
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-linked polyfuran networks with elastomeric behaviour based on humins biorefinery by-products†
Roxana
Dinu
and
Alice
Mija
 *
*
Université Côte d'Azur, Université Nice-Sophia Antipolis, Institut de Chimie de Nice, UMR CNRS 7272, 06108 Nice Cedex 02, France. E-mail: Alice.Mija@unice.fr
First published on 4th October 2019
Abstract
Biorefinery by-products have received much attention in the last few years. Humins are one of these candidates. Nevertheless, one common feature of humins, as a polyfuranic thermoset material, is their inherent brittleness which is a direct consequence of the network's structure. Auto-crosslinked humins networks exhibit only minor deformation and break very easily. Consequently, this behavior limits their use in many industrial applications. For this reason, we used in this work the copolymerization strategy, by combining humins with epoxide based aliphatic ethers, as a toughening approach. To gain a fundamental understanding of the humins based copolymers we thoroughly investigated their structure, behavior and properties by FT-IR, rheology, DSC, TGA, DMA and tensile tests. These investigations show one of the most important results: the humins copolymers have a ductile and elastomeric character. The tensile strain at break of the copolymers reaches ≈60% which is a significant advantage in terms of applications of humins as structural materials. To our knowledge, these are the first data reported on the synthesis of elastomeric humins based copolymers paving the way for the utilization of these emergent materials in industrial applications.
Introduction
In the last few years, particular attention has been paid to the use of renewable resources in order to replace fossil derivatives, which increasingly affect the environment. A continuous challenge for researchers is to find effective ways of conversion of abundant or cheap bioresources into fuels, chemicals or materials. Lignocellulosic biomass is a renewable resource composed mainly of three principal constituents e.g. lignin, cellulose, and hemicellulose, each of them having different compositions and reactivities. Lignin is an amorphous, branched polymer of aromatic alcohols while cellulose and hemicellulose are polysaccharides consisting of hexoses and pentoses.1 Generally, 35–50% of lignocellulosic biomass is represented by cellulose, 20–35% by hemicellulose, and 10–25% by lignins and the remaining fraction is composed of oils, ash and proteins.2Under acid-catalyzed hydrolysis, the cellulosic biomass can be converted into various platform molecules such as 5-hydroxymethylfurfural (HMF), levulinic acid or formic acid in biorefineries but the processing is sometimes accompanied by the formation of a dark-colored by-product.3,4 It has been revealed that this by-product, called humins, is formed more rapidly from fructose than from glucose.5 The formation of this by-product has been reported in almost all the papers linked to the synthesis of levulinic acid, HMF or furfural. The yield of humins depends much on process parameters (catalysts, temperature, duration, etc.).6 Although many efforts have been focused on reducing the amount of this side-product from biorefineries, the yield of humins still remains high, between 14 and 30 wt%.7,8 Considering these factors, the valorization of humins is the optimal method for improving the economic value of a biorefinery chain, but above all, it is the best way to improve the environmental impact of the entire technological process. Another environmentally friendly process in which this side-product is formed is represented by the hydrothermal conversion of different biomass and derivatives.9–11
Since the 90s, the production and composition of the caramel of sugars have been a subject of great interest to the scientists.12–15 Caramel is produced by heating sucrose and, from the compositional point of view, appears as a mixture of humins and iso-saccharosan in various proportions.5,16,17 Despite numerous investigations regarding caramel formation, the compositions of caramel and humins are not yet fully understood.5 Following the investigations, humins have been found to consist of about 50–65% C, 29–46% O and 4–5.5% H and contain in their structure aromatic rings, most of them furanics.5–7 The structure of humins is known to depend on the reaction time, temperature, feedstock and solvent (water and alcohol).10,18–20
Van Zandvoort et al.18 have analyzed in detail the formation, morphology and molecular structure of laboratory made humins samples, prepared from different feedstocks using various processing parameters (Fig. 1). Also, Hoang et al.21,22 have investigated the chemical structure of humins prepared in a laboratory by dehydration of D-glucose in order to produce levulinic acid. Based on these studies, the authors revealed that humins present a variable furanic structure formed by a dehydration pathway and contain alcohol, acid, ketone, and aldehyde functional groups. In their study, Constant et al.23 have quantified and classified the ketone and aldehyde carbonyl functional groups contained in industrial humins and lignins by 19F NMR. Following these investigations, they found that the industrial humins contains 6.6 wt% carbonyl functions. The abundance of such functional groups provides opportunities for the use of these humins by-products in various domains of applications.
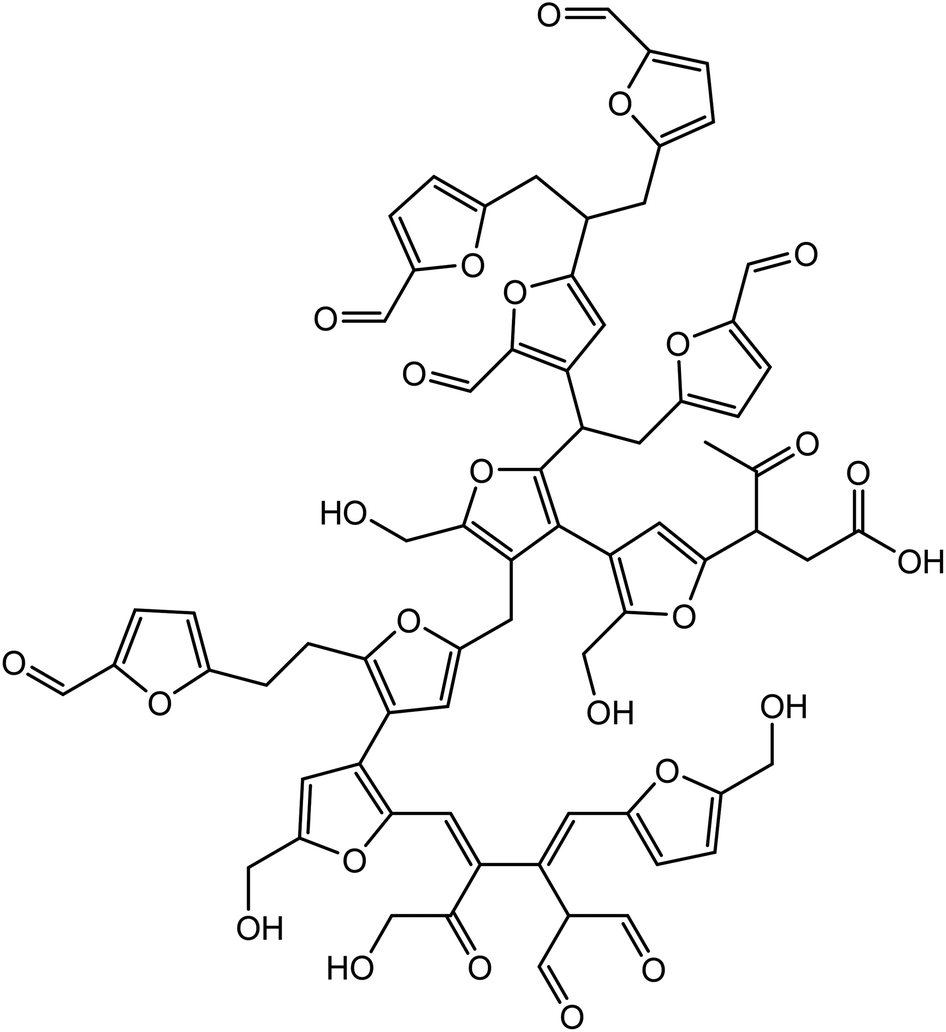 | ||
| Fig. 1 Proposed molecular structure of a glucose-derived humins fragment.18 | ||
Recently, researchers started to explore possible routes to valorize this by-product derived from biomass conversion processes in order to turn them into high-value products. Kang et al.24 have completely degraded humins by alkaline-catalytic hydrothermal treatment followed by wet oxidation. By this technology, the authors managed to convert humins into acetic acid obtaining a yield of 25.6% and a purity of 46.2%. Hoang et al.6 studied the gasification and steam reforming of humins at 750 °C over a variety of alkali- and alkaline-earth based catalysts in order to obtain syngas (a mixture of hydrogen and COx). The authors found that Na2CO3 was the most effective catalyst enabling complete conversion of humins (81% of H2 yield). The liquefaction of humins has been reported by Rasrendra et al.8 through fast pyrolysis resulting in low molecular furanics such as furfural and 2,5 dimethylfuran.
