
Freestanding 1T MoS2/graphene heterostructures as a highly efficient electrocatalyst for lithium polysulfides in Li–S batteries†
Jiarui
He
ab,
Gregory
Hartmann
c,
Myungsuk
Lee
c,
Gyeong S.
Hwang
 c,
Yuanfu
Chen
*b and
Arumugam
Manthiram
*a
c,
Yuanfu
Chen
*b and
Arumugam
Manthiram
*a
aMaterials Science and Engineering Program & Texas Materials Institute, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: manth@austin.utexas.edu
bState Key Laboratory of Electronic Thin Films and Integrated Devices, University of Electronic Science and Technology of China, Chengdu 610054, P. R. China. E-mail: yfchen@uestc.edu.cn
cMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, TX 78712, USA
First published on 27th November 2018
Abstract
A novel approach to effectively suppress the “polysulfide shuttle” in Li–S batteries is presented by designing a freestanding, three-dimensional graphene/1T MoS2 (3DG/TM) heterostructure with highly efficient electrocatalysis properties for lithium polysulfides (LiPSs). The 3DG/TM heterostructure is constructed by few-layered graphene nanosheets sandwiched by hydrophilic, metallic, few-layered 1T MoS2 nanosheets with abundant active sites. The porous 3D structure and the hydrophilic feature of 1T-MoS2 are beneficial for electrolyte penetration and Li-ion transfer, and the high conductivities of both graphene and the 1T MoS2 nanosheets facilitate electron transfer. These merits lead to a high electrocatalytic efficiency for LiPSs due to excellent ion/electron transfer and the presence of sufficient electrocatalytic active sites. Therefore, the cells with 3DG/TM exhibit outstanding electrochemical performance, with a high reversible discharge capacity of 1181 mA h g−1 and a capacity retention of 96.3% after 200 cycles. The electrocatalysis mechanism of LiPSs is further experimentally and theoretically revealed, which provides new insights and opportunities to develop advanced Li–S batteries with highly efficient electrocatalysts for LiPS conversion.
Broader contextAdvanced energy-storage technologies are urgently needed to satisfy the energy demands of society. Lithium–sulfur (Li–S) batteries are one of the most attractive candidates for next-generation energy-storage technology. However, it is extremely crucial to suppress the inherent polysulfide shuttle. Although electrocatalysis has been shown to enhance cell stability, further performance improvement of Li–S cells is seriously hindered due to the lack of understanding of the electrocatalysis mechanism of the sulfur redox process. Herein, for the first time, we design and synthesize a novel freestanding, three-dimensional graphene/1T MoS2 (3DG/TM) heterostructure to improve the electrochemical performance and reveal the electrocatalysis mechanism. The well-designed 3DG/TM heterostructure can be used as a highly efficient electrocatalyst for the conversion of lithium polysulfides (LiPSs): the unique 3DG/TM nanoarchitecture, constructed by few-layered 2D MoS2 nanosheets in situ grown on a porous 3D graphene network, guarantees abundant active sites, thus ensuring sufficient catalytic activity for the LiPSs; the high conductivities of both the 3D graphene skeleton and the metallic 1T MoS2 nanosheets (with conductivity six orders higher than that of 2H MoS2) greatly accelerate the electron transfer; the rich porosity of 3DG/TM, along with the hydrophilic nature of both 1T MoS2 and 3DG, facilitates the electrolyte penetration and Li-ion transfer. Benefitting from these merits and synergistic effects, the cells with 3DG/TM exhibit a large reversible specific capacity (1181 mA h g−1) and an outstanding long-term cycling stability. Furthermore, by combining post-mortem SEM and XPS characterizations and first-principles calculations, the electrocatalysis mechanism of LiPSs is clearly revealed, and the enhanced catalytic activity of the MoS2-supported systems is mainly attributed to the enhanced binding and availability of LiPSs at the electrode interface. This work provides new insights and opportunities to develop advanced Li–S batteries with highly efficient electrocatalysts for LiPS conversion. |
Introduction
The spread of portable electronics and electric vehicles has prompted the development of energy storage systems with high-energy density and long-cycle life.1–4 Of the many alternatives, rechargeable lithium–sulfur (Li–S) batteries are one of the most attractive candidates for next-generation energy storage technology, owing to their advantages, such as high theoretical energy density, nontoxicity, and low cost.5–8 However, the practical application of Li–S batteries is still hindered by several technical problems. For example, the insulating nature of sulfur and sulfides9–13 limits electron transport in the cathode and leads to low-active material utilization; in addition, the dissolution and migration of intermediate lithium polysulfides (LiPSs) result in a loss of the active material and shuttling of LiPSs.14–18Recently, intensive research efforts, such as the fabrication of nanostructured carbon/sulfur composites, have led to an improvement in conductivity and cycling performance.19–22 However, since carbon materials provide poor adsorption toward LiPSs, limited loading of the sulfur active material has been realized, which cannot meet the requirement for the practical application of Li–S batteries.23–26 In order to improve the adsorption of LiPSs, polar hosts, such as metal compounds and metal–organic frameworks, have been developed.27–32 On the one hand, polar hosts possess poor conductivity, which is detrimental to the kinetics of sulfur electrochemistry during cell operation, thus compromising the rate capability. On the other hand, most reports based on these hosts lack a deep exploration of the mechanism of the electrochemical-performance enhancement. More recently, the electrocatalytic effect has been recognized in metal- (e.g., Pt, Au) and metal sulfide (e.g., CoS2, WS2)-based sulfur hosts, and it was found to play a vital role in improving the reaction kinetics and suppressing the LiPS shuttle process.33–36 However, as the catalysis of polysulfide conversion is still in a very early phase of research, the deep impact of the catalytic activity of the catalyst is still unclear, and thus the electrochemical performance based on the above-mentioned cathodes is still far from realizing its full potential in Li–S batteries.
