
Diallyl disulphide suppresses the cannonical Wnt signaling pathway and reverses the fibronectin-induced epithelial mesenchymal transition of A549 lung cancer cells†
Bornita
Das
and
Dona
Sinha
 *
*
Receptor Biology and Tumor Metastasis, Chittaranjan National Cancer Institute, Kolkata 700026, India. E-mail: donasinha2012@gmail.com; Fax: +91 33 2475 7606; Tel: +91 33 2476 5101 ext. 327
First published on 13th November 2018
Abstract
Globally, non-small cell lung cancer is a leading cause of cancer-related mortality and about 40% of these cancers are detected in the metastatic stage. Epithelial mesenchymal transition (EMT) plays a critical role during malignant transformation, and the extracellular matrix component, fibronectin (FN), is a known inducer of invasion and metastasis. Diallyl disulphide (DADS), a bioactive component of garlic, exhibits a wide spectrum of biological activities including the inhibition of cancer cell migration and invasion. The present study was aimed at deciphering the effect of DADS on the regulation of FN-induced EMT in A549 lung cancer cells. DADS suppressed the FN-induced invasion and migration potential of A549 cells which may be attributed to the reduced activity of gelatinases. DADS suppressed the FN-aggravated EMT of A549 cells by the upregulation of the epithelial markers, E-cadherin and cytokeratin-18, and the downregulation of the mesenchymal markers, N-cadherin and vimentin, and the transcription factors, snail, slug and twist. DADS was effective in inhibiting the nuclear translocation of β-catenin and the phosphorylation of glycogen synthase kinase-3β and in suppressing the activity of dishevelled homolog 2 and T-cell-factor/lymphoid enhancer factor in FN-induced A549 cells. Cumulatively, this study indicated that DADS might be able to reverse FN-induced EMT in A549 cells via the suppression of Wnt signaling.
1. Introduction
Lung cancer is the most common cause of death from cancer in men worldwide. According to GLOBOCAN 2012, it is the leading cause of cancer-related mortality, accounting for 1.58 million cancer deaths per year worldwide. The estimated incidence of lung cancer in India was 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 275 in 2012 among all ages and both sexes. Between the two main types of lung cancer – small cell lung cancer and non-small cell lung cancer (NSCLC), the latter accounts for 80–85% of the lung cancer cases with a survival rate of 1%.1 NSCLC related mortality is associated with the development of the metastatic potential of the primary tumor.2 Considering this high rate of incidence and mortality, it is crucial to understand the mechanisms behind metastasis and identify new therapeutic targets against cancer metastasis. Cancer metastasis is a multistep process consisting of intravasation, attachment to a vessel, extravasation, angiogenesis and growth in distal tissues.3 Increasing scientific literature hints at the role of higher mRNA expression of EMT markers in NSCLC specimens, indicating the involvement of EMT in NSCLC metastasis.4 During EMT, cobblestone-like epithelial cells change into spindle-like mesenchymal cells with the subsequent downregulation of epithelial markers like E-cadherin and cytokeratin (CK) and the upregulation of mesenchymal markers such as N-cadherin and vimentin. In addition, EMT also endows the incipient cancer cell with invasive and metastatic properties and leads to the generation of cancer cells with increased self-renewal property, tumor-initiating capabilities, and resistance to apoptosis and chemotherapy. Hence targeting EMT could be an attractive strategy in the treatment of cancer metastasis.5
275 in 2012 among all ages and both sexes. Between the two main types of lung cancer – small cell lung cancer and non-small cell lung cancer (NSCLC), the latter accounts for 80–85% of the lung cancer cases with a survival rate of 1%.1 NSCLC related mortality is associated with the development of the metastatic potential of the primary tumor.2 Considering this high rate of incidence and mortality, it is crucial to understand the mechanisms behind metastasis and identify new therapeutic targets against cancer metastasis. Cancer metastasis is a multistep process consisting of intravasation, attachment to a vessel, extravasation, angiogenesis and growth in distal tissues.3 Increasing scientific literature hints at the role of higher mRNA expression of EMT markers in NSCLC specimens, indicating the involvement of EMT in NSCLC metastasis.4 During EMT, cobblestone-like epithelial cells change into spindle-like mesenchymal cells with the subsequent downregulation of epithelial markers like E-cadherin and cytokeratin (CK) and the upregulation of mesenchymal markers such as N-cadherin and vimentin. In addition, EMT also endows the incipient cancer cell with invasive and metastatic properties and leads to the generation of cancer cells with increased self-renewal property, tumor-initiating capabilities, and resistance to apoptosis and chemotherapy. Hence targeting EMT could be an attractive strategy in the treatment of cancer metastasis.5
Cellular fibronectin (FN), one of the main components of the extracellular matrix (ECM), is well documented for its function in regulating normal cell adhesion and migration. However, there are also few studies implicating the role of FN in EMT and cancer progression.6 Over-expression of FN, found in several solid carcinomas, has been postulated to be beneficial for the establishment of an ideal microenvironment for tumor growth.7 An increased expression of FN is associated with an increased risk of mortality in breast cancer and could be a marker for predicting poor prognosis in breast cancer patients.8 A study by Li et al. showed the induction of EMT by FN in MCF-7 cells via the activation of calpain.9 However, to the best of our knowledge, the induction of EMT in NSCLC by FN treatment has not been reported to date.
Canonical Wnt signaling has been associated with a range of diseases including cancer. The Wnt/β-catenin pathway has been reported to be one of the molecular pathways that plays a critical role in promoting EMT.10 The binding of the Wnt ligand to the frizzled receptor at the cell surface leads to the activation of dishevelled homolog 2 (dvl2) followed by phosphorylation of glycogen synthase kinase (GSK)-3β and inhibits the degradation of β-catenin. The accumulated cytosolic β-catenin is subsequently translocated to the nucleus where it binds to the T-cell-factor (TCF)/lymphoid enhancer factor (LEF) and activates the target genes, leading to the induction of EMT.11
Recently, the use of phytochemicals to impede tumor metastasis via multiple targets that regulate the invasion and migration potential of tumor cells has gained enormous importance. Garlic, a common household spice rich in sulphur compounds, is well known for its anticancer property.12 Among the organosulphur compounds of garlic, diallyl disulphide (DADS) representing about 60% of garlic oil is reported to be the most stable antioxidant exhibiting anti-metastatic property.13 DADS has been reported to suppress cell migration and the invasion of human triple negative breast cancer, human colorectal cancer, human colon cancer and human prostate cancer cells.14–17 Kuttan et al.18 also showed that DADS inhibited the lung metastasis of B16-F10 melanoma cells in mice. A recent report suggested that DADS could suppress EMT in gastric cancer cells by the down-regulation of LIM kinase-1.19 Thus, DADS possesses great potential for use in the development of a novel anti-metastatic drug. However, studies involving the role of DADS in the regulation of FN-induced EMT in NSCLC are still elusive.
Approximately 85% of the lung cancer cases are diagnosed as non-small-cell lung cancer, and of these, roughly 50% are adenocarcinomas. Therefore, we have chosen A549 which is the most commonly used model for non-small cell adenocarcinoma. Our present work aimed at the induction of EMT in A549 lung cancer cells by FN and an investigation of the role of DADS in the modulation of FN-induced EMT in this cell line. The experimental results indicated that FN-induced cell adhesion, migration and invasion were effectively counteracted by DADS. The expressions of FN-induced EMT markers were also modulated by DADS treatment. Further study of the molecular circuitries revealed that DADS regulated FN-induced EMT in A549 cells by the inhibition of the nuclear translocation of β-catenin by the suppression of GSK-3β phosphorylation and the inhibition of TCF/LEF activity and dvl2 expression.
