
Occurrence and formation of incidental metallic Cu and CuS nanoparticles in organic-rich contaminated surface soils in Timmins, Ontario†
Haley
Mantha
a,
Michael
Schindler
 *ab and
Michael F.
Hochella
cd
*ab and
Michael F.
Hochella
cd
aDepartment of Chemistry, Laurentian University, Sudbury, ON, Canada. E-mail: mschindler@laurentian.ca
bDepartment of Earth Sciences, Laurentian University, Sudbury, ON, Canada
cNanoEarth, Department of Geosciences, Virginia Tech, Blacksburg, VA 24061, USA
dEnergy and Environment Directorate, Pacific Northwest National Laboratory, Richland, WA 99352, USA
First published on 23rd November 2018
Abstract
This study investigates the fate of Cu in organic-rich soils contaminated by mining related activities at the Timmins Kidd Creek metallurgical site, Ontario, Canada. The surficial soil layers (top 5 cm) contain on average 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm Cu, predominantly in the form of Cu–Fe-sulfide particulate matter (PM) attached to organic residues. The sequestration of Cu by organic material (OM) is investigated with a combination of focused ion beam (FIB) technology and transmission electron microscopy (TEM). FIB sections are extracted from the interior of OM as well as along the interface between OM and a Cu–Fe sulfide grain. Nanoscale analyses show that Cu occurs as incidental metallic Cu and covellite (CuS) nanoparticles (NPs) within the OM. These NPs can occur along the interface towards attached Cu–Fe sulfide grains (e.g. mooihoekite Cu9Fe9S16, talnakhite Cu9(Fe,Ni)8S16), in remote pore spaces in the interior of the OM, and in association with magnetite precipitates within the OM. These occurrences indicate that Cu is sequestered by OM through various redox and precipitation mechanisms, induced by ferrous iron or humic substances. This sequestration process is thus similar to the formation of supergene Cu-ore deposits where the dissolution of primary ore minerals (e.g. chalcopyrite CuFeS2) and the subsequent precipitation of secondary sulfides (chalcocite Cu2S, covellite) result in an enrichment of Cu. Furthermore, the extraction of the colloidal (mobile) fraction of the upper 1 cm of the studied soil (through column experiments and ultra-centrifugation) indicate that Cu-bearing colloids are predominantly composed of incidental covellite NPs embedded within colloidal OM. The speciation of Cu within organic residues and colloids indicates that Cu is recycled in surficial organic-rich soil through repetition of (a) sequestration processes within OM, (b) decomposition of OM resulting in the release of Cu-bearing incidental NPs, and (c) re-adsorption to newly formed OM as solutes or incidental NPs.
000 ppm Cu, predominantly in the form of Cu–Fe-sulfide particulate matter (PM) attached to organic residues. The sequestration of Cu by organic material (OM) is investigated with a combination of focused ion beam (FIB) technology and transmission electron microscopy (TEM). FIB sections are extracted from the interior of OM as well as along the interface between OM and a Cu–Fe sulfide grain. Nanoscale analyses show that Cu occurs as incidental metallic Cu and covellite (CuS) nanoparticles (NPs) within the OM. These NPs can occur along the interface towards attached Cu–Fe sulfide grains (e.g. mooihoekite Cu9Fe9S16, talnakhite Cu9(Fe,Ni)8S16), in remote pore spaces in the interior of the OM, and in association with magnetite precipitates within the OM. These occurrences indicate that Cu is sequestered by OM through various redox and precipitation mechanisms, induced by ferrous iron or humic substances. This sequestration process is thus similar to the formation of supergene Cu-ore deposits where the dissolution of primary ore minerals (e.g. chalcopyrite CuFeS2) and the subsequent precipitation of secondary sulfides (chalcocite Cu2S, covellite) result in an enrichment of Cu. Furthermore, the extraction of the colloidal (mobile) fraction of the upper 1 cm of the studied soil (through column experiments and ultra-centrifugation) indicate that Cu-bearing colloids are predominantly composed of incidental covellite NPs embedded within colloidal OM. The speciation of Cu within organic residues and colloids indicates that Cu is recycled in surficial organic-rich soil through repetition of (a) sequestration processes within OM, (b) decomposition of OM resulting in the release of Cu-bearing incidental NPs, and (c) re-adsorption to newly formed OM as solutes or incidental NPs.
Environmental significanceThe deposition of metal(loid)-bearing particulate matter (PM) on soils adjacent to and downwind from a contaminant source is a world-wide environmental issue. The exponential decrease in the concentration of metal(loid)s with depth in these soils suggests that they are retained by inorganic and organic constituents in the surficial layers of these soils. An in-depth nanoscale investigation of the interaction of Cu-bearing PM with organic material (OM) in Cu-contaminated soils reveals for the first time that even under oxic conditions in surficial soil layers, Cu can be sequestrated by OM in the form of metallic Cu and covellite (CuS) incidental nanoparticles (NPs). Furthermore, this study provides insights into potential mobilization mechanisms for Cu in organic-rich soils as it identifies covellite NPs embedded within mobile organic colloids. |
Introduction
Metal-bearing particulate matter (PM) released from mining related activities are often deposited onto surrounding soils, resulting in elevated concentrations of metals. For example, in 2015, industrial activities just within Canada resulted in the release of approximately 3 million tonnes of PM and aerosols to the atmosphere.1 Upon deposition, particulates typically remain in the surficial layers of soil, however environmental conditions induce weathering and subsequently the release of metal(loid)s from altered PM surfaces. Processes that drive PM weathering are dependent on the atmospheric and soil matrix conditions, pore water volume, and composition as well as the chemical and mineralogical composition of the particulates.2 This study reveals insights into previously unknown sequestration and transport processes of Cu released during the weathering of Cu-bearing PM in an organic-rich surficial soil layer in proximity to the former Kidd Creek metallurgical site in Timmins, Ontario, Canada. The ore processed at this location predominantly contained chalcopyrite (CuFeS2), bornite (Cu5FeS4), pyrite (FeS2), pyrrhotite (Fe(1−x)S x = 0–0.2), sphalerite (ZnS), and galena (PbS).3The majority of studies assessing deposition and fate of PM are primarily focused on spatial distributions or total and bioavailable metal concentrations within soils surrounding a specific contaminant source.4–9 Although these studies are important, they often do not specifically characterize the PM or determine the speciation of the metals within particulates or secondary phases. This characterization is required to understand how metal(loid)s are sequestered and transported within soils, allowing for a better comprehension on the level of contamination as well as providing the capability of designing targeted reclamation strategies. Specific, in-depth characterization of the fate of metal(loid)s in surficial layers of soils and altered rocks can be accomplished using electron beam techniques such as scanning electron microscopy (SEM) or transmission electron microscopy (TEM). These methods were successfully applied to identify and characterize emitted particulate matter,5,6,10–12 its weathering products,5,6 and sequestration mechanisms of released metal(loids) by mineral surface coatings and mineralized organic matter.11,13–16 Classical sequential-extraction methods of organic-rich soils have suggested that organic material can be an important (even dominant) sorbent for metals in the surface horizons of soils, dependent on the type of organic matter, and the ratio between the total surface areas of organic matter and minerals.8,17,18 However, synchrotron-based spectroscopy and diffraction studies have not provided a uniform picture of the fate of metal(loid)s in surficial soils containing minerals and organic material (OM). For example, some studies state that Cu predominantly sorbs to OM,19 whereas other work reports a close association of Cu with Fe-(hydr)oxides.20 The latter observations indicate an uncertainty with respect to the sequestration of metal(loid)s by OM in soil horizons containing both organic and inorganic material.
To address this gap in knowledge, the objectives of this study are to characterize the speciation of Cu within organic matter around the Kidd Creek site using a combination of focused ion beam technology (FIB) and TEM, rather than bulk analytical methods such as sequential extraction and synchrotron-based spectroscopy. This study demonstrates that the former analytical methods allow the characterization of nanometer-size phases within organic matter. More specifically it shows for the first time that Cu-bearing PM is transformed on the surface and within pore spaces of organic material into secondary incidental nanoparticles (NPs) that are composed of metallic Cu and Cu-sulfides. It further shows that Cu can be transported via the colloidal fraction as incidental Cu-sulfide NPs embedded within larger organic colloids.
In this study, organic material is used to describe the overall organic fraction (i.e. organic residues and matter) whereas the term organic matter defines decomposed organic material (i.e. humic substances). Similarly, dissolved organic matter (DOM) is defined as the fraction of organic matter that is capable of passing through a 0.45 μm filter, and therefore is no longer considered particulate organic matter. Additionally, in this study, the colloidal fraction is defined as the fraction that deposits during centrifugation, colloids are considered particles with diameters between 50 and 450 nm and the term nanoparticle is used when particles have diameters smaller than 50 nm.
