
New renewably-sourced polyesters from limonene-derived monomers†
Megan R.
Thomsett
 a,
Jonathan C.
Moore
a,
Antoine
Buchard
b,
Robert A.
Stockman
*a and
Steven M.
Howdle
*a
a,
Jonathan C.
Moore
a,
Antoine
Buchard
b,
Robert A.
Stockman
*a and
Steven M.
Howdle
*a
aSchool of Chemistry, University Park, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: robert.stockman@nottingham.ac.uk; Fax: +44 (0)115 951356
bDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 26th November 2018
Abstract
The functionalisation of limonene has enabled the synthesis of two renewably-sourced monomers for the formation of terpene-derived polyesters. Three methods for the synthesis of the novel hydroxy-acid 6 are reported and their green-credentials scrutinised through comparison of their sustainability-metrics. Step-growth homo-polymerisation of 6 is demonstrated to yield a low molecular weight (2.6 kDa) novel polyester with 100% of its carbon content originating from the terpene starting material. The corresponding diol 2 is shown to act as a co-monomer with a renewable diacid. The resultant polyesters display impressive Mns of up to 30 kDa with Tgs between −6 and 24 °C. These materials have been shown to depolymerise under basic conditions for reclamation of the diol monomer 2.
Introduction
If the present consumption of fossil fuels continues at the current rate, the reserves of oil feedstocks will be expended within the next century. This development combined with the associated environmental issues has resulted in a driving force for scientific research to alleviate the dependence on polymers derived from fossil fuels.1 The utilisation of fossil fuels in the manufacture of plastics currently accounts for about 7% of worldwide oil and gas usage.2 Of the 300 million tonnes of plastics produced annually worldwide, renewable polymers make up just 1%.1,3 The comparatively high cost of polymers from renewable feedstocks has likely contributed to their small share in the market, and severely detracts from their commercial viability.4–6 Renewable raw materials have the potential to provide a wide range of monomers arguably as comprehensive as those provided by the petrochemical industry.7,8 In recent years considerable research has been devoted to finding renewable monomers for the synthesis of sustainable polymers.Terpenes, terpenoids and resin acids are a group of non-polar small molecules typically produced biosynthetically by many classes of trees and plants.9 Terpenes are an abundant waste stream, are inexpensive, and do not directly compete with food sources; this makes them ideal small molecule building blocks for many applications.9 The many desirable properties of terpenes have recently made them very attractive for polymer research and this is highlighted by reviews which have been published within the past five years.10–13 Much of the research into the polymerisation of terpenes in their raw form has resulted only in polymers which lack the desired properties to compete with those currently used commercially.14–21 It is clear that new approaches are required to unlock the potential of terpenes as monomers for renewable polymers. These observations have engendered a nascent field of research into the controlled and sustainable functionalisation of terpenes to produce monomers that can be “dropped-in” to established and efficient polymerisation processes. Synthetic methods have been developed to derive terpene and terpenoid based monomers which can readily undergo free radical polymerisation.22–24 We recently reported a series of acryloyl and methacryloyl terpene derivatives that can be readily polymerised under standard free-radical conditions to produce materials with a diverse and attractive range of properties.24 There are also multiple examples of the ring expansion of oxygen containing terpenes to produce lactones and lactams for use in ring opening polymerisation (ROP) to form polyesters and polyamides, respectively.25–31
Limonene (1) is one of the most abundant terpenes (present in more than 300 plants) and is a prominent waste stream of the citrus industry, with the (R)-enantiomer being produced on a scale of over 70 KTA.32 With two double bonds, methods for its functionalisation are broad and well established in organic chemistry, as summarised by Bessière and co-workers.33 In the field of polymer chemistry however, the majority of research has stemmed from the epoxidation of the endocyclic double bond to produce cis- and trans-limonene oxide. Coates and co-workers performed seminal studies on the copolymerisation of these epoxides with CO2 to produce linear polycarbonates.34 They went on to show that the (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) combination of amorphous (S)- and (R)-poly(trans-limonene oxide) produces a stereocomplexed, semi-crystalline polymer with enhanced mechanical properties beyond those observed for its individual components.35 Greiner and co-workers developed a stereoselective synthesis of limonene oxide, which was shown to be more efficient and to produce high molecular weight polymers (Mn > 100 kDa) with attractive thermal properties (e.g. glass transition temperature (Tg) = 130 °C).36
:1) combination of amorphous (S)- and (R)-poly(trans-limonene oxide) produces a stereocomplexed, semi-crystalline polymer with enhanced mechanical properties beyond those observed for its individual components.35 Greiner and co-workers developed a stereoselective synthesis of limonene oxide, which was shown to be more efficient and to produce high molecular weight polymers (Mn > 100 kDa) with attractive thermal properties (e.g. glass transition temperature (Tg) = 130 °C).36
However, approaches using step-growth polymerisation are few. Currently, the only method to incorporate limonene into a step-growth polymer uses thiol–ene chemistry and has been used to produce polyesters,37 polyamides and polyurethanes.38 Whilst highly atom-efficient, this approach relies on the coupling of terpenes with polymerisable functionalities, which are not necessarily derived from renewable sources. In recent years, a wide range of terpene derived polymers have been reported, but some of the synthetic strategies employed might be considered step-intensive and not sustainable. There is a lack of terpene derived monomers for step-growth polymerisation, and studies into end of life solutions are few. Designing a closed-loop thermoplastic which can be depolymerised and then re-polymerised to feed back into the system would capture the embodied energy and value currently contained in polymers, and could also reduce leakage into natural systems. This is crucial to the concepts of a circular economy and allows us to move away from the “take, make, dispose” status quo.39
Herein we report the efficient synthesis of two terpene-derived monomers, which are accessed in three steps or less from limonene, to produce bio-derived polyesters via step-growth polymerisation. These materials have been shown to depolymerise under basic conditions to allow reclamation of the terpene derived component, opening up the potential for recyclability of this novel bio-derived polymer and creating a closed, circular system.