2. Materials and methods
2.1. Chemicals
Roswell Park Memorial Institute (RPMI)-1640, fetal bovine serum (FBS), 0.25% trypsin-EDTA, LHC-9 and penicillin–streptomycin were purchased from Gibco, Life Technologies (Grand Island, NY). 3-(4,5-Dimethylthiazol-2-yl)-2-5-diphenyl-2H-tetrazolium bromide (MTT) was purchased from Amresco (Solon, OH). DADS and phenylmethylsulfonyl fluoride (PMSF) were purchased from Sigma (St Louis, MO). The primary antibodies to E-cadherin, vimentin, twist, β-catenin, GSK-3β, β-actin, matrix metalloproteinase-2 (MMP-2), matrix metalloproteinase-9 (MMP-9) and proliferating cell nuclear antigen (PCNA) were obtained from Santa Cruz Biotechnology (Dallas, TX). The primary antibodies to N-cadherin, snail, slug, TCF, LEF, phospho-GSK3β and dvl2 were purchased from Cell Signaling Technology (Beverly, MA). The primary antibody to cytokeratin-18 was purchased from R&D Systems (Minneapolis, MN). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was purchased from Life Technologies (Eugene, OR). Nitro blue tetrazolium (NBT)/5-bromo-4-chloro-3-indolyl-phosphate (BCIP) stock solution was purchased from Roche Diagnostics GmbH (Mannheim, Germany). Polyvinylidene difluoride (PVDF) membrane was procured from Millipore, Sigma-Aldrich (St Louis, MO). RNAqueousR-4PCR (DNA-free™ RNA isolation for RT-PCR) kit, RETROscript kit (for RT-PCR) and Super Taq Plus polymerase were purchased from Ambion, Life Technologies (Grand Island, NY). The primers for E-cadherin and N-cadherin and β actin were synthesized by GCC Biotech (Kolkata, India). The Wnt antagonist, C59, was purchased from Abcam (CA, USA).2.2. Cell culture
The lung epithelial adenocarcinoma cell line A549 was procured from the National Centre for Cell Science (Pune, India). The BEAS-2B cell line was a gift from Dr Mainak Sengupta, University of Calcutta, India. The A549 cells were routinely cultured in RPMI-1640 medium supplemented with 10% heat inactivated FBS and the BEAS-2B cells were maintained in LHC-9 medium with 5% FBS along with gentamycin (40 μg ml−1), penicillin (100 units per ml) and streptomycin (10 μg ml−1) at 37 °C under a humidified atmosphere of 5% CO2/95% air.2.3. Cell viability assay
Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2-tetrazolium-bromide (MTT) assay. Exponentially growing cells (1 × 104) were seeded in 96-well plates. After 24 h of growth, the cells were treated with various concentrations of DADS (1, 2.5, 5, 7.5 and 10 μM) and incubated at 37 °C for 24 h. MTT solution (1.2 mg ml−1) was added to each well and incubated for 4 h. The formazan product formed was dissolved in DMSO and the absorbance was measured at 570 nm in a multiwell microplate reader (Infinite M200, TECAN, Mannedorf, Switzerland). The percent viability was calculated as: viability = [Atest/Acontrol] × 100, where Atest and Acontrol are the absorbance of the test sample and control sample, respectively.2.4. Treatment of cells
Cells were allowed to reach 70% confluency before treatment. Four sets of treatment were followed for each experiment. (i) Control: cells (5 × 104) were seeded without any treatment; (ii) cells (5 × 104) were seeded in pre-coated (cellular FN 20 μg ml−1 for 2 h at 37 °C) Petri dishes and further incubated for 24 h in the presence of 5% FBS for the induction of EMT; (iii) cells (5 × 104) were seeded in pre-coated (cellular FN 20 μg ml−1 for 2 h at 37 °C) Petri dishes and treated with a non-cytotoxic dose of DADS (7.5 μM) for 24 h (determined from the cell viability assay); and (iv) cells (5 × 104) were seeded and subjected to DADS (7.5 μM) treatment for 24 h. For the inhibitor study, the following four sets of treatment were given for each experiment: (i) control: cells (5 × 104) were seeded and treated with an equivalent amount of DMSO used as a vehicle for C59; (ii) cells (5 × 104) were seeded in pre-coated (cellular FN 20 μg ml−1 for 2 h at 37 °C) Petri dishes and further incubated for 24 h in the presence of 5% FBS for the induction of EMT; (iii) cells (5 × 104) were seeded in pre-coated (cellular FN 20 μg ml−1 for 2 h at 37 °C) Petri dishes and subjected to a non-cytotoxic dose of C59 (5 μM) treatment for 24 h; and (iv) cells (5 × 104) were seeded and subjected to C59 (5 μM) treatment for 24 h.2.5. Cell adhesion assay
Cell adhesion assay was performed with FN (10 or 20 μg mL−1) coated microtiter plates both in the presence and absence of DADS. The procedure followed was the same as described earlier.202.6. Cell migration assay
Cell migration was assessed using modified Boyden's chamber system. Briefly, both treated and control cells were added to Boyden's invasion chamber (2 × 105 cells per chamber in triplicate) and grown for 24 h in a serum-free culture medium (SFCM) with the chamber inserted in RPMI containing 2% FBS as the chemoattractant. After 24 h the medium was pipetted out from the inserts and the membrane was washed thrice with PBS. The cells were then fixed with methanol and stained with Gill's haematoxylin for 10 min. The cells which migrated towards the opposite side of the membrane were viewed under an inverted microscope. Cells from at least ten different fields were counted under a microscope (Olympus cx40, Tokyo, Japan) equipped with the Magnus Pro 3.7 software at 40× magnification and the mean was calculated.2.7. Wound healing assay
Wound-healing assay was performed to assess the motility of FN-induced cells both in the presence and absence of DADS according to a protocol described earlier.202.8. Gelatin zymography
Gelatin zymography was performed to evaluate the activities of matrix metalloproteinases (MMPs)-2/9 in FN-induced cells both in the presence and absence of DADS according to a protocol described earlier.202.9. ELISA
The wells of the microtitre plate were coated with 10 μg of protein isolated from treated and untreated cells. The plate was covered with Southern Wrap to prevent evaporation and incubated overnight at 4 °C. The wells were then washed thrice with blocking buffer (1% BSA in PBS) and incubated with blocking buffer for 1 h at 37 °C. Then the wells were washed thrice with washing buffer (0.5% NP-40 and 0.5% BSA in PBS) and incubated with anti-E-cadherin or anti-N-cadherin or anti-vimentin or anti-snail or anti-slug or anti-twist antibody (1:1000 dilution) for 1 h. The wells were again washed thrice with washing buffer and the respective alkaline phosphatase conjugated secondary antibodies (1:1000 dilution) were added and incubated at 37 °C for 1 h. After incubation, the wells were washed 6 times with washing buffer and the substrate was added and incubated in the dark until the color developed. Then the stop solution was added to each well and the absorbance was measured at 450 nm using an ELISA reader.
2.10. Immunocytochemistry
A549 cells were seeded on coverslips to 70% confluency and then treated with FN alone or FN along with DADS or left untreated for 24 h. After the treatment, the cells were washed with cold PBS and fixed with ice cold methanol for 10 min at −20 °C. The non-specific binding sites were blocked with blocking buffer for 1 h at 37 °C. The cells were then immunolabelled with anti-E-cadherin, anti-N-cadherin and anti-CK-18 antibody for 24 h, washed with cold PBS and incubated for 1 h with the respective secondary antibodies for 1 h at room temperature. The cells were again washed with PBS and the nuclei were counterstained with DAPI for 10 min. The coverslips were mounted on glass slides and the images were acquired under a fluorescence microscope using the software Fluorescent Workstation 500 (Nikon, Tokyo, Japan).2.11. Immunoprecipitation
After the treatment, the cells were washed with PBS and lysed with radioimmunoprecipitation assay (RIPA) buffer [20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg ml−1 leupeptin]. An equal amount of protein (250 μg) from the cell lysate was precleared using protein A-Sepharose beads and then incubated with the anti-MMP-2 or anti-MMP-9 antibody overnight. The immune complexes were captured using protein A Sepharose beads. The immunoprecipitates were washed with TBST buffer and mixed with the Laemmli sample buffer and western blot was performed.2.12. Western blot
Equal amounts of the protein (70 μg) isolated from each treatment mode or eluted from immunoprecipitates were suspended in Laemmli's buffer containing β-mercaptoethanol and heated for 10 min at 90 °C. The samples were run on SDS–PAGE and blotted onto polyvinylidene difluoride membranes. The membranes were blocked with blocking buffer and incubated with blocking buffer containing 5% BSA in TBST and incubated with anti-E-cadherin, anti-CK-18, anti-N-cadherin, anti-vimentin, anti-snail, anti-slug, anti-twist, anti-DSH, anti-GSK3β, anti-pGSK3β, anti-β-catenin, anti-TCF and anti-LEF-1 antibodies [1:1000 (1 μg antibody in 1 ml buffer) dilution] overnight at 4 °C. Then the secondary antibodies conjugated with alkaline phosphatase were added at 1:1000 dilution for 1 h at room temperature. Protein bands were developed using BCIP/NBT. The band intensity was measured using the Image J Launcher (version 1.4.3.67) software.
