
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic oxidative desulfurization of a 4,6-DMDBT containing model fuel by metal-free activated carbons: the key role of surface chemistry†
Zoi Christina
Kampouraki
,
Dimitrios A.
Giannakoudakis
 *,
Konstantinos S.
Triantafyllidis
and
Eleni A.
Deliyanni
*
*,
Konstantinos S.
Triantafyllidis
and
Eleni A.
Deliyanni
*
Laboratory of Chemical and Environmental Technology, Department of Chemistry, Aristotle University of Thessaloniki, GR-541 24 Thessaloniki, Greece. E-mail: DaGchem@gmail.com; lenadj@chem.auth.gr
First published on 25th October 2019
Abstract
Commercial micro/mesoporous activated carbons were utilized as metal-free catalysts for the desulfurization of a model fuel, i.e. 4,6-dimethyldibenzothiophene (4,6-DMDBT) in hexadecane under ambient conditions. Both adsorption and catalytic oxidation were investigated as means of 4,6-DMDBT removal. The effect of chemical modification/oxidation of the carbon surface via treatment with two different acids (HNO3 or H2SO4) aiming to introduce additional functional groups was also investigated. The catalysts were characterized by FT-IR spectroscopy, N2 porosimetry, potentiometric titration, Boehm titration, and SEM-EDX, while adsorption and catalytic oxidation activity towards sufloxides and sulfones were assessed by GC-MS and UV-Vis analysis. The surface chemistry of the carbons, expressed by the density of the acidic functional groups, was found to be the most critical parameter with regard to adsorption or to catalytic oxidative performance. The surface modification of carbons by oxidation had a positive impact on the catalytic oxidation activity, leading to a 100% conversion of 4,6-DMDBT towards the corresponding sulfoxide and sulfone, compared to 67% with the parent non-oxidized carbon. Reusability tests showed that the oxidation activity of the carbons can be maintained for at least 5 cycles.
1. Introduction
Nowadays, great interest has been focused on the mitigation of sulfur compounds in fuels in order to comply with stringent regulations, as they cause environmental problems and human health issues.1–3 According to the Environmental Protection Agency of USA (EPA), the admissible sulfur concentration in diesel fuel has been 15 ppm since 2006.1,4 The European Parliament and the Council of the European Union have established in 2009 an even lower concentration of sulfur in fuels, 10 ppm.5The industrial process used for decades to remove sulfur from fuels is hydrodesulfurization (HDS), a catalytic process in which organic sulfur compounds are converted to hydrogen sulfide and sulfur-free hydrocarbons by reaction with hydrogen over CoMo/Al2O3 or NiMo/Al2O3 catalysts.6,7 An important drawback of HDS is the required high hydrogen pressure,4 leading to a very costly process, especially when deep desulfurization is aimed.8,9 Furthermore, HDS is not effective in the removal of sulfur heterocyclic hydrocarbons such as dibenzothiophene (DBT) and its derivatives, especially 4,6-dimethyldibenzothiophene (4,6-DMDBT), which is not so reactive due to steric hindrance effects.4,9–13 Within this context, the development of more efficient and greener/sustainable desulfurization methods which have the potential to produce extremely ultra-low-sulfur fuels, so as to replace or complement the HDS process, is of great importance. Adsorption,14,15 biodesulfurization,16,17 extraction by ionic-liquids,18 photocatalytic oxidation19 and oxidative desulfurization (ODS)20,21 are some new processes that have been introduced for efficient fuel desulfurization. Among these approaches, oxidative desulfurization constitutes a promising method, as it is simple and of higher efficiency compared to HDS.22,23 In addition, 4,6-DMDBT is expected to exhibit higher reactivity in ODS.24,25
The ODS method involves the oxidation of the sulfur-containing compounds, followed by the extraction/removal of the oxidized products from the fuel due to their polarity. In the ODS process, sulfur-containing compounds are oxidized using selective oxidants such as nitric acid,26 nitrogen oxides,27 organic hydroperoxides,28 hydrogen peroxide24,29,30 or/and ozone31 in the presence of a catalyst to produce sulfone compounds which can be extracted preferably due to their increased polarity.32 The most commonly used oxidant is hydrogen peroxide (H2O2), due to the fact that it is environmentally friendly, it has low cost and it is commercially available.29 Various homogeneous and heterogeneous catalysts like polyoxometalates (POMs),33 mono- or bimetallic alloys,34 organic acids,35 ionic liquids,18,36 multi-walled carbon nanotubes37 and activated carbons38 have been shown to be active in the desulfurization reactions. In particular, the latter ones have been previously utilized as supports for active phases like metal oxide, metal or bimetallic nanoparticles,39–43 as for example in the deposition of molybdenum cobalt nanocatalysts on a carbon support which led to 34% increment of dibenzothiophene hydrodesulfurization from a model oil, compared to pure MoCo.44
Even though carbons are often used as catalyst supports, they can also be utilized as catalysts on their own, because of their physicochemical properties, such as the rich surface chemistry due to the presence of oxygen containing functional groups, the large micro/mesopore surface area, and their relatively stable structure and morphology at high temperatures and/or various liquid reaction media.45–47 One of the most known reactions catalyzed by carbonaceous materials, the decomposition of hydrogen peroxide, is highly dependent on the nature of the surface functional groups. Chemical reactions on carbon's surfaces usually follow a free radical mechanism.48,49 The produced radicals are stabilized on the surface of the carbon so that they can act as adsorption/oxidation sites of sulfur compounds.48,50
Despite the wide application of carbons in environment related reactions and processes, limited investigation has been devoted to the use of activated carbons as metal-free catalysts in desulfurization processes and especially for ODS of 4,6-DMDBT. In most cases, the carbons were studied as adsorbents for the removal of smaller DBT or other derivatives.22,50 Considering that the production of carbons with different morphology, texture and surface properties can be achieved by utilizing the most abundant and renewable source, biomass,51–54 the use of biomass derived carbocatalysts will be of great importance towards a sustainable future. In the present study, we examined five activated carbons, with or without prior treatment with acids, for the oxidation of 4,6-DMDBT within the process of fuel desulfurization. Emphasis was given to the investigation of the role of carbon's textural characteristics and surface chemistry features towards maximization of desulfurization activity.
2. Experimental methods
2.1 Materials and reagents
Five commercial activated carbons, obtained from Cabot Norit activated carbon and CPL activated carbons, were chosen to be tested as carbocatalysts: (i) Norit SX-PLUS, (ii) Norit SAE-SUPER, (iii) Norit D-10, (iv) Norit SAE-2, and (v) CPL. 4,6-DMDBT (4,6-dimethyldibenzothiophene), hexadecane, commercially available 30% wt% H2O2, methanol, HNO3, and H2SO4 99.999% purity were purchased from Sigma Aldrich.2.2 Modification of the carbon catalyst
The SX PLUS carbon was chemically treated either with HNO3 or H2SO4, targeting to modify its surface chemistry.2.3. Materials characterization methods
where CA and CB (mol L−1) are the acid (HCl) and base (NaOH) concentrations (mol L−1), respectively, [H+] and [OH−] are the equilibrium concentrations of these ions (mol L−1) and W is the concentration of the solid (g L−1).

2.4. Adsorption and catalytic oxidation experiments
where C0 and Ce (ppmw of sulfur) are the initial and the equilibrium concentrations, respectively.61

Kinetic experiments were performed by dispersing 0.01 g L−1 of carbon in 20 mL of 4,6-DMDBT solution in hexadecane (C0 = 20 ppmw of S), under stirring at 60 °C for different intervals of time.62 For each separate experiment, filtration through a 50 μm pore size membrane was performed and the 4,6-DMDBT concentration was measured by using a UV spectrophotometer as mentioned above.
The experimental results were fitted by Lagergren's pseudo-first order kinetic model,63 given by the equation
ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − k1t qe − k1t |

The liquid products were also analyzed by GC-MS on a 7890A/5975C system by Agilent (electron energy 70 eV, helium flow rate: 0.7 cc min−1, Column: HP-5MS 30 m × 0.25 mm ID × 0.25 μm). Identification of mass spectra peaks was performed by the use of the scientific library NIST11s. Extraction solutions of the spent samples were also analyzed with the same instrument.
