
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtending the chemical product tree: a novel value chain for the production of N-vinyl-2-pyrrolidones from biogenic acids†
Moritz Otto
Haus
 ,
Yannik
Louven
and
Regina
Palkovits
*
,
Yannik
Louven
and
Regina
Palkovits
*
Institut für Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, Worringerweg 2, Aachen, DE-52074, Germany. E-mail: palkovits@rwth-aachen.de
First published on 16th October 2019
Abstract
The sustainable production of polymers from biogenic platform chemicals shows great promise to reduce the chemical industry's dependence on fossil resources. In this context, we propose a new two-step process leading from dicarboxylic acids, such as succinic and itaconic acid, to N-vinyl-2-pyrrolidone monomers. Firstly, the biogenic acid is reacted with ethanolamine and hydrogen using small amounts of water as solvent together with solid catalysts. For effective conversion, the optimal catalyst (carbon supported ruthenium) has to hold the ability of activating H2 as well as (imide) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The obtained products, N-(2-hydroxyethyl)-2-pyrrolidones, are subsequently converted in a continuous gas phase dehydration over simple sodium-doped silica, with excellent selectivity of above 96 mol% and water as the sole by-product. With a final product yield of ≥72 mol% over two process steps and very little waste due to the use of heterogeneous catalysis, the proposed route appears promising – commercially as well as in terms of Green Chemistry.
O bonds. The obtained products, N-(2-hydroxyethyl)-2-pyrrolidones, are subsequently converted in a continuous gas phase dehydration over simple sodium-doped silica, with excellent selectivity of above 96 mol% and water as the sole by-product. With a final product yield of ≥72 mol% over two process steps and very little waste due to the use of heterogeneous catalysis, the proposed route appears promising – commercially as well as in terms of Green Chemistry.
Introduction
Several carboxylic acids have been proposed as valuable platform chemicals for the future bio-economy.1 However, their potential is often hampered by a misfit between their highly oxygenated nature and the available processing technologies, which are optimized for the introduction of functionalities into non-polar, oil-derived feedstocks. The design of tailored conversion technologies, which target a partial deoxygenation, while effectively utilizing the (acid) functionalities of the platform, is thus an integral step towards the valorization of alternative carbon sources.2The reduction of biogenic acids to valuable chemicals, such as diols, is well researched.3–9 However, selective transformations are often hard to achieve and mixtures of reduced products are obtained. Another approach that effectively utilizes the acid functionality presents the conversion into pyrrolidones by reaction with amines. In this context, the reductive amination of levulinic acid has found substantial attention.10–13 Here, the carbonyl functionality of the substrate readily forms an imine intermediate with the added amine. Consecutive reduction, even at comparably low hydrogen pressure and temperature, yields cyclic pyrrolidones, which may find applications as solvents or pharmaceutical intermediates. Touchy et al.10 tested the conversion of levulinic acid with n-octylamine and found promoted platinum catalysts holding superior performance in the synthesis of N-alkylpyrrolidones. Their finding that the acid properties of the promoting metal oxide play a prominent role in determining catalyst activity agrees well with the assumption that imine formation is the rate determining step. This hypothesis was later supported by research on TiO2-supported platinum catalysts, where decoration of the metal surface with TiOx proved to enhance catalyst acidity and activity.12 Using ethyl levulinate as substrate, a plethora of amines, including alkyl-, phenyl-, ether- and hydroxyl-functionalities, was shown to be suitable for reductive amination with good yields.
Dicarboxylic acids, such as succinic and itaconic acid, are not susceptible to reductive amination as they lack the necessary carbonyl group. Instead they form amides, diamides and imides, when reacted with amines at elevated temperatures. Amide reduction has long been considered a major challenge for heterogeneous catalysis.14 Consequently, there are few publications on the successful production of pyrrolidones from dicarboxylic acids. Budroni et al.15 have elaborated on the reduction of succinic anhydride to γ-butyrolactone, a main intermediate in today's pyrrolidone production, over Au/TiO2. The one pot conversion with phenylamine to phenylpyrrolidone was also demonstrated. However, the yield was limited to <60% due to the sequential formation of the respective pyrrolidine. Other studies concerned the production of N-methyl-2-pyrrolidone (NMP) from succinate containing fermentation broth.16 Here, N-methyl-succinimide was distilled from the broth to be reduced over a commercial rhodium catalyst, yielding up to 90% NMP at 220 °C and 100 bar H2. However, the substrate scope was not expanded to allow for the production of more valuable chemicals.
Our group has recently shown the efficient conversion (90 mol% yield) of itaconic acid and ammonia to N-unsubstituted 3- and 4-methyl-2-pyrrolidones over carbon-supported ruthenium (conditions: 200 °C, 150 bar H2).17 The products of this conversion may subsequently be transformed into N-vinyl-2-pyrrolidone monomers by reaction with pressurized acetylene over a molecular potassium catalyst. The second reaction is known as part of the fossil-based value chain running from acetylene, over γ-butyrolactone and 2-pyrrolidone, to N-vinyl-2-pyrrolidone (NVP), pioneered by W. Reppe.18 NVP is polymerized to yield polyvinylpyrrolidone, a water-soluble, non-toxic polymer with growing application and demand, especially in the pharmaceutical industries.19
Despite its long-standing use, vinylation with pressurized acetylene raises concerns about its sustainability and safety. Specifically, the use of a hardly reusable catalyst and the necessity to remove traces of water to achieve effective conversion are undesirable process specifications.20,21 The most promising alternative to produce NVP is presented by the thermal reaction of ethanolamine and γ-butyrolactone. The product, N-(2-hydroxyethyl)-2-pyrrolidone (HEP) can be dehydrated in a gas-phase reaction leading to a simple, continuous production process, which is now in commercial application.22 Using conventional γ-butyrolactone as feedstock, this process remains fossil resource-based.
We herein present the reductive transformation of dicarboxylic acids with ethanolamine, leading to N-(2-hydroxyethyl)-2-pyrrolidones (reductive amidation), which are dehydrated to yield bio-based monomers analogous to NVP. Specifically, succinic acid is chosen as a model compound to study the underlying reaction network and kinetics of this transformation. Catalysts are screened for activity in the rate-determining step of the overall transformation and process conditions are optimized to show the potential for obtaining high yields of valuable products. Due to the spectrum of dicarboxylic acids obtainable from biomass, the substrate scope of the reductive transformation as well as of the continuous gas phase dehydration is explored. Overall, the envisioned two-step process (Fig. 1) yields NVP-like monomers on the basis of biogenic acids with water as sole by-product and, at the same time, solvent of the heterogeneously catalyzed reactions. Given the excellent yields obtained and the use of simple and/or commercially available catalysts, this seems to offer real potential for an applicable Green Chemistry value chain.
