
Metal- and base-free regioselective thiolation of the methyl C(sp3)–H bond in 2-picoline N-oxides†
Dong
Wang
 *,
Zhenlin
Liu
,
Zhentao
Wang
,
Xinyue
Ma
and
Peng
Yu
*
*,
Zhenlin
Liu
,
Zhentao
Wang
,
Xinyue
Ma
and
Peng
Yu
*
China International Science and Technology Cooperation Base of Food Nutrition/Safety and Medicinal Chemistry, College of Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, China. E-mail: wangdong@tust.edu.cn; yupeng@tust.edu.cn
First published on 21st November 2018
Abstract
A one-pot, two-step synthesis of pyridine-2-ylmethyl thioethers is developed through a TFAA-mediated [3,3]-sigmatropic rearrangement of pyridine N-oxides and TBAB-catalyzed direct conversion of trifluoroacetates into thioethers under metal- and base-free conditions. This methodology enables thiolation of the unactivated methyl C(sp3)–H bond in 2-picolines with thiols. Remarkable features of the method include high regioselectivity, step- and atom-economy, mild conditions, simple operation, wide substrate scope and scalability. Furthermore, the method has been successfully applied to the synthesis of omeprazole sulfide and rabeprazole sulfide without the need for TBAB catalysis. A comprehensive green chemistry metrics analysis indicated that this method is much more efficient and greener than the reported synthesis of rabeprazole sulfide.
Introduction
Sulfides, also known as thioethers, are versatile intermediates for the synthesis of biologically active natural products,1 and many synthetic and biosynthetic pathways involve the formation and transformation of sulfides.2 Thioethers are important in biology, notably in the amino acid methionine and the cofactor biotin. The conventional method to access thioethers relies upon SN2 reactions between organic halides and thiols under basic conditions, which is like the Williamson ether synthesis. However, this method has critical issues, such as the use of highly prefunctionalized organic halides and the generated toxic halide waste. Alternatively, thiolation of the C–H bond seems to be a promising solution. Surprisingly, it has only recently gained increasing attention. This direct thiolation is commonly realized by metal-mediated protocols utilizing various thiolating reagents, including disulfides, thiols, elemental sulfur, tosyl chlorides, etc.3Pyridine-2-ylmethyl thioethers are a very important type of thioether, not only because they are key scaffolds for a variety of biologically active compounds,4 but also because several on-market hot drugs, such as omeprazole, pantoprazole, lansoprazole and rabeprazole (Fig. 1), contain this core structure. These compounds were found to be powerful therapeutic agents for treating gastric and duodenal ulcer disease.5 Several methods for the synthesis of these active pharmaceutical ingredients (APIs) have been reported.6 The most common strategy involves the oxidation of the corresponding pyridine-2-ylmethyl thioethers (3), which were prepared in four steps (Scheme 1A). Firstly, 2-picoline N-oxides (1) treated with a large excess of acetic anhydride provided 4, which was then hydrolyzed to give 5. The reaction of 5 with thionyl chloride provided chloride 6. Finally, alkylation of 6 with appropriate thiols afforded 3. However, this synthetic route is far from satisfactory, not only because of the tedious reaction procedures, the use of a large excess of acetic anhydride and the highly corrosive thionyl chloride as reactants, but also because of the generation of too much hazardous waste, including sulfur dioxide, hydrogen chloride, sodium acetate, etc. A major cause of waste, particularly in the manufacture of fine chemicals and pharmaceuticals, is the use of stoichiometric, mainly inorganic, reagents in organic synthesis.7 However, the use of an inorganic base, such as NaOH, is required in both the ester hydrolysis reaction (from 4 to 5) and the last alkylation reaction (from 6 to 3). Although this synthetic route has been reported for thirty years, during which several improved synthetic methods have been reported,6a,8 no significant progress has been made. For example, the Mitsunobu reaction utilizing 5 to react with thiophenols seems to be attractive,8a but the atom economy is low considering the generated hydrazine and triphenylphosphine oxide byproducts. Moreover, 5 needs a two-step synthesis. Therefore, a concise, more efficient and greener method is demanded.
