 30 mL of resulting reactant was used for small scale purification. After adding 30 mL brine, liquid–liquid extraction was performed 3 times with 30 mL ethyl acetate in a 250 mL separation funnel. The organic phase was pooled and concentrated by rotary evaporation. The crude sample dissolved in 10 mL DCM was subjected to flash chromatography using a column (2 × 14 cm) packed with 20 g silica (Merck) equilibrated with DCM. The column was washed with 150 mL DCM, and elution and fractionation was performed using a mixture of DCM/ethyl acetate (1/2) as the mobile phase. The HMF fractions were collected and concentrated by evaporation (246 g, 82%).Optimized synthetic protocol of Monomer S
To a well-stirred partially dissolved solution of HMF (10.1 g, 79.7 mmol) and pentaerythritol (5.41 g, 39.7 mmol) in 2-PrOH (30 mL) was added at room temperature a solution of p-TsOH (150 mg, 0.79 mmol) in 2-PrOH (30 mL). The reaction mixture was stirred at room temperature overnight under N2. Afterward, the reaction was quenched by the addition of solid NaHCO3 (150 mg) and continued stirring for 30 minutes. The precipitate was then collected by vacuum filtration and washed with 2-PrOH (2 × 20 mL), NaHCO3 (20 mL, 0.10 M), and distilled water (2 × 20 mL) to yield a white solid as the first fraction of crude Monomer S (8.83 g, 25.1 mmol).The mother liquor and the 2-PrOH used to wash the crude product were combined and evaporated to yield a yellow oil (5.30 g), which was re-dissolved in 2-PrOH (10 mL). To this solution was then added pentaerythritol (1.43 g, 10.5 mmol) and a solution of p-TsOH (40 mg, 0.21 mmol) in 2-PrOH (5 mL), and the resulting mixture was stirred at room temperature overnight under N2. Next, the formed precipitate was collected by vacuum filtration and washed with 2-PrOH (2 × 10 mL), NaHCO3 (10 mL, 0.1 M), and distilled water (2 × 10 mL) to yield a second fraction of crude Monomer S (3.41 g, 9.66 mmol). The two fractions of the obtained crude Monomer S were then combined and recrystallized in 2-PrOH to yield a white solid as the final product Monomer S (9.09 g, 65%). Tm: 185 °C (DSC), 1H NMR (400.13 MHz, DMSO-d6, δ, ppm): 3.63 (d, 2H, J = 11.5 Hz, CH2C(CH2)3), 3.74 (dd, 2H, J = 11.5 Hz, J = 2.2 Hz, CH2C(CH2)3), 3.86 (d, 2H, J = 11.5 Hz, CH2C(CH2)3), 4.36 (d, 4H, J = 3.7 Hz, ArCH2), 4.44 (d, 2H, J = 11.5 Hz, CH2C(CH2)3), 5.24 (t, 2H, J = 5.4 Hz, J = 7.8 Hz, –OH), 5.53 (s, 2H, ArCH), 6.25 (d, 2H, J = 3.2 Hz, Ar), 6.38 (d, 2H, J = 3.2 Hz, Ar), 13C NMR (100.61 MHz, DMSO-d6, δ, ppm) 32.2, 55.6, 69.0, 69.6, 95.6, 107.3, 108.2, 149.8, 155.3, FTIR: 1469, 1398, 1337, 1201, 1156, 1075, 1045, 1033, 996, 983, 967, 943, 911, 792, 748, 701, 653, 622, 578, 534, 484, 439, 422, 401. HRMS (ESI+, m/z): exact mass calcd for C17H21O8+: 353.1231, found 353.1238. Elemental analysis: Calcd for C17H20O8 (%) C 57.95, H 5.72. Found: C 57.88, H 5.70.
Life cycle assessment (LCA)
The LCA was developed following the methodology standardized in the ISO 14040-14044 series by the International Organization of Standardization (ISO 14040-14044).65,66 The method that covers the impact category of greenhouse gas (GHG) emissions is the ‘Greenhouse Gas Protocol (GGP)’ (v1.01) method which is based on the GWP100 (100-year timeframe) (IPCC, 2007).67 The characterization factors needed to quantify how much impact the new monomer has in GHG emissions are taken from this GGP method. The system boundary, for this preliminary LCA, was set up following a cradle-to-gate approach. The resource extraction, transportation to the raw building blocks factory, production of building blocks (i.e. HMF and pentaerythritol) and Monomer S production were included. Within the system boundary, the contributions of HMF and bio-based pentaerythritol and the contribution of the synthesis process were analyzed separately up to the step of the new rigid Monomer S production. Transportation of the building blocks from one factory to the other for the Monomer S production was excluded. The LCA on HMF from sugar beet was based on Garcia Gonzalez's work.68 The LCA on bio-based pentaerythritol (commercially named Voxtar M100) was obtained from Perstorp AB.69 The synthesis simulation was developed within the Ecoinvent database version 3.5. The functional unit was defined as 1 kg of Monomer S. Finally, the data used were incorporated into the SimaPro LCA software.Synthesis of polyesters
A typical polymerization protocol (PHST-10) is described. Dimethyl terephthalate (15.0 g, 77.4 mmol), HD (8.72 g, 73.8 mmol), Monomer S (4.00 g, 11.4 mmol), and DBTO catalyst (150 mg, 0.8 mol%) were added to a two necked 50 mL round bottom flask, which was equipped with a mechanical stirrer and a vacuum outlet (Fig. S8†). The flask was degassed and purged with nitrogen three times. The reaction mixture was heated to 145 °C to form a homogeneous melt, which was stirred for 3 h. Next, the reaction mixture was stirred for 16 h at 180 °C under vacuum (10–20 mbar), before it was cooled to room temperature. Subsequently, the reaction mixture was dissolved in dichloromethane (70 mL), and precipitated into methanol (700 mL). The precipitate was washed twice with methanol to give the final polymer PHST-10 (18.8 g, 86%).Synthesis of poly(urethane-urea)s
PSU, PSU-5 and PSU-10 were synthesized similarly. A typical polymerization protocol for PSU-10 is described. HD (6.53 g, 55.2 mmol), Monomer S (2.16 g, 6.13 mmol), IPDI (15.0 g, 67.5 mmol) and 2-butanone (8 mL) were added to a 100 mL round bottom flask with a magnetic stirrer. The reaction was heated with reflux (∼100 °C) for 4 h. Every hour, one droplet of the reaction mixture was taken out and analyzed by FTIR to monitor the isocyanate content. After each time a droplet was taken out, 2-butanone (5 mL) was added to the reaction mixture to decrease the viscosity. When the isocyanate content was reduced to 10% of the starting value, a solution of EDA (3 mL, 10% in 2-butanone) was added dropwise, followed by the addition of 2-butanone (10 mL) to reduce the viscosity. After being stirred at room temperature for 15 min, a second batch of EDA solution (3 mL, 10% in 2-butanone) was added dropwise, followed by additional 2-butanone (10 mL). After the addition of all EDA solutions, a droplet of the reaction mixture was taken out and measured by FTIR, showing the complete disappearance of isocyanate signal. Afterward, the reaction mixture was cooled to room temperature, diluted in THF (50 mL), and precipitated in heptane (750 mL). The precipitate was dried under vacuum overnight to give a pale yellow solid as PSU-10 (20.7 g, 86%).PSU-18 was synthesized according to a modified procedure, because of the low solubility of Monomer S in 2-butanone. HD (2.90 g, 24.5 mmol), Monomer S (2.16 g, 6.13 mmol), IPDI (7.50 g, 33.7 mmol) and 2-butanone (15 mL) were added to a 50 mL round bottom flask with a magnetic stirrer. The reaction was heated with reflux for 5.5 h. Afterward, dibutyltin laurate (0.5 mL, 10% in 2-butanone) was added, and the reaction solution mixture immediately turned transparent. After 30 min, EDA (2 mL, 10% in 2-butanone) was added. A single droplet of the reaction mixture was taken out and measured by FTIR, showing complete disappearance of the isocyanate signal. Afterward, the reaction mixture was cooled to room temperature, diluted in THF (25 mL), and precipitated in heptane (400 mL). The precipitate was dried under vacuum overnight to give a pale yellow solid as PSU-18 (9.27 g, 73%).
PSU-43 and PSU-62 was synthesized by a further optimized procedure. A typical polymerization protocol for PSU-62 is described. HD (1.45 g, 12.3 mmol), Monomer S (6.48 g, 18.4 mmol), IPDI (7.50 g, 33.7 mmol) and 2-butanone (15 mL) were added to a 50 mL round bottom flask with a magnetic stirrer and then heated to 30 °C. As Monomer S was not fully soluble in 2-butanone, a heterogeneous slurry was formed. Afterward, dibutyltin laurate solution (0.2 mL, 10% in 2-butanone) was added dropwise into the polymerization mixture, and then the temperature was raised to 40 °C. After the reaction stirred for 10 min, a second fraction of dibutyltin laurate solution (0.2 mL, 10% in 2-butanone) was added, followed by a further increase of the reaction temperature (to 50 °C). After 10 min stirring, a third fraction of dibutyltin laurate solution (0.1 mL, 10% in 2-butanone) was added, followed by an increase of reaction temperature (to 60 °C). After 60 min stirring, 2-butanone (5 mL) was added to reduce the viscosity. After another 25 min stirring, another 5 mL 2-butanone was added to reduce the viscosity. After 10 min additional stirring, the reaction mixture turned transparent. After another 35 min stirring, 2-butanone (5 mL) was added, followed immediately by dropwise addition of EDA (0.5 mL, 10% in 2-butanone). After 15 min, the viscosity of the reaction mixture became high, so 2-butanone (5 mL) was added. After 15 min stirring, a single droplet was taken out and analyzed by FTIR to show full depletion of isocyanates. Afterward, 1.5 mL of EDA (10% in 2-butanone) was added into the reaction in order to ensure that there was no isocyanate group left. Afterward, the reaction mixture was cooled to room temperature, diluted (1:1) with THF, and precipitated into tert-butyl methyl ether. The precipitate was dried under vacuum at 50 °C overnight, followed by 8 hours at 120 °C, and finally 2 hours at 150 °C, giving a pale yellow solid as PSU-62 (11.8 g, 76%).
PSU-43 was synthesized by a similar procedure, yielding a pale yellow solid with 11.1 g (78%).