2.13. RT-PCR
Total RNA from FN-induced A549 cells treated with or without DADS was isolated using TRIzol reagent and the isolated RNA was quantified spectrophotometrically. cDNA was synthesized from 2 μg of total RNA using a RetroScript kit. The cDNAs were amplified with specific primers for E-cadherin (forward-TTGACGCCGAGAGCTACAC, reverse-AATTCACTCTGCCCAGGACG), N-cadherin (forward-CCTTTCAAACACAGCCACGG, reverse-TGTTTGGGTCGGTCTGGATG) and β-actin (forward-GTCCACCTTCCAGCAGATGTG, reverse-GCATTTGCGGTGGACGAT). The PCR conditions included an initial denaturation step for 7 min at 95 °C and 35 cycles of 40 s at 95 °C, annealing at 49.7 °C for E-cadherin, 49.5 °C for N-cadherin and 52.5 °C for β-actin and extension at 72 °C for 30 s. After the last cycle, a final extension was performed at 72 °C for 7 min. β-Actin was used as an internal control. Densitometric quantitation of the products was determined using the Image-J (version 1.4.3.67) software. The relative intensity was expressed as the ratio of the intensity of the band of the object gene to that of β-actin.2.14. Statistical analysis
All quantitative data are reported as mean ± SEM obtained from at least three independent experiments (unless stated otherwise). Comparisons between the two groups of samples were performed using unpaired Student's t-test, whereas multiple comparisons of more than two groups of samples were performed using one-way analysis of variance (ANOVA) with Dunnett's post-hoc test. P values less than 0.05 were considered statistically significant.3. Results
3.1. Effect of DADS on the viability and adhesion of A549 cells
The MTT assay, performed to evaluate the effect of DADS on the viability of A549 cells, revealed that with 7.5 μM and 10 μM DADS treatment for 24 h, the cell viability was 94.45% and 78.93%, respectively (Fig. 1a). Hence 7.5 μM DADS with a cytotoxicity of 5.55% was used for further studies. The selected dose of DADS did not induce any cytotoxicity in normal bronchial epithelial BEAS-2B cells (ESI, Fig. 1a†).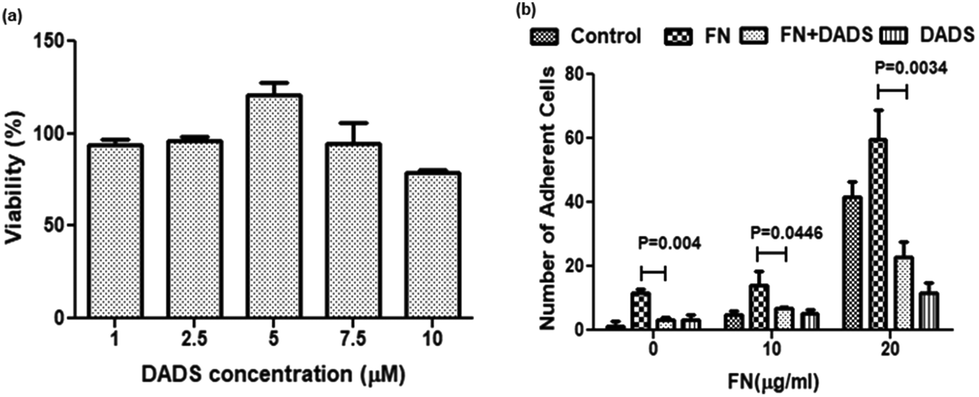 | ||
| Fig. 1 Effect of DADS on the viability and FN-induced adhesion of A549 cells. (a) Cells were treated with different concentrations of DADS (1, 2.5, 5, 7.5, 10 μM) for 24 h and the viability was assessed by the MTT assay. (b) Inhibition of FN-induced cell adhesion by DADS (7.5 μM) treatment for 24 h. Data are shown as the mean ± SD from three replicate measurements. | ||
Cell adhesion assay showed that different concentrations of FN (10 and 20 μg ml−1) acting as extracellular ligand treatment enhanced the cell adhesion of A549 cells as compared to the control. Interestingly in the presence of 10 and 20 μg ml−1 of FN, the cell adhesion decreased by 2.3- and 2.6-fold, respectively (Fig. 1b), when the same cells were co-treated with DADS (7.5 μM). However, DADS (7.5 μM) did not have any effect on the adhesion of normal BEAS-2B cells either with or without FN (ESI, Fig. 1b†).
3.2. DADS inhibited the FN-induced migration and invasion in A549 cells by the inhibition of gelatinases
In normal tissues, the dormant EMT program is usually reactivated during wound repair and tissue regeneration. However, the non-metastatic cancer cells undergo the EMT program to attain the migratory and invasive potential required for metastatic progression.21 Hence, we sought to assess the effect of DADS on FN-induced cell migration by the wound healing assay. The results showed that the wound (scratch) in the cell monolayer healed slowly in A549 cells treated with both FN and DADS as compared to those treated with FN alone (Fig. 2a and b). FN-treated cells showed 60.60, 70.170, and 82.630% of wound closure at 8, 16 and 24 h time points, respectively. However, the cells treated with both FN and DADS showed 5.31, 43.00 and 57.41% of wound closure at 8, 16 and 24 h time points, respectively.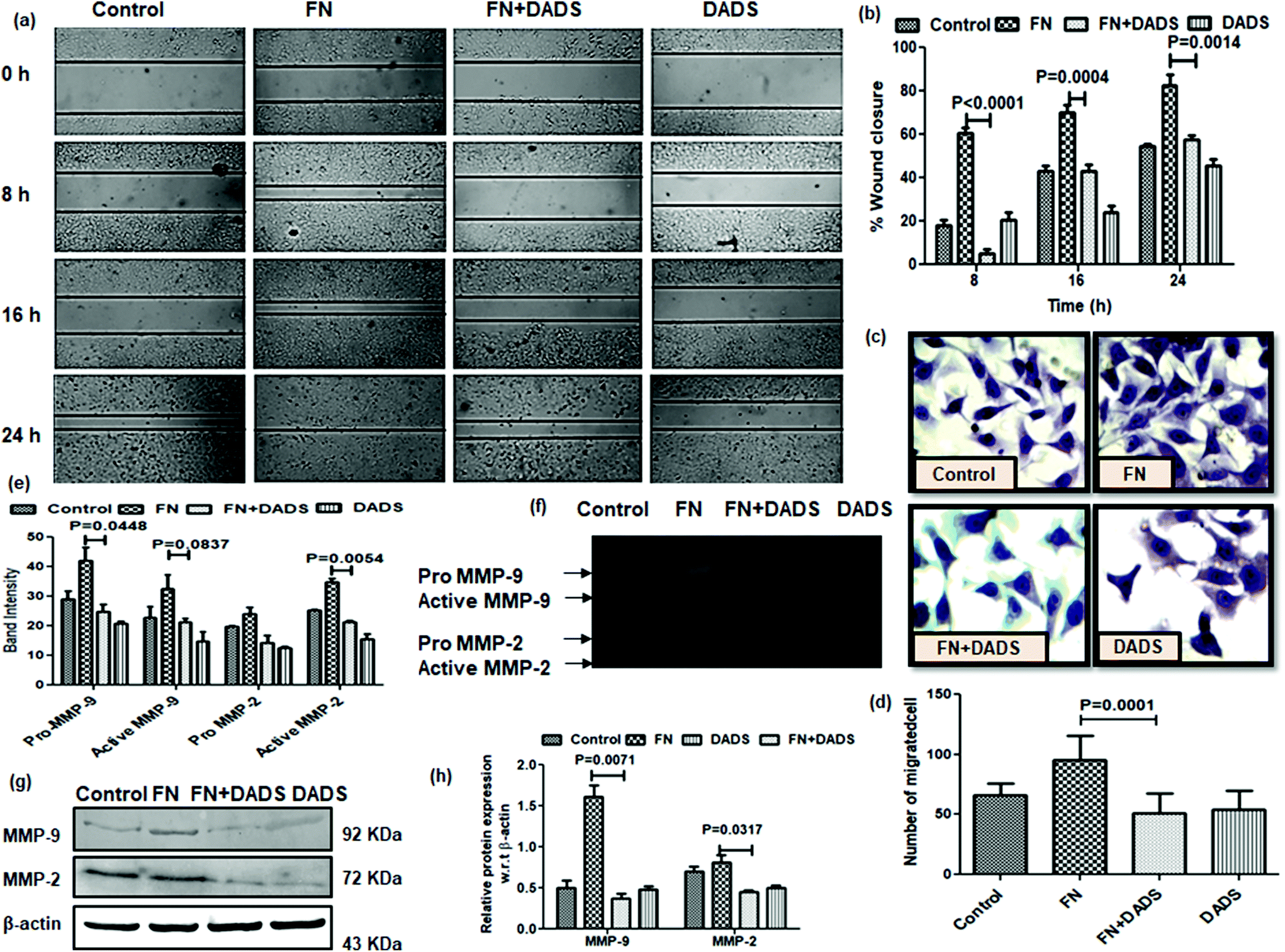 | ||
| Fig. 2 Effect of DADS on the migration, invasion and activities of MMPs in A549 cells. (a) Inhibition of FN-induced cell migration by treatment with DADS for 24 h as evidenced at different time intervals (0, 8, 16 and 24 h); (b) graphical representation of the percentage of wound closure; (c) inhibition of FN-induced cell invasion by treatment with DADS (7.5 μM) for 24 h as compared to the untreated control; (d) graphical representation showing significant reduction in the number of invading cells with DADS (7.5 μM) for 24 h compared to the untreated control; (e) gelatin zymography showing the inhibition of FN-induced pro and active MMP2/9 activities by DADS treatment for 24 h; (f) band intensities of MMP2/9(pro and active) showed significant inhibition of FN-induced MMP2/9 activities by DADS treatment for 24 h. Data are shown as the mean ± SD from three replicate measurements. | ||
Metastatic progression of tumors occurs through the invasion of the basement membrane by cancer cells and their migration to distant sites from the primary tumor site to form metastatic lesions.22 As NSCLC is associated with an increased risk of metastasis,23 inhibiting the powerful metastatic activity of NSCLC cells is essential for improving the prognosis of NSCLC patients. Upon the examination of the effect of DADS on the FN-induced invasion potential of A549 cells, we found that DADS (7.5 μM) could significantly inhibit the FN-induced migration of lung cancer cells 1.85-fold (Fig. 2c and d).
MMPs (particularly MMP2/9) play a pivotal role in degrading the extracellular matrix and thus facilitating cell migration by the detachment of epithelial cells from the surrounding tissue. The efficacy of DADS in the suppression of FN-induced cell migration instigated us to look into the effect of DADS on the activities of MMP2/9 in FN-treated A549 cells by gelatin zymography. The band intensities reflected that both pro and active MMP-2/MMP-9 were significantly suppressed when FN-induced A549 cells were co-treated with DADS (Fig. 2e). Experimental results suggested that FN enhanced the activities of MMP2/9 which was efficiently counteracted by DADS (7.5 μM) treatment (Fig. 2f). Furthermore, immunoprecipitation followed by western blot was also performed to check the expression of MMP2/9 in the protein level. The immunoblot data indicated that DADS treatment significantly downregulated the FN-induced expression of both MMP2/9 (Fig. 2g and h).