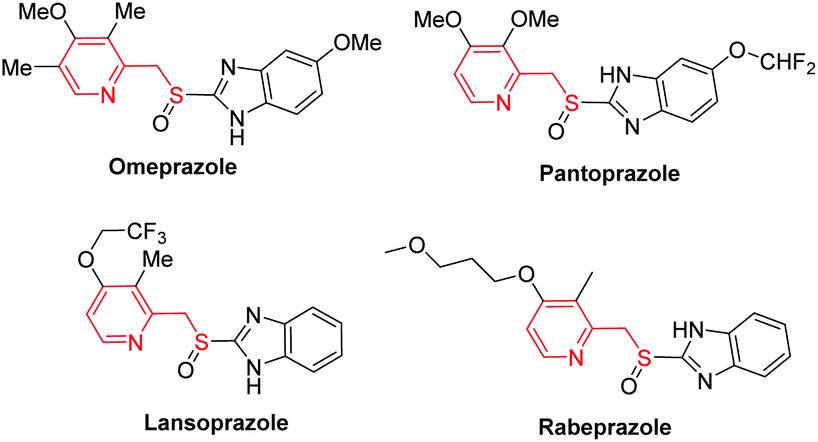 | ||
| Fig. 1 Structures of the thioether-derived drugs. | ||
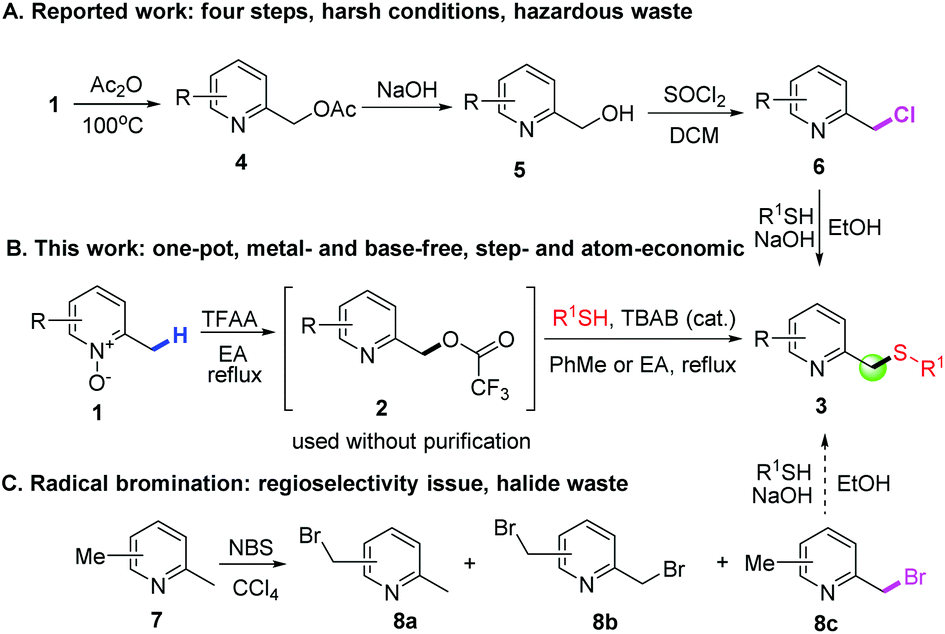 | ||
| Scheme 1 Approaches to pyridine-2-ylmethyl thioethers. | ||
Our group has been exploring efficient and concise synthetic methods to prepare complex pyridine derivatives, utilizing easily accessible pyridine N-oxides as substrates.9 Although functionalization of the C(sp3)–H bond in 2-alkylpyridines has received lots of attention recently, such as acyloxylation,10 phosphorylation11 and sulfonyloxylation,12 thiolation of the C(sp3)–H bond in 2-alkylpyridines has not yet been reported. In fact, direct thiolation of the C(sp3)–H bond or a simple, one-pot operation remains challenging. The reported methods, using disulfides as thiolation agents, are only suitable for specific types of compounds, such as aliphatic amides13 and ethers.14 Herein, we disclose a two-step, one-pot thiolation of the C(sp3)–H bond to prepare pyridine-2-ylmethyl thioethers from 2-picoline N-oxides under metal- and base-free conditions (Scheme 1B). In contrast to the conventional methods, 1 was converted into trifluoroacetate 2 using only a stoichiometric amount of TFAA in refluxing ethyl acetate. After rotary evaporation of the volatiles, 2 reacts with R1SH in refluxing toluene or EA under the catalysis of TBAB to afford the desired product 3 with a good yield. The remarkable features of the method include several points. First, the reaction is both step- and atom-economical, as trifluoroacetic acid is the only byproduct for the first step, and trifluoromethane and carbon dioxide are the only byproducts for the second step. Second, the use of TBAB catalysis significantly reduces base waste, which is usually required in thioether or ether formation reactions. Third, instead of using an environmentally problematic solvent, only the environmentally friendly solvent EA is used.15 Finally, this C–H functionalization is highly regioselective as only the pyridine-2-ylmethyl position can be functionalized, and alkyl groups at other positions are unreactive. This feature is important as alternative approaches such as radical bromination (Scheme 1C) have the regioselectivity problem,16 not to mention the use of a large excess of toxic CCl4 as the solvent and the associated halide waste.
Results and discussion
The first-step reaction (from 1 to 4), also known as the Boekelheide reaction, is a well-established and frequently used procedure in heterocyclic chemistry for the introduction of oxygen on an alkyl group α to a ring nitrogen atom.17 Although several improved methodologies of this transformation have been reported,6a,18 no systematic study with a satisfactory green standard has been yet established. We first focused on developing a more efficient and eco-friendly reaction process of this transformation. It is expected that 2-picoline N-oxide (1a) would be converted into its corresponding pyridine-2-ylmethyl oxygenated product 2 under the activation of various acylating reagents through a [3,3]-sigmatropic rearrangement. In order for 2 to react directly with R1SH, 2 must carry a good leaving group (Table 1). Therefore, we are especially interested in using those acylating agents with strong electron-withdrawing groups, such as TFAA and Ts2O. After the screening of the acylating agents, solvents and concentrations (Table 1), it was found that TFAA was the best activation reagent, using EA as the solvent, at reflux temperature (entry 6).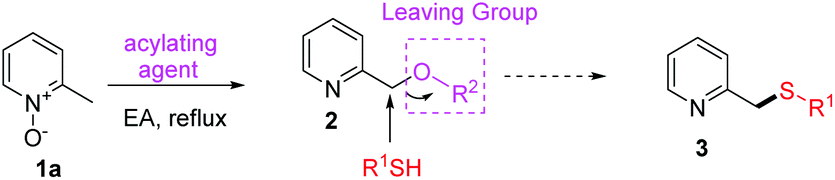
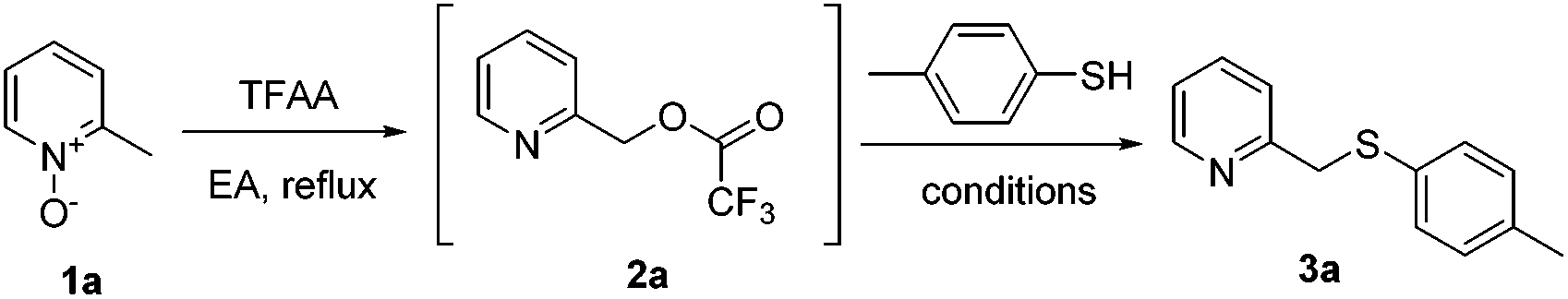
Next, instead of hydrolyzing the resulting trifluoroacetate 2a, direct conversion of the trifluoroacetate into the thioether product was attempted. As trifluoroacetyloxy is a good leaving group, the SN2 reaction between 2a and p-toluenethiol under neutral or basic conditions was expected. However, little conversion was observed (entries 1–3, Table 2), although slightly better yields were obtained under the catalysis of KI (entry 4) or TBAI (entry 5). We then focused on exploring the reaction under either protonic acid (entries 6–9) or Lewis acid (entries 10–17) conditions. To our delight, 3a was delivered in 51% yield by mixing 2a and p-toluenethiol in refluxing aqueous HBr (entry 6), and the improved yield was obtained by using more p-toluenethiol under more concentrated conditions (72% yield, entry 8). Although the yield is good, the harsh strong acidic condition is not satisfactory. It was reported that thioethers could be prepared from carboxylates and thiols under the catalysis of FeCl3 in acetonitrile.19 However, no reaction was detected for our case utilizing this method (entry 10); probably the reported method is only suitable for those carboxylates with acetyloxy, pivalyloxy, benzoyloxy or p-methoxybenzoyloxy as the leaving group. Screening of more Lewis acids led us to identify that BBr3 (entry 12) or LiBr (entries 13–17) could catalyze the desired reaction. Furthermore, it was found that LiBr in refluxing toluene gave the best yield (73%, entry 16). However, on considering that LiBr is not easy to handle (highly hygroscopic), the tedious long reaction time (typically 15 hours), and the failed reaction in a greener solvent, such as EA (entry 17), an alternative bromide reagent, TBAB, was tried. To our delight, the best condition was identified using this reagent in refluxing toluene (77%, entry 19). More concentrated or diluted conditions led to decreased product yields (entries 20 and 21). Moreover, the use of alternative green solvents, including ethyl acetate (entry 22), ethanol (entry 23) and anisole (entry 24), was screened, and it was found out that the reaction worked well in hot EA, although a slightly lower yield was obtained (71%, entry 22). Therefore, the condition in entry 19 is the optimum reaction condition, which not only affords the highest yield, but also reacts quickly (typically 5 hours) and is compatible with EA.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Catalyst | Eq./R-SH | Solvent | [M] | Yield, % |
| a Unless otherwise noted, all reactions were conducted with N-oxide (1.0 equiv.), catalyst (0.1 eq.), and additive (2.0 equiv.) under reflux conditions. b Reaction temperature was 100 °C. c 0.2 eq. of catalyst was used. d Reaction temperature was 120 °C. | ||||||
| 1 | N/A | N/A | 1.0 | Toluene | 0.8 | 10 |
| 2 | Et3N | N/A | 1.2 | DCM | 0.3 | 4 |
| 3 | K2CO3 | N/A | 1.2 | DMF | 0.3 | 11 |
| 4 | K2CO3 | KI | 1.2 | Acetone | 0.3 | 17 |
| 5 | Et3N | TBAI | 1.2 | DCM | 0.3 | 23 |
| 6 | N/A | N/A | 2.0 | HBr | 0.8 | 51 |
| 7 | N/A | N/A | 1.2 | HBr | 1.6 | 34 |
| 8 | N/A | N/A | 2.0 | HBr | 1.6 | 72 |
| 9 | N/A | N/A | 2.0 | HCl | 1.6 | 0 |
| 10 | N/A | FeCl3 | 1.1 | CH3CN | 0.4 | 0 |
| 11 | N/A | AlCl3 | 1.1 | DCM | 0.4 | 0 |
| 12b | N/A | BBr3 | 1.1 | CH3CN | 0.4 | 42 |
| 13 | N/A | LiBr | 1.1 | THF | 0.4 | 14 |
| 14 | N/A | LiBr | 1.1 | Dioxane | 0.4 | 32 |
| 15 | N/A | LiBr | 1.1 | Toluene | 0.4 | 65 |
| 16c | N/A | LiBr | 1.0 | Toluene | 0.8 | 73 |
| 17 | N/A | LiBr | 1.1 | EA | 0.8 | 0 |
| 18 | N/A | TBAB | 1.0 | Toluene | 0.8 | 37 |
| 19c | N/A | TBAB | 1.0 | Toluene | 0.8 | 77 |
| 20c | N/A | TBAB | 1.0 | Toluene | 1.0 | 66 |
| 21c | N/A | TBAB | 1.0 | Toluene | 0.6 | 71 |
| 22c,d | N/A | TBAB | 1.0 | EA | 0.8 | 71 |
| 23c,d | N/A | TBAB | 1.0 | EtOH | 0.8 | 0 |
| 24c,d | N/A | TBAB | 1.0 | Anisole | 0.8 | 16 |
With the optimized reaction conditions in hand, we surveyed the substrate scope of 2-picoline N-oxides and thiols. We found that they can be successfully converted into the desired thioether products in modest to good yields (up to 77% yield, Scheme 2), and the reaction is fast (typically one to five hours). It is worth noting that similar yields were obtained for most reactions run in toluene or EA with the same substrate. Among the three reactions (3a, 3g & 3j) run in both toluene and EA, similar yields were obtained for 3a and 3g (Scheme 2).
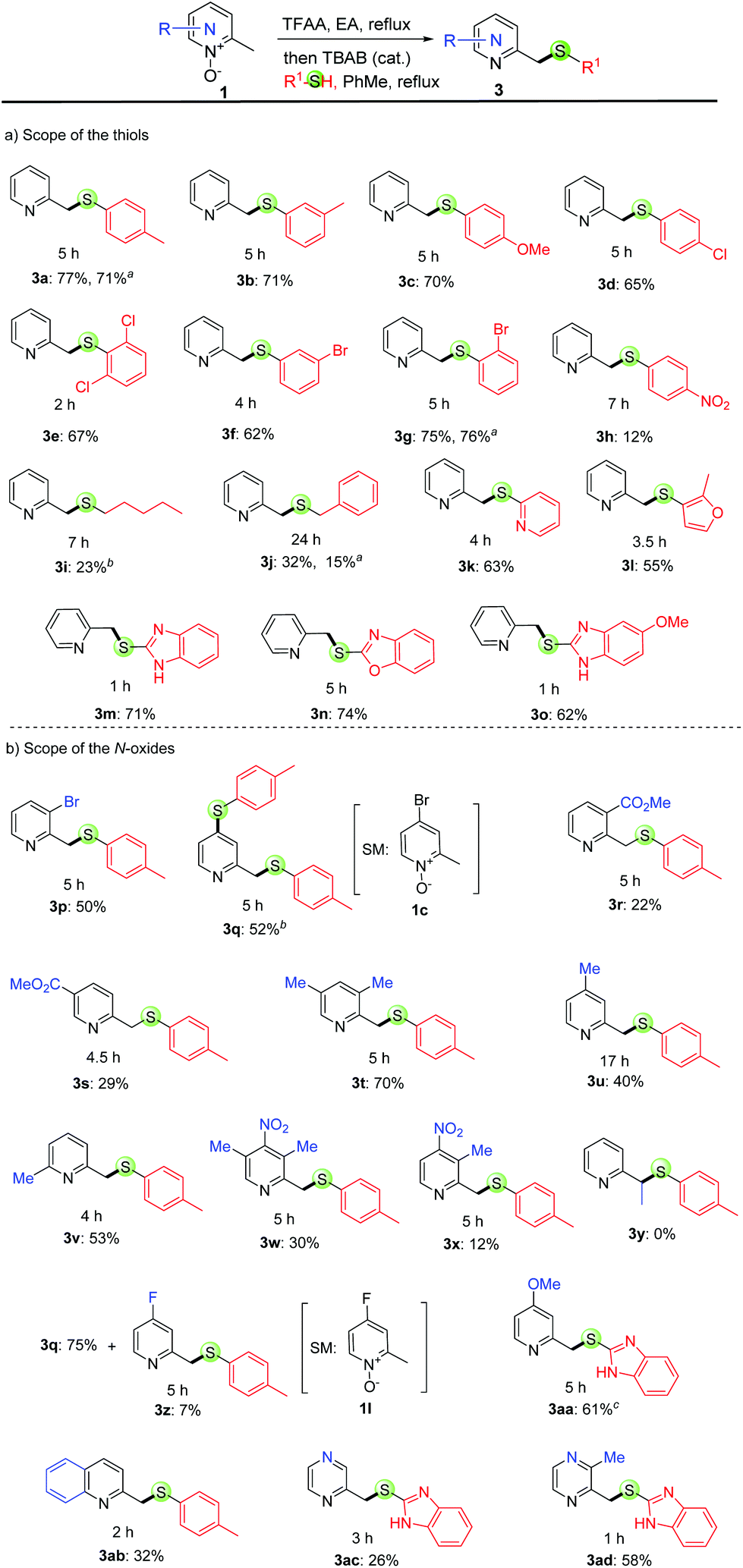 | ||
Scheme 2 Substrate scope for the thiolation of the methyl C(sp3)–H bond in 2-picoline N-oxides. The reaction time of the second step is listed under the structure. Unless otherwise noted, all reactions were conducted at 0.8 M concentration with N-oxide (1.0 equiv.), R1SH (1.0 equiv.), and TBAB (0.2 eq.) in toluene at reflux. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reaction was conducted at 0.8 M concentration with N-oxide (1.0 equiv.), p-toluenethiol (1.0 equiv.), and TBAB (0.2 eq.) in EA at 120 °C. bR1SH (2.0 equiv.) was employed. cThe reaction was run without TBAB. The reaction was conducted at 0.8 M concentration with N-oxide (1.0 equiv.), p-toluenethiol (1.0 equiv.), and TBAB (0.2 eq.) in EA at 120 °C. bR1SH (2.0 equiv.) was employed. cThe reaction was run without TBAB. | ||
With regard to the scope of thiols, both thiophenols (3a–h) and other arylthiols (3k–o), such as mercaptopyridine (3k), furanthiol (3l) and mercaptobenzimidazole (3m), are all good substrates. Furthermore, the reaction works for alkylthiols (3i & 3j) although lower yields were observed. It is found that 2a is not very reactive with these alkylthiols, and the hydrolyzed compound, 2-(hydroxymethyl)pyridine, was separated as the major product. These electron-rich thiophenols afforded similar yields to those of electron-poor thiophenols (i.e., 3avs.3cvs.3e), except for 4-nitrothiophenol (3h, 12% yield), which is probably due to the very poor nucleophilicity. We next studied the scope of pyridine N-oxides. Notably, the electron-rich substrates afforded better yields than those of electron-poor substrates (i.e., 3t > 3u > 3s ≈ 3r > 3x), probably due to the stronger reactivity of these electron-rich substrates in nucleophilic substitution reactions. Pyridine rings carrying 3-bromo (3p), C3- or C5- (3t, 3w & 3x), C4- (3u) and C6-methyl (3v) substituents are compatible with the reaction, thus providing additional handles for further functionalization at the halogenated or benzylic positions using cross-coupling reactions. Furthermore, this reaction is highly regioselective as only the pyridine-2-ylmethyl position can be functionalized; the methyl groups at other positions, such as the C3- or C5- (3t, 3w & 3x) and C4- (3u) are unreactive. On the other hand, this reaction is not effective with other 2-alkylpyridine N-oxides as 3y was not formed, which is probably due to the steric hindrance. Interestingly, 4-bromo-2-picoline N-oxide (1c) afforded disulfide product 3q in 52% yield, which was formed through the desired reaction at the pyridine-2-ylmethyl position and the SNAr reaction between the bromide and p-toluenethiol. A similar result was obtained for 4-fluoro-2-picoline N-oxide (1l), with 3q as the major product.
We then assessed the reaction with other azine N-oxides, including 2-methylquinoline N-oxides, 2-methylpyrazine N-oxides and 2,3-dimethylpyrazine N-oxides, and found that the desired products were obtained in medium yields (3ab–ad).
To further demonstrate the synthetic utility of this newly developed method, we next applied this developed protocol to the synthesis of omeprazole sulfide (3ae, Scheme 3) and rabeprazole sulfide (3af, Scheme 4). However, instead of the desired product 3ae, only the demethylated product 3ae′ was obtained in 71% yield using our developed method (Table 3, entry 1). This undesired 3ae′ might be produced from the reaction with the bromide anion of TBAB. Gratefully, the desired product 3ae was produced in 40% yield when 0.1 equiv. of TBAB was used (entry 2). More reactions were carried out with less quantity of TBAB, and we found out that the smaller the amount of TBAB, the higher the yield of the desired product. In fact, the optimized condition for this reaction is not to use any TBAB (60% yield for 3ae, entry 5). Therefore, we proposed that those highly electron-rich substrates can react with arylthiols directly without the need for any TBAB catalyst. Indeed, this hypothesis was verified in the synthesis of 3aa (61% yield, Scheme 2) and 3af, which was obtained in 80% yield by mixing equal equiv. of 2r with mercaptobenzimidazole in refluxing toluene (Scheme 4, Process A). It is worth mentioning that the demethylation is common for these 4-methoxy substituted N-oxides as the yield was decreased a lot for 3aa but not for 3af with TBAB conditions. In comparison with Process A, the known protocol for 3af (Process B) comprises 4 steps in 48% overall yield, which uses the same approach as in Scheme 1A and is the best known process for the production of rabeprazole sulfide so far to the best of our knowledge.20 Another remarkable feature of our developed method is scalability. 3m, 3ae and 3af were prepared on two different scales (∼5 mmol and ∼50 mmol), and similar yields were obtained.
 | ||
| Scheme 3 Synthesis of omeprazole sulfide. | ||
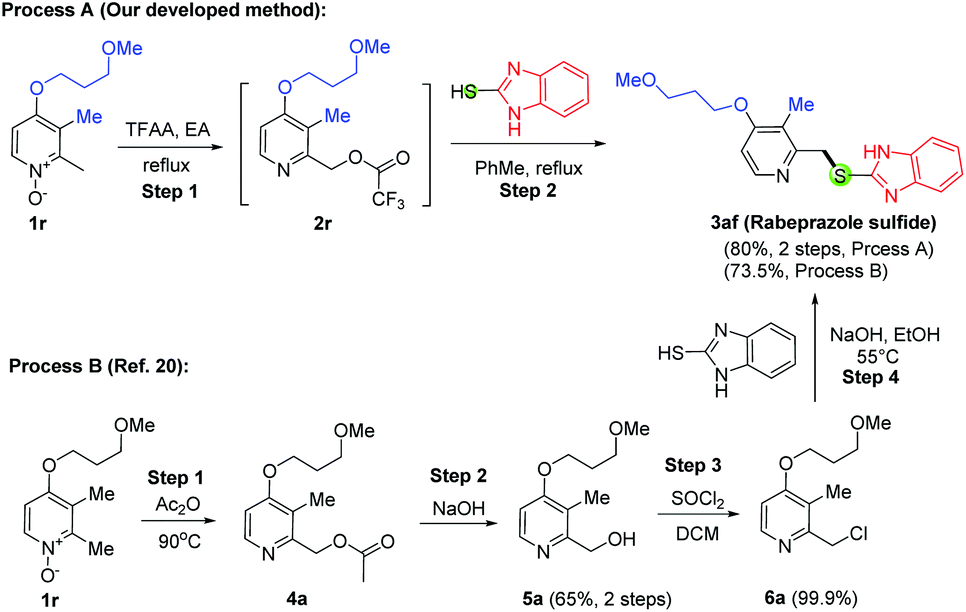 | ||
| Scheme 4 Synthesis of rabeprazole sulfide. | ||
| Entry | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Eq. of TBAB | 0.2 | 0.1 | 0.05 | 0.01 | 0 |
| Yield % of 3y | 0 | 40 | 49 | 55 | 60 |
| Yield % of 3y′ | 71 | 18 | 13 | 10 | 5 |
Evaluation of the green chemistry credentials of the two processes in Scheme 4 was done by performing a series of green metrics calculations.21 Atom economy (AE) and atom efficiency (AEf) were calculated to estimate the amount of the reactants that will remain in the product. However, these metrics ignore the inorganic reagents and solvents that are not incorporated into the final product. Moreover, significant stoichiometric excess is also not considered in AE and AEf. Therefore, carbon efficiency (CE), reaction mass efficiency (RME) and overall efficiency (OE) were also calculated (Table 4). Furthermore, as waste production is a major concern in the modern pharmaceutical industry, the E-factor, mass of waste/mass of product, was also calculated (Table 5).7 These green metrics of Process A are significantly better than those of Process B, especially the AEf, CE, OE and E-factor. Therefore, Process A is a greener and much more efficient process than Process B.
| Process | Isolation steps | Yield (%) | AE (%) | AEf (%) | CE (%) | RME (%) | OE (%) |
|---|---|---|---|---|---|---|---|
| A | 1 | 80 | 60 | 48 | 51.4 | 31.0 | 51.6 |
| B20 | 3 | 48 | 55.2 | 26.3 | 34.4 | 21.6 | 39.2 |
Based on our preliminary experimental results, three possible reaction pathways leading to 3 are outlined in Scheme 5. Pathway 1 predominates for highly electron-rich 2-picoline N-oxides, which can react with arylthiols directly by a SN2 reaction through a six-membered ring transition state. In comparison, pathway 2 or pathway 3 predominates for other types of 2-picoline N-oxides, whose reactivity is poorer than those of electron-rich N-oxides, and needs the catalysis of TBAB. In pathway 2, TBAB reacts with 2 by a SN2 reaction to afford the bromide intermediate 9. Then, the bromo atom of 9 is replaced with the thiol by the second SN2 reaction to afford 3 and bromide, which proceeds to the next catalytic cycle. However, this pathway is ruled out as 9 was not detected in the reaction mixture. Besides, no 3a was formed by subjecting 9 under the developed reaction conditions (Scheme 6). In pathway 3, the trifluromethyl group of 2 is replaced with a bromo atom to give the more reactive bromoformate intermediate 10 and CF3−, which reacts with R1SH to produce R1S− and trifluoromethane. This reaction explains why alkylthiols give lower yields than arylthiols. Arylthiols are more acidic than alkylthiols, leading to a more favorable equilibrium for this acid–base reaction. Finally, a nucleophilic attack on 10 by R1S− affords 3 and carbon dioxide and the regeneration of bromide completes the catalytic cycle.
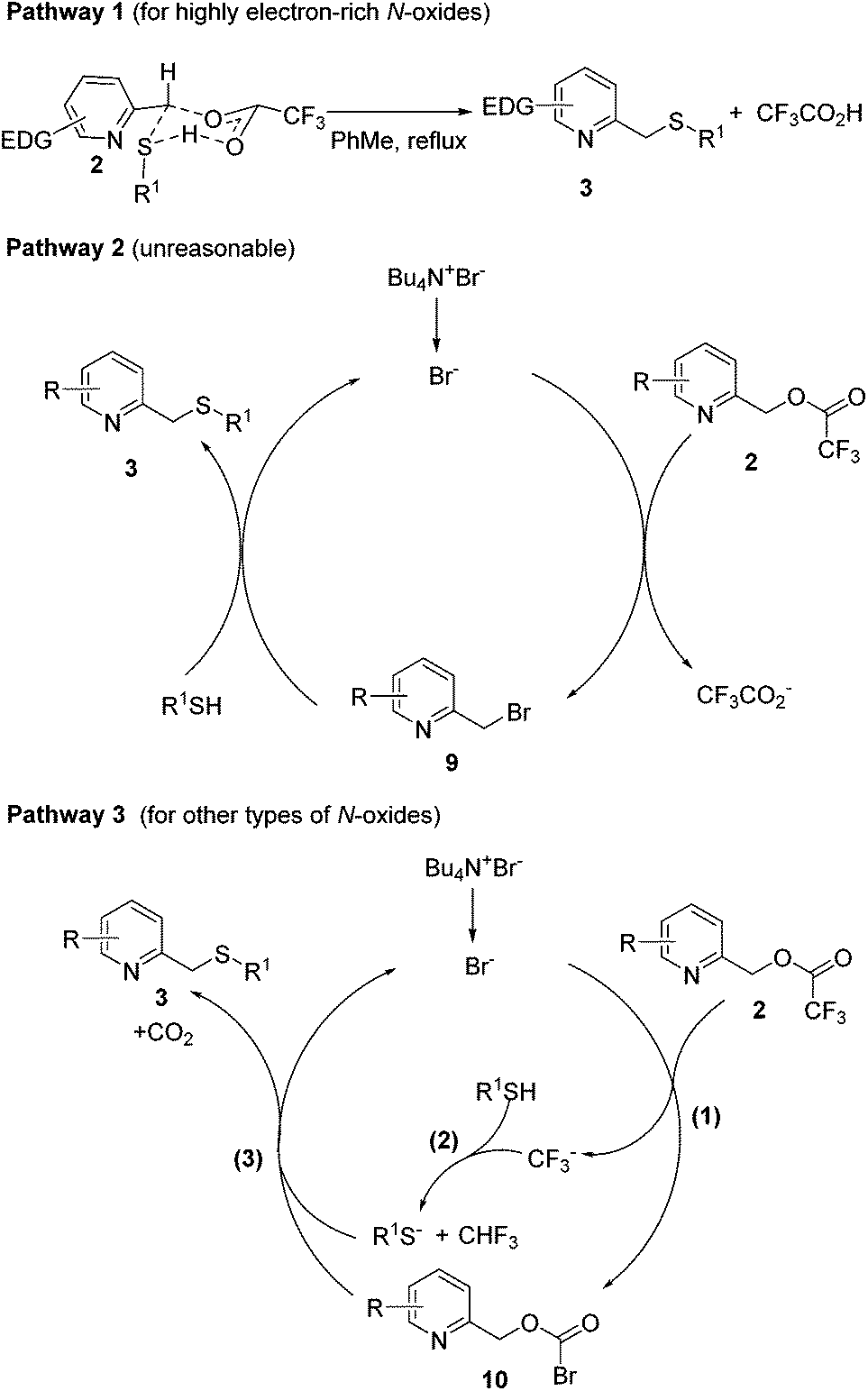 | ||
| Scheme 5 Proposed reaction mechanism. | ||
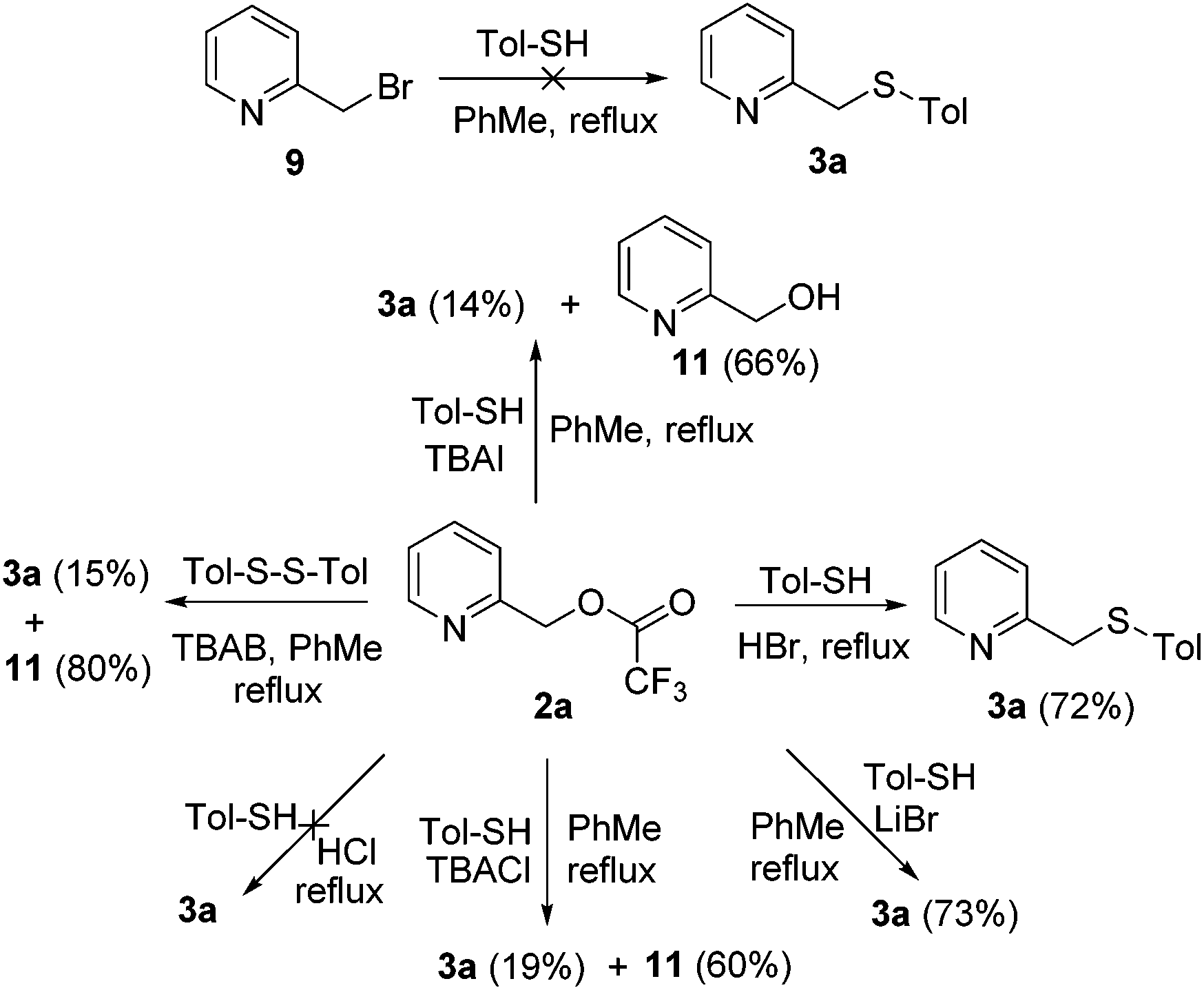 | ||
| Scheme 6 Mechanistic studies. | ||
Pathway 3 was further evidenced by the reactions given in Scheme 6. First, similar yields were obtained when subjecting 2a to HBr or LiBr conditions, whereas no product was formed under HCl conditions, indicating that a bromide anion is necessary and sufficient for this conversion. Indeed, much lower yields were observed under TBAI or TBACl conditions, and the hydrolyzed compound 11 was obtained as the major product (Scheme 6). The last