
Nanomaterial identification of powders: comparing volume specific surface area, X-ray diffraction and scanning electron microscopy methods†
Claire
Dazon
 a,
Olivier
Witschger
*a,
Sébastien
Bau
a,
Vanessa
Fierro
b and
Philip L.
Llewellyn
c
a,
Olivier
Witschger
*a,
Sébastien
Bau
a,
Vanessa
Fierro
b and
Philip L.
Llewellyn
c
aLaboratoire de Métrologie des Aérosols, Institut National de Recherche et de Sécurité (INRS), Vandœuvre, France. E-mail: olivier.witschger@inrs.fr; Tel: +33 83 50 20
bInstitut Jean Lamour – UMR CNRS 7198, Epinal, France
cLaboratoire MADIREL, CNRS, Aix Marseille University, Marseille, France
First published on 26th October 2018
Abstract
Nanomaterials in powder forms are widely produced and used in numerous applications. Nanomaterial identification is a growing concern for several areas encountering these substances. The current reference criterion of the European Commission (EC) for nanoparticle identification is the number size distribution of the constituent particles. One example of how the latter can be obtained is by the electron microscopy (EM) method. However, this method is not widely available and is time-consuming to perform and use for analysis. Alternative methods, such as the volume specific surface area (VSSA), also allow nanomaterial identification. The VSSA is the product of the external specific surface area of the powder and its skeletal density, and appears to be adapted more specifically for powders. The techniques required to measure the two parameters used to calculate the VSSA are more widely available than EM, but sample preparation can be delicate. Furthermore, deeper evaluation of the reliability of VSSA for nanomaterial classification as well as a more detailed characterization methodology for its implementation and discussion about the relative merits of this method versus EM are still necessary. Here, we determined, through a detailed and operational characterization strategy, the VSSA for seven metal oxide powders (4 TiO2, 1 SiO2, and 2 CaCO3) and an activated carbon, with all of them produced on an industrial scale. These eight samples covered a range of constituent particle sizes between 10 nm and 18 μm. Equivalent particle sizes determined by the VSSA, X-ray diffraction (XRD) (another method giving access to an equivalent particle size and integrated into our characterization methodology) and scanning electron microscopy (SEM) (reference method) were compared. The results showed that the VSSA can robustly identify nanomaterials in the form of powders (−12% mean bias on equivalent particle sizes relative to SEM).
Environmental significanceNanoparticulate powders can be released in any environment, and constitute sources of occupational exposure throughout their life cycle. Nanoparticles can entail adverse effects on human health, or have an impact on the environment. Therefore, the responsible development of these nanomaterials involves identifying their “nano” nature to further propose safer design and use. In this article, the volume specific surface area (VSSA) of eight industrial powders covering a large range of properties was compared to the primary particle size determined by scanning electron microscopy and X-ray diffraction. Determining the VSSA of a powder is claimed to be more accessible than measuring the primary particle size. It is shown that the VSSA is a reliable criterion allowing relevant nanomaterial identification. |
1. Introduction
Nanomaterials are now a large industry.1–3 The improvement and increasing control of synthesis processes as well as the new properties of materials developed on the nanoscale suggest that this area is expected to continue to grow.Regardless of the application and the context, the identification of the “nano” nature of substances is an essential step. For example, when implementing the chemical registration dossiers in Europe (REACH regulation),4 nanomaterials match the definition of “substances” and therefore, identification and information provisions for them are applicable. Similarly, the responsible management of risks associated with nanomaterials in the workplace requires their identification to propose preventative measures.5,6
Boverhof et al.7 focused on available nanomaterial definitions in the regulatory context worldwide and pointed out a lack of harmonization. In consequence, it is important to indicate the nanomaterial definition used when characterizing any substance. In 2011, the European Commission (EC) issued a recommendation on the definition of nanomaterials related to legal classification.8 As explained by Gao and Lowry,9 this recommended EC definition “provides the most specific measurable parameters of nanomaterials compared to other definitions”, that is why it appears that several areas rely on this recommendation solely for the nanomaterial identification. A substance is considered as a nanomaterial when at least 50% of the constituent particles in the number size distribution have one or more external dimensions between 1 nm and 100 nm. To determine this distribution, the EC recommends implementing the best available methods and the most appropriate and harmonized protocols possible. Electron microscopy (EM) methods are widely used in this view since they enable direct characterization of particle size and shape.10,11 However, these methods are time-consuming and require significant human and material resources for both imaging and post-hoc analysis.