Materials and methods
Background information on the Kidd Creek metallurgical site
The Kidd Creek metallurgical site, located within the city limits of Timmins, Canada, was in operation for 30 years, closing in 2010. The ore was shipped by train from the mine site (27 km away) to the metallurgical site for processing. The Mitsubishi copper smelting process was employed at start-up with furnaces operating around 1200–1300 °C.21 From 2002–2009, the average amount of total PM (<100 μm) released to the atmosphere was 844 tonnes per year.22Sample collection
Surficial soil samples (∼upper 5 cm) were collected in October 2016 from various sites surrounding the former Kidd Creek smelter. The selected sites exhibited well-developed and undisturbed soil profiles. The surficial sample from site 1 was thoroughly characterized on the nano- to micro-meter scale (Fig. 1). Site 1, located 10 m from the highway within a birch stand with a grassy understory, exhibits 5 cm of an LFH layer with a 10 cm Aheg layer (Fig. S1†). Site 2 and 3 were used as control sites, with site 2 exhibiting a 4 cm LFH layer, 6 cm Aheg layer, and a 5 cm Aeg layer and site 3 exhibiting a 15 cm Ah layer (Fig. S2 and S3†). Sampling at site 1 continued in May 2017, when samples were collected at 1 cm increments to a depth 10 cm. All samples were transported to the lab in a cooler, dried at 80 °C, sieved (<1.4 mm), and stored under dry conditions.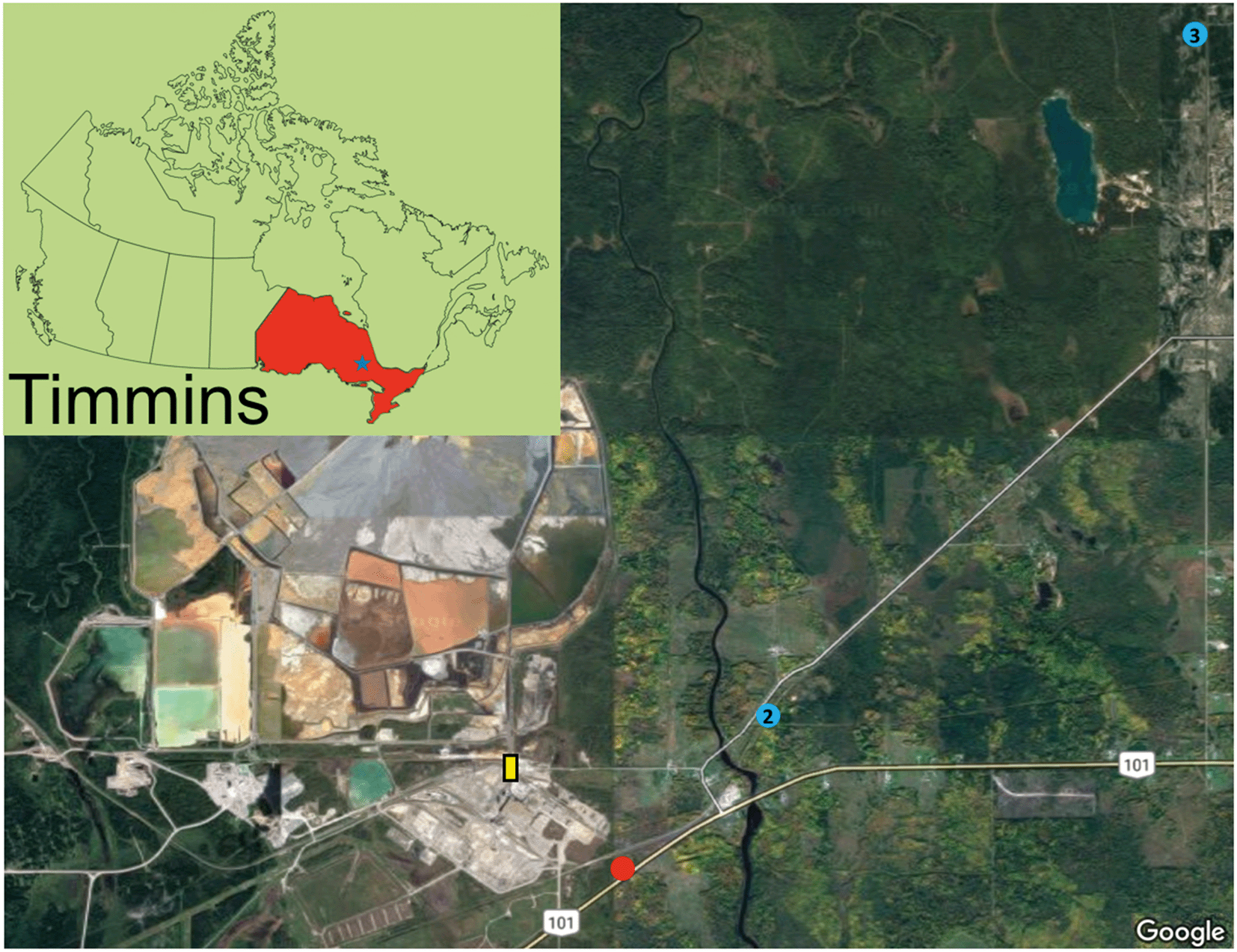 | ||
| Fig. 1 Satellite image of the area surrounding the Kidd Creek metallurgical site; the main sample location is indicated by the red dot and the two control sites are designated and labelled by the blue dots; the yellow rectangle indicates where the copper smelter was located prior to its removal; an inset image shows the location of Timmins within Ontario. | ||
Chemical analysis
Soil pH was measured by mixing 2 g of soil with 10 mL of a 0.01 M CaCl2 solution (1:5 soil:solution ratio). After stirring multiple times over a 30 minutes period, the mixture was allowed to settle for 15 minutes before the pH measurement. Oxidation–Reduction potential (ORP) was measured in triplicate analysis from five separate wet surficial samples, taken within a 10 × 10 m area at site 1. For the control sites (site 2 and 3), the ORP was measured on dried and sieved samples. Samples were prepared by mixing 2 g of soil with 10 mL of distilled water, stirred for 5 minutes and allowed to settle for 30 minutes. Samples were then measured using a Ag/AgCl reference electrode which was calibrated with an ORP standard. The uncertainty of the measured soil pH and reduction potential are 0.2 units and 10 mV, respectively.
Total carbon and sulfur within the 0–5 cm fraction of site 1 was measured in the Ontario Geological Survey Geoscience Laboratories (Geo Labs) with a LECO CS844. In short, approximately 0.2 g of soil was combusted in a stream of purified O2 gas and passed over a heated catalyst, oxidizing total S and C to SO2 and CO2, respectively, which are then detected by two non-dispersive infrared cells.23 Total soil digestion and inductively coupled plasma-mass spectrometry (ICP-MS) measurements of the 0–5 cm fractions were completed by Testmark Laboratories with a ELAN DRC II (Perkin Elmer) instrument. Soils were digested using a concentrated HNO3/HCl mixture, as per the BC SALM protocol24 and then analyzed using the SW846-6020A method for ICP-MS. Chemical analyses of the samples collected in the depth profiles (ten samples for the first 10 cm and one sample for the 10–15 cm depth) were analyzed at the Geo Labs, Sudbury, ON, Canada. Samples were prepared in a reverse aqua-regia digest following the method of Burnham25 and analyzed with an iCAP Q ICP-MS (Thermo Scientific).
Preparation of soils for electron microscopy and focused ion beam extraction
The interaction between PM and OM occurs at the nanoscale and can be studied best through the extraction of focused ion beam (FIB) sections along their interfaces. However, extraction and subsequent thinning of a FIB section requires that the area to be extracted is free of fractures or larger pore spaces. From our experience, these topographic homogenous areas can be best located with SEM on surfaces of sand-size grains. Hence, the interaction of PM with OM, including the chemical characterization of PM, were mainly investigated in the fine to coarse sand size fraction (180 μm–1.4 mm) of the surficial soil layer of site 1. Samples of both the clay to sand size fraction (<180) and fine sand to coarse sand size fraction (180 μm–1.4 mm) were embedded into epoxy pucks and then carefully polished to prevent damage or loss of OM.The diffusion of epoxy into pore spaces stabilizes interfaces between OM and attached minerals which allow efficient extraction of sections along these interfaces with FIB technology (see below). Some mineral phases may alter under ambient conditions during storage, which may have been limited through the storage of the samples under dry conditions. Significant mineralogical and chemical transformations can occur under high vacuum and exposure to electron- and ion-beams during FIB extraction and TEM usage. Examples of these alterations include the potential for either the removal of phases, phase transformation (e.g. dehydration of hydrous minerals), or deposition of (semi)metallic Ga and Pt NPs in soft materials. These processes have been studied in depth26–28 and therefore artifact fabrication is carefully considered and caution is taken so that the significance of results is not overstated.
X-ray powder diffraction, scanning electron microscopy, and electron microprobe analysis
Powder X-ray diffraction (XRD) was done with an automated Philips PW3050/60 and PW 1729 X-ray diffractometers using Co Kα radiation (1.79 Å) at a voltage and current of 40 kV and 30 mA, respectively. X-ray diffraction patterns from well-mounted powdered samples were collected over a scan range of 5–65° 2θ with a step size of 0.02° 2θ and a dwell time of 2 s. Chemical analyses of PM were performed with a Cameca SX50 electron microprobe. Data was collected by wavelength-dispersion spectroscopy (WDS) with counting times of 30 s on the peak and 10 s on the background. The operating voltage was 20 kV, beam current of 20 nA, and a focused beam of 20 nA. Data reduction was done with a PAP routine29 using the XMAS software package. Chalcopyrite was used as the standard for Cu (Lα), Fe (Kα), and S (Kα). Characterization of the (semi-quantitative) chemical composition of organic material was accomplished using scanning electron microscopy (SEM) with a JEOL 6400 SEM, operating with an accelerating voltage of 20 kV and a beam current of 1 nA, in combination with energy dispersive X-ray spectrometry (EDS).Focused ion beam technology and transmission electron microscopy
An interface between a sulfide grain and organic residue (Fig. 2a and b) and the interior of a Cu-rich organic residue (Fig. 2c and d) were selected for extraction of FIB sections with a FEI Helios 600 NanoLab FIB (March, July 2017). The sections were subsequently lifted using a platinum gas-glue, thinned to electron transparency by ion gas milling (Ga+ ions) and mounted on a molybdenum holder. Transmission electron microscopy was conducted with a JEOL 2100 transmission electron microscope (a field thermionic emission analytical electron microscope) at the Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure (NanoEarth) (April, August 2017). Measurements were taken with an accelerating voltage of 200 kV and a beam current of approximately 107 mA. EDS point analyses and maps were acquired in STEM mode with JEOL bright field (BF) and JED-2300T EDS detectors. Selected area electron diffraction (SAED) patterns were acquired using a Gatan Orius SC200D detector. Nanoparticles and larger crystals were identified using a combination of SAED, fast Fourier transformations (FFT) of lattice fringes, and EDS-STEM chemical distribution maps. | ||
| Fig. 2 SEM images in backscattering mode (BSE) of organic residues from site 1 within a 0–5 cm coarse fraction chosen for nanoscale analysis. (a and b) Extraction site of FIB section 1 (c and d) extraction site of FIB section 2; open rectangles in (a) and (c) indicate the locations of the areas shown in (b) and (d); filled rectangles in (c) and (d) indicate the locations of the FIB sections. | ||
Colloid preparation and transmission electron microscopy
Soil leachates were collected during a column leaching experiment. Hollow, plastic tubes were filled with approximately 0.8 g of the dried and sieved (<1.4 mm) 1 cm fraction with a small layer of inert and plastic beads on the top and bottom of the soil sample. A 0.01 μM CaCl2 solution was passed through the column at an approximate rate of 3 mL h−1 until 30 mL were collected in a tube below the column. A fraction of the leachate was centrifuged using a Beckman Coulter JA-17 centrifuge rotor 8000 rpm at 480 minutes which, according to the Stokes equation, will remove all spherical particles from solution with a density similar to metallic Cu (8.96 g cm−3) and a diameter greater than 12 nm. A second fraction (neat) was filtered using a 0.45 μm membrane. Neat and centrifuged leachates were acidified prior to analysis by ICP-MS to determine the total and dissolved metal concentration. Samples were acidified with a 1% HNO3 and analyzed on an ELAN DRC II (Perkin Elmer) instrument at the Geo Labs. Concentrations of the metal(loid)s in the colloidal fraction (>12 nm) were calculated by subtracting their concentrations in the solutions after centrifugation from the total metal(loid) concentration (which has not been centrifuged).To prepare the colloids for TEM analysis, soil leachates were centrifuged using a Sorvall ST16 centrifuge equip with a TX-400 swing-bucket rotor. A molybdenum TEM grid (400 mesh lacey carbon, 100 μm) was fixed to an epoxy support at the bottom of the centrifuge tube and the tube was then filled with 2 mL of the leachate. According to Stokes equation, spherical particles greater than 16 nm and a density similar to metallic Cu (8.96 g cm−3) deposit onto the grid during centrifugation for 8 hours at a speed of 5000 rpm. The colloidal fraction on the TEM grid was examined with a field emission TEM FEI Talos F200x at the Manitoba Institute of Materials. Imaging and SAED was performed with an accelerating voltage of 200 kV in bright and dark field mode with a 16 MB Ceta camera and a Fischicone high angle annular dark field (HAADF) detector. Compositional analysis was performed in STEM-EDS mode with 4 SDD detectors.