substitution reaction would be very unfavorable for TBAI as iodoformate is less reactive than bromoformate. For TBACl, although the last step is more favorable, the first step is less favorable, as chloride is less nucleophilic than bromide. Second, 3a was obtained in only 15% yield by using 4-methylphenyl disulfide as the thiolation source, which further proves the necessity of the acid–base reaction in pathway 3, requiring the participation of the thiol hydrogen.
Conclusions
In summary, we have disclosed the first protocol for the thiolation of the unactivated methyl C(sp3)–H bond in 2-picolines. The protocol comprises a one-pot, two-step reaction through a TFAA-mediated [3,3]-sigmatropic rearrangement of pyridine N-oxides and TBAB-catalyzed direct conversion of trifluoroacetates into thioethers under metal- and base-free conditions. The present method is mild, scalable, highly regioselective, and step- and atom-economical. The TBAB catalyst is not required for those highly electron-rich 2-picoline N-oxides. On the other hand, the method doesn't work well for electron-poor N-oxides or alkylthiols. Furthermore, the method has been successfully applied to the synthesis of omeprazole sulfide and rabeprazole sulfide, which are the advanced intermediates of two on-market hot drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Foundation of Tianjin Educational Committee (no. 2017KJ008) is greatly acknowledged. The authors are thankful to the Research Center of Modern Analytical Technology, Tianjin University of Science and Technology for NMR measurements and high resolution mass analysis.Notes and references
- (a) H. Hagiwara, K. Kobayashi, S. Miya, T. Hoshi, T. Suzuki, M. Ando, T. Okamoto, M. Kobayashi, I. Yamamoto, S. Ohtsubo, M. Kato and H. Uda, J. Org. Chem., 2002, 67, 5969–5976 CrossRef CAS PubMed; (b) J. Bonjoch, J. Catena and N. Valls, J. Org. Chem., 1996, 61, 7106–7115 CrossRef CAS PubMed.
- K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521–5577 CrossRef CAS PubMed.
- For reviews, see: (a) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (b) S. Liang, S. Shaaban, N.-W. Liu, K. Hofman and G. Manolikakes, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2018, ch. 3, vol. 69, pp. 135–207 Search PubMed.