Results and discussion
On the stability of HMF
The rationally designed rigid Monomer S was synthesized by an acid-catalyzed double acetalation of HMF and pentaerythritol (Scheme 1).70,71 Because HMF becomes unstable at elevated temperatures under acidic conditions, it is important to minimize its degradation during the synthesis. Therefore, the stability of HMF dissolved in different solvents (and in the pure state) was screened at 25, 40 and 70 °C respectively in the presence of p-toluene sulfonic acid (p-TsOH). A broad range of green or non-green solvents (according to the CHEM21 solvent recommendation) were screened, including the six recommended environmentally friendly solvents (water, EtOH, 2-PrOH, t-BuOH, acetone and ethyl acetate), and the six non-recommended solvents (MeCN, DCM, chloroform, DMF, DMSO, and 1-PrOH). After being treated for 5 h, the color of the HMF samples was visually inspected. Since the degradation of HMF usually yielded dark polymeric materials (i.e. humins),72–74 the solutions with darker color than bright yellow were excluded from further investigations. As shown in Fig. 1A, HMF solutions were only stable (light yellow) when the 4 protic solvents (i.e., water, EtOH, 2-PrOH and t-BuOH) were used at 25 or 40 °C. For the rest of the tested conditions, coloration was observed, indicating degradation of HMF. Among the “non-green” solvents (Fig. 1B), the protic solvent 1-PrOH showed similar behavior as the “green” alcohols (Fig. 1A). In addition, the solutions in DMF and DMSO remained bright yellow after the treatment at all temperatures, indicating that HMF was stable under these conditions. The rest of the evaluated solutions with the 3 aprotic solvents (i.e. MeCN, CHCl3, and DCM) all showed significant degradation at all temperatures (25–70 °C).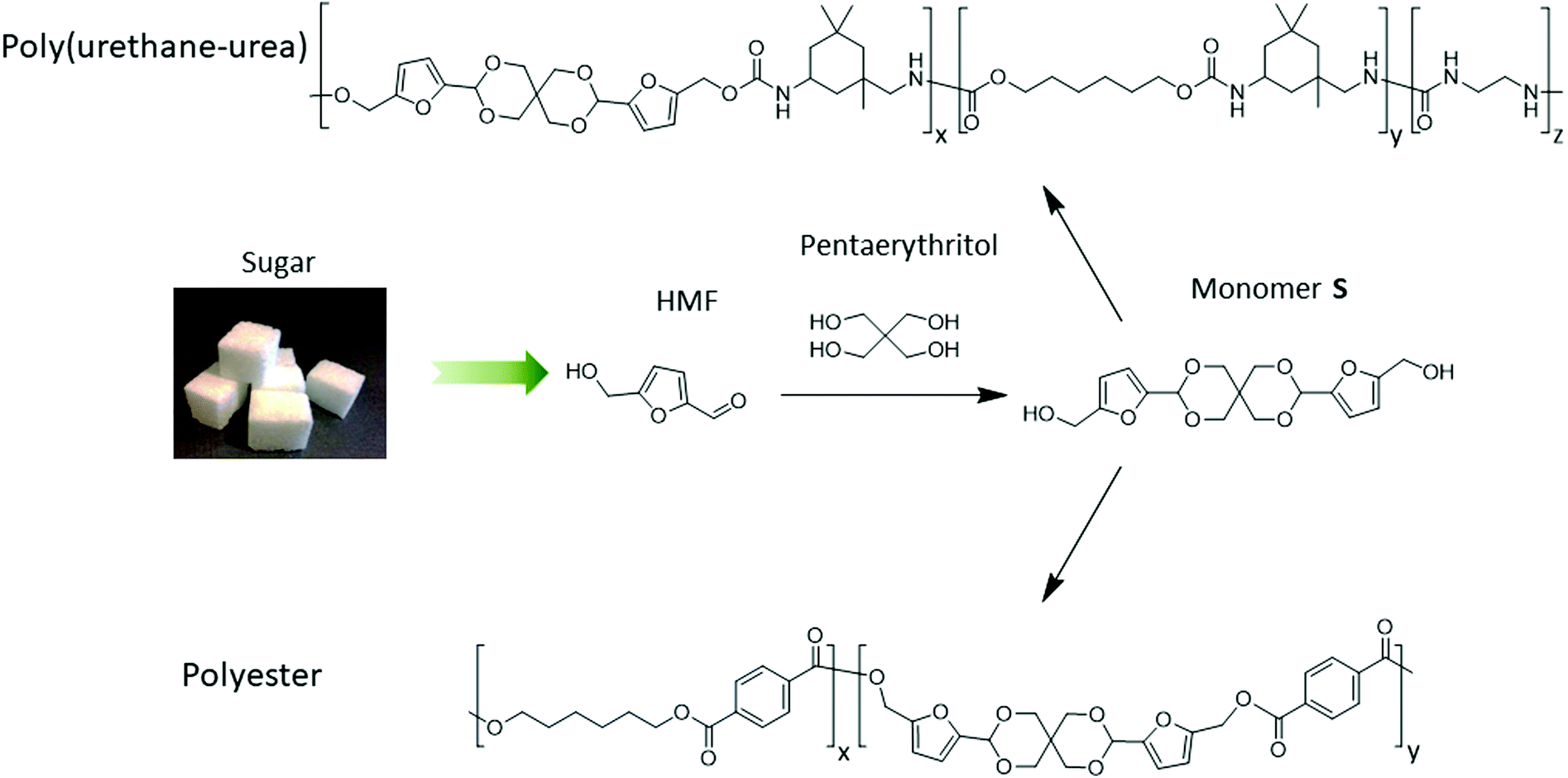 | ||
| Scheme 1 Synthesis of the HMF-based rigid diol (Monomer S, indicating its spirocyclic structure) as well as the polymerization towards copolyesters and copoly(urethane-urea)s. | ||
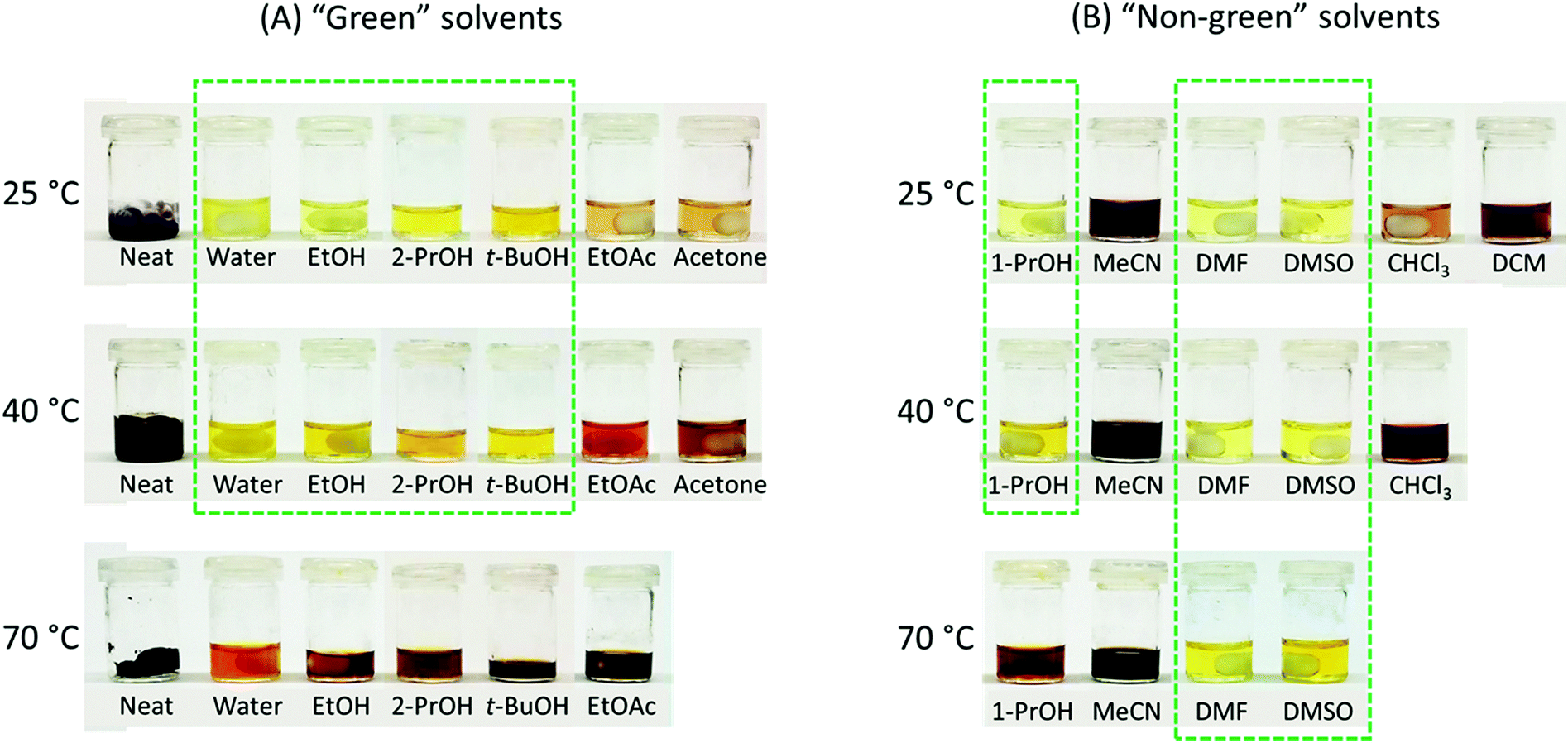 | ||
| Fig. 1 Stability evaluation of HMF in various solvents. According to the CHEM21 recommendation, the 12 tested solvents were categorized as “Green” and “Non-green”.54 Neat condition (solvent-free) was categorized as “Green”. The photos were taken after the solutions of HMF had been stirred for 5 h with p-TsOH at 25, 40, and 70 °C. The solutions without significant HMF degradation (coloration) are highlighted by green dashed boxes. | ||
Synthesis and LCA of Monomer S
Based on the results from the solvent screening, the conditions under which HMF was stable (highlighted in Fig. 1 in green boxes) were all evaluated for the monomer synthesis. We studied both the four CHEM21-recommended solvents (i.e. water, EtOH, 2-PrOH, and t-BuOH) and the non-recommended ones (DMF, DMSO and 1-PrOH) to gain comprehensive understanding of this reaction. As shown in Table 1, Monomer S was formed with different conversions under the investigated conditions, as shown by 1H NMR analyses of the crude reaction mixtures. The use of water, t-BuOH, DMF and DMSO-solutions resulted in low conversions (≤40%) at the temperatures used. Employing EtOH, 1-PrOH and 2-PrOH resulted in significantly higher conversions (60–80%). It was also noted that the highest conversions were achieved at 25 °C when the three high-conversion solvents (EtOH, 2-PrOH, and 1-PrOH) were used. As seen in Table 1, EtOH gave the highest conversion at 25 °C, followed by 1-PrOH. However, 1H NMR investigations revealed that the reactions in EtOH and 1-PrOH gave rise to new signals due to the occurrence of side reactions (Fig. S1†). These signals were not observed in the 1H NMR spectrum of the crude reaction mixture containing 2-PrOH. Fortunately, 2-PrOH is one of the CHEM21-recommended environmentally-friendly solvents.75 Therefore, the use of 2-PrOH at room temperature was chosen for the monomer synthesis. In addition, different acid catalysts were also evaluated for the synthesis of Monomer S in 2-PrOH, 25 °C. We found that strong mineral acid catalysts, such as sulfuric acid and hydrochloric acid, gave very similar results as p-TsOH with respect to the conversion of Monomer S (65–72%). However, relatively weak organic acids (e.g. oxalic acid, formic acid, acetic acid, or citric acid) failed to yield the desired product, likely due to their insufficient protonating ability for aldehydes. Based on the results of the solvent-screening, the optimized monomer synthesis was carried out in 2-PrOH at room temperature with p-TsOH catalyst. To facilitate our study, commercial HMF (fossil-based) was initially used in order to produce Monomer S on a scale up to ∼300 g per batch. After the reaction, NaHCO3 was added to quench the acid catalyst. The base quenching was essential because trace amounts of acid residues (even after excessive water washing) in the solid monomer led to slow decomposition of the monomer back to HMF and pentaerythritol, followed by the formation of dark humins. This was demonstrated by the deep coloration of a monomer sample prepared without base-quenching (Fig. S2†). After the reaction, Monomer S was precipitated and collected by filtration to yield a white solid as a crude sample with ∼30% yield. This yield was further improved by the utilization of the mother liquor from the filtration. The mother liquor contained unreacted HMF, which was further converted by the addition of pentaerythritol and p-TsOH. The additional step improved the yield of Monomer S to 65% (calculated based on HMF).| Recommendedb | Not recommendedb | ||||||
|---|---|---|---|---|---|---|---|
| Water | EtOH | 2-PrOH | t-BuOH | 1-PrOH | DMF | DMSO | |
| a The conversion was estimated by 1H NMR spectroscopy analysis of the crude reaction mixtures. Note that the conversions given are not the isolated yields of the product (after these experiments, Monomer S was not isolated). b According to the recommendation of CHEM21 solvent guide.53 c This experiment was carried out at 30 °C instead of 25 °C in order to be above the melting point of t-BuOH (∼25–26 °C). | |||||||
| 25 °C | 16 | 80 | 72 | 38c | 75 | 30 | 23 |
| 40 °C | 15 | 60 | 62 | 44 | 65 | 40 | 30 |
| 70 °C | — | — | — | — | — | 23 | 20 |
In the meantime, we also synthesized Monomer S using a bio-sourced HMF prepared by the dehydration of fructose (see Experimental for synthetic details). The fructose-based HMF had slightly lower purity (88%) compared with the commercial one (95%, Fig. S3†). Nevertheless, after performing the synthesis of Monomer S using the fructose-based HMF under the optimized reaction conditions, the crude conversion was found to be just slightly lower (∼70%) than that of the reaction using commercial HMF (∼75%, Fig. S4†). The further purification and upscaling of the fructose-based HMF production is currently being investigated, and will be communicated elsewhere.