3. Results and discussion
3.1. Characterization of activated carbon
The porosity characteristics and surface chemical features of the different activated carbons were initially determined and correlated with the adsorption and catalytic oxidation of 4,6-DMDBT. The N2 adsorption–desorption isotherms of all the carbon samples (Fig. s1a†) showed a bimodal type of shape based on IUPAC classification.65 At low relative pressures, there is a clear typical type I adsorption isotherm for all parent commercial carbons, revealing the abundance of micropores. At intermediate and higher relative pressures, the adsorption isotherms tend to adopt a type IV shape with the corresponding hysteresis loop, which was more pronounced for the CPL and SAE SUPER carbons, due to capillary condensation in narrow slit-shaped mesopores.66,67 The pore size distribution curves (Fig. s1b†) revealed that there is a relatively narrow distribution in the low micropore range, i.e. <1 nm, and a broader distribution for pores >1 nm, except for the case of CPL which showed a relatively narrow distribution between 1 and 2 nm. The BET surface area values and porosity parameters for the activated carbons are presented in Table s1.† It is seen that CPL (1702 m2 g−1) presented the highest surface area, followed by SX PLUS (1280 m2 g−1), SAE SUPER (1182 m2 g−1), and SAE2 (892 m2 g−1), while D10 showed a relatively low surface area (515 m2 g−1). The total pore volumes (Table s1†) were found to follow the same trend as the SSABET. The values of the micro- and meso-pore volume are shown in Fig. 1a (also listed in Table s1†). CPL revealed also the highest ratio of mesopore to micropore or total pore volume, while SX PLUS the lowest. Considering that the molecular size of 4,6-DMDBT is 0.59 × 0.89 nm,4 it could be expected that micropores of ≤1 nm, as well as bigger micropores and small mesopores (up to ca. 2–3 nm), would play an important role regarding the adsorption and oxidation activity of the carbons.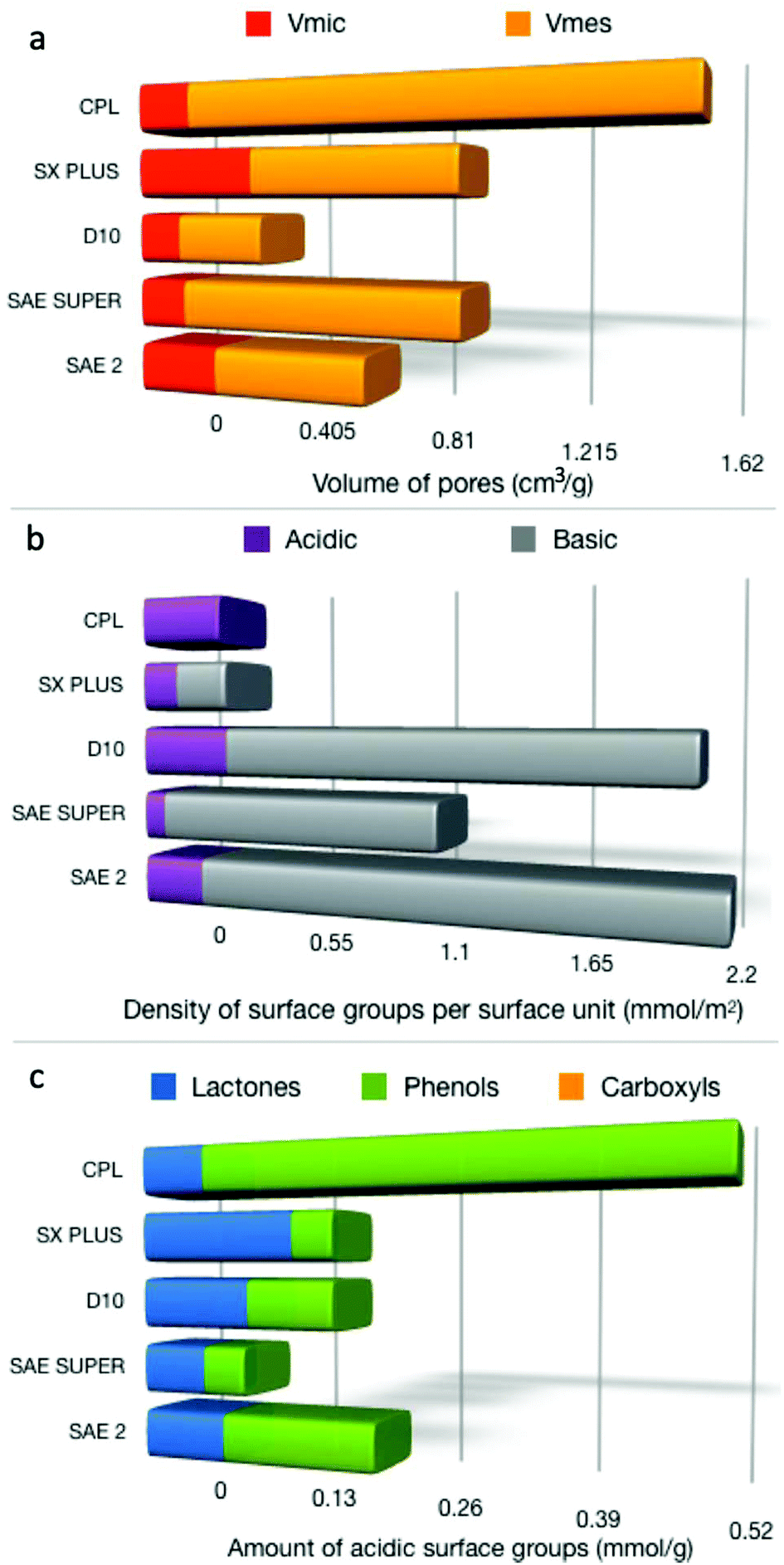 | ||
| Fig. 1 Micro- and mesopore volumes (a), the density of acidic and basic surface functional groups per SBET (b), and the distribution of the acidic groups (c). | ||
The surface chemistry characteristics of the activated carbons are reported in Table s2.† From the surface pH measurements, it can be seen that D10, SAE SUPER, and SAE 2 activated carbons possess a relatively basic surface, SX PLUS almost neutral, while CPL exhibits a more acidic nature. The results from the point of zero charge (pzc) determination (Fig. s2†) are in-line with the proton binding curve potentiometric titration results (Fig. s3†). The most interesting observation can be derived from the ratio of acidic to basic groups, with CPL having only acidic groups, followed by SX PLUS (0.70), while SAE SUPER has the lowest ratio (0.08). The calculated density of acidic surface functional groups (dASFG) and of the basic (dBSFG) per surface area is presented in Fig. 1b. The Boehm titration results revealed mainly two kinds of oxygen functional groups on carbon's surface, i.e. lactonic and phenolic, while no carboxyl groups were detected. The amount of each type of group (mmol g−1) as well as their density per specific surface area unit (μmol m−2) is collected in Table s2.† SX PLUS shows the highest number of lactones, while SAE2 the highest amount of phenol groups.
3.2. Desulfurization – adsorption and catalytic oxidation results for the commercial carbons
The 4,6 DMDBT (20 ppmw of sulfur) adsorption results in hexadecane as a solvent in the dark, at 60 °C, without H2O2 are presented in Fig. 2a. SX PLUS showed by far the best performance, achieving 60% removal while the lowest adsorptive capability was observed for the carbon with the lowest surface area and pore volume, D10. On the other hand, the activated carbon CPL with the highest BET surface area exhibited a moderate adsorption efficiency. Furthermore, SX PLUS and SAE SUPER have a similar surface area, but the former showed substantially higher adsorption of 4,6-DMDBT. It can thus be concluded that the BET surface area of the carbons is not the sole critical parameter that would define the adsorption performance. If one considers the correlation between the size of 4,6-DMDBT and the size of the pores, it appears that the carbons with a higher abundance of smaller micropores, i.e. with a size of ≤1 nm, such as SX-PLUS, SAE-2 and SAE SUPER, are more efficient possibly due to increased confinement effects due to size similarity. | ||
| Fig. 2 Adsorptive and catalytic removal (conversion of 4,6-DMDBT) (a), and correlation between the adsorption capacity and the density of the acidic surface functional groups per unit surface area (dASFG) of the activated carbons (b); adsorption tests: 25 mg carbon, 10 ml hexadecane, 20 ppmw of sulfur, T = 60 °C, t = 24 h, catalytic tests: the same as the adsorption tests plus 1 ml H2O2 solution. | ||
With regard to the effect of the functional groups and correlation with the surface pH, SX PLUS and CPL provided a neutral/slightly acidic pH, compared to SAE-2 and SAE SUPER which were basic, and furthermore, they exhibited the highest ratio of acidic to basic surface groups, i.e. 1.0 for CPL and 0.7 for SX PLUS. In order to further investigate the possible combined effects of the surface area and acidic sites, the density of the acidic surface functional groups, per surface area unit, was estimated and it was plotted against the removal efficiently (Fig. 2b). As can be seen in Fig. 2b, the maximum adsorption capacity increases with the increment of the density of acidic surface functional groups (dASFG) up to an optimum value followed by a decrement upon further increase of dASFG. This fact can be assigned to an enhanced steric effect when the active surface functional groups are close to each other, as well as to a blockage of the pores’ entrance by adsorbed molecules, hindering the penetration of other molecules towards the active adsorption sites at the interior of the pores. This behavior leads to the suggestion that the density and dispersion of the active sites may be an important parameter for the adsorption efficiency of the carbons. However, when comparing the properties and performance of SX PLUS and CPL, it can be seen that the former contains a high portion of lactones while phenolic groups are predominant in the latter (Fig. 1c). This may also lead to the assumption that the more acidic lactones compared to phenols are more prone to interact with 4,5-DMDBT, considering also the relatively (i.e. compared to pyrrole for example) moderate Lewis basicity strength and enhanced aromatic stabilization of DMDBT. Overall, it can be suggested that both the type of acidic functional group and their density are key factors towards increasing the adsorption efficiency of the carbons.
