
Cardioprotective effects of dihydroquercetin against ischemia reperfusion injury by inhibiting oxidative stress and endoplasmic reticulum stress-induced apoptosis via the PI3K/Akt pathway
Zunpeng
Shu
 a,
Yanni
Yang
a,
Liu
Yang
b,
Hai
Jiang
*b,
Xiaojin
Yu
*b and
Yi
Wang
*a
a,
Yanni
Yang
a,
Liu
Yang
b,
Hai
Jiang
*b,
Xiaojin
Yu
*b and
Yi
Wang
*a
aGuangdong Pharmaceutical University, Guangzhou 510006, P.R. China. E-mail: 784789997@qq.com
bKey Laboratory of Chinese Materia Medica (Ministry of Education), Heilongjiang University of Chinese Medicine, Harbin 150040, P.R. China. E-mail: yuxiaojin1979@126.com; jianghai_777@126.com
First published on 20th November 2018
Abstract
Dihydroquercetin (DHQ), a dihydroxyflavone, possesses potent antioxidant properties and is proposed to be useful in the prevention and treatment of cardiovascular disease. In this study, we aimed to investigate whether DHQ has protective effects against MIRI and to elucidate the mechanism of attenuation of oxidative stress-and ERS-induced apoptosis via the PI3K/Akt pathway. Isolated rat hearts and H9c2 cardiomyocytes were treated with or without DHQ, and then subjected to I/R and H/R, respectively. Our results showed that DHQ pretreatment markedly alleviated cardiac dysfunction, scavenged free radicals, reduced lipid peroxidation, and increased the activity of antioxidant enzymes ex vivo and in vitro. The result of western blot analysis showed that DHQ inhibited the apoptotic pathway and the expression of pro-apoptotic proteins CHOP, Caspase-12, and p-JNK. In addition, DHQ delayed the onset of ERs by reducing GRP78, p-PERK, and p-eif2α expression levels and by increasing HO-1 expression and Nrf2 binding to antioxidant response elements. These cardioprotective effects of DHQ were partially counteracted by the PI3K/Akt inhibitor LY294002. The protective effects of DHQ on the inhibition of MIRI may be mediated by activating the PI3K/Akt pathway to reduce oxidative stress-and ERS-induced apoptosis.
1. Introduction
Ischemic heart disease is a common multiple cardiovascular disease in clinical settings. It is a major contributor to morbidity and mortality worldwide and poses a serious threat to human life and health.1 I/R is a pathological phenomenon that causes structural damage and dysfunction or metabolic disturbance. These changes are caused by the recovery of blood flow to the ischemic myocardium. The mechanism of I/R injury is very complex, and oxidative stress and ERS are the hot spots of the current research. Excessive ROS generated at the onset of myocardial reperfusion has been reported to enhance oxidative stress, which is known to cause detrimental changes.2,3 HO-1 is a major antioxidant/cytoprotective enzyme. Enhanced HO-1 expression can reduce the incidence of oxidant-induced cell damage. Nrf2, a redox-sensitive transcription factor capable of interacting with AREs, plays a key role in the transcriptional activation of HO-1 gene expression. Recently, emerging evidence has revealed that the induction of HO-1 through Nrf2 activation confers protection against oxidative stress in the heart.4–7 ROS is a key regulator of oxidative stress, which is closely linked to ERS. ROS accumulation in the ER leads to the increased expression of CHOP, which eventually results in the induction of cell apoptosis.8–10 The ERS-initiated apoptotic signaling pathway, mitochondrial pathway, death receptor-associated pathway of another major apoptotic pathway, and ERS-induced apoptosis play crucial roles in MIRI.11,12Flavonoids are the major active ingredients in traditional Chinese medicine used for the treatment and prevention of cardiovascular disease. Epidemiological studies have shown that a high intake of dietary flavonoids can possibly reduce the risk of acute myocardial infarction and reduce mortality due to cardiovascular disease.13 DHQ (Fig. 1A) is a flavonoid that can exert a wide range of pharmacological effects such as antioxidant, anti-inflammatory, anti-viral, anti-tumor, free radical scavenging, and other biological activities.14–16 Owing to the diversity of its pharmacological activities, the bioavailability and biological characteristics of DHQ have attracted widespread attention. DHQ exerts effective antioxidant effects, which contribute to its cardiovascular protective effects. Studies have shown that DHQ not only decreased the peroxidase activity of cytochrome C with dioleyl cardiolipin and lipid peroxidation/free radical production, but also regulated the activity of oxidative enzymes and the overproduction of ROS for improving I/R injury.16,17 However, the mechanism underlying the myocardial protective effects of DHQ has not yet been fully elucidated. Therefore, in this study, we aimed to investigate the potential effects of DHQ on cardioprotection against I/R injury.
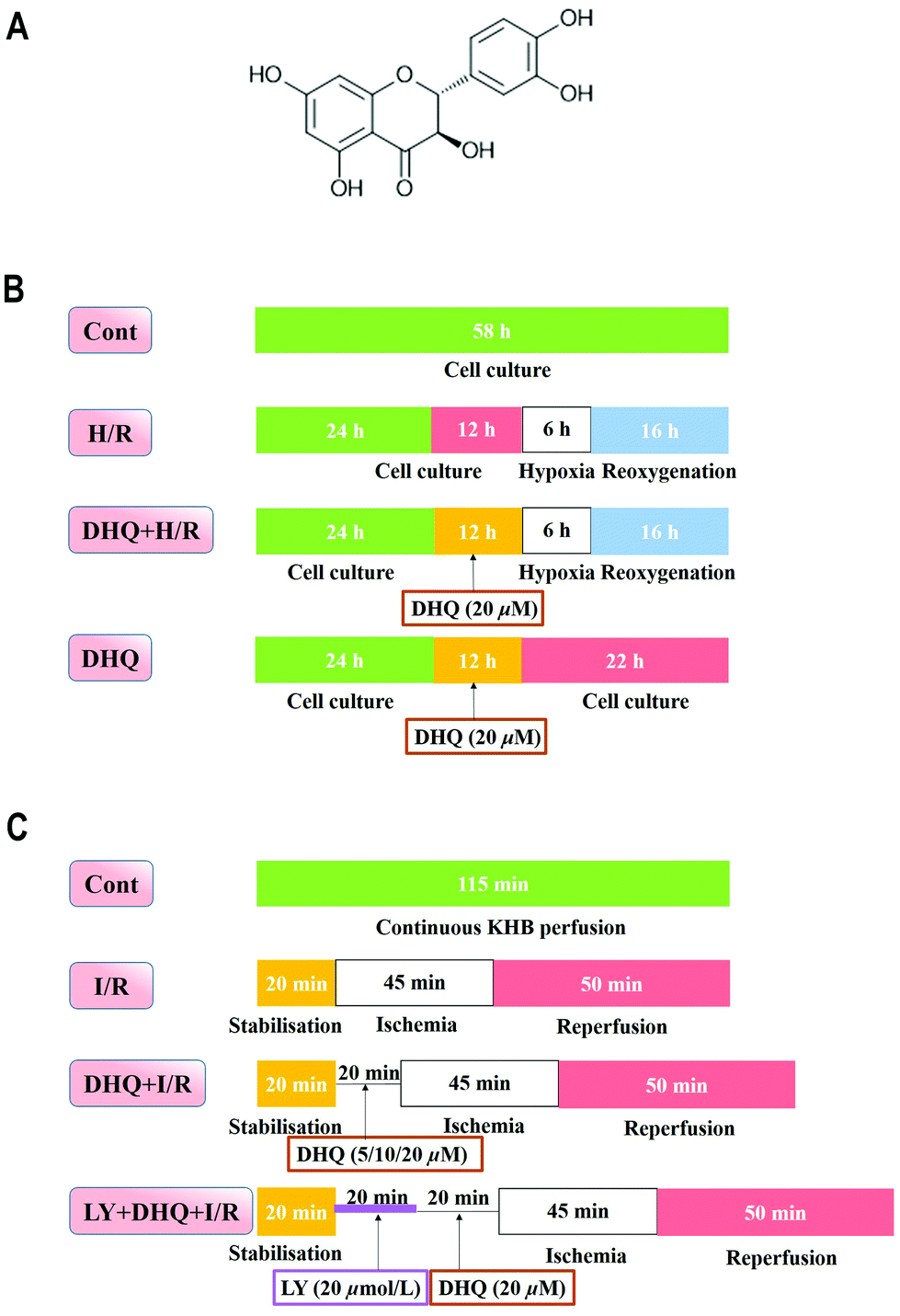 | ||
| Fig. 1 The chemical structure of dihydroquercetin (A) and the experimental designs of in vitro (B) and ex vivo (C) experiments. | ||
2. Materials and methods
2.1. Materials
All cell culture materials, Dulbecco's modified Eagle's medium (DMEM), foetal bovine serum (FBS), and penicillin/streptomycin were purchased from Gibco (Grand Island, NY, USA). The kits for determining the MDA content and the activity of CK, CAT, AST, GSH-Px, and SOD were acquired from Jiancheng Bioengineering Institute (Nanjing, China). ROS fluorimetric assay kits were purchased from Bio Vision Inc. (CA, USA). JC-1 fluorescent dyes were purchased from Sigma-Aldrich (St Louis, USA). TUNEL assay kit was purchased from Roche Diagnostics GmbH (Mannheim, Germany). Protein extraction kits were obtained from Co Win Bioscience Co., Ltd, Beijing, China. BCA kit (Pierce Corporation, Rockford, USA) and primary antibodies against PI3K, p-PI3K, Akt, p-Akt, JNK, p-JNK, CHOP, GRP78, ATF6, p-PERK, PERK, IRE1, eIf2α, p-eIf2α, Caspase-12, HO-1, Nrf2, Bcl-2, Bax, Bad, and β-actin were obtained from Santa Cruz Biotechnology, Inc. (CA, USA). Dimethyl sulfoxide (DMSO), MTT, LY294002, and other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA).2.2. Cell culture and experimental protocols
Clonal cell lines of BDIX rat embryonic heart tissue-derived H9c2 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured as previously described. Briefly, H9c2 cells were cultured in high glucose DMEM supplemented with 10% (v/v) fetal bovine serum, 1% penicillin/streptomycin (v/v) and 2 mM L-glutamine, and maintained in a humidified incubator at 37 °C in a 5% CO2 atmosphere.18,19 For all the experiments, cells were plated at an appropriate density according to the experimental design and were grown for 36 h to reach 70% to 80% confluence before experimentation.The H9c2 cells were randomly divided into different groups (Fig. 1B): the control (Cont) group and the H/R group, which was modified from a previous study.20 Briefly, high glucose DMEM medium was changed to glucose-free DMEM to mimic ischemia and the cells were incubated at 37 °C for 6 h in the environment of 5% CO2, 5% H2, and 90% N2 in an anaerobic glove box (Coy Laboratory, USA). Then it was removed and the medium was replaced with high glucose medium and maintained for 16 h to mimic reperfusion. In the DHQ-treated group (DHQ + H/R), H9c2 cells were pretreated with DHQ (2.5, 5, 10, 20, 40, 80 μM) for 12 h before H/R. However, in the DHQ group, H9c2 cells were pretreated only with different concentrations of DHQ for 12 h. DHQ was freshly dissolved in DMSO as a stock solution and diluted with DMEM when used.