The potential of humins to generate materials has also been tested. Pin et al.25 combined furfuryl alcohol (FA) with humins to produce a fully bio-based furanic resin tested in the development of thermoset furanic composites where cellulose fibers act as reinforcement. An original way to valorize the humins was their transformation into polymeric foams with potential applications in substrates for plant growth, absorbents for wastewater treatment, insulation materials, supports for solid catalysts, etc.26–30 In our previous studies26,27 we succeeded in developing macroporous foam materials based on an auto-crosslinking reaction of industrial humins. By adjusting the parameters of the humins-foam preparation process, we achieved a good control of the morphology and cell dimensions. The obtained foams show open and/or closed cells with cell diameters between 0.2 and 3.5 mm making possible the use of these materials in different application sectors. Humins were used by Kang et al.28 in the production of activated carbon as a sorbent for wastewater treatment. These activated carbons were developed through KOH activation in a temperature range of 500–900 °C to investigate the optimal adsorption of methylene blue (MB) and phenol in aqueous solutions. The authors obtained very good results; the methylene blue adsorption capacity was about 40% while the phenol adsorption was around 39.7%. These results show the capacity of humins to produce porous materials with potential use as membranes for water purification, for the building sector or for use in green houses.
Following our previous work, the objective of this study is not only the valorization of industrial humins by-products obtained in biorefineries but also the development of emergent materials for different application sectors using non-toxic, low cost and environmentally friendly co-monomers. To do this, we had to decrease the inherent brittleness of reticulated humins, as it limits their use in many industrial applications. Auto-crosslinked humins networks allow only minor deformation and break very easily. To toughen brittle polyfuranic humins resins the strategy used in this work was chemical modifications of their backbone structure. The idea was to include more flexible moieties by copolymerizing humins with more flexible chains such as aliphatic diglycidyl ethers. Moreover, this pathway will possibly also result in lowering the crosslinking density intrinsic to humins. To produce a toughened network there are some requirements. Firstly, good compatibility between the humins matrix and the diglycidyls must exist, meaning the two components need to be totally miscible prior to curing. Subsequently, the humins should react with the diglycidyls in order to form chemical linkages between the modifier and the matrix.
To chemically modify the humins backbone we chose to copolymerize it with two aliphatic diglycidyl ethers: the polyethylene glycol diglycidyl ether (PEGDE) and the glycerol-1,3-diglycidyl ether (GDE). Polyethylene glycol (PEG) is a synthetic polyether which can be easily synthesized with a wide range of molecular weights. This polymer is soluble in water and also in many organic solvents.31 PEG has been found to be nontoxic and is approved by the U.S. Food and Drug Administration; therefore it is used in many different applications such as biomedical research, drug deliveries, tissue engineering scaffolds, surface functionalization, etc.31–34 For example, Kono35 crosslinked a carboxymethyl cellulose sodium salt with PEGDE obtaining new hydrogels which could be used as carriers of drug delivery systems for protein-based drugs.
Glycerol, a by-product obtained by biodiesel refining, can be used as a monomer in the synthesis of polymers and materials in its original form or after chemical modifications.36 Zhao et al.37 developed a biodegradable microgel system based on glycerol-1,3-diglycidyl ether cross-linked with oxidized potato starch for controlled uptake and release of proteins. In another study, ur Rehman et al.38 reported the preparation of a new microgel from tris(2-aminoethyl)amine and glycerol diglycidyl ether by using L-α lecithin as a surfactant and gasoline as the organic phase. The prepared microgels were found to be biocompatible against L929 fibroblast cells, showing antibacterial characteristics against common bacteria and possessing great potential in the biomedical fields.
In consequence, the two chosen chemical modifiers for the humins structure, the PEGDE and GDE, are non-toxic, have biomedical applications, and have good potential to produce materials by crosslinking in copolymerization with targeted structures. Together with their low viscosity and relatively low molar mass, they were ideal candidates for humins modification. The reactivity study of these novel formulations was realized by differential scanning calorimetry (DSC), chemo-rheology and in situ Fourier Transform Infrared Spectroscopy (FTIR). The physical and thermo-mechanical properties of the obtained thermoset materials were analyzed by thermogravimetric analysis (TGA), dynamic mechanical analysis (DMA), Shore hardness test and tensile tests.
Experimental
Materials
The main component of formulations is represented by humins, which are an industrial polymeric by-product produced by Avantium Chemicals at their Pilot Plant in Geleen, The Netherlands, formed during acid-catalyzed processing of carbohydrates. As already reported, the humins are carbonaceous, heterogeneous, polydisperse structures whose molecular structures remain largely unknown. To obtain thermoset materials, the humins were copolymerized with diglycidyl comonomers such as the poly(ethylene glycol) diglycidyl ether (PEGDE) and glycerol diglycidyl ether (GDE), with N,N-dimethylbenzylamine (BDMA) being used as an initiator. The comonomers and the initiator were purchased from Sigma-Aldrich and used as received.| Compound name | Molecular weight (g mol−1) | Density at 25 °C (g cm−3) |
|---|---|---|
| N,N-Dimethylbenzylamine (BDMA) | 135.21 | 0.9 |
| Glycerol diglycidyl ether (GDE) | 204.22 | 1.23 |
| Poly(ethylene glycol) diglycidyl ether (PEGDE) | Average Mn 500–650 | 1.14 |
Experimental techniques
The spectrum of air was recorded as a background before each sample. All spectra were acquired with a spectral resolution of 4 cm−1 and 32 scans in the range of 4000–500 cm−1.
Three different Tg values can be determined by the measurement according to AITM 1-0003 and ASTM D7028-07.39 The three different Tg values are (i) Tg-onset, defined as the temperature of extrapolated tangents drawn from points on the storage modulus curve before and after the start of the glass transition; (ii) Tg-loss, defined as the maximum temperature on the loss modulus versus temperature curve; and (iii) Tg-peak, defined as the maximum of tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ (damping). The Tg-peak value is generally higher than the Tg-loss value and corresponds more closely to the transition midpoint while the Tg-onset value more closely signifies the initial drop from the glassy state into the transition state. To unify the notation, the Tg-peak will be noted as tanδ assigned to the temperature of the maximum of the loss factor peak and attributed to the α transitions.
δ (damping). The Tg-peak value is generally higher than the Tg-loss value and corresponds more closely to the transition midpoint while the Tg-onset value more closely signifies the initial drop from the glassy state into the transition state. To unify the notation, the Tg-peak will be noted as tanδ assigned to the temperature of the maximum of the loss factor peak and attributed to the α transitions.
The crosslinking densities for the humins-based copolymers were calculated using Flory's theory.40 According to Flory the value of the storage modulus at the rubber plateau allows the calculation of the crosslinking density of the resins:
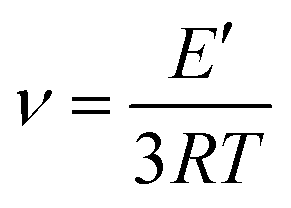 δ are constant, namely at Tg + 80 °C.
δ are constant, namely at Tg + 80 °C.