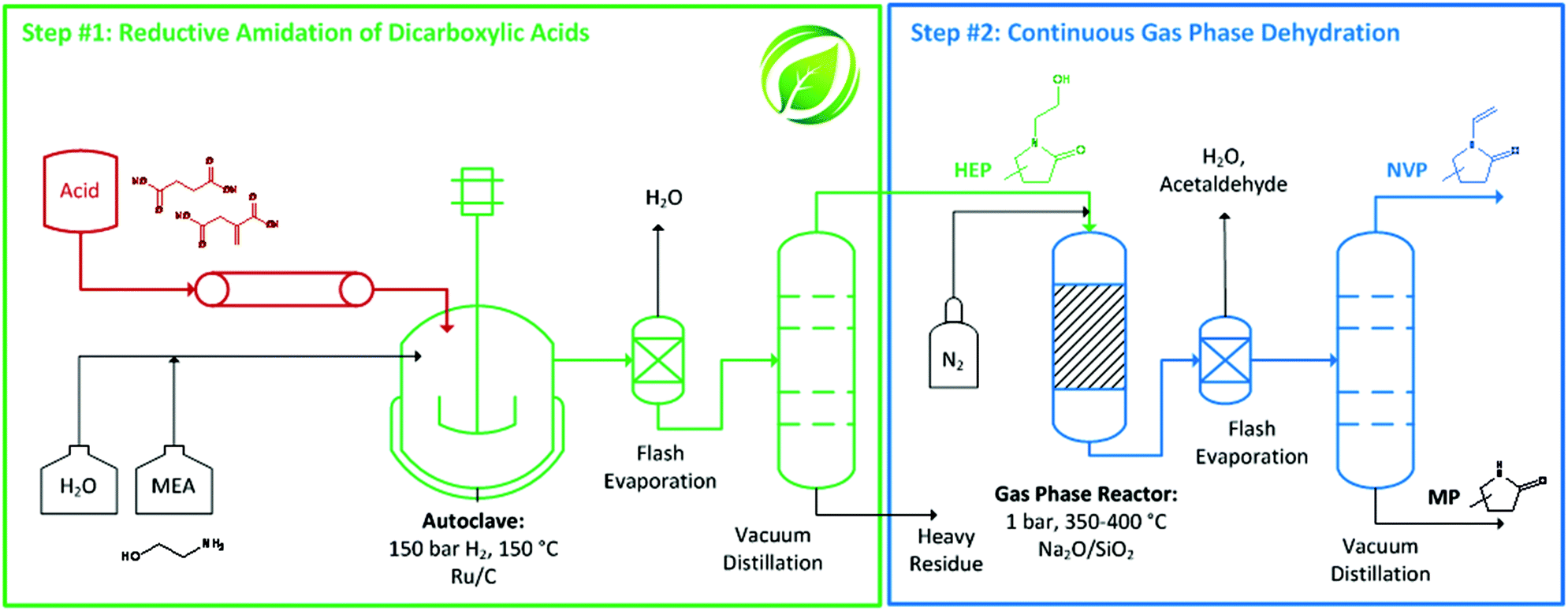 | ||
| Fig. 1 Schematic of the green value chain proposed herein. Two steps lead from dicarboxylic acids over N-(2-hydroxyethyl)-2-pyrrolidones (HEP) to valuable N-vinyl-2-pyrrolidone monomers (NVP). (MP = methyl-2-pyrrolidone).36 | ||
Experimental
Catalytic materials and chemicals
Hydrogenation reactions with ethanolamine
Reductive amidations were conducted with hydrogen as a reducing agent in 50 ml stainless steel autoclaves. Glass inlays were used to protect the autoclaves against corrosive solutions. For a typical experiment, 1.5 g of dicarboxylic acid was added to 1.5 g of de-ionized water and a stoichiometric amount of ethanolamine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). 2.5 wt% of catalyst (37.5 mg) were added and the resulting slurry was pre-mixed using a magnetic stirrer bar. Subsequently, the autoclave was sealed and purged three times with hydrogen before finally raising the pressure to 150 bar. For experiments at lower p(H2), nitrogen was added to reach 150 bar total pressure. The so-prepared autoclave was inserted into a pre-heated heating jacket (150 °C). Homogeneous mixing was accomplished with 750 rpm of the magnetic stirrer. After a pre-set time, the autoclave was removed from its jacket, to be quenched in ice. Following cool down to room temperature, the autoclave was depressurized and opened. Solids were removed from the product solution by a polyamide syringe filter before running HPLC analysis on an Organic Acid Resin column (CS – Chromatography Services) with dilute phosphoric acid as mobile phase and a refractive index detector.
:1). 2.5 wt% of catalyst (37.5 mg) were added and the resulting slurry was pre-mixed using a magnetic stirrer bar. Subsequently, the autoclave was sealed and purged three times with hydrogen before finally raising the pressure to 150 bar. For experiments at lower p(H2), nitrogen was added to reach 150 bar total pressure. The so-prepared autoclave was inserted into a pre-heated heating jacket (150 °C). Homogeneous mixing was accomplished with 750 rpm of the magnetic stirrer. After a pre-set time, the autoclave was removed from its jacket, to be quenched in ice. Following cool down to room temperature, the autoclave was depressurized and opened. Solids were removed from the product solution by a polyamide syringe filter before running HPLC analysis on an Organic Acid Resin column (CS – Chromatography Services) with dilute phosphoric acid as mobile phase and a refractive index detector.
Conversion and yield are calculated based on concentrations determined via HPLC through the assumption of mass conservation within the autoclave. For reasons that will shortly become evident, the following definitions are applied for the conversion X, yield Yi and selectivity Si:
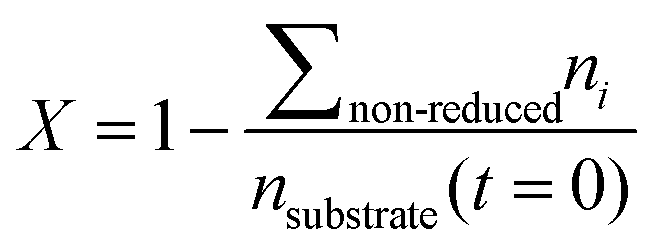 | (1) |
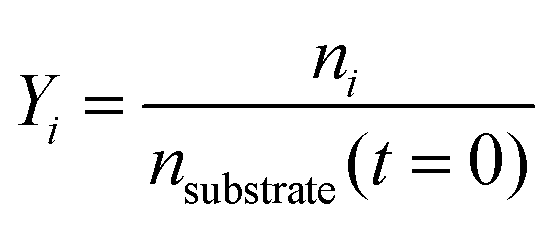 | (2) |
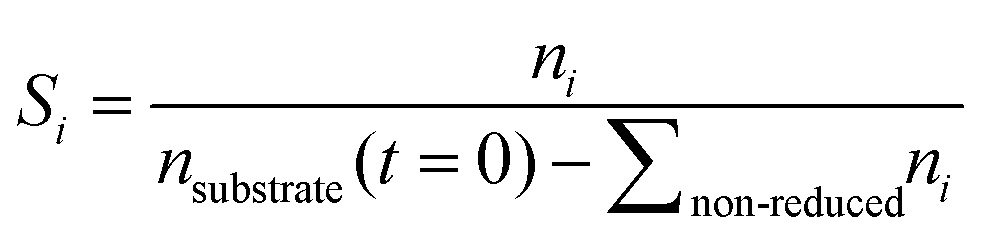 | (3) |
For recycling experiments the catalyst was retrieved by use of polycarbonate filter membranes after the reaction. Before re-use it was washed with 60 ml of deionized water and subsequently dried in a vacuum oven (60 °C, 14 h). These experiments were conducted with a slightly upscaled batch size (corresponding to 50 mg Ru/C) to allow for easier handling in the recycle process. Liquid samples of the reaction mixtures were diluted in 0.05 M HNO3 (1:1000) and analyzed by ICP-MS (Agilent 8800 ICP-MS Triple Quad) to quantify Ru leaching.