2.3. Analysis of cell viability
Cell viability was measured by the MTT assay as described previously.19 H9c2 cells were plated in 96 well plates at a density of 5 × 104 cells per well with 100 μL DMEM medium. After the designated treatment, 20 μL MTT (5 mg mL−1) solution was added to each well and incubated for 4 h at 37 °C. Then the medium with MTT was removed, and DMSO (150 mL per well) was added to dissolve the formazan crystals. The absorbance value was detected at 570 nm on a microplate reader (TECAN Infinite M1000, Austria).2.4. Measurement of ROS production, LDH & MDA levels and SOD, CAT, and GSH-Px activities
The level of intracellular ROS production was analyzed using a total ROS detection kit according to the manufacturer's instructions. Briefly, H9c2 cells were cultured in 6-well plates for 36 h. Then after all indicated treatments, the cells were harvested, washed with 1× washing buffer, and centrifuged for 5 min at 1000 rpm at room temperature, and the supernatant was discarded. The cells were resuspended and then incubated with (25 μM) 5-(and-6)-carboxy-20,70-dichlorodihydrofluorescein diacetate in the dark for 30 min at 37 °C and cleaned two times with PBS buffer. The fluorescence was analyzed using a FACS Calibur flow cytometer (BD, Biosciences, CA, USA).After different treatments, the supernatant was used to measure the level of LDH, and then the cells were collected, ultrasonicated and centrifuged at 1000 rpm for 5 min at 4 °C, and the supernatants were used to measure the levels of MDA, and the activities of SOD, CAT and GSH-Px. All indicators were measured with the corresponding detection kit according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
2.5. Detection of mitochondrial membrane potential (ΔΨm)
We used 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) (Sigma-Aldrich, St Louis, USA) to analyze the changes in the mitochondrial transmembrane potential. After treatment, the cells were removed and washed two times with the PBS solution. Then 500 μL JC-1 (the final concentration of 2 mmol L−1) working fluid was added and incubated for 30 min at 37 °C in the dark, and PBS solution was used to clean two times. The cells labeled with JC-1 were observed under an inverted fluorescence microscope.2.6. Isolated rat hearts Langendorff perfusion and experiment grouping design
The SD rats were anesthetized with 20% urethane (i.p.). Then, the heart was rapidly excised and the aorta was mounted on a Langendorff perfusion apparatus with oxygenated Krebs–Henseleit (K–H) solution including 120 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 1.25 mM CaCl2, and 11 mM glucose that saturated with 95% O2–5% CO2, pH 7.4 at 37 °C. The heart and the left ventricular pressure was measured using a pressure transducer (AD Instruments, Sydney, NSW, Australia) that was connected to a water-filled wrap balloon inserted into the left ventricle achieving a LVEDP between 2 and 8 mmHg. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Guangdong Pharmaceutical University and approved by the Animal Ethics Committee of Guangdong Pharmaceutical University. All data were recorded using Power Lab and analysed using Chart V 7.3.3 (AD Instruments). The index of the myocardial function was determined from the LVSP, the maximum derivatives of the ventricular pressure (±dp/dtmax) and the heart rate. The perfusion liquid was collected for detecting the level of LDH using the kits. All of the hearts were equilibrated with K–H solution for 20 min before the application of experimental protocols.The following experimental treatments were performed (n = 12 hearts per group) (Fig. 1C): (1) in the control group (Cont), using K–H solution with oxygenation to perfuse the isolated heart for 120 min; (2) in the ischemia/reperfusion group (I/R), after 15 min of stabilisation, perfusion was stopped to induce cardiac ischemia for 45 min and then reperfusion was performed for another 60 min; (3) in the DHQ treatment group (DHQ + I/R), the isolated hearts were perfused with K–H solution containing DHQ (5, 10, 20 μM) for 20 min and then subjected to I/R; (4) in the PI3K inhibitor LY294002 group (LY + DHQ + I/R), the isolated hearts were perfused with KH solution containing 20 μM LY and 20 μM DHQ for 20 min each and then subjected to I/R, and (5) in the DHQ group (DHQ), after 15 min of stabilisation, the hearts were perfused 20 μM DHQ for 20 min. According to the preliminary experiment and with reference to the relevant literature, the concentration of the inhibitor used had no effect on the cardiac function of the heart I/R injury.
2.7. Preparation of samples and measurement of biochemical variables
The heart tissues after the Langendorff processing were homogenized (10% w/v) with phosphate buffer (pH 7.4) and centrifuged at 5000 rpm for 15 min. The supernatant was used to estimate the reactive oxygen species levels. For the in vitro sample, the cells were washed twice in ice-cold PBS, resuspended in 1 mL of 0.1 M phosphate buffer (pH 7.4) and homogenized. Homogenized renal samples were tested for CK, AST, SOD, CAT, T-AOC, GSH-Px, ROS, and MDA activities using specific assay kits (NJJC Bio, Nanjing, China) according to the manufacturer's instructions.2.8. Heart histopathological examination
The rat hearts were fixed with 4% paraformaldehyde for more than 48 h. Then, the left ventricles of the hearts were dissected and embedded in paraffin blocks, sectioned, stained with hematoxylin and eosin (HE), and examined under a light microscope (CKX41, 170 Olympus, Tokyo, Japan) by a pathologist, who was blinded to the groups under study.2.9. Western blot analysis
Cultured H9c2 cells were harvested and washed with PBS, lysed with cell lysis buffer containing 1% phenylmethylsulfonyl fluoride, and myocardial tissue protein was homogenated and extracted from the left ventricle of the hearts using extraction buffer containing 1% phenylmethylsulfonyl fluoride. Then the supernatant was collected after centrifugation for 15 min at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and 4 °C, and the protein concentration was measured using a BCA kit. Equal amounts of protein from each sample were separated by 10% SDS-PAGE, and transferred onto nitrocellulose membranes (Millipore Corporation, Bedford, MD, USA) in Tris–glycine buffer at 100 V for 55 min in an ice box. The membranes were blocked in 5% (w/v) non-fat milk powder in Tris–buffered saline containing 0.1% (v/v) Tween-20 (TBST) for 2 h at room temperature and incubated overnight at 4 °C with primary antibodies from Santa Cruz Biotechnology (PI3K, p-PI3K, AKT, p-AKT, GRP78, PERK, p-PERK, ATF6, IRE1, CHOP, JNK, p-JNK, Caspase-12, HO-1, Nrf2, Bcl-2, and Bax and all ratios were 1:500). The membranes were washed thrice with TBST, incubated with appropriate secondary HRP-conjugated antibodies (1:1000) for 2 h on a shaking table, and washed with TBST three times for 45 min. An enhanced chemiluminescence solution was used to develop the blots for 5 min. The results were observed using Image Lab software (Bio-Rad, USA). The different protein levels were expressed as a percentage of the control, as calculated by Gel-Pro Analyzer software.