In order to verify whether the production of bio-based Monomer S was indeed eco-friendly, a life cycle assessment (LCA) was performed to identify both the positive and negative aspects in the development of Monomer S and to explore possible alternatives to enhance environmental quality.76 The evaluation of the total impact of GHG emissions by LCA for Monomer S was assessed on the basis of a cradle-to-gate approach. Fig. 2 shows the total impact of the GHG emissions for Monomer S with a very low value of 1.18 kg CO2eq per kg S, of which 0.67 kg CO2eq per kg S was derived from the HMF production and 0.41 kg CO2eq per kg S originated from the bio-based pentaerythritol production (according to Perstorp AB). The synthetic process contributed to the total emissions with a small value of 0.10 kg CO2eq per kg S (8.5% of the total). Subsequently, Monomer S was compared with bio- and fossil-based 1,3-propanediol (1,3-PD).77 As shown in Fig. 2, Monomer S showed a remarkably lower GHG emission value compared with both the bio- and fossil-based 1,3-PD. The GHG value of Monomer S equals to only 54% of the value of bio-based 1,3-PD (2.18 kg CO2eq per kg bio-1,3-PD), and even as low as 24% of the value of fossil-based 1,3-PD (5.00 kg CO2eq per kg fossil-1,3-PD). This important result suggests that Monomer S tends to be more environmentally favorable in terms of GHG emissions. Admittedly, this assessment was only intended to provide first-hand information regarding the environmental impact of Monomer S. To our knowledge, this kind of assessment during the synthetic development of new bio-based monomers or polymers is very rare.78 In order to gain a deeper insight into the environmental impacts of new building blocks and polymers, more thorough LCA investigations (e.g., estimations of other critical environmental impact categories) will be needed. It will also be highly interesting to compare the LCA results of Monomer S with other bio-based (e.g. mannitol, isosorbide, galactitol, isoidide)79 or fossil-based (e.g. CHDM, CBDO) diols, of which the LCA investigation is lacking in the literature currently. These investigations are currently being conducted and will be communicated separately.
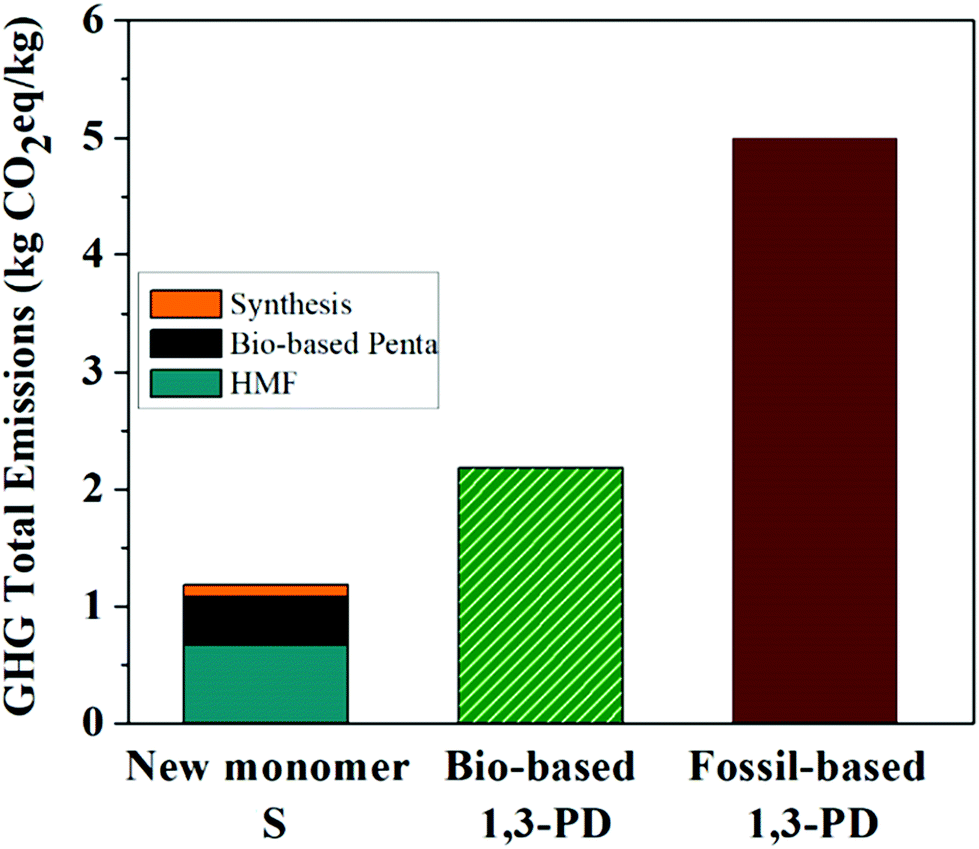 | ||
| Fig. 2 Total impact of the GHG emissions for the new Monomer S compared to bio- and fossil-based 1,3-propanediol. | ||
Synthesis and characterization of polyesters
The effect of the incorporation of the rigid Monomer S into polyester structures was investigated by copolymerizations with HD and dimethyl terephthalate, yielding a benchmark homopolymer PHT (using HD and dimethyl terephthalate) and four copolymers PHST (using HD, Monomer S, and dimethyl terephthalate). A modified two-step melt polycondensation protocol was used.80 Up to 19 mol% of Monomer S with respect to terephthalate was used in the polymerizations, and the resulting polymers were designated PHST-x (x is the mol% of spirocyclic structures with respect to terephthalate in the copolymer backbone). Since HD partially evaporated during the polymerization, a 10% excess of this monomer was used. The reaction mixture was first stirred during 3 h at 145 °C under nitrogen to form oligomers by transesterification, and then it was kept at 180 °C under vacuum for 15–17 h to increase the molecular weight by polycondensation. It was observed that longer reaction times (e.g., 2–3 days) led to partial gelation for PHST-19, and even longer reaction times (4 days) gave completely insoluble gels. In order to reduce the degree of crosslinking, we added a small amount of xylene and mesitylene as solvents, and lowered the reaction temperature to 160 °C (for the polycondensation step). As a result, the polydispersity value (Đ) was reduced to 2.0 and a reduced coloration was observed (Fig. S5, ESI†). However, the molecular weight was low (Mn ∼6.5 kDa, ∼60% lower compared to the original vacuum-based procedure). In addition, we also investigated whether we could reduce the crosslinking by the addition of an antioxidant in the polymerization of PHST-19.81 As a result, when the polymerization with antioxidant tris(nonylphenyl) phosphite (TNPP) was carried out without solvents (xylene and mesitylene) under vacuum, significant foaming was observed during the polymerization, which blocked the vacuum outlet (Fig. S6†). When the polymerization with TNPP was carried out with solvents (xylene and mesitylene) under nitrogen, complete gelation was observed, which prevented NMR or SEC analysis.The Mn values of PHT, PHST-3, PHST-10 and PHST-16 were similar (∼9100–10900 kDa), but their Mw values increased with the increased S content, leading to increased molecular weight distribution. However, the molecular weight and polydispersity for PHST-19 were significantly higher, which was consistent with the observation that the measured hydrodynamic radius of PHST-19 (Rh ∼7 nm) was significantly larger than that of the other samples (Rh ∼5 nm). The intrinsic viscosity ([η]) values of the obtained polyesters were in the range of 0.38–0.57 dL g−1 (measured by SEC with a triple detector system) without a significant decreasing trend as the S-content increased. This result is different from a previously reported series of copolyesters with rigid-diol units, where the [η] values and molecular weights decreased as the rigid-diol content increased.41 In our case, the molecular weights and [η] values of the PHST series were not significantly reduced by the use of rigid Monomer S, which may be explained by the presence of partial branching/crosslinking that increased the molecular weight.