3.3. DADS suppressed the characteristic markers of EMT in A549 cells
The potency of DADS in the modulation of cell migration and invasion prompted us to examine whether this organosulphur compound could reverse the process of EMT in FN-treated A549 cells.FN-induced A549 cells exhibited suppressed epithelial markers, E cadherin and CK-18, in comparison with the control. The same epithelial markers showed increased expression when FN-induced cells were administered with DADS (7.5 μM). The results were evident from both immunocytochemistry (Fig. 3a–c) and western blot (Fig. 3d and e). The mRNA profile further validated that DADS (7.5 μM) was effective in increasing the expression of E-cadherin (Fig. 3f and g).
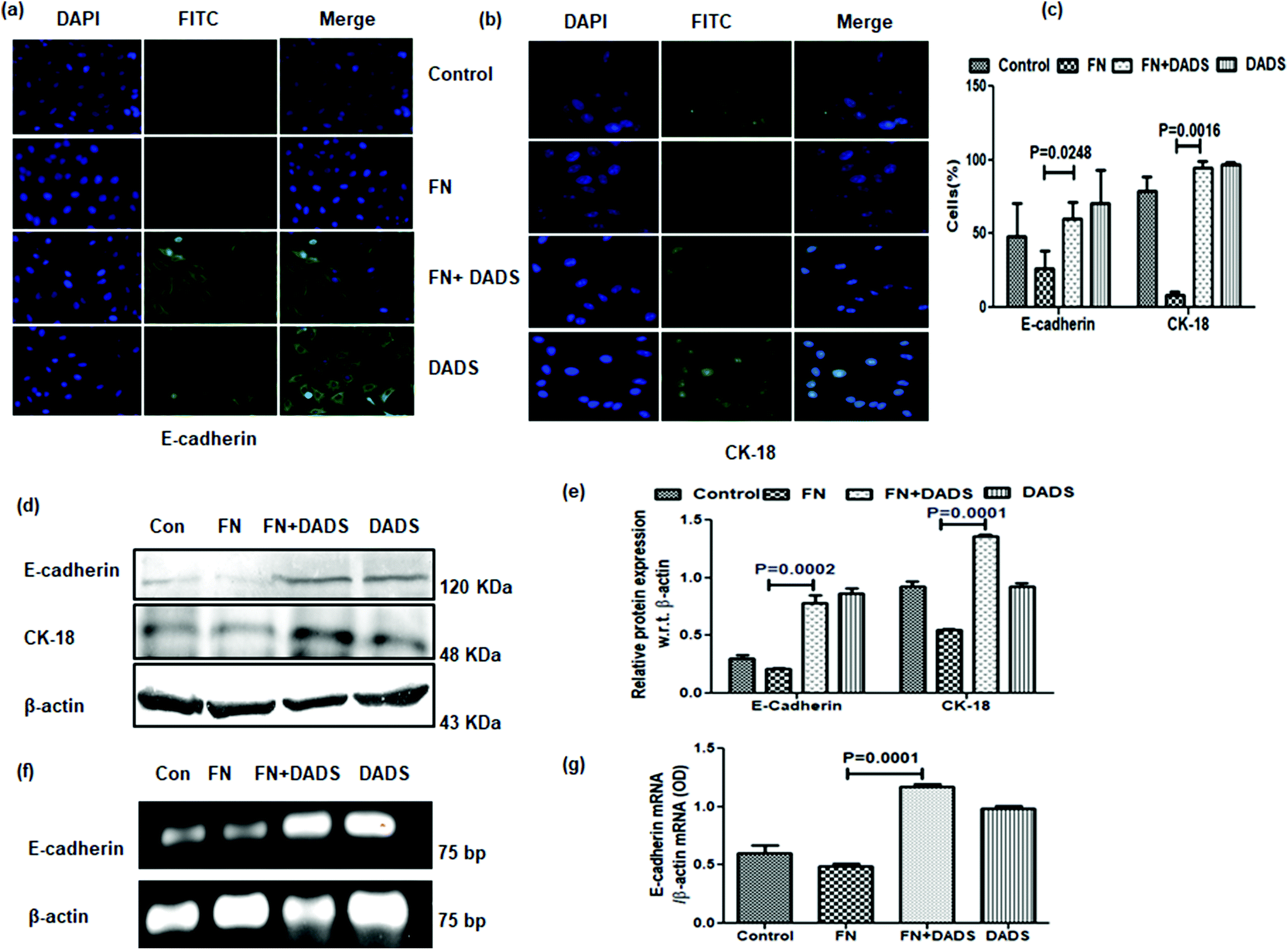 | ||
| Fig. 3 Effect of DADS on epithelial markers in A549 cells. Immunocytochemistry showing the modulation of the FN-induced down-regulation of (a) E-cadherin and (b) CK-18 by 7.5 μM DADS; (c) graphical representation of the percentage of cells expressing E-cadherin and CK-18 in each treatment mode. (d) Western blot showing the effect of DADS (7.5 μM) on the FN-induced expression of E-cadherin and CK-18; (e) the respective band intensities of the blots reflected similar profiles of significant upregulation in the expression of E-cadherin and CK-18 by DADS (7.5 μM) treatment for 24 h; (f) RT-PCR showing the up-regulation of FN-induced E-cadherin expression by 7.5 μM DADS in A549 cells as compared to only FN treated cells; (g) graphical representation of the respective band intensities of the PCR products. Data are shown as the mean ± SD from three replicate measurements. | ||
FN-induced A549 cells showed overexpressed mesenchymal markers, N-cadherin and vimentin, which were significantly reduced with DADS (7.5 μM) as evident from both immunocytochemistry (Fig. 4a–c) and western blot (Fig. 4d and e) analyses. The efficacy of DADS in reducing the N-cadherin expression of FN-induced A549 cells was also reflected in the mRNA profile (Fig. 4f and g).
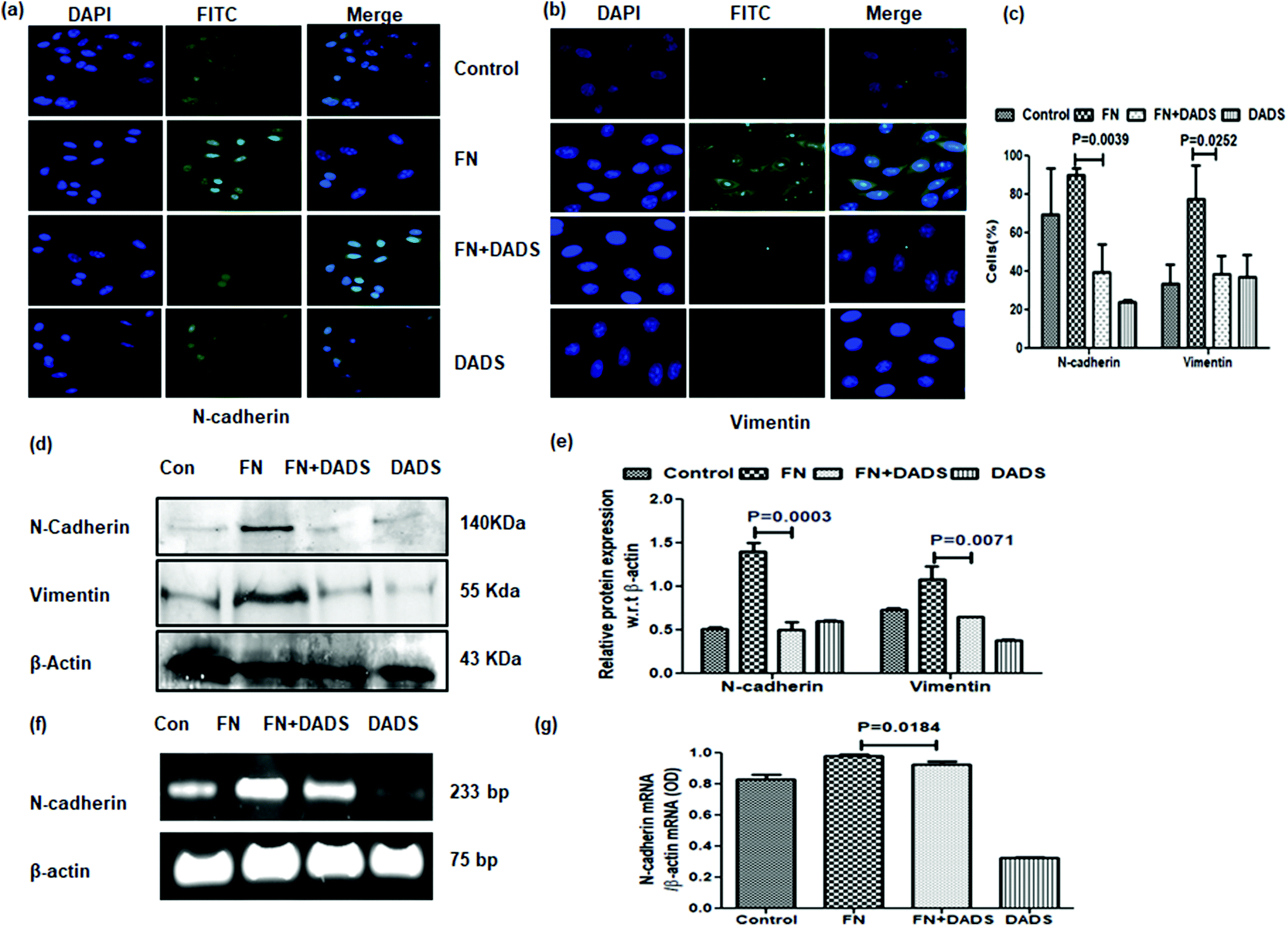 | ||
| Fig. 4 Effect of DADS on mesenchymal markers in A549 cells. Immunocytochemistry showing the downregulation of FN-induced expression of (a) N-cadherin and (b) vimentin by 7.5 μM DADS; (c) graphical representation of the percentage of cells expressing N-cadherin and vimentin in each treatment mode; (d) western blot showing the down-regulation of FN-induced N-cadherin and vimentin expression by DADS (7.5 μM); (e) the respective band intensities of the blots reflected similar profiles of significant down-regulation in the expression of N-cadherin and vimentin by DADS (7.5 μM) treatment for 24 h; (f) RT-PCR showing the down-regulation of FN-induced N-cadherin expression by DADS (7.5 μM) in A549 cells as compared to only FN treated cells; (g) graphical representation of the respective band intensities of the PCR products. Data are shown as the mean ± SD from three replicate measurements. | ||
3.4. DADS inhibited the expression of transcription factors associated with EMT
The upregulation of the E-cadherin level by DADS instigated us to investigate the effect of DADS on the transcription factors, snail, slug, and twist, involved with EMT. ELISA and western blot results showed that FN treatment in A549 cells upregulated the expressions of snail, slug and twist as compared to the control (Fig. 5a–c). However, when the FN-induced cells were co-treated with DADS (7.5 μM), the expression of snail, slug and twist was significantly downregulated (Fig. 5a–c).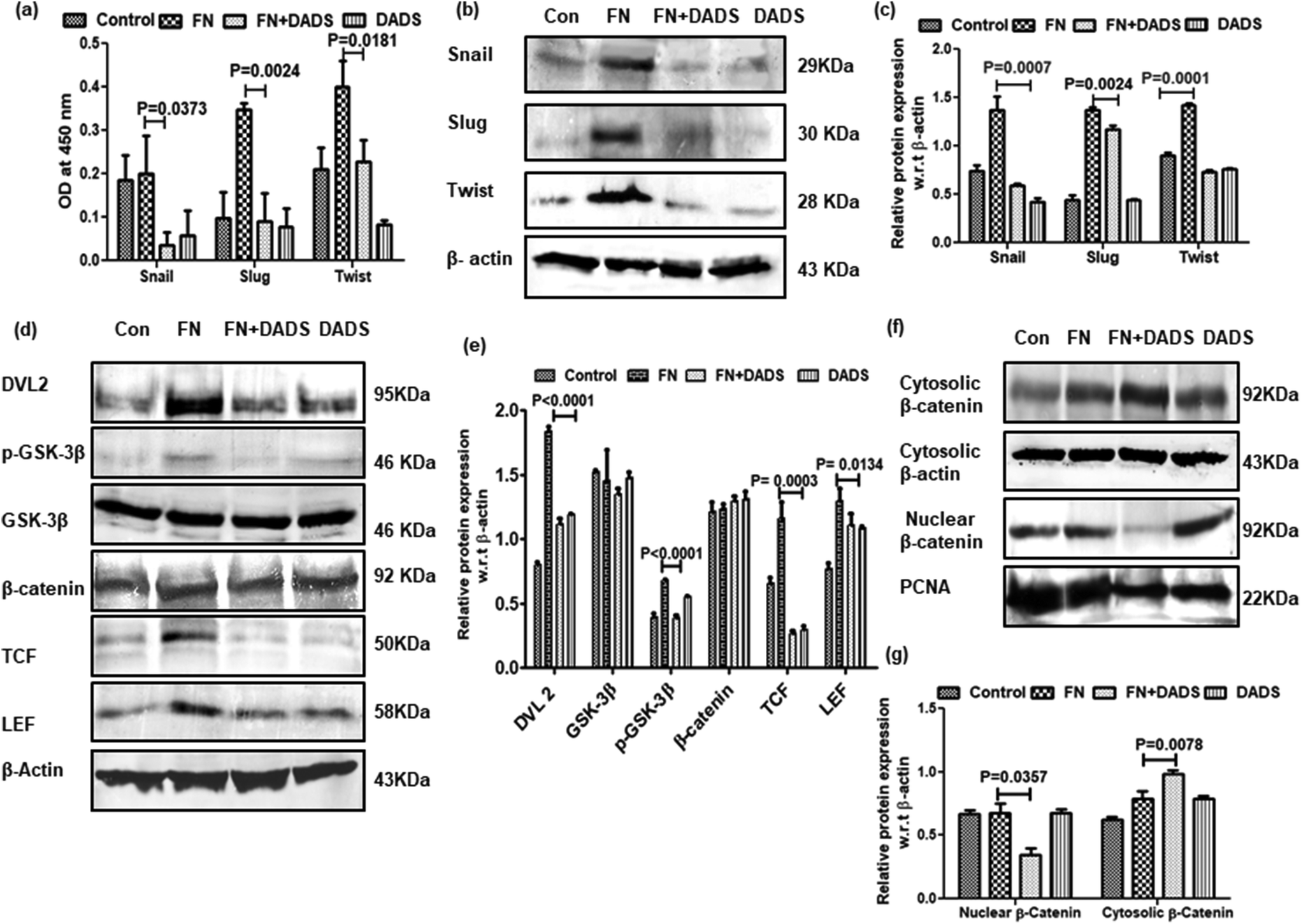 | ||