000 rpm and 4 °C, and the protein concentration was measured using a BCA kit. Equal amounts of protein from each sample were separated by 10% SDS-PAGE, and transferred onto nitrocellulose membranes (Millipore Corporation, Bedford, MD, USA) in Tris–glycine buffer at 100 V for 55 min in an ice box. The membranes were blocked in 5% (w/v) non-fat milk powder in Tris–buffered saline containing 0.1% (v/v) Tween-20 (TBST) for 2 h at room temperature and incubated overnight at 4 °C with primary antibodies from Santa Cruz Biotechnology (PI3K, p-PI3K, AKT, p-AKT, GRP78, PERK, p-PERK, ATF6, IRE1, CHOP, JNK, p-JNK, Caspase-12, HO-1, Nrf2, Bcl-2, and Bax and all ratios were 1:500). The membranes were washed thrice with TBST, incubated with appropriate secondary HRP-conjugated antibodies (1:1000) for 2 h on a shaking table, and washed with TBST three times for 45 min. An enhanced chemiluminescence solution was used to develop the blots for 5 min. The results were observed using Image Lab software (Bio-Rad, USA). The different protein levels were expressed as a percentage of the control, as calculated by Gel-Pro Analyzer software.
2.10. Statistical analysis
All experiments were repeated three times at least. The experimental results are expressed as mean ± SD. A one-way analysis of variance followed by a Student–Newman–Keuls test for pairwise comparisons was applied in the intergroup analysis. P < 0.05 was considered statistically significant.3. Results
3.1. Effects of DHQ against H/R-induced H9c2 cell injury
The MTT assay was used to detect the protective effects of DHQ on cell viability. We initially evaluated the general toxicity or cell proliferation of DHQ. The cells were treated with a series of concentrations (2.5, 5, 10, 20, 40, and 80 μM) of DHQ for 24 h. As shown in Fig. 2A, cell viability had no significant differences between these groups. Then, the ability of DHQ to inhibit H/R-induced H9c2 cell injury was assessed. H/R significantly reduced cell viability. Pretreatment with 20 μM DHQ significantly maintained cell viability at approximately 75% (Fig. 2B) and reduced LDH release (Fig. 2C). Other DHQ concentrations showed no additional benefits. Therefore, we used a concentration of 20 μM DHQ for subsequent experiments in vitro.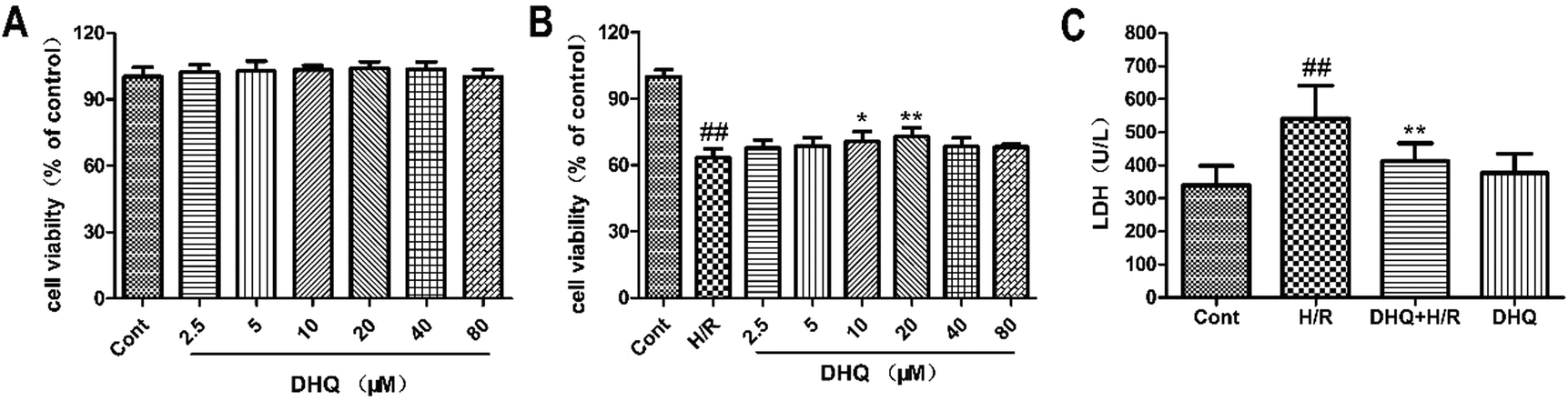 | ||
| Fig. 2 Effects of DHQ on cell viability and LDH release induced by H/R in H9c2 cardiomyocytes. (A). H9c2 cells were treated with different concentrations of DHQ for 24 h, and cell viability was measured by MTT assay. (B). H9c2 cardiomyocytes were treated for 12 h with DHQ (2.5, 5, 10, 20, 40, 80 μM), and then exposed to 6 h of hypoxia and 16 h of reoxygenation. The cell viability was measured by MTT assay. (C). LDH release was measured using the corresponding detection kit. The data are expressed as means ± SD from three independent experiments. ##P < 0.01 versus control group; *P < 0.05, **P < 0.01 versus H/R group. | ||
3.2. DHQ enhanced antioxidant capacity in H9c2 cells