In this report, we present the synthesis of well-designed, freestanding, three-dimensional graphene/1T MoS2 (3DG/TM) heterostructures as a highly efficient electrocatalyst for LiPS conversion. The metallic 1T MoS2 nanosheets are hydrophilic (Fig. S1a and b, ESI†) with rich active sites and a high electronic conductivity that is six orders of magnitude higher than that of 2H MoS2. The high electronic conductivity facilitates fast electron transfer, the hydrophilic property benefits ion diffusion, and the dense active sites ensure sufficient catalytic activity for LiPSs. The 3DG/TM heterostructures of our designed material can maximize the aspect ratio of the active catalytic sites. The freestanding, porous morphology of the 3DG/TM material facilitates electrolyte accessibility (Fig. S1c and d, ESI†), enabling better ion transport. Benefitting from these synergistic effects (Scheme 1), the cells with 3DG/TM exhibit excellent specific capacity and outstanding cycling stability. Furthermore, a fundamental understanding of the enhanced catalytic activity in the MoS2-supported systems is achieved via experimental characterizations and theoretical calculations, which provide new insights and opportunities to develop advanced Li–S batteries with highly efficient electrocatalysts for LiPSs. Based on the deep understanding of the electrocatalysis of LiPS conversion, the well-designed 3DG/TM heterostructures with rich electrocatalytically active sites ensure a high catalytic activity and thus significantly improve the electrochemical performance of Li–S batteries. Even with a very high sulfur loading (10 mg cm−2), the 3DG/TM cathode not only effectively mitigates the LiPS shuttling, but also delivers an excellent specific capacity, outstanding rate capability, and pronounced cycling stability for an impressive number of 500 cycles.
 | ||
| Scheme 1 The conversion process of LiPSs on a graphene surface with 1T MoS2. The 3DG/TM heterostructures work as a highly efficient electrocatalyst for LiPS conversion. | ||
It is believed that the structure of the electrocatalyst plays a vital role in catalytic activity. Therefore, we present a well-designed electrocatalyst composed of three-dimensional graphene/1T MoS2 (3DG/TM) heterostructures synthesized through a facile one-pot hydrothermal process. The graphene/MoS2 hydrogel was formed by a hydrothermal reaction using MoO3, thioacetamide, and graphene oxide as starting materials; during the hydrothermal reaction, 1T MoS2 was in situ formed on the surface of graphene oxide owing to its abundant functional groups and defects.37–39 Then, a self-assembled 3DG/TM aerogel with graphene/1T MoS2 heterostructures was obtained by simply freeze-drying the hydrogel to remove water, as further described in the ESI.†
As shown in Fig. S2 (ESI†), the 3DG/TM aerogel is a freestanding electrode with high flexibility that can be cut and compressed easily. In order to reveal the structure of 3DG/TM, scanning electron microscopy (SEM) was performed. The morphology of 3DG/TM is shown in Fig. 1. Fig. 1a and b show that 3DG/TM is composed of graphene/1T MoS2 nanosheets, which cross-link to form a 3D interconnected network with rich pores. The SEM images in Fig. S3 (ESI†) show that 3DG/HM and 3DG also have a porous architecture. Such a porous structure can not only facilitate electrolyte accessibility, which benefits ion transport, but also provide a large number of active sites for the reaction. The TEM image in Fig. 1c further confirms that the graphene/1T MoS2 nanosheets in 3DG/TM are very thin. The high-resolution TEM image in Fig. 1d shows the graphene/1T MoS2 heterostructures in 3DG/TM.40 Such a unique structure can provide a high aspect ratio of edge sites and accessibility to a large catalytic surface, which is beneficial for the electrocatalytic reaction. The elemental mappings of C, S, and Mo in Fig. 1f and g illustrate the uniform distribution of MoS2 on the graphene surface.