Moreover, the Langmuir68 and Freundlich69 isotherms were fitted to the experimental results, as presented in Fig. s4,† and models’ parameters are presented in Table s3.† Both models exhibit relatively good fitting indicating the initial monolayer formation of sorbed 4,6-DMDBT as well as the formation of secondary layers, due to the presence of meso/macroporosity as well as due to the inhomogeneity of the adsorption sites. The models’ parameters are presented in Table s3,† from where it is seen that SX PLUS presented the highest adsorption capacity between all carbons.
The potential of fuel desulfurization via catalytic oxidation was investigated by the use of H2O2 as the oxidation agent with the parent commercial activated carbons as catalysts (Fig. 2a). Control oxidation experiments were initially performed without any activated carbon, showing negligible conversion of 4,6-DMDBT. The catalytic oxidation tests (use of H2O2 in the dark) supported further the superior performance of SX PLUS, with the difference from the second performing sample (SAE 2) to be more pronounced in comparison with the adsorption tests. The 60% adsorptive removal of 4,6-DMDBT for SX PLUS was increased by 28% for the catalytic removal with the same carbon in the presence of H2O2 (with the rest of the conditions being the same), showing the clear additional benefits of oxidation. An interesting observation is related to the observed decrease of 4,6-DMDBT removal by adding H2O2 for the three basic carbon catalysts, i.e. D-10, SAE 2 and SAE SUPER (Fig. 2a).
3.3. Effect of oxidation reaction parameters
The effect of different parameters i.e. catalyst's dose, reaction temperature, and H2O2 relative amount, on the catalytic oxidation was also studied, using SX PLUS being identified above as the most efficient. As mentioned above, no conversion of 4,6-DMDBT was observed in the presence of H2O2 without using carbons at 60 °C, showing that the activated carbon actually acts as a catalyst in the reaction.70 The 4,6-DMDBT removal efficiency with different amounts of catalysts is shown in Fig. 3a. It is observed that by increasing the amount of carbocatalyst, the removal extent is enhanced. More than 90% removal is achieved by a relatively low amount of carbon (100 mg). 25 mg can be assumed as the optimum one, since the removal extent by a further increase of the carbon amount is minimal. | ||
| Fig. 3 The effect of the amount of the catalyst (SX PLUS) on 4,6-DMDBT removal; T = 60 °C, t = 24 h, 20 ppmw of sulfur in 10 mL of hexadecane + 1 mL of H2O2 (a), the effect of temperature on 4,6-DMDBT removal; 20 ppmw of sulfur in 10 mL of hexadecane + 1 mL of H2O2, t = 24 h (b), and the effect of H2O2; 25 mg of catalyst, 20 ppmw of sulfur in 10 mL of hexadecane + x mL of H2O2 + y mL of water where x + y = 1 mL, T = 60 °C, t = 24 h (c). | ||
The effect of temperature was also investigated by carrying out catalytic experiments at three different temperatures, 25, 60 and 90 °C and the results are shown in Fig. 3b. From 25 to 60 °C, a clear improvement in the removal efficiency can be observed from ∼39% to ∼76%. This can be linked to a variety of factors, like activation of the acidic surface functional groups and increase of the thermodynamics of the system, resulting in enhanced mass transfer (diffusion inside the pores towards the active sites) and decrement of adsorption/decomposition activation energy. In contrast, a further temperature rise to 90 °C had a negative effect, decreasing the removal performance to ∼47%. This can be assigned to an increase of the hydrogen peroxide decomposition rate, which may also lead to undesirable secondary species other than the targeted hydroxyl radicals. Besides, oxidation of useful components in the fuel may also occur at elevated temperatures. This result is in agreement with other research results reported in the literature for the same desulfurization via adsorption/oxidation methods.30,70–72 In view of these results, the optimum reaction temperature was set at 60 °C.
The results of the effect of the amount of H2O2 on the removal of 4,6-DMDBT are shown in Fig. 3c. It is observed that by increasing the volume ratio, and therefore the molar ratio of H2O2, the removal percentage of 4,6-DMDBT is enhanced during the catalytic oxidation. The maximum removal was for 1 mL of H2O2 solution (25 mg SX PLUS, 20 ppmw of sulfur PLUS) and the results are consistent with the literature.70 An important conclusion was gained for the test in which only one 1 mL of water was used (no H2O2 addition); the removal efficiency was limited to ∼44%, a value even lower than the maximum adsorptive removal without hydrogen peroxide (adsorption tests, Fig. 2a). This is strong evidence that the presence of water has a negative impact on the removal of 4,6-DMDBT from hexadecane (or a real fuel) by blocking/hindering the active adsorptive/reactive centers.
The above experiments (referring to the results of Fig. 3) were also carried out in the dark in order to explore if the presence of ambient light has an effect of the adsorption/oxidation capability. The catalytic oxidation removal under ambient light exposure was 10% higher than that in the dark. This can be linked either to the fact that the light plays a positive role in the reactive and/or adsorptive sites or that light power can enhance the formation of reactive oxygen species during the decomposition of H2O2 on the carbon's surface. These two aspects will be discussed herein after in more detail.
3.4. Reusability cycles
To investigate the possibility of regeneration and reusability of the catalyst, the carbocatalyst SX PLUS was washed with water and methanol (dried for 2 h at 60 °C) after the oxidation experiments, in order to remove any product or reactant from its surface. From the results presented in Fig. 4, a small decrease of 4.5% in the removal efficiency occurred after the first cycle of reuse, while after 5 cycles the removal efficiency loss was not higher than 10.8%. It can be concluded that the catalytically active sites are maintained during reaction/regeneration. It is worth mentioning that the removal extent (∼63%) even after 5 cycles is higher than the maximum adsorptive capability of the rest of the commercial carbons tested (Fig. 2a).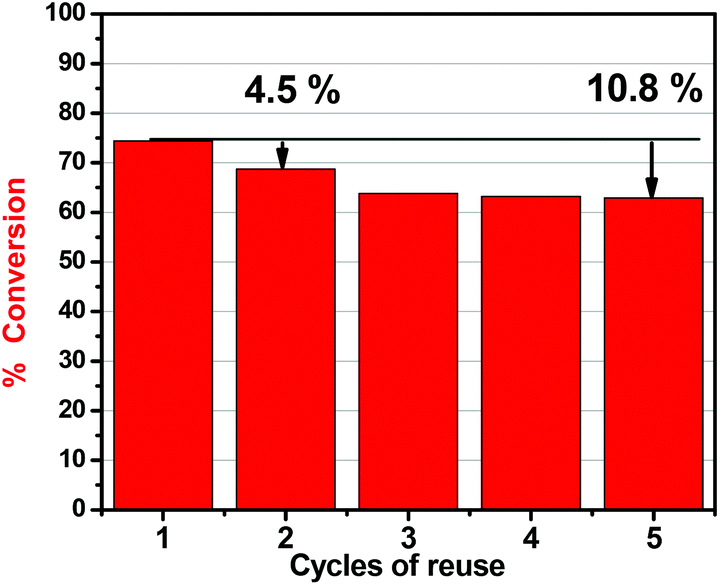 | ||
| Fig. 4 Reusability cycles of the catalytic desulfurization capability of SX PLUS; 25 mg carbon, 10 ml hexadecane, 20 ppmw of sulfur, 1 ml H2O2 solution, T = 60 °C, t = 24 h. | ||
3.5. Adsorptive/catalytic performance of oxidized carbons
Taking into account the high adsorption and catalytic oxidation efficiency of the parent commercial SX PLUS carbon, this carbon was further used to investigate the effect of chemical modification of its surface by increasing the density of the acidic surface functional groups. To this end, two different counterparts of modified SX PLUS were prepared. The first one was obtained after treatment with HNO3, herein-after referring to as SX PLUS-Nox, while for the second one H2SO4 was used, with the prepared sample named SX PLUS-Sox. The catalytic oxidation results of the HNO3 and H2SO4 oxidized counterparts, as well as of the adsorption data (included for the sake of comparison), are presented in Fig. 5. A significant increase of the removal efficiency via catalytic oxidation can be observed for both treated SX PLUS samples, reaching 100% removal, being an additional indication of the positive effect of the surface acidity on the catalytic oxidation by activated carbons as metal-free catalysts. A small positive effect can also be identified in the adsorption removal, verifying further the enhanced interaction between 4,6-DMDBT and the surface acidic sites of carbon. | ||
| Fig. 5 Comparison of 4,6-DMDBT adsorptive and catalytic oxidation removal for the parent commercial activated carbon SX PLUS and its counterparts oxidized by HNO3 (SX PLUS-Nox) and H2SO4 (SX PLUS-Sox); adsorption tests: 25 mg carbon, 10 ml hexadecane, 20 ppmw of sulfur, T = 60 °C, t = 24 h, catalytic tests: the same as the adsorption tests plus 1 ml H2O2 solution. | ||
3.6. Characterization of oxidized carbons
The porosity characteristics of the parent and oxidized SX PLUS carbons are shown in Table 1. Both the BET surface area and total pore volume were decreased upon oxidation. The decrement of the SBET was slightly more pronounced after treatment with HNO3 (−27%) rather than after H2SO4 treatment (−22%). Similar trends of decrease upon oxidation were found for the pore volumes. It is worth pointing out that although the oxidized counterparts showed inferior porosity characteristics in comparison with the parent SX PLUS, their adsorptive performance was superior, a fact that confirms a more crucial role of carbon's surface chemistry against porosity.| Activated carbons | S BET, m2 g−1 | V tot, cm3 g−1 | V mic, cm3 g−1 | V mes, cm3 g−1 |
|---|---|---|---|---|
| SX PLUS | 1280 | 0.95 | 0.35 | 0.60 |
| SX PLUS-Nox | 929 (−27%) | 0.67 (−30%) | 0.25 (−30%) | 0.42 (−29%) |
| SX PLUS-Sox | 1012 (−21%) | 0.74 (−22%) | 0.28 (−20%) | 0.46 (−23%) |
The surface pH as well as the amount of acidic and basic surface groups of the SX PLUS carbons is shown in Fig. 6, while the densities per surface area unit can be seen in Table s4.† The general observation is that the amount of the acidic surface groups was increased upon oxidation and the amount of the basic surface groups was decreased, thus resulting in a decrease of surface pH. The increment of the acidic groups was substantially more pronounced in the case of SX PLUS-Nox, while the decrement of the basic groups was more pronounced for SX PLUS-Sox. Boehm titration experiments (results not shown) revealed that the phenol groups were totally eliminated, lactones were increased, and carboxylic acids were formed by the oxidative treatment.