| Fig. 5 Effect of DADS on the regulation of EMT-related Wnt signaling network in A549 cells. (a) Graphical representation of ELISA showing significant reduction of FN-induced snail, slug and twist expression by DADS treatment for 24 h; (b) western blot analyses showing the down-regulation of FN-induced snail, slug and twist expression by DADS (7.5 μM); (c) the respective band intensities of the blots reflected similar profiles of significant inhibition in the expression of snail, slug and twist by DADS (7.5 μM); (d) western blot analyses showing the effect of DADS (7.5 μM) on FN-induced dvl2, GSK-3β, β-catenin, TCF, and LEF expression; (e) the respective band intensities exhibiting significant inhibition of dvl2, GSK-3β, β-catenin, TCF, and LEF in FN-induced A549 cells treated with DADS for 24 h; (f) western blot depicting the inhibition of nuclear localization of β-catenin following DADS treatment; (g) the respective band intensities of the nuclear and cytosolic fraction of β-catenin exhibiting significant inhibition of β-catenin in the nuclear fraction of FN-induced A549 cells treated with DADS (7.5 μM) for 24 h with respect to the untreated control. Data are shown as the mean ± SD from three replicate measurements. | ||
3.5. Suppression of EMT in A549 cells by DADS is associated with the modulation of Wnt/β-catenin signaling
Several reports delineate the role of Wnt/β-catenin pathway activation in triggering EMT and cancer cell invasion.24 In the absence of Wnt signaling, β-catenin remains associated with membrane-bound E-cadherin. However, if unbound with membrane E-cadherin, β-catenin translocates to the nucleus and transcriptionally activates several pro-migratory genes necessary for EMT in association with the TCF/LEF transcription factor.25 On investigation of Wnt/β-catenin signaling in FN-induced A549 cells, it was observed that TCF/LEF, dvl2 and pGSK3β were significantly higher in FN-treated cells as compared to the control. DADS administration was effective in downregulating the expression of TCF/LEF, Dvl2 and pGSK3β in FN-induced A549 cells (Fig. 5d and e). The expression of total β-catenin remained unaltered even with the co-treatment of DADS in FN-induced cells (Fig. 5d and e). However, the nuclear expression of β-catenin was efficiently reduced by DADS (7.5 μM) in FN-treated A549 cells (Fig. 5f and g).To further validate the participation of the Wnt pathway in the regulation of FN-induced EMT by DADS, we antagonized the Wnt signaling in A549 cells with C59, which is a potent Wnt pathway inhibitor. C59 inhibits porcupine required for the secretion and biological activity of Wnt. Western blot performed with various non-cytotoxic doses of C59 (1, 5, 10 μM) showed that 5 μM C59 with a cytotoxicity of 6.051% was able to block FN-induced Wnt signaling in A549 cells (data not shown). Furthermore, the immunoblot data also revealed that FN-induced cells when treated with C59 (5 μM) for 24 h elicited a downregulated expression of Wnt response elements (Dvl2, p-GSK3β, TCF and LEF) confirming the inhibition of the Wnt pathway by C59 (Fig. 6a and b). Moreover, the blocking of the Wnt pathway by C59 resulted in an upregulated expression of E-cadherin and the downregulation of N-cadherin as compared to only FN-treated cells (Fig. 6c and d). Taken together these data established a pivotal role of the Wnt signaling pathway in the modulation of FN-induced EMT in A549 cells.