Another possibility proposed by the EC to identify nanomaterials relies on the determination of the volume specific surface area (VSSA) when possible. This parameter was suggested as an alternative when defining nanomaterials to overcome disadvantages related to size measurements. The VSSA (eqn (1)) of a material is calculated from the external specific surface area (AEx) and the material's skeletal density ρ:
| VSSA (m2 cm−3) = AEx m2 g−1 × ρ g cm−3 | (1) |
Based on its VSSA, a substance is considered as a nanomaterial when it presents a volume specific surface area greater than 60 m2 cm−3.8 This limit of 60 m2 cm−3, also named VSSACutoff, corresponds to the VSSA of monodisperse spherical non-porous particles with a density of 1 g cm−3 and a diameter of 100 nm. The VSSACutoff has been recently discussed for adapting the threshold value according to the particle shape (40 m2 cm−3 for fiber-like particles or 20 m2 cm−3 for platelets12). However, in the absence of consensus, the threshold of 60 m2 cm−3 remains the VSSACutoff to consider when applying the EC recommendation for nanomaterial identification. Moreover, if a material is identified as a non-nanomaterial with its VSSA, particle sizing remains mandatory to confirm the previous statement.
The VSSA method is a particularly attractive approach since it is relatively well adapted for powders. Indeed, for a powder, the AEx can be determined by the relevant gas adsorption method13–15 and the skeletal density of the material can be obtained by helium pycnometry.16 These two methods are less expensive overall and more readily available than EM. Above all, powders are increasingly encountered in industry and laboratories (synthesis and/or use) as they are included in the composition of products across a very wide range of activity sectors: building, food and agriculture, cosmetics, energy, and so forth. The 2016 public R-Nano report indicates that almost 475![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons of nanomaterials were manufactured, distributed or imported in France.17 Thus, there is an increasing need for nanomaterial identification for regulation purposes, process control, and risk assessment. Therefore, the relatively easier accessibility of the VSSA could be an interesting solution to identify nanomaterials.
000 tons of nanomaterials were manufactured, distributed or imported in France.17 Thus, there is an increasing need for nanomaterial identification for regulation purposes, process control, and risk assessment. Therefore, the relatively easier accessibility of the VSSA could be an interesting solution to identify nanomaterials.
Although gas adsorption and helium pycnometry are relatively widely available, precise sample preparation and rigorous data analysis are necessary. Both techniques need sample outgassing to limit underestimation of measured values.18–22 The AEx determination requires an accurate selection of the models (BET for non-porous, macroporous or mesoporous powders and the t-plot model for microporous powders) as explained by Lecloux23 and Wohlleben et al.24 in their works about the use of the VSSA for nanomaterial classification. Besides, the skeletal density measured by helium pycnometry can be validated by comparing the measured value with the material's theoretical density, based on its theoretical chemical and crystallographic composition.
To date, the Nanodefine European project25 allowed advances in the assessment of the relevance of the VSSA for the classification of powders as nanomaterials. This project particularly focused on industrial powders representative of the most frequently encountered materials. However, it is still necessary to evaluate the reliability of the VSSA for nanomaterial powder identification for promoting this parameter as an alternative approach when EM methods are not available. Besides, in spite of the availability of SOPs and guides, an operational strategy for VSSA implementation, notably accessible for non-specialists of material characterization, is needed. Furthermore, it would be interesting to discuss the relative merits of the VSSA versus EM methods.
In this context, our study aimed to bring new results supplementing those of the Nanodefine European project for nanomaterial identification. In particular, a detailed operational approach was applied to a series of representative industrial powders to determine their VSSA and state whether they have to be considered as nanomaterials. The relevance of the classification of powders by the VSSA method is evaluated by comparing the measured particle size with the corresponding equivalent particle size obtained with the scanning electron microscopy (SEM) reference method and with the equivalent particle size derived from X-ray diffraction (XRD).
2. Materials and methods
2.1. Materials
The eight powders used in this study are presented in Table 1. The metal oxides TiO2, SiO2 and CaCO3, seven in total, represent the most frequently encountered materials in France according to the latest R-Nano report (2016), with recorded quantities exceeding 1000 tons. That is why we selected them as we considered these materials as representative of powders potentially found in workplaces. These metal oxides are produced on an industrial scale and are used in products with applications in engineering, food and agriculture, cosmetics, paints and varnishes, energy and even textiles. In addition to the metal oxides, a powdered activated carbon (Maxsorb®), which is used for its high specific surface area (>3000 m2 g−1) mainly for gas storage by adsorption,26 was chosen. These powders were selected based on the At (total surface area) indicated by their de-identified suppliers, which covered a broad range from a few tens of m2 g−1 to more than 3000 m2 g−1.| Powder | Code | Supplier | A t supplier (m2 g−1) | Purity (wt%) |
|---|---|---|---|---|
| TiO2 | LSSA | A | 10 | 99 |
| MSSA | 90 | 99 | ||
| HSSA | 350 | 99.5 | ||
| P25 | B | 50 | 99 | |
| SiO2 | SiO2 | C | >600 | 99.5 |
| CaCO3 | CaCO3 A | D | n.a | 97.5 |
| CaCO3 B | 94.5 | |||
| Activated carbon (Maxsorb®) | MSC-30 | E | >3000 | 99 |
2.2. Characterization approach
The industrial powders were characterised by the approach schematized in Fig. 1. | ||
| Fig. 1 Diagram schematizing the general approach used in this work to determine the VSSA of powders and comparison of the XRD and SEM (reference) methods for nanomaterial identification. | ||
This strategy can be broken down into three phases.