 | ||
| Fig. 1 Morphological characterizations of 1T MoS2 on the graphene aerogel. (a and b) SEM images of the porous 3DG/TM. (c) Low-magnification TEM images of 3DG/TM. (d) High-resolution TEM images of 3DG/TM. (e) TEM morphology of 3DG/TM, and the corresponding elemental mapping images of (f) carbon, (g) sulfur, and (h) molybdenum. | ||
In order to identify the influence of structure on catalytic activity, three dimensional graphene/2H MoS2 (3DG/HM) and three-dimensional graphene (3DG) were prepared as well. Fig. 2a shows the X-ray diffraction (XRD) pattern of 3DG/TM in comparison with those of 3DG/HM and 3DG. As the XRD patterns in Fig. 2a show, in contrast to the (002) peak at 14.18° for 3DG/HM, the XRD pattern of 3DG/TM shows two peaks at 8.7° and 17.4° corresponding to the (002) and (004) planes of 1T MoS2, which matches well with a previous report on 1T MoS2.41 Interestingly, the (002) peak of 3DG cannot be detected in the pattern of 3DG/TM, indicating that the graphene nanosheets do not stack during the hydrothermal synthesis process. Fig. 2b shows the Raman spectra of 3DG/TM, 3DG/HM, and 3DG. A strong Raman band is observed at 144 cm−1, which corresponds to the Mo–Mo stretching vibrations in 1T MoS2 in 3DG/TM. The intense peaks at 218, 282, and 328 cm−1 are attributed to the phonon modes of 1T MoS2.42 3DG/HM exhibits typical Raman shifts at 376 and 405 cm−1 corresponding to the E12g and A1g modes, respectively, which are substantially different from those of 3DG/TM.42 All samples show peaks at 1325 and 1596 cm−1 corresponding to the D band and G band of graphene.43
 | ||
| Fig. 2 (a) XRD patterns and (b) Raman shifts of 3DG/TM, 3DG/HM, and 3DG. High-resolution XPS spectra of Mo 3d (c) and S 2p (d) for 3DG/TM and 3DG/HM. | ||
The phase identification of the prepared 3DG/TM and 3DG/HM was further investigated by X-ray photoelectron spectroscopy (XPS). Fig. 2c shows the high-resolution XPS spectra of Mo 3d, in which the peaks observed at 228.7 and 231.8 eV correspond to the 3d5/2 and 3d3/2 components of Mo4+ of 1T MoS2 in 3DG/TM. The peaks of Mo 3d in 3DG/HM are shifted to higher binding energies by 1 eV compared to those of 3DG/TM. Such results match well with previous reports.42 Similarly, as shown in Fig. 2d, the S 2p spectra of 3DG/TM consist of peaks at 161.8 and 162.9 eV, associated with S 2p3/2 and 2p1/2, which are lower than the corresponding peaks in 3DG/HM. These spectra indicate the presence of pure 1T MoS2 in 3DG/TM. Thermogravimetric analysis (TGA), shown in Fig. S4 (ESI†), revealed that the content of 1T MoS2 in 3DG/TM is 25.2 wt%. For a fair comparison, the content of MoS2 in 3DG/HM was controlled the same as that of 3DG/TM in Fig. S4 (ESI†).
In order to demonstrate the advantages of 3DG/TM as an electrocatalyst towards LiPS conversion, electrochemical tests were performed with a three-electrode system. The electrocatalyst was coated onto glassy carbon (GC) as the working electrode, lithium foil was used as the counter electrode, Pt foil was used as the reference electrode, and 10 mM Li2S6 solution was used as the electrolyte. The electrocatalyst (3DG/TM, 3DG/HM, or 3DG) was loaded onto GC with 5 wt% Nafion solution as reported previously in the literature.1Fig. 3a–c show the representative cyclic voltammetry (CV) curves and lithium-ion diffusion characteristics based on the three-electrode system. The corresponding parameters, such as the open-circuit voltage (OCV), anodic peak potential (Epa), cathodic peak potential (Epc), anodic peak current density (Ipa), cathodic peak current density (Ipc), and lithium-ion diffusivity (DLi+), are given in Table S1 (ESI†). The Epa related to the oxidation of LiPSs with the 3DG/TM electrode is 2.40 V, showing a negative shift compared to the values of 2.43 V for 3DG/HM and 2.51 V for 3DG, under identical experimental conditions. Similarly, for 3DG/TM, the observed Epc corresponding to the reduction of LiPSs is 2.29 V, showing a positive shift compared to that of 3DG/HM (2.28 V). Importantly, the Epc of 3DG is missing, which can be explained as follows: during the oxidation process, the LiPS converts to S8 on the surface of the electrocatalyst. Therefore, the electrocatalysts must have a strong affinity to LiPSs/Li2S/S8 for the reversible transformation to be realized. In addition, all the electrocatalysts (3DG, 3DG/HM, and 3DG/TM) have a similar specific surface area and pore size distribution (Fig. S5, ESI†). Therefore, differences in the electrocatalytic performance mainly come from the catalytic activity of the materials. Owing to the poor interaction between 3DG and S8/LiPSs, the oxidation product of LiPSs (S8) desorbs from the surface of 3DG. Thus, the reduction peak of 3DG cannot be detected. The LiPS adsorption tests shown in Fig. S6 (ESI†) confirm the strong interaction of 3DG/TM with LiPSs. Furthermore, the CV curves of the three-electrode system with 3DG/TM remain stable, as shown in Fig. S7 (ESI†), suggesting the outstanding durability of 3DG/TM.