It has been reported that oxidative stress damage plays a key role in the initiation of injury or apoptosis during MIRI. Therefore, the effects of DHQ against H/R-induced oxidative stress damage were further evaluated in vitro. The experimental results are shown in Fig. 3A–D. H/R-induced oxidative stress damage in H9c2 cells is demonstrated by an increase in MDA levels and a decrease in SOD, CAT, and GSH-Px activities. However, DHQ pretreatment significantly ameliorated these effects. ROS production was examined by flow cytometry in DCFHDA-labeled H9c2 cells (Fig. 3E). ROS significantly accumulated in H/R-damaged H9c2 cells. DHQ pretreatment effectively reduced H/R-induced ROS production. DHQ treatment alone had no effect on these indicators when compared with the control treatment. Based on these results, we concluded that DHQ could effectively alleviate H/R-induced oxidative stress injury in H9c2 cells.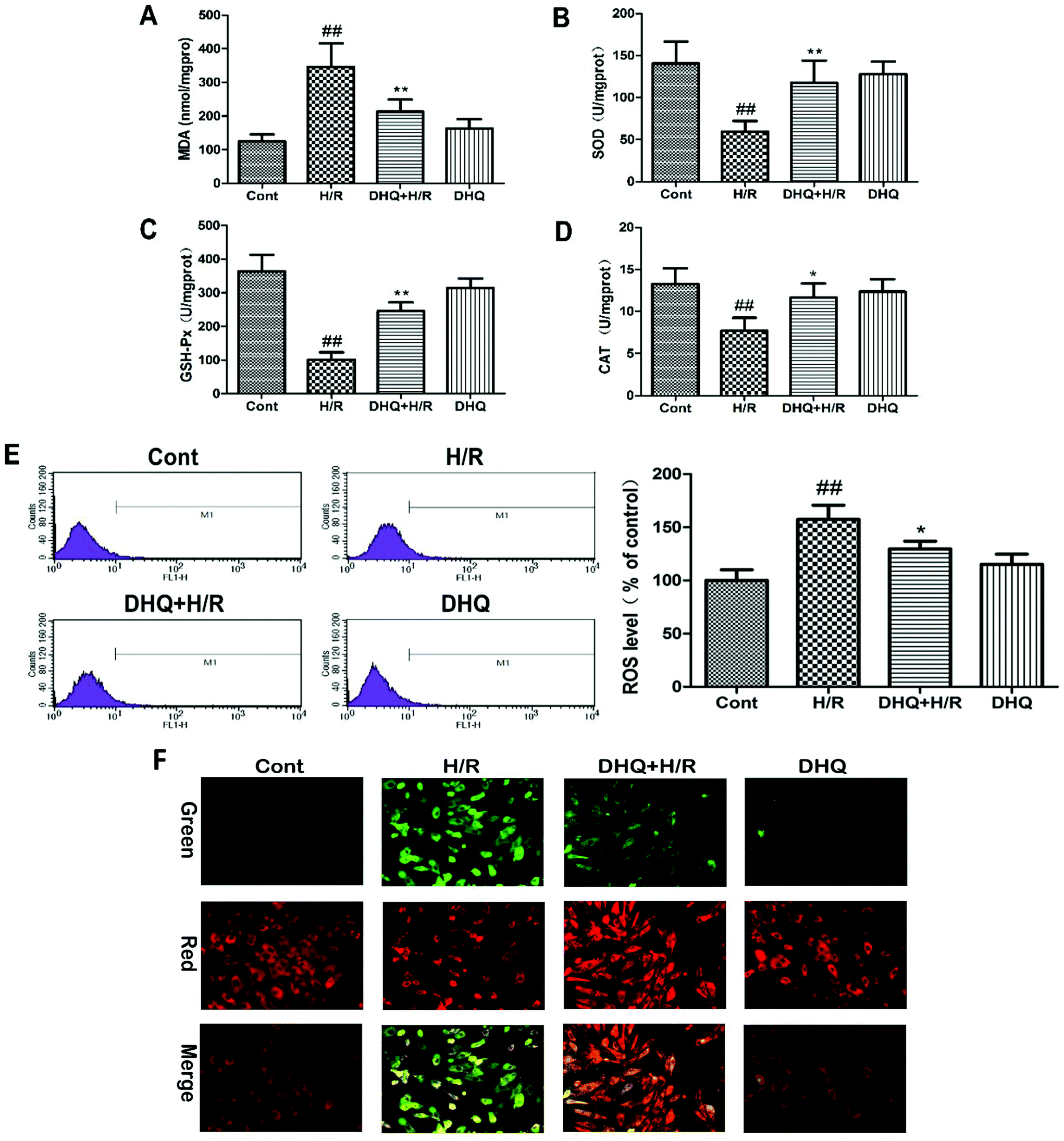 | ||
| Fig. 3 Effects of DHQ on the activation of antioxidant enzymes, intracellular ROS production, and ΔΨm induced by H/R in H9c2 cells in vitro. (A) The effects of DHQ on the MDA level; (B) the effects of DHQ on SOD activity; (C) the effects of DHQ on GSH-Px activity; (D) the effects of DHQ on CAT activity; (E) the effects of DHQ on intracellular ROS levels evaluated using a FACSCalibur flow cytometer; (F) the representative images of JC-1 red/green cells and merges showed that DHQ decreased green fluorescence and increased the ratio of red to green fluorescence intensity. The data are expressed as means ± SD from three independent experiments. ##P < 0.01 versus control group; *P < 0.05, **P < 0.01 versus the H/R group. | ||
3.3. Effects of DHQ on mitochondrial transmembrane potential (ΔΨm)
The mitochondrion is the major organelle related to cell energy supply and cellular apoptosis. The loss of ΔΨm leads to membrane depolarization and triggers the apoptosis cascade. We evaluated the effect of DHQ on ΔΨm by JC-1 staining. As shown in Fig. 3F, H/R disrupted ΔΨm of H9c2 cells as observed by an increase in green fluorescence in cells. Pretreatment with DHQ protected myocardial cells against H/R-induced ΔΨm loss in a dose-dependent manner.3.4. DHQ prevents I/R-induced myocardial injury
The levels of two myocardial enzymes, namely AST and CK, were detected. As shown in Fig. 4A, I/R-induced myocardial damage resulted in a significant increase in the levels of myocardial enzymes. However, DHQ pretreatment before I/R significantly ameliorated these effects.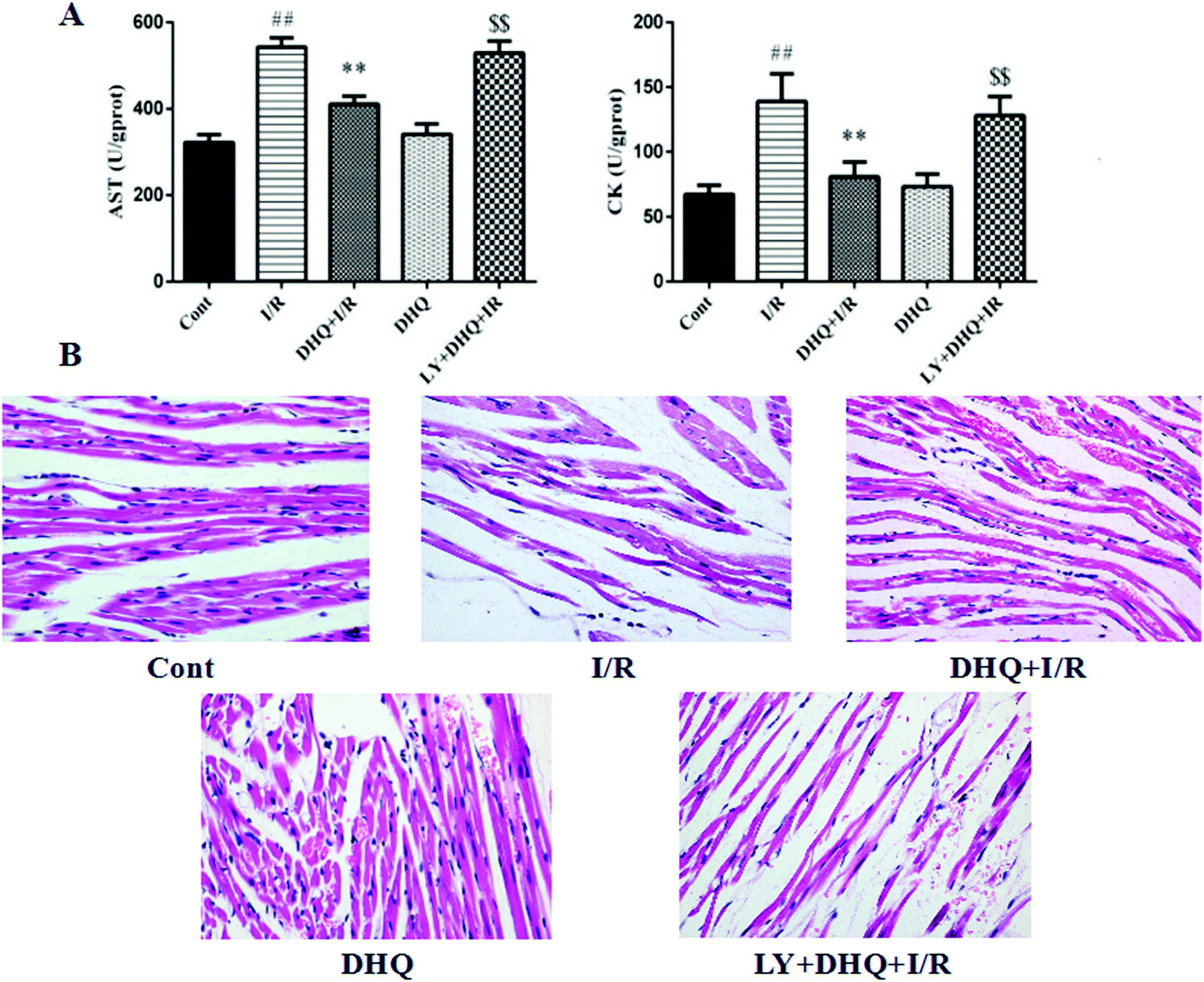 | ||
| Fig. 4 Effects of DHQ on I/R induced myocardial injury ex vivo. A. Effects of DHQ treatment on the AST and CK level; B. histological changes in the rat hearts were measured by HE staining (200×). The data are expressed as means ± SD from three independent experiments. ##P < 0.01 versus control group; **P < 0.01versus the I/R group; $$P < 0.01 versus DHQ + I/R. | ||
After H&E staining, the pathological changes of myocardial tissue were observed by light microscopy. As shown in Fig. 4B, no obvious abnormalities were observed in the control group, whereas severe myocardial damage characterized by myocardial fiber fracture, a widened interstitium, the local wavy cytoplasm dissolution phenomenon, and membrane rupture was observed in the I/R group. Pretreatment with DHQ significantly improved the above pathological changes. Notably, the specific inhibitor of Akt, LY294002, inhibited the cardioprotective effects of DHQ to a large extent, indicating that the Akt signaling pathway is involved in the effects of DHQ.