• TGA allows an optimised outgassing protocol to be selected for sample preparation before the gas adsorption and helium pycnometry measurements when seeking to calculate the powders' VSSAs.
• Chemical analysis by X-ray fluorescence, ICP-MS or ICP-OES, or alternatively the supplier's data allow the material's composition to be included when selecting the theoretical density ρth to be compared to the ρ measured by helium pycnometry, for validation of the experimental results.
• XRD allows the determination of the crystal structure of the particles making up the powder as well as their proportions. Moreover, it is possible to calculate the mean particle size dXRD by applying the Scherrer formula (eqn (2)):
 | (2) |
This formula is based on the hypothesis that the particles are spherical or cubic and monocrystalline and appears to be appropriate for crystal sizes greater than 10 nm.27
The crystal structure is also considered when selecting the theoretical density of the material to be compared to the density measured by helium pycnometry for validation of the experimental results.
• SEM can be used to determine the main shape of the constituent particles in the powder, making it possible to choose a D factor from which a cutoff VSSA value can be defined (VSSACutoff) as proposed by Wohlleben et al.24 In addition, SEM can be used to establish the size distribution of the constituent particles in the powder when the number of identifiable and isolated particles in the acquired images is sufficient, i.e., at least 100 to 200 particles.28,29 The size of the measured particles corresponds, most of the time, to the mean Feret diameter29,30 or to the projected equivalent surface diameter dS of a disc of area S according eqn (3) for spherical particles:
 | (3) |
For particles in the form of fibres (D = 2), the diameter, rather than the length of the fibre, is measured. For particles in the form of platelets (D = 1), the thickness is measured. In this work, dSEM corresponds to dS for spherical particles, the diameter of a fibre for fibre-like particles or the thickness of platelets.
2.2.3.1. Gas adsorption. The gas adsorption isotherms are compared to the typical isotherms proposed by IUPAC14 so as to apply:
• The BET model when a type II (non-porous or macroporous material) or type IV (mesoporous material) isotherm is obtained, for which AEx = At. This model is the easiest to apply and the most relevant for these types of isotherms.31
• The t-plot model is applied when a type I (microporous model) isotherm is obtained, and it provides the AEx from the At for microporous materials. Rouquerol et al.32,33 described how the calculation for the t-plot method should be applied from the values for the Boer “universal t curve” obtained for a non-porous alumina. This reference is the most frequently used to determine the AEx for microporous materials using the t-plot model and is the one used in this work.
2.2.3.2. Helium pycnometry. Measurements of the skeletal density ρ by helium pycnometry were compared to the theoretical values ρth selected based on the chemical and crystallographic data available for the powders. The measurements were validated by verifying that the relative difference between ρ and ρth was less than 10%, which corresponds to the uncertainty of the pycnometry measurement.
2.2.3.3. Determining the VSSA and comparing equivalent particle diameters. The VSSA was calculated (eqn (1)) from validated AEx and skeletal density data and compared to the VSSACutoff, which was selected on the basis of the particle shapes observed in SEM images (D factor).
The equivalent mean diameter dVSSA of the particles making up the powder was calculated from the value of the VSSA and the D factor (eqn (4)):
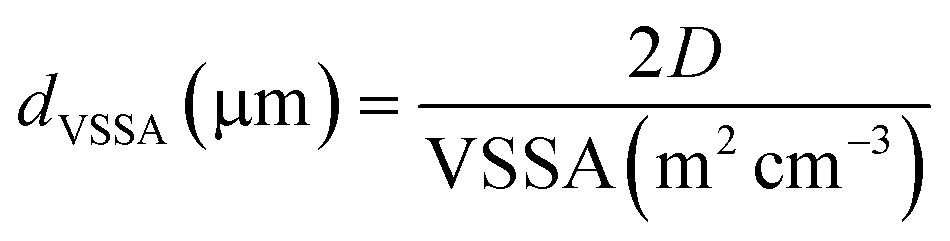 | (4) |
The mean diameters obtained by the three characterisation methods for size measurement: VSSA (dVSSA), XRD (dXRD), and SEM (dSEM) were then compared to the size determined by SEM, with an arbitrarily determined relative bias tolerance of ±15% corresponding to the maximum rounded absolute average bias we obtained in the comparison of VSSA and SEM. As the dSEM is the reference diameter, the ultimate classification of the powder as a nanomaterial or not was based on that value. The comparison of equivalent particle size allows the validation of the nanomaterial classification based solely on the VSSA value.