Results
Bulk soil chemistry
The brownish black humic gley soils are oxic, slightly acidic and organic-rich soils with a pH/Eh of 5.0/726 mV at the time of sampling (Table 1). At site 1 the concentrations of Cu, Pb, and, Zn exponentially decrease with depth from 9220 ppm (Cu), 447 ppm (Pb), and 3280 ppm (Zn) at depth 0–1 cm to 265 ppm, 38 ppm, and 964 ppm at depth 9–10 cm, respectively (Table S1 and Fig. S4†). The concentrations of these metals also decrease with distance from the contaminant source; the concentrations of Cu, Pb, and Zn in the surface layers at the two control sites (sites 2 and 3) are approximately 100 ppm (Table 1). These significant drops in metal concentrations with depth and distance (Table 1, S1 and Fig. S4†) are typical for soils contaminated by atmospheric depositions from a point source, that being the former Kidd Creek smelter complex.8,30| Site | Trace metal concentrationa (ppm) | Physiochemical parameters | Distance to former smelter complex (km) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cu | Zn | Pb | Fe | pH | Eh (mV) | Total C (wt%) | Total S (wt%) | ||
| a Concentration reported in ppm except where noted. | |||||||||
| 1 | 13000 |
8210 | 685 | 2.07% | 5.0 | 726 | >110 | 0.411 | 1.3 SE |
| 2 | 117 | 123 | 31 | 4135 | 4.8 | 626 | — | — | 2.5 ENE |
| 3 | 92 | 163 | 15 | 5720 | 5.1 | 613 | — | — | 7.5 NE |
Mineralogical composition
The fine to coarse sand size fraction (>180 μm) of the surficial soil layer at site 1 (0–5 cm) is composed predominantly of OM with minor amounts of quartz (SiO2) and traces of digenite (Cu9S5), talnakhite (Cu9(Fe,Ni)8S16), and magnetite (Fe3O4, Fig. S5†). The finer clay to sand size fraction (<180 μm) contains mainly quartz, hematite (Fe2O3), and albite (NaAlSi3O8) (Fig. S6†). Scanning electron microscope analyses indicate that many organic residues in the coarse fraction contain dispersed Cu- and Si-bearing particles on their surfaces and within their pore system (Fig. S7†). Electron microprobe analyses (n = 63) indicate that the majority of grains attached to organic residues are composed of Fe- and Cu–Fe-sulfides with minor amounts of Fe-oxides (Fig. S8†). The fine fraction lacks organic residues containing Cu-bearing phases but contains a higher proportion of silicified organic grains, spherical smelter-derived particulates and angular silicate-based particles (Fig. S9†).Chemical and mineralogical features along the interface of a Cu–Fe-sulfide and an organic residue
The first FIB section was extracted along the interface of a Cu–Fe-sulfide (mooihoekite, Cu9Fe9S16) grain attached to a highly porous organic residue (Fig. 2a, Fig. S10 and S11†). The organic material around the sulfide grain contains elevated concentrations of Cu in the range of 0.5–2.0 wt%, which can be recognized as brighter areas in backscattering electron images (BSE, Fig. 2b).Fig. 3 indicates that the FIB section has been severely damaged but contains an intact sulfide grain and residuals of the organic material. It also indicates the locations of the areas described in greater detail below.
 | ||
| Fig. 3 Bright field (BF) STEM image of the entire FIB section 1. Numbered rectangles indicate the locations of the TEM images shown in the Fig. 4–7. | ||
Chemical analyses, STEM imaging, and electron diffraction patterns indicate the presence of a porous magnetite/maghemite (Fe3O4/Fe2O3) layer that formed along the exterior of mooihoekite and occurs along the interface of the organic material (Fig. 4 and S12†). Magnetite and maghemite are isostructural and therefore cannot be distinguished with electron diffraction. The diameter of the magnetite/maghemite layer varies between 30 and 160 nm. Elongated pore spaces occur throughout the layer with diameters in the range of 3–9 nm and with orientations parallel and perpendicular to the interface (Fig. 4b). Fragments of magnetite/maghemite also occur within OM in close proximity to the interface between sulfide and organic residue (Fig. 5b and S13†).
 | ||
| Fig. 4 (a) STEM-EDS chemical distribution map for Fe, O and S along the interface between the sulfide grain (blue) and organic material (red), indicating the occurrence of an iron oxide layer in between (yellow); (b) dark field (DF) STEM image of the Fe oxide layer displaying its high porosity; red arrows indicate pore spaces in different orientations and orange dashed lines indicate interfaces between mooihoekite and magnetite/maghemite and magnetite/maghemite and organic material. | ||
 | ||
| Fig. 5 (a, c and d) BF TEM images of Cu-bearing magnetite/maghemite fragments near the interface towards the sulfide, (b) STEM-EDS chemical distribution maps of Cu (green), Fe (blue) and C (red). | ||
A Cu-rich area containing minor S and Fe occurs on the edge of the sulfide grain where no magnetite/maghemite surface layer occurs. Fast Fourier transformation patterns of lattice fringes and SAED patterns indicate the presence of the prominent d-spacings of 2.11 and 1.83 Å, however more than 10 other d-spacings occur in the FFT and SAED patterns suggesting the presence of an unknown structural derivative of a Cu-rich sulfide (Fig. S14†). Adjacent to the Cu-rich area occur fragments of boehmite (γ-AlO(OH)) and diaspore (α-AlO(OH)) which are embedded in the organic material (Fig. S15†). Copper-bearing NPs < 2 nm occur within the organic material along the interface of the Cu-rich area and the aluminum hydroxide fragments (Fig. S16†). Fast Fourier transformations of lattice fringes of these NPs indicate interplanar spacings of 2.10, 1.80, and 1.25 Å, matching the prominent (111), (200), and (220) d-spacings of metallic copper. However, various other d-spacings also occur suggesting that some of the NPs might be composed of a Cu-oxide or -sulfide.
In the interior of the FIB section occur numerous porous, carbon-based features which are often associated with magnetite/maghemite. Copper occurs as NPs within both organic material and magnetite/maghemite (Fig. 5a–d). These NPs are a mixture of covellite (CuS), a Cu2+/1+ bearing phase, and metallic Cu NPs. Covellite (yellow) and metallic Cu (green) are also closely associated in pore spaces of the OM further from the interface and as such, not in association with magnetite/maghemite (Fig. 6). Here, metallic Cu appears to be attached to spherical NPs of covellite (Fig. 6).
 | ||
| Fig. 6 (a) DF STEM image of an inclusion of covellite and metallic copper within a pore space in organic material approximately 2 μm from the sulfide grain (b) corresponding STEM-EDS chemical distribution map of Cu (green) and S (red); the covellite and metallic Cu inclusions are coloured in yellow and green and are labelled as Ct and Cu, respectively. | ||
Approximately 1 μm from the Cu–Fe-sulfide-OM interface, a layer of magnetite/maghemite is embedded within the OM (Fig. 7a and S17†). On both sides of the iron oxide layer, Cu-bearing NPs occur predominantly in the surrounding OM, and also in a few cases on the surface of the magnetite. Selected area electron diffraction patterns indicate the presence of both tenorite (CuO) and metallic Cu NPs (Fig. S18†).
 | ||
| Fig. 7 (a) BF TEM image of a magnetite/maghemite layer within organic material (b) corresponding STEM-EDS chemical distribution map of Fe (blue), Cu (green) and C (red); magnetite/maghemite, porous organic material and metallic Cu NPs are indicated in blue, red and green, respectively; an arrow indicates the direction where the main sulfide is located. | ||
Copper NPs within the interior of an organic residue in a second FIB section
A second FIB section was extracted from the interior of an organic residue (Fig. 2c and d) as the organic matrix in the first FIB section was severely damaged during the extraction. Numerous Cu-sulfide and silicate grains are attached to the organic residue (Fig. 2c). The area around the FIB extraction contains higher Cu concentrations than its surroundings. The organic matrix of the extracted FIB is more homogeneous in terms of mineralogical and chemical features, as well as texture relative to the matrix in FIB section 1 (Fig. 3 and 8a). The average Cu-concentrations in the section is 2 wt% (on the basis of STEM-EDS). Scanning-TEM images, SAED patterns, and STEM-EDS chemical distribution maps for Cu (green) and C (red) indicate the occurrence of metallic Cu NPs (Fig. 8b–d and S19†). These NPs are relatively uniform in size (6–20 nm) and have an average diameter of 9 ± 3 nm (n = 330). These size ranges do not include aggregates of NPs, which can be composed of up to five spherical NPs (Fig. 8e). | ||
| Fig. 8 (a) BF STEM overview of FIB section 2 extracted from the interior of an organic residue; (b and c) BF-TEM and corresponding STEM-EDS chemical distribution maps for Cu (green) and C (red) indicating the occurrence of metallic Cu NPs (green) within organic material (red); (d and e) BF high resolution TEM images of individual and agglomerated metallic Cu nanoparticles; the numbers in (e) indicate the number of agglomerated NPs. | ||
Chemical and mineralogical composition of the colloidal material
The total concentration of Cu in the soil leachate from site 1 (0–1 cm depth) is 6630 ppb. The proportions of the dissolved and colloidal fraction of Cu in the leachate are 88.6% (5872 ppb) and 11.4% (758 ppb), respectively. TEM analysis of the colloidal fraction indicates that the bulk of the colloidal material is composed of carbon-based material with particle sizes often greater than 200 nm. These larger colloids often contain Copper-bearing NPs (Fig. 9a and b). Colloids composed exclusively of inorganic based NPs include unidentified titanium oxide NPs with surface precipitates containing Cu and S (Fig. 9c and d and S20†). | ||
| Fig. 9 Dark field STEM images and corresponding STEM-EDS chemical distribution maps (a and b) covellite NPs embedded in carbon-based material with Cu in green, S in red and C in blue (c and d) Cu–S-bearing surface precipitates on rounded titanium oxide crystals with Cu in red and Ti in blue. | ||
Scanning-TEM-EDS chemical distribution maps and fast Fourier transformations of lattice fringes indicate that the majority of Cu-bearing NPs in carbon-based colloids (Fig. 10a and b) are composed of covellite with two NPs identified as anilite (Cu7S4) and chalcocite (Cu2S) (Fig. S21†). Nanoparticles of covellite are mainly spherical and have diameters in the range of 15–35 nm. They are often composed of smaller NPs which depict lattice fringes with d spacings of 3.08 (102), 2.73 (006), 1.88 (110), and 1.66 (200) Å. In some cases, the NPs are agglomerated in a random fashion with lattice fringes in different orientations whereas in others, fringes depict the same orientation (100, 200), with the exception of newly attaching NPs (Fig. 10c and d). In addition to lattice fringes with different orientations, attachment of NPs with diameters of approximately 1–4 nm to larger particles cause a distortion of their spherical shapes (Fig. 10e). Spherical NPs identified as anilite and chalcocite occur in close proximity (<5 nm) to each other (Fig. 10f) and have diameters of approximately 26 and 16 nm, respectively.