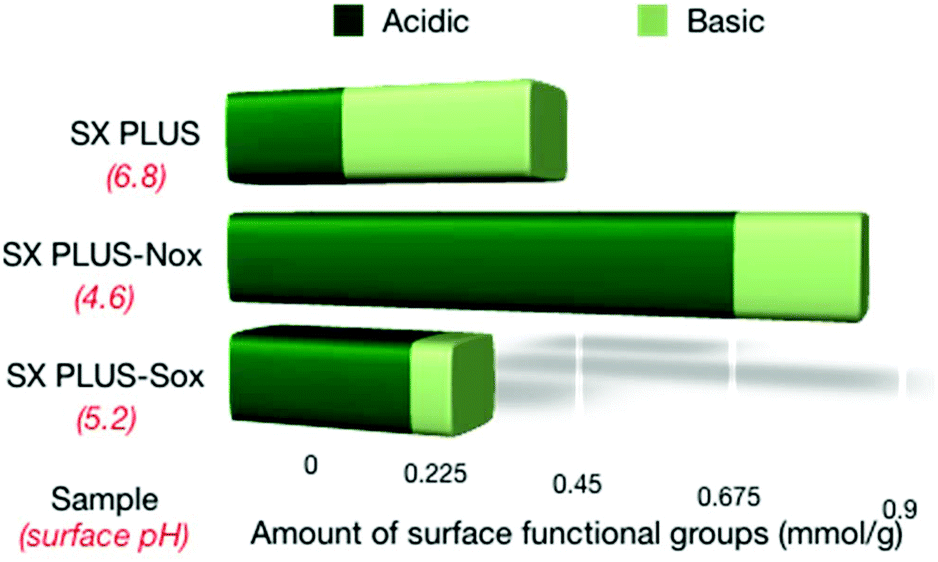 | ||
| Fig. 6 The amounts of the acidic and basic surface functional groups of the parent commercial SX PLUS and its counterparts oxidized by HNO3 and H2SO4. | ||
With regard to the correlation between adsorptive/catalytic performance and the surface acidity, the above derived volcano-type trend between adsorption of 4,6-DMDBT and the density of surface acid groups (Fig. 2b) was found valid also for the parent SX PLUS and its oxidized counterparts, since SX PLUS-Nox showed a significantly higher dASFG but a slightly lower adsorption performance compared to SX PLUS-Sox. On the other hand, in the case of catalytic oxidation performance, both treated carbons reached the maximum removal efficiency, as shown in Fig. 5, at least for the experimental conditions used in this study.
3.7. Effect of contact time – kinetics of the 4,6-DMDBT removal
In order to examine the effect of the adsorption/oxidation reaction time on the removal efficiency of the carbon catalysts, the remaining sulfur in the solution, expressed as C/C0, was plotted against the contact time and the derived curves are presented in Fig. 7. A different 4,6-DMDBT removal rate can be observed between the adsorption and the catalytic oxidation process. In the adsorption experiments, 4,6-DMDBT removal increased rapidly in the first 5–10 min followed by a gradual equilibration and a plateau after ca. 50 min, for both the parent SX PLUS and the two oxidized counterparts. The pseudo-first and the pseudo-second order kinetic models in their linear form were applied for the fitting of the experimental results. The linear fitting curves and the corresponding kinetic parameters are shown in Fig. s5 and Table s5,† respectively. The pseudo-second kinetic model found to fit better the adsorptive removal results, as concluded by the R2 values presented in Table s5,† indicative of a physical adsorption onto the carbon surface, results consistent with findings by other researchers.73,74 | ||
| Fig. 7 Kinetic experiments for the adsorptive and catalytic oxidation removal of 4,6-DMDBT with the parent commercial SX PLUS activated carbon and its oxidized counterparts. | ||
The kinetic experiments of the 4,6-DMDBT catalytic oxidation showed that 100% removal was achieved for the SX PLUS-Nox and SX PLUS-Sox carbons within 10 and 16 min, respectively. Interestingly, more than half of the 4,6-DMDBT was converted within less than 2 min. Since two separate slopes were observed (a steep one for 0 to 2 min and one for 2 to 10 or 16 min), it can be suggested that initially the catalytic oxidation/removal occurs on the large internal surface of the micropores where the majority of the active sites exist, and is being enhanced by the favorable confinement effects due to size similarity, as discussed above. As the micropores are filled by the first reactant molecules, in combination with the decrease of the concentration and abundance of H2O2, the reaction takes place at the meso/macropore and external surface of the carbons at lower rates. This theory can be supported from the fact that the catalytic oxidative removal follows both the pseudo-first and the pseudo-second order kinetics (Table s5†), although with a slightly better fitting indicative of an oxidation process.3,30,70,75
The importance of the oxidation treatment and enrichment of the carbon's surface with acidic groups can be also concluded from the fact that in the case of pristine SX PLUS, 4,6-DMDBT was not completely eliminated (Fig. 7). Taking also into account that the adsorptive performance between the parent and the oxidized SX PLUS carbons is more or less similar, then it can be further suggested that the formed acidic groups, i.e. carboxyls and lactones, have a higher impact on the oxidation mechanism of 4,6-DMDBT than on just increasing the chemical interaction/sorption.
3.8. Characterization of the carbons after reaction
In order to examine the surface chemistry alterations and the possibility of strongly sorbed reaction products on the carbon catalysts’ surface, FTIR spectra analysis was conducted (Fig. 8a–c). The comparative study of the parent and oxidized SX PLUS samples, as well as of the corresponding carbons after exposure to 4,6-DMDBT, shed light on the chemical interactions between 4,6 DMDBT and the catalysts’ surface functional groups and on the related oxidation mechanism. In all spectra, the bands presented at about 1610–1630 cm−1 can be attributed to C–C stretching vibrations of the aromatic rings. The bands at 1100–1150 cm−1 can be attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–H bonds of alcoholic, phenolic and carboxyl oxygen groups while the peak at about 1700–1725 cm−1 can be attributed to CO stretching vibrations of the carboxyl groups. For the modified activated carbons, (SX PLUS-Nox and SX PLUS-Sox) prior to the exposure to 4,6-DMDBT in hexadecane, an increase in the intensity of the bands at 1720 and 1100 cm−1 was presented, representing carboxylic acids and lactones, due to their oxidation with HNO3 or H2SO4 acids.