The Mark–Houwink–Sakurada parameters for the polyesters were given directly by SEC analysis. As shown in Table 2, the a parameter showed a general decreasing trend with the increased S-content. For PHT, a = 0.75, indicating that this polymer formed a random coil conformation in chloroform. For copolyester PHST-3, the a value decreased to 0.67, indicating that chloroform was a less favorable solvent for this polymer. For the polyesters with higher S-content (PHST-10, PHST-16 and PHST-19), the a parameter was slightly below 0.5 (0.44–0.48), indicating that these polymers attained compact spherical conformations in chloroform.82 This observation was consistent with the presence of branching in these polymers. Compared with hyperbranched polymers with a ∼0.16–0.40,83 the PHST samples had a values closer to 0.5, which qualitatively suggested that they had a lower degree of branching compared with hyperbranched polymers.84–86
| Feed S (%) | Incorporated S (%) | M n (kDa) | M w (kDa) | Đ | [η] (dL g−1) | R h (nm) | −logK |
a | |
|---|---|---|---|---|---|---|---|---|---|
| a Feed S is the is the mol% of Monomer S in the monomer mixture. Incorporated S is the mol% of Monomer S in the polymer as estimated by NMR. Mn, Mw, Đ, intrinsic viscosity [η], hydrodynamic radius Rh, and Mark–Houwink–Sakurada parameters K and a were determined by SEC. | |||||||||
| PHT | — | — | 9.4 | 18.1 | 1.93 | 0.57 | 5.3 | 3.4 | 0.75 |
| PHST-3 | 3.7 | 3 | 9.1 | 18.5 | 2.03 | 0.52 | 5.1 | 3.1 | 0.67 |
| PHST-10 | 14.7 | 10 | 10.9 | 27.5 | 2.57 | 0.38 | 5.0 | 2.4 | 0.47 |
| PHST-16 | 18.3 | 16 | 9.8 | 37.2 | 3.80 | 0.40 | 5.1 | 2.4 | 0.48 |
| PHST-19 | 21.8 | 19 | 15.4 | 113.0 | 7.37 | 0.47 | 7.4 | 2.3 | 0.44 |
The presence of partial branching/crosslinking in PHST-10, PHST-16 and PHST-19 might be attributed to a ring-opening process of the spirocyclic acetal structures (Fig. S7, ESI†), as well as furfural alcohol condensation and subsequent branching reactions.87 In order to gain some insight into ring-opening possibility, we analyzed the condensed yellow solid in the vacuum outlet of the reaction flask (Fig. S8†). According to the 1H NMR spectrum, the condensed yellow solid from the polymerization mixture contained HD and dimethyl terephthalate monomers, as well as HMF (Fig. S9†). Because HMF was not present in the starting polymerization mixture, we assumed that it was formed through the degradation of Monomer S, possibly giving HMF and a polyol (Fig. S7†). These polyols can participate in the condensation polymerization to form branched or cross-linked structures. In addition, the possible side reaction caused by furfural alcohol condensation was checked by the 1H NMR spectra. As shown in Fig. S10 (ESI†), a small signal at ∼3.94 ppm was observed, which could potentially correspond to the methylene bridge between two furan rings, as a product formed by furfural condensation.88
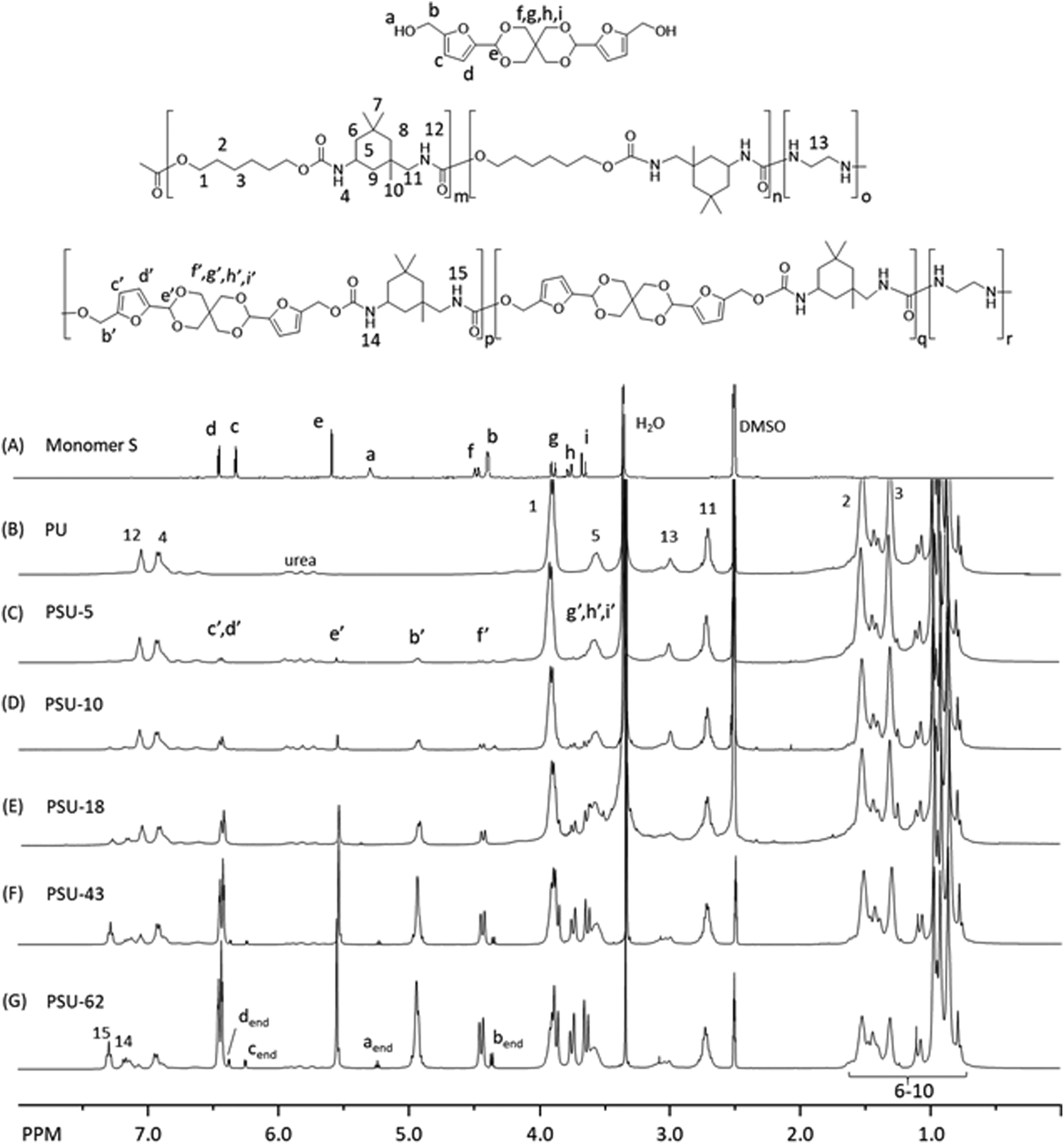
The molecular structure of the polyesters was investigated by 1H NMR spectroscopy. First, all the signals in the spectrum of Monomer S were unambiguously assigned (Fig. 3A), including the two doublets at 6.25 (c) and 6.38 ppm (d) (the aromatic protons of furan), the singlet at 5.53 ppm (e, acetal proton), the triplet at 5.24 ppm (a, the OH proton), and the doublet at 4.44 ppm (b, the CH2 groups α to the furan rings). Interestingly, the CH2 protons on the spiroacetal units showed four discernable peaks, which was due to the different axial and equatorial C–H orientation on the rigid spirocyclic structure. With the help of 2D NMR investigations (i.e. COSY, HMQC, HMBC and NOESY, see Fig. S11–S26†), we assigned the two doublets at 3.63 (i) and 3.86 ppm (f) to the axial protons, and the other two doublets at 3.74 (g) and 4.36 ppm (h) to the equatorial protons. Such an assignment was consistent with other similar reported compounds with spirocyclic acetal structures.71,89 After the polymerizations, the resulting polyesters (Fig. 3B–F) showed broadened 1H NMR signals, indicating the formation of the polymers. In all these spectra, a CH2 signal (α to the ester bond) at 4.38 ppm (1), two broad aliphatic signals at 1.85 ppm (2) and 1.56 ppm (3), and an aromatic singlet at 8.10 ppm (4) were observed, which confirmed the PHT structures in all polyesters. The incorporation of Monomer S residues in the copolyesters was confirmed by the observation of the corresponding signals (b′–i′ in Fig. 3C–F). Furthermore, the OH signal of Monomer S (a) disappeared in the spectra of copolyesters, and the signal of the CH2 attached to the furan ring significantly shifted toward lower field (from 4.44 to 5.55 ppm). These observations also confirmed that Monomer S residues were incorporated by the formation of ester bonds. In addition, the furan signals (c′, d′) moved slightly downfield, which was consistent with the formation of electron withdrawing ester bonds. The intensity of the signals indicated that the spirocyclic units consistently increased as the Monomer S content increased. It was noted that there were small shifts for the spirocyclic CH2 signals (i.e. f′, g′, h′ and i′) compared with the corresponding signals of the monomer (i.e. f, g, h and i), which could be attributed to the different NMR solvents used for the monomer and the polymers. Unfortunately, Monomer S was only soluble in DMSO-d6 (among commonly used NMR-solvents), while the polyesters were only soluble in chloroform-d. Hence, the NMR spectra of monomer and polymers were recorded in different solvents.
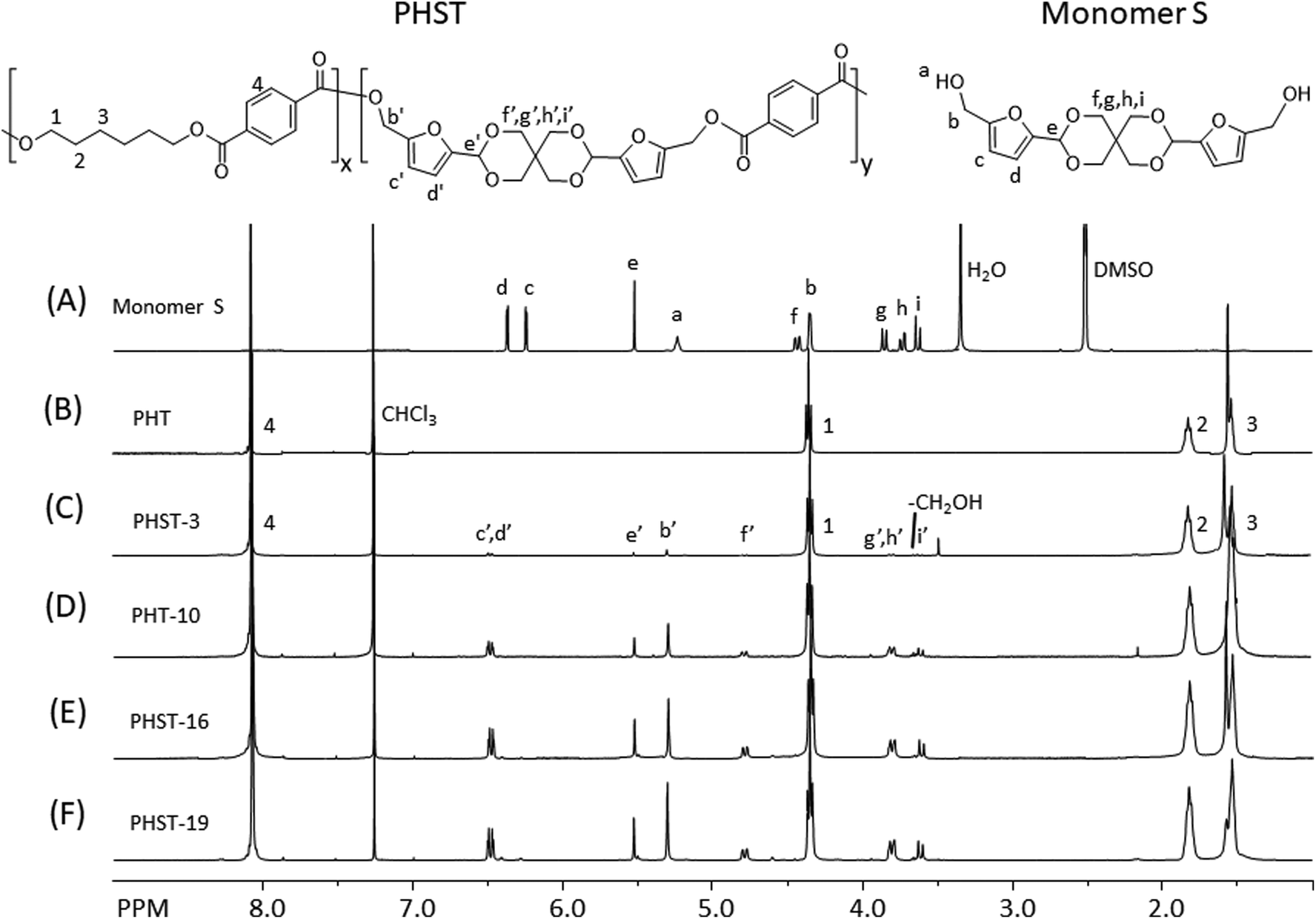 | ||
| Fig. 3 1H NMR spectra of (A) Monomer S and (B–F) the polyesters. | ||
The integration of the 1H NMR signals provided valuable information regarding the chemical composition of the resulting polyesters. By comparing the integrals of the furan signals (c′ and d′) with that of the terephthalate aromatic signals (4), we calculated the S content (taking terephthalate as 100%) for each copolyester (Table 2). Compared with the monomer feed, the S content of the copolyester was lower in percentage (68–87% of the fed Monomer S). This may be attributed to a lower reactivity of the sterically hindered Monomer S compared with that of HD. Another explanation may be that the degradation of Monomer S (or incorporated S units in the polyesters) during the polymerization (Fig. S7†) could lower the observed S-content in the resulting polymers. The latter was consistent with the observation of HMF in the vacuum outlet of the reaction vessel (Fig. S8 and 9†).