3.5. DHQ enhances the antioxidant capacity in cardiac tissues
During I/R, oxygen-derived free radicals are thought to play an important role in the genesis of tissue injury. The effects of I/R treatment on antioxidant enzyme activities were further evaluated in Langendorff-perfused rat hearts. As shown in Fig. 5A, I/R significantly reduced the activities of GSH-Px, SOD, and CAT in the rat myocardium (P < 0.01), and the expression levels of T-AOC in the heart compared with those of normal control rats. DHQ pretreatment before I/R significantly ameliorated these effects. Furthermore, I/R induced an increase in MDA production and intracellular ROS levels (Fig. 5B), which were decreased by pretreatment with DHQ. DHQ-induced protective effects were largely inhibited by LY294002. These results suggested that DHQ could considerably enhance the antioxidant capacity of cells against I/R-induced oxidative stress. | ||
| Fig. 5 Effects of TAB on anti-oxidant enzyme activity and MDA and ROS levels in isolated hearts. I/R treatment down-regulates the expression of antioxidative components (A. T-AOC, SOD, CAT, GSH-PX) and up-regulates the levels of MDA and ROS (B. MDA, ROS), and these effects were reversed by DHQ pre-treatment. The data are expressed as means ± SD from three independent experiments. ##P < 0.01 versus control group; **P < 0.01versus the I/R group; $$P < 0.01 versus DHQ + I/R. | ||
3.6. DHQ improved cardiac function of I/R hearts
We detected whether DHQ pretreatment improved systolic and diastolic function by using LY294002, a specific inhibitor of PI3K/AKT. As shown in Fig. 6A–D, compared with the I/R group, pretreatment with 20 μM DHQ showed the best effect in improving functional recovery, which was demonstrated by significant increases in LVSP, ±dP/dtmax, and heart rate throughout the reperfusion period. However, the baseline mechanical parameters in the DHQ group were not significantly different from those in the control group. Treatment with the PI3K/Akt inhibitor LY294002 showed that LY294002 could inhibit the ameliorating effects of DHQ on the myocardial contractile function of I/R hearts. | ||
| Fig. 6 Effects of different concentrations of DHQ on cardiac function in isolated rat hearts. Hearts were exposed to DHQ at concentrations of 5–20 μM for 20 min prior to ischemia. (A–D) Time-course of cardiac functional indexes in isolated hearts during I/R. (A) Left ventricular systolic pressure (LVSP); (B) heart rate; (C) maximum rate of LV pressure development (+dP/dtmax); (D) maximum rate of LV pressure decline (−dP/dtmax). The data are expressed as means ± SD from three independent experiments. n > 10 in each group. *P < 0.05, **P < 0.01 versus I/R, $P < 0.05, $$P < 0.01 versus DHQ + I/R. | ||
3.7. DHQ increased the expression level of phosphorylation of PI3K and Akt, thereby providing cardioprotection
To investigate the cardioprotective effect of DHQ on MIRI and elucidate the underlying mechanism, we used H/R-induced H9c2 cardiomyocytes and Langendorff-perfused rat hearts in vitro to mimic MIRI in clinical settings. The results showed that DHQ pretreatment increased the expression level of PI3K and Akt phosphorylation compared with H/R and I/R groups (Fig. 7A and B). Analytical results of the Bcl-2 family of apoptosis-related proteins showed that the Bcl-2 protein level was decreased and the Bax level was increased in H/R and I/R groups, which in turn resulted in a lower Bcl-2/Bax ratio than that in the normal control group. Pretreatment with DHQ significantly increased the Bcl-2/Bax ratio by increasing Bcl-2 expression and decreasing Bax expression (Fig. 7C and D).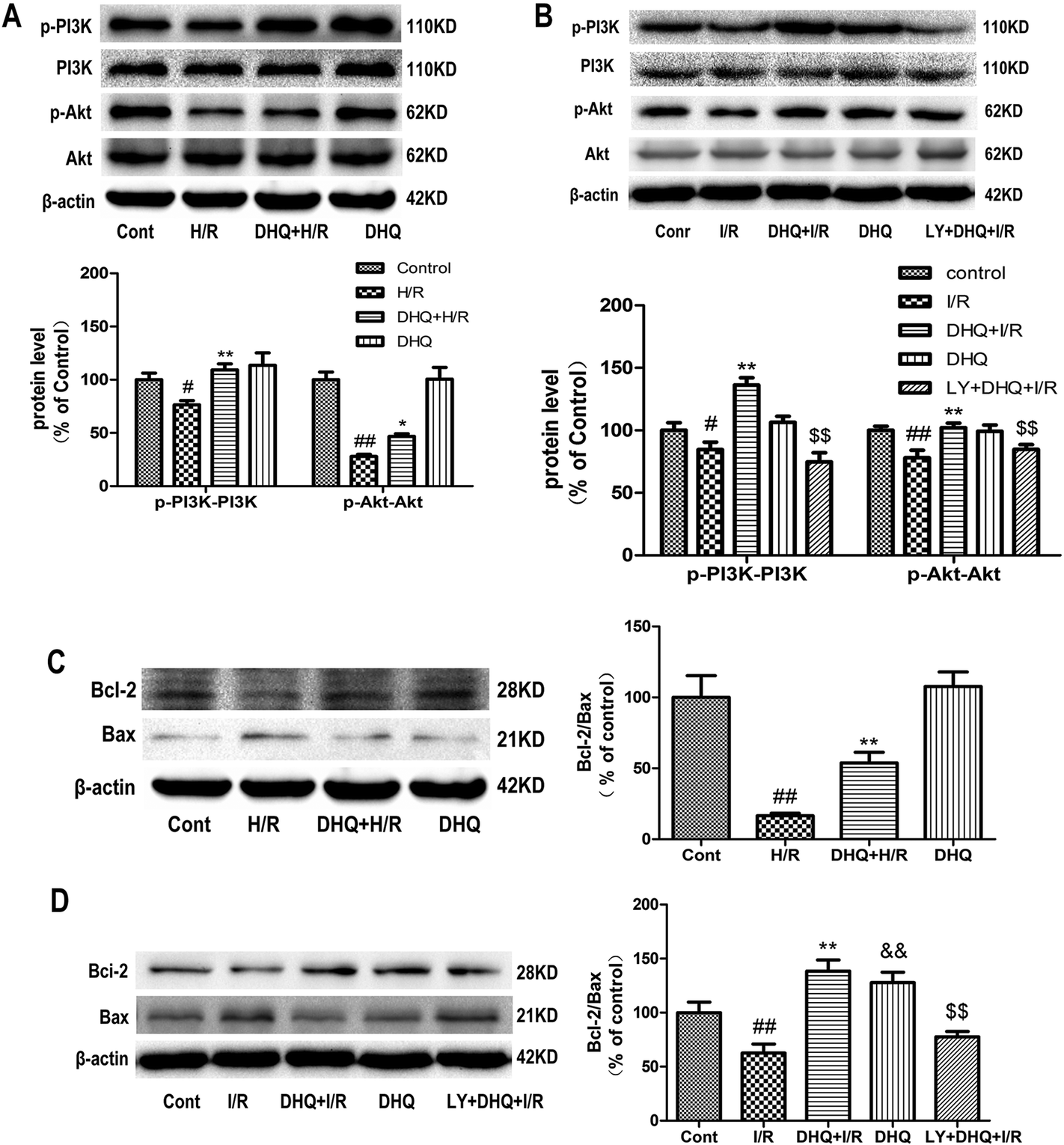 | ||
| Fig. 7 Effects of DHQ or LY294002 on apoptosis via the PI3K/Akt pathways and Bcl-2/Bax pathways in vitro and ex vivo. The expression levels of PI3K, p-PI3K, Akt, p-Akt, Bcl-2, Bax and β-actin were detected using an immunoblotting assay in vitro (A, C) and ex vivo (B, D). The relative protein expression of p-PI3K/PI3K, p-Akt/Akt and Bcl-2/Bax is expressed in the bar graphs. The data are expressed as the means ± SD from three independent experiments. #P < 0.05, ##P < 0.01 versus the control group; *P < 0.05, **P < 0.01 versus the H/R or I/R group; $$P < 0.01 versus DHQ + I/R; &&P < 0.01 versus the control group. | ||
3.8. Effects of DHQ against myocardial I/R injury both in Langendorff-perfused rat hearts and H9c2 cardiomyocytes
To evaluate the role of ERS in the cardioprotective effects of DHQ, the ERS-responsive marker GRP78 and the ERS sensors ATF6, PERK, eif2α, and IRE1 were investigated both in Langendorff-perfused rat hearts and in H9c2 cardiomyocytes. As shown in Fig. 8A and B, the expression level of GRP78 increased in the I/R-treated group compared with that in the control group. Significant increases were also observed in the phosphorylation of PERK and eif2α, and in the protein expression of IRE1 and ATF6 to varying degrees. However, both Langendorff-perfused rat hearts and H9c2 cardiomyocytes pretreated with DHQ showed a significant reduction in the expression levels of GRP78, p-PERK, p-eif2α, IRE1, and ATF6 compared with the I/R group. Excessive ERS is a key mediator in determining cellular apoptosis, and it results in the activation of ERS-related apoptosis proteins such as CHOP, Caspase-12, and p-JNK. The results showed that CHOP, Caspase-12, and p-JNK protein levels increased in the H/R- or I/R-treated group compared with the levels in the control group. Langendorff-perfused rat hearts and H9c2 cardiomyocytes pretreated with DHQ showed a significant reduction in the expression levels of CHOP, Caspase-12, and p-JNK compared with the I/R and H/R groups, respectively (Fig. 8A and B). Nevertheless, the effects of DHQ against I/R-induced cardiomyocyte apoptosis in isolated hearts were almost abolished by pretreatment with LY294002. Collectively, these results indicated that ERS was involved in I/R-induced myocardium injury and mediated cardiomyocyte apoptosis. DHQ preconditioning markedly suppressed ERS and ERS-associated apoptosis and activated the PI3K/Akt pathway. | ||