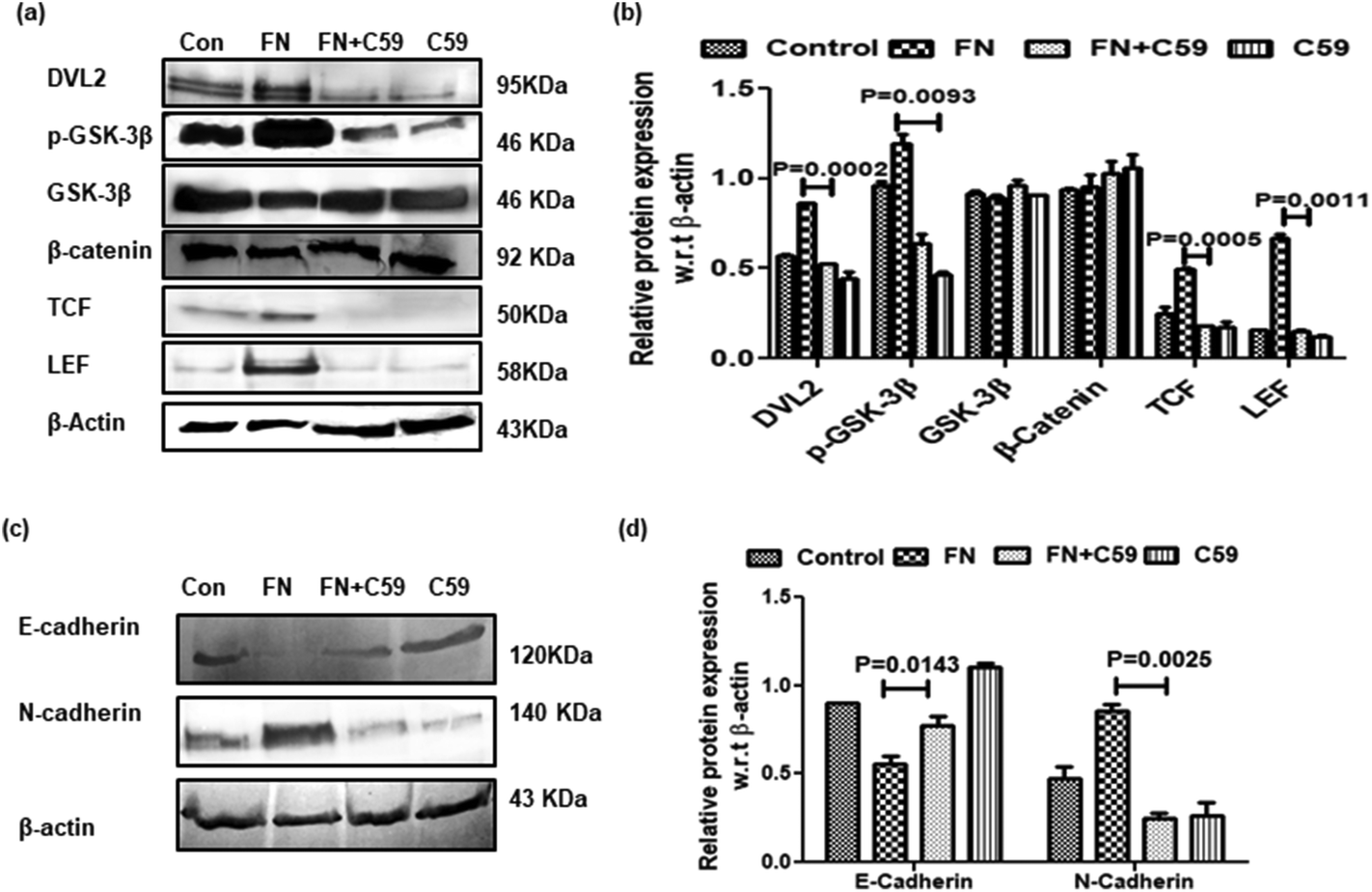 | ||
| Fig. 6 Effect of the Wnt inhibitor C59 on the regulation of the Wnt signaling network and EMT-related proteins in A549 cells. (a) Western blot analyses showing the effect of C59 (5 μM) on FN-induced dvl2, pGSK-3β, GSK-3β, β-catenin, TCF, and LEF expressions; (b) the respective band intensities exhibiting significant inhibition of dvl2, pGSK-3β, GSK-3β, β-catenin, TCF, and LEF in FN-induced A549 cells treated with C59 for 24 h; (c) western blot showing the effect of C59 (μM) on the FN-induced expression of E-cadherin and N-cadherin; (d) the respective band intensities of the blots reflected similar profiles of significant upregulation in the expression of E-cadherin and the downregulation of N-cadherin by C59 (5 μM) treatment for 24 h. | ||
4. Discussion
The presence of metastasis is the main cause of morbidity and mortality in millions of lung cancer patients. Type III EMT occurring during metastasis plays a critical role during malignant transformation, cancer progression, increased resistance to apoptosis and chemotherapy. Hence exploration of agents that could suppress EMT at their sub-toxic doses and understanding the related mechanism of action are important for finding the therapeutic implication of natural compounds against EMT. DADS, an oil soluble bioactive component of garlic, has been reported to exert its anti-cancer effects in multiple ways, including the negative regulation of cancer metastasis, angiogenesis, apoptosis, cell-cycle progression and inflammation.12 However, the study involving the effect of DADS on the modulation of FN-induced EMT in A549 cells is still elusive. Hence our present study aimed at delineating the role of DADS in the regulation of FN-induced EMT in A549 cells.FN, an extracellular matrix component, has been reported to induce cell migration, invasion and also EMT which are believed to prelude metastasis in cancer cells.26 In normal tissues, FN forms the provisional matrix that provides the framework for the assembly of fibrillar collagens and other ECM proteins. Classical wound healing studies indicated that matrix deposition follows a temporal sequence that begins with the assembly of FN, followed by cell invasion, deposition of type III and then type I collagen and eventually loss of FN. However, in tumor tissues, often referred to as non-healing wound tissues, it was observed that the upregulated expression of FN was sustained.27 Cultivation of cells in the presence of FN leads to increased activities of extracellular proteases which are capable of degrading different components of the ECM and basement membrane.28 Tumor cells interact with the ECM by specific cell surface receptors and these interactions influence cell adhesion, migration, etc.29 FN has well-characterized binding sites for α5β1 integrin receptor on the cell surface. Integrin-mediated responses to FN include adhesion, downstream signaling to cause cell migration, assembly of cytoskeleton, invasion, etc.30 Therefore, it might be presumed that FN acts differentially in normal and tumor tissues. This might have been the reason why DADS reduced the adhesion of FN-induced A549 cells but had no effect on the adhesion of normal bronchial epithelial BEAS-2B cells. In congruence with the fact that FN promotes EMT and metastasis in tumor cells, Knowles et al., in 2013, showed that plasma FN promotes lung metastasis by facilitating tumor cell invasion in blood clots.26 Li et al., in 2017,9 demonstrated that FN stimulation caused EMT in MCF-7 breast cancer cells by the downregulation of epithelial markers like E-cadherin and the upregulation of the mesenchymal markers, N-cadherin and vimentin. A study by Gopal et al.31 indicated that the overexpression of FN in head and neck squamous cell carcinomas is associated with poor clinical outcome. FN also plays a major role in cell adhesion.32 Cancer cell interaction with the endothelium in distant tissues facilitates the metastatic spread. The present study also revealed that cellular FN-induced cell invasion, migration and adhesion in A549 cells were significantly subdued by DADS. Our results are in line with prior reports where DADS was an effective inhibitor of lung metastasis of B16F-10 melanoma cells in mice.18
MMPs, a family of zinc-dependent endopeptidases, are capable of cleaving virtually all ECM substrates. The degradation of the ECM is a key event in the progression, invasion, and metastasis of potentially malignant lesions.33,34 The elevated expression of different MMPs have been reported to be associated with different metastatic stages in the progression of various types of tumors. Das et al. showed the FN-mediated activation of MMP-2/9 in MCF-7 human breast cancer cells.35 Another communication by Pal et al. indicated that FN-induced signaling through α5β1 integrin leads to the upregulation of MMP-9 and MMP-1 in PC-3 prostate cancer cells.36 Hence the inhibition of the activity of MMPs holds great promise for the prevention or the inhibition of metastasis. Among various MMPs, MMP-2 and -9 (gelatinases A and B) in particular play a pivotal role in tumor invasion and metastasis and are highly expressed in epithelial cancer cells.37,38 The transcription factor snail1 has been reported to upregulate MMP-2 and -9 and initiate EMT and, subsequently, the synchronized effect of snail1 and slug continually triggered MMP-9 and promoted EMT.39 There is evidence showing that DADS suppressed the invasion in human prostate and gastric cancer cells by the inhibition of MMP2/9 activities.17,40 Hence in the present study, decreased MMP2/9 activities might contribute to the anti-metastatic effect of DADS treatment in A549 cells.
FN stimulated A549 cells to undergo EMT, characterized by the acquisition of a mesenchymal phenotype, an increase in cellular adhesion, migration, and upregulation of mesenchymal markers (N-cadherin and vimentin) along with the subsequent downregulation of epithelial markers (E-cadherin and CK-18). The loss of E-cadherin in metastatic cells is often amalgamated with a high expression of N-cadherin, a phenomenon referred to as ‘cadherin switch’. Cadherin switching has a profound effect on the EMT process. The cadherin switch is essential for increased cell motility; however, it is not always required for the morphological changes associated with EMT.41 The literature suggests that N-cadherin expression is more important for cancer metastasis than the downregulation of E-cadherin and other EMT inducers.42 N-Cadherin expression in epithelial cells actively destabilizes cell–cell adhesion, which allows the cells to detach from the primary tumor and associate selectively with the stroma and/or endothelium and thus promotes motility.42,43 High N-cadherin expression is also associated with the increased secretion of MMP-9.44,45 In most of the metastatic cancer cells, E-cadherin loss is associated with the elevated expression of vimentin. Apart from E-cadherin, CK-18 is another epithelial marker which is an intermediate filament protein involved in the cell structure, cell signaling, and the cell cycle. Snail1 inhibits CK-18 during the induction of EMT.46 In consonance with other reports, this study also revealed that DADS was able to block FN-induced EMT by the upregulation of E-cadherin and CK-18 with the concomitant down-regulation of N-cadherin and vimentin.