 | ||
| Fig. 3 (a) Comparative cyclic voltammograms of 3DG/TM and 3DG electrodes vs. Li/Li+ in a catholyte solution. (b) LSV curves of the 3DG/TM electrode with a 10 mM Li2S6-based electrolyte at various scan rates. (c) Peak current for the 3DG/TM electrode in comparison with the 3DG/HM and 3DG electrodes vs. the square root of the scan rates and (d) CV of Li–S batteries with Li as the anode and 3DG/TM, 3DG/HM and 3DG with a catholyte consisting of 1 M Li2S6 as the cathode at a scan rate of 0.1 mV s−1 between 1.8 and 2.8 V. (e) Potentiostatic polarization curves of 3DG/TM, 3DG/HM, and 3DG electrodes at a scan rate of 0.1 mV s−1 for charging and discharging processes. (f) Tafel plots derived from the potentiostatic polarization curves. | ||
In addition, 3DG/TM shows the highest current densities (Ipa and Ipc), suggesting that 3DG/TM has a superior catalytic ability, as shown in Table S1 (ESI†). To investigate the kinetic behavior of each electrocatalyst, linear sweep voltammograms (LSVs) at different scan rates were recorded (Fig. 3b, Fig. S8, and Fig. S9, ESI†). The peak current densities for the 3DG/TM electrode in comparison with the 3DG/HM and 3DG electrodes vs. the square root of the scan rates are shown in Fig. 3c. The lithium-ion diffusivity can be determined utilizing the classical Randles–Sevcik equation: 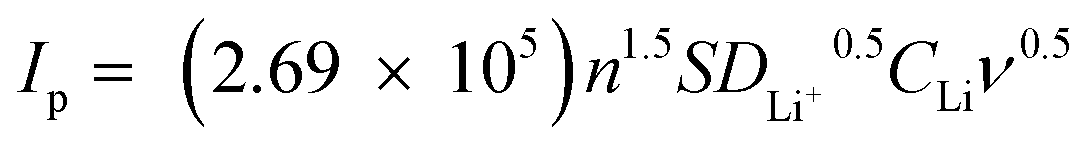 , where IP is the peak current, n is the charge-transfer number, S is the geometric area of the active electrode, DLi+ is the lithium-ion diffusion coefficient, CLi is the concentration of the lithium ions in the cathode, and ν is the potential scan rate.44 According to the calculated DLi+, it can be clearly seen that 3DG/TM exhibits the highest lithium-ion diffusivity, which is further evidence of the high catalytic activity of 3DG/TM.
, where IP is the peak current, n is the charge-transfer number, S is the geometric area of the active electrode, DLi+ is the lithium-ion diffusion coefficient, CLi is the concentration of the lithium ions in the cathode, and ν is the potential scan rate.44 According to the calculated DLi+, it can be clearly seen that 3DG/TM exhibits the highest lithium-ion diffusivity, which is further evidence of the high catalytic activity of 3DG/TM.
The binding strength of LiPSs on free-standing and MoS2-supported graphene was evaluated by dispersion-corrected DFT (DFT-D3) calculations. The details of the computational methods and model structures (Fig. S10–S12, ESI†) employed are described in the ESI.† We assume the following route for the redox reaction between S8 and Li2S during the charge cycle: 8Li2S ⇌ 16Li+ + 16e− + S8. The binding energy (Eb) is obtained by subtracting the total energy of the LiPS/slab system (ELiPS/slab) from the sum of the slab (Eslab) and LiPS species (ELiPS) energies (i.e., Eb = Eslab + ELiPS − ELiPS/slab). As shown in Fig. 4, the results clearly demonstrate that LiPSs is more strongly bound to graphene supported on MoS2 than on free-standing graphene, while the increase in Eb tends to consistently be larger in the case of G/1T compared to G/2H. The Eb of Li2S and S8 in G/1T compared to G is enhanced by 0.13 eV and 0.16 eV, respectively. Other intermediate species also show an increase in Eb by a similar order of magnitude. The Eb difference between the G/1T and G/2H systems tends to become distinct for lower molecular weight LiPSs (Li2Sn, n ≤ 4); for Li2S, the Eb is enhanced by 0.08 eV in G/1T compared to G/2H. In each case, the binding of LiPSs to bare MoS2 is significantly stronger compared to that for the graphene basal planes, and the binding energy of each system is in agreement with previously reported values.45,46
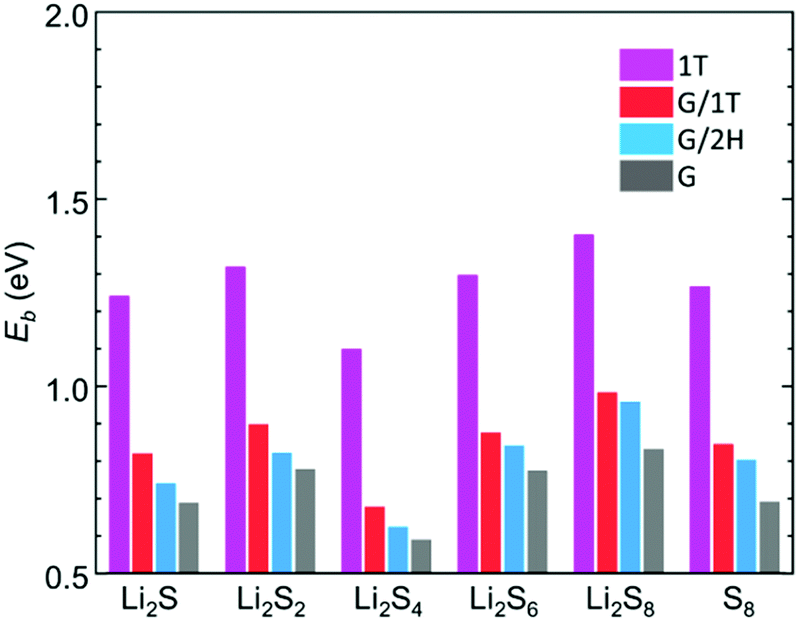 | ||
| Fig. 4 Binding energies of LiPS on MoS2, MoS2-supported graphene, and pristine graphene. | ||
Classical force field calculations were also performed to evaluate the adhesion of MoS2 on corrugated graphene (see the ESI† for details). As illustrated in Fig. S13 (ESI†), MoS2 exhibits a high-fidelity conformation to graphene topographic features, although it may not conform well to the relatively small jagged features of the graphene surface. This may serve not only to possibly produce distorted and activated MoS2 surfaces, but it can also possibly increase the interlayer spacing of few-layered MoS2, allowing LiPSs to diffuse between the layers and dramatically increasing the available surface area. Together, experimental and theoretical results suggest that the enhanced catalytic activity in the MoS2-supported systems is primarily due to the increased MoS2 surface area and the availability of LiPSs at the electrode interface.