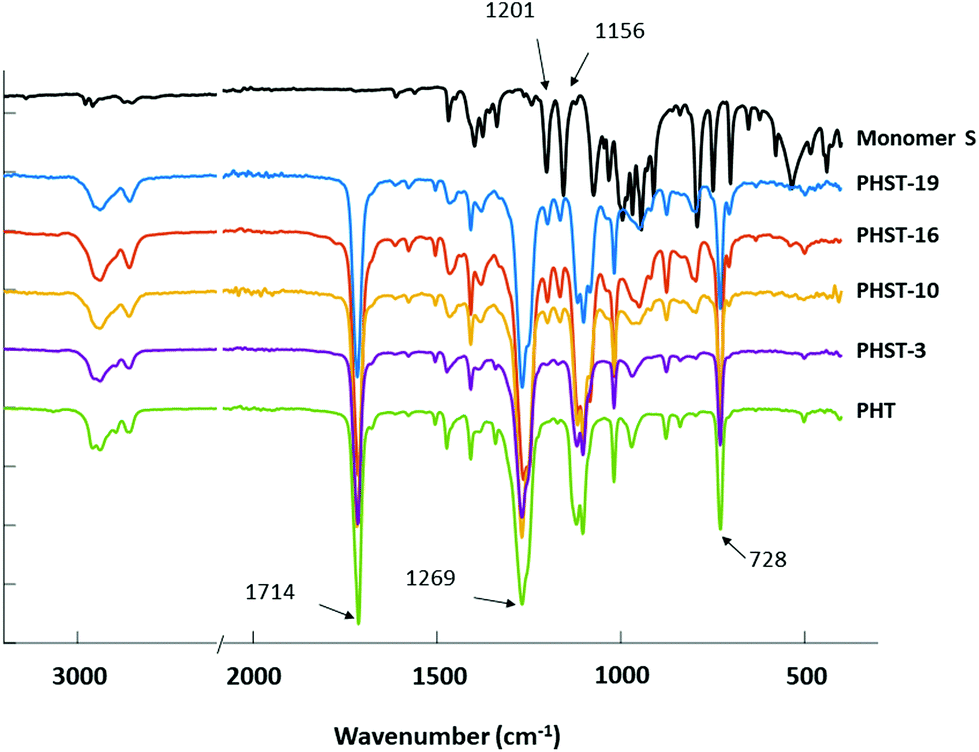
The chemical structures of the polyesters were also confirmed by FTIR analyses (Fig. 4). All the polyesters’ spectra showed characteristic ester signals, including C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1714 cm−1 and C–O stretching at 1269 cm−1. The signal at 728 cm−1 originated from the out of plane C–H bending.90 The aromatic and aliphatic C–H stretching was also observed at 2800–3000 cm−1,91 which was consistent with the presence of PHT residues in all polymers. Furthermore, the characteristic spiroacetal signals at 1201 and 1156 cm−1 (shown in the spectrum of Monomer S) were also observed in the spectra of all polyesters with increasing intensity along with the increasing S content.71,92 This confirmed the incorporation of the spiroacetal S residues in the copolyesters.
O stretching at 1714 cm−1 and C–O stretching at 1269 cm−1. The signal at 728 cm−1 originated from the out of plane C–H bending.90 The aromatic and aliphatic C–H stretching was also observed at 2800–3000 cm−1,91 which was consistent with the presence of PHT residues in all polymers. Furthermore, the characteristic spiroacetal signals at 1201 and 1156 cm−1 (shown in the spectrum of Monomer S) were also observed in the spectra of all polyesters with increasing intensity along with the increasing S content.71,92 This confirmed the incorporation of the spiroacetal S residues in the copolyesters.
 | ||
| Fig. 4 FTIR-spectra of Monomer S and polyesters. | ||
The thermal stability of Monomer S and the different polyesters was evaluated by TGA (Fig. 5 and Table 3). For the homopolymer PHT, a single decomposition rate maximum was observed at 400 °C (Td). All copolymers showed three decomposition rate maxima (Td ∼320 °C, 400 °C, and 450 °C). The first decomposition rate maximum at ∼320 °C was attributed to the degradation of the spirocyclic acetal units in the PHST backbone, which was similar as the thermal decomposition profile of Monomer S (Td ∼320 °C). The second decomposition rate maximum at ∼400 °C was attributed to the degradation of PHT units in the copolymers, presumably through a 6-membered cyclic β-hydrogen transfer mechanism.93 The third decomposition rate maximum at 450 °C was more prominent for PHST samples with higher Monomer S content. This may be explained by the formation of degradation-induced crosslinking (Fig. S7†) during the TGA measurements. To further verify this explanation, a PHST-19 sample was purposely prepared by extending the reaction time to 4 days, which resulted in the formation of completely insoluble polymer gel (designated PHST-19gel). Comparing the TGA profile of this sample with that of the original PHST-19 (Fig. S27†), a much more pronounced decomposition rate maximum was observed for PHST-19gel at 450 °C. This supported that the decomposition rate maximum at 450 °C was due to crosslinking. Among the polyester series, the T5 and Td values showed a general decreasing trend as more Monomer S units were incorporated. The char yields (CY) increased with the Monomer S contents, which was consistent with the results for polyesters with rigid building blocks.54
 | ||
| Fig. 5 (A) TGA thermograms and (B) first derivative curves of Monomer S and the polyesters. | ||
| TGA | DSC | DMA | ||||||
|---|---|---|---|---|---|---|---|---|
| T 5 (°C) | T d (°C) | CY (%) | T g (°C) | T m (°C) | T c (°C) | T g (°C) | E′ (MPa) | |
| a T 5 is the temperature at 5% weight loss. Td is the temperature with the maximal degradation rate. T5, Td, and char yield (CY) were all obtained by TGA. Tg and Tm were measured from the second heating DSC curves, and Tc was measured from the second cooling DSC curves. Note that there were three decomposition rate maxima in the TGA curves (Fig. 5), but only the highest rate maxima were listed in the table as the Td values. | ||||||||
| PHT | 368 | 401 | 4 | 17 | 140, 146 | 116 | 29 | 1279 |
| PHST-3 | 349 | 402 | 6 | 25 | 133, 141 | 106, 122 | 42 | 1453 |
| PHST-10 | 312 | 302, 389, 432 | 12 | 34 | 123 | — | 32 | 1143 |
| PHST-16 | 324 | 317, 402, 446 | 15 | 42 | — | — | 42 | 1615 |
| PHST-19 | 304 | 305, 388, 435 | 15 | 47 | — | — | 43 | 1488 |
| Monomer S | 267 | 323 | 20 | — | 185 | — | — | — |
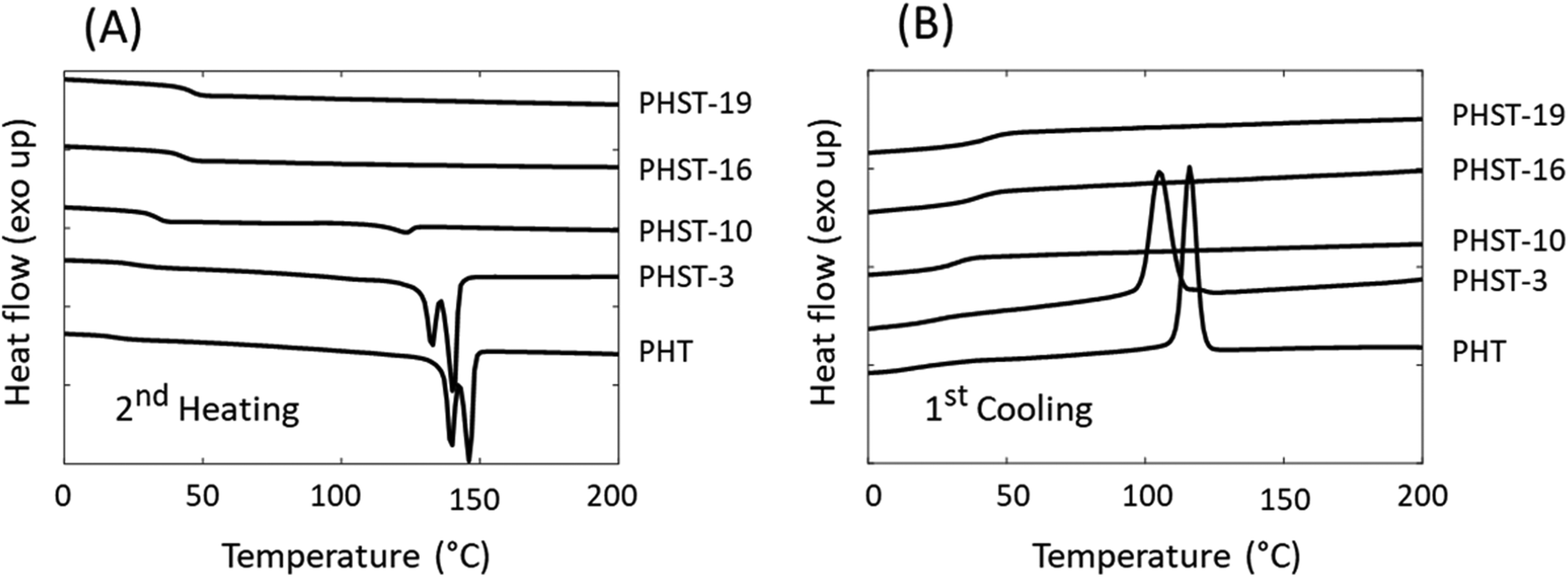
The thermal transitions of the polymers were investigated by DSC (Fig. 6 and Table 3). First, it was noted that the incorporation of rigid S units into PHT significantly increased the Tg of the polyesters (from 17 °C for PHT to 47 °C for PHST-19). This was consistent with the previous reports on polyesters containing bicyclic diacetalized glucitol units.54 It should be noted that the increase in Tg may be partially ascribed to crosslinking.94 Furthermore, the incorporation of S units also affected the melting/crystallization behavior, similar to the incorporation of other rigid structures in polyesters.54,80,95 The DSC results showed that PHT and PHST-3 were semicrystalline, which was indicated by the presence of melting endotherms during heating (Fig. 6A) and crystallization exotherms during cooling (Fig. 6B). The existence of two melting peaks was consistent with the multiple crystalline forms reported for PHT.96,97 The melting (Tm) and crystallization temperature (Tc) both decreased with the incorporation of S units (3 mol%). When more than 10% S units were incorporated, the copolyesters became fully amorphous. PHST-10 showed a minor melting endotherm during the heating cycle most likely caused by cold crystallization, since no visible crystallization peak was observed during the cooling cycle. PHST-16 and PHST-19 did not show any melting or crystallization peaks.
 | ||
| Fig. 6 DSC (A) second heating and (B) first cooling curves of the polyesters. | ||
Finally, the obtained polyesters were characterized by DMA, and results were compared with that of commercially available Akestra90 (ESI Fig. S28†). The Tg values were measured as the temperature for the peak loss modulus of each sample (Fig. S28B, ESI†), which showed consistent growing trend for PHST-10, PHST-16 and PHST-19. The other two polymers PHT and PHST-3 showed some deviation in their Tg values compared with the DSC results, which could be attributed to the broad peaks on the E′′ curves. It was also noted that the storage modulus at the glassy plateau (20 °C) for the obtained polyesters was in the range of ∼1143 to 1615 MPa, which is close to the value of Akestra (∼1428 MPa, under the same measurement conditions).