O and O–H bonds of alcoholic, phenolic and carboxyl oxygen groups while the peak at about 1700–1725 cm−1 can be attributed to CO stretching vibrations of the carboxyl groups. For the modified activated carbons, (SX PLUS-Nox and SX PLUS-Sox) prior to the exposure to 4,6-DMDBT in hexadecane, an increase in the intensity of the bands at 1720 and 1100 cm−1 was presented, representing carboxylic acids and lactones, due to their oxidation with HNO3 or H2SO4 acids.
 | ||
| Fig. 8 FTIR (a–c) and of EDX (d and e) spectra for the parent and oxidized SX PLUS carbon samples before and after the catalytic oxidation of 4,6-DMDBT. | ||
The bands at 1330–1370 cm−1, which appeared on the spectra for the carbons after catalytic oxidation tests and after heating to 280 °C to remove the solvent, can be attributed to the sulfur compounds formed on the carbon surface, i.e. to sulfoxides, sulfones or sulfonic acid. The sulfones are specified by absorption at 1300–1350 cm−1 and 1120–1160 cm−1. The bands at 1166, 1076 and 1020 cm−1 are characteristic of the molecular vibrations of C–S bonds. The increase in intensity of the bands at 1730 cm−1 may be due to hydrogen bond interactions of the 4,6-DMDBT oxidation products with carboxyl groups.
The FTIR results indicated that 4,6-DMDBT was oxidized onto the carbon surface to the corresponding sulfones, sulfoxides or/and sulfonic acids as well as that these products were strongly adsorbed on the carbon's surface. Besides, EDX analysis, presented in Fig. 8d and e, supported further the FTIR results and proved the presence of sulfur on the surface of the carbocatalysts’ after the catalytic experiment. The slight decrease of oxidation/removal performance observed in the reusability tests can be linked to the formation and strong retention of these sulfur compounds which were not totally removed/desorbed by washing from the active sites.
3.9. UV-Vis and GC-MS analysis of the liquid products and extracts from the used catalysts
At the end of the catalytic oxidation tests, the hexadecane solution was spectroscopically (UV-vis) analyzed, while the oxidation products accumulated on the carbons’ surface were extracted with methanol for analysis. Methanol was selected because it is a polar solvent and could dissolve the formed sulfones and sulfoxides, compounds with enhanced polarity. The UV-Vis spectra of the liquid phase as well as the spectrum of the methanol extract after a catalytic oxidation experiment with SX PLUS-Nox as a catalyst are shown in Fig. s6.† In the spectra of the reaction product, the intensity decrement of the bands attributed to 4,6 DMDBT was obvious (Fig. s6a†), while in the spectrum of the methanol extract (Fig. s6b†), new peaks appeared corresponding to 4,6-DMDBT derived sulfoxides and/or sulfones.76 From the spectra it can be concluded that the oxidation products were accumulated/strongly sorbed on the carbon surface after the oxidation of 4,6-DMDBT.The liquid phases were also analyzed by GC-MS. From the results presented in Fig. 9a, the decrease of the peak's intensity corresponding to 4,6-DMDBT (at a retention time of 31.996 min) by the use of the parent activated carbon SX PLUS and the disappearance of this peak for SX PLUS-Nox and SX PLUS-Sox verify the UV-Vis analysis results and the obtained high or 100% removal of 4,6-DMDBT (Fig. 5 and 7). GC-MS analysis of the extracts of the used carbon with another polar solvent (acetonitrile) also showed the presence of 4,6-DMDBT products, such as sulfone and sulfoxide, as can be seen in Fig. 9b.
 | ||
| Fig. 9 GC-MS analysis of the initial/reactant solution of 4,6-DMDBT in hexadecane, of the liquid phase of the catalytic oxidation experiment with the parent SX PLUS and oxidized counterparts SX PLUS-Nox, and SX PLUS-Sox (a), and of the extract of the used SX PLUS catalyst with acetonitrile (b). | ||
3.10. Mechanistic insights
From the above results it can be suggested that the aromatic ring of 4,6-DMDBT interacts with the carbon surface through the π–π stacking and/or through the donor acceptor mechanism, and these interactions are responsible for the high adsorptive capability of the studied activated carbons. The activity of the carbons as oxidation catalysts is associated with the concentration of oxygen surface functional groups, i.e. lactones or/and carbonyl groups.50 These surface groups can also be responsible for the strong retention of the formed oxidation products, like sulfoxide and sulfones. The most crucial and still not clearly elucidated aspect is the catalytic role of the carbon surface. The major and most well-established pathway is that the above functional groups on the carbon's surface catalyze the decomposition of H2O2 to free radicals such as ˙OH or ˙OOH, which are powerful oxidants for the oxidation of sulfur compounds to their corresponding sulfoxide and/or sulfones. These radicals can also be responsible for the formation of active molecular oxygen anions that also can act as an oxidant of 4,6-DMDBT. And all these reactions occur inside the micropores of carbon that act as “microreactors”.In order to conclude if the catalytic capability can be linked to the formation of free radicals, methanol was used as a scavenger (Fig. 10). In the presence of methanol, the conversion/removal percentage of 4,6-DMDBT was decreased for all carbons. In the case of SX PLUS-Sox, the decrement of the removal was 40% by the addition of 10% methanol per volume of the solvent. This removal efficiency is even lower from the measured adsorptive performance of this sample, a fact that suggests the blockage of both adsorption and catalytically active sites. This can be assigned to the water moieties, which remain adsorbed on the surface of the carbon by polar forces or/and hydrogen bonds, forming a film that hinders the interaction with 4,6-DMDBT (Fig. 11a). This is in perfect agreement with the above presented results when pure water was added, leading to a decrement of the oxidation/removal efficiency. The negative effect upon methanol addition may be attributed to the reaction of methanol with the free radicals, which are generated by the decomposition of hydrogen peroxide on the surface of the activated carbon,50,77,78 and these reactions are shown in Fig. 11. Additionally, methanol reacts faster with free radicals (kMeOH/˙OH = 1.109 M−1 s−1) than with H2O2 (kH2O2/MeOH = 2.7103 M−1 s−1). Therefore, it is concluded that activated carbon acts as a catalyst in the decomposition of H2O2, via free radical formation that contributes to 4,6-DMDBT oxidation. However, for both the adsorptive and catalytic oxidation tests, the decrement of removal due to antagonistic adsorption between 4,6-DMDBT and MeOH, or as mentioned above due to the occupation of the active sites by MeOH (as by water), cannot be excluded.
 | ||
| Fig. 10 Effect of MeOH on catalytic oxidation/removal of 4,6-DMDBT with the pristine and oxidized SX PLUS carbons (25 mg carbon, 10 ml hexadecane, 20 ppmw of sulfur, 1 mL H2O2 solution, T = 60 °C, t = 24 h). | ||
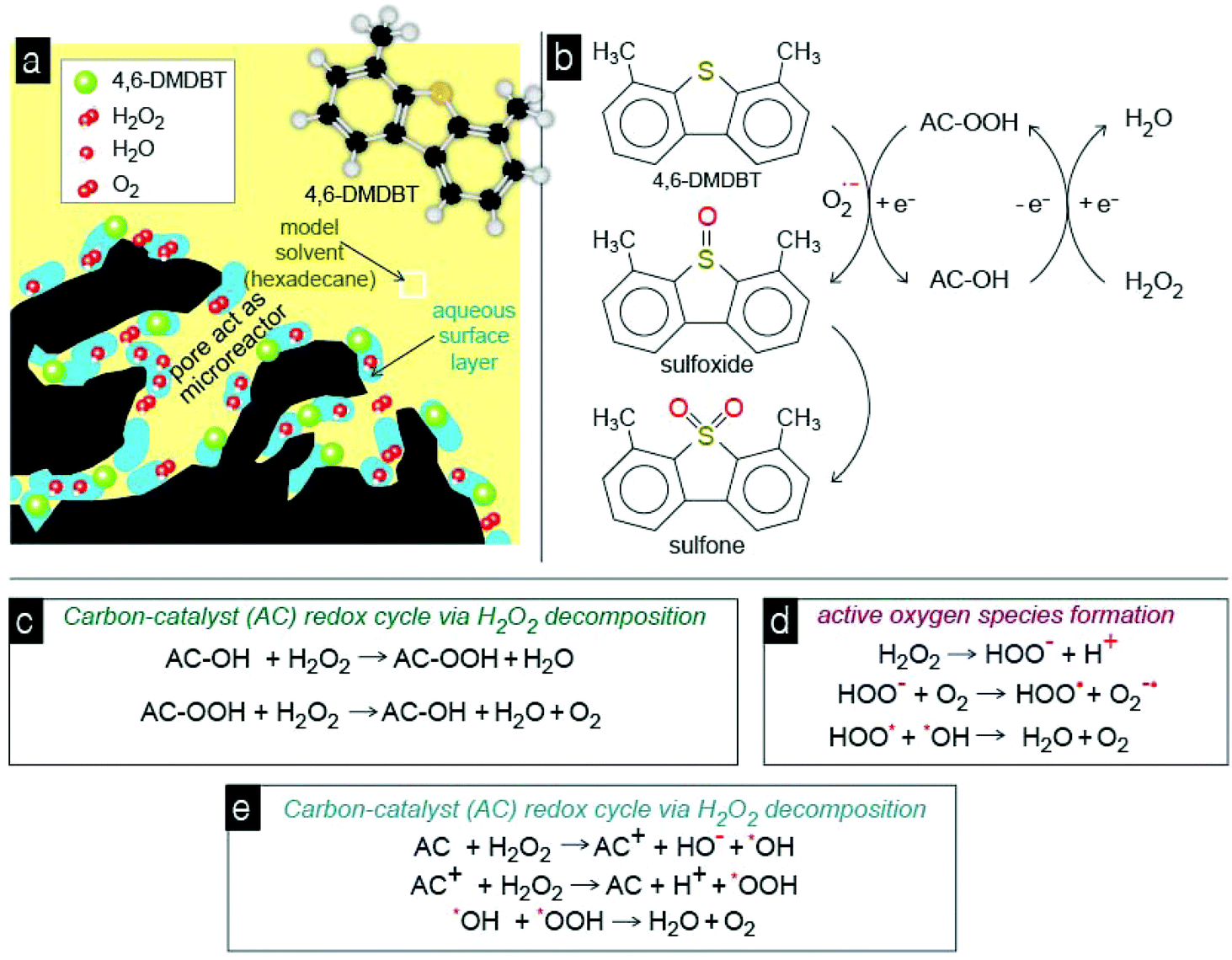 | ||
| Fig. 11 All the proposed interactions, mechanisms, and reactions (a–e) during the catalytic oxidation of 4,6-DMDBT by the acidic activated porous carbon-catalyst. | ||
Since hydrogen peroxide can undergo a fast decomposition in the presence of activated carbon, the most crucial aspect is the simultaneous presence of 4,6-DMDBT and H2O2 inside the pores in order for all the consecutive reactions to occur (Fig. 11a). The presence of water in a high amount can decrease the reaction kinetics by blocking/occupying the adsorption/reaction sites. The oxidation of 4,6-DMDBT undergoes through a redox cycle of carbon with the participation of superoxide anionic radicals and H2O2 (Fig. 11b). Analogous redox cycle based catalytic oxidation pathways were reported for metal oxides.79 In general, peroxide has a triple role. The first one is to activate carbon's surface (Fig. 11c), the second to act as a pool of molecular oxygen (Fig. 11d), and the third to participate in the formation of superoxide anionic radicals (Fig. 11e).