For experiments on imide reduction, the amount of substrate was adjusted to reflect the molar equivalent of 1.5 g of the respective acid. The amount of solvent was increased to ∼2 g, reflecting the water formed upon imide formation from the acid and ethanolamine.
Continuous gas phase dehydration
The continuous gas phase dehydration was conducted at close-to-ambient pressure in a stainless steel lab-scale setup, consisting of dosing units, an evaporator, a tubular reactor (9 mm i.d.) and a condenser with liquid sample collection. Previous to an experiment, 1.0 g of catalyst was pelletized (mesh size 180–250 μm) and mixed with 4.0 g of inert material (SiC, 46grit abcr). The mixture was filled into the reactor tube and topped off with another 2.0 g of inert packing, before the setup was sealed. Experiments were conducted with a 90:10 (vol%) mixture of nitrogen and substrate gas. Substrate flow rates were varied between 1.2 and 3.6 g h−1. Samples were collected at defined time intervals after the setup reached a steady operating point. Conversion and yield were calculated based on GC-determined (50 m CP-Wax-57 CB, ID-0.25 mm + DF-0.20 μm) concentrations and sample weights.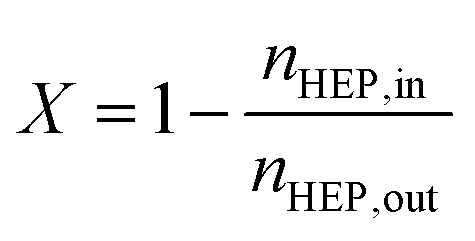 | (4) |
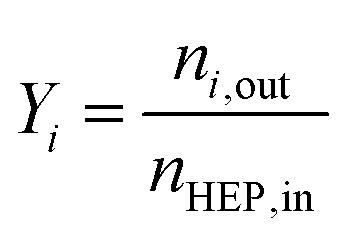 | (5) |
Stability tests were performed with a reduced amount of catalyst (0.1 g), thus limiting conversion to intermediate levels.
Results
Reaction network analysis
Ruthenium catalysts of different types have previously been shown to be effective in the reduction of biomass-derived chemicals such as sugars,24 acids25–27 and amides.28 Most importantly, Ru/C has been utilized for the reductive conversion of itaconic acid and ammonia to 3-/4-methyl-2-pyrrolidone.17 It is therefore sensible to test for the activity of Ru/C in the reductive amidation of the green value chain proposed herein.Results for the reaction of succinic acid and ethanolamine (Fig. 2) underline the fundamental suitability of carbon-supported ruthenium for the reductive formation of N-(2-hydroxyethyl)-2-pyrrolidone (HEP). Moreover, the sequence of intermediates and products observed, allows for the construction of a consistent reaction network (Fig. 3): with increasing reaction time, the amount of acid in the solution rapidly declines, giving way to mono- and, to a lesser extent, diamides. Their subsequent cyclization to the imide limits the maximum concentration of amides observed during the course of reaction. N-(2-hydroxyethyl)succinimide shows good stability in aqueous solution and thus reaches a maximum intermediate yield close to 60% after 2 h. Its amount declines only as more and more pyrrolidone is formed by reduction.
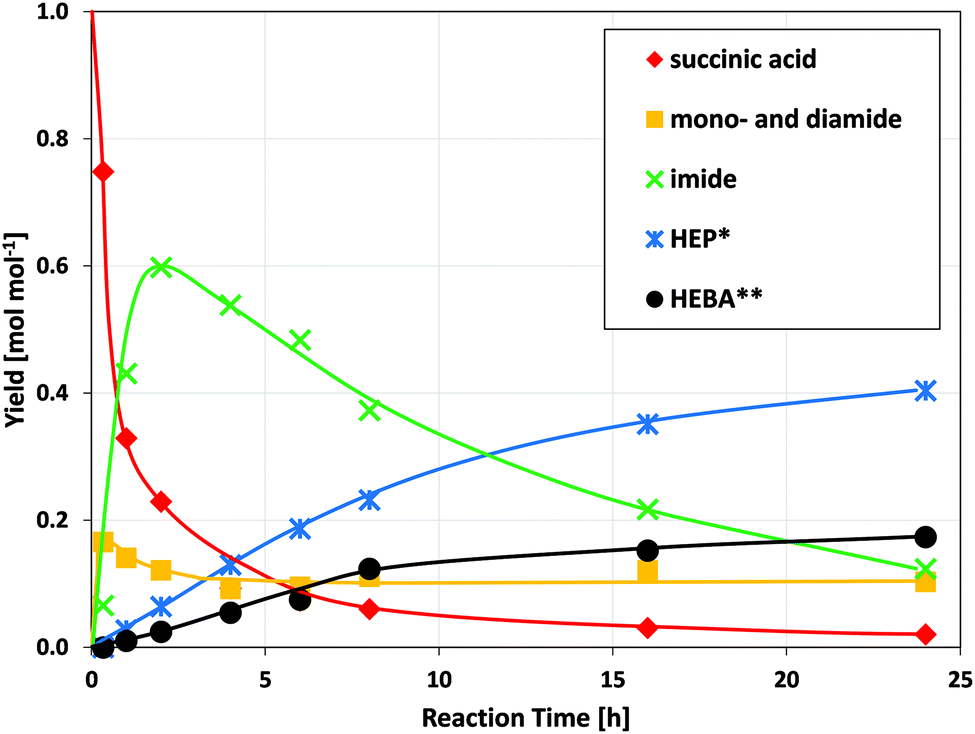 | ||
| Fig. 2 Concentration-time-profile of the reductive amidation of succinic acid with ethanolamine. (Conditions: 150 °C, 150 bar H2, 750 rpm, 1.5 g acid, 1.5 g deionized water, 1 mol. equivalent of amine, 37.5 mg Ru/C) The corresponding reaction network is outlined in Fig. 3. *HEP = N-(2-hydroxyethyl)-2-pyrrolidone, **HEBA = N-(2-hydroxyethyl)-4-hydroxybutanamide. | ||
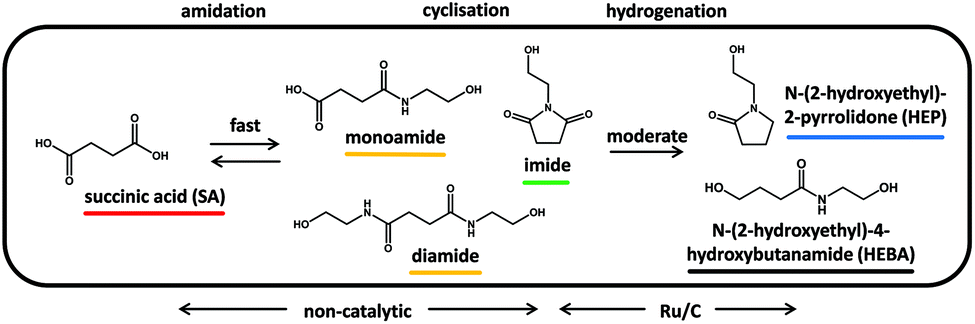 | ||
| Fig. 3 Proposed reaction network for the reductive amidation of succinic acid with ethanolamine. Water and ethanolamine, which are consumed and/or formed, are implicit in the network. | ||
Given the fast initial rate of amide and imide formation, which is also observed in the absence of catalyst, and the comparably slow conversion to reduced products, the latter step emerges as rate-determining. N-(2-hydroxyethyl)succinimide, due to its abundance and cyclic structure, is identified as the most likely substrate to be hydrogenated on the ruthenium surface.16 Moreover, N-(2-hydroxyethyl)-4-hydroxybutanamide (HEBA) – the acyclic equivalent of HEP – is observed as a second product, which initially forms at a rate proportional to that of HEP. This is tentatively assigned to their formation from a joint substrate (N-(2-hydroxyethyl)succinimide) via two competing reactions on the ruthenium surface (hydrogenolysis of CO vs. C–N bond).14 On the same note, previous publications have reported that the catalytic hydrogenation of imides may lead to lactams or hydroxyl carboxamides depending on catalyst selectivity and process conditions.29
At any rate, HEBA formation from HEP via hydrolysis was excluded in separate experiments on product stability under reaction conditions. On the other hand, the apparent decline in N-(2-hydroxyethyl)-4-hydroxybutanamide formation in later reaction stages is rationalized by the conversion to HEP through intramolecular condensation. This reaction is known to proceed without catalyst at elevated temperatures,30 wherefore the HEBA formed here is not lost to the production of HEP and may be considered a valuable product.