Synthesis and characterization of poly(urethane-urea)s
Monomer S was also used as the rigid diol in the synthesis of thermoplastic poly(urethane-urea)s in combination with a flexible diol (HD) and an aliphatic diisocyanate (IPDI). A series of six poly(urethane-urea)s were prepared using different contents of rigid diol S in the diol mixture. The resulting polymers were named as PU (only using HD and IPDI) or PSU-x (number x indicates the actually incorporated S units in the copolymers with respect to IPDI units, according to the NMR analysis). The polymerizations for PU, PSU-5 and PSU-10 were carried out following a conventional two-step synthetic protocol.98 First, a mixture of the diols (i.e. HD and Monomer S) was reacted with an excess (10 mol% excess) of IPDI in a refluxing solution of 2-butanone. Additional solvent (2-butanone) was added during the polymerization when the viscosity of the reaction mixture significantly increased and the stirring stopped. After ∼2 h, a prepolymer (containing primarily isocyanate end groups) with relatively low molecular weight was formed, which was directly used in the next step. For the second step, ethylene diamine (EDA) as a chain extender was added dropwise, and the content of residual isocyanate groups was monitored by FTIR analysis of the reaction mixture. After the depletion of the isocyanate groups, the resulting polymer was purified by dissolution in THF and precipitation in heptane to yield a pale yellow poly(urethane-urea) product. For the synthesis of PU, PSU-5 and PSU-10, this procedure resulted in decent molecular weights (Mn >10 kDa, Table 4) without noticeable side reaction. However, for PSU-18, the same procedure resulted in relatively low molecular weight (Mn ∼3500 Da, not shown). This was due to the low solubility of Monomer S in the reaction mixture, which prevented the polymerization from achieving higher conversion and thus degree of polymerization. Therefore, further modification of the polymerization procedure was investigated for the preparation of PSU-18. First, we increased the amount of solvent (3× the starting volume) for the polymerization of PSU-18, but it was observed that Monomer S was still not completely dissolved after 5.5 h under polymerization conditions. Further addition of solvent was expected to significantly reduce the polymerization rate, so it was not carried out. Alternatively, since it is Monomer S that has low solubility not the polymers, it is expected that acceleration of the polymerization rate may have a positive impact on the molecular weight. Therefore, the addition of an effective catalyst, dibutyltin laurate, was investigated. It was observed that the reaction mixture immediately turned transparent right after the addition of the catalyst, indicating that most of Monomer S was consumed quickly. By this procedure, PSU-18 was prepared with decent molecular weight without significant gelation (Table 4). However, when this procedure was used for PSUs with even higher S content (e.g. PSU-43 and PSU-62), it was observed that the accelerated polymerization was associated with an uncontrolled increase of reaction temperature, which could lead to increased extent of side reactions and gelation. In order to control the reaction rate and side reactions of PSU-43 and PSU-62, we further modified the reaction conditions by adding the catalyst in 3 portions over 30 minutes at lower temperatures (30–50 °C). After the addition of catalyst, the reaction temperature was raised to 60 °C and kept for the remaining reaction time. In addition, EDA was also added in two portions (0.75 mmol at the beginning of chain extension, and 2.25 mmol at the end). This could avoid oversaturation of EDA that would cause mono-reaction of the NH2 groups on EDA and result in low molecular weights. By the modified procedure, PSU-43 and PSU-62 were prepared without significant gelation. The molecular weight and polydispersity index values were decent (Table 4). All the obtained poly(urethane-urea) samples were insoluble in EtOAc, heptane, Et2O, toluene, but soluble in DMSO, DM Ac and THF. The molecular weights were determined by SEC analysis in DMAc with LiBr (5 g L−1).| Feed S (%) | Incorporated S (%) | M n (kDa) | M w (kDa) | Đ | T g (°C) | T d (°C) | T 5 (°C) | CY (%) | |
|---|---|---|---|---|---|---|---|---|---|
| a Feed S is the mol% of Monomer S in the monomer mixture. Incorporated S is the mol% of Monomer S in the polymer as estimated by NMR. Mn, Mw and Đ were determined by SEC in DMF. T5 is the temperature at 5% weight loss. Td is the temperature with the maximal degradation rate. T5, Td, and the char yield (CY) were obtained by TGA. Tg was measured from the second heating DSC curves. | |||||||||
| PU | — | — | 12 | 22.8 | 1.9 | 79 | 323 | 276 | 2 |
| PSU-5 | 5.0 | 5.4 | 16 | 43.8 | 2.7 | 90 | 321 | 279 | 5 |
| PSU-10 | 10 | 10 | 17 | 78.5 | 4.6 | 97 | 315 | 270 | 9 |
| PSU-18 | 20 | 18 | 21 | 75.0 | 3.5 | 104 | 306 | 266 | 8 |
| PSU-43 | 40 | 43 | 53 | 137 | 2.6 | 120 | — | 283 | 14 |
| PSU-62 | 60 | 62 | 37 | 125 | 3.4 | 131 | — | 284 | 19 |
The molecular structure of the poly(urethane-urea)s was analyzed using 1H NMR spectroscopy. As shown in Fig. 7B–E, the signals for the polymers were broadened compared to that of Monomer S, which indicated the formation of polymers. Furthermore, two new signals at 6.8–7.1 ppm (signals 12 and 4, in Fig. 7B–E) were observed after the polymerizations, which corresponded to the N–H protons in urethane bonds formed between IPDI and HD. Furthermore, two new signals at 7.16 (14) and 7.30 ppm (15) were observed with increasing intensity from PSU-10 and PSU-62, which corresponded to the N–H protons in urethane bonds formed between IPDI and Monomer S. In addition, the S content was determined by comparing the integral of the signal corresponding to the α-protons of the primary isocyanate on IPDI (11) with that of the signal corresponding to the aromatic protons on the furan units (c′ and d′). As a result, the S content in the copolymers was consistent with the initial monomer feed ratio (Table 4). It was also noted that in the 1H NMR spectra of PSU-43 and PSU-62, signals corresponded to the S structure at the chain ends were clearly visible (designated as aend, bend, cend and dend in Fig. 7G). By comparing the integrals of these chain end groups with that of the backbone signals (e.g. the methylene signal 11), the molecular weights of PSU-43 and PSU-62 were estimated as 22 and 13 kDa, which were significantly lower than the results from SEC in DMAc. This discrepancy could be related to the relatively large error range of the NMR integrals of small end groups and the assumption that every polymer has two end groups (neglecting branching, crosslinking and cyclic structures), as well as the reported overestimation tendency for SEC for rigid polymers using flexible PS standards.99–102 Finally, the 1H NMR spectrum of PSU-62 was examined in order to evaluate whether there were furfural condensation side reactions (Fig. S60, ESI†). Unfortunately, the area where the peak for the methylene CH2 group (in connection with two furan rings) is severely overlapped with the backbone peaks, which prevented this assessment.
 | ||
| Fig. 7 1H NMR spectra of (A) Monomer S and (B–G) poly(urethane-urea)s. | ||
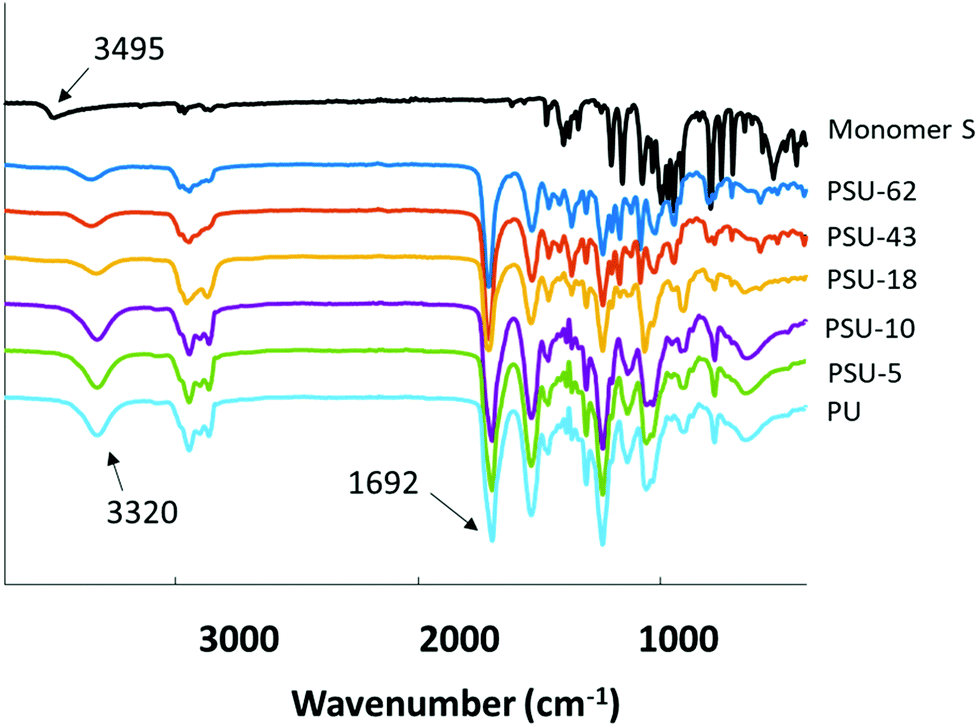
The molecular structures of the obtained poly(urethane-urea)s were further confirmed by FTIR spectra. As shown in Fig. 8, the absorption band at 3495 cm−1 for Monomer S was attributed to the –OH stretching vibrations.103 This band was not observed in the spectra of any of the poly(urethane-urea)s. Furthermore, a broad absorption band at 3320 cm−1 was observed in the spectra of all the obtained polymers, which was attributed to the N–H stretching band in the urethane units. These observations indicated that the reaction between the isocyanate and the diols was complete.103 Finally, the complete disappearance of the isocyanate absorption bands (2250–2270 cm−1) and the appearance of characteristic carbonyl absorption band at 1692 cm−1 also confirmed the formation of the target poly(urethane-urea)s.103,104
 | ||
| Fig. 8 FTIR-spectra of Monomer S and poly(urethane-urea)s. | ||
The thermal stability of the poly(urethane-urea)s was evaluated using TGA. As shown in Fig. 9 and Table 4, all these polymers showed good thermal stability (T5 > 270 °C). A single decomposition rate maximum was observed for polymers with relatively low S content (<10%), which was in agreement with other reported IPDI-based polyurethanes.105 However, the copolymers with higher S content (≥18%) showed a complex decomposition pattern in their derivative weight loss curves (Fig. 9B). For PSU-18, a small shoulder was observed at ∼350 °C on the derivative weight loss curve. For PSU-43 and PSU-62, the corresponding curves look even more complicated. For PSU-43, there was a local peak at ∼300 °C, followed by a broad peak at 320–380 °C. For PSU-62, there was a shoulder at ∼300 °C, and a broad peak at higher temperature. Therefore, no Td value could be meaningfully given (Table 4). This complicated thermal decomposition behavior when higher S content was incorporated in PSUs may be related to the thermal decomposition of the spirocyclic acetal or furan structures in PSU, which can cause subsequent crosslinking during TGA measurements. The exact mechanism remains to be unraveled. The char yield (CY) increased with the increased S content in the polymers, which was consistent with the observed trend in the PHST polyesters discussed above. This may be attributed to the increased aromatic content in the polymers, which has been reported to increase the char yields under nitrogen.106 The thermal behavior of the poly(urethane-urea)s was further investigated using DSC. As shown in Fig. 10 and Table 4, all the obtained polymers were completely amorphous without any melting endotherm, which was expected due the asymmetric structure of IPDI. The Tg values increased significantly with the increased S content (from 79 °C for PU to 131 °C for PSU-62).