To further confirm the advantages of 3DG/TM as an electrocatalyst for the conversion of LiPSs, standard 2032 coin cells were assembled with the freestanding 3DG/TM, 3DG/HM, and 3DG aerogels, a catholyte consisting of 1 M Li2S6 as the cathode, lithium foil as the anode, and Celgard membrane as the separator. As shown in Fig. 3d, the CV curve of the cells with 3DG/TM shows two cathodic peaks at 2.32 and 1.97 V, which can be attributed to the conversion of long-chain LiPSs to short-chain LiPSs and finally to Li2S. In the subsequent anodic scan, the anodic peak at 2.50 V corresponds to the reversible transformation of Li2S to LiPSs and ultimately to elemental sulfur. Such a reversible conversion further illustrates the excellent catalytic activity. It is noted that 3DG/TM displays a distinguishable positive shift in the cathodic peak and a negative shift in the anodic peak along with a much higher current density with respect to 3DG/HM and 3DG, in agreement with the phenomena observed in the three-electrode system and further confirming a significant improvement in the catalytic activity of 3DG/TM towards LiPS conversion. Accordingly, the onset potential for the oxidation of long-chain LiPSs to short-chain LiPSs in 3DG/TM is reduced compared to those of 3DG/HM and 3DG (Fig. 3e). Tafel plots were obtained from Fig. 3e for the identification of the electrocatalytic effect on the charge-transfer kinetics during LiPS conversion, as shown in Fig. 3f. 3DG/TM displays a lower Tafel slope (76 mV dec−1) compared to those of 3DG/HM (81 mV dec−1) and 3DG (87 mV dec−1). The significant decrease in the Tafel slope values of 3DG/TM confirms the accelerated rate of Li–S redox conversion.
The electrocatalysts of 3DG/TM and 3DG coated on the glassy carbon after scanning from 1.5 to 3 V were visualized using SEM. The SEM images of 3DG/TM and 3DG before and after scanning are shown in Fig. 5a–d. The nanosheets in 3DG/TM become thick, which is attributed to the conversion and accumulation of sulfur at the electrocatalytically active sites. In sharp contrast, the morphologies of 3DG before and after scanning remain unchanged. The unchanged morphology indicates that few sulfur particles deposit on the surface of 3DG, due to the weak interactions with LiPSs/sulfur. The XPS results shown in Fig. 5e and f support this explanation. The high-resolution S 2p spectra of 3DG/TM at different states are shown in Fig. 5e. When the cell was scanned to 3.0 V, the peaks at 164.9 and 163.6 eV can be attributed to the sulfur on the nanosheets of 3DG/TM. The new features between 168 and 172 eV are due to the thiosulfate and polythionate species formed by the redox reaction between LiPSs and 1T MoS2 in 3DG/TM, as is consistent with previous reports.42 When the cell was scanned to the Epc, the sulfur on 3DG/TM can be efficiently reduced to LiPSs; therefore, the sulfur peaks diminish. On the other hand, limited sulfur deposited on the surface of 3DG when the cell was scanned to 3.0 V, as indicated by the weak intensity of sulfur peaks observed in Fig. 5f. When the cell was returned to 2.29 V from 3.0 V, the weak peak detected at 165–161 eV can be attributed to the minor residual LiPSs in 3DG. The XPS analysis results agree with the results of CV, and they further reveal the excellent electrocatalytic effect of 3DG/TM towards LiPS conversion.
 | ||
| Fig. 5 SEM images of (a) the pristine 3DG/TM electrode and (b) the 3DG/TM working electrode removed from the three-electrode system after scanning to 2.4 V; (c) the pristine 3DG electrode and (d) the 3DG working electrode after scanning to 3.0 V. XPS spectra of the (e) 3DG/TM and (f) 3DG working electrodes of the three-electrode system after scanning to 3.0 V, or after scanning to 3.0 V and returning to 2.29 V. | ||
The long-term cyclability is a key factor to evaluate the catalytic stability, which was further examined in 2032 coin cells with metallic Li as the anode and the freestanding 3DG/TM, 3DG/HM, and 3DG with a catholyte consisting of 1 M Li2S6 as the cathode. As shown in Fig. 6a, the cell with 3DG/TM exhibits superior performance, with a highly reversible discharge capacity of 1181 mA h g−1 and a capacity retention of 96.3% after 200 cycles. In contrast, the cells with 3DG/HM and 3DG display poor cyclic performance, with a low capacity retention of 72.6% and 42.6%, due to the limited catalytically active sites. It is noted that the coulombic efficiency of 3DG/TM is the highest compared to those of 3DG/HM and 3DG, which further confirms that the high electrocatalytic ability of 3DG/TM can significantly mitigate the LiPS shuttling. The photographs of the separators and lithium foils extracted from cells with 3DG, 3DG/HM, and 3DG/TM after 200 cycles show that the LiPS dissolution is remarkably suppressed in Li–S cells with 3DG/TM (Fig. S14a–f, ESI†). The corrosion depths on these lithium foils were also obtained from the cross-sectional SEM morphologies of the cycled lithium foil paired with 3DG, 3DG/HM, and 3DG/TM (Fig. S14g–I, ESI†). The lithium foil paired with 3DG/TM shows the smallest corrosion depth, which clearly demonstrates the significant mitigation of LiPS shuttling.