2.3. Experimental
For each characterisation method in the approach, the experimental details and additional explanations are provided hereafter.Before performing gas adsorption measurements, it is important to outgas the samples so as to eliminate the chemical species that may be adsorbed on the (external and internal) surfaces of the particles as water or atmospheric pollutants. Otherwise, At measurements might be underestimated due to the presence of previously adsorbed molecules.13 Outgassing is also required prior to measurement of the skeletal density by helium pycnometry. The appropriate outgassing protocol can be selected by thermogravimetric analysis of the powder, whereby the powder sample is subjected to a temperature range, with addition of a neutral sweeping gas such as nitrogen or argon (to avoid chemical reactions between molecules and the powder). The sample's mass is monitored during the heating programme. The sample is considered “cleaned” when the mass variation as a function of time reaches a plateau that is maintained for at least 30 minutes. Before gas adsorption, the powders are generally outgassed for at least 1 hour in a given vacuum, at the temperature corresponding to the one at which the mass variation plateau was attained in thermogravimetry. When elaborating the outgassing protocol before gas adsorption, one can chose, for instance, the outgassing temperature corresponding to the beginning of plateau observation obtained with TGA.
However, thermogravimetry does not exactly reproduce the vacuum conditions implemented when outgassing the powders before gas adsorption. In addition, although recommended, it is important to ensure that the samples are not degraded by the vacuum and the temperature applied during outgassing. A visual inspection is generally sufficient. A change in colour or appearance of the powder indicates that the sample has become degraded and that the outgassing procedure must be reviewed, such as by reducing the degassing temperature or extending its duration.
000 and ×500000. The mean particle size dSEM was calculated to be fewer than 50 particles which could be counted on all the images obtained for each powder. The images were treated using ImageJ software (Java version 1.8.0).
3. Results and discussion
The results of this work are presented in the order of the characterisation approach used (see Fig. 1).3.1. XRD, chemical composition and densities
The XRD and helium pycnometry results are presented together in Table 2.| Powder | Code | Crystal structure | Proportions (% weight) | ρ (g cm−3) | ρ th (g cm−3) | Bias ρ (%) |
|---|---|---|---|---|---|---|
| TiO2 | LSSA | Anatase | 100% | 3.87 ± 0.01 | 3.92 | −0.7 |
| MSSA | 3.71 ± 0.01 | −4.8 | ||||
| HSSA | 3.37 ± 0.01 | −13.5 | ||||
| P25 | Anatase | 87% | 3.83 ± 0.01 | 3.94 | −2.7 | |
| Rutile | 13% | |||||
| SiO2 | SiO2 | Amorphous | 100% | 2.07 ± 0.02 | 2.2 | −5.7 |
| CaCO3 | CaCO3 A | Calcite | 2.71 ± 0.01 | 2.71 | 0 | |
| CaCO3 B | 2.57 ± 0.01 | −0.7 | ||||
| Activated carbon (Maxsorb®) | MSC-30 | Amorphous | 2.58 ± 0.01 | 2.2 | 17.3 |
The crystal phases of the powders are indicated along with their mass proportions. TiO2 powders LSSA, MSSA and HSSA have a 100% anatase structure, TiO2 P25 is an anatase/rutile mixture with mass proportions of 87/13. The two CaCO3 have a 100% calcite structure. The crystal phases detected were those expected for these materials. SiO2 and activated carbon, as MSC-30, powders are amorphous materials. By combining the crystallographic results with the chemical composition of the powders indicated by the suppliers, the theoretical densities can be selected for comparison with the skeletal densities measured by helium pycnometry for validation. The powders studied here were considered to be pure between 94.5 and 100% (Table 1). The theoretical densities of each powder therefore corresponded to the density of the corresponding raw materials. In the case of TiO2 P25, the theoretical density is a combination of those of anatase and rutile, taking the mass ratio of each crystal phase into consideration. The theoretical densities used for the comparison were taken from the Handbook of Chemistry and Physics (96th edition).
Compared to the theoretical densities, most of the skeletal densities measured by helium pycnometry displayed biases of less than 10%. These skeletal densities were therefore validated and used to calculate the VSSA. The bias in the skeletal densities measured was greater for two powders – TiO2 HSSA (−13.5%) and MSC-30 (+17.3%) – and these bias values were attributed to insufficient degassing prior to analysis, perhaps due to the fact that the powder samples were not placed under vacuum before the helium pycnometry measurements with the Accupyc 1340 device. Thus, for these two powders, the theoretical density was used to calculate the VSSA.
3.2. SEM
Fig. 2 shows representative SEM images for each of the powders at different magnifications for comparison on the 100 nm scale except for the activated carbon and CaCO3 A. For all the powders, we observed that the constituent particles were extensively agglomerated, but individual particles remained sufficiently distinguishable to allow at least 50 particles to be measured, although in many cases, measurements may have been imprecise. TiO2 P25 was made up of spherical particles corresponding to the anatase phase. The rutile particles would have been characterised by a rod-like shape on the SEM images, but they were impossible to identify. Given the mass proportions of the crystal phases of anatase (87%) and rutile (13%) for TiO2 P25, it was assumed that only the anatase particles were counted. The CaCO3 A and MSC-30 powders have particles in the form of platelets, while the other powders are composed of spherical particles. Consequently, the value D = 1 gives a VSSACutoff of 20 m2 cm−3 for CaCO3 A and MSC-30, whereas D = 3 gives a VSSACutoff of 60 m2 cm−3 for the other powders. | ||
| Fig. 2 SEM images of the powders studied in this work. | ||
3.3. Specific surface area and VSSA
Fig. 3 shows the nitrogen adsorption isotherms of TiO2 MSSA and SiO2. These adsorption isotherms were selected to demonstrate the most operational approach for analysis of the nitrogen adsorption isotherms of industrial powders. These adsorption isotherms were determined to be of type II for TiSO2 MSSA and composite type I (b) + II for SiO2, based on the reference isotherms proposed by IUPAC.14 | ||
| Fig. 3 Characteristic type II nitrogen adsorption isotherm obtained for TiO2 MSSA and mixed isotherm obtained for SiO2. | ||
The BET model was used to calculate AEx for the TiO2 MSSA sample. This choice is obvious as the TiO2 MSSA isotherm is equivalent to the IUPAC type II isotherm. In contrast, the calculation is more difficult for SiO2 as its isotherm combines a type I (b) and a type II form. Fig. 4 shows, for relative pressures P/P° between 10−6 and 10−3, that nitrogen adsorption for silica varies between 0 and 6 mmol g−1. This represents a significant gas adsorption (almost 30% of the total) covering three orders of magnitude, as a result, we applied the t-plot model to calculate the AEx.33,34 From these observations, we can conclude that the microporous surface of this silica represents 85% of the total surface area.
 | ||
| Fig. 4 SiO2 displays a mixed nitrogen adsorption isotherm on a semi-log scale. | ||
The other nitrogen adsorption isotherms can be found in the ESI.† The isotherms obtained for TiO2 LSSA and P25, and those for CaCO3 A and B were type II (non-porous or macroporous), whereas that for TiO2 HSSA corresponded to a composite I (b) and II isotherm. MSC-30 presented a type I (b) adsorption isotherm. The BET model was therefore applied for TiO2 LSSA and P25, and CaCO3 A and B powders, whereas the t-plot model was selected for TiO2 HSSA and MSC-30.
Table 3 lists the AEx, the D factor determined by SEM, the VSSACutoff and the VSSA for the powders studied. The values are indicated with ±1 standard deviation. The powders for which the VSSA is greater than the VSSACutoff are considered as nanomaterials according to this method, and they are indicated by bold text. The powders not identified as nanomaterials are indicated by italic text.
| Powder | Code | A Ex (m2 g−1) | D | VSSACutoff (m2 cm−3) | VSSA (m2 cm−3) |
|---|---|---|---|---|---|
| TiO2 | LSSA | 9 ± 0.3 | 3 | 60 | 35 |
| P25 | 55 ± 0.5 | 210 | |||
| MSSA | 83 ± 0.7 | 307 | |||
| HSSA | 179 ± 0.4 | 701 | |||
| SiO2 | SiO2 | 87 ± 0.4 | 180 | ||
| CaCO3 | CaCO3 A | 5 ± 0.1 | 1 | 20 | 13 |
| CaCO3 B | 25 ± 0.3 | 3 | 60 | 64 | |
| Activated carbon (Maxsorb®) | MSC-30 | 358 ± 41 | 1 | 20 | 788 |
The powders studied had a large diversity of surface areas, with AEx values between 5 m2 g−1 and 358 m2 g−1, spanning almost three orders of magnitude.
The VSSA values obtained were between 13 m2 cm−3 and 923 m2 cm−3. When compared to the VSSACutoff values, these values identify the following six powders as nanomaterials: TiO2 MSSA, HSSA and P25, SiO2, and CaCO3 B. The remaining two powders (TiO2 LSSA and CaCO3 A) were classed as non-nanomaterials. It should be noted that the VSSA for CaCO3 B (64 m2 cm−3) is very close to the VSSACutoff value (60 m2 cm−3).
Interestingly, when we compare all the VSSA values to the VSSACutoff of 60 m2 cm−3 (hypothesis D = 3), which we would do if the shape of the particles was unknown, the classification remains the same.
3.4. Comparison of VSSA, XRD and SEM methods for measurement of constituent particle size
The equivalent mean diameters of the constituent particles determined for the powders, dVSSA, from their VSSA were compared to the mean particle diameters dXRD and dSEM determined by XRD and SEM, respectively (Fig. 5). Amorphous materials were not compared in terms of dXRD as their diffractograms were impossible to exploit. Similarly, the rutile phase of TiO2 P25 was not used in the comparison as the VSSA could not distinguish the crystal phases of the particles and their corresponding sizes and because no rutile was visible (rod-shaped particles) in the SEM images. The sizes determined by the three methods and the bias between the results can be found in Table S4 of the ESI.† | ||
| Fig. 5 Equivalent particle diameters obtained by VSSA, XRD and SEM methods. The error bars correspond to the standard deviation. | ||
The lowest bias (0%) was obtained for TiO2 MSSA, which had a constituent particle size of 19 nm, and SiO2, with a constituent particle size of 10 nm. The greatest bias was obtained with MSC-30 (−100%). The other biases were between −19% and +31%. The mean bias for all experimental points combined was −12%. Generally, XRD underestimates the particle sizes, whereas VSSA biases do not display a trend in the discrepancies. The XRD underestimation could be attributed to the Scherrer formula application as the particle shapes are not perfectly spherical. Based on this comparison, MSC-30 is falsely identified as a nanomaterial by the VSSA. Indeed, the VSSA classes this activated carbon as a nanomaterial based on its porosity and the shape of the particles (D = 1 for the platelets and a VSSACutoff of 20 m2 cm−3). However, the dSEM for carbon is 18 μm with a high standard deviation attributed to the difficulty in achieving repeatable measurement of the platelet thickness in the SEM images. This difference can be attributed to the use of the t-plot model which may not be appropriate for this type of material. In contrast, the t-plot model works well for the other microporous powders, TiO2 HSSA and SiO2, for which the bias on particle size was −15% and 6%, respectively. This bias is within the tolerance range ±15% set. For the other materials in this study, the VSSA (m2 cm−3) correctly classified the powders as nanomaterials or not, as determined by comparison with the reference dSEM. Both the VSSA and the SEM methods indicate that TiO2 LSSA and CaCO3 A are not nanomaterials, whereas all the other powders are nanomaterials according to the dSEM or the VSSA. For CaCO3 B, the VSSA (64 m2 cm−3) was close to the VSSACutoff (60 m2 cm−3), and its classification as a nanomaterial was confirmed by the dSEM (94 nm).
These results show that the VSSA could have been used in a reliable way for the studied powders without requiring EM analysis. If only the VSSACutoff of 60 m2 cm−3 (hypothesis D = 3) is used for comparison with the experimental VSSA data, the classification of the powders as nanomaterials remains correct, except for MSC-30. TiO2 LSSA and CaCO3 A are not nanomaterials and the other powders are nanomaterials according to their dSEM. These results corroborate the observations of Wohlleben et al.24 who correctly classified 12 powders as nanomaterials based on their VSSA but could have avoided EM analysis to confirm this classification for the materials they tested. These new data should be considered with caution however because of the broad discrepancies observed. These latter might have been reduced with more replication of experiments or supplementary EM characterization (transmission electron microscopy for instance).
The dXRD values were also compared to the dSEM, and a mean bias of −25% was found. Once again, the lowest bias (0%) was obtained for TiO2 MSSA, whereas the greatest bias (−63%) was obtained with CaCO3 A. The other biases were between 22% and 57%. These results indicate that XRD can be used in the sole case of spherical and monocrystalline particles to calculate an equivalent particle size. These results agree with the previous study of Weibel et al.35 Therefore, one can consider XRD as a complementary method for particle size determination. In contrast to VSSA, XRD cannot be used without EM analysis for a reliable classification of powders as nanomaterials because of the necessary shape determination (spherical particles) to apply the Scherrer equation.
4. Conclusion
In the context of the identification of nanomaterials, this study proposes to compare equivalent particle sizes of eight industrial powders determined by the VSSA, XRD and SEM (reference) methods through a detailed and operational characterisation approach. The results highlight the importance of sample preparation before the gas adsorption and helium pycnometry measurements, and the careful analysis of the data to determine the VSSA. In particular, the proposed data treatment involves a precise analysis of the gas adsorption isotherms to calculate the external specific surface area for the particles making up the powders, and validation of the helium pycnometry measurements by comparison with the theoretical density. These two parameters are used to calculate the VSSA for the powder. The comparison of results obtained by the VSSA and SEM methods shows a good correlation in terms of equivalent particle sizes, leading us to conclude that the VSSA nanomaterial identification is reliable, although one microporous activated carbon was falsely classified as a nanomaterial based on its VSSA values. In contrast, the comparison of the XRD method with the SEM method reveals that XRD can only reasonably be used to calculate an equivalent particle size when the particles being studied are spherical and monocrystalline.This work provides additional details on VSSA determination (gas adsorption and helium pycnometry) complementary to the recent NanoDefine European project examining the relevance of the VSSA for the classification of nanomaterials. The notable results were that no false negatives were obtained with the VSSA method, and that this parameter appeared to be potentially used for the powder studied without confirmation of the nanomaterial classification with size measurement by EM.