 | ||
| Fig. 10 Cu-bearing NPs in organic colloids within the leachate of the 0–1 cm fraction of the soils at site 1 (a and b) BF high resolution TEM image and corresponding STEM-EDS chemical distribution maps with Cu in red, S in green and C in blue of a covellite nanoparticle (in yellow) partially embedded into organic material (blue), (c) and (d) BF high resolution TEM images of covellite nanoparticles with nano-domains of different orientation, (e) TEM image depicting the distortion of the spherical shape of a covellite nanoparticle most likely due to the attachment of new nanoparticles; (f) high resolution TEM image of an anilite (An) nanoparticle in close proximity to a chalcocite (Ch) nanoparticle with nano-domains of different orientations. In (a), (c), (d) and (f) white parallel lines indicate lattice fringes and white curved lines indicate boundaries between NPs or nano-domains of different orientations; the fringes are labelled with their corresponding (hkl) notations. | ||
Discussion
Examinations of a soil sample at the nanoscale using a combination of FIB and TEM allows for a site-specific, in-depth chemical and mineralogical analysis of an area of interest. For example, the Cu–Fe-sulfide in FIB section 1 (Fig. 2a) was initially identified as chalcopyrite and assumed to attach directly onto the OM (using SEM). However, the nanoscale investigations in this study indicate the occurrence of the sulfide mooihoekite (with a similar composition relative to chalcopyrite) and the attachment of the mooihoekite grain to OM via a magnetite/maghemite surface layer. The TEM studies show that sequestration of Cu by OM can occur through metallic Cu and covellite formation, though both phases were not identified using SEM and EMPA.However, care must be taken to not overstate the significance of findings discovered at the nanoscale. For example, a FIB section is typically only 5 × 20 μm, and the high costs and skills required to prepare FIB sections and operate the TEM limit the preparation of many subsamples at the nanoscale. Furthermore, the soil sample collected for this TEM study originates from only one location in close proximity to the former Kidd Creek smelter. This area was chosen to ensure the presence of anthropogenic Cu-bearing phases. Therefore, it is not realistic to claim that the results presented in this study are representative for the speciation and mobilization of Cu in all organic-rich soils in the Timmins area. For example, the speciation of Cu may differ at sites which are (a) located farther from the former smelter (lower Cu concentrations) or (b) characterized by different environmental conditions (i.e. wetlands or non-forested areas). However, this study provides for the first-time insights into complex nanoscale dissolution–precipitation–reduction processes along the interfaces of PM, Cu-bearing pore solutions, and OM; processes that cannot be visualized using bulk analytical methods such as sequential extraction or synchrotron-based spectroscopy.
Origin of the particulate matter observed in the surficial soil layer of site 1
Atmospheric deposition of PM derived from mining-related activities onto surficial soils can be detrimental to the environment. In this study, Cu-bearing PM is found to be associated with organic residues in the 0–5 cm coarse fraction of soils sampled near the former Kidd Creek metallurgical site (Site 1). The observed particulates have angular shapes, are heterogeneous in terms of their mineralogical and chemical composition, and lack cooling or crystallization features such as rims or idiomorphic crystals in a glass matrix.31 Contrary, smelter-derived PM commonly have a spherical shape and display high-temperature features such as dendritic, skeletal, tabular, and porphyritic patterns.6 The latter particulates occur predominantly in the fine fraction (<180 μm) but have not been identified on the surface or within organic residues in the coarse fraction (Fig. S6†).The minerals mooihoekite (identified with TEM) and talnakhite (identified with XRD) can occur in close association with chalcopyrite,32 one of the major ore minerals at the Kidd Creek complex. The former two sulfides are metal-rich structural derivatives of chalcopyrite and form a solid solution with the latter mineral at high T (≳500 °C).33 At low T (≲500 °C), talnakhite, mooihoekite, and haycockite (Cu4Fe5S8) form stable minerals with distinct stoichiometric compositions and well-defined structures.34 These minerals are, however, difficult to distinguish from chalcopyrite using optical microscopy.32 Hence, the textural and mineralogical composition of PM on the surfaces of organic residues suggest that particulates composed of Cu–Fe-sulfides originated from waste piles and were wind-blown to site 1. Here, talnakhite and mooihoekite may have been either (overlooked) ore-forming minerals or phases formed during processing of the chalcopyrite-bearing ore.
Grains of digenite (traces identified with XRD) have not been observed on the surface of the organic matter (Fig. S8†). The mineral can form at lower T than Cu–Fe sulfides (as low as T = 105 °C)35 and may have formed during enrichment of Cu during alteration processes within the ore body or during processing of the chalcopyrite-bearing ore.
Alteration and attachment of the Cu–Fe-sulfide grains to organic material
The majority of the grains present in the <1.4 mm soil fraction are composed of partially decomposed organic material with attached Cu–Fe-sulfides. The latter phases predominantly occur as clay to silt-size grains (<75 μm) and therefore are expected to occur in the fine fraction after sieving and in the colloidal fraction after extraction. The predominant occurrence of these particles in the coarse sand-size fraction and their absence in the colloidal fraction suggests that (a) attachment is based on electrostatic forces rather than being a result of forced compression and (b) nanometer-size airborne Cu–Fe-sulfides are not stable in the oxic pore water at site 1.The attachment of sulfide grains to the organic residue via electrostatic forces can be understood when considering the point of zero charge (pzc) of these compounds. The typical pzc value of most organic based compounds in soil is 2 to 3.36,37 If the surfaces of the organic grains are not modified through mineralization processes, their surface should be negatively charged in the pH environment of the Timmins soils (pH = 5.0). Similarly, Cu–Fe-sulfides such as chalcopyrite have a pzc value of approximately 2.38 and their surface would be also negatively charged. Hence, the attachment of the sulfide grains to the negatively-charged organic residues required a positively-charged interlayer, which was identified in this study as a nanometer thick surface layer of magnetite/maghemite. The latter minerals or their Fe-(hydr)oxide precursors have commonly pzc values above 6.539 and are thus positively-charged at pH = 5. That being said, surfaces of minerals with pzc > 6.5 can be also negatively charged in weak acidic, organic-rich soils due to the presence of adsorbed organic molecules.40
Formation and properties of the magnetite/maghemite alteration layer
Sulfides are known to readily oxidize in the environment. The oxidation of sulfides exposed by mining activities can be accomplished through atmospheric oxidation prior to soil deposition or by electrochemical oxidation (pH/Eh dependent) after deposition. A combination of the two processes likely contributed to the formation of the magnetite/maghemite surface layer, as suggested by the lower solubility of Cu–Fe-sulfides relative to other sulfides.41,42 Studies on oxidation mechanisms of chalcopyrite indicate the formation of Fe-(hydr)oxide surface layers on its surface, though the Fe-(hydr)oxide minerals in these studies are not unequivocally identified.41,43 The first generation of alteration products on the surface of Fe-sulfide minerals is commonly composed of schwertmannite (under acidic conditions) and ferrihydrite (under weak acidic to alkaline pH conditions).44 The formation of these nanocrystalline phases follows Ostwald's rule that a solid phase with the lowest interfacial free energy forms first although it may not be the most stable by bulk thermodynamics. The second generation of Fe-sulfate or hydroxide phases may include jarosite at pH < 4 and goethite, magnetite, maghemite, and hematite at pH > 5.44 These phases commonly form through the re-arrangement of the Fe-oxide polyhedra in the precursor structure or through dissolution re-precipitation processes.45For example, the transformation of ferrihydrite to magnetite requires the dissolution of the former mineral, the partial reduction of Fe3+ under reducing conditions and the re-precipitation of magnetite.44,45 As such, a magnetite surface layer on mooihoekite could not have formed from a ferrihydrite precursor in contact with the observed oxic pore water and most likely developed after attachment to the organic residue. The solution that percolated through the ferrihydrite (or another precursor) layer before and after attachment was likely different in terms of composition and reduction potential. Prior to attachment the solution was most likely acidic and oxic (Table 1). After attachment parts of the solution had to pass through pore spaces of the organic residue and contained thus less dissolved O2 and some dissolved organic matter (DOM). The influx of electron donors such as DOM most likely induced the reduction of Fe3+ to Fe2+, promoting the transformation of the precursor into magnetite. This conclusion is also supported by the observation that a layer of magnetite/maghemite occurs within the OM (Fig. 7). Maghemite commonly forms through oxidation of magnetite, though it can also form from a ferrihydrite precursor under high temperature (>150 °C) or through the transformation sequence ferrihydrite–goethite–maghemite under alkaline conditions.46,47 However, the latter two conditions can be ruled out for the surficial weakly acidic soils at site 1.
As such, we propose the following sequences for the formation/transformation processes on the surface of the sulfide: (1) sulfide oxidation followed by the precipitation of ferrihydrite, (2) transformation of ferrihydrite to magnetite under reducing conditions, and (3) perhaps partial transformation of magnetite to maghemite through the oxidation of Fe2+.
During a mineral replacement reaction, coupling between the dissolution of a parent and precipitation of a daughter can be achieved when the rate-controlling step is the dissolution of the parent and when the activation energy barrier for the nucleation of the daughter is low.48 This is most likely the case during the alteration of mooihoekite (parent) to potentially ferrihydrite (daughter) as Cu–Fe-sulfides are commonly less soluble than other sulfides and as ferrihydrite has a lower interfacial free energy than other Fe-oxides (see above). Putnis48 emphasizes that (a) porosity in the product phase is commonly generated when there is a volume deficit reaction and (b) the porosity of the replacing phase allows the mass transfer of material from the solution reservoir to the reaction interface. The observed porosity in the alteration rim (Fig. 4b) suggests that the replacing phases of the first and second generation (ferrihydrite and magnetite) have smaller molar volumes than the phases to be replaced (sulfide and ferrihydrite). This is indeed the case: the molar volumes decrease from mooihoekite (Vm = 363 cm3 mol−1) to ferrihydrite (Vm = 167 cm3 mol−1) to magnetite (Vm = 44 cm3 mol−1). Additionally, molar ratios of parent to daughter minerals confirm this volume deficit (Table S2†).
Occurrence and formation of the observed Cu-bearing nanoparticles
Metallic Cu rarely occurs under natural environmental conditions. It occurs as metallic Cu NPs within the interlayer structure of biotite samples within oxidized rock from various copper porphyry deposits.49,50 Experimental studies show that metallic Cu NPs form in the interlayer of Fe-bearing micas such as biotite and illite through a reductive alteration processes involving Fe2+.51,52The formation of metallic Cu in the presence of OM occurs in bogs, wetlands, and floodplains. For example, metallic Cu can form as sub-micron particles and centimeter-sized masses in permanently flooded parts of a bog under anaerobic conditions.53,54 Experimental studies show that metallic Cu NPs form in contaminated floodplain soils along and within roots of two common wetland plant species through a combination of limited sulfur availability, copper-stressed bacteria (Gram-positive Clostridium sp.), Fe2+ in clays, or by endomycorrhizal fungi under anoxic conditions.49–54 Upon soil drainage (i.e. exposure to higher FO2) or the addition of biogenic sulfide, metallic Cu is commonly converted to Cu-bearing aqueous species or Cu-sulfide phases such as CuxS phases.55
In this study, we propose that the metallic Cu NPs identified in OM are products of the mobilization of Cu in the form of Cu1+/2+ aqueous species released during the alteration of the Cu–Fe-sulfide grains. The reduction of Cu is induced by Fe2+ or by redox-active functional groups present on the surface of humic substances (Fig. 11). In the following sections, possible mechanisms for the mobilization Cu1+/2+ aqueous species and formation of metallic Cu and covellite NPs are discussed.
Diffusion of Cu into the organic material
The replacement of the Cu–Fe-sulfide by Fe-(hydr)oxides after attachment of the sulfide grain may involve an influx of hydrogen and DOM-species and an outflux of H2S and Cu-bearing species. As such, the continuous mineral replacement reaction may be seen as the driving force for the diffusion of Cu through the porous Fe-(hydr)oxide layer into the porous OM. The transport of Cu through both porous entities might be facilitated by DOM as humic and fulvic acids are known to complex Cu.56 The results above show that metallic Cu is incorporated or attached to both OM and magnetite/maghemite within the organic residue (Fig. 5–7). This requires porosity along the surface of the organic residue, a diffusion gradient between the interface and interior of the organic residue, and that inner-sphere adsorption complexes between Cu1+/2+-bearing species and negatively-charged functional groups on the surface of the residue did not limit the diffusion of Cu into the interior of the residue. The latter requirement may have been achieved through neutralization of the negatively-charged surface sites via the formation of inner-sphere Cu1+/2+-OM complexes.An alternative mechanism of Cu transport may be the diffusion of negatively charged Cu-DOM species into the organic residue. In this case, immobilization of the Cu-DOM species within the OM and subsequent reduction of Cu1+/2+ to Cu0 would require the removal or reduction of electrostatic forces to allow bridging between available DOM functional groups and the organic residue. A reduction of electrostatic forces can be achieved by an increase in ionic strength57 which seems, however, an unlikely scenario for a diffusion pathway into the interior of an organic residue. Alternatively, the presence of positively-charged surface groups in the OM could facilitate the adsorption of the negatively-charged Cu-DOM species. Although the surface charge of OM is predominantly negative at a pH of 5, a small fraction of positively-charged amine and amide groups could facilitate the deposition of Cu-DOM complexes within the OM.
Diffusion of Cu into OM at the location of the second FIB section was likely not a result of the weathering of an attached Cu-bearing particulate as the closest attached Cu–Fe sulfides are approximately 500 μm from the location of the FIB section extraction (Fig. 2c). Hence, the Cu observed within this section likely originated from surrounding soil pore solutions and its diffusion into the OM required porosity, a diffusion gradient, and the absence of attractive electrostatic forces between the Cu-bearing aqueous species (Cu1+, Cu2+, Cu-DOM) and the surface functional groups of the residue.
Reduction mechanisms for Cu1+/2+ ions
Occurrence of metallic Cu NPs around and within the magnetite/maghemite layer in the OM (Fig. 7) is best explained by the reduction of adsorbed Cu1+/2+ species by Fe2+-bearing surface terminations as octahedrally coordinated Fe2+ can induce the reduction of redox-sensitive metal ions such as Cu2+ into their respective elemental species.51,58,59 This process led to the oxidation of Fe2+ to Fe3+ and most likely to the partial transformation of magnetite into maghemite. This conclusion seems to contradict, however, the absence of metallic Cu NPs in the magnetite/maghemite layer along the interface between mooihoekite and the organic residue (Fig. 5b). Absence and presence of Cu NPs in the magnetite/maghemite layers on the surface of mooihoekite and within the OM, respectively, may have been controlled by the following mineralogical, chemical, and textural features:(1) A higher porosity of the surface layer on mooihoekite relative to the layer in the OM (Fig. 4bvs.7a) as this allowed the more efficient removal of metallic Cu NPs formed along adsorption sites in the former layer;
(2) A higher positive charge of the surface layer on mooihoekite relative to the layer within the OM as the former layer was likely more exposed to pore-water of higher acidity than the latter one. This greater positive surface charge coupled with the positive surface charge of the Cu NPs (pzc = 9.460), facilitated a higher mobilization of the metallic Cu NPs relative to the layer in the OM.
(3) A larger proportion of DOM around the layer within the OM relative to the layer on the surface of mooihoekite as this allowed a more efficient reduction of (a) newly formed Fe3+ to Fe2+ or (b) Cu1+ to Cu0.
The reduction of Cu1+/2+ by exclusively DOM had to take place within the organic residue of FIB section 2 where layers of magnetite were absent (Fig. 8). Experimental studies show that metallic Cu can form through the direct reduction of Cu ions by reduced functional groups on soil HS.61 Here, HS can act as electron shuttles where redox-active functional groups gain electrons from soil microorganisms and subsequently donate those electrons to oxidized metal species. This process is effective for the reduction of various metals by soil or aquatic fulvic and humic acids.61–64 A variety of functional groups have been identified in HS, however only those capable of accepting and transferring electrons may induce metal reduction. This redox activity of HS can be primarily attributed to the quinone–hydroquione and phenolic moieties.65 Although soil HA cannot induce the reduction of Ag+ to metallic Ag NPs at room temperture62 and aquatic FA can only reduce Cu2+ ions to the monovalent species,66 Fulda et al.61 shows that soil HA are capable of reducing Cu2+ to metallic Cu NPs. As such, the vast amount of Cu NPs dispersed throughout the OM in FIB section 2 (Fig. 8) suggests that the corresponding OM has experienced significant reduction prior to the diffusion of Cu ions, subsequently enabling functional groups such as quinone or phenolic groups to reduce Cu1+/2+ into Cu0.
Previous studies demonstrate that metallic copper NPs can form in Cu-rich, S-limited, reducing environments through microbial-mediated reduction of Cu1+/2+ species.55,67–69 Although it is possible that metallic Cu NPs formed through biomineralization in the soils around the Kidd Creek site, this pathway does not explain the high abundance of NPs within confined (and thus not bioavailable) pore spaces of the organic matter (Fig. 5–8). Additionally, any Cu NPs formed through biomineralization under reducing conditions would be oxidized to Cu1+/2+ species upon exposure to oxidizing conditions55 unless the NPs occur in confined pore spaces in surface coatings on bacteria (e.g. through formation of clay mineral surface coatings on bacteria).70
The formation of covellite
The predominant Cu-bearing NPs identified in the colloidal fraction of the surficial 1 cm soil layer are composed of covellite, however covellite NPs have rarely been reported to form in a soil environment. One can consider here two approaches that can lead to the formation of the covellite NPs: a top-down or bottom-up approach. The top-down approach would involve the formation of covellite NPs through the breakdown of larger (micrometer-size) grains of covellite. However, three observations point against this top-down approach:(1) Covellite is not one of the major Cu-bearing ore minerals processed at the Kidd Creek metallurgical site;
(2) Neither electron microprobe nor SEM analyses indicate the presence of larger covellite grains in the silt to sand-size fractions of the examined soil sample (Fig. S9†); the absence of micrometer-size covellite attached to the organic residues is hereby expected as their surface charge is most likely negative (pzc71 < pHsoil) and as a positively-charged Fe-(hydr)oxide layer cannot form through non-stoichiometric dissolution of covellite;
(3) No Cu–Fe-sulfide NPs are observed within the leachate, which would be expected if the breakdown of larger sulfides grains contributes to the formation of mobile NPs.
An alternative explanation for the occurrence of the covellite NPs can be explained with a bottom-up approach. Gramp et al.72 show for example that covellite and chalcocite precipitate from a CuSO4 solution in the presence of sulfate-reducing bacteria whereby the formation of the former over the latter mineral is favored at higher activities of Cu than sulfate-bearing species. Furthermore, Weber et al.68 show that the formation of covellite colloids (<150 nm) occurs through the reaction of reduced Cu species with biogenic sulfide. The authors argue that the formation of covellite NPs is environmentally relevant for redox-variable soils with low concentrations of Cu and high sulfide gradients.68 Here the reduction of Cu2+ into Cu1+ or Cu0 and the formation of biogenic sulfide species can occur under reducing conditions such as for example during the temporarily flooding of the soils. Upon re-oxidation of the surrounding environment, a fraction of the covellite NPs would undergo oxidative dissolution.55
Although the mobile covellite NPs observed in our study may have formed through the reactions indicated by Weber et al.,68 they may also originate from organic residues similar to those analyzed in FIB section 1. In the latter residue, covellite most likely formed through the reaction between Cu1+/2+ and H2S released from the attached sulfide or from dissolved sulfur present in the pore spaces of the OM (Fig. 11).