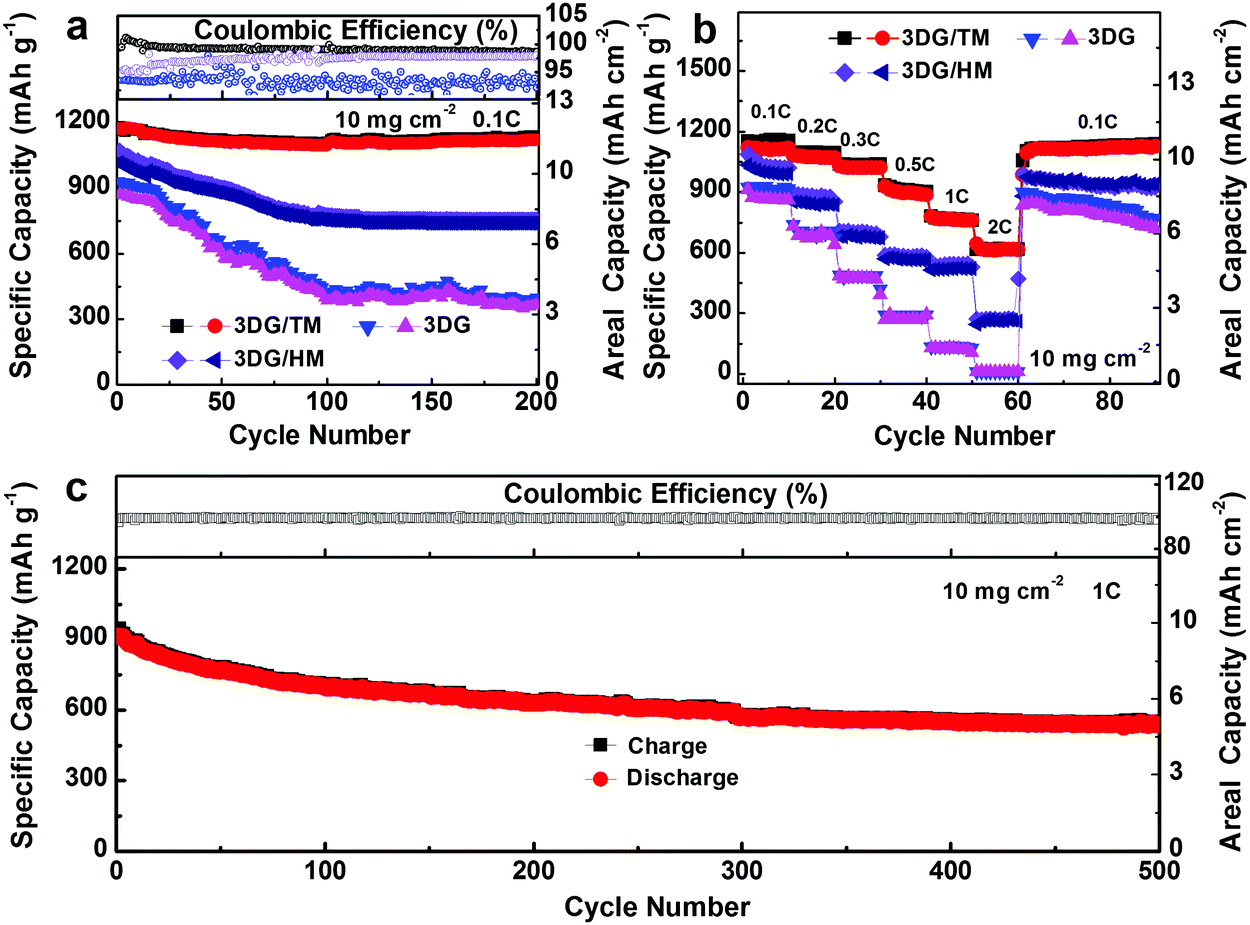 | ||
| Fig. 6 (a) Cyclic stability of electrocatalytically active 3DG, 3DG/HM, and 3DG/TM as working electrodes vs. Li/Li+ with a catholyte consisting of 1 M Li2S6 at 0.1C rate. (b) Rate performances at various cycling rates of 3DG/TM with the catholyte. (c) Long-term cycling cyclability of 3DG/TM with the catholyte at 1C rate. | ||
The rate performance reflects the redox reaction kinetics of the Li–S battery. The cells with 3DG/TM, 3DG/HM, and 3DG were also cycled under various C-rates. As shown in Fig. 6b and Fig. S15–S17 (ESI†), the high capacity of 613 mA h g−1 at the rate of 2C of the cell with 3DG/TM directly illustrates the accelerated rate of Li–S redox conversion in 3DG/TM. In contrast, the cells with 3DG/HM and 3DG exhibit inferior rate performance, owing to the slow kinetics towards LiPS conversion. More importantly, compared to 3DG/HM and 3DG, the EIS spectrum of 3DG/TM after 200 cycles (Fig. S18, ESI†) suggests that the cell with 3DG/TM has high redox kinetics, which matches well with the rate capability results. To further determine whether the 3DG/TM can retain the high redox kinetics during long-term cycling, the prolonged cycling performance of the cells with 3DG/TM was evaluated at 1C rate for 500 cycles (Fig. 6c). The cell with 3DG/TM exhibits outstanding cyclic performance at a high rate of 1C for 500 cycles with a low capacity fading of 0.08% per cycle. Such an appreciable cyclic performance directly demonstrates that 3DG/TM can maintain fast redox kinetics and remain stable even under super long-term cycles. The excellent electrochemical performance of the cells with 3DG/TM complements the electrocatalytic analysis, which further confirms the outstanding electrocatalytic activity of 3DG/TM.