In more recent publications, free radical species (˙OH/˙OOH) have been postulated as the main intermediates in the reaction mechanism, whose formation would take place through an electron-transfer reaction similar to the Fenton mechanism, with AC and AC+ as the reduced and oxidized catalyst states. The recombination of free radical species (˙OH/˙OOH) in the liquid phase and/or onto the activated carbon surface will produce water and oxygen according to reactions in Fig. 11. When 4,6-DMDBT is adsorbed on the surface of activated carbon, in which also H2O2 is decomposed into radicals H+ and HOOt, the latter reacts with O2 to superoxide, which further oxidizes 4,6-DMDBT to sulfoxides and/or sulfones. It is also feasible that the free ˙OH or ˙OOH radicals attack directly and oxidize 4,6-DMDBT.
4. Conclusions
In the current work, we presented the potential use of activated carbons not only as adsorbents of sulfur compounds but also as metal-free oxidation catalysts for the desulfurization of fuels via oxidation of 4,6-DMDBT to the corresponding sulfones and sulfoxides in a reusable manner and under ambient conditions. The comparison of five commercial porous carbons revealed that the removal capability depends predominately on the density of acidic surface functional groups, while the adsorption is governed by the formation of donor–acceptor complexes between the adsorbent and the adsorbate. Further chemical modification of the carbon's surface via treatment/oxidation with HNO3 or H2SO4 was also investigated. In both cases, the basic surface groups were decreased, the phenolics were eliminated, while the lactones were increased, and carboxyl groups were formed. The modification with the former acid had a more pronounced effect on the formation of acidic groups and induced faster catalytic oxidation/removal kinetics, while treatment with the latter acid led to a significant decrease of the basic sites and to the highest adsorptive capability. Although the oxidation of carbons had a relatively moderate negative effect on porosity characteristics, the oxidized counterparts showed a 100% desulfurization capability, a value being 33% higher compared to the performance of the parent non-modified commercial carbon, confirming the predominant role of the surface chemistry. In addition, it was shown that the abundance of micropores with a size of ≤1 nm is beneficial for the adsorptive/catalytic reactivity due to confinement effects considering the similar size of 4,6-DMDBT. The reaction of 4,6-DMDBT in the presence of H2O2 led to the formation of the corresponding sulfoxide and sulfone, as a result of the formation of superoxide and free ˙OH and ˙OOH radicals upon the decomposition of H2O2 on the carbocatalyst surface. These results can boost the research activities and exploitation of activated porous carbons, without the use of any external metals with redox activity, as highly efficient green catalysts in oxidative deep desulfurization of fuels or other related processes.Conflicts of interest
There are no conflicts to declare.References
- M. T. Timko, J. An, J. Burgess, P. Kracke, L. Gonzalez, C. Jaye and D. A. Fischer, Roles of surface chemistry and structural defects of activated carbons in the oxidative desulfurization of benzothiophenes, Fuel, 2016, 163, 223–231, DOI:10.1016/j.fuel.2015.09.075.
- C. Song, An overview of new approaches to deep desulfurization for ultra-clean gasoline, diesel fuel and jet fuel, Catal. Today, 2003, 86, 211–263, DOI:10.1016/S0920-5861(03)00412-7.
- M. Zhu, G. Luo, L. Kang and B. Dai, Novel catalyst by immobilizing a phosphotungstic acid on polymer brushes and its application in oxidative desulfurization, RSC Adv., 2014, 4, 16769–16776, 10.1039/c4ra01367k.
- K. S. Triantafyllidis and E. A. Deliyanni, Desulfurization of diesel fuels: Adsorption of 4,6-DMDBT on different origin and surface chemistry nanoporous activated carbons, Chem. Eng. J., 2014, 236, 406–414, DOI:10.1016/j.cej.2013.09.099.
- European Environment Agency, European Union emission inventory report 1990–2014 under the UNECE Convention on Long-range Transboundary Air Pollution (LRTAP), 2016. DOI:10.2800/18374.
- R. Shafi and G. J. Hutchings, Hydrodesulfurization of hindered dibenzothiophenes: an overview, Catal. Today, 2000, 59, 423–442, DOI:10.1016/S0920-5861(00)00308-4.
- T. A. Saleh, K. O. Sulaiman, S. A. Al-hammadi, H. Dafalla and G. I. Danmaliki, Adsorptive desulfurization of thiophene, benzothiophene and dibenzothiophene over activated carbon manganese oxide nanocomposite: With column system evaluation, J. Cleaner Prod., 2017, 154, 401–412, DOI:10.1016/j.jclepro.2017.03.169.
- M. Seredych, J. Lison, U. Jans and T. J. Bandosz, Textural and chemical factors affecting adsorption capacity of activated carbon in highly efficient desulfurization of diesel fuel, Carbon, 2009, 47, 2491–2500, DOI:10.1016/j.carbon.2009.05.001.
- V. C. Srivastava, An evaluation of desulfurization technologies for sulfur removal from liquid fuels, RSC Adv., 2012, 2, 759–783, 10.1039/c1ra00309g.
- M. Muzic and K. Sertic-Bionda, Alternative Processes for Removing Organic Sulfur Compounds from Petroleum Fractions, Chem. Biochem. Eng. Q., 2013, 27, 101–108 CAS.
- H. Lü, W. Ren, H. Wang, Y. Wang, W. Chen and Z. Suo, General Deep desulfurization of diesel by ionic liquid extraction coupled with catalytic oxidation using an Anderson-type catalyst, Appl. Catal., A, 2013, 453, 376–382, DOI:10.1016/j.apcata.2012.12.047.
- J. Qiu, G. Wang, Y. Zhang, D. Zeng and Y. Chen, Direct synthesis of mesoporous H 3 PMo 12 O 40/SiO 2 and its catalytic performance in oxidative desulfurization of fuel oil, Fuel, 2015, 147, 195–202, DOI:10.1016/j.fuel.2015.01.064.
- L. Yang, X. Li, A. Wang, R. Prins, Y. Wang, Y. Chen and X. Duan, Hydrodesulfurization of 4,6-dimethyldibenzothiophene and its hydrogenated intermediates over bulk Ni2P, J. Catal., 2014, 317, 144–152, DOI:10.1016/j.jcat.2014.06.020.
- C. O. Ania and T. J. Bandosz, Importance of structural and chemical heterogeneity of activated carbon surfaces for adsorption of dibenzothiophene, Langmuir, 2005, 21, 7752–7759, DOI:10.1021/la050772e.
- T. A. Saleh and G. I. Danmaliki, Influence of acidic and basic treatments of activated carbon derived from waste rubber tires on adsorptive desulfurization of thiophenes, J. Taiwan Inst. Chem. Eng., 2015, 000, 1–9, DOI:10.1016/j.jtice.2015.11.008.