Several studies indicate the loss of E-cadherin to be a hallmark of EMT, confirming E-cadherin to be a caretaker of the epithelial state of the cell. The cadherin–catenin based adhesion system plays a major role in cell–cell adhesion. Findings suggest that slug and snail, both of which are transcriptional targets of β-catenin, are involved in E-cadherin repression.25 Twist also represses E-cadherin indirectly.47 These transcriptional repressors of E-cadherin are essential for the onset of EMT and their expression is associated with the tumor stage and clinical–pathological features. However, according to the findings of Howard et al.,48 a mere loss of E-cadherin is not sufficient to activate β-catenin signaling. Our study showed that DADS treatment significantly abrogated the FN-induced expression of snail, slug and twist in A459 cells.
β-Catenin, the central structural component of the cell–cell adhesion complex, acts as a transcriptional co-activator in the Wnt signaling pathway which is activated during cancer progression in many tissues.48 Wnt receptor activation inhibits the degradation of newly synthesized β-catenin, allowing the cadherin-free form of β-catenin to enter the nucleus, bind with the LEF/TCF family of DNA binding protein and act as a transcriptional co-activator of specific target genes associated with Wnt signaling.48 It has been reported that LEF-1 interacts with snail1 via the Wnt, PI3K and TGF-β1 pathways, and both snail1 and LEF-1 are essential for the propagation of EMT. Snail-1 interacts with LEF-1 to upregulate its expression.46 Interestingly, Gottardi et al.49 indicated that Wnt signaling generates a form of β-catenin that binds with TCF but not cadherin, suggesting that the β-catenin used for transcription is molecularly distinct from that used for adhesion. Signaling from the Wnt receptor proceeds via the phosphorylation of dvl at its DIX and PDZ domain, leading to the destruction of the axin complex, comprising the scaffolding protein axin, the tumor suppressor adenomatous polyposis coli gene product, casein kinase 1, and GSK-3β, which catalyzes the degradation of β-catenin.50–52 However, in the absence of Wnt signaling, cytosolic β-catenin is phosphorylated and subjected to proteosomal degradation by the axin complex. Consistent with these findings, we found that FN treatment in A549 cells showed an increased nuclear localization of β-catenin along with the consequent activation of LEF/TCF transcription factors which were suppressed by DADS treatment. GSK-3β, a serine-threonine kinase, is activated by auto-phosphorylation at Tyr216.53–55 However, GSK-3β is reported to be inactivated by phosphorylation at the N-terminal Ser9 by several protein kinases.56 GSK-3β, phosphorylated at Ser9, binds to a pocket for a priming phosphorylation in the substrate-binding region to reduce its binding affinity for the substrate β-catenin.57 DADS treatment inhibited the FN-induced phosphorylation of GSK-3β at ser9 and thus played a crucial role in the degradation of cytosolic β-catenin and abrogated the activation of Wnt signaling. Furthermore, blocking of the Wnt pathway by the C59 [potent Wnt antagonist that inhibits porcupine required for the biological activity of Wnt] exhibited the same signaling pattern as that of DADS in A549 cells. C59-induced the downregulation of N-cadherin and the upregulation of E-cadherin expression was similar to that of the DADS-mediated modulation of cadherins in the FN-induced EMT model of lung cancer. The inhibition of Wnt by C59 abrogated the expression of dvl2/pGSK-3β/TCF/LEF similar to that of DADS. This finding therefore validated that DADS inhibited FN-induced EMT by suppressing the Wnt/β-catenin signaling pathway. Cumulatively, the present study demonstrated that the modulation of Wnt/β-catenin signaling may be a possible mode of action by which DADS may elicit the EMT reversal effect in A549 cells.
5. Conclusion
We reported for the first time, to the best of our knowledge, that DADS suppressed the FN-induced migration and invasion potential of A549 cells to an appreciable extent by perturbing the commencement of EMT via the Wnt signaling pathway. However, the study had a limitation of using a single lung cancer line, A549, but in-depth analysis contributed towards a better understanding of the mechanistic basis of action of DADS against EMT in this model. Further preclinical studies are warranted to validate the present findings in other in vitro and in vivo metastatic models to establish the efficacy of DADS against EMT in lung cancer. In the long run, this might help open a new arena of therapeutic strategies for the inhibition of EMT and better management of lung cancer.Author contributions
BD performed the experiments and wrote the manuscript. DS designed the plan of work and edited the manuscript.Conflicts of interest
The authors declare that they have no conflict of interests.Acknowledgements
The authors are indebted to the Science and Engineering Research Board, New Delhi, India (Grant No. PDF/2016/000248) for funding the work and providing a post-doctoral fellowship to BD. The authors are grateful to the Director of Chittaranjan National Cancer Institute, Kolkata for providing the infrastructural facilities.References
- GLOBOCAN 2012: estimated cancer incidence, mortality and prevalence worldwide in 2012, 2012.
- J. R. Molina, P. Yang, S. D. Cassivi, S. E. Schild and A. A. Adjei, Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship, Mayo Clin. Proc., 2008, 83, 584–594 CrossRef PubMed.
- F. Perlikos, K. J. Harrington and K. N. Syrigos, Key molecular mechanisms in lung cancer invasion and metastasis: a comprehensive review, Crit. Rev. Oncol. Hematol., 2013, 87, 1–11 CrossRef PubMed.
- D. Xiao and J. He, Epithelial mesenchymal transition and lung cancer, J. Thorac. Dis., 2010, 2, 154 CAS.
- V. Daniele, S. Pasquale, J. Frank, T. Marco, G. Anna, C. Loredana, T. Andrea, B. Claudia, F. Isabelle and A. Gaballo, Translating epithelial mesenchymal transition markers into the clinic: Novel insights from proteomics, EuPA Open Proteomics, 2016, 10, 31–41 CrossRef CAS PubMed.
- J. Park and J. E. Schwarzbauer, Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition, Oncogene, 2014, 33, 1649 CrossRef CAS PubMed.
- A. Lal, A. E. Lash, S. F. Altschul, V. Velculescu, L. Zhang, R. E. McLendon, M. A. Marra, C. Prange, P. J. Morin and K. Polyak, A public database for gene expression in human cancers, Cancer Res., 1999, 59, 5403–5407 CAS.
- Y. K. Bae, A. Kim, M. K. Kim, J. E. Choi, S. H. Kang and S. J. Lee, Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer, Hum. Pathol., 2013, 44, 2028–2037 CrossRef CAS PubMed.
- C.-L. Li, D. Yang, X. Cao, F. Wang, D.-Y. Hong, J. Wang, X.-C. Shen and Y. Chen, Fibronectin induces epithelial-mesenchymal transition in human breast cancer MCF-7 cells via activation of calpain, Oncol. Lett., 2017, 13, 3889–3895 CrossRef CAS PubMed.
- A. Wodarz and R. Nusse, Mechanisms of Wnt signaling in development, Annu. Rev. Cell Dev. Biol., 1998, 14, 59–88 CrossRef CAS PubMed.
- R. T. Moon, A. D. Kohn, G. V. De Ferrari and A. Kaykas, WNT and β-catenin signalling: diseases and therapies, Nat. Rev. Genet., 2004, 5, 691–701 CrossRef CAS PubMed.
- L. Yi and Q. Su, Molecular mechanisms for the anti-cancer effects of diallyl disulfide, Food Chem. Toxicol., 2013, 57, 362–370 CrossRef CAS.
- P. Sujatha, V. T. Siddhartha, M. N. Suma, K. Gowthamarajan and V. M. Subba Rao, Understanding the Mechanism of Breast and Prostate Cancer Cell Killing By Diallyldisulfide-An Over View, J. Chem. Applied Biochem., 2015, 2, 112 Search PubMed.
- J. Huang, B. Yang, T. Xiang, W. Peng, Z. Qiu, J. Wan, L. Zhang, H. Li, H. Li and G. Ren, Diallyl disulfide inhibits growth and metastatic potential of human triple-negative breast cancer cells through inactivation of the β-catenin signaling pathway, Mol. Nutr. Food Res., 2015, 59, 1063–1075 CrossRef CAS PubMed.
- Y. Zhou, J. Su, L. Shi, Q. Liao and Q. Su, DADS downregulates the Rac1-ROCK1/PAK1-LIMK1-ADF/cofilin signaling pathway, inhibiting cell migration and invasion, Oncol. Rep., 2013, 29, 605–612 CrossRef CAS.
- K.-C. Lai, S.-C. Hsu, C.-L. Kuo, J.-S. Yang, C.-Y. Ma, H.-F. Lu, N.-Y. Tang, T.-C. Hsia, H.-C. Ho and J.-G. Chung, Diallyl sulfide, diallyl disulfide, and diallyl trisulfide inhibit migration and invasion in human colon cancer colo 205 cells through the inhibition of matrix metalloproteinase-2, -7, and-9 expressions, Environ. Toxicol., 2011, 28, 479–488 CrossRef.
- D. Y. Shin, G.-Y. Kim, J.-I. Kim, M. K. Yoon, T. K. Kwon, S. J. Lee, Y.-W. Choi, H. S. Kang, Y. H. Yoo and Y. H. Choi, Anti-invasive activity of diallyl disulfide through tightening of tight junctions and inhibition of matrix metalloproteinase activities in LNCaP prostate cancer cells, Toxicol. in Vitro, 2010, 24, 1569–1576 CrossRef CAS PubMed.
- G. Kuttan and R. Kuttan, Effect of diallyl sulphide, diallyl disulphide, and allyl methyl sulphide on the inhibition of lung metastasis of B16F-10 melanoma cells in mice, J. Clin. Biochem. Nutr., 1999, 27, 131–139 CrossRef CAS.