The mobilization of covellite NPs within the 1 cm fraction at site 1 is clearly facilitated by organic material (Fig. 9a, b and 10b). The following two observations strongly suggest that the covellite NPs formed within the organic material:
(1) The covellite NPs are most commonly embedded in organic material (Fig. 9a, b and 10b). Although it is possible that minor concentrations of covellite could be stable in oxic pore water,73 nanoscale analysis of the leachate have not indicated the presence of covellite particles in the absence of organic matter.
(2) The negative surface charges of covellite and the organic material at pH = 5 do not favor the attachment of covellite NPs to the surface of the large organic colloids (see above).
Formation and agglomeration of the metallic Cu and covellite NPs
The formation of NPs requires a decrease in total free energy and the ability for many nuclei to develop which can occur by sudden changes in supersaturation, an increase in reaction sites, or Eh/pH conditions.15,74,75 Hence, the formation of spherical metallic Cu and covellite NPs is most likely induced by a sudden change in the environmental conditions upon the adsorption and reduction of Cu species by OM or magnetite. Their formation in the organic residues is most likely promoted by a high number of reductive sorption sites present on or within organic material or magnetite. Through an increase in porosity around surface rims and larger pores, these sorption sites become available for the adsorption of Cu or S species, the reduction of Cu1+/2+-bearing species and the subsequent nucleation of metallic Cu or covellite NPs.Upon the formation of metallic Cu or covellite, the NPs either attach or repel one another upon collision which is controlled by weak attractive van der Waals forces or attraction–repulsion forces such electrostatic interactions.76 Nanoparticles naturally agglomerate in order to reduce the free energy and thus to increase their stability. However, a limit can be reached where the agglomeration of particles no longer increases stability, but rather decreases it. At this point, the process of agglomeration would require minimization of repulsion forces, and therefore the charged particles must be screened or exhibit a neutral surface charge. Crystalline NPs can attach to a surface of a growing aggregate or crystal either in a specific crystallographic orientation (i.e., orientated attachment), or in a random orientation. The orientated attachment requires the rotation of NPs upon attachment and is controlled by Coulombic and Lewis acid/base interactions between the surface atoms of the NPs and aggregate.77 Attachment of a single NP in a random orientation requires subsequent structural re-organization for their full integration into the aggregate or bulk crystal.47
This study shows that the agglomeration of spherical metallic Cu NPs results in the formation of larger aggregates (<30 nm) in non-spherical moieties (Fig. 8). Smaller covellite NPs agglomerate into larger (<20 nm) particles within organic material (Fig. 10). The observed aggregations are likely related to charge screening by DOM or effective van der Walls forces. In the case of the growing aggregates of covellite NPs, attachment of the NPs occurs in an orientated (Fig. 10d) and non-orientated fashion (Fig. 10a).
Colloidal versus solute transport of Cu and its recycling in surficial soil layers
During their transport in the soil column, the negatively-charged covellite-bearing organic colloids may become immobilized through sorption to positively-charged Fe-(hydr)oxides surfaces. Alternatively, microbial activity could degrade their organic material78 which would result in the release and oxidative–dissolution of the covellite NPs by the oxic pore water (Fig. 11). In the latter case, CuSO4 or Cu1+/2+ could be recycled by sulfur-reducing bacteria (see above) or through their adsorption and diffusion into organic residues and their subsequent precipitation as covellite or metallic Cu within organic residues (Fig. 11).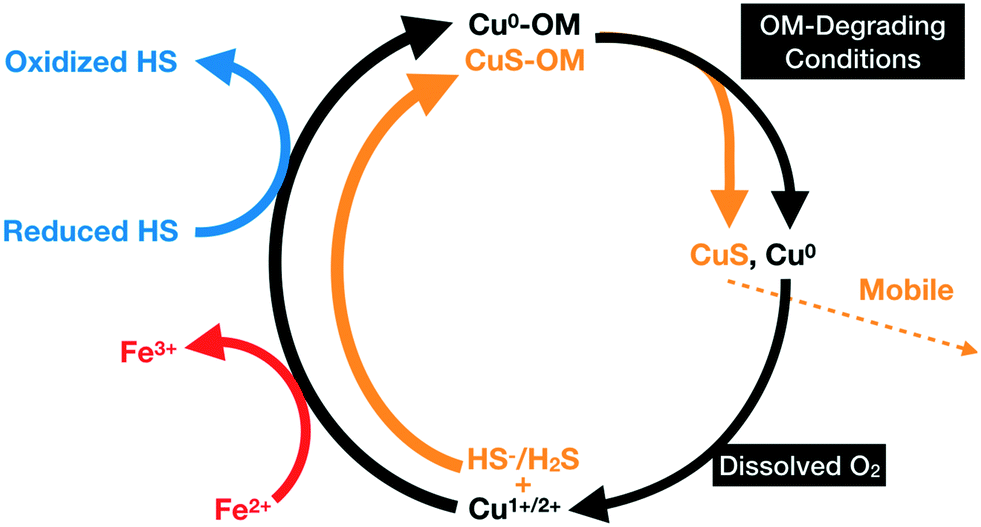 | ||
| Fig. 11 Schematics of the proposed recycling of Cu (black) in organic-rich soils; Cu2+ species become reduced by either ferrous iron (red) or reduced humic substances (blue) to metallic copper, which forms NPs in organic residue. Upon degradation of the organic residue, NPs are released. Metallic copper can be re-oxidized by the oxic pore water to form Cu1+/2+, allowing its diffusion into the organic material. The cycle for sulfur (orange) includes the formation of covellite NPs by the reaction of ionic Cu and dissolved hydrogen sulfide or bisulfide within organic matter. The particles are released during degradation of the residue and their dissolution results in the formation of ionic Cu which can then re-enter an organic residue. | ||
However, the covellite-bearing colloids likely have a small impact on the transport of Cu to greater depths as (a) the colloidal fraction of Cu represents only 11.4% of the total Cu in the leachate, and (b) larger organic colloids (>200 nm) commonly have a lower mobility than Cu-bearing aqueous species. The observed exponential decrease of the Cu concentration with depth suggests that the strength of the Cu-OM adsorption complexes significantly limits the transport of Cu to greater depth (Fig. S4†). As such, the observed low mobility of Cu in the organic rich soils in Timmins and smelter-impacted areas elsewhere79,80 is most likely a result of (a) the low solubility of most Cu-bearing PM,81 (b) the attachment of Cu-bearing PM onto organic residues, (c) the recycling of Cu through complex sequestration processes involving oxidation, reduction and adsorption of Cu ions and the nucleation and agglomeration of metallic Cu and Cu-sulfide NPs (Fig. 11), (d) the minor proportion of Cu-bearing NPs in the colloidal fraction, (e) the occurrence of Cu-bearing NPs embedded into larger less mobile organic colloids, and (f) complexation of Cu aqueous species by OM.
Summary
The majority of information gathered within this study revolves around the application of nanoscale techniques. Without these precise, in-depth analytical methods, data gathered from the microscale would have been generalized and therefore approximated. For instance, Cu detected throughout the organic grains with SEM likely would have been noted to occur as absorbed species. Additionally, the Cu–Fe-sulfides found to be attached to organic grains would have been mainly identified as chalcopyrite. From our work, we know that neither of those two situations are correct, highlighting the importance of nanoscale analyses.The presence of metallic Cu NPs and covellite within surficial, oxic soils has not been observed until this study. Their occurrence indicates that even within a soil solution that has a high oxidative-reduction potential, Cu can remain in its reduced, native form or in association with S as Cu1+.
The sequestration of Cu by organic residues through the attachment of Cu–Fe-sulfide particles and the transformation into metallic Cu and covellite is similar to the formation of supergene Cu-ore deposits where the dissolution of primary ore minerals (e.g. chalcopyrite) and the subsequent precipitation of secondary sulfides (e.g. chalcocite, covellite) result in an enrichment of Cu.82 The presence of metallic Cu and covellite NPs in organic residues without attached micrometer-size Cu–Fe-sulfides particulates indicates that the future cycling of Cu (Fig. 11) will be independent from the weathering of Cu-bearing PM and will solely depend on the diffusion of Cu1+, Cu2+, and oxidized or reduced S-species into the organic residues and their subsequent sequestration.
The apparent stability of reduced Cu-bearing NPs in the examined soils is of great importance for remediation strategies of Cu-contaminated soils using organic-rich substances. For example, Kumpiene et al.83 show that the addition of coal fly ash and peat to contaminated soils decreases the amount of leachable metals by 98.2 and 99.9% for Cu and Pb, respectively. The authors indicate that this reduction of exchangeable metals is due in part to the formation of Cu- and Pb-bearing inorganic phases as well as an increase in metal sorption to additional surface sites provided by the fly ash and peat. While these explanations are likely true, the results of our study suggest that the observed Cu retention could also be attributed to the absorption of Cu by the porous organic materials and its subsequent reduction and sequestration to its native form.
On a broader scale, this study may help to fill in potential gaps in research centered on interactions between Cu and organic material. The majority of previous studies focused on the interaction between ionic Cu and its sorption to organics or the formation of stable inorganic compounds, independent of the organic-rich environment it may reside in. Adsorption of metals is predominantly thought of as a transient mechanism of retention where ionic metal species are weakly retained until stronger inner-sphere complexes occur. This study shows that Cu can be absorbed by organic material through the in situ formation of Cu-bearing NPs which leads subsequently to its immobilization within the organic host. Hence, future research studies on the retention and sequestration of Cu in organic-rich environments using bulk analytical methods such as sequential extraction and synchrotron-based spectroscopy should consider the formation of Cu-bearing NPs in confined pore spaces as an additional mechanism when interpreting their results.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a NSERC (RGPIN-2018-04678) Discovery grant to MS, as well as by the Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure (NanoEarth), a member of the National Nanotechnology Coordinated Infrastructure (NNCI) funded by NSF (ECCS 1542100). Additional support was also provided by NSF Cooperative Agreement EF-0830093 (Center for the Environmental Implications of Nanotechnology). We also thank Associate Editor Joel Pedersen for handling the paper, R. Mathur and an anonymous reviewer for comments on an earlier version of this paper and Christopher Winkler, James Tuggle, Remy Poulin and Abdul Khan, the Nanoscale Fabrication and Characterization Laboratory at Virginia Tech and the Manitoba Institute of Materials.References
- National Pollutant Release Inventory Summary Report: Reviewed Facility-Reported Data 2015, Environment and Climate Change Canada, 2016 Search PubMed.