Conclusions
In summary, we have designed and synthesized freestanding, three-dimensional graphene/1T MoS2 (3DG/TM) heterostructures as a highly efficient electrocatalyst for polysulfides. The metallic 1T MoS2 nanosheets are hydrophilic with rich active sites. The high electronic conductivity of 1T MoS2 facilitates electron transfer, while the dense active sites ensure sufficient catalytic activity for LiPSs. The 3DG/TM heterostructures can maximize the aspect ratio of active catalytic sites, and the freestanding, hydrophilic feature of the 3DG/TM porous nanoarchitecture can facilitate electrolyte accessibility, thus enhancing ion transport. Benefiting from these synergistic effects, the cells with 3DG/TM exhibit excellent specific capacity and outstanding cycling stability. Moreover, the experimental and theoretical results suggest that the enhanced catalytic activity in the MoS2-supported systems is primarily due to the enhanced binding and availability of LiPSs at the electrode interface. A fundamental understanding of the electrocatalysis of polysulfides can provide new insights and opportunities to develop advanced Li–S batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering under award number DE-SC0005397. The TEM work was supported by the National Natural Science Foundation of China (Grant No. 21773024). The computational work was supported by the Welch Foundation Grant F-1535. The authors thank the Texas Advanced Computing Center (TACC) for providing HPC resources. The authors thank Prof. Yang Xia and Ms Martha Gross for helpful discussion.Notes and references
- L. Li, S. Chai, S. Dai and A. Manthiram, Energy Environ. Sci., 2014, 7, 2630 RSC.
- F. Wu, T. P. Pollard, E. Zhao, Y. Xiao, M. Olguin, O. Borodin and G. Yushin, Energy Environ. Sci., 2018, 11, 807 RSC.
- Y. You, X. Wu, Y. Yin and Y. Guo, Energy Environ. Sci., 2014, 7, 1643 RSC.
- Y. Fu and A. Manthiram, J. Phys. Chem. C, 2012, 116, 8910 CrossRef CAS.
- J. Zhou, R. Li, X. Fan, Y. Chen, R. Han, W. Li, J. Zheng, B. Wang and X. Li, Energy Environ. Sci., 2014, 7, 2715 RSC.
- Y. Yang, G. Zheng and Y. Cui, Energy Environ. Sci., 2013, 6, 1552 RSC.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS.
- Y. Fu, Y. Su and A. Manthiram, Adv. Energy Mater., 2014, 4, 201300655 Search PubMed.
- Z. Li, J. Zhang, Y. Lu and X. W. D. Lou, Sci. Adv., 2018, 4, t1687 CrossRef PubMed.
- M. J. Klein, G. M. Veith and A. Manthiram, J. Am. Chem. Soc., 2017, 139, 9229 CrossRef CAS PubMed.
- M. J. Klein, G. M. Veith and A. Manthiram, J. Am. Chem. Soc., 2017, 139, 10669 CrossRef CAS PubMed.
- Y. Fu, C. Zu and A. Manthiram, J. Am. Chem. Soc., 2013, 135, 18044 CrossRef CAS PubMed.
- H. Xu, S. Wang and A. Manthiram, Adv. Energy Mater., 2018, 1800813 CrossRef.
- C. Nan, Z. Lin, H. Liao, M. Song, Y. Li and E. J. Cairns, J. Am. Chem. Soc., 2014, 136, 4659 CrossRef CAS PubMed.
- J. He, Y. Chen, W. Lv, K. Wen, C. Xu, W. Zhang, Y. Li, W. Qin and W. He, ACS Nano, 2016, 10, 10981 CrossRef CAS.
- J. He, Y. Chen, P. Li, F. Fu, Z. Wang and W. Zhang, J. Mater. Chem. A, 2015, 3, 18605 RSC.
- T. Lei, W. Chen, J. Huang, C. Yan, H. Sun, C. Wang, W. Zhang, Y. Li and J. Xiong, Adv. Energy Mater., 2016, 1601843 Search PubMed.