- B. Su, J. Su, Y. Zeng, F. Liu, H. Xia, Y.-H. Ma, Z.-G. Zhou, S. Zhang, B.-M. Yang and Y.-H. Wu, Diallyl disulfide suppresses epithelial-mesenchymal transition, invasion and proliferation by downregulation of LIMK1 in gastric cancer, Oncotarget, 2016, 7, 10498 Search PubMed.
- D. Sinha, K. Dutta, K. K. Ganguly, J. Biswas and A. Bishayee, A novel synthetic oleanane triterpenoid suppresses adhesion, migration, and invasion of highly metastatic melanoma cells by modulating gelatinase signaling axis, Mol. Carcinog., 2015, 54, 654–667 CrossRef CAS.
- J. P. Thiery, H. Acloque, R. Y. J. Huang and M. A. Nieto, Epithelial-mesenchymal transitions in development and disease, Cell, 2009, 139, 871–890 CrossRef CAS.
- T. A. Martin, L. Ye, A. J. Sanders, J. Lane and W. G. Jiang, Cancer invasion and metastasis: molecular and cellular perspective, 2013 Search PubMed.
- A. Ali, J. R. Goffin, A. Arnold and P. M. Ellis, Survival of patients with non-small-cell lung cancer after a diagnosis of brain metastases, Curr. Oncol., 2013, 20, e300 CrossRef CAS.
- S. A. O'Toole, J. M. Beith, E. K. A. Millar, R. West, A. McLean, A. Cazet, A. Swarbrick and S. R. Oakes, Therapeutic targets in triple negative breast cancer, J. Clin. Pathol., 2013, 66, 530–542 CrossRef.
- R. Thakur and D. P. Mishra, Pharmacological modulation of beta-catenin and its applications in cancer therapy, J. Cell. Mol. Med., 2013, 17, 449–456 CrossRef CAS PubMed.
- L. M. Knowles, L. A. Gurski, C. Engel, J. R. Gnarra, J. K. Maranchie and J. Pilch, Integrin αvβ3 and fibronectin upregulate Slug in cancer cells to promote clot invasion and metastasis, Cancer Res., 2013, 73, 6175–6184 CrossRef CAS.
- R. O. Hynes, Fibronectins, Sci. Am., 1986, 254, 42–51 CrossRef CAS.
- H. G. Munshi and M. S. Stack, Reciprocal interactions between adhesion receptor signaling and MMP regulation, Cancer Metastasis Rev., 2006, 25, 45–56 CrossRef CAS.
- R. L. Juliano, Signal transduction by cell adhesion receptors and the cytoskeleton: functions of integrins, cadherins, selectins, and immunoglobulin-superfamily members, Annu. Rev. Pharmacol. Toxicol., 2002, 42, 283–323 CrossRef CAS PubMed.
- T. Sen, A. Dutta, G. Maity and A. Chatterjee, Fibronectin induces matrix metalloproteinase-9 (MMP-9) in human laryngeal carcinoma cells by involving multiple signaling pathways, Biochimie, 2010, 92, 1422–1434 CrossRef CAS PubMed.
- S. Gopal, L. Veracini, D. Grall, C. Butori, S. Schaub, S. Audebert, L. Camoin, E. Baudelet, A. Radwanska and S. Beghelli-de la Forest Divonne, Fibronectin-guided migration of carcinoma collectives, Nat. Commun., 2017, 8, 14105 CrossRef CAS PubMed.
- E. Ruoslahti, Fibronectin in cell adhesion and invasion, Cancer Metastasis Rev., 1984, 3, 43–51 CrossRef CAS PubMed.
- M. J. Duffy, T. M. Maguire, A. Hill, E. McDermott and N. O'Higgins, Metalloproteinases: role in breast carcinogenesis, invasion and metastasis, Breast Cancer Res., 2000, 2, 252 CrossRef CAS.
- P. Vihinen, R. Ala-aho and V.-M. Kahari, Matrix metalloproteinases as therapeutic targets in cancer, Curr. Cancer Drug Targets, 2005, 5, 203–220 CrossRef CAS.
- S. Das, A. Banerji, E. Frei and A. Chatterjee, Rapid expression and activation of MMP-2 and MMP-9 upon exposure of human breast cancer cells (MCF-7) to fibronectin in serum free medium, Life Sci., 2008, 82, 467–476 CrossRef CAS PubMed.
- S. Pal, K. K. Ganguly and A. Chatterjee, Extracellular matrix protein fibronectin induces matrix metalloproteinases in human prostate adenocarcinoma cells PC-3, Cell Commun. Adhes., 2013, 20, 105–114 CrossRef CAS PubMed.
- L. M. Matrisian, The matrix-degrading metalloproteinases, Bioessays, 1992, 14, 455–463 CrossRef CAS PubMed.
- O. R. F. Mook, W. M. Frederiks and C. J. F. Van Noorden, The role of gelatinases in colorectal cancer progression and metastasis, Biochim. Biophys. Acta, Rev. Cancer, 2004, 1705, 69–89 CrossRef CAS PubMed.
- B. Qiao, N. W. Johnson and J. Gao, Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-β11 is Snail family-dependent and correlates with matrix metalloproteinase-2 and-9 expressions, Int. J. Oncol., 2010, 37, 663–668 CAS.
- H. S. Park, G.-Y. Kim, I.-W. Choi, N. D. Kim, H. J. Hwang, Y.-W. Choi and Y. H. Choi, Inhibition of matrix metalloproteinase activities and tightening of tight junctions by diallyl disulfide in AGS human gastric carcinoma cells, J. Food Sci., 2011, 76, T105–T111 CrossRef CAS PubMed.
- M. Maeda, K. R. Johnson and M. J. Wheelock, Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition, J. Cell Sci., 2005, 118, 873–887 CrossRef CAS.
- S. Nakajima, R. Doi, E. Toyoda, S. Tsuji, M. Wada, M. Koizumi, S. S. Tulachan, D. Ito, K. Kami and T. Mori, N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma, Clin. Cancer Res., 2004, 10, 4125–4133 CrossRef CAS PubMed.
- T. Tsukamoto and S. K. Nigam, Cell-cell dissociation upon epithelial cell scattering requires a step mediated by the proteasome, J. Biol. Chem., 1999, 274, 24579–24584 CrossRef CAS.
- K. Suyama, I. Shapiro, M. Guttman and R. B. Hazan, A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor, Cancer Cell, 2002, 2, 301–314 CrossRef CAS.
- R. B. Hazan, R. U. I. Qiao, R. Keren, I. Badano and K. Suyama, Cadherin switch in tumor progression, Ann. N. Y. Acad. Sci., 2004, 1014, 155–163 CrossRef CAS PubMed.
- S. Kaufhold and B. Bonavida, Central role of Snail1 in the regulation of EMT and resistance in cancer: a target for therapeutic intervention, J. Exp. Clin. Cancer Res., 2014, 33, 62 CrossRef PubMed.
- V. Pozharskaya, E. Torres-González, M. Rojas, A. Gal, M. Amin, S. Dollard, J. Roman, A. A. Stecenko and A. L. Mora, Twist: a regulator of epithelial-mesenchymal transition in lung fibrosis, PLoS One, 2009, 4, e7559 CrossRef PubMed.
- S. Howard, T. Deroo, Y. Fujita and N. Itasaki, A positive role of cadherin in Wnt/β-catenin signalling during epithelial-mesenchymal transition, PLoS One, 2011, 6, e23899 CrossRef CAS PubMed.
- C. J. Gottardi and B. M. Gumbiner, Distinct molecular forms of β-catenin are targeted to adhesive or transcriptional complexes, J. Cell Biol., 2004, 167, 339–349 CrossRef CAS PubMed.
- M. Boutros and M. Mlodzik, Dishevelled: at the crossroads of divergent intracellular signaling pathways, Mech. Dev., 1999, 83, 27–37 CrossRef CAS PubMed.
- P. Polakis, Wnt signaling and cancer, Genes Dev., 2000, 14, 1837–1851 CAS.
- J. R. Miller, B. Baker, B. Thomas, S. W. Singer and J. Banfield, Genome Biol, Wnts, 2002, 3, 3001 Search PubMed.
- K. Hughes, E. Nikolakaki, S. E. Plyte, N. F. Totty and J. R. Woodgett, Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation, EMBO J., 1993, 12, 803–808 CrossRef CAS PubMed.
- P. A. Lochhead, R. Kinstrie, G. Sibbet, T. Rawjee, N. Morrice and V. Cleghon, A chaperone-dependent GSK3β transitional intermediate mediates activation-loop autophosphorylation, Mol. Cell, 2006, 24, 627–633 CrossRef CAS PubMed.
- A. Cole, S. Frame and P. Cohen, Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3β (GSK3β) in mammalian cells is an autophosphorylation event, Biochem. J., 2004, 377, 249 CrossRef CAS PubMed.
- C. Sutherland, I. A. Leighton and P. Cohen, Inactivation of glycogen synthase kinase-3β by phosphorylation: new kinase connections in insulin and growth-factor signalling, Biochem. J., 1993, 296, 15–19 CrossRef CAS PubMed.
- R. Dajani, E. Fraser, S. M. Roe, N. Young, V. Good, T. C. Dale and L. H. Pearl, Crystal structure of glycogen synthase kinase 3β: structural basis for phosphate-primed substrate specificity and autoinhibition, Cell, 2001, 105, 721–732 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8fo00246k |
| This journal is © The Royal Society of Chemistry 2019 |