- V. Ettler, Soil contamination near non-ferrous metal smelters: A review, Appl. Geochem., 2016, 64, 56–74 CrossRef CAS.
- M. D. Hannington, W. Bleeker and I. Kjarsgaard, Sulfide Mineralogy, Geochemistry, and Ore Genesis of the Kidd Creek Deposit: Part I. North, Central, and South Orebodies, In The Giant Kidd Creek Volcanogenic Massive Sulfide Deposit, Western Abitibi Subprovince, Canada, ed. M. D. Hannington and C. T. Barrie, Society of Economic Geologists, 1999 Search PubMed.
- V. Ettler, M. Mihaljevič and M. Komárek, ICP-MS measurements of lead isotopic ratios in soils heavily contaminated by lead smelting: tracing the sources of pollution, Anal. Bioanal. Chem., 2004, 378(2), 311–317 CrossRef CAS PubMed.
- S. Lanteigne, M. Schindler and A. McDonald, Distribution of metals and metalloids in smelter-derived particulate matter in soils and mineralogical insights into their retention and release in a low-T environment, Can. Mineral., 2014, 52(3), 453–471 CrossRef CAS.
- S. Lanteigne, M. Schindler, A. M. McDonald, K. Skeries, Y. Abdu, N. M. Mantha, M. Murayama, F. C. Hawthorne and M. F. Hochella, Mineralogy and Weathering of Smelter-Derived Spherical Particles in Soils: Implications for the Mobility of Ni and Cu in the Surficial Environment, Water, Air, Soil Pollut., 2012, 223(7), 3619–3641 CrossRef CAS.
- B. Qasim, M. Motelica-Heino, E. Joussein, M. Soubrand and A. Gauthier, Potentially toxic element phytoavailability assessment in Technosols from former smelting and mining areas, Environ. Sci. Pollut. Res., 2015, 22(8), 5961–5974 CrossRef CAS PubMed.
- P. Adamo, S. Dudka, M. J. Wilson and W. J. McHardy, Chemical and mineralogical forms of Cu and Ni in contaminated soils from the Sudbury mining and smelting region, Canada, Environ. Pollut., 1996, 91(1), 11–19 CrossRef CAS PubMed.
- R. Knight and P. J. Henderson, Smelter dust in humus around Rouyn-Noranda, Quebec, Geochem.: Explor., Environ., Anal., 2006, 6(2–3), 203–214 CrossRef CAS.
- M. D. Buatier, S. Sobanska and F. Elsass, TEM-EDX investigation on Zn- and Pb-contaminated soils, Appl. Geochem., 2001, 16(9), 1165–1177 CrossRef CAS.
- J. N. Caplette, M. Schindler and T. K. Kyser, The black rock coatings in Rouyn-Noranda, Québec: fingerprints of historical smelter emissions and the local ore, Can. J. Earth Sci., 2015, 52(11), 952–965 CrossRef CAS.
- N. M. Mantha, M. Schindler, M. Murayama and M. F. Hochella, Silica- and sulfate-bearing rock coatings in smelter areas: Products of chemical weathering and atmospheric pollution I, Formation and mineralogical composition, Geochim. Cosmochim. Acta, 2012, 85, 254–274 CrossRef CAS.
- M. Schindler and M. F. Hochella, Soil memory in mineral surface coatings: Environmental processes recorded at the nanoscale, Geology, 2015, 43(5), 415–418 CrossRef CAS.
- M. Schindler and M. F. Hochella, Nanomineralogy as a new dimension in understanding elusive geochemical processes in soils: The case of low-solubility-index elements, Geology, 2016, 44(7), 515–518 CrossRef CAS.
- M. Schindler and M. F. Hochella, Sequestration of Pb-Zn-Sb- and As-bearing incidental nanoparticles by mineral surface coatings and mineralized organic matter in soils, Environ. Sci.: Processes Impacts, 2017, 19(8), 1016–1027 RSC.
- M. Schindler, S. Lanteigne, A. McDonald and M. F. Hochella, Evidence of Cu- and Ni-bearing Surface Precipitates and Adsorption Complexes in Remediated Soils At the Nanoscale: a Tem, Micro-raman, and Laser-ablation Icp-ms Study of Mineral Surface Coatings, Can. Mineral., 2016, 54(1), 285–309 CrossRef CAS.
- J. P. Gustafsson, P. Pechová and D. Berggren, Modeling Metal Binding to Soils: The Role of Natural Organic Matter, Environ. Sci. Technol., 2003, 37(12), 2767–2774 CrossRef CAS PubMed.
- A. Neagoe, V. Iordache and I. C. Fărcăşanu, The Role of Organic Matter in the Mobility of Metals in Contaminated Catchments, in Bio-Geo Interactions in Metal-Contaminated Soils, ed. E. Kothe and A. Varma, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 297–325 Search PubMed.
- D. G. Strawn and L. L. Baker, Speciation of Cu in a Contaminated Agricultural Soil Measured by XAFS, μ-XAFS, and μ-XRF, Environ. Sci. Technol., 2008, 42(1), 37–42 CrossRef CAS PubMed.
- J. Yang, J. Liu, J. J. Dynes, D. Peak, T. Regier, J. Wang, S. Zhu, J. Shi and J. S. Tse, Speciation and distribution of copper in a mining soil using multiple synchrotron-based bulk and microscopic techniques, Environ. Sci. Pollut. Res., 2014, 21(4), 2943–2954 CrossRef CAS PubMed.
- P. Coursol, N. Tripathi, P. Mackey, T. Leggett and A. Salomon de Friedberg, Slag Chemistry of the Mitsubishi S and Cl Furnaces at the Xstrata Copper-Kidd Metallurgical Site, Can. Metall. Q., 2010, 49(3), 255–262 CrossRef CAS.
- National Pollutant Release, Inventory Historical Substance Reports: Falconbridge Ltd-Kidd Metallurgical Div./Kidd Metallurgical Site: Total Particulate Matter, Environment and Climate Change Canada, 2017 Search PubMed.
- F. Amirault and O. M. Burnham, Carbon and sulphur analysis in geological samples by combustion–infrared: Verifying method capabilities on new instrumentation, in Summary of Field Work and Other Activities 2013, Open File Report 6290, Ontario Geological Survey, 2013, p. 43 Search PubMed.
- Strong Acid Leachable Metals (SALM) in Soil – Prescriptive, in British Columbia Environmental Laboratory Manual, ed. J. Austin, Environmental Monitoring, Reporting & Economics Section, Knowledge Management Branch, B.C. Ministry of Environment, Victoria, BC, 2015 Search PubMed.
- O. M. Burnham, Inductively coupled plasma mass spectrometry analysis of trace elements in geological samples after aqua regia extraction at the Geoscience Laboratories: Revised capabilities resulting from new instrumentation, in Summary of Field Work and Other Activities, Open File Report 6333, Ontario Geological Survey, 2017, p. 31 Search PubMed.
- N. D. Bassim, B. T. De Gregorio, A. L. D. Kilcoyne, K. Scott, T. Chou, S. Wirick, G. Cody and R. M. Stroud, Minimizing damage during FIB sample preparation of soft materials, J. Microsc., 2012, 245(3), 288–301 CrossRef CAS.
- J. Mayer, L. A. Giannuzzi, T. Kamino and J. Michael, TEM Sample Preparation and FIB-Induced Damage, MRS Bull., 2011, 32(5), 400–407 CrossRef.
- J. Yu, J. Liu, J. Zhang and J. Wu, TEM investigation of FIB induced damages in preparation of metal material TEM specimens by FIB, Mater. Lett., 2006, 60(2), 206–209 CrossRef CAS.
- J. L. Pouchou and F. Pichoir, New model for quantitative X-ray microanalysis, Part I: Application to the analysis of homogeneous samples, English edn, 1984, vol. 3, pp. 13–38 Search PubMed.
- K. Winterhalder, Early History of Human Activities in the Sudbury Area and Ecological Damage to the Landscape, 1995, pp. 17–31 Search PubMed.
- E. I. B. Chopin and B. J. Alloway, Trace element partitioning and soil particle characterisation around mining and smelting areas at Tharsis, Ríotinto and Huelva, SW Spain, Sci. Total Environ., 2007, 373(2), 488–500 CrossRef CAS PubMed.
- P. M. J. Gray, G. J. Bowyer and J. F. Castle, Sulphide deposits—their origin and processing, Springer, Netherlands, 2012 Search PubMed.
- L. Cabri, New data on Phase Relations in the Cu-Fe-S System, Econ. Geol., 1973, 68(4), 443–454 CrossRef CAS.
- A. Putnis, Talnakhite and mooihoekite; the accessibility of ordered structures in the metal-rich region around chalcopyrite, Can. Mineral., 1978, 16(1), 23–30 CAS.
- D. T. Rickard, Copper sulphide formation chemistry at low temperatures, TMPM, Tschermaks Mineral. Petrogr. Mitt., 1973, 19(1), 60–76 CrossRef CAS.
- O. Selinus and B. J. Alloway, Essentials of Medical Geology: Impacts of the Natural Environment on Public Health, Elsevier Academic Press, 2005 Search PubMed.
- D. L. Sparks, Environmental Soil Chemistry, Elsevier Science & Technology Books, 2nd edn, 2003 Search PubMed.
- M. Kosmulski, Surface Charging and Points of Zero Charge, CRC Press, 2009 Search PubMed.
- H. I. Adegoke, F. A. Adekola, O. S. Fatoki and B. J. Ximba, Sorptive interaction of oxyanions with iron oxides: A review, Pol. J. Environ. Stud., 2013, 22(1), 7–24 CAS.
- P. M. Bertsch and J. C. Seaman, Characterization of complex mineral assemblages: Implications for contaminant transport and environmental remediation, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(7), 3350 CrossRef CAS.
- Y. Li, N. Kawashima, J. Li, A. P. Chandra and A. R. Gerson, A review of the structure, and fundamental mechanisms and kinetics of the leaching of chalcopyrite, Adv. Colloid Interface Sci., 2013, 197–198, 1–32 CAS.
- S. W. Goh, A. N. Buckley, R. N. Lamb, R. A. Rosenberg and D. Moran, The oxidation states of copper and iron in mineral sulfides, and the oxides formed on initial exposure of chalcopyrite and bornite to air, Geochim. Cosmochim. Acta, 2006, 70(9), 2210–2228 CrossRef CAS.