Experimental
Materials
Benzyl alcohol (BnOH) was distilled from CaH2 prior to use; THF was freshly distilled from sodium metal and benzophenone; water was deionised before use. Limonene was purchased from Sigma Aldrich. All other reagents were purchased from a chemical supplier and used without further purification. Experiments carried out under inert atmosphere used argon or nitrogen, and glassware was flame dried prior to use. All solutions are saturated unless stated otherwise.General methods and instrumentation
1H and 13C NMR spectra were recorded in CDCl3, at ambient temperature using Bruker AV400 (400 MHz), AV3400 (400 MHz), AV3400HD (400 MHz) and AV3500 (500 MHz) spectrometers. Gel permeation chromatography (GPC) was performed using an Agilent 1260 Infinity Series HPLC (Agilent Technologies, USA) in THF (HPLC grade, Fisher Scientific) as eluent at room temperature using two Agilent PL-gel mixed-E columns in series with a flow rate of 1 mL min−1. A differential refractometer (DRI), was used for sample detection. Infra-red spectra were recorded using a Bruker Tensor 27 FT-IR spectrophotometer using either an ATR attachment or a solution cell using CHCl3 as the solvent (νmax is reported in cm−1). High-resolution mass spectra were obtained using a Bruker MicroTOF mass spectrometer operating in electrospray ionisation (ESI) mode. Dynamic mechanical analysis (DMA) was used to determine the Tg of the polymers. Measurements were performed on a Triton Technologies DMA (now Mettler Toledo DMA1) using the powder pocket accessory. The sample (40 mg ± 5 mg) was weighed into a powder pocket. Samples were measured at 1 and 10 Hz in single cantilever bending geometry between −100–100 °C. The value of the Tg was taken as the peak of the tan delta (tanδ) curve. DFT calculations details (methods, data and output files) can be found in the ESI.†
Synthesis of 2a and 2b
To a solution of limonene (1) (16.1 mL, 100 mmol) in THF (30 mL) cooled to 0 °C was added borane (100 mL, 100 mmol, 1 M in THF) dropwise over 25 minutes. Once the addition was complete the reaction was allowed to warm to room temperature and stirred (2 h). The reaction was cooled to 0 °C and a solution of NaOH(aq) (350 mL, 350 mmol, 1 M) was added followed by H2O2 (34 mL, 350 mmol, 30% w/v in H2O). The reaction mixture was stirred (30 min) at 0 °C and then at room temperature (30 min). EtOAc (400 mL) was added to the reaction mixture and the organic layer separated and washed with water (200 mL), Na2CO3(aq) (200 mL), NaCl(aq) (200 mL), dried over Na2SO4, and concentrated under reduced pressure to give the crude material as a yellow oil. Purification by silica gel column chromatography (40% EtOAc in petroleum ether) afforded 2a and 2b (3:1) as a pale yellow oil (16.7 g, 97.0 mmol, 97%).
Synthesis of 4
To a solution of 2a and 2b (3:1) (17.95 g, 104 mmol) in CH2Cl2 (1000 mL) was added a solution of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) (6.5 g, 41.7 mmol) in CH2Cl2 (20 mL) dropwise, bisacetoxyiodobenzene (BAIB) (100.4 g, 312 mmol) was subsequently added in portions over 15 min. The reaction was stirred at room temperature (16 h). 5% Na2S2O3(aq) (540 mL) then NaHCO3(aq) (1050 mL) was added and the mixture was stirred vigorously (15 min). The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (3 × 500 mL). The organic extracts were combined, washed with NaCl(aq) (500 mL), dried over Na2SO4 and concentrated under reduced pressure. Purification by silica gel column chromatography (5–7% EtOAc in petroleum ether) afforded 4 as a pale-yellow oil (5.8 g, 34.5 mmol, 33%).
Synthesis 5a and 5b
To a solution of 2a and 2b (3:1) (5.00 g, 29.1 mmol) in MeCN (300 mL) was added [Cu(MeCN)4]OTf (546 mg, 1.4 mmol). A solution of TEMPO (210 mg, 1.4 mmol), 2,2-bipyridyl (210 mg, 1.4 mmol) and 1-methylimidazole (240 mg, 2.91 mmol) in MeCN (9 mL) was added dropwise, and the mixture stirred at room temperature (20 h). The reaction mixture was passed through a plug of silica and the product eluted with Et2O. The solvent was removed under reduced pressure to give the crude product, purification by silica gel chromatography (15% EtOAc in petroleum ether) afforded 5a and 5b (3:1) as a colourless oil (3.83 g, 22.5 mmol, 77%).
Synthesis of 6a and 6b
To a solution of 5a and 5b (3:1) (3.00 g, 17.5 mmol) in tBuOH/H2O (2:1, 80 mL) was added NaH2PO4 (8.19 g, 52.5 mmol) and the resulting solution was cooled to 0 °C. H2O2 (6.83 mL, 87.5 mmol, 30% w/v in H2O) was added followed by NaClO2 (11.87 g, 105 mmol, 80%) in portions over 45 min. The reaction was allowed to stir at 0 °C (30 min) and then extracted into EtOAc (80 mL). The organic extract was washed with H2O (5 × 50 mL), NaCl(aq) (50 mL), dried over Na2SO4 and concentrated under reduced pressure to yield 6a and 6b (3:1) as a white semi-solid (2.2 g, 11.8 mmol, 68%).
Synthesis of 7
6a and 6b (3:1) (500 mg, 2.68 mmol) and Sn(oct)2 (8.5 μL, 1 mol%) were added to a flame dried flask under a flow of N2, which was heated to either 120 or 180 °C and stirred for the required time period. The temperature was then set to 180 °C and the system put under vacuum for 24 h. The polymerisation was stopped by cooling to −5 °C and the resulting polymer dried under vacuum for 24 h.
Synthesis of 10
All reagents were dried under vacuum at 25 °C for at least 24 h prior to use. The mixture of 2a and 2b (3:1) (1.50 g, 8.7 mmol) was added to a 50 mL round bottomed flask equipped with a condenser and trap cooled to 0 °C. Succinic acid (9) (1.13 g, 9.57 mmol) was added, and the reaction heated to 190 °C (6 h). The metal catalyst (Sn(oct)2 or Ti(OBu)4) was added (1 mol%), the temperature increased and the system put under vacuum. After an elapsed time the polymerisation was stopped by cooling to −5 °C and the polymer washed with methanol.
Degradation studies
10 (96 mg) was added to a mixture of NaOH(aq) (4 mL, 3 M) and THF (4 mL) and heated in a 95 °C oil bath (10 days). The solvent was removed under reduced pressure and the residue acidified with HCl(aq) (1 M), the aqueous phase was extracted with CH2Cl2 (4 × 15 mL), dried over Na2SO4 and concentrated under reduced pressure to yield 2a and 2b as a yellow oil (59 mg, 0.43 mmol).Results and discussion
We initially set out to investigate the formation of a limonene derived monomer that could homo-polymerise. The target was the polymerisation of hydroxy-acid 6 to yield the polyester 7, which contains an exclusively terpene-derived backbone. To the best of our knowledge a material of this nature is yet to be reported. Step-growth polycondensation of hydroxy-acids is well known. However, there are very few examples where a (carbocyclic) ring is incorporated into the backbone of the polymer. We hypothesised that this feature might impart structural rigidity and perhaps lead to desirable properties such as a high Tg. Using Brown's conditions,40 limonene (1) was converted to its corresponding 1,5-syn and 1,5-anti diol derivatives 2a and 2b, respectively (Scheme 1). The reaction proceeded smoothly in an excellent yield of 97%, yielding 2a and 2b in a 3:1 ratio, each as a 1:1 mixture of C-8 epimers. Three synthetic routes (labelled as Route A, B and C) were investigated for the conversion of these diols to the targeted hydroxy-acid 6 (Fig. 1).