Results and discussion
A first series of studies were carried out to determine the optimal formulations for the development of humins based thermosets by its copolymerization with the PEGDE and GDE in the presence of BDMA as the initiator. The formulations were analyzed using differential scanning calorimetry (DSC), rheology analysis and FTIR to determine the temperature range and the reactivity of the mixtures. We studied at the beginning 8 formulations by increasing from 20 to 50% the weight ratio of each comonomer in the humins mix in order to determine the optimum amounts of compounds required for the development of each of the systems humins/PEGDE and humins/GDE. Thereafter, we complexified the system by combining the humins with both GDE and PEGDE. Again, these formulations were analyzed, and the optimal formulations retained. So, in total, we decided to select 3 formulations: humins/PEGDE, humins/GDE, and humins/PEGDE/GDE.DSC studies of reactivity
Humins are materials with peculiar compositions and reactivities. To gain insights into their reactivity with the selected diglycidyl structures, dynamic DSC was conducted during thermally induced crosslinking via copolymerization. To select the optimum ratios of humins/PEGDE and humins/GDE copolymerization mixtures the reactivity between the humins and the comonomers without an initiator was analysed first by DSC. The collected results are shown in Fig. 2. As can be seen from the DSC analysis, the curing mixtures with low PEGDE contents (20 and 30 wt%) have a complex and wide temperature interval of reactivity, ranging between 115 °C and 260 °C. On increasing the PEGDE content (40 and 50 wt%), an evident double reaction can be observed, with the first exotherm observed between 120 °C and 180 °C followed by a second one between 190 °C and 250 °C.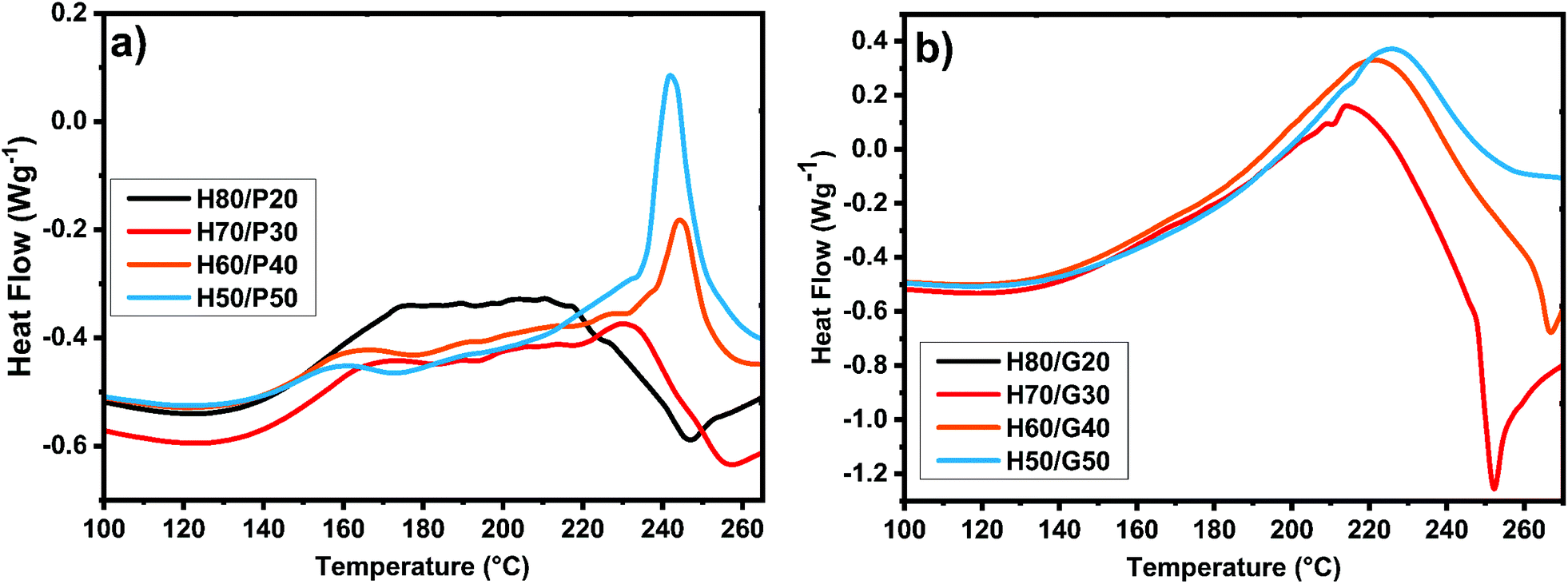 | ||
| Fig. 2 DSC analysis of curing mixtures without an initiator at 10 °C min−1 between (a) humins and different amounts of PEGDE; and (b) humins and different amounts of GDE. | ||
With the increase in the amount of PEGDE, a decrease in the enthalpy of reaction can be observed which could be a sign that the PEGDE interrupts the auto-crosslinking of humins and that the copolymerization needs activation by an initiator. In the case of humins/GDE mixtures, the double reaction peak is no longer observed, because a single, long temperature range, reaction occurs.
Regardless of the amount of GDE, the maximum reactivity of the mixtures is observed around a temperature of 220 °C, the temperature range of reactivity ranging between 110 °C and 260 °C. With the increase in the amount of GDE, the total heat release of the reaction rises from ≈240 J g−1 to ≈300 J g−1, except for the mixture with 50% comonomer, where the enthalpy of the reaction decreased considerably.
Following this study, these formulations were developed and cured in an oven to analyse if the amounts of compounds influence the physicochemical and mechanical properties of the materials. Practically, the thermosets obtained with less than 20 wt% PEGDE or GDE were rigid, brittle and thermally unstable, starting to foam at 130 °C, while the materials obtained with more than 30 wt% PEGDE or GDE were elastic and thermally stable when heated for curing at 150 °C. Then, the resins with a 50 wt% comonomer ratio were not selected because the aim of the study is valorization of the industrial humins by-product which should be the major part of the formulation composition. Following these rationales, the optimal amount of comonomers was determined to be 40 wt%. Therefore, three formulations were selected: HP40B5, HG40B5 and the combination HP20G20B5.
To analyze properly the copolymerization reactions between humins and PEGDE or GDE, the reactivity of selected formulations in the presence of BDMA (5 wt%) as the initiator was studied. Normalized dynamic DSC thermograms of humins-based resin polymerizations are shown in Fig. 3 and data summarized in Table 2.
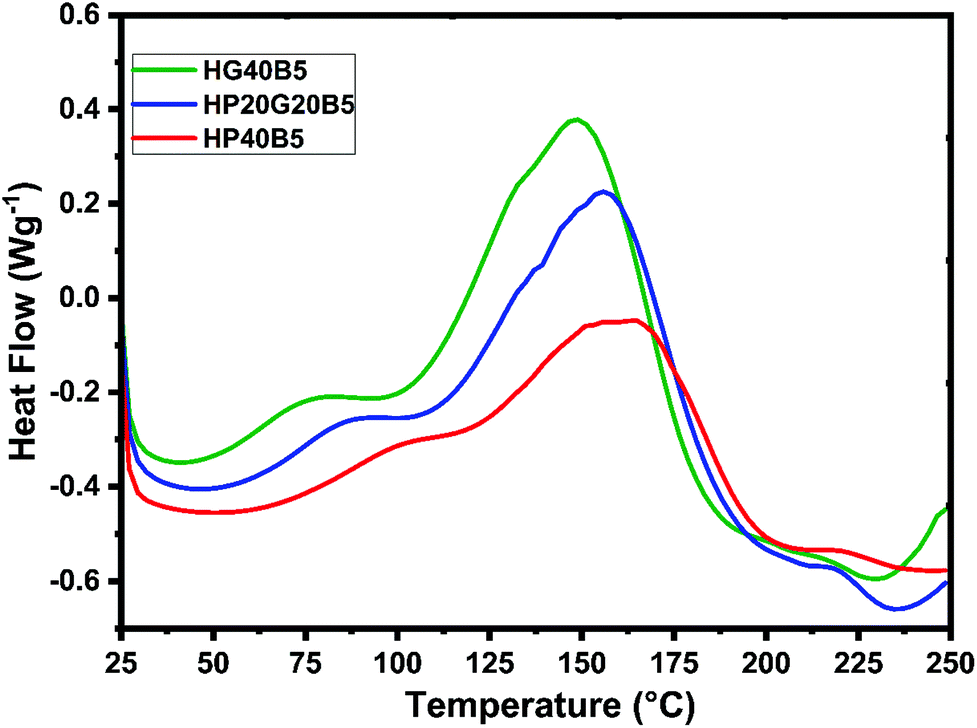 | ||
| Fig. 3 Comparison between humins copolymerization reactions by dynamic DSC at 10 °C min−1. | ||
| HP40B5 | HP20G20B5 | HG40B5 | |
|---|---|---|---|
| T peak (°C) (reaction interval) | 164 ± 1 (42–215) | 157 ± 1 (40–230) | 149 ± 1 (38–228) |
| Δ r H (J g−1) | 212 ± 4 | 319 ± 4 | 333 ± 4 |
When BDMA is used as the initiator a strong decrease in the starting reaction temperature occurs, whatever the type of diglycidyl type comonomer. If the systems without an initiator start to react at temperatures higher than ≈100 °C, the presence of BDMA allows reactions to start at around 40 °C. Then, the reactivities remain complex, with overlapped thermal events indicating the multiple reactions that could occur between the functional groups present in humins and the diglycidyls. The three thermograms follow the same trend, with a first small exotherm with a peak at ≈80–100 °C followed by the main exotherm of the reaction with the maximum at 150–160 °C. At around 230 °C the DSC heat flows return to a quasi-linear response which represents the completion of curing. The areas under the exothermal peaks were integrated to obtain the heat of copolymerization. Among these three formulations, the greatest reaction enthalpy was shown by the mixture in which GDE was used and it is around 333 J g−1. Secondary DSC scans at 250 °C reveal that no residual reactions occur.
As presented in Scheme 1 the copolymerization reactions of the humins with the diglycidyls could occur via an anionic mechanism. The first step is the initiation reaction with the participation of the tertiary amine in the ring opening of epoxy groups and formation of the zwitterions and then of the alkoxides which are the active species in anionic propagation reactions. We can suppose that this reaction of initiation appears in the three DSC thermograms as the small exotherms in the 80–100 °C temperature range. Then, once the alkoxides are formed, the propagation reactions occur faster, with a high enthalpy of reaction. The propagation reactions of humins–diglycidyl copolymerization are the main exothermic phenomena appearing in the DSC thermograms (Fig. 3) in the ∼100–200 °C interval. By comparing the reactions of humins with PEGDE and GDE, it is found that the copolymerization with the glycerol diglycidyl ether is more exothermic, with a higher reaction rate as shown by the higher slope of the peak. This higher reactivity could be explained by the presence of –OH in the GDE structure. The hydroxyl group of GDE could have a dual contribution: (i) via the interaction with the –COOH functions of the humins, and/or (ii) via the formation of ternary systems in the initiation steps. GDE's contribution also appears in terms of enthalpy and the reaction rate in the HP20G20B5 copolymerization system.