- M. L. Farquhar, P. L. Wincott, R. A. Wogelius and D. J. Vaughan, Electrochemical oxidation of the chalcopyrite surface: an XPS and AFM study in solution at pH 4, Appl. Surf. Sci., 2003, 218(1), 34–43 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrence and Uses, 2003 Search PubMed.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nucleation and growth of magnetite from solution, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- V. Barrn and J. Torrent, Evidence for a simple pathway to maghemite in Earth and Mars soils, Geochim. Cosmochim. Acta, 2002, 66(15), 2801–2806 CrossRef CAS.
- X. Gong, J. Li, Y. Lin, X. Liu, L. Chen, J. Li and D. Li, Formation of highly crystalline maghemite nanoparticles from ferrihydrite in the liquid phase, Chin. Sci. Bull., 2014, 59(29), 3904–3911 CrossRef CAS.
- A. Putnis, Mineral Replacement Reactions, Rev. Mineral. Geochem., 2009, 70(1), 87–124 CrossRef CAS.
- E. S. Ilton and D. R. Veblen, Copper inclusions in sheet silicates from porphyry Cu deposits, Nature, 1988, 334, 516 CrossRef CAS.
- E. S. Ilton and D. R. Veblen, Origin and mode of copper enrichment in biotite from rocks associated with porphyry copper deposits; a transmission electron microscopy investigation, Econ. Geol., 1993, 88(4), 885–900 CrossRef CAS.
- E. S. Ilton, D. Earley, D. C. Marozas and D. R. Veblen, Reaction of some trioctahedral micas with copper sulfate solutions at 25 degrees C and 1 atmosphere; an electron microprobe and TEM investigation, Econ. Geol., 1992, 87(7), 1813–1829 CrossRef CAS.
- J. H. Ahn, H. Xu and P. R. Buseck, Transmission electron microscopy of native copper inclusions in illite, Clays Clay Miner., 1997, 45(2), 295–297 CrossRef CAS.
- R. E. W. Lett and W. K. Fletcher, Syngenetic sulphide minerals in a copper-rich bog, Miner. Deposita, 1980, 15(1), 61–67 CAS.
- T. S. Lovering, Organic precipitation of metallic copper: Chapter C in Contributions to economic geology (short papers and preliminary reports), 1927: Part I - Metals and nonmetals except fuels; 795C, Washington, D.C., 1927, pp. 45–52 Search PubMed.
- B. Fulda, A. Voegelin, K. Ehlert and R. Kretzschmar, Redox transformation, solid phase speciation and solution dynamics of copper during soil reduction and reoxidation as affected by sulfate availability, Geochim. Cosmochim. Acta, 2013, 123, 385–402 CrossRef CAS.
- K. Yang, G. Miao, W. Wu, D. Lin, B. Pan, F. Wu and B. Xing, Sorption of Cu2+ on humic acids sequentially extracted from a sediment, Chemosphere, 2015, 138, 657–663 CrossRef CAS PubMed.
- R. Kretzschmar and T. Schäfer, Metal Retention and Transport on Colloidal Particles in the Environment, Elements, 2005, 1(4), 205–210 CrossRef CAS.
- E. J. O'Loughlin, S. D. Kelly, K. M. Kemner, R. Csencsits and R. E. Cook, Reduction of AgI, AuIII, CuII, and HgII by FeII/FeIII hydroxysulfate green rust, Chemosphere, 2003, 53(5), 437–446 CrossRef.
- A. F. White and M. L. Peterson, Reduction of aqueous transition metal species on the surfaces of Fe(II) -containing oxides, Geochim. Cosmochim. Acta, 1996, 60(20), 3799–3814 CrossRef CAS.
- D. Collins, T. Luxton, N. Kumar, S. Shah, V. K. Walker and V. Shah, Assessing the Impact of Copper and Zinc Oxide Nanoparticles on Soil: A Field Study, PLoS One, 2012, 7(8), e42663 CrossRef CAS PubMed.
- B. Fulda, A. Voegelin, F. Maurer, I. Christl and R. Kretzschmar, Copper Redox Transformation and Complexation by Reduced and Oxidized Soil Humic Acid. 1. X-ray Absorption Spectroscopy Study, Environ. Sci. Technol., 2013, 47(19), 10903–10911 CrossRef CAS PubMed.
- N. Akaighe, R. I. MacCuspie, D. A. Navarro, D. S. Aga, S. Banerjee, M. Sohn and V. K. Sharma, Humic Acid-Induced Silver Nanoparticle Formation Under Environmentally Relevant Conditions, Environ. Sci. Technol., 2011, 45(9), 3895–3901 CrossRef CAS PubMed.
- A. Manceau, C. Lemouchi, M. Enescu, A.-C. Gaillot, M. Lanson, V. Magnin, P. Glatzel, B. A. Poulin, J. N. Ryan, G. R. Aiken, I. Gautier-Luneau and K. L. Nagy, Formation of Mercury Sulfide from Hg(II)–Thiolate Complexes in Natural Organic Matter, Environ. Sci. Technol., 2015, 49(16), 9787–9796 CrossRef CAS PubMed.
- D. T. Scott, D. M. McKnight, E. L. Blunt-Harris, S. E. Kolesar and D. R. Lovley, Quinone Moieties Act as Electron Acceptors in the Reduction of Humic Substances by Humics-Reducing Microorganisms, Environ. Sci. Technol., 1998, 32(19), 2984–2989 CrossRef CAS.
- Z. Struyk and G. Sposito, Redox properties of standard humic acids, Geoderma, 2001, 102(3), 329–346 CrossRef CAS.
- A. N. Pham, A. L. Rose and T. D. Waite, Kinetics of Cu(II) Reduction by Natural Organic Matter, J. Phys. Chem. A, 2012, 116(25), 6590–6599 CrossRef CAS PubMed.
- A. F. Hofacker, S. Behrens, A. Voegelin, R. Kaegi, T. Losekann-Behrens, A. Kappler and R. Kretzschmar, Clostridium Species as Metallic Copper-Forming Bacteria in Soil under Reducing Conditions, Geomicrobiol. J., 2015, 32(2), 130–139 CrossRef CAS.
- F.-A. Weber, A. Voegelin, R. Kaegi and R. Kretzschmar, Contaminant mobilization by metallic copper and metal sulphide colloids in flooded soil, Nat. Geosci., 2009, 2(4), 267–271 CrossRef CAS.
- F.-A. Weber, A. Voegelin and R. Kretzschmar, Multi-metal contaminant dynamics in temporarily flooded soil under sulfate limitation, Geochim. Cosmochim. Acta, 2009, 73(19), 5513–5527 CrossRef CAS.
- K. Konhauser, Introduction to Geomicrobiology, Blackwell Science Ltd, 2007 Search PubMed.
- M. K. Nduna, A. E. Lewis and P. Nortier, A model for the zeta potential of copper sulphide, Colloids Surf., A, 2014, 441, 643–652 CrossRef CAS.
- J. P. Gramp, K. Sasaki, J. M. Bigham, O. V. Karnachuk and O. H. Tuovinen, Formation of Covellite (CuS) Under Biological Sulfate-Reducing Conditions, Geomicrobiol. J., 2006, 23(8), 613–619 CrossRef CAS.
- T. F. Rozan, M. E. Lassman, D. P. Ridge and G. W. Luther III, Evidence for iron, copper and zinc complexation as multinuclear sulphide clusters in oxic rivers, Nature, 2000, 406, 879–882 CrossRef CAS PubMed.
- Y.-S. Jun, D. Kim and C. W. Neil, Heterogeneous Nucleation and Growth of Nanoparticles at Environmental Interfaces, Acc. Chem. Res., 2016, 49(9), 1681–1690 CrossRef CAS PubMed.
- G. A. Waychunas, C. S. Kim and J. F. Banfield, Nanoparticulate Iron Oxide Minerals in Soils and Sediments: Unique Properties and Contaminant Scavenging Mechanisms, J. Nanopart. Res., 2005, 7(4), 409–433 CrossRef CAS.
- E. M. Hotze, T. Phenrat and G. Lowry, Nanoparticle Aggregation: Challenges to Understanding Transport and Reactivity in the Environment, J. Environ. Qual., 2010, 39, 1909–1924 CrossRef CAS PubMed.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Crystallization by particle attachment in synthetic, biogenic, and geologic environments, Science, 2015, 349(6247), aaa6760 CrossRef PubMed.
- J. Hur, B.-M. Lee and H.-S. Shin, Microbial degradation of dissolved organic matter (DOM) and its influence on phenanthrene–DOM interactions, Chemosphere, 2011, 85(8), 1360–1367 CrossRef CAS PubMed.
- G. Spiers, C. Hawson and F. PrevostMetal Levels in the Soils of the Sudbury Smelter Footprint, Sudbury Regional Soils Project, 2004 Search PubMed.
- T. Sterckeman, F. Douay, N. Proix and H. Fourrier, Vertical distribution of Cd, Pb and Zn in soils near smelters in the North of France, Environ. Pollut., 2000, 107(3), 377–389 CrossRef CAS PubMed.
- V. Ettler, Z. Johan, B. Kříbek, F. Veselovský, M. Mihaljevič, A. Vaněk, V. Penížek, V. Majer, O. Sracek, B. Mapani, F. Kamona and I. Nyambe, Composition and fate of mine- and smelter-derived particles in soils of humid subtropical and hot semi-arid areas, Sci. Total Environ., 2016, 563–564, 329–339 CrossRef CAS PubMed.
- M. Reich and P. M. Vasconcelos, Geological and Economic Significance of Supergene Metal Deposits, Elements, 2015, 11(5), 305–310 CrossRef CAS.
- J. Kumpiene, A. Lagerkvist and C. Maurice, Stabilization of Pb- and Cu-contaminated soil using coal fly ash and peat, Environ. Pollut., 2007, 145(1), 365–373 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: bulk chemical composition of the soil samples; Table S2: calculation of molar volumes; Fig. S1–S3: photographic images of the sampling sites; Fig. S4: change in the concentrations of Cu, Pb and, Zn with depth; Fig. S5 and S6: XRD pattern; Fig. S7: SEM images of the coarser fraction of the soil (>180 μm); Fig. S8: composition of sulfides and oxides attached to organic residues: Fig. S9: SEM images of the finer fraction of the soils (<180 μm); Fig. S10–S21: TEM and STEM images, SAED pattern, and d-spacings. See DOI: 10.1039/c8en00994e |
| This journal is © The Royal Society of Chemistry 2019 |