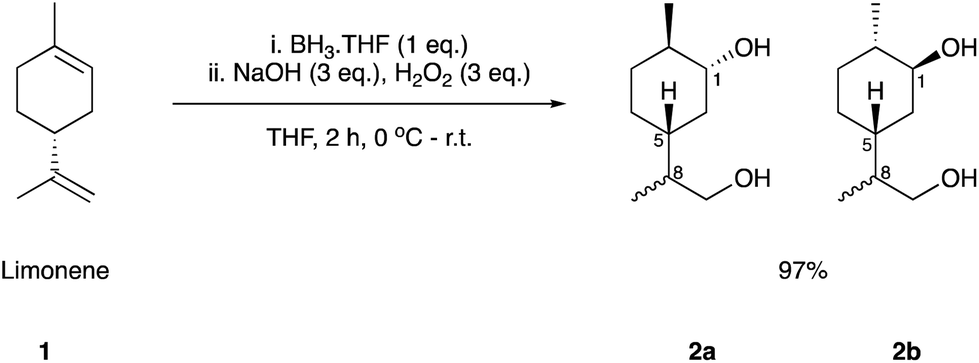 | ||
| Scheme 1 Synthesis of diols 2a and 2b from limonene (1) using a Brown hydroboration/oxidation sequence. | ||
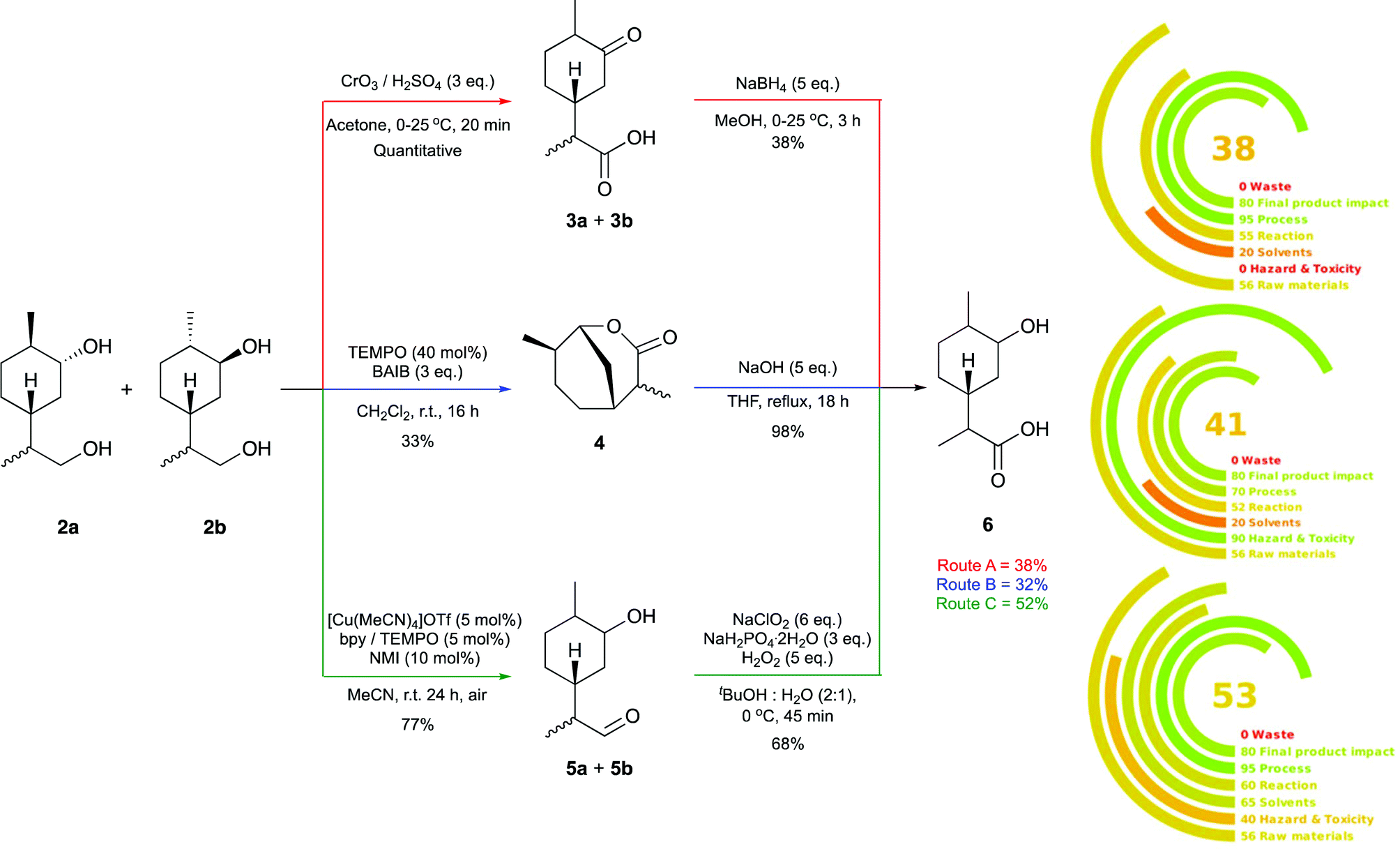 | ||
| Fig. 1 Comparison of Route A (top), Route B (middle) and Route C (bottom) for the synthesis of hydroxy-acid 6 and subsequent analysis of the three routes using the metric tool Green Motion™. | ||
Route A involved global oxidation followed by ketone reduction, Route B comprised oxidative lactonisation and subsequent hydrolytic ring opening, and Route C utilised a two-step chemoselective primary alcohol to carboxylic acid oxidation. The optimised two-step routes were compared in terms of yield, operational simplicity and sustainability. The latter was investigated using the metric tool Green Motion™ (Fig. 1).41 One benefit of Route A is its operational simplicity; the synthetic sequence could be carried out in a single working day, and following aqueous work up, no further purification of the products was required. However, this route did require the use of super-stoichiometric Jones’ reagent, and produced a mixture of eight diastereomers, complicating analysis of the reaction mixtures considerably.
Route B had a similar overall yield (32% vs. 38%), but purification of the lactone 4via chromatography was required and each step took longer than a day for completion. Route C was the highest yielding, affording the hydroxy-acid 6 in 52% over two steps. Other benefits to Route C are the incorporation of a catalytic protocol for aldehyde formation that utilises molecular oxygen as the terminal oxidant and the mild conditions employed for the Pinnick oxidation. One drawback of this route was that it required purification of 5a and 5b using silica gel chromatography.
Using Green Motion™ we evaluated each route with respect to overall sustainability; the lower the impact to the environment the higher the rating (100 being ideal). This metric combines the twelve principles of green chemistry with a penalty point system. The use of the toxic Jones’ reagent in Route A resulted in a score of zero with respect to “hazard and toxicity” and subsequently, the lowest overall score of 38. Route B scores a slightly higher 41 however falls down due to the use of halogenated solvents for the oxidative lactonisation. The highest yielding approach (Route C), which employs a catalytic protocol and benign reagents, also scored the highest on Green Motion™ with a score of 53. According to the guidelines laid out within the metric,41 this score validates that the route can be considered “green”. For this reason, Route C was adopted for further investigations.
With the hydroxy-acid 6 in hand, initial studies into its polymerisation were performed. These were carried out using bulk polymerisation, with Sn(oct)2 as a catalyst at 180 °C or 120 °C followed by heating at 180 °C under vacuum (Table 1). The consumption of the monomer was followed by 1H NMR spectroscopy, using the resonances obtained from the respective C-1 environments. Initially the polymerisation was attempted using a (3:1) mixture of 6a and 6b (Table 1, entry 1). 1H NMR analysis after 20 min indicated that the concentration of 6a had decreased and that the major component was the lactone 4. After 3 hours, no significant change could be observed. However, after 24 h the relative concentration of 6b began to decrease and a resonance at approximately 4.65–4.90 ppm emerged, indicating formation of the polyester 7. At this stage vacuum was applied to drive the polymerisation to high conversion and remove lactone 4 from the reaction mixture. The broad peaks at approximately 4.40–4.55 and 4.65–4.90 ppm were tentatively assigned to the respective axial and equatorial protons alpha to the oxygen in the ester bond (Fig. 2).
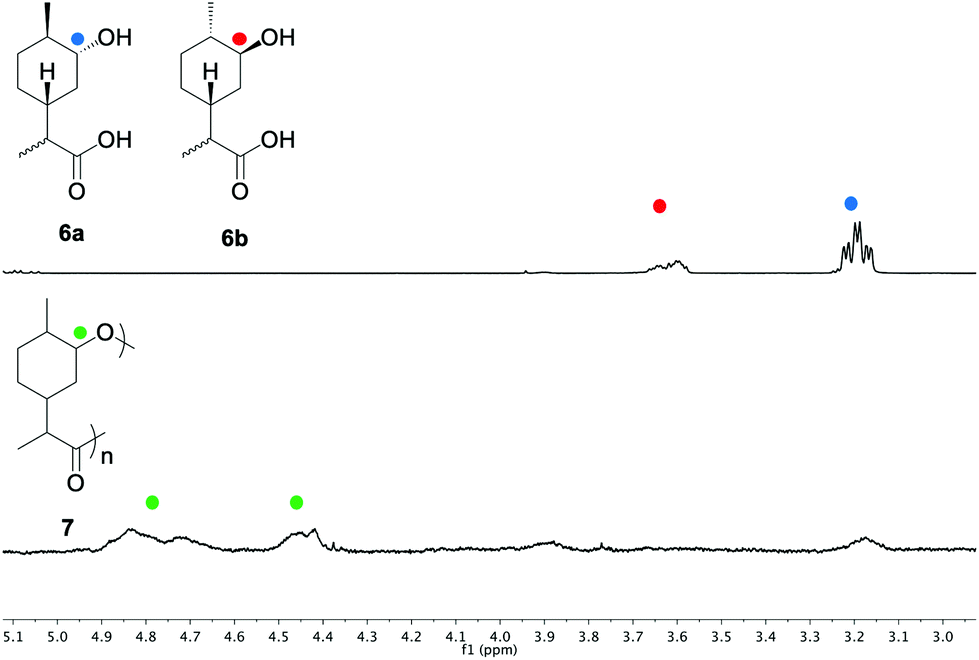 | ||
| Fig. 2 1H NMR of the monomers 6a and 6b and the resulting polymer 7. | ||
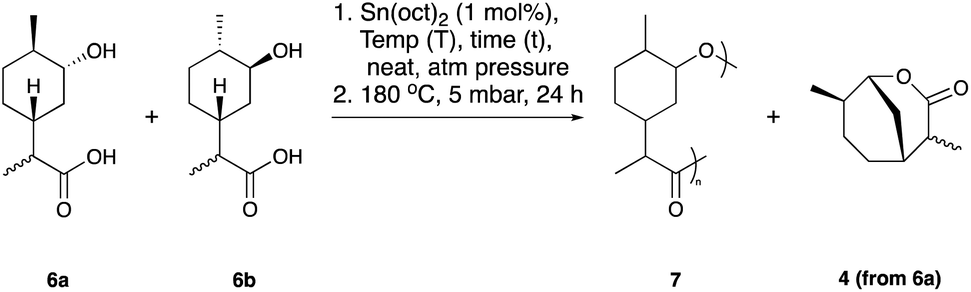
GPC analysis revealed molecular weights of up to 2.6 kDa, but with a large proportion of the reaction mixture being comprised of low molecular weight oligomers. It is well established that sterically hindered secondary alcohols can be slow to propagate in polyesterifications.42 We infer that lactonisation occurs through the disfavoured 1,5-diaxial conformer of 6a, whereby (unlike 6b) the close proximity of the endo-alcohol functionality with the carboxylic acid would be sufficient to allow ring closing. To test our hypothesis, the hydroxy-acid 6a (accessed through the hydrolysis of 4) was subjected to the polymerisation conditions in the absence of 6b (Table 1, entry 2). Quantitative conversion to the lactone 4 was observed, with no polymeric species being detected. In an effort to promote the formation of linear polymers, the polymerisation was repeated at 120 °C (Table 1, entry 3). Again, GPC analysis revealed that the material was mainly composed of low molecular weight oligomers. Since the reduced temperature was likely contributing to a slower rate of reaction, the polymerisation was subsequently repeated for a much longer period of 1 month (Table 1, entry 4). 1H NMR analysis suggested that the reaction had gone to full conversion and consumption of the monomer was confirmed by high resolution mass spectrometry (HRMS). GPC analysis showed a broad peak (typical of step growth polymerisation) and a significantly greater proportion of higher molecular weight species (Mn = 2.2 kDa) albeit with a higher dispersity (Đ = 2.4) than the polymer synthesised at 180 °C. Both polymers were investigated using DMA to study their thermal properties. The analysis indicated that 7 is an amorphous polymer with a Tg of 44 °C, which is relatively high for polyesters with molecular weights in the region of 2.2–2.6 kDa. This lends support to our prior hypothesis that the presence of a carbocyclic ring in the backbone would impart rigidity to the polymer.
One potential explanation for the slow rate of polymerisation observed with the hydroxy-acids 6a and 6b is the somewhat sterically hindered environment of the secondary alcohols. Using DFT analysis it could be inferred that the formation of the lactone via6a was favoured (ΔG = −8.3 kcal mol−1) over homopolymerisation (ΔG −0.9 kcal mol−1) or copolymerisation with 6b (ΔG = as low as −3.9 kcal mol−1). The polymerisation of 6b is marginally favoured thermodynamically (ΔG = between −0.8 and −1.9 kcal mol−1 depending on the conformation(s) of 6b units in the polymer linkage – e.g. axial/axial, equatorial/equatorial, axial/equatorial or equatorial/axial). This somewhat explains the long reaction times and low molecular weights obtained from the polymerisation of 6a and 6b. One potential solution to this is to utilise 6a and 6b as comonomers in the synthesis of copolyesters.
It was also intriguing to investigate whether the lactone 4, which had served as an intermediate in the synthesis of 6a, could itself be utilised as a monomer for synthesis of the polyester 7via ring opening polymerisation (ROP). It is well known that six membered (delta) lactones are typically stable to ring opening, due in part to low ring strain.43 It is also recognised that substituents on the ring decrease the equilibrium monomer conversion. This is especially true for lactones substituted in the alpha and delta positions.44,45 The terpene derived delta-lactone 4 is substituted in both the alpha and delta positions suggesting that its ring opening polymerisation may be thermodynamically unfavourable. However, 4 is also a bridged bicyclic system (oxabicyclo[3.3.1]nonane) and we had speculated that this may provide the additional ring strain required to facilitate efficient ROP.
The polymerisation of 4 was attempted under a wide range of conditions that are well precedented for the ROP of lactones. Various Brønsted46 and Lewis47 acids, organocatalysts,48,49 and metal alkoxides47 were investigated under a range of temperatures (see ESI†). Disappointingly however, even with extended reaction times of up to 6 months, only initiation to the benzyl ester 8 could be observed and no evidence of propagation could be detected (Scheme 2).
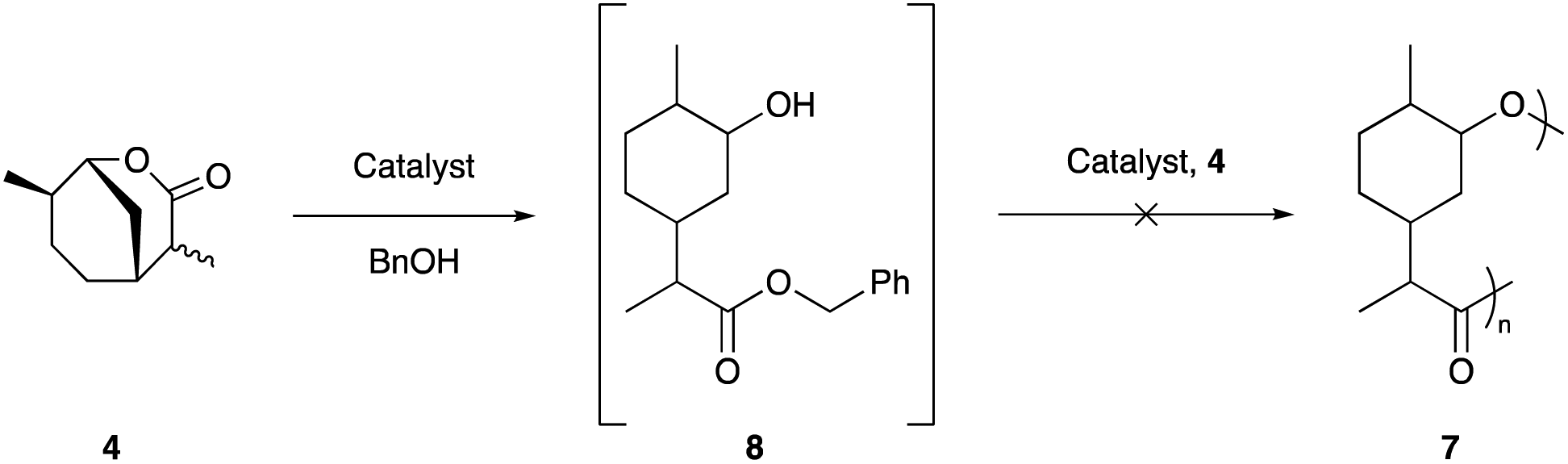 | ||
| Scheme 2 Attempted ROP of lactone 4. | ||
To better understand why lactone 4 was not undergoing ring opening polymerisation DFT calculations were conducted (protocol rM062X-D3/6-311++g(2d,p)/cpcm = ethylethanoate). DFT analysis indicated that the most stable conformation of 4 occurs when the cyclohexane ring is in a chair conformation. The driving force for most ROP is the release of ring strain. The enthalpy of polymerisation (ΔH) can be used as a measure of the ring strain and in general for polymerisation to be favourable, it is required that ΔH < 0.50 The ring strain for the predicted structure was calculated to be at least −4.7 kcal mol−1 which implied that polymerisation may be enthalpically favourable. However, the overall ΔG of the polymerisation was calculated as at best +1.4 kcal mol−1. This indicates the entropic factor of the ROP is counteracting the ring opening, and that the enthalpic driving force is not large enough to overcome it.
Following our studies into the synthesis of an entirely terpene derived polymer, we moved to investigate the potential of the limonene derived diol 2 as a comonomer. An analogous approach has been reported recently by Roth and co-workers whereby a borneol derived diol was employed in step-growth polymerisations.51 We envisaged that the copolymerisation of 2 with a suitable diacid would produce polymers with a higher degree of flexibility than 7, which may possess lower Tgs and be suitable for different applications. Succinic acid (9) was selected as co-monomer due to its established availability from renewable resources.52 The co-polymerisation of 2 and 9 was anticipated to yield the entirely bio-based polyester 10 (Table 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | T/°C | t/h | M n/kDa | T g/°C | Đ |
| 1 | Ti(OBu)4 | 190 | 24 | 18.4 | 6 | 2.4 |
| 2 | Sn(oct)2 | 190 | 24 | 20.7 | 21 | 2.5 |
| 3 | N/A | 190 | 24 | 9 | −7 | 1.7 |
| 4 | Ti(OBu)4 | 230 | 24 | 11.2 | 4 | 1.5 |
| 5 | Sn(oct)2 | 230 | 24 | 10.1 | 15 | 1.5 |
| 6 | N/A | 230 | 24 | 9.2 | 19 | 1.4 |
| 7 | Ti(OBu)4 | 230 | 3 | 30.4 | 16 | 2.5 |
| 8 | N/A | 230 | 3 | 9.3 | 6 | 1.3 |
| 9 | Sn(oct)2 | 230 | 3 | 28.3 | 23 | 2.3 |
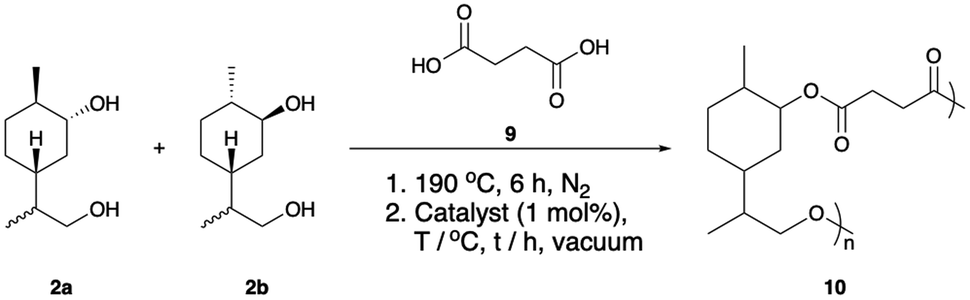
One of the most commonly employed catalysts for condensation polymerisation is Sn(oct)2, however the use of this species raises concerns with respect to sustainability. One class of catalyst that can be considered a greener alternative are the titanium alkoxide complexes,7,53 whose environmental benefits arise predominantly from the fact that titanium is generally non-toxic and earth abundant.54 The polymerisation of 2a and 2b with succinic acid (9) was investigated using Sn(oct)2 and Ti(OBu)4 and the results were compared by GPC analysis (Table 2). Initially, esterification was performed for 6 hours under an inert atmosphere at 190 °C. At this stage the catalyst was added and polymerisation was induced via the application of vacuum. Pleasingly, GPC analysis revealed that similar results could be obtained using both Sn(oct)2 and Ti(OBu)4; in each case high molecular weight species were produced. Furthermore, in the absence of catalyst, polymerisation was still observed to occur. However, in this case significantly lower molecular weight species (ca. 9 kDa) were obtained (Fig. 3). By optimising the polymerisation, we found that at a temperature of 230 °C in the presence of catalyst (Table 2, entries 4–9), significantly higher molecular weights (30 kDa) were achieved and the materials also exhibited low Tgs (6–23 °C).
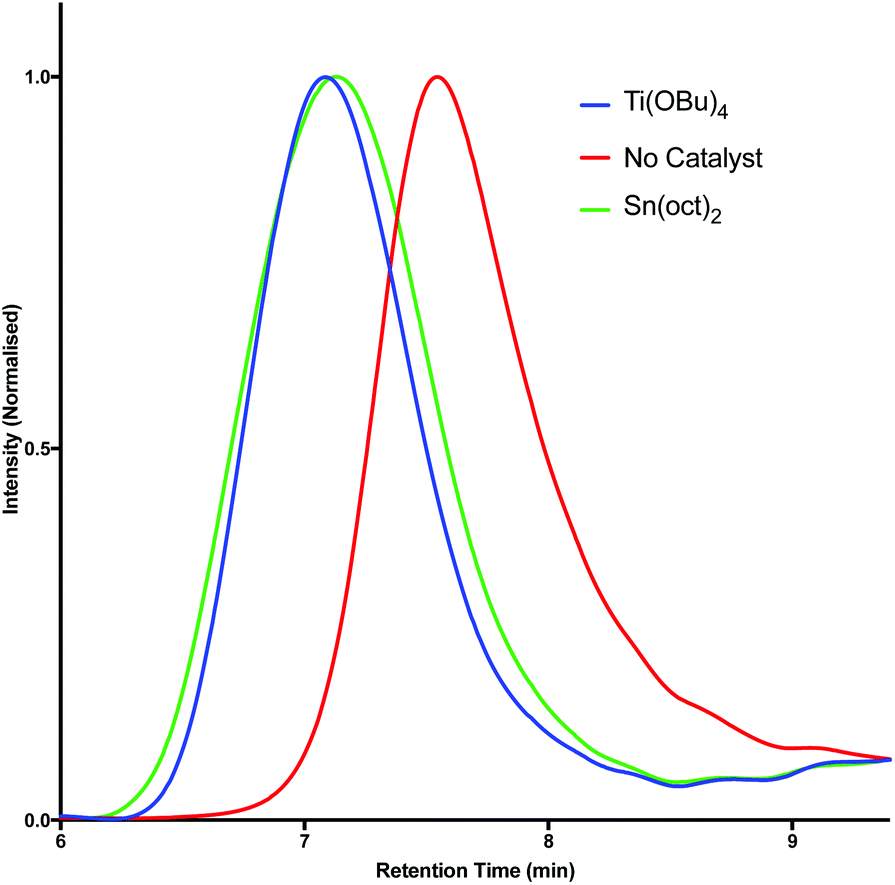 | ||
| Fig. 3 GPC traces for the synthesis of polymer 10 (Table 2, entries 7–9). | ||
It is becoming increasingly important that chemists design end of life solutions into their molecules. The polyesters synthesised from limonene diol (2) and succinic acid (9) should be susceptible to hydrolytic degradation at a variety of pHs and temperatures. Three samples of polymer 10 were submerged in aqueous buffer solutions at pH 3, 7 and 11 and heated to 50 °C for two weeks. Analysis by GPC indicated no observable change in the molecular weight and it was hypothesised that this was due to the observed insolubility of the polymer in aqueous media. In an attempt to encourage degradation, the reaction was repeated using a mixture of THF and NaOH(aq) (3 M) (1:1) and heated in a 95 °C oil bath for 10 days (Scheme 3). Analysis by GPC revealed that the crude mixture extracted from the degradation study contained no high-molecular weight species, as demonstrated by the disappearance of the peak at a retention time of ∼32 min (Fig. 4). 1H NMR analysis indicated that the polymer had fully depolymerised back to monomer (2a and 2b), which was recovered in excellent purity and with an identical d.r. of 3:1 by simple extraction into CH2Cl2 (Fig. 5). This opens up the possibility that polymer 10 could be easily and continuously recycled via a closed, circular system.