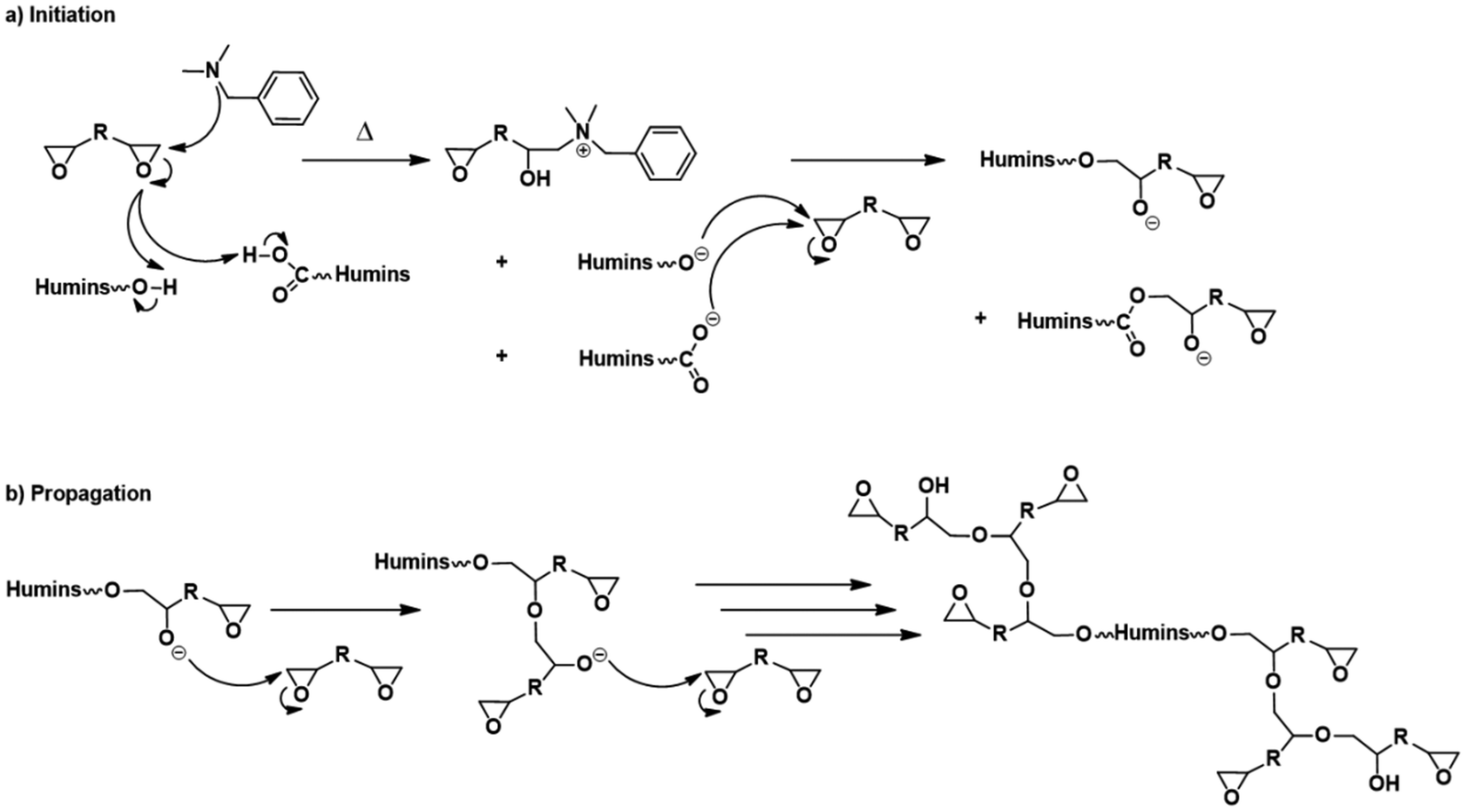 | ||
| Scheme 1 Summary of possible interactions among humins, aliphatic diepoxides and BDMA. | ||
Evolution of the structure during copolymerization reactions
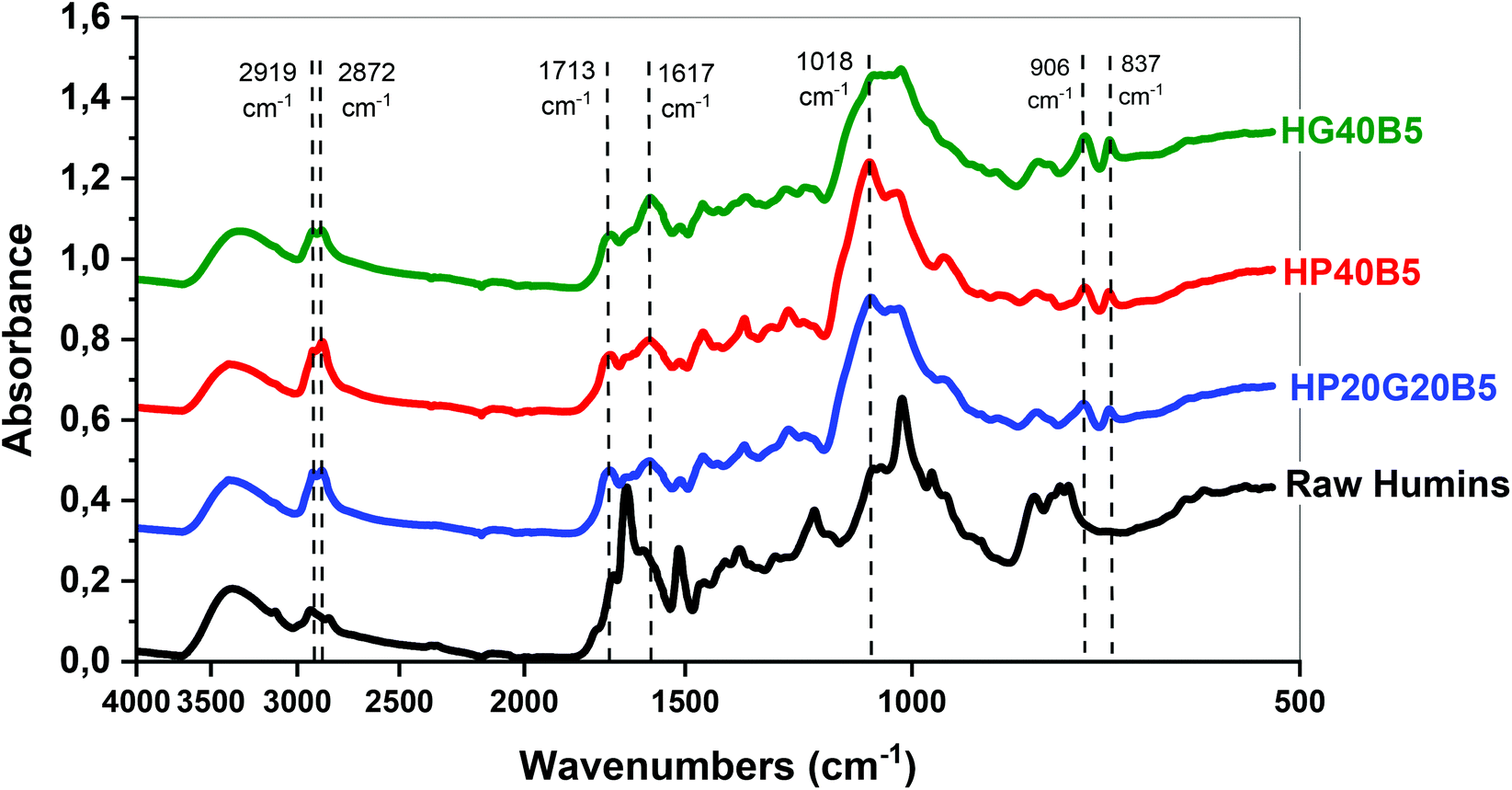
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the aldehyde groups of HMF and MMF, while the CO asymmetric stretching vibrations are presented at 1702 cm−1. FT-IR absorbtion signals that can be attributed to the substituted furan rings are at 1617 cm−1 a CC stretching vibration, at 1018 cm−1 a C–O stretching vibration or furan ring deformation, and also the signals from C–H out-of-plane deformation vibrations at 804 cm−1, 768 cm−1 and 756 cm−1. In the case of PEGDE and GDE spectra, the most important signals are CH2–O–CH2 asymmetric stretching vibrations of the ether groups at 1092 cm−1 for PEGDE and at 1089 cm−1 for GDE, and also the asymmetric stretching vibrations for the epoxy ring which appear at 994, 946, 910, and 840 cm−1 for PEGDE and at 984, 906, and 837 cm−1 for GDE. After analyzing the spectra of precursors, the three crosslinked resins were examined. For example, in the HG40B5 resin case we can observe that the peaks corresponding to –OH of humins and –OH present in the GDE structure are still present in the thermoset, meaning that the main reactions are not the etherification through epoxy ring additions to these groups (Fig. 4b). Then at 2919 and 2872 cm−1 a well-defined doublet corresponding to p3 C–H stretching could indicate that during the copolymerization alkyl connections were formed inside the network. The CO asymmetric stretching vibration at 1702 cm−1 in the spectrum of humins is shifted slightly to the left (1713 cm−1) and the shape is wider in the spectrum of the thermoset. This peak is not observed for GDE. The CC stretching peak for the disubstituted furans is no longer visible in the spectrum of the resin, and the peak of the polysubstituted furans shifts from 1517 cm−1 to 1596 cm−1, this being larger and lower in intensity. The C–O–C bonding of the humins are present as a multiplet with a maximum at 1018 cm−1, and in the case of GDE there is an intense and well-defined peak at 1090 cm−1.