As this work was carried out on a restricted number of materials, this trend should be confirmed by testing more substances with the same approach.
Conflicts of interest
The authors confirm that there are no conflicts of interest to declare.Acknowledgements
We would like to thank E. Jankowska, a former member of the Central Institute for Labour Protection (CIOP – Poland), for coordinating with the Nanostruktur laboratory where the scanning electron microscopy images were produced.Notes and references
- M. C. Roco, The long view of nanotechnology development: the National Nanotechnology Initiative at 10 years, J. Nanopart. Res., 2011, 13, 427–445 CrossRef.
- D. T. Leong, Nanosafety Issues of Nanomaterials, in Reference Module in: Materials Science and Materials Engineering, Elsevier, 2017 Search PubMed.
- S. Foss Hansen, L. R. Heggelund, P. Revilla Besora, A. Mackevica, A. Boldrin and A. Baun, Nanoproducts - what is actually available to European consumers?, Environ. Sci.: Nano, 2016, 3, 169–180 RSC.
- ECHA, https://echa.europa.eu/fr/regulations/nanomaterials.
- O. Witschger, O. Le Bihan, M. Reynier, C. Durand, A. Marchetto, E. Zimmermann and D. Charpentier, Recommendations for characterizing potential emissions and exposure to aerosols released from nanomaterials in workplace operations, Hyg. Secur. Trav., 2012, 2355, 41–55 Search PubMed , ND.
- INRS, Aide au repérage des nanomatériaux en entreprise, ED6174, 2014 Search PubMed.
- D. R. Boverhof, C. M. Bramante, J. H. Butala, S. F. Clancy, M. Lafranconi, J. West and S. C. Gordon, Comparative assessment of nanomaterial definitions and safety evaluation considerations, Regul. Toxicol. Pharmacol., 2015, 73, 137–150 CrossRef CAS PubMed.
- EC, Commission recommendation of 18 October 2011 on the definition of nanomaterial, Official Journal of European Union, 2011, L275, pp. 38–40 Search PubMed.
- X. Gao and G. V. Lowry, Progress towards standardized and validated characterizations for measuring physicochemical properties of manufactured nanomaterials relevant to nano health and safety risks, NanoImpact, 2018, 9, 14–30 CrossRef.
- J. Mielke, F. Babick, C. Ullmann and V.-D. Hodoroaba, Evaluation of particle sizing techniques for the implementation of the EC definition of a nanomaterial, presented in the International Congress on Particle Technology, Nuremberg, 2016 Search PubMed.
- F. Babick, J. Mielke, W. Wohlleben, S. Weigel and V.-D. Hodoroaba, How reliably can a material be classified as a nanomaterial? Available particle-sizing techniques at work, J. Nanopart. Res., 2016, 18, 158 CrossRef PubMed.
- G. R. Hubert Rauscher, A. B. Sanfeliu, N. G. Hendrik Emons, R. Koeber, K. R. Thomas Linsinger, J. R. Sintes and H. S. Birgit Sokull-Klüttgen, Towards a review of the EC Recommendation for a definition of the term “nanomaterial”. Part 3 Scientific-technical evaluation of options to clarify the definition and to facilitate its implementation, European Commission, Joint Research Centre, Institute for Health and Consumer Protection, Italy, 2015 Search PubMed.
- J. Rouquerol and F. Rouquerol, Methodology of Gas adsorption, in: Adsorption by Powders and Porous Solids, Academic Press, Oxford, 2nd edn, 2014, pp. 57–104 Search PubMed.
- M. Thommes, K. Kaneko, V. Neimark Alexander, P. Olivier James, F. Rodriguez-Reinoso, J. Rouquerol and S. W. Sing Kenneth, Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87, 1051 CAS.
- V. A. Hackley and A. B. Stefaniak, “Real-world” precision, bias, and between-laboratory variation for surface area measurement of a titanium dioxide nanomaterial in powder form, J. Nanopart. Res., 2013, 15, 1742 CrossRef PubMed.
- S. Lowell, J. E. Shields, M. A. Thomas and A. M. Thommes, Density measurement, in: Characterization of porous solids and powders: surface area, pore size and density, ed. S. S. B. Media, Kluwer Academic Publisher, 2004, ch. 19 Search PubMed.