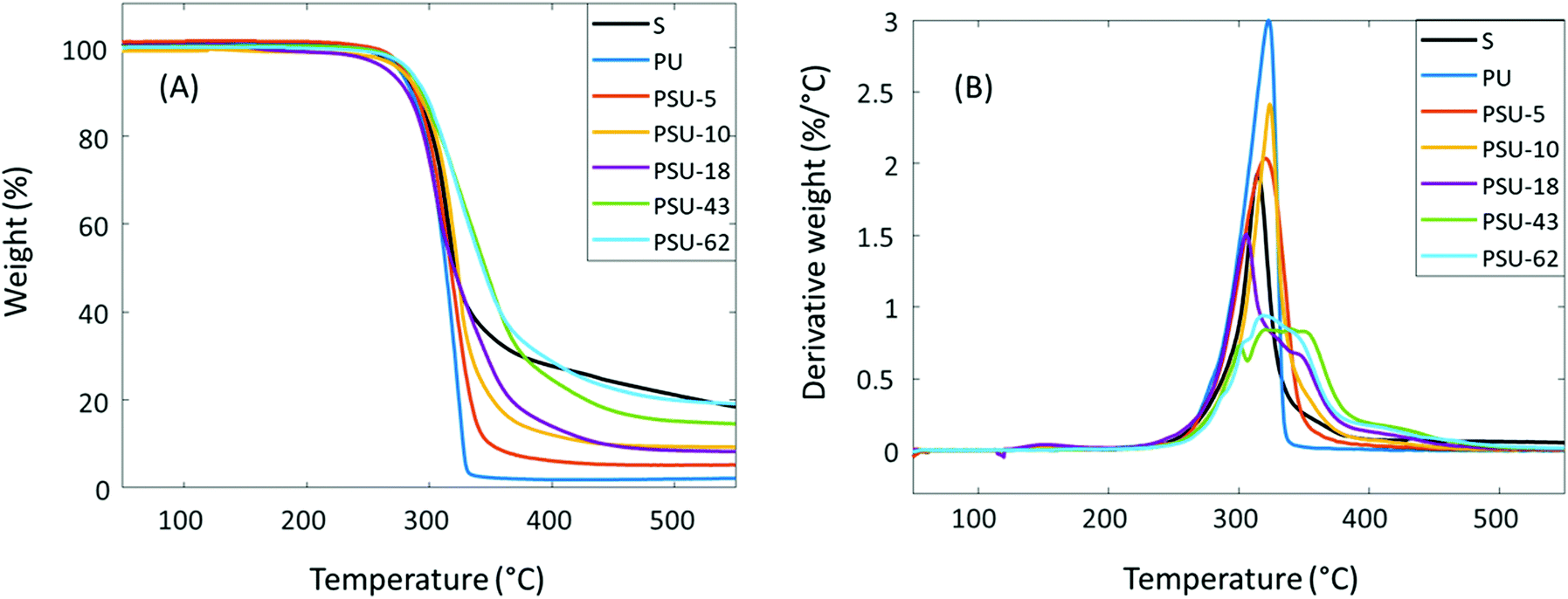 | ||
| Fig. 9 TGA (A) thermograms and (B) first derivative curves of Monomer S and the poly(urethane-urea)s. | ||
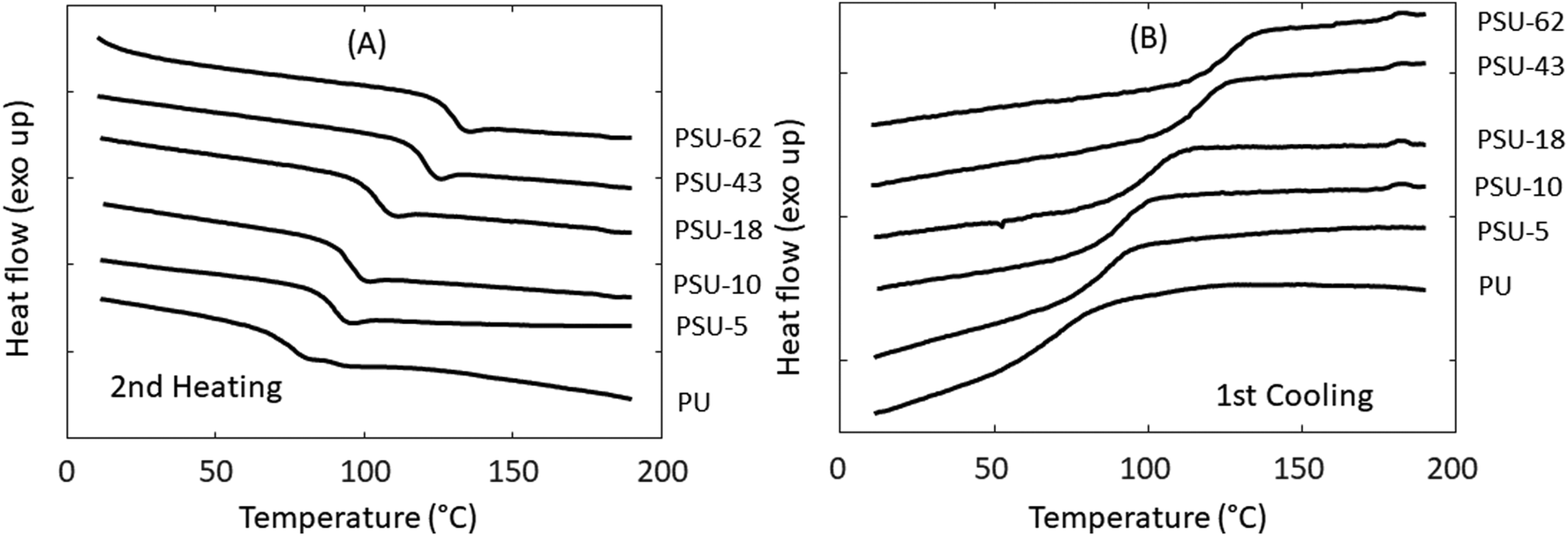 | ||
| Fig. 10 DSC (A) second heating and (B) first cooling curves of the poly(urethane-urea)s. | ||
Finally, the obtained poly(urethane-urea) samples were dissolved in THF (50 mg mL−1) for film-casting. The homogeneous polymer solution was spread evenly across a glass slide, followed by evaporation at room temperature under a glass conical funnel overnight. The use of glass conical funnel enabled the production of smooth polymer films without significant wrinkles, cracks and bubbles. Afterward, the glass slides were placed under vacuum for 24 h to completely remove the solvent THF, yielding almost colorless transparent films (Fig. 11). The water contact angle (θ) on the films was seemingly unaffected by the S content (74–79°, Fig. S61, ESI†).
 | ||
| Fig. 11 Poly(urethane-urea) samples cast on glass slides. | ||
Conclusions
A novel sugar-based and eco-friendly spiro-diol (Monomer S) was prepared by a convenient reaction of two bio-based molecules (HMF and pentaerythritol) in a green solvent (2-propanol). Life cycle analysis of this diol indicated that its production may generate significantly lower greenhouse gas emissions, compared with fossil-based and even bio-based 1,3-propanediol, which is a well-recognized green diol for polymer production. Monomer S was then used to produce two series of polyester and poly(urethane-urea) copolymers. The polyester series had a varying S content up to 19 mol%. Decent molecular weights and intrinsic viscosity values were achieved. The incorporation of the HMF-based spirocyclic units effectively increased the Tg of the copolyesters. However, partial branching or crosslinking was observed for the polyesters with >10 mol% S content due to side reactions at high temperatures, which may cause challenges in the processing and application. The poly(urethane-urea) series had a varying S content up to 62 mol% and high molecular weights. The Tg values were effectively increased from 79 to 131 °C upon the incorporation of the spirocyclic structures in the backbones. No significant coloration was observed for the poly(urethane-urea)s series, which suggested promising application potential toward various bio-based coating materials. Development of thermosetting coating products derived from the reported sugar-based poly(urethane-urea)s is currently being investigated. Regarding polyesters, it is also highly interesting to investigate the possibility of using Monomer S to polymerize with aliphatic dicarboxylates like adipate or succinate, which in general require lower polymerization temperatures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Mistra Foundation (the “STEPS” project, no. 2016/1489), the Crafoord Foundation (no. 20160774 and 20180939), and the Royal Physiographic Society in Lund. NilsonGroup AB is acknowledged for the kind financial support. We thank Åsa Halldén Björklund and Linda Zellner from Perstorp AB for valuable discussions and supplying the bio-based pentaerythritol, Lars J. Nilsson for assisting the LCA analysis, Bartosz Schmidt for the SEC analysis of the poly(urethane-urea)s, Sofia Essén for the mass spectrometry measurements, and John P. Jensen from Nordzucker Technology for supplying fructose.References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- J. Tomaszewska, D. Bieliński, M. Binczarski, J. Berlowska, P. Dziugan, J. Piotrowski, A. Stanishevskye and I. A. Witońska, RSC Adv., 2018, 8, 3161–3177 RSC.
- X. Ge, S. Zhang, X. Chen, X. Liu and C. Qian, Green Chem., 2019, 21, 2771–2776 RSC.
- K. Neimert-andersson, S. Sauer, O. Panknin, T. Borg and E. So, J. Org. Chem., 2006, 71, 3623–3626 CrossRef CAS PubMed.
- I. La, S. Sproules and E. R. Draper, ChemNanoMat, 2018, 4, 776–780 CrossRef.
- J. A. Galbis, M. D. G. Garc, M. V. De Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- W. C. Shearouse, L. M. Lillie, T. M. Reineke and W. B. Tolman, ACS Macro Lett., 2015, 4, 284–288 CrossRef CAS.
- S. M. McKenna, P. Mines, P. Law, K. Kovacs-Schreiner, W. R. Birmingham, N. J. Turner, S. Leimkühler and A. J. Carnell, Green Chem., 2017, 19, 4660–4665 RSC.
- T. Gao, T. Gao, W. Fang and Q. Cao, Mol. Catal., 2017, 439, 171–179 CrossRef CAS.
- T. Pan, J. Deng, Q. Xu, Y. Zuo, Q. X. Guo and Y. Fu, ChemSusChem, 2013, 6, 47–50 CrossRef CAS PubMed.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- A. Gandini, D. Coelho, M. Gomes, B. Reis and A. Silvestre, J. Mater. Chem., 2009, 19, 8656–8664 RSC.
- G. Z. Papageorgiou, V. Tsanaktsis, D. G. Papageorgiou, K. Chrissafis, S. Exarhopoulos and D. N. Bikiaris, Eur. Polym. J., 2015, 67, 383–396 CrossRef CAS.
- J. G. Rosenboom, D. K. Hohl, P. Fleckenstein, G. Storti and M. Morbidelli, Nat. Commun., 2018, 9, 2701 CrossRef PubMed.
- S. K. Burgess, O. Karvan, J. R. Johnson, R. M. Kriegel and W. J. Koros, Polymer, 2014, 55, 4748–4756 CrossRef CAS.
- S. K. Burgess, R. M. Kriegel and W. J. Koros, Macromolecules, 2015, 48, 2184–2193 CrossRef CAS.
- N. Hernández, R. C. Williams and E. W. Cochran, Org. Biomol. Chem., 2014, 12, 2834–2849 RSC.
- N. Kasmi, M. Majdoub, G. Z. Papageorgiou and D. N. Bikiaris, Polym. Degrad. Stab., 2018, 152, 177–190 CrossRef CAS.
- A. Celli, P. Marchese, L. Sisti, D. Dumand and G. Totaro, Polym. Int., 2013, 62, 1210–1217 CrossRef CAS.
- Y. Hu, Z. Zhao, Y. Liu, G. Li, A. Wang, Y. Cong, T. Zhang, F. Wang and N. Li, Angew. Chem., Int. Ed., 2018, 57, 6901–6905 CrossRef CAS PubMed.
- T. Chen, W. Zhang and J. Zhang, Polym. Degrad. Stab., 2015, 120, 232–243 CrossRef CAS.
- D. R. Kelsey, B. M. Scardino, J. S. Grebowicz and H. H. Chuah, Macromolecules, 2000, 33, 5810–5818 CrossRef CAS.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2019, 1–18 Search PubMed.
- A. Hufendiek, S. Lingier and F. E. Du Prez, Polym. Chem., 2019, 10, 9–33 RSC.