- (a) R. A. Daines, P. A. Chambers, J. J. Foley, D. E. Griswold, W. D. Kingsbury, L. D. Martin, D. B. Schmidt, K. K. C. Sham and H. M. Sarau, J. Med. Chem., 1996, 39, 3837–3841 CrossRef CAS PubMed; (b) J. J. Li, L. G. Hamann, Z. Ruan, C. B. Cooper, S. C. Wu, L. M. Simpkins, H. Wang, A. Nayeem and S. R. Krystek, US20060235028A1, 2006; (c) F. Haviv, R. W. DeNet, R. J. Michaels, J. D. Ratajczyk, G. W. Carter and P. R. Young, J. Med. Chem., 1983, 26, 218–222 CrossRef CAS; (d) H. Schickaneder, H. Engler and I. Szelenyi, J. Med. Chem., 1987, 30, 547–551 CrossRef CAS PubMed.
- (a) P. Lindberg, A. Braendstroem, B. Wallmark, H. Mattsson, L. Rikner and K. J. Hoffmann, Med. Res. Rev., 1990, 10, 1–54 CrossRef CAS; (b) C. I. Carswell and K. L. Goa, Drugs, 2001, 61, 2327–2356 CrossRef CAS PubMed.
- (a) S. Rádl, O. Klecán and J. Havlíček, J. Heterocycl. Chem., 2006, 43, 1447–1453 CrossRef; (b) P. R. Reddy, V. Himabindu, L. Jaydeepkumar, G. M. Reddy, J. V. Kumar and G. M. Reddy, Org. Process Res. Dev., 2009, 13, 896–899 CrossRef CAS; (c) B. Kohl, E. Sturm, J. Senn-Bilfinger, W. A. Simon, U. Krueger, H. Schaefer, G. Rainer, V. Figala and K. Klemm, et al. , J. Med. Chem., 1992, 35, 1049–1057 CrossRef CAS PubMed; (d) S. Souda, N. Ueda, S. Miyazawa, K. Tagami, S. Nomoto, M. Okita, N. Shimomura, T. Kaneko, M. Fujimoto, et al., EP268956A2, 1988; (e) R. L. M. Broeckx, D. De Smaele and S. M. H. Leurs, WO2003008406A1, 2003.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- (a) D. Wei, W. Song, D. Gao, L. Dong, Z. Jia, L. Wang, W. Wang, C. Yang, Y. Wang and C. Liu, CN106674198A, 2017; (b) K. M. Joseph and I. Larraza-Sanchez, Tetrahedron Lett., 2011, 52, 13–16 CrossRef CAS.
- (a) D. Wang, Y. Wang, J. Zhao, M. Shen, J. Hu, Z. Liu, L. Li, F. Xue and P. Yu, Org. Lett., 2017, 19, 984–987 CrossRef CAS PubMed; (b) D. Wang, H. Feng, L. Li, Z. Liu, Z. Yan and P. Yu, J. Org. Chem., 2017, 82, 11275–11287 CrossRef CAS PubMed; (c) D. Wang, M. Shen, Y. Wang, J. Hu, J. Zhao and P. Yu, Asian J. Org. Chem., 2018, 7, 879–882 CrossRef CAS; (d) D. Wang, J. Hu, J. Zhao, M. Shen, Y. Wang and P. Yu, Tetrahedron, 2018, 74, 4100–4110 CrossRef CAS.
- C.-S. Wang, T. Roisnel, P. H. Dixneuf and J.-F. Soulé, Org. Lett., 2017, 19, 6720–6723 CrossRef CAS PubMed.
- Y. Yang, C. Qu, X. Chen, K. Sun, L. Qu, W. Bi, H. Hu, R. Li, C. Jing, D. Wei, S. Wei, Y. Sun, H. Liu and Y. Zhao, Org. Lett., 2017, 19, 5864–5867 CrossRef CAS PubMed.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Org. Biomol. Chem., 2018, 16, 4954–4957 RSC.
- X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Yin, Org. Lett., 2015, 17, 1970–1973 CrossRef CAS PubMed.
- S.-r. Guo, Y.-q. Yuan and J.-n. Xiang, Org. Lett., 2013, 15, 4654–4657 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- (a) A. Mobinikhaledi and N. Foroughifar, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 405–412 CrossRef CAS; (b) G.-H. Chu, C. T. Saeui, K. Worm, D. G. Weaver, A. J. Goodman, R. L. Broadrup, J. A. Cassel, R. N. DeHaven, C. J. La Buda, M. Koblish, B. Brogdon, S. Smith, B. Le Bourdonnec and R. E. Dolle, Bioorg. Med. Chem. Lett., 2009, 19, 5931–5935 CrossRef CAS PubMed; (c) S. Pal, J. Hatai, M. Samanta, A. Shaurya and S. Bandyopadhyay, Org. Biomol. Chem., 2014, 12, 1072–1078 RSC.
- (a) V. Boekelheide and W. J. Linn, J. Am. Chem. Soc., 1954, 76, 1286–1291 CrossRef CAS; (b) T. Cohen and G. L. Deets, J. Am. Chem. Soc., 1972, 94, 932–938 CrossRef CAS.
- C. Fontenas, E. Bejan, H. A. Haddou and G. G. A. Galavoine, Synth. Commun., 1995, 25, 629–633 CrossRef CAS.
- K. Venkatesham, C. Bhujanga Rao, C. B. Dokuburra, R. A. Bunce and Y. Venkateswarlu, J. Org. Chem., 2015, 80, 11611–11617 CrossRef CAS PubMed.
- A. Kumar, K. R. More, P. M. Gounder, P. N. Patil, T. D. Bhor, S. S. Chikane and S. N. Darunde, WO2009116072A2, 2009.
- (a) F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC; (b) D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC; (c) C. Jimenez-Gonzalez, D. J. C. Constable and C. S. Ponder, Chem. Soc. Rev., 2012, 41, 1485–1498 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc03072c |
| This journal is © The Royal Society of Chemistry 2019 |