- Report on the declaration of substances imported, manufactured or distributed in France in 2015, 2016, available to http://www.r-nano.fr.
- E. Olson, The importance of sample preparation when measuring specific surface area, Journal of GXP Compliance, 2012, 16, 52–62 Search PubMed.
- J. Rouquerol, F. Rouquerol, M. Triaca and O. Cerclier, The use of thermal analysis to select the standard outgassing conditions of a set of reference adsorbents, Thermochim. Acta, 1985, 85, 311–314 CrossRef.
- L. Clausen and I. Fabricius, BET Measurements: Outgassing of Minerals, J. Colloid Interface Sci., 2000, 227, 7–15 CrossRef CAS PubMed.
- ASTM.B923-10, Standard Test Method for Metal Powder Skeletal Density by Helium or Nitrogen Pycnometry, 2010 Search PubMed.
- ISO.12154, Determination of density by volumetric displacement — Skeleton density by gas pycnometry, 2014 Search PubMed.
- A. J. Lecloux, Discussion about the use of the volume-specific surface area (VSSA) as criteria to identify nanomaterials according to the EU definition, J. Nanopart. Res., 2015, 17, 447 CrossRef.
- W. Wohlleben, J. Mielke, A. Bianchin, A. Ghanem, H. Freiberger, H. Rauscher, M. Gemeinert and V.-D. Hodoroaba, Reliable nanomaterial classification of powders using the volume-specific surface area method, J. Nanopart. Res., 2017, 19, 61 CrossRef PubMed.
- S. Weigel, P. Müller, K. Löschner, V. D. Hodoroaba, M. Stintz, F. von der Kammer, R. Köber and H. Rauscher, NanoDefine - An integrated analytical approach to implement the EC definition of nanomaterial, presented in the International Congress on Particle Technology, Nuremberg, 2016 Search PubMed.
- T. Otowa, R. Tanibata and M. Itoh, Production and adsorption characteristics of MAXSORB: High-surface-area active carbon, Gas Sep. Purif., 1993, 7, 241–245 CrossRef CAS.
- R. Sharma, D. P. Bisen, U. Shukla and A. B. G. Sharma, X-ray diffraction: a powerful method of characterizing nanomaterials, Recent Res. Sci. Technol., 2012, 4, 77–79 CAS.
- S. Bau, O. Witschger, F. Gensdarmes, O. Rastoix and D. Thomas, A TEM-based method as an alternative to the BET method for measuring off-line the specific surface area of nanoaerosols, Powder Technol., 2010, 200, 190–201 CrossRef CAS.
- P.-J. De Temmerman, J. Lammertyn, B. De Ketelaere, V. Kestens, G. Roebben, E. Verleysen and J. Mast, Measurement uncertainties of size, shape, and surface measurements using transmission electron microscopy of near-monodisperse, near-spherical nanoparticles, J. Nanopart. Res., 2013, 16, 2177 CrossRef.
- P.-J. De Temmerman, E. Van Doren, E. Verleysen, Y. Van der Stede, M. A. D. Francisco and J. Mast, Quantitative characterization of agglomerates and aggregates of pyrogenic and precipitated amorphous silica nanomaterials by transmission electron microscopy, J. Nanobiotechnol., 2012, 10, 24 CrossRef CAS PubMed.
- K. S. W. Sing, F. Rouquerol and J. Rouquerol, Classical Interpretation of Physisorption Isotherms at the Gas–Solid Interface, in Adsorption by Powders and Porous Solids, Academic Press, Oxford, 2nd edn, 2014, pp. 159–189 Search PubMed.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Is the bet equation applicable to microporous adsorbents?, in Studies in Surface Science and Catalysis, ed. P. L. Llewellyn, F. Rodriquez-Reinoso, J. Rouqerol and N. Seaton, Elsevier, 2007, vol. 160, pp. 49–56 Search PubMed.
- F. Rouquerol, L. Luciani, P. Llewellyn, R. Denoyel and J. Rouquerol, Techniques de l'ingénieur Surfaces et structures fonctionnelles, 2003, TIB534DUO Search PubMed.
- K. S. W. Sing, F. Rouquerol, P. Llewellyn and J. Rouquerol, Assessment of Microporosity, in: Adsorption by Powders and Porous Solids, Academic Press, Oxford, 2nd edn, 2014, pp. 303–320 Search PubMed.
- A. Weibel, R. Bouchet, F. Boulc and P. Knauth, The Big Problem of Small Particles: A Comparison of Methods for Determination of Particle Size in Nanocrystalline Anatase Powders, Chem. Mater., 2005, 17, 2378–2385 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8en00760h |
| This journal is © The Royal Society of Chemistry 2019 |