- D. J. Monticello, Biodesulfurization and the upgrading of petroleum distillates, Curr. Opin. Biotechnol., 2000, 11, 540–546 CrossRef CAS PubMed.
- M. Soleimani, A. Bassi and A. Margaritis, Biodesulfurization of refractory organic sulfur compounds in fossil fuels, Biotechnol. Adv., 2007, 25, 570–596, DOI:10.1016/j.biotechadv.2007.07.003.
- M. H. Ibrahim, M. Hayyan, M. A. Hashim and A. Hayyan, The role of ionic liquids in desulfurization of fuels: A review, Renewable Sustainable Energy Rev., 2017, 76, 1534–1549, DOI:10.1016/j.rser.2016.11.194.
- X. Wang, F. Li, J. Liu, C. Kou, Y. Zhao and Y. Hao, Preparation of TiO2 in Ionic Liquid via Microwave Radiation and in Situ Photocatalytic Oxidative Desulfurization of Diesel Oil, Energy Fuels, 2012, 26, 6777–6782 CrossRef CAS.
- M. Feng, Review on Recent Patents in Sulfur Removal from Liquid Fuels by Oxidative Desulfurization (ODS) Process, Recent Pat. Chem. Eng., 2010, 3, 30–37, DOI:10.2174/1874478811003010030.
- B. R. Fox, B. L. Brinich, J. L. Male, R. L. Hubbard, M. N. Siddiqui, T. A. Saleh and D. R. Tyler, Enhanced oxidative desulfurization in a film-shear reactor, Fuel, 2015, 156, 142–147 CrossRef CAS.
- L. Rivoira, J. Juárez, H. Falcón, M. Gómez Costa, O. Anunziata and A. Beltramone, Vanadium and titanium oxide supported on mesoporous CMK-3 as new catalysts for oxidative desulfurization, Catal. Today, 2017, 282, 123–132, DOI:10.1016/j.cattod.2016.07.003.
- W. Jin, Y. Tian, G. Wang, D. Zeng, Q. Xu and J. Cui, Ultra-deep oxidative desulfurization of fuel with H2O2 catalyzed by molybdenum oxide supported on alumina modified by Ca2+, RSC Adv., 2017, 7, 48208–48213, 10.1039/c7ra08900g.
- S. Otsuki, T. Nonaka, N. Takashima, W. Qian, A. Ishihara, T. Imai and T. Kabe, Oxidative Desulfurization of Light Gas Oil and Vacuum Gas Oil by Oxidation and Solvent Extraction, Energy Fuels, 2000, 14, 1232–1239, DOI:10.1021/ef000096i.
- Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Desulfurization and denitrogenation process for light oils based on chemical oxidation followed by liquid-liquid extraction, Ind. Eng. Chem. Res., 2002, 41, 4362–4375, DOI:10.1021/ie010618x.
- P. S. Tam, J. R. Kittrell and J. W. Eldridge, Desulfurization of Fuel Oil by Oxidation and Extraction. 1.Enhancement of Extraction Oil Yield, Ind. Eng. Chem. Res., 1990, 29, 321–324 CrossRef CAS.
- F. Yu and R. Wang, Deep Oxidative Desulfurization of Dibenzothiophene in Simulated Oil and Real Diesel Using Heteropolyanion-Substituted Hydrotalcite-Like Compounds as Catalysts, Molecules, 2013, 13691–13704, DOI:10.3390/molecules181113691.
- A. Chica, A. Corma and M. E. Dómine, Catalytic oxidative desulfurization (ODS) of diesel fuel on a continuous fixed-bed reactor, J. Catal., 2006, 242, 299–308, DOI:10.1016/j.jcat.2006.06.013.
- A. Teimouri, M. Mahmoudsalehi and H. Salavati, Catalytic oxidative desulfurization of dibenzothiophene utilizing molybdenum and vanadium oxides supported on MCM-41, Int. J. Hydrogen Energy, 2018, 1–18, DOI:10.1016/j.ijhydene.2018.05.102.
- S. Xun, W. Zhu, F. Zhu, Y. Chang, D. Zheng, Y. Qin, M. Zhang, W. Jiang and H. Li, Design and synthesis of W-containing mesoporous material with excellent catalytic activity for the oxidation of 4,6-DMDBT in fuels, Chem. Eng. J., 2015, 280, 256–264, DOI:10.1016/j.cej.2015.05.092.
- J. Wang, D. Zhao and K. Li, Oxidative Desulfurization of Dibenzothiophene Using Ozone and Hydrogen Peroxide in Ionic Liquid, Energy Fuels, 2010, 2527–2529, DOI:10.1021/ef901324p.
- J. M. Campos-Martin, M. Capel-Sanchez, P. Perez-Presas and J. L. G. Fierro, Oxidative processes of desulfurization of liquid fuels, J. Chem. Technol. Biotechnol., 2010, 85, 879–890, DOI:10.1002/jctb.2371.
- C. Zou, P. Zhao, J. Ge, Y. Qin and P. Luo, Oxidation/adsorption desulfurization of natural gas by bridged cyclodextrins dimer encapsulating polyoxometalate, Fuel, 2013, 104, 635–640, DOI:10.1016/j.fuel.2012.06.096.
- T. A. Saleha, S. A. AL-Hammadi and A. M. Al-Amerb, Effect of boron on the efficiency of MoCo catalysts supported on alumina for the hydrodesulfurization of liquid fuels, Process Saf. Environ. Prot., 2019, 121, 165–174 CrossRef.
- H. Song, J. Gao, X. Chen, J. He and C. Li, Catalytic oxidation-extractive desulfurization for model oil using inorganic oxysalts as an oxidant and a Lewis acid-organic acid mixture as a catalyst and an extractant, Appl. Catal., A, 2013, 456, 67–74, DOI:10.1016/j.apcata.2013.02.017.
- T. A. Saleh, The influence of treatment temperature on the acidity of MWCNT oxidized by HNO3 or a mixture of HNO3 /H2SO4, Appl. Surf. Sci., 2011, 257, 7746–7751 CrossRef CAS.
- F. Rodriguez-Reinoso, The Role of Carbon Materials in Heterogeneous Catalysis, Carbon, 1998, 36, 159–175, DOI:10.1016/j.na.2009.11.013.
- T. A. Saleh and G. I. Danmaliki, Adsorptive desulfurization of dibenzothiophene from fuels by rubber tyre-derived carbons: Kinetics and isotherm evaluation, Process Saf. Environ. Prot., 2016, 102, 9–19, DOI:10.1016/j.psep.2016.02.005.
- T. A. Saleh, Simultaneous adsorptive desulfurization of diesel fuel over bimetallic nanoparticles loaded on activated carbon, J. Cleaner Prod., 2017, 1–10 CAS.
- J. C. Colmenares, R. S. Varmab and P. Lisowski, Sustainable hybrid photocatalysts: Titania immobilized on carbon materials derived from renewable and biodegradable resources, Green Chem., 2016, 18, 5736–5750 RSC.
- P. Lisowski, J. C. Colmenares, O. Mašek, D. Łomot, O. Chernyayevaa and D. Lisovytskiya, Novel biomass-derived hybrid TiO2/carbon material using tar-derived secondary char to improve TiO2 bonding to carbon matrix, J. Anal. Appl. Pyrolysis, 2018, 131, 35–41 CrossRef CAS.
- G. I. Danmaliki, T. A. Saleh and A. A. Shamsuddeen, Response surface methodology optimization of adsorptive desulfurization on nickel/activated carbon, Chem. Eng. J., 2017, 313, 993–1003 CrossRef CAS.
- T. A. Saleh, Nanocomposite of carbon nanotubes/silica nanoparticles and their use for adsorption of Pb(II): from surface properties to sorption mechanism, Desalin. Water Treat., 2015, 1–15, DOI:10.1080/19443994.2015.1036784.
- T. A. Saleh, S. A. AL-Hammadi, I. M. Abdullahi and M. Mustaqeem, Synthesis of molybdenum cobalt nanocatalysts supported on carbon for hydrodesulfurization of liquid fuels, J. Mol. Liq., 2018, 272, 715–721, DOI:10.1016/j.molliq.2018.09.118.
- W. Jiang, W. Zhu, Y. Chang, H. Li, Y. Chao, J. Xiong and H. Liu, Oxidation of Aromatic Sulfur Compounds Catalyzed by Organic Hexacyanoferrates in Ionic Liquids with a Low Concentration of H2O2 as an Oxidant, Energy Fuels, 2014, 28, 3–9 Search PubMed.
- D. A. Giannakoudakis, M. Barczak, M. Florent and T. J. Bandosz, Analysis of interactions of mustard gas surrogate vapors with porous carbon textiles, Chem. Eng. J., 2019, 362, 758–766, DOI:10.1016/j.cej.2019.01.064.