- W. Chen, T. Qian, J. Xiong, N. Xu, X. Liu, J. Liu, J. Zhou, X. Shen, T. Yang, Y. Chen and C. Yan, Adv. Mater., 2017, 29, 1605160 CrossRef.
- R. Fang, S. Zhao, P. Hou, M. Cheng, S. Wang, H. Cheng, C. Liu and F. Li, Adv. Mater., 2016, 28, 3374 CrossRef CAS PubMed.
- G. Zhou, S. Pei, L. Li, D. Wang, S. Wang, K. Huang, L. Yin, F. Li and H. Cheng, Adv. Mater., 2014, 26, 625 CrossRef CAS PubMed.
- J. Zhang, C. Yang, Y. Yin, L. Wan and Y. Guo, Adv. Mater., 2016, 28, 9539 CrossRef CAS.
- S. Xin, L. Gu, N. Zhao, Y. Yin, L. Zhou, Y. Guo and L. Wan, J. Am. Chem. Soc., 2012, 134, 18510 CrossRef CAS.
- X. Liu, J. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef.
- A. Manthiram, S. Chung and C. Zu, Adv. Mater., 2015, 27, 1980 CrossRef CAS.
- C. Milroy and A. Manthiram, Adv. Mater., 2016, 28, 9744 CrossRef CAS.
- J. He, Y. Chen and A. Manthiram, Energy Environ. Sci., 2018, 11, 2560 RSC.
- J. He, L. Luo, Y. Chen and A. Manthiram, Adv. Mater., 2017, 29, 1702707 CrossRef PubMed.
- Z. Xiao, Z. Yang, L. Wang, H. Nie, M. Zhong, Q. Lai, X. Xu, L. Zhang and S. Huang, Adv. Mater., 2015, 27, 2891 CrossRef CAS.
- Z. Cui, C. Zu, W. Zhou, A. Manthiram and J. B. Goodenough, Adv. Mater., 2016, 28, 6926 CrossRef CAS.
- Y. Zhang, Z. Mu, C. Yang, Z. Xu, S. Zhang, X. Zhang, Y. Li, J. Lai, Z. Sun, Y. Yang, Y. Chao, C. Li, X. Ge, W. Yang and S. Guo, Adv. Funct. Mater., 2018, 28, 1707578 CrossRef.
- M. Wang, L. Fan, D. Tian, X. Wu, Y. Qiu, C. Zhao, B. Guan, Y. Wang, N. Zhang and K. Sun, ACS Energy Lett., 2018, 3, 1627 CrossRef CAS.
- Y. C. Jeong, J. H. Kim, S. H. Kwon, J. Y. Oh, J. Park, Y. Jung, S. G. Lee, S. J. Yang and C. R. Park, J. Mater. Chem. A, 2017, 5, 23909 RSC.
- G. Babu, K. Ababtain, K. Y. S. Ng and L. M. R. Arava, Sci. Rep., 2015, 5 Search PubMed.
- H. Al Salem, G. Babu, C. V. Rao and L. M. R. Arava, J. Am. Chem. Soc., 2015, 137, 11542 CrossRef CAS PubMed.
- Z. Yuan, H. Peng, T. Hou, J. Huang, C. Chen, D. Wang, X. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519 CrossRef CAS.
- G. Babu, N. Masurkar, H. Al Salem and L. M. R. Arava, J. Am. Chem. Soc., 2016, 139, 171 CrossRef.
- K. Chang, D. Geng, X. Li, J. Yang, Y. Tang, M. Cai, R. Li and X. Sun, Adv. Energy Mater., 2013, 3, 839 CrossRef CAS.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef CAS PubMed.
- J. He, P. Li, W. Lv, K. Wen, Y. Chen, W. Zhang, Y. Li, W. Qin and W. He, Electrochim. Acta, 2016, 215, 12 CrossRef CAS.
- X. Geng, Y. Jiao, Y. Han, A. Mukhopadhyay, L. Yang and H. Zhu, Adv. Funct. Mater., 2017, 1702998 CrossRef.
- Q. Liu, X. Li, Q. He, A. Khalil, D. Liu, T. Xiang, X. Wu and L. Song, Small, 2015, 11, 5556 CrossRef CAS.
- X. Geng, W. Sun, W. Wu, B. Chen, A. Al-Hilo, M. Benamara, H. Zhu, F. Watanabe, J. Cui and T. Chen, Nat. Commun., 2016, 7, 10672 CrossRef CAS.
- J. He, Y. Chen, W. Lv, K. Wen, Z. Wang, W. Zhang, Y. Li, W. Qin and W. He, ACS Nano, 2016, 10, 8837 CrossRef CAS.
- G. Zhou, H. Tian, Y. Jin, X. Tao, B. Liu, R. Zhang, Z. W. Seh, D. Zhuo, Y. Liu, J. Sun, J. Zhao, C. Zu, D. S. Wu, Q. Zhang and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 840 CrossRef CAS.
- Y. C. Jeong, J. H. Kim, S. H. Kwon, J. Y. Oh, J. Park, Y. Jung, S. G. Lee, S. J. Yang and C. R. Park, J. Mater. Chem. A, 2017, 5, 23909 RSC.
- S. P. Jand, Y. Chen and P. Kaghazchi, J. Power Sources, 2016, 308, 166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee03252a |
| This journal is © The Royal Society of Chemistry 2019 |