Choice of catalyst
Given the above, any catalyst suitable for the formation of HEP from succinic acid requires high activity in the selective hydrogenation of imide functionalities. We therefore tested a range of noble and non-noble metals for their performance in the hydrogenation of N-(2-hydroxyethyl)succinimide (Fig. 4). With the exception of RANEY® Ni, which leads to the production of small HEP quantities, none of the non-noble metal catalysts facilitates the targeted hydrogenation. Even common reduction catalysts such as Pt/C, Pd/C and Rh/C show low yield under the applied conditions. Carbon-supported ruthenium, on the other hand, stands out with an order of magnitude increase in valuable product yield (HEBA and HEP) as compared to the next best catalyst in the series.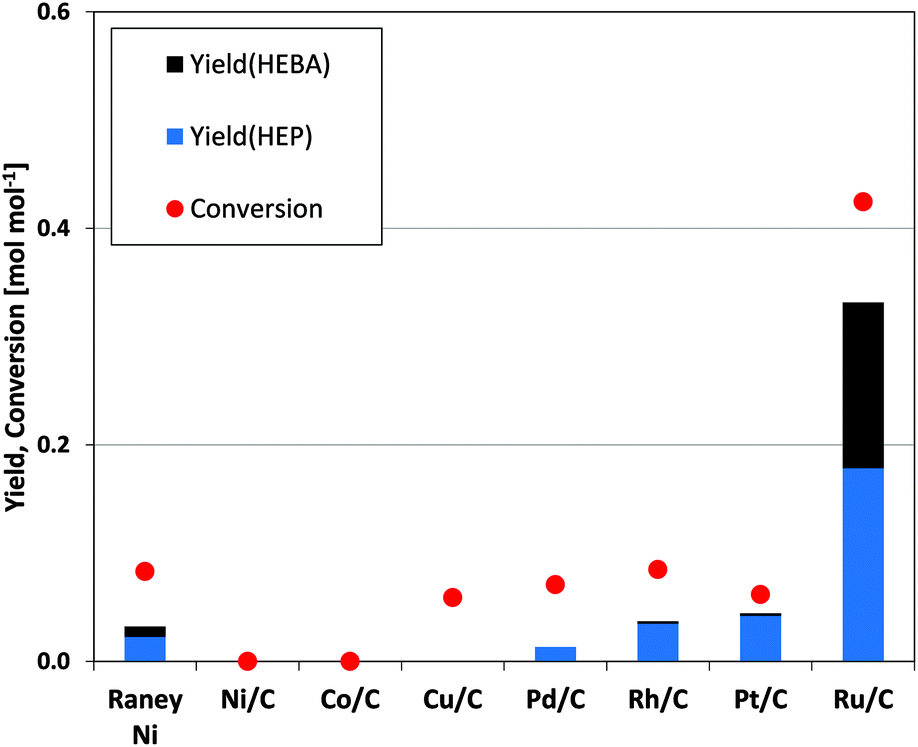 | ||
| Fig. 4 Screening of catalytic metals for N-(2-hydroxyethyl)succinimide hydrogenation. (Cond.: 150 °C, 150 bar H2, 6 h, 750 rpm, 37.5 mg catalyst). | ||
Carbonyl groups in amides and, by analogy, also in imides are known to be extremely stable against reduction due to their low electrophilicity.14,31 Consequently, any metal with the ability to activate the imide CO functionality would be superior for the tested reaction. While platinum for example has an outstanding ability to activate hydrogen, ruthenium combines the potential for hydrogen activation with that for CO-bond activation, as has recently been underlined by experimental32 and theoretical35 considerations. This combination then proves beneficial for the reduction of imides, as is underlined by the presented data.
Ruthenium as the most promising metal may then be supported on different carrier materials, possibly influencing the catalytic performance (Table 1).26 It is observed that zirconia and alumina lead to an increase in the overall yield of valuable products (HEP and HEBA), whereas yields stay the same over Ru/TiO2 and decrease for Ru/SiO2. Since differences such as the above often originate from metal dispersion and/or support acidity, these catalyst properties were subjected to further study (see ESI,† p. 5). While slight variations of both properties were evident in our set of oxide-supported catalysts, a simple correlation with catalyst activity was not found. Further studies to elucidate these aspects are ongoing. In the meantime, commercial Ru/C already offers promising catalytic activity and is thus used for the further development of the targeted value chain. The obtained trends are expected to be transferable to a range of support materials, excluding specific cases of strong metal-support-interaction and bifunctionality due to a direct participation of the support in the ongoing reaction.31
Optimal process parameters
Since amide reductions traditionally require harsh conditions, it is interesting to test the dependence of reductive amidation on process parameters. Raising the temperature above 150 °C leads to an increase in conversion and valuable product yield (Fig. 5a). At the same time, the overall selectivity towards valuable products is lowered, due to (i) the reduction of hydroxyl entities in the N-(2-hydroxyethyl)-substituent and (ii) the formation of pure reduction products, such as butanols. A significant decline in the amount of N-(2-hydroxyethyl)-4-hydroxybutanamide (HEBA) product underlines the effect of thermally activated condensation to HEP.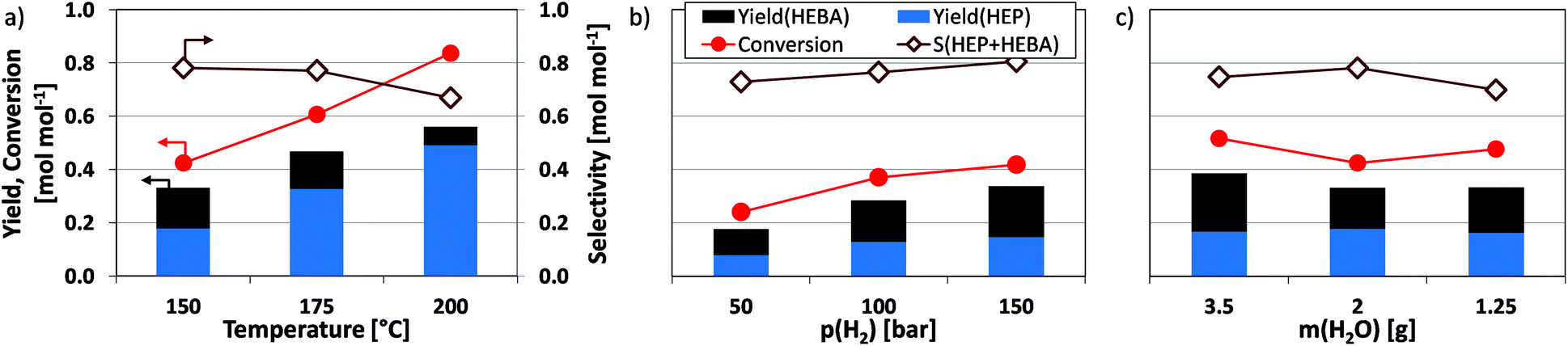 | ||
| Fig. 5 Influence of process conditions on the reduction of N-(2-hydroxyethyl)succinimide using Ru/C. (Center point for all variations are standard conditions: 150 °C, 150 bar H2, 6 h, 750 rpm, 37.5 mg catalyst, 2 g H2O solvent.) | ||
Furthermore, a positive and linear correlation is found between hydrogen pressure (50–150 bar) and the desired product yield (Fig. 5b). While this effect might be attributed to gas-liquid mass transfer limitations, results obtained for the reduction of itaconic acid in the same setup show that hydrogen mass transfer is orders of magnitude faster than the reductive amidation studied herein.17 The results from experiments conducted at different stirring intensities and catalyst loadings further support this (see Fig. S2†). It is therefore possible that p(H2) exerts its influence on the reaction by inducing a higher coverage of the catalysts metal surface with activated hydrogen (θH), which benefits the ongoing reduction. A slight change in product selectivity hints towards the different degree of hydrogen influence in desired and undesired reaction pathways.
Further experiments focused on the dependence of reductive amidation on substrate concentration cS, which was modified through the amount of water added to the reaction mixture. Following common adsorption theory, lower amounts of water (higher cS) increase the substrate surface coverage θS of the catalyst and thus influence the rate of surface reactions. Assuming the latter to be rate-determining, product yields after a specified time vary accordingly. However, no significant change in product yields is observed in the respective data (Fig. 5c). Notwithstanding future in depth analysis, this may be associated with a decoupling of cS and θS, i.e. saturation of the substrate adsorption isotherm. In the case at hand, this means that the chosen solvent (water) does not compete effectively with the organic substrate for the adsorption sites on Ru/C, wherefore the surface is likely covered with organic moieties. This, in turn, agrees well with the behavior of carbon-supported Ru in the aqueous phase hydrogenation of acetic acid with molecular hydrogen.32
To further elucidate the role of substrate adsorption on Ru/C, the imide structure to be reduced in our experiments was systematically varied (Table 2). It is interesting to see that methyl substituents attached to the carbon atoms of the succinimide cycle hardly have any influence on the yield of pyrrolidone (compare entries 1 and 2 or 3 and 4). However, adding an ethyl-substituent to the imide N-atom (entries 4 and 5), which greatly increases the thermodynamic stability of the succinimide in an aqueous environment, doubles the pyrrolidone yield obtained under otherwise equivalent conditions. This may further underline the importance of the respective imides in the rate determining step of reductive amidation. Finally, the fastest conversion rates are obtained for substrates containing an additional hydroxyl group derived from ethanolamine. Since the hydroxyl group, due to its position, is unlikely to have a direct electronic effect on the carbonyl to be reduced, the effect is tentatively assigned to a change in substrate adsorption.
| Substrate/structure | Y (Pyrrolidone) [mol mol−1] |
|---|---|
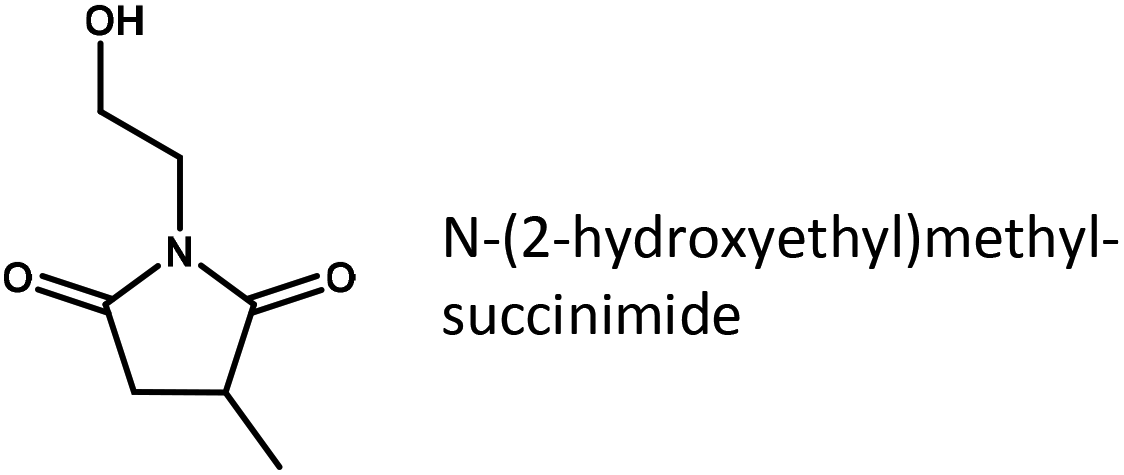
|
0.18 |
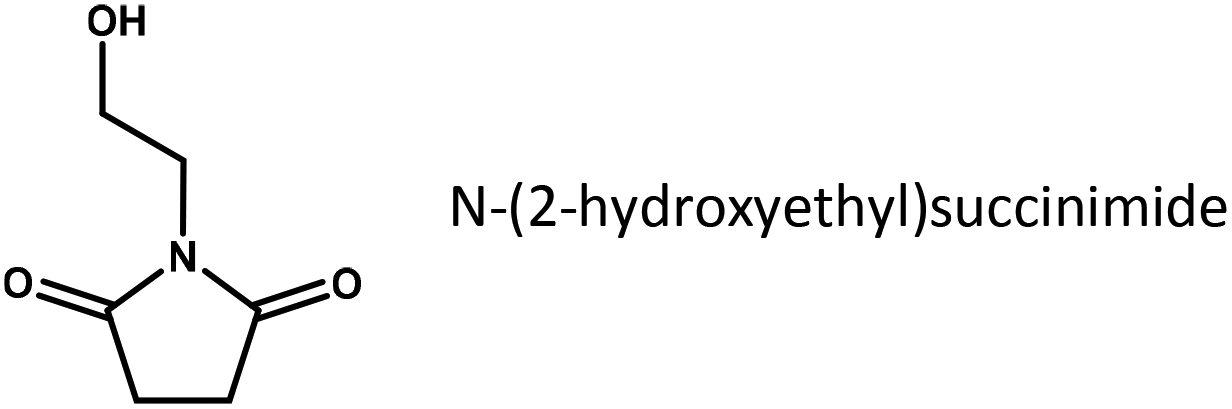
|
0.18 |

|
0.10 |
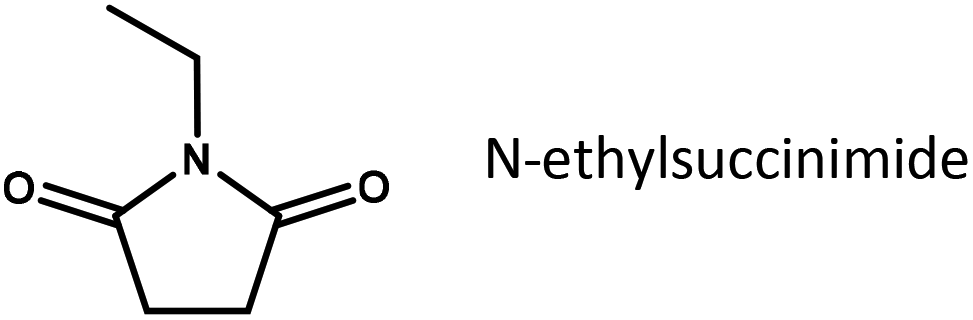
|
0.12 |
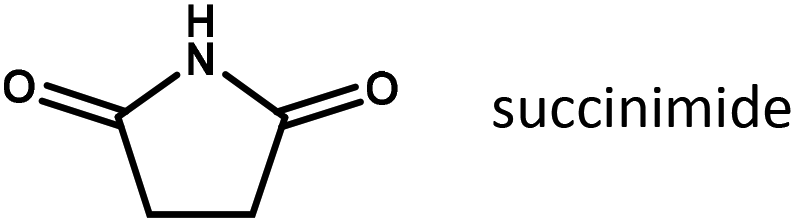
|
0.04 |
Substrate scope and catalyst stability in reductive amidation
The target of the reductive transformations discussed so far is to convert biogenic acids into N-(2-hydroxyethyl)2-pyrrolidones serving as substrates for the gas phase dehydration to vinyl monomers. In this context, high product yields must be achieved, so as to guarantee the economic viability of the proposed sustainable value chain. Therefore, the conversion of ethanolamine with the suggested platform chemicals – succinic and itaconic acid – was tested under optimized reaction conditions. Methylsuccinic acid was added to the set, in order to reveal the influence of the double bond contained in itaconic acid (Table 3).| Dicarboxylic acid substrate | Conversion [mol mol−1] | Y (Pyrrolidone)a [mol mol−1] | Y (Butanamide)b [mol mol−1] |
|---|---|---|---|
| a Equivalent of HEP in the case of succinic acid. b Equivalent of HEBA in the case of succinic acid. | |||
| Succinic acid (150 °C) | 0.75 | 0.40 | 0.17 |
| Succinic acid (200 °C) | 0.98 | 0.74 | 0.01 |
| Methylsuccinic acid (150 °C) | 0.81 | 0.50 | 0.10 |
| Methylsuccinic acid (200 °C) | 0.88 | 0.74 | 0.00 |
| Itaconic acid (150 °C) | 0.73 | 0.39 | 0.06 |
| Itaconic acid (200 °C) | 0.96 | 0.39 | 0.02 |
The conversion of succinic acid is especially successful, yielding more than 60 mol% of valuable products at 150 °C. Utilizing the improved rates of imide hydrogenation and HEBA condensation at 200 °C this value is increased to 75 mol%, while maximizing the final HEP content at the same time. Further enhancements are then limited by the chemoselectivity of the catalyst, which still allows for the formation of N-ethyl-substituted by-products and substrate hydrogenation without nitrogen incorporation (e.g. formation of γ-butyrolactone and butanols).
These results are well transferable to methylsuccinic acid, reaching up to 74 mol% yield of N-(2-hydroxyethyl)-methylpyrrolidones. For itaconic acid, however, yields are consistently lower than for the other two acids, despite comparable conversion levels. This is rationalized by the possibility of additional reactions taking place on the methylene functionality prior to its reduction. More specifically, ethanolamine is known to react with itaconic acid in a thermally-activated aza-Michael addition yielding N-(2-hydroxyethyl)-2-pyrrolidone-4-carboxylic acid – another monomer from biomass.33 The resulting undesired competition between addition and reductive amidation may, however, be circumvented by the selective reduction of itaconic acid to methylsuccinic acid at mild conditions previous to the introduction of ethanolamine. For example, the electrochemical production of methylsuccinic acid from itaconic acid containing fermentation broth has been reported.34
Finally, the viability of reductive amidation as a production tool depends on the stability of the noble metal catalyst. In this context, no signs of catalyst deactivation are observed during five consecutive batch reactions (Fig. 6). After a slight increase in the yield of desired products from the first to the second use cycle of a catalyst batch, further recycling experiments show constant catalytic performance. ICP-MS analysis of the filtered product solution indicates a small and decreasing fraction of ruthenium leaching from the catalyst over sequential use cycles. However, the observed stability of catalyst performance would indicate a negligible contribution of the leached species to the ongoing reduction. Finally, it was tested whether the removal of Ru/C from an ongoing reaction precludes further substrate conversion (Fig. S3†). Since hydrogenation reactions are completely suppressed after the filtration step, the heterogeneous nature of the applied catalyst is verified.
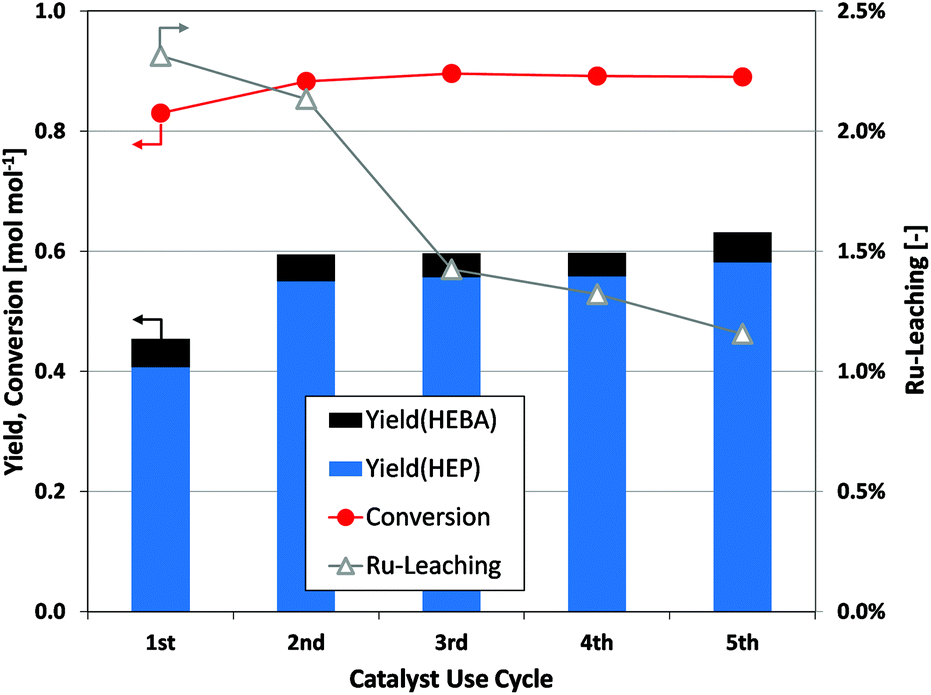 | ||
| Fig. 6 Assessment of Ru/C catalyst stability by recycling through several N-(2-hydroxyethyl)succinimide reduction batch experiments. (Cond.: 200 °C, 150 bar H2, 6 h, 750 rpm; dry catalyst weight for runs 1–5: 50.0 mg, 56.7 mg, 53.5 mg, 52.8 mg and 49.0 mg). | ||
Gas phase dehydration of N-(2-hydroxyethyl)-2-pyrrolidones
Having achieved high yields of N-(2-hydroxyethyl)-2-pyrrolidones via reductive amidation, gas phase dehydration (Fig. 7) offers a simple means to produce N-vinyl-2-pyrrolidones, which serve as valuable monomers. With water as the only by-product and the possibility of achieving excellent selectivity over inexpensive, non-toxic, solid catalysts it was chosen as second step of the here-presented value chain.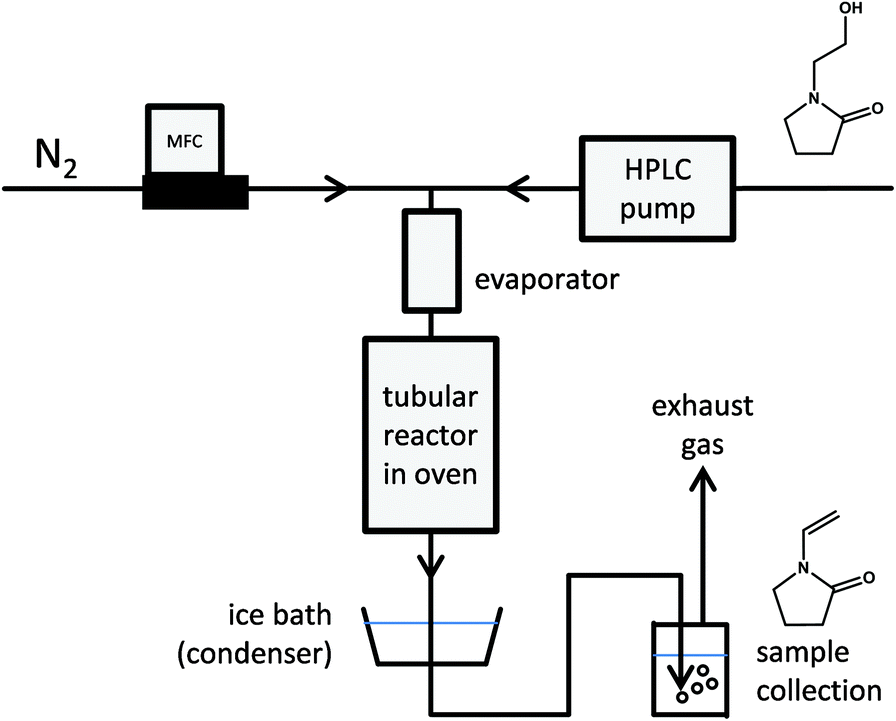 | ||
| Fig. 7 Simplified schematic of the gas phase dehydration of N-(2-hydroxyethyl)pyrrolidones in lab-scale. Vinylpyrrolidone monomers are obtained after condensing the product stream. | ||
Firstly, the functionality of a simple Na2O/SiO2 catalyst (1:20 molar ratio)21 was verified using commercially available N-(2-hydroxyethyl)-2-pyrrolidone as test substrate (Table 4). Here, 2-pyrrolidone appears as main side-product, due to the undesired cleavage of the C–N bond. However, 2-pyrrolidone formation can be mitigated by the choice of reaction conditions. In detail, the right balance between residence time and reaction temperature has to be achieved, so that the gas mixture leaves the catalyst bed immediately after full conversion is reached. A positive example of this is obtained at WHSV = 2.4 gsubstrate gcat−1 h−1 and T = 350 °C, where the reaction yields 95 mol% N-vinyl-2-pyrrolidone. The stability of the catalytic material at reaction conditions was further assessed by running the setup for 8 h at constant operating conditions (Fig. S6†). No obvious deactivation is observed in line with a very limited amount of catalyst coking evidenced by thermogravimetric analysis.
:20 molar ratio) in the gas phase dehydration of commercial N-(2-hydroxyethyl)-2-pyrrolidone at different conditions (Cond.: 90 vol% N2, 10 vol% substrate in gas flow, 1.0 g catalyst 180–250 μm mesh)
| Temperature [°C] | WHSVa [h−1] | Conversion [mol mol−1] | Y (NVP) [mol mol−1] | Y (2-pyrrolidone) [mol mol−1] |
|---|---|---|---|---|
| a Weight hourly space velocity on a gsubstrate gcatalyst−1 basis. | ||||
| 350 | 1.2 | 1.00 | 0.83 | 0.16 |
| 350 | 2.4 | 0.99 | 0.95 | 0.03 |
| 350 | 3.6 | 0.89 | 0.86 | 0.01 |
| 400 | 1.2 | 1.00 | 0.78 | 0.15 |
| 400 | 2.4 | 1.00 | 0.93 | 0.05 |
| 400 | 3.6 | 1.00 | 0.96 | 0.02 |
Further tests concern the applicability of the same catalyst and process conditions to the dehydration of methyl-N-(2-hydroxyethyl)-2-pyrrolidones, which are obtained by the conversion of itaconic and methylsuccinic acids. To satisfy the need for relatively large substrate quantities in order to operate the continuous setup, 3-methyl-N-(2-hydroxyethyl)-2-pyrrolidone was synthesized by thermally reacting 3-methyl-γ-buytrolactone and ethanolamine (see ESI†). The desired product (≥95% NMR, see Fig. S7†) is obtained after vacuum distillation. Subsequent gas phase dehydration on the continuously operated setup yields 75 mol% of 3-methyl-N-vinyl-2-pyrrolidone at previously optimized conditions (WHSV = 2.4 gsubstrate gcat−1 h−1 and T = 350 °C). Since the main by-product is 3-methyl-2-pyrrolidone, a reduction in residence time through an increase in substrate flow rate (WHSV = 3.6 gsubstrate gcat−1 h−1) increases yields to 90 mol% of vinylpyrrolidone. Overall, the setup was operated continuously for 7 h converting 20 gsubstrate gcatalyst−1 (see Fig. S10†). The collected product mixture was vacuum distilled leading to the facile separation of 3-methyl-N-vinyl-2-pyrrolidone (≥95% NMR, see Fig. S8†), which may be used for polymer synthesis. It thus appears that an industrial process could make use of rather simple and efficient separation techniques.
Conclusions
The reaction network for the reductive amidation of dicarboxylic acids with ethanolamine has been elucidated, clearly showing the rate-determining nature of the hydrogenation step. Based thereon, the challenge of this transformation lies in (i) the low reducibility of the intermediate imide functionality and (ii) the possibility for over-reduction, e.g. of the hydroxyl-substituent introduced with ethanolamine. Ru/C stands out from the set of common reduction catalysts tested herein, due to its ability to activate not only H2, but also CO bonds. It could be shown that, using optimal process conditions, the reductive amidation of succinic and itaconic acid can yield up to 75 mol% of valuable products. In order to conclude the sustainable value chain leading from biogenic acids to monomers, N-(2-hydroxyethyl)-2-pyrrolidones were dehydrated in a continuous gas phase reactor leading to NVP monomers. With water as the only by-product and the use of solid catalysts, this process shows promise to overcome the drawbacks of fossil-based NVP production. The achievable yield of NVP from succinic acid over the whole two-step process is above 72 mol% and could be further optimized by the design of improved chemoselective catalysts for reductive amidation. As a potential for future research, it is noted that the applied Ru/C catalyst requires long reaction times to achieve high conversion and product yield. It would thus be interesting to develop alternative materials with a more pronounced ability to activate CO bonds. Investigations in this direction are ongoing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the German Federal Ministry of Education and Research (BMBF) for funding of the projects BioPyrr and BioPVP (FKZ IBÖ-03 031B0249 and 031B0487 A) in the framework of “New Products for the Bioeconomy” (Neue Produkte für die Bioökonomie). Furthermore, Mr Haus likes to thank the German Chemical Industry Fund (Fonds der chemischen Industrie, FCI) for the generous support of his PhD project. Lastly, Saint-Gobain NorPro is acknowledged for providing the catalyst support materials mentioned above.Notes and references
- (a) T. Werpy, G. Petersen, A. Aden, J. Bozell, J. Holladay, J. White and A. Manheim, Top Value Added Chemicals from Biomass (Volume I), U.S.: Department of Energy, 2004 Search PubMed; (b) J. Holladay, J. Bozell, J. White and D. Johnson, Top Value Added Chemicals from Biomass (Volume II), U.S: Department of Energy, 2007 Search PubMed.