- M. Florent, D. A. Giannakoudakis and T. J. Bandosz, Mustard Gas Surrogate Interactions with Modified Porous Carbon Fabrics: Effect of Oxidative Treatment, Langmuir, 2017, 33, 11475–11483, DOI:10.1021/acs.langmuir.7b02047.
- D. Trimm, Carbon as a Catalyst and Reactions of Carbon, 1981 Search PubMed.
- H. Sun, B. Shi, D. A. Lytle, Y. Bai and D. Wang, Formation and release behavior of iron corrosion products under the influence of bacterial communities in a simulated water distribution system, Environ. Sci.: Processes Impacts, 2014, 16, 576–585, 10.1039/c3em00544e.
- G. Yu, S. Lu, H. Chen and Z. Zhu, Diesel fuel desulfurization with hydrogen peroxide promoted by formic acid and catalyzed by activated carbon, Carbon, 2005, 43, 2285–2294, DOI:10.1016/j.carbon.2005.04.008.
- I. Anastopoulos, I. Pashalidis, A. Hosseini-Bandegharaei, D. A. Giannakoudakis, A. Robalds, M. Usman, L. B. Escudero, Y. Zhou, J. C. Colmenares, A. Núñez-Delgado and É. C. Lima, Agricultural biomass/waste as adsorbents for toxic metal decontamination of aqueous solutions, J. Mol. Liq., 2019, 111684, DOI:10.1016/j.molliq.2019.111684.
- D. A. Giannakoudakis, A. Hosseini-Bandegharaei, P. Tsafrakidou, K. S. Triantafyllidis, M. Kornaros and I. Anastopoulos, Aloe vera waste biomass-based adsorbents for the removal of aquatic pollutants: A review, J. Environ. Manage., 2018, 227, 354–364, DOI:10.1016/j.jenvman.2018.08.064.
- I. Anastopoulos, A. Robalds, H. N. Tran, D. Mitrogiannis, D. A. Giannakoudakis, A. Hosseini-Bandegharaei and G. L. Dotto, Removal of heavy metals by leaves-derived biosorbents, Environ. Chem. Lett., 2019, 17, 755–766, DOI:10.1007/s10311-018-00829-x.
- A. A. Spagnoli, D. A. Giannakoudakis and S. Bashkova, Adsorption of methylene blue on cashew nut shell based carbons activated with zinc chloride: The role of surface and structural parameters, J. Mol. Liq., 2017, 229, 465–471, DOI:10.1016/j.molliq.2016.12.106.
- P. A. Lazaridis, S. A. Karakoulia, C. Teodorescu, N. Apostol, D. Macovei, A. Panteli, A. Delimitis, S. M. Coman, V. I. Parvulescu and K. S. Triantafyllidis, High hexitol selectivity in cellulose hydrolytic hydrogenation over platinum (Pt) vs. ruthenium (Ru) catalysts supported on micro/mesoporous carbon, Appl. Catal., B, 2017, 214, 1–14, DOI:10.1016/j.apcatb.2017.05.031.
- B. Boulinguiez and P. Le Cloirec, Adsorption/desorption of tetrahydrothiophene from natural gas onto granular and fiber-cloth activated carbon for fuel cell applications, Energy Fuels, 2009, 23, 912–919, DOI:10.1021/ef800757u.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60, 309–319, DOI:10.1021/ja01269a023.
- H. P. Boehm, Surface oxides on carbon and their analysis: A critical assessment, Carbon, 2002, 40, 145–149, DOI:10.1016/S0008-6223(01)00165-8.
- J. S. Mattson and H. B. Mark Jr., Activated carbon: surface chemistry and adsorption from solution, M. Dekker, New York, 1971 Search PubMed.
- G. Newcombe, R. Hayes and M. Drikas, Granular activated carbon: Importance of surface properties in the adsorption of naturally occurring organics, Colloids Surf., A, 1993, 78, 65–71, DOI:10.1016/0927-7757(93)80311-2.
- D. Pathania, S. Sharma and P. Singh, Removal of methylene blue by adsorption onto activated carbon developed from Ficus carica bast, Arabian J. Chem., 2017, 10, S1445–S1451, DOI:10.1016/j.arabjc.2013.04.021.
- Test No. 106: Adsorption–Desorption Using a Batch Equilibrium Method, OECD Guideline for the testing of Chemicals, 2000, 1–44.
- S. Langergren and B. Svenska, The theory of adsorption of solutes, K. Sven. Vetenskapsakad. Handl., 1898, 24, 1–39 Search PubMed.
- Y. S. Ho, Review of second-order models for adsorption systems, J. Hazard. Mater., 2006, 136, 681–689 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. S. Pierotti, J. Rouquerol and T. Siemieniewska, International union of pure commission on colloid and surface chemistry including catalysis-Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity, Pure Appl. Chem., 1985, 57, 603–619, DOI:10.1351/pac198557040603.
- D. Angin, Utilization of activated carbon produced from fruit juice industry solid waste for the adsorption of Yellow 18 from aqueous solutions, Bioresour. Technol., 2014, 168, 259–266, DOI:10.1016/j.biortech.2014.02.100.
- K. Y. Foo and B. H. Hameed, Preparation of activated carbon from date stones by microwave induced chemical activation: Application for methylene blue adsorption, Chem. Eng. J., 2011, 170, 338–341, DOI:10.1016/j.cej.2011.02.068.
- I. Langmuir, The adsorption of gases on the plane surface of glass, mica and platinum, J. Am. Chem. Soc., 1918, 40(9), 1361–1403 CrossRef CAS.
- H. Freundlich, Uber Dye Adsorption in hosungen, Z. Phys. Chem., 1906, 57, 387–470 Search PubMed.
- L.-p. Hou, R.-x. Zhao, X.-p. Li and X.-h. Gao, Preparation of MoO2/g-C3N4composites with a high surface area and its application in deep desulfurization from model oil, Appl. Surf. Sci., 2018, 434, 1200–1209, DOI:10.1016/j.apsusc.2017.10.076.
- Y. Zhang and R. Wang, Nanocarbon materials for oxidative-adsorptive desulfurization using air oxygen under mild conditions, Diamond Relat. Mater., 2017, 73, 161–168, DOI:10.1016/j.diamond.2016.09.006.
- K. G. Haw, W. A. W. A. Bakar, R. Ali, J. F. Chong and A. A. A. Kadir, Catalytic oxidative desulfurization of diesel utilizing hydrogen peroxide and functionalized-activated carbon in a biphasic diesel-acetonitrile system, Fuel Process. Technol., 2010, 91, 1105–1112, DOI:10.1016/j.fuproc.2010.03.021.
- X. Sun, A. H. M. Hussain, M. Chi, X. Cheng and B. J. Tatarchuk, Persistent adsorptive desulfurization enhancement of TiO2 after one-time ex-situ UV-treatment, Fuel, 2017, 193, 95–100 CrossRef CAS.
- Y. N. Prajapati and N. Verma, Adsorptive desulfurization of diesel oil using nickel nanoparticle-doped activated carbon beads with/without carbon nanofibers: effects of adsorbate size and adsorbent texture, Fuel, 2017, 189, 186–194 CrossRef CAS.
- M. Zhang, W. Zhu, S. Xun, H. Li, Q. Gu, Z. Zhao and Q. Wang, Deep oxidative desulfurization of dibenzothiophene with POM-based hybrid materials in ionic liquids, Chem. Eng. J., 2013, 220, 328–336, DOI:10.1016/j.cej.2012.11.138.
- G. Abdi, M. Ashokkumar and A. Alizadeh, Ultrasound-assisted oxidative-adsorptive desulfurization using highly acidic graphene oxide as a catalyst-adsorbent, Fuel, 2017, 210, 639–645, DOI:10.1016/j.fuel.2017.09.024.
- A. Rey, J. A. Zazo, J. A. Casas, A. Bahamonde and J. J. Rodriguez, Influence of the structural and surface characteristics of activated carbon on the catalytic decomposition of hydrogen peroxide, Appl. Catal., A, 2011, 402, 146–155, DOI:10.1016/j.apcata.2011.05.040.
- E. Vega and H. Valdés, New evidence of the effect of the chemical structure of activated carbon on the activity to promote radical generation in an advanced oxidation process using hydrogen peroxide, Microporous Mesoporous Mater., 2018, 259, 1–8, DOI:10.1016/j.micromeso.2017.09.018.
- D. A. Giannakoudakis, V. Nair, A. Khan, E. A. Deliyanni, J. C. Colmenares and K. S. Triantafyllidis, Additive-free photo-assisted selective partial oxidation at ambient conditions of 5-hydroxymethylfurfural by manganese(IV) oxide nanorods, Appl. Catal., B, 2019, 256, 117803, DOI:10.1016/j.apcatb.2019.117803.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc03234g |
| This journal is © The Royal Society of Chemistry 2019 |