| Fig. 8 Effects of DHQ and LY294002 on ERS and the ERS-mediated apoptosis signaling pathway in H9c2 cells and Langendorff-perfused rat hearts. The expression levels of PERK, p-PERK, eif2α, p-eif2α ATF6, IRE1, GRP78, CHOP, Caspase12, JNK, p-JNK and β-actin were detected using an immunoblotting assay in vitro (A) and ex vivo (B). The relative protein expression levels of p-PERK/PERK, p-eif2α/eif2α, ATF6/β-actin, IRE1/β-actin, GRP78/β-actin, CHOP/β-actin, Caspase12/β-actin and p-JNK/JNK are expressed in the bar graphs. The data are expressed as the means ± SD from three independent experiments. #P < 0.05, ##P < 0.01 versus control group; *P < 0.05, **P < 0.01 versus the H/R and I/R group; $P < 0.05, $$P < 0.01 versus DHQ + I/R; &&P < 0.01 versus control group. | ||
3.9. DHQ-induced HO-1 expression and nuclear accumulation of Nrf2 proteins both in Langendorff-perfused rat hearts and H9c2 cardiomyocytes
The upregulation of HO-1 expression plays a key role in protecting cells against toxicity caused by various oxidative insults. Nrf2 is the major transcription factor that binds to ARE and enhances ARE-driven expression of target genes. Therefore, we attempted to examine HO-1 expression and the nuclear accumulation of Nrf2 proteins in H9c2 cells and Langendorff-perfused rat hearts. As shown in Fig. 9A and B, the expression level of HO-1 and the nuclear levels of Nrf2 increased in the DHQ-treated group compared with those in the H/R and I/R groups. However, LY294002 effectively suppressed the DHQ-mediated upregulation of HO-1 and Nrf2 expression.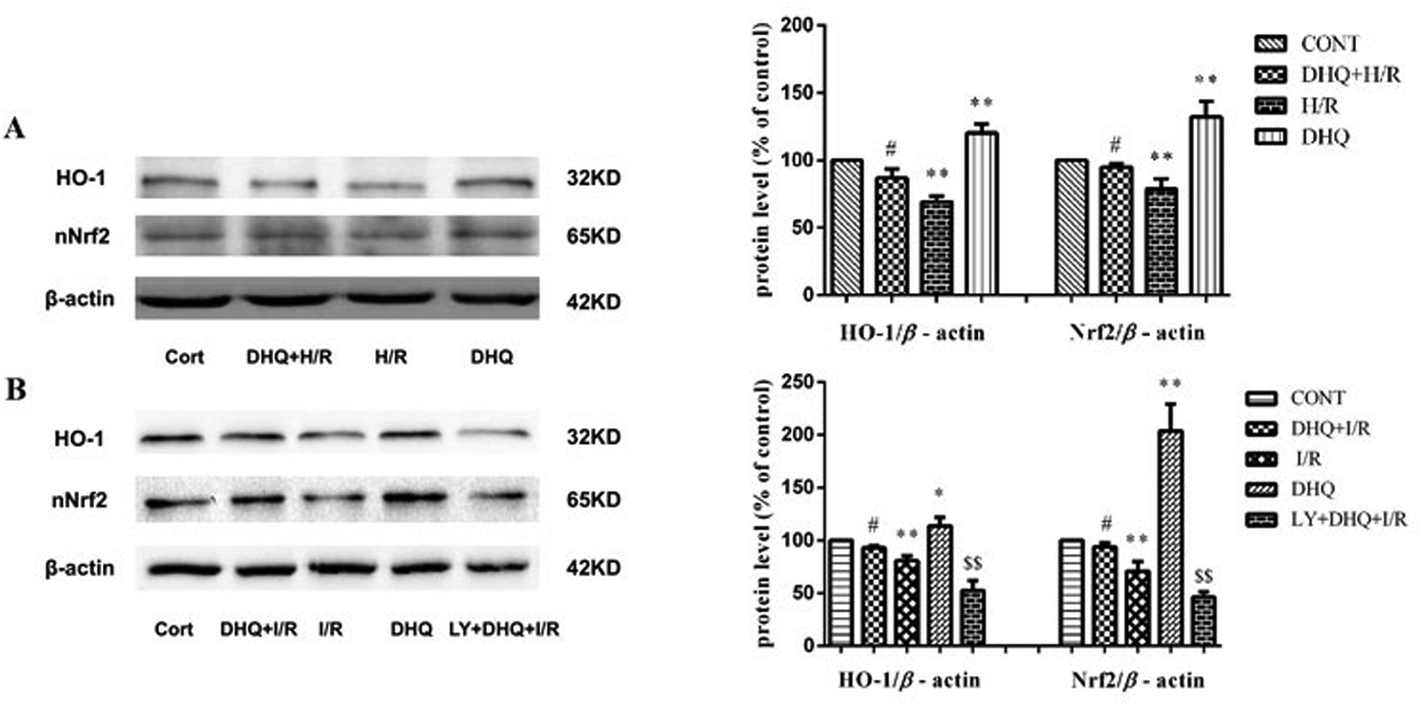 | ||
| Fig. 9 Effect of DHQ treatment on HO-1/Nrf2 activation in H9c2 cells and Langendorff-perfused rat hearts. (A). The expression levels of HO-1 and Nrf2 proteins were detected in H9c2 cells. The relative protein expression levels of HO-1/β-actin and Nrf2/β-actin are expressed in the bar graphs. (B). The expression levels of HO-1 and Nrf2 proteins were detected in the Langendorff-perfused rat hearts. The relative protein expression levels of HO-1/β-actin and Nrf2/β-actin are expressed in the bar graphs. The data are expressed as the means ± SD from three independent experiments. *P < 0.05, **P < 0.01 versus control group; #P < 0.05 versus the H/R or I/R group; $$P < 0.01 versus DHQ + I/R. | ||
4. Discussion
Apoptosis of cardiac myocytes induced by oxidative stress and ERS plays a pivotal role in the pathogenesis of cardiovascular disease, including ischemic heart disease, I/R injury, and heart failure.21 Therefore, the prevention of oxidative stress and ERS-induced apoptosis may serve as a beneficial intervention for the treatment of these diseases.22,23Flavonoids, a well-known component of traditional Chinese herbal medicine used to prevent myocardial injury, have potent anti-apoptotic effects.17,18 DHQ is a major ingredient of flavonoids and it belongs to the flavanonol subclass. Many plants, including “medicinal and edible” vegetables and fruits, have been reported to contain DHQ. Our laboratory previously showed that DHQ has an effective role in treating coronary heart disease by inhibiting ISO-induced myocardial tissue ischemia.24 However, the mechanism underlying the myocardial protection of DHQ has not yet been fully elucidated. Therefore, we explored the protective effects and the detailed molecular mechanisms of DHQ on the myocardium.
Oxidative stress is a phenomenon where an imbalance of ROS and free radicals leads to detrimental effects on the body. It is widely recognized as a major mechanism in the development of I/R injury.25 ROS are the main factors that induce oxidative stress, which in turn results in cell damage to biological macromolecules, affects the normal physiological state of cells, and initiates oxidative stress reaction. The present study showed that H/R and I/R significantly increased ROS levels, caused H9c2 cell damage and pathological changes, and increased MDA production, the end-product of lipid peroxidation.26 Treatment with DHQ reduced ROS levels and alleviated H9c2 cell and cardiac muscle tissue injuries. In addition, DHQ treatment enhanced SOD activity. Our results are consistent with previous reports that DHQ has an effective role in the treatment of coronary heart disease in ISO-induced rat myocardial ischemia.27
Nrf2, a ubiquitously expressed mammalian transcription factor, is a key component in cellular defense against harmful stressors. HO-1 upregulation is mediated by the activation, nuclear translocation, and selective ARE binding of Nrf2. We demonstrated that DHQ pretreatment protects H9c2 cells and Langendorff-perfused rat hearts against H/R- and I/R-induced toxicity through the upregulation of HO-1 expression. In addition, DHQ significantly upregulated Nrf2 activation, which was correlated with a significant upregulation of HO-1 expression. In agreement with our findings, a previous study demonstrated that DHQ protected cells from oxidative stress by regulating the Nrf2 activity and the protein expression of the antioxidant enzyme NQO1 via the Akt, ERK, and JNK signaling pathways.28 Other reports also demonstrated that DHQ exerted hepatoprotective effects against ConA-induced liver injury via the upregulation of HO-1 activity through the MAPK/Nrf2 signaling pathway in macrophages/Kupffer cells.29 DHQ has been reported to induce HO-1 expression in several cell types and therefore, understanding the molecular mechanisms of DHQ-induced HO-1 activation is important for the therapeutic application of DHQ.