- H. T. H. Nguyen, P. Qi, M. Rostagno, A. Feteha and S. A. Miller, J. Mater. Chem. A, 2018, 6, 9298–9331 RSC.
- S. Lingier, Y. Spiesschaert, B. Dhanis, S. De Wildeman and F. E. Du Prez, Macromolecules, 2017, 50, 5346–5352 CrossRef CAS.
- G.-H. Choi, D. Y. Hwang and D. H. Suh, Macromolecules, 2015, 48, 6839–6845 CrossRef CAS.
- P. Wang, C. R. Arza and B. Zhang, Polym. Chem., 2018, 9, 4706–4710 RSC.
- S. Curia, A. Biundo, I. Fischer, V. Braunschmid, M. G. Georg and J. F. Stanzione, ChemSusChem, 2018, 11, 2529–2539 CrossRef CAS PubMed.
- C. Gioia, M. B. Banella, P. Marchese, M. Vannini, M. Colonna and A. Celli, Polym. Chem., 2016, 7, 5396–5406 RSC.
- M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol and B. Boutevin, Green Chem., 2014, 16, 1987–1998 RSC.
- S. Thiyagarajan, J. Wu, R. J. I. Knoop, J. Van Haveren, M. Lutz and D. S. Van Es, RSC Adv., 2014, 4, 47937–47950 RSC.
- D. Juais, A. F. Naves, C. Li, R. A. Gross and L. H. Catalani, Macromolecules, 2010, 43, 10315–10319 CrossRef CAS.
- I. S. Ristić, N. Vukić, S. Cakić, V. Simendić, O. Ristić and J. Budinski-Simendić, J. Polym. Environ., 2012, 20, 519–527 CrossRef.
- W. J. Yoon, K. S. Oh, J. M. Koo, J. R. Kim, K. J. Lee and S. S. Im, Macromolecules, 2013, 46, 2930–2940 CrossRef CAS.
- A. Saadaoui, R. Medimagh, S. Marque, D. Prim, H. Casabianca and M. S. Zina, Des. Monomers Polym., 2017, 20, 221–223 CrossRef CAS PubMed.
- H. Blache, F. Méchin, A. Rousseau, É. Fleury, J. Pascault, P. Alcou, N. Jacquel and R. Saint-loup, Ind. Crops Prod., 2018, 121, 303–312 CrossRef CAS.
- R. Marín, A. Alla, A. Martínez de Ilarduya and S. Muñoz-Guerra, J. Appl. Polym. Sci., 2011, 123, 986–994 CrossRef.
- F. Zamora, E. Benito, M. D. G. Garcı and J. A. Galbis, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4638–4646 CrossRef.
- C. Lavilla, A. M. De Ilarduya, A. Alla, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, Macromolecules, 2012, 45, 8257–8266 CrossRef CAS.
- C. Lavilla, A. Alla, A. Martínez De Ilarduya and S. Muñoz-Guerra, Biomacromolecules, 2013, 14, 781–793 CrossRef CAS PubMed.
- A. Díaz, R. Katsarava and J. Puiggalí, Int. J. Mol. Sci., 2014, 15, 7064–7123 CrossRef PubMed.
- B. Begines, F. Zamora, M. V. de Paz, I. Roffé, M. Mancera and J. A. Galbis, J. Renewable Mater., 2013, 1, 212–221 CrossRef CAS.
- C. Lavilla, A. Alla, A. M. De Ilarduya, E. Benito, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1591–1604 CrossRef CAS.
- C. Lavilla, A. Alla, A. Martínez De Ilarduya, E. Benito, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, Biomacromolecules, 2011, 12, 2642–2652 CrossRef CAS PubMed.
- C. Japu, A. M. De Ilarduya, A. Alla and S. Muñoz-guerra, Polymer, 2013, 54, 1573–1582 CrossRef CAS.
- C. Japu, A. M. De Ilarduya, A. Alla and S. Muñoz-guerra, Polymer, 2014, 55, 2294–2304 CrossRef CAS.
- C. Lavilla, E. Gubbels, A. Alla, B. A. J. Noordover and C. E. Koning, Green Chem., 2014, 16, 1789–1798 RSC.
- J. Besset, A. T. Lonnecker, J. M. Stre and K. L. Wooley, Biomacromolecules, 2011, 12, 2512–2517 CrossRef PubMed.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- J. Thiem and H. Luders, Polym. Bull., 1984, 11, 365–369 CrossRef CAS.
- W. A. P. Black, E. T. Dewar and J. B. Hare, J. Chem. Soc., 1963, 5724–5727 RSC.
- C. Japu, A. Alla, A. Martínez de Ilarduya, M. G. García-Martín, E. Benito, J. A. Galbis and S. Muñoz-Guerra, Polym. Chem., 2012, 3, 2092–2101 RSC.
- Y. Xu, X. Jia, J. Ma, J. Gao, F. Xia, X. Li and J. Xu, Green Chem., 2018, 20, 2697–2701 RSC.
- N. Yoshida, N. Kasuya, N. Haga and K. Fukuda, Polym. J., 2008, 40, 1164–1169 CrossRef CAS.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- F. Hu, J. J. La Scala, J. M. Sadler and G. R. Palmese, Macromolecules, 2014, 47, 3332–3342 CrossRef CAS.
- J. A. Moore and J. E. Kelly, Macromolecules, 1978, 11, 568–573 CrossRef CAS.
- M. Gomes, A. Gandini, A. Silvestre and B. Reis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3759–3768 CrossRef CAS.
- T. Ikezaki, R. Matsuoka, K. Hatanaka and N. Yoshie, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 216–222 CrossRef CAS.
- R. M. West, Z. Y. Liu, M. Peter and J. A. Dumesic, ChemSusChem, 2008, 1, 417–424 CrossRef CAS PubMed.
- P. V. Bonsignore, WO2008022287, 2008.
- S. Pyo, M. Sayed and R. Hatti-kaul, Org. Process Res. Dev., 2019, 23, 952–960 CrossRef CAS.
- ISO 14044:2006 – Environmental management – Life cycle assessment – Requirements and guidelines, 2006.
- ISO 14040:2006 – Environmental management – Life cycle assessment – Principles and framework, 2006.
- IPPC Intergovernmental Panel on Climate, IPCC Fourth Assessment Report: Climate Change, 2007 Search PubMed.
- M. N. García González, P. Börjesson, M. Levi and S. Turri, J. Polym. Environ., 2018, 26, 3626–3637 CrossRef.
- Perstorp AB, Proof of Sustainability for Perstorp's Pro-Environment Polyols, 2007 Search PubMed.
- H. R. Shaterian and F. Rigi, Chin. J. Chem., 2012, 30, 695–698 CrossRef CAS.
- C. Ummadisetti, B. N. P. Rachapudi and L. A. P. D. Bethala, Eur. J. Chem., 2014, 5, 536–540 CrossRef.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Green Chem., 2006, 8, 701–709 RSC.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef CAS.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- F. Brentrup, J. Küsters, H. Kuhlmann and J. Lammel, Eur. J. Agron., 2004, 20, 247–264 CrossRef.
- DuPont Tale and Lyle Bio Products (DT&L), Life Cycle Analysis Overview-Susterra ® Propanediol, 2009 Search PubMed.
- Q. Wang, J. Cai, L. Zhang, M. Xu, H. Cheng, C. C. Han, S. Kuga, J. Xiao and R. Xiao, J. Mater. Chem. A, 2013, 1, 6678 RSC.
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS.
- C. Lavilla, A. Alla, A. Martínez De Ilarduya, E. Benito, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3393–3406 CrossRef CAS.
- C. Lavilla, A. Martínez De Ilarduya, A. Alla and S. Muñoz-Guerra, Polym. Chem., 2013, 4, 282–289 RSC.
- V. M. De Benedictis, G. Soloperto and C. Demitri, Polymers, 2016, 8, 1–17 CrossRef PubMed.
- A. B. Cook, R. Barbey, J. A. Burns and S. Perrier, Macromolecules, 2016, 49, 1296–1304 CrossRef CAS.
- D. Hölter, A. Burgath and H. Frey, Acta Polym., 1997, 48, 30–35 CrossRef.
- V. T. Wyatt and G. D. Strahan, Polymers, 2012, 4, 396–407 CrossRef.
- C. J. Hawker, R. Lee and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4583–4588 CrossRef CAS.
- M. Choura, N. M. Belgacem and A. Gandini, Macromolecules, 1996, 29, 3839–3850 CrossRef CAS.
- S. Sun, R. Yang, P. Sun, C. Ma and J. Chen, Energy, 2017, 135, 577–584 CrossRef CAS.
- T. S. Jin, H. X. Wang, K. F. Wang and T. S. Li, Synth. Commun., 2004, 34, 2993–2999 CrossRef CAS.
- Z. Chen, J. N. Hay and M. J. Jenkins, Eur. Polym. J., 2012, 48, 1586–1610 CrossRef CAS.
- Z. Chen, J. N. Hay and M. J. Jenkins, Thermochim. Acta, 2013, 552, 123–130 CrossRef CAS.
- U. Chudasama and S. Ghodke, IJATES, 2015, 2, 340–353 Search PubMed.
- H. Ohtani, T. Kimura and S. Tsuge, Anal. Sci., 1986, 2, 179–182 CrossRef CAS.
- H. Stutz, K.-H. Illers and J. Mertes, J. Polym. Sci., Part B: Polym. Phys., 1990, 28, 1483–1498 CrossRef CAS.
- C. Japu, A. M. De Ilarduya, A. Alla, M. G. García-martín, J. A. Galbis and S. Muñoz-guerra, Macromol. Chem. Phys., 2014, 215, 2048–2059 CrossRef CAS.
- I. H. Hall and B. A. Ibrahim, Polymer, 1982, 23, 805–816 CrossRef CAS.
- K. Inomata and S. Sasaki, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 83–92 CrossRef CAS.
- T. J. Touchet and E. M. Cosgriff-Hernandez, in Advances in Polyurethane Biomaterials, Elsevier Ltd, 2016, pp. 1–22 Search PubMed.
- M. Al-Hashimi, Y. Han, J. Smith, H. S. Bazzi, S. Y. A. Alqaradawi, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Chem. Sci., 2016, 7, 1093–1099 RSC.
- F. P. V. Koch, P. Smith and M. Heeney, J. Am. Chem. Soc., 2013, 135, 13695–13698 CrossRef CAS PubMed.
- J. Liu, R. S. Loewe and R. D. McCullough, Macromolecules, 1999, 32, 5777–5785 CrossRef CAS.
- M. Rehahn, A. Schlüter and G. Wegner, Makromol. Chem., 1990, 191, 1991–2003 CrossRef CAS.
- W.-Y. Chang, Y.-W. Pan, C.-N. Chuang, J.-J. Guo, S.-H. Chen, C.-K. Wang and K.-H. Hsieh, J. Polym. Res., 2015, 22, 243 CrossRef.
- V. Chiaradia, A. Valério, P. E. Feuser, D. de Oliveira, P. H. H. Araújo and C. Sayer, Colloids Surf., A, 2015, 482, 596–603 CrossRef CAS.
- Y. Zhang, Z. Xia, H. Huang and H. Chen, J. Anal. Appl. Pyrolysis, 2009, 84, 89–94 CrossRef CAS.
- D. van Krevelen, Polymer, 1975, 16, 615–620 CrossRef CAS.
- C. R. Arza and B. Zhang, ACS Omega, 2019, 4, 15012–15021 CrossRef CAS PubMed.
- T. S. Hansen, K. Barta, P. T. Anastas, P. C. Ford and A. Riisager, Green Chem., 2012, 14, 2457–2461 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03055g |
| This journal is © The Royal Society of Chemistry 2019 |