O stretching vibrations of the aldehyde groups of HMF and MMF, while the CO asymmetric stretching vibrations are presented at 1702 cm−1. FT-IR absorbtion signals that can be attributed to the substituted furan rings are at 1617 cm−1 a CC stretching vibration, at 1018 cm−1 a C–O stretching vibration or furan ring deformation, and also the signals from C–H out-of-plane deformation vibrations at 804 cm−1, 768 cm−1 and 756 cm−1. In the case of PEGDE and GDE spectra, the most important signals are CH2–O–CH2 asymmetric stretching vibrations of the ether groups at 1092 cm−1 for PEGDE and at 1089 cm−1 for GDE, and also the asymmetric stretching vibrations for the epoxy ring which appear at 994, 946, 910, and 840 cm−1 for PEGDE and at 984, 906, and 837 cm−1 for GDE. After analyzing the spectra of precursors, the three crosslinked resins were examined. For example, in the HG40B5 resin case we can observe that the peaks corresponding to –OH of humins and –OH present in the GDE structure are still present in the thermoset, meaning that the main reactions are not the etherification through epoxy ring additions to these groups (Fig. 4b). Then at 2919 and 2872 cm−1 a well-defined doublet corresponding to p3 C–H stretching could indicate that during the copolymerization alkyl connections were formed inside the network. The CO asymmetric stretching vibration at 1702 cm−1 in the spectrum of humins is shifted slightly to the left (1713 cm−1) and the shape is wider in the spectrum of the thermoset. This peak is not observed for GDE. The CC stretching peak for the disubstituted furans is no longer visible in the spectrum of the resin, and the peak of the polysubstituted furans shifts from 1517 cm−1 to 1596 cm−1, this being larger and lower in intensity. The C–O–C bonding of the humins are present as a multiplet with a maximum at 1018 cm−1, and in the case of GDE there is an intense and well-defined peak at 1090 cm−1.
 | ||
| Fig. 4 FT-IR spectra of cured humins-based resins. | ||
| Sample | Wavenumber (cm−1) | Assignments | Sample | Wavenumber (cm−1) | Assignments |
|---|---|---|---|---|---|
| Humins | 3369 | O–H stretching | GDE | 3482 | O–H stretching |
| 2930, 2837 | Alkyl C–H stretching | 3000, 2872 | C–H stretching | ||
| 1702 | CO stretching |
1457, 1432 | CH2 δ-deformation vibration, coincides with CH3 δ asymmetric stretching | ||
| 1666 | CO stretching in furan |
||||
| 1617, 1517 | CC stretching |
1339 | O–H δ-deformation, in plane | ||
| 1396, 1360 | O–H δ-deformation, in plane | 1253 | C–O stretching | ||
| 1277, 1190, 1058, 1018 | C–O stretching | 1089 | CH2–O–CH2 asymmetric stretching (ether group) | ||
| C–O stretching in furan | |||||
| 965 | C–H δ deformation |
984, 906, 837 | Asymmetric stretching vibration for the epoxy ring | ||
| 804, 768, 756 | C–H out-of-plane deformation | 755, 702 | CH2 γ-skeletal deformation | ||
| PEGDE | 2865 | C–H stretching vibration | BDMA | 3086, 3063, 3027, 2973, 2941, 2854, 2814, 2763 | C–H stretching |
| 1456 | CH2 δ-deformation, coincides with CH3 δ as | 1601 | CC stretching vibration (benzene ring) |
||
| 1349, 1295, 1251 | C–O stretching | 1495, 1452 | CH2 δ-deformation, coincides with CH3 δ asymmetric | ||
| 1092 | CH2–O–CH2 asymmetric stretching (ether group) | 1363, 1317 | CH3 δ-symmetric deformation (from –N(CH3)2) | ||
| 994, 946, 910, 840 | Asymmetric stretching for the epoxy ring | 1259, 1174, 1146, 1097, 1075, 1025 | C–N stretching | ||
| 758 | CH2 γ-skeletal | 975 | C–H deformation |
||
| 908, 850, 734, 696, 608 | C–H out-of-plane deformation (benzene ring) |
||||
| System | Tanδ |
Storage modulus, E′ (MPa) | ν (mmol cm−3) | ||||
|---|---|---|---|---|---|---|---|
| Peak (°C) | Interval of tanδ (°C) |
Peak height | In glassy plateau (at −100 °C) | At tanδ |
In rubbery plateau (at 150 °C) | ||
| HP40B5 | 30 | 100 | 0.76 | 5473 | 27 | 1 | 0.07 |
| HP20G20B5 | 49 | 123 | 0.5 | 5916 | 25 | 4.5 | 0.43 |
| HG40B5 | 71 | 142 | 0.4 | 6777 | 96 | 6.6 | 0.56 |
In the resin spectrum, the peak is located at 1020 cm−1, being larger compared to those of the other two compounds, and its absorbance is lower. The disappearance of the peak of oxirane ring stretching which absorbs in GDE at 980–840 cm−1 confirms the consumption of epoxy functions during humins/GDE copolymerization (Table 3).
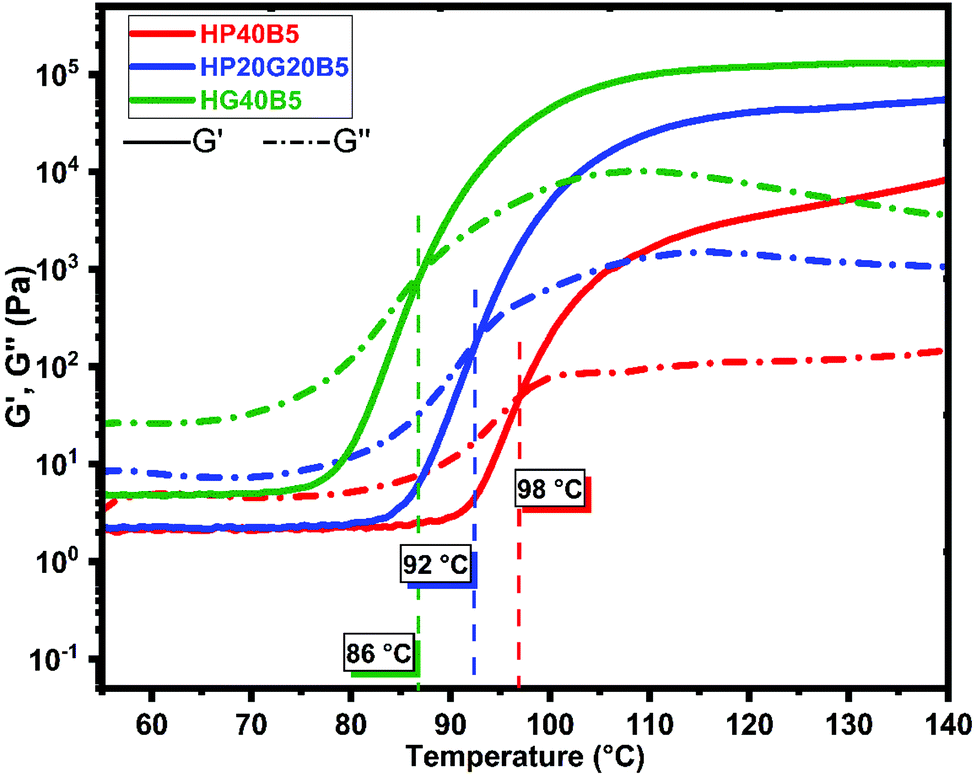 | ||
| Fig. 5 Rheology study of the humins-based systems for copolymerization. Evolution of moduli and viscosity during heating at 50–200 °C at 0.2 °C min−1. Comparison between formulations. | ||
From the rheology analyses it can be observed that the systems start to react, when heated at 0.2 °C min−1, in the interval 65–75 °C depending on the composition. The onset of reaction for the three formulations takes place at different temperatures, decreasing from 75 °C for the formulation with PEGDE till 65 °C for the system with humins and GDE. These results are in good agreement with the DSC analysis results (Fig. 3) which show a higher reactivity for the system with GDE. The hardening of resin is given by the gel point of the system, taken as a crossover between G′ and G′′. For the resin formulation composed of humins and PEGDE the gelation process takes place at around 98 °C, while for the humins/GDE resin the gelling point is at around 86 °C. It can also be seen that the viscosity at 25 °C of the formulation which contains PEGDE is low, around ≈0.35 Pa s, around ten times lower compared with the viscosity of the formulation with GDE with an approximate value of 2.5 Pa s.
Physicochemical characterization of humins-based copolymers
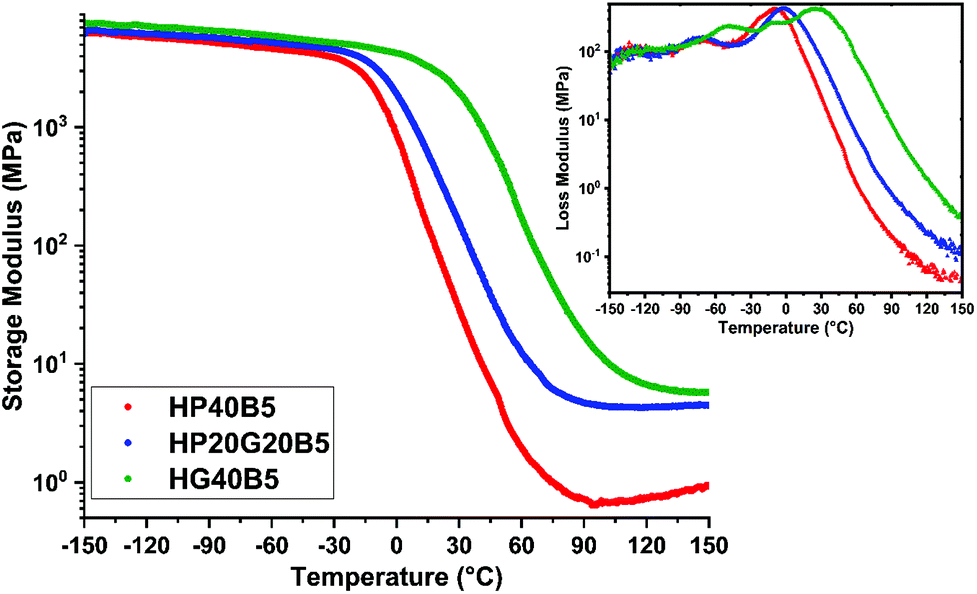
The DMA results obtained over the interval temperature range −150 to 150 °C are given in Fig. 6 and the main parameters are listed in Table 4. The values of maximum of tanδ are assigned to the α relaxation (Tα) phenomenon related to cooperative chain motions and associated with the macroscopic Tg. Fig. 6 shows the evolution of the storage and loss moduli of the three humins-based resins as a function of temperature.
 | ||
| Fig. 6 Evolution of the storage modulus and loss modulus vs. temperature for humins-based copolymers. | ||
At temperatures below the glass transition, in the glassy region of the materials, high storage modulus values were measured, of about 5400–6700 MPa. We can notice that the humins/PEGDE copolymer presents the smallest value of the E′ modulus (≈5500 MPa), emphasizing the contribution of aliphatic flexible chains inside the copolymer network, giving an elastic resin.
A higher value of modulus (≈6800 MPa) was measured in the humins/GDE copolymer showing a stiffer network, which was initially intended to achieve by the choice of a shorter aliphatic ether chain. Obviously, the copolymer with mixed glycidyls has an intermediate behavior.
With the increase of temperature, in the transition zone, we can observe a substantial decrease in the E′ values. A clear shift of the E′ drop between the three resins can be observed. The temperature at which the E′ of the flexible humins-based resin starts to decrease is around −26 °C while for the GDE based resin this drop starts at around 10 °C.
The domain above the glass transition has a width dependent on the molecular weight of the chains between entanglements or crosslinks.44,48 By increasing the molecular weight of chain segments the transition to the rubbery plateau appears at higher temperatures. In the prepared humins/diglycidyl copolymers the zone appears in the order HP40B5 < HP20G20B5 < HG40B5.
In the rubbery plateau, the storage modulus value is directly correlated with the crosslinking density of the networks (Flory's theory).40 The bigger the crosslinking density of the polymer, the smaller the drop of the storage modulus value.45,46 The crosslinking densities for the three humins-based copolymers were calculated and are given in Table 4.
Moreover, the α, β and γ transitions of copolymers were also investigated by DMA. Fig. 7 shows the evolution of tanδ for the three humins based copolymer networks. In our previous work27 we have shown by dynamic DSC that the crude humins have a Tg at −15 °C, while humins auto-crosslinked at 250 °C have a Tg around 66 °C. Compared with the humins thermoset we can observe that the copolymerization with tailored diglycidyl aliphatic ethers allowed the tailoring of the mechanical properties. The shorter aliphatic ether chains from GDE gave a more rigid network, with a Tα ≈ 71 °C and a smaller tanδ peak. In contrast, by increasing the length of the aliphatic ether chains, copolymers with PEGDE, the Tα decreases to ≈30 °C. The copolymer prepared with the combination of diglycidyls (PEGDE + GDE) and humins has an intermediate trend, with a Tα ≈ 49 °C and a peak intensity more determined by the GDE contribution. Interestingly, all the three copolymer networks have similar ranges of the α relaxation peaks which are very large (between 100 and 140 °C) indicating a large distribution of relaxation times, characteristic of long polymeric chains. The amplitude of tanδ provides information on the crosslinking density of the materials. For the HG40B5 resin, the small amplitude of the tanδ peak indicates a low damping capacity of the material and consequently indicates that HG40B5 is a less elastic thermoset. This observation is in accord with the fact that the HG40B5 resin presents the highest crosslinking density. In contrast, HP40B5 shows a relaxation at lower temperature, as a consequence of a much more flexible network due to the PEGDE aliphatic ether contribution. The corresponding tanδ peak exhibits an intensity that indicates higher damping capacity through energy dissipation, and consequently a flexible network, in good accord with its low value of crosslinking density. As illustrated in Fig. 7b, all the three copolymers present a small γ relaxation at around Tγ ≈ −130 °C which could be associated with the rotation of hydroxyl or other functional groups from humins or from diglycidyls. Then, at ≈−73 °C for HP40B5 and at ≈−71 °C for HP20G20B5 beta relaxations corresponding to local motions of copolymers occur. For the HG40B5 system two beta transitions emerge at Tβ ≈ −47 °C and Tβ′ ≈ −12 °C. The free moving dangling of segments of chains present in the network after cross-linking and the local breathing of furanic entities could be the origin and could explain these relaxations.49
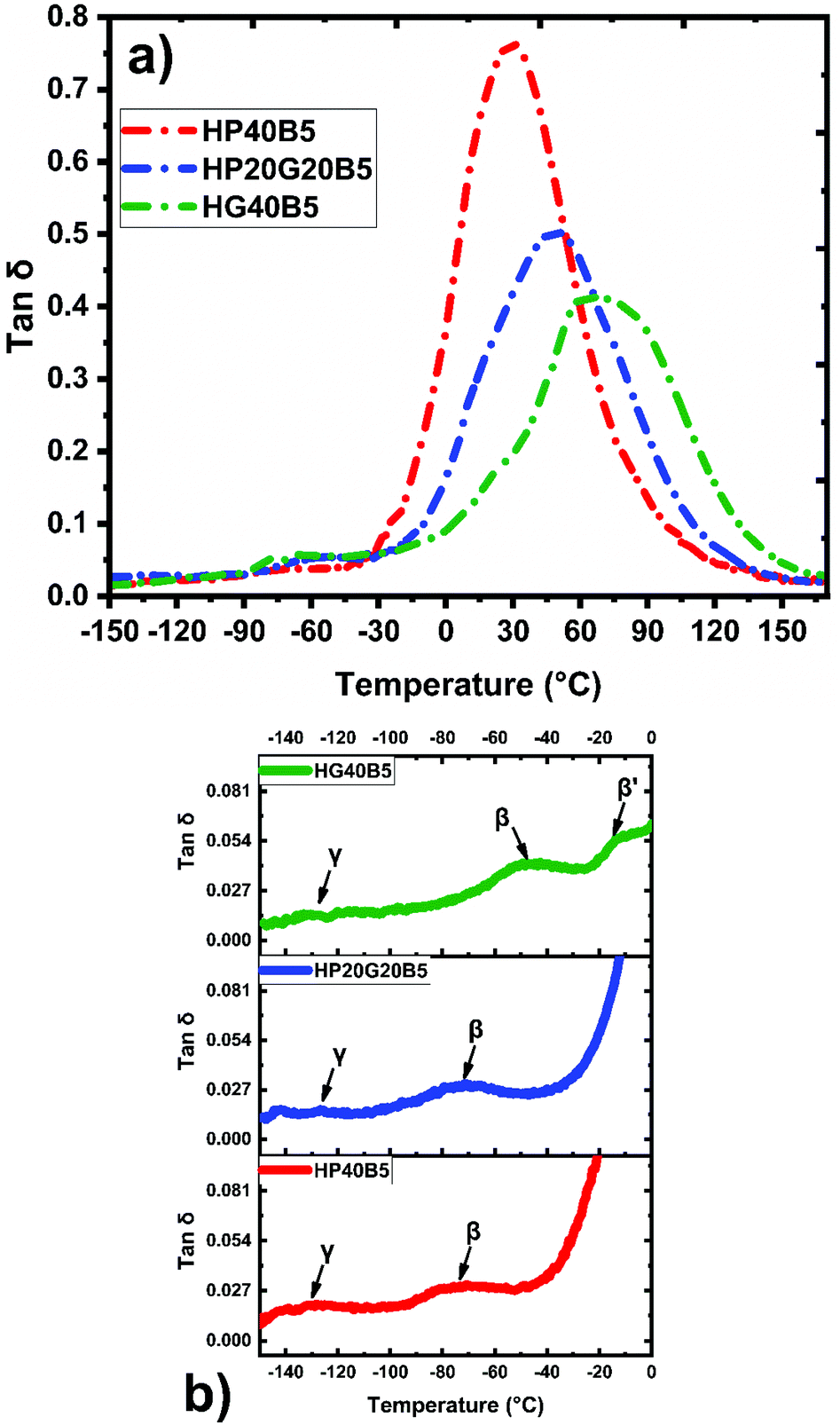 | ||
| Fig. 7 Evolution of relaxation processes with temperature on the humins-based copolymers: (a) tanδ; and (b) β and γ transitions. | ||
The glass transition phenomenon was also studied with the help of DSC. The Tg values of cured materials were analyzed using 2 heating/cooling cycles at a temperature range of −50 to 220 °C and a 10 °C min−1 heating rate.
The obtained results (not presented here) corroborate very well with those measured by DMA (Tg-loss) and are summarized in Table 5. In perfect accord with previously reported DMA results, the DSC results indicate the difference between the three humins based copolymers in terms of Tg. Accordingly, the presence of PEGDE in the copolymer network has a strong effect on its thermomechanical properties. The copolymer with a higher content on PEGDE – HP40B5 – has negative Tg values ≈−10 to −8 °C.
|
|
|
|
|
|---|---|---|---|
| Tg from DSC/°C | −8 | 1 | 27 |
| T g-loss/°C (DMA) | −10 | −2 | 26 |
| T g-onset/°C (DMA) | −11 | −4 | 27 |
| Shore hardness*/SD | 44 | 59 | 72 |



Its combination with a shorter aliphatic ether (GDE) gives a less elastic material, with a higher Tg at around 10 °C, Tg ≈ −2 to −1 °C. In contrast, the resin with GDE was considered a more rigid material, having a Tg of ≈27 °C. These results were corroborated with the hardness test results where the hardness of the materials follow the same trend as the Tg values. Using the Shore hardness tests, the resistance of the humins-based resins was measured at the penetration of a spring-loaded needle-like indenter. Hardness of hard elastomers and most polymer materials such as thermoplastics and thermosets is measured with a Shore D scale. The Shore hardness test of the three copolymers revealed values from 44SD to 72SD. According to the Shore hardness scale, the first formulation corresponds to a medium hard material which can be compared with a shoe heel, door seal, or automotive tire tread, while the hardness of the other two resins can be compared with ebonite rubber, solid truck tires, hard wheels of roller skates and skateboard, computer casing, etc.
Fig. 8 shows the tensile test results with stress–strain curves of the three humins based copolymers investigated in this work. We can notice that the combination of humins–diglycidyls gave copolymers with different mechanical responses which vary coherently with the values obtained by DMA, DSC and hardness tests. A significant increase in the maximum strength of the resins with the GDE content can be noticed. In agreement with the DMA results, the HP40B5 copolymer presents low Tα and high ductility, the tensile strength for this resin being around 0.4 MPa, while for the rigid HG40B5 copolymer the strength reaches around 2.2 MPa. The elastic character of the HP40B5 resin is also shown by its elongation at break, the maximum strain being at around 60%.
 | ||
| Fig. 8 Tensile profiles for the three humins-based resins. | ||
As the HG40B5 resin matrix is crosslinked more densely, its capability to undergo plastic deformation is reduced. The HG40B5 resin presents a low elongation at break (≈7%) which means that this resin is more rigid and brittle than the HP40B5 resin (Table 6). Compared with the commercial resins it appears that the HP40B5 resin could be included in the category of elastomers having a tensile strength value close to that of butyl rubber (0.001–0.002 GPa), or of silicone elastomers (0.005–0.2 GPa).50
| Young's modulus (MPa) | Tensile stress (MPa) | Strain (%) | |
|---|---|---|---|
| HP40B5 | 1.01 ± 0.08 | 0.38 ± 0.12 | 62.4 ± 6.18 |
| HP20G20B5 | 8.29 ± 0.61 | 1.33 ± 0.19 | 23.2 ± 6.76 |
| HG40B5 | 106.1 ± 39 | 2.14 ± 0.55 | 6.4 ± 5.23 |
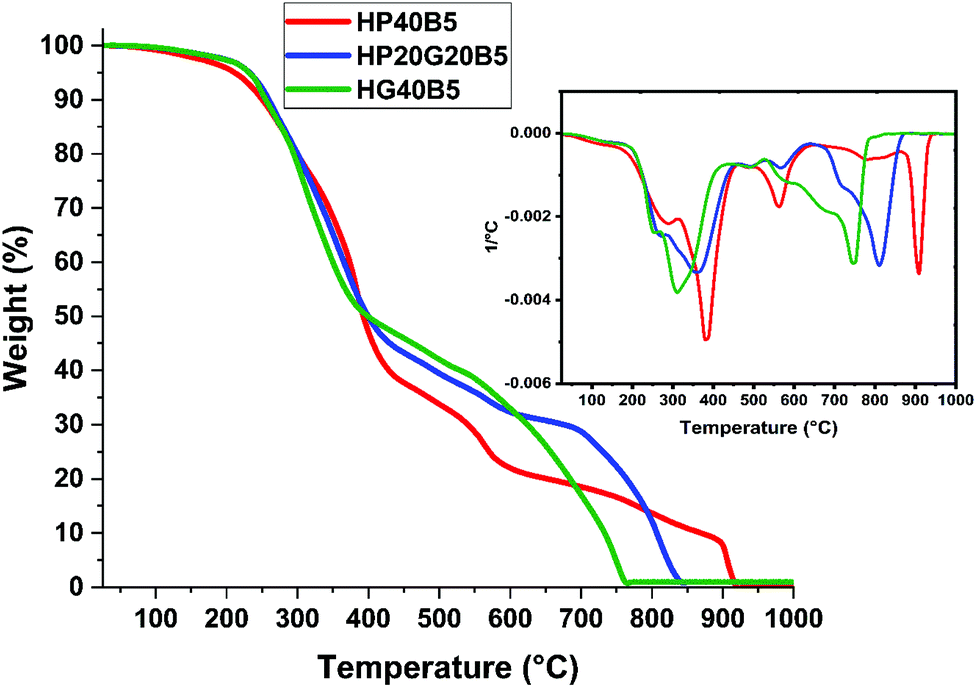 | ||
| Fig. 9 Thermogravimetric analysis of humins based copolymers at 10 °C min−1 under air flow. Inset: weight loss of the derivative. | ||
| Temperature (°C) | |||
|---|---|---|---|
| HP40B5 | HP20G20B5 | HG40B5 | |
| T 5% under air | 212 | 232 | 233 |
| T 5% under N2 | 244 | 251 | 246 |
According to previous studies performed on industrial humins produced by Avantium,29,55 the carbon content of the crude humins is around 54%. On the basis of weight, the %C in PEGDE is 51.69%, in GDE it is 52.88% and in BDMA it is 79.88%. The HP40B5 and HG40B5 systems contain 55 g humins, 40 g PEGDE or GDE and 5 g BDMA. The HP20G20B5 system contains 55 g humins, 20 g PEGDE, 20 g GDE and 5 g BDMA. Theoretically, when only carbon is considered, the biobased carbon content in each formulation is: ≈54.63% for HP40B5, ≈54.14% for HG40B5 and ≈54.39% for HP20G20B5. Hence, the prepared formulations are biomass materials satisfying the criteria for biobased carbon contents.
Conclusions
In conclusion three thermoset resins with a biobased carbon content >54% were obtained from humins and aliphatic diglycidyl ethers. The effect of the formulation composition on the reactivity of the systems were studied by various methods, DSC, FT-IR and rheometry. All systems showed good reactivity during the copolymerization between the humins and the diglycidyls. The properties of the obtained materials were analyzed by DMA, tensile and Shore tests. These analyses proved the macroscopic homogeneity of the three copolymer materials and their ductile character. Resins with longer aliphatic chains, HP40B5, reached ≈60% elongation at break. All these properties together with the low curing time and temperature, and also the reduced numbers of components make these results feasible for the industry. To our knowledge, these are the first data reported on the synthesis of elastomeric humins based copolymers paving the way for their utilization, for example, as matrices for composites for industrial applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the KaRMA2020 project. This project has received funding from the European Union's Horizon 2020 Research and Innovation program under Grant Agreement no. 723268.Notes and references
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- S. K. R. Patil, J. Heltzel and C. R. F. Lund, Energy Fuels, 2012, 26, 5281–5293 CrossRef CAS.
- A. Schweizer, Recl. Trav. Chim. Pays-Bas, 1938, 57, 345–382 CrossRef CAS.
- T. M. C. Hoang, L. Lefferts and K. Seshan, ChemSusChem, 2013, 6, 1651–1658 CrossRef CAS PubMed.
- X. Hu, C. Lievens, A. Larcher and C. Z. Li, Bioresour. Technol., 2011, 102, 10104–10113 CrossRef CAS PubMed.
- C. B. Rasrendra, M. Windt, Y. Wang, S. Adisasmito, I. G. B. N. Makertihartha, E. R. H. Van Eck, D. Meier and H. J. Heeres, J. Anal. Appl. Pyrolysis, 2013, 104, 299–307 CrossRef CAS.
- M. M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204–1212 RSC.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289 CrossRef CAS.
- M. Zheng, Y. Liu, K. Jiang, Y. Xiao and D. Yuan, Carbon, 2010, 48, 1224–1233 CrossRef CAS.
- M. D. P. Buera, J. Chirife, S. L. Resnik and G. Wetzler, J. Food Sci., 1987, 52, 1063–1067 CrossRef.
- L. W. Kroh, Food Chem., 1994, 51, 373–379 CrossRef CAS.
- B. Cämmerer, B. L. Wedzicha and L. W. Kroh, Eur. Food Res. Technol., 1999, 209, 261–265 CrossRef.
- M. A. C. Quintas, T. R. S. Brandão and C. L. M. Silva, J. Food Eng., 2007, 83, 483–491 CrossRef CAS.
- M. del Pi. Buera, J. Chirife, S. L. Resnik and R. D. Lozano, J. Food Sci., 1987, 52, 1059–1062 CrossRef.
- A. Campanella, M. Zhan, P. Watt, A. T. Grous, C. Shena and R. P. Wool, Composites, Part A, 2015, 72, 192–199 CrossRef CAS.
- I. Van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. Van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- M. Sevilla and A. B. Fuertes, Chem. – Eur. J., 2009, 15, 4195–4203 CrossRef CAS.
- L. Filiciotto, A. M. Balu, J. C. Van der Waal and R. Luque, Catal. Today, 2018, 302, 2–15 CrossRef CAS.
- H. T. M. Châu, PhD Thesis, University of Twente, 2014.
- T. M. C. Hoang, E. R. H. van Eck, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Green Chem., 2015, 17, 959–972 RSC.
- S. Constant, C. S. Lancefield, B. M. Weckhuysen and P. C. A. Bruijnincx, ACS Sustainable Chem. Eng., 2017, 5, 965–972 CrossRef CAS.
- S. Kang, G. Zhang, Q. Yang, J. Tu, X. Guo, F. G. F. Qin and Y. Xu, BioResources, 2017, 11, 9496–9505 Search PubMed.
- J. M. Pin, N. Guigo, A. Mija, L. Vincent, N. Sbirrazzuoli, J. C. Van Der Waal and E. De Jong, ACS Sustainable Chem. Eng., 2014, 2, 2182–2190 CrossRef CAS.
- A. Mija, E. de Jong, J. C. van der Waal and G. van Klink, Humins containing foam, PCT Int. Appl., WO 2017074183 A1 20170504, 2017.
- P. Tosi, G. P. M. van Klink, A. Celzard, V. Fierro, L. Vincent, E. de Jong and A. Mija, ChemSusChem, 2018, 11, 2797–2809 CrossRef CAS PubMed.
- S. Kang, J. Fu, Z. Deng, S. Jiang, G. Zhong, Y. Xu, J. Guo and J. Zhou, Sustain., 2018, 10, 16–19 Search PubMed.
- A. Muralidhara, P. Tosi, A. Mija, N. Sbirrazzuoli, C. Len, V. Engelen, E. De Jong and G. Marlair, ACS Sustainable Chem. Eng., 2018, 6, 16692–16701 CrossRef CAS.
- A. Mija, E. de Jong, J. C. Van der Waal and G. Klink, Process for the Modification of Humins, WO 2018/062995 A1, 2018.
- N. S. V. Capanema, A. A. P. Mansur, A. C. de Jesus, S. M. Carvalho, L. C. de Oliveira and H. S. Mansur, Int. J. Biol. Macromol., 2018, 106, 1218–1234 CrossRef CAS PubMed.
- V. S. Ghorpade, A. V. Yadav, R. J. Dias, K. K. Mali, S. S. Pargaonkar, P. V. Shinde and N. S. Dhane, Int. J. Biol. Macromol., 2018, 118, 783–791 CrossRef CAS PubMed.
- E. M. Van Wagner, A. C. Sagle, M. M. Sharma, Y. H. La and B. D. Freeman, J. Membr. Sci., 2011, 367, 273–287 CrossRef CAS.
- N. Vasylieva, B. Barnych, A. Meiller, C. Maucler, L. Pollegioni, J. S. Lin, D. Barbier and S. Marinesco, Biosens. Bioelectron., 2011, 26, 3993–4000 CrossRef CAS PubMed.
- H. Kono, Carbohydr. Polym., 2014, 106, 84–93 CrossRef CAS PubMed.
- S. Kasetaite, J. Ostrauskaite, V. Grazuleviciene, J. Svediene and D. Bridziuviene, Polym. Bull., 2015, 72, 3191–3208 CrossRef CAS.
- L. Zhao, Y. Chen, W. Li, M. Lu, S. Wang, X. Chen, M. Shi, J. Wu, Q. Yuan and Y. Li, Carbohydr. Polym., 2015, 121, 276–283 CrossRef CAS PubMed.
- S. ur Rehman, M. Sahiner, K. Sel, M. Siddiq and N. Sahiner, Colloids Surf., B, 2015, 136, 1156–1165 CrossRef CAS PubMed.
- ASTM-D7028, 2008.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, New York, 1953 Search PubMed.
- ISO 527-1 - Tensile test, 1996.
- ASTM-D638-14, 2015.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- M. S. Carvalho, M. Padmanabhan and C. W. Macosko, J. Rheol., 2002, 38, 1925–1936 CrossRef.
- V. B. Gupta, L. T. Drzal and C. Y.-C. Lee, Polym. Eng. Sci., 1985, 25, 812–823 CrossRef CAS.
- A. Ullah and J. Wu, Macromol. Mater. Eng., 2013, 298, 153–162 CrossRef CAS.
- K. P. Menard, Dynamic Mechanical Analysis - A Practical Introduction, CRC Press LLC, Boca Raton, Florida, 1999 Search PubMed.
- J. Pascault, H. Sautereau, J. Verdu and R. J. J. Williams, Thermosseting polymers, MarcelDekker, Inc., 2002 Search PubMed.
- J. M. Pin, N. Guigo, L. Vincent, N. Sbirrazzuoli and A. Mija, ChemSusChem, 2015, 8, 4149–4161 CrossRef CAS PubMed.
- Materials Data Book, Cambridge University Engineering Department, 2003 Search PubMed.
- G. A. Norton and S. L. Devlin, Bioresour. Technol., 2006, 97, 2084–2090 CrossRef CAS PubMed.
- M. Kunioka, Radioisotopes, 2013, 62, 901–925 CrossRef CAS.
- NCS-16785, Bio-based content certification scheme, 2016.
- X. Pan, P. Sengupta and D. C. Webster, Biomacromolecules, 2011, 12, 2416–2428 CrossRef CAS PubMed.
- A. Muralidhara, A. Bado-Nilles, G. Marlair, V. Engelen, C. Len and P. Pandard, Biofuels, Bioprod. Biorefin., 2018, 1–7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc01813a |
| This journal is © The Royal Society of Chemistry 2019 |