Lewen found that the ER calcium pump is sensitive to oxidative damage in a mouse model of I/R and the proteins in the ER were oxidized, which indicated that the ER may be an important target of ROS attack.30 In addition, ERS-related apoptosis is an important pathway for the progress of I/R.31 Our study showed that DHQ significantly reduced the relative protein levels of the ERS-responsive marker GRP78 and ERS sensors ATF6, PERK, eiF2α, and IRE1. Moreover, the downstream apoptotic proteins, including CHOP, Caspase-12, p-JNK, and Bax, were inhibited, and the expression of Bcl-2 both in H9c2 cells and Langendorff-perfused rat hearts was increased, suggesting that DHQ significantly inhibited ERS and its related apoptosis in I/R injury.
In this study, we found that the DHQ-induced HO-1 expression and ERS pathway in H9c2 cells and Langendorff-perfused rat hearts are regulated by PI3K/Akt signaling. The PI3K/Akt signaling pathway is a significant signal transduction pathway for survival, and it is necessary for providing protective effects on ischemic preconditioning and various pharmacological mediators.32,33 In the present study, the PI3K/Akt signaling pathway inhibitor LY294002 abolished the cardioprotective effects of DHQ, suggesting that the cardioprotective effects of DHQ were related to the activation of the PI3K/Akt pathway. Moreover, the DHQ-induced HO-1 expression and ERS pathway are attenuated by the pharmacological inhibition of PI3K/Akt. Similar results were also observed in other pharmacological mediator studies.34,35 Therefore, we suggest that the DHQ-induced HO-1 expression and ERS pathway are likely to be mediated by the activation of PI3K/Akt.
In conclusion, MIRI is mainly caused by oxidative stress- and ER stress-induced apoptosis. We determined the protective effects of DHQ against I/R injury and demonstrated the suppression of ER stress and oxidative stress by DHQ via the PI3K/AKT pathway in vitro and ex vivo (Fig. 10). DHQ could be a promising therapeutic agent against ischemic cardiovascular disease. However, further studies are warranted to elucidate the mechanisms involved in the cardioprotective action of DHQ in detail.
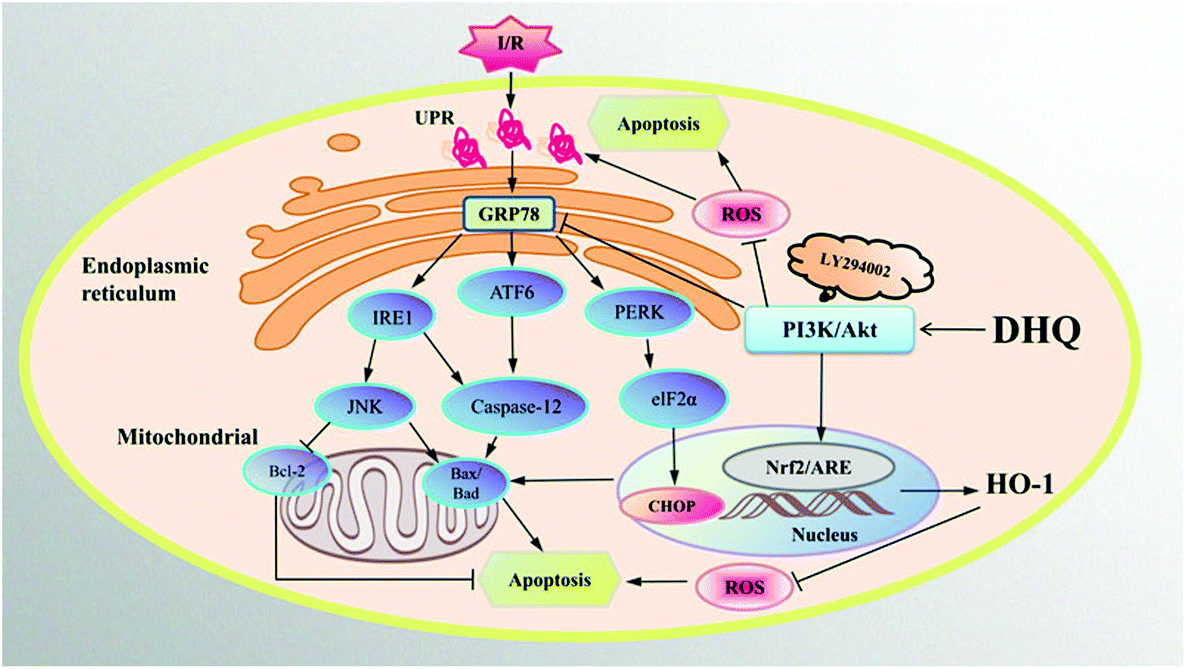 | ||
| Fig. 10 Summary scheme of the mechanisms underlying DHQ's inhibition of I/R or H/R-induced oxidative stress and ER stress. I/R or H/R treatment induces the overexpression of GRP78 and the accumulation of reactive oxygen species. The activation of GRP78 is able to induce a cascade of events that include the upregulation of ERS sensors ATF6, IRE1, and phosphorylation of PERK, resulting in the induction of apoptosis through CHOP, Caspase-12, and JNK-dependent pathways. DHQ attenuates the upregulated GRP78 expressions and increases the HO-1 expression through activating the phosphorylation of PI3K and Akt, thus blocking the signalling pathways of ERS and the associated apoptosis. | ||
Abbreviations
| TCM | Traditional Chinese medicine |
| DHQ | Dihydroquercetin |
| MIRI | Myocardial ischemia reperfusion injury |
| I/R | Ischemia/reperfusion |
| H/R | Hypoxia/reoxygenation |
| ER | Endoplasmic reticulum |
| ERS | Endoplasmic reticulum stress |
| LDH | Lactate dehydrogenase |
| HO-1 | Heme oxygenase-1 |
| Nrf2 | NF-E2-related factor |
| ARE | Antioxidant response elements |
| ROS | Reactive oxygen species |
| MDA | Malondialdehyde |
| SOD | Superoxide dismutase |
| CK | Creatine kinase |
| CAT | Catalase |
| AST | Aspartate aminotransferase |
| GSH-Px | Glutathione peroxidase |
| UPR | Unfolded protein response |
| PERK | Pancreatic ER kinase-like |
| ATF6 | Activating transcription factor 6 |
| IRE1α | Inositol-requiring enzyme-1α |
| JNK | c-JunN-terminal kinase |
| PI3K | Phosphoinositide 3-kinase |
| Akt | Protein kinase B |
| CHOP | C/EBP homologous protein |
| GRP78 | Glucose-regulated protein 78 kDa |
| eIf2α | The α subunit of eukaryotic initiation factor 2 |
| Bcl-2 | B cell lymphoma 2 |
| Bax | BCL2-associated X protein |
| Bad | Boxing after dark |
| LVEDP | Left ventricular end-diastolic pressure |
| LVSP | Left ventricular systolic pressure |
| HE | Hematoxylin and eosin |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling |
| JC-1 | 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide |
| MTT | 3-(4,5-Dimethylthiazol-2yl-)-2,5-diphenyl tetrazolium bromide |
| DMSO | Dimethylsulfoxide |
| ANOVA | Analysis of variance |
Conflicts of interest
We declare no conflict of interests.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 81603366) and the Natural Science Foundation of Heilongjiang (Grant No. H2015041).References
- P. Z. Gerczuk and R. A. Kloner, An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials, J. Am. Coll. Cardiol., 2012, 59(11), 969–978 CrossRef PubMed.
- S. Bertuglia, A. Giusti and S. P. Del, Antioxidant activity of a nitro derivative of aspirin against ischemia-reperfusion in hamster cheek pouch microcirculation, Am. J. Physiol.: Gastrointest. Liver Physiol., 2004, 286(3), G437–G443 CrossRef CAS PubMed.
- C. C. Winterbourn, Reconciling the chemistry and biology of reactive oxygen species, Nat. Chem. Biol., 2008, 4(5), 278–286 CrossRef CAS PubMed.
- H. Mizutani, S. Tada-Oikawa, Y. Hiraku, M. Kojima and S. Kawanishi, Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide, Life Sci., 2005, 76(13), 1439–1453 CrossRef CAS PubMed.
- L. Cheng, Z. Jin, R. Zhao, K. Ren, C. Deng and S. Yu, Resveratrol attenuates inflammation and oxidative stress induced by myocardial ischemia-reperfusion injury: role of nrf2/are pathway, Int. J. Clin. Exp. Med., 2015, 8(7), 10420–10428 CAS.
- Z. A. Shah, R. C. Li, R. K. Thimmulappa, T. W. Kensler, M. Yamamoto, S. Biswal and S. Doré, Role of reactive oxygen species in modulation of nrf2 following ischemic reperfusion injury, Neuroscience, 2007, 147(1), 53–59 CrossRef CAS PubMed.
- B. Xu, J. Zhang, J. Strom, L. Sang and Q. M. Chen, Myocardial ischemic reperfusion induces de novo nrf2 protein translation, Biochim. Biophys. Acta, 2014, 1842(9), 1638–1647 CrossRef CAS PubMed.