- R. Palkovits, Chem. Ing. Tech., 2018, 90, 1699–1708 CrossRef CAS.
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- (a) X. Di, C. Li, G. Lafaye, C. Especel, F. Epron and C. Liang, Catal. Sci. Technol., 2017, 7, 5212–5223 RSC; (b) X. Di, C. Li, B. Zhang, J. Qi, W. Li, D. Su and C. Liang, Ind. Eng. Chem. Res., 2017, 56, 4672–4683 CrossRef CAS.
- K. H. Kang, U. G. Hong, Y. Bang, J. H. Choi, J. K. Kim, J. K. Lee, S. J. Han and I. K. Song, Appl. Catal., A, 2015, 490, 153–162 CrossRef CAS.
- T. Toyao, S. M. A. Hakim Siddiki, A. S. Touchy, W. Onodera, K. Kon, Y. Morita, T. Kamachi, K. Yoshizawa and K. Shimizu, Chem. – Eur. J., 2017, 23, 1001–1006 CrossRef CAS.
- L. Corbel-Demailly, B.-K. Ly, D.-P. Minh, B. Tapin, C. Especel, F. Epron, A. Cabiac, E. Guillon, M. Besson and C. Pinel, ChemSusChem, 2013, 6, 2388–2395 CrossRef CAS.
- C. S. Spanjers, D. K. Schneiderman, J. Z. Wang, J. Wang, M. A. Hillmyer, K. Zhang and P. J. Dauenhauer, ChemCatChem, 2016, 8, 3031–3035 CrossRef CAS.
- J. Ullrich and B. Breit, ACS Catal., 2018, 8, 785–789 CrossRef CAS.
- A. S. Touchy, S. M. A. Hakim Siddiki, K. Kon and K. Shimizu, ACS Catal., 2014, 4, 3045–3050 CrossRef CAS.
- X.-L. Du, L. He, S. Zhao, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., Int. Ed., 2011, 50, 7815–7819 CrossRef CAS PubMed.
- (a) J. D. Vidal, M. J. Climent, P. Concepción, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2015, 5, 5812–5821 CrossRef CAS; (b) J. D. Vidal, M. J. Climent, A. Corma, P. Concepción and S. Iborra, ChemSusChem, 2017, 10, 119–128 CrossRef CAS PubMed.
- G. Gao, P. Sun, Y. Li, F. Wang, Z. Zhao, Y. Qin and F. Li, ACS Catal., 2017, 7, 4927–4935 CrossRef CAS.
- A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS.
- G. Budroni and A. Corma, J. Catal., 2008, 257, 403–408 CrossRef CAS.
- J. F. White, J. E. Holladay, A. A. Zacher, J. G. Frye and T. A. Werpy, Top. Catal., 2014, 57, 1325–1334 CrossRef CAS.
- Y. Louven, K. Schute and R. Palkovits, ChemCatChem, 2019, 11, 439–442 CrossRef CAS.
- (a) W. Reppe, Experientia, 1949, 5, 93–132 CrossRef CAS; (b) W. Reppe, et al. , Liebigs Ann. Chem., 1956, 601, 81–137 CrossRef CAS; (c) A. L. Harreus, R. Backes, J.-O. Eichler, R. Feuerhake, C. Jäkel, U. Mahn, R. Pinkos and R. Vogelsang, 2-Pyrrolidone, in Ullmann's Encyclopedia of Industrial Chemistry, 2011 Search PubMed.
- (a) M. L. Hallensleben, R. Fuss and F. Mummy, Polyvinyl Compounds, Others, in Ullmann's Encyclopedia of Industrial Chemistry, 2015 Search PubMed; (b) Grand View Research Inc., Polyvinylpyrrolidone (PVP) Market Worth $2.75 Billion by 2024, PR Newswire, 2016, (last accessed 15.10.2017) Search PubMed.
- Y. Shimasaki, S. Otsu-Shishi, H. Yano and O. Suita-Shishi, Process for production of tertiary n-alkenyl carboxylic acids, EP0701998B1, 1995 Search PubMed.
- Y. Shimasaki, H. Yano, K. Ariyoshi and H. Kambe, J. Mol. Catal. A: Chem., 2005, 239, 125–129 CrossRef CAS.
- Y. Shimasaki and H. Yano, Catal. Surv. Asia, 2010, 14, 132–139 CrossRef CAS.
- A. S. Piskun, J. Ftouni, Z. Tang, B. M. Weckhuysen, P. C. A. Bruijnincx and H. J. Herres, Appl. Catal., A, 2018, 549, 197–206 CrossRef CAS.
- C. Hernandez-Mejia, E. S. Gnanakumar, A. Olivos-Suarez, J. Gascon, H. F. Greer, W. Zhou, G. Rothenberg and N. R. Shiju, Catal. Sci. Technol., 2016, 6, 577–582 RSC.
- K. Liu, X. Huang, E. A. Pidko and E. J. M. Hensen, ChemCatChem, 2018, 10, 810–817 CrossRef CAS PubMed.
- L. Chen, Y. Li, X. Zhang, Q. Zhang, T. Wang and L. Ma, Appl. Catal., A, 2014, 478, 117–128 CrossRef CAS.
- A. S. Piskun, J. Ftouni, Z. Tang, B. M. Weckhuysen, P. C. A. Bruijnincx and H. J. Heeres, Appl. Catal., A, 2018, 549, 197–206 CrossRef CAS.
- G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, J. Catal., 2011, 278, 228–238 CrossRef CAS.
- A. M. Maj, I. Suisse, N. Pinault, N. Robert and F. Agbossou-Niedercorn, ChemCatChem, 2014, 6, 2621–2625 CrossRef CAS.
- Y. Shimasaki, H. Yano, H. Sugiura and H. Kambe, Bull. Chem. Soc. Jpn., 2008, 4, 449–459 CrossRef.
- K. Shimizu, W. Onodera, A. S. Touchy, S. M. A. Hakim Siddiki, T. Toyao and K. Kon, ChemistrySelect, 2016, 4, 736–740 CrossRef.
- J. Shangguan, M. V. Olarte and Y. H. Chin, J. Catal., 2016, 340, 107–121 CrossRef CAS.
- P. Qi, H.-L. Chen, H. T. H. Hguyen, C.-C. Lin and S. A. Miller, Green Chem., 2016, 18, 4170 RSC.
- F. J. Holzhäuser, J. Artz, S. Palkovits, D. Kreyenschulte, J. Büchs and R. Palkovits, Green Chem., 2017, 19, 2390 RSC.
- R. Burch, C. Paun, X.-M. Cao, P. Crawford, P. Goodrich, C. Hardacre, P. Hu, L. McLaughlin, J. Sá and J. M. Thompson, J. Catal., 2011, 283, 89–97 CrossRef CAS.
- Imbedded image of leaf designed by Mudassir101 - Freepik.com.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc01488h |
| This journal is © The Royal Society of Chemistry 2019 |