 | ||
| Scheme 3 Degradation of 10 under basic conditions. | ||
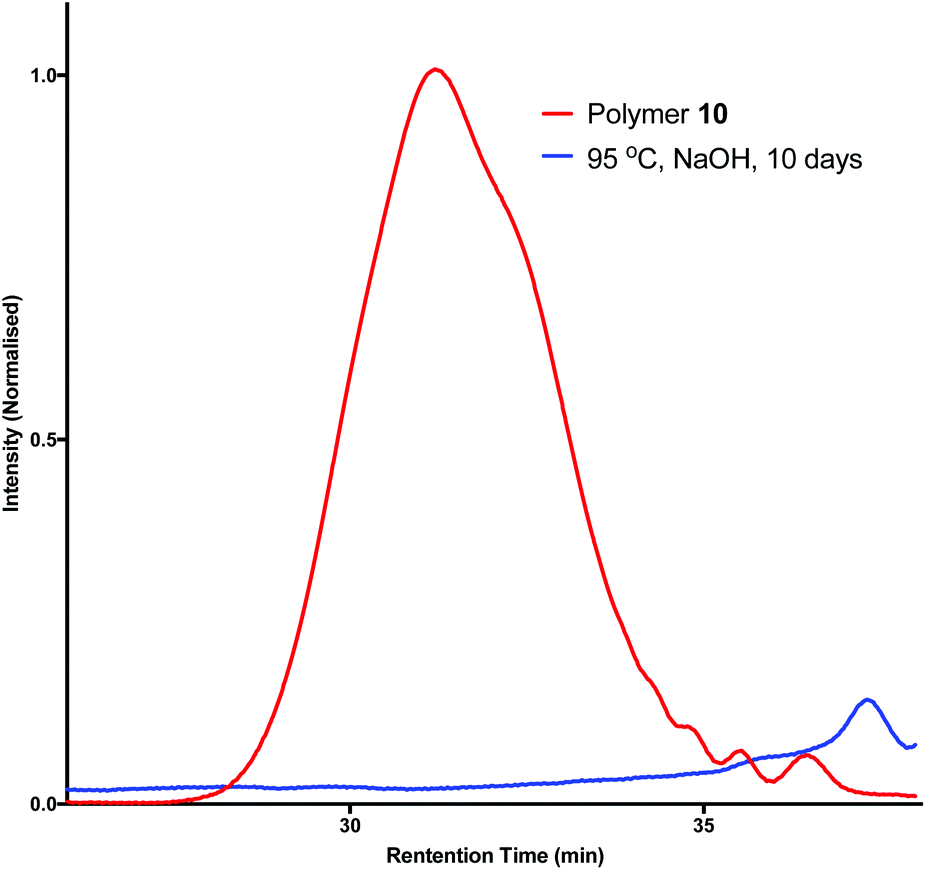 | ||
| Fig. 4 GPC traces showing the degradation of polymer 10 through the disappearance of the peak representing high molecular weight species. | ||
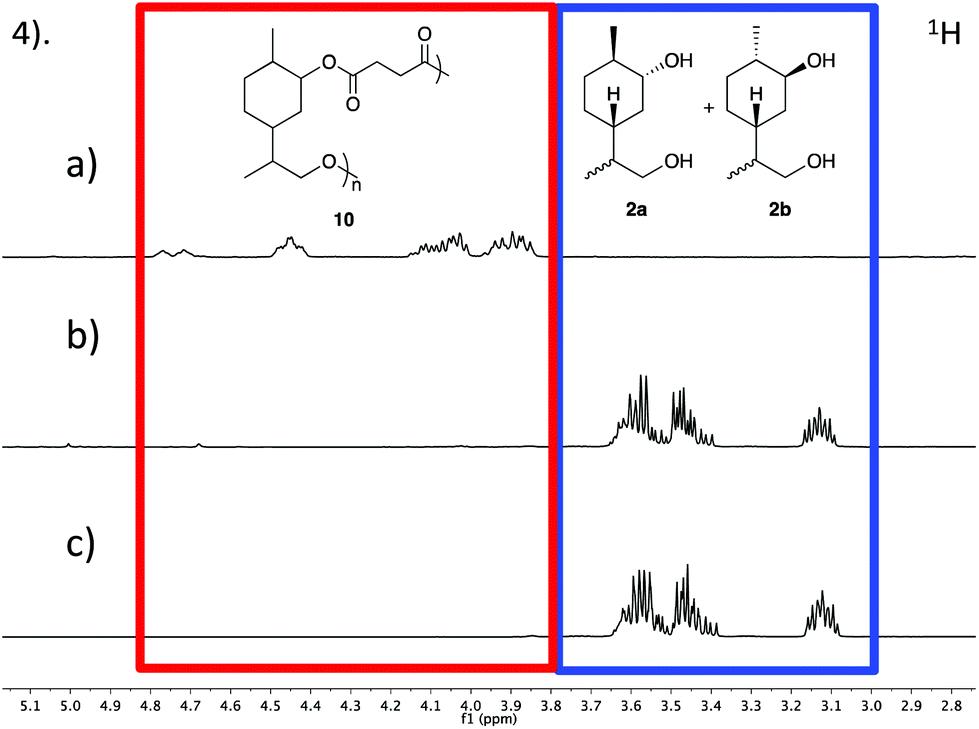 | ||
| Fig. 5 1H NMR spectra showing the decomposition of polymer 10 into the monomer 2. (a) The polymer 10 before degradation studies (b) the crude mixture from the extraction after the degradation (c) pure monomer 2 synthesised from 1. | ||
Conclusions
Two terpene derived monomers (2 and 6) have been synthesised from limonene (1) and their polymerisation potentials investigated. The unusual bridged bicyclic lactone 4 was also studied both experimentally and computationally but was observed not to undergo ring opening polymerisation. Polymerisation of the hydroxy-acid 6 using a polycondensation technique yielded low molecular weight polymers (Mns ca. 2.6 kDa) with Tgs of ca. 44 °C. However, co-polymerisation of the diol 2 and succinic acid produced the novel polyester 10 with considerably higher Mns of up to 30 kDa. This material is derived completely from renewable resources and a high percentage of its carbon content can be sourced from terpene waste streams. Finally, the polymer 10 was shown to degrade under basic conditions into its substituent monomer 2, demonstrating that the system has a high potential for recyclability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the EPSRC for funding this work (MT, Centre for Doctoral Training in Sustainable Chemistry); EP/L015633/1 and (JM) EP/N019784/1. Ms Alice Haddleton for assistance with thermal analysis. AB thanks the University of Bath HPC for computing resources and the Royal Society for funding (Fellowship UF140207).References
- C. K. Williams and M. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- A. L. Holmberg, K. H. Reno, R. P. Wool and T. H. Epps, Soft Matter, 2014, 10, 7405–7424 RSC.
- B. P. Mooney, Biochem. J., 2009, 418, 219–232 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- R. T. Mathers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1–15 CrossRef CAS.
- A. J. D. Silvestre and A. Gandini, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, 2008, pp. 17–38 Search PubMed.
- M. Winnacker and B. Rieger, ChemSusChem, 2015, 8, 2455–2471 CrossRef CAS.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS.
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115–134 CrossRef.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- W. J. Roberts and A. R. Day, J. Am. Chem. Soc., 1950, 72, 1226–1230 CrossRef CAS.
- H. Pietila, A. Sivola and H. Sheffer, J. Polym. Sci., Part A: Polym. Chem., 1970, 8, 727–737 CrossRef CAS.
- C. Snyder, W. McIver and H. Sheffer, J. Appl. Polym. Sci., 1977, 21, 131–139 CrossRef CAS.
- J. Lu, H. Liang, R.-J. Zhang and Y.-X. Deng, Chem. J. Chin. Univ., 1995, 17, 1314–1318 Search PubMed.
- J. Lu, M. Kamigaito, M. Sawamoto, T. Higashimura and Y.-X. Deng, J. Appl. Polym. Sci., 1996, 61, 1011–1016 CrossRef CAS.
- J. Lu, M. Kamigaito, M. Sawamoto, T. Higashimura and Y. Deng, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1423–1430 CrossRef CAS.
- J. Lu, M. Kamigaito, M. Sawamoto, T. Higashimura and Y.-X. Deng, Macromolecules, 1997, 30, 22–26 CrossRef CAS.
- J. Lu, M. Kamigaito, M. Sawamoto, T. Higashimura and Y.-X. Deng, Macromolecules, 1997, 30, 27–31 CrossRef CAS.
- H. Miyaji, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2016, 55, 1372–1376 CrossRef CAS PubMed.
- Y. Zheng, K. Yao, J. Lee, D. Chandler, J. Wang, C. Wang, F. Chu and C. Tang, Macromolecules, 2010, 43, 5922–5924 CrossRef CAS.
- M. F. Sainz, J. A. Souto, D. Regentova, M. K. G. Johansson, S. T. Timhagen, D. J. Irvine, P. Buijsen, C. E. Koning, R. A. Stockman and S. M. Howdle, Polym. Chem., 2016, 7, 2882–2887 RSC.
- D. Zhang, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2005, 6, 2091–2095 CrossRef CAS.
- J. R. Lowe, M. T. Martello, W. B. Tolman and M. A. Hillmyer, Polym. Chem., 2011, 2, 702–708 RSC.
- M. Winnacker, M. Neumeier, X. Zhang, C. M. Papadakis and B. Rieger, Macromol. Rapid Commun., 2016, 37, 851–857 CrossRef CAS.
- M. Winnacker, A. Tischner, M. Neumeier and B. Rieger, RSC Adv., 2015, 5, 77699–77705 RSC.
- M. Winnacker, S. Vagin, V. Auer and B. Rieger, Macromol. Chem. Phys., 2014, 215, 1654–1660 CrossRef CAS.
- H. C. Quilter, M. Hutchby, M. G. Davidson and M. D. Jones, Polym. Chem., 2017, 8, 833–837 RSC.
- M. Winnacker and J. Sag, Chem. Commun., 2018, 54, 841–844 RSC.
- M. Bahr, A. Bitto and R. Mulhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- A. F. Thomas and Y. Bessière, Nat. Prod. Rep., 1989, 6, 291–309 RSC.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chemie Int. Ed., 2015, 54, 1215–1218 CrossRef CAS PubMed.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- M. Firdaus and M. A. R. Meier, Green Chem., 2013, 15, 370–380 RSC.
- J. Korhonen, A. Honkasalo and J. Seppälä, Ecol. Econ., 2018, 143, 37–46 CrossRef.
- H. C. Brown and C. D. Pfaffenberger, Tetrahedron, 1975, 31, 925–928 CrossRef CAS.
- For an in-depth explantation of the Green Motion tool, see: T. V. T. Phan, C. Gallardo and J. Mane, Green Chem., 2015, 17, 2846–2852 RSC.
- H. Zhang and M. W. Grinstaff, Macromol. Rapid Commun., 2014, 35, 1906–1924 CrossRef CAS.
- C. Alemán, O. Betran, J. Casanovas, K. N. Houk and H. K. Hall, J. Org. Chem., 2009, 74, 6237–6244 CrossRef.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef.
- E. N. Stepurko and G. N. Roganov, Fibre Chem., 2014, 46, 80–89 CrossRef CAS.
- X. Lou, C. Detrembleur and R. Jérôme, Macromolecules, 2002, 35, 1190–1195 CrossRef CAS.
- B. M. Cooper, D. Chan-Seng, D. Samanta, X. Zhang, S. Parelkar and T. Emrick, Chem. Commun., 2009, 815–817 RSC.
- L. Simón and J. M. Goodman, J. Org. Chem., 2007, 72, 9656–9662 CrossRef PubMed.
- M. Xiong, D. K. Schneiderman, F. S. Bates, M. A. Hillmyer and K. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8357–8362 CrossRef CAS PubMed.
- A. Duda and A. Kowalski, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J. Raquez, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009, pp. 1–51 Search PubMed.
- S. Roth, I. Funk, M. Hofer and V. Sieber, ChemSusChem, 2017, 10, 3574–3580 CrossRef CAS PubMed.
- M. Jiang, J. Ma, M. Wu, R. Liu, L. Liang, F. Xin, W. Zhang, H. Jia and W. Dong, Bioresour. Technol., 2017, 245, 1710–1717 CrossRef CAS.
- E. Gubbels, L. Jasinska-Walc and C. E. Koning, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 890–898 CrossRef CAS.
- V. P. Ananikov, Sustainable Catalysis, Royal Society of Chemistry, Cambridge, 2015, vol. 55 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8gc02957a |
| This journal is © The Royal Society of Chemistry 2019 |