- G. Zhou, L. A. Dada, M. Wu, A. Kelly, H. Trejo, Q. Zhou, J. Varga and J. I. Sznajder, Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1, Am. J. Physiol.: Lung Cell Mol. Physiol., 2009, 297(6), 1120–1130 CrossRef PubMed.
- X. Song, Z. Ning, X. U. Hongde, C. Liu and H. Zhang, Combined preconditioning and postconditioning provides synergistic protection against liver ischemic reperfusion injur, Int. J. Biol. Sci., 2012, 8(5), 707–718 CrossRef CAS PubMed.
- D. O. Moon, S. Y. Park, Y. H. Choi, J. S. Ahn and G. Y. Kim, Guggulsterone sensitizes hepatoma cells to trail-induced apoptosis through the induction of chop-dependent DR5: involvement of ros-dependent ER-stress, Biochem. Pharmacol., 2011, 82(11), 1641–1650 CrossRef CAS PubMed.
- S. Chen, F. F. He, H. Wang, Z. Fang, N. Shao, X. J. Tian, J. S. Liu, Z. H. Zhu, Y. M. Wang, S. Wang, K. Huang and C. Zhang, Calcium entry via trpc6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes, Cell Calcium, 2011, 50(6), 523–529 CrossRef CAS PubMed.
- A. Griciuc, L. Aron and M. Ueffing, ER stress in retinal degeneration: a target for rational therapy?, Trends Mol. Med., 2011, 17(8), 442–451 CrossRef CAS PubMed.
- P. J. Mink, C. G. Scrafford, L. M. Barraj, L. Harnack, C. P. Hong, J. A. Nettleton and D. R. Jacobs Jr., Flavonoid intake and cardiovascular disease mortality: a prospective study in postmenopausal women, Am. J. Clin. Nutr., 2007, 85(3), 895–909 CrossRef CAS PubMed.
- N. Oi, H. Chen, K. M. Ok, R. A. Lubet, A. M. Bode and Z. Dong, Taxifolin suppresses UV-induced skin carcinogenesis by targeting EGFR and PI3K, Cancer Prev. Res., 2012, 5(9), 1103–1114 CrossRef CAS PubMed.
- M. Satué, M. M. Arriero, M. Monjo and J. M. Ramis, Quercitrin and taxifolin stimulate osteoblast differentiation in MC3T3-E1 cells and inhibit osteoclastogenesis in RAW264.7 cells, Biochem. Pharmacol., 2013, 86(10), 1476–1486 CrossRef PubMed.
- T. V. Arutyunyan, A. F. Korystova, L. N. Kublik, M. K. Levitman, V. V. Shaposhnikova and Y. N. Korystov, Effects of taxifolin on the activity of angiotensin-converting enzyme and reactive oxygen and nitrogen species in the aorta of aging rats and rats treated with the nitric oxide synthase inhibitor and dexamethasone, Age, 2013, 35(6), 2089–2097 CrossRef CAS PubMed.
- Y. A. Vladimirov, E. V. Proskurnina, E. M. Demin, N. S. Matveeva, O. B. Lubitskiy, A. A. Novikov, D. Y. Izmailov, A. N. Osipov, V. P. Tikhonov and V. E. Kagan, Dihydroquercetin (taxifolin) and other flavonoids as inhibitors of free radical formation at key stages of apoptosis, Biochemistry, 2009, 74(3), 301–307 CAS.
- X. Sun, R. C. Chen, Z. H. Yang, G. B. Sun, M. Wang, X. J. Ma, L. J. Yang and X. B. Sun, Food Chem. Toxicol., 2014, 63(1), 221–232 CrossRef CAS.
- G. B. Sun, X. Sun, M. Wang, J. X. Ye, J. Y. Si, H. B. Xu, X. B. Meng, M. Qin, J. Sun, H. W. Wang and X. B. Sun, Oxidative stress suppression by luteolin-induced heme oxygenase-1 expression, Toxicol. Appl. Pharmacol., 2012, 265(2), 229–240 CrossRef CAS PubMed.
- J. Sun, G. Sun, X. Meng, H. Wang, M. Wang, M. Qin, B. Ma, Y. Luo, Y. Yu, R. Chen, Q. Ai and X. Sun, Ginsenoside rk3 prevents hypoxia-reoxygenation induced apoptosis in h9c2 cardiomyocytes via akt and mapk pathway, J. Evidence-Based Complementary Altern. Med., 2013, 2013(4), 690190–690202 Search PubMed.
- Y. Tagawa, N. Hiramatsu, A. Kasai, K. Hayakawa, M. Okamura, J. Yao and M. Kitamura, Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP), Free Radicals Biol. Med., 2008, 45(1), 50–59 CrossRef CAS PubMed.
- S. Yasuda and H. Shimokawa, Acute myocardial infarction: the enduring challenge for cardiac protection and survival, Circ. J., 2009, 73(11), 2000–2008 CrossRef PubMed.
- L. M. Buja, Myocardial ischemia and reperfusion injury, Cardiovasc. Pathol., 2005, 14(4), 170–175 CrossRef CAS PubMed.
- Q. H. Wang, H. X. Kuang, L. Wu, L. Yang and Z. B. Wang, Protective Effect of Dihydroquercetin on Acute Myocardial Ischemia Induced by Isoproterenol in Rats, Chin. J. Exp. Tradit. Med. Formulae., 2011, 17(17), 177–180 CAS.
- C. L. Rayner, S. E. Bottle, G. A. Gole, M. S. Ward and N. L. Barnett, Real-time quantification of oxidative stress and the protective effect of nitroxide antioxidants, Neurochem. Int., 2015, 92, 1–12 CrossRef PubMed.
- O. Sehirli, A. Tozan, G. Z. Omurtag and S. Cetinel, Protective effect of resveratrol against naphthalene-induced oxidative stress in mice, Ecotoxicol. Environ. Saf., 2008, 71(1), 301–308 CrossRef CAS PubMed.
- S. Prabhu, M. Jainu, K. E. Sabitha and C. S. Devi, Role of mangiferin on biochemical alterations and antioxidant status in isoproterenol-induced myocardial infarction in rats, J. Ethnopharmacol., 2006, 107(1), 126–133 CrossRef CAS PubMed.
- L. Liang, C. Gao, M. Luo, W. Wang, C. Zhao, Y. Zu, T. Efferth and Y. Fu, Dihydroquercetin (DHQ) induced HO-1 and NQO1 expression against oxidative stress through the Nrf2-dependent antioxidant pathway, J. Agric. Food Chem., 2013, 61(11), 2755–2761 CrossRef CAS PubMed.
- M. Zhao, J. Chen, P. Zhu, M. Fujino, T. Takahara, S. Toyama, A. Tomita, L. Zhao, Z. Yang, M. Hei, L. Zhong, J. Zhuang, S. Kimura and X. K. Li, Dihydroquercetin (DHQ) ameliorated concanavalin A-induced mouse experimental fulminant hepatitis and enhanced HO-1 expression through MAPK/Nrf2 antioxidant pathway in RAW cells, Int. Immunopharmacol., 2015, 28(2), 938–944 CrossRef CAS PubMed.
- A. Lewén, P. Matz and P. H. Chan, Free radical pathways in CNS injury, J. Neurotrauma, 2000, 17(10), 871–890 CrossRef.
- S. Y. Kim, J. S. Hwang and I. O. Han, Tunicamycin inhibits Toll-like receptor-activated inflammation in RAW264.7 cells by suppression of NF-κB and c-Jun activity via a mechanism that is independent of ER-stress and N-glycosylation, Eur. J. Pharmacol., 2013, 721(1–3), 294–300 CrossRef CAS PubMed.
- B. Ebner, S. A. Lange, D. Hollenbach, N. Steinbronn, A. Ebner, C. Fischaleck, R. Braun-Dullaeus, C. Weinbrenner and R. H. Strasser, In situ postconditioning with neuregulin-1β is mediated by a pi3k/akt-dependent pathway, Can. J. Cardiol., 2015, 31(1), 76–83 CrossRef PubMed.
- D. J. Hausenloy, S. Lecour and D. M. Yellon, Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin, Antioxid. Redox Signaling, 2011, 14(5), 893–907 CrossRef CAS PubMed.
- H. Yu, H. Zhang, W. Zhao, L. Guo, X. Li, Y. Li, X. Zhang and Y. Sun, Gypenoside protects against myocardial ischemia-reperfusion injury by inhibiting cardiomyocytes apoptosis via inhibition of chop pathway and activation of pi3k/akt pathway in vivo and in vitro, Cell. Physiol. Biochem., 2016, 39(1), 123–136 CrossRef CAS PubMed.
- S. J. Somers, M. Frias, L. Lacerda, L. H. Opie and S. Lecour, Interplay between SAFE and RISK pathways in sphingosine-1-phosphate-induced cardioprotection, Cardiovasc, Cardiovasc. Drugs Ther., 2012, 26(3), 227–237 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |