
A biomass based free radical scavenger binder endowing a compatible cathode interface for 5 V lithium-ion batteries†
Yue
Ma‡
ab,
Kai
Chen‡
a,
Jun
Ma
a,
Gaojie
Xu
a,
Shanmu
Dong
a,
Bingbing
Chen
a,
Jiedong
Li
a,
Zheng
Chen
a,
Xinhong
Zhou
c and
Guanglei
Cui
 *a
*a
aQingdao Industrial Energy Storage Research Institute, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao, 266101, P. R. China. E-mail: cuigl@qibebt.ac.cn; Fax: +86-532-80662746; Tel: +86-532-80662746
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cCollege of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao, 266042, P. R. China
First published on 17th November 2018
Abstract
5 V lithium ion batteries (LIBs) are promising candidates for high energy density batteries. However, conventional carbonate-based liquid electrolyte is vulnerable to oxidative decomposition caused by free radical attack, which leads to poor cycling performance of the 5 V LIBs. Herein, we present a novel strategy based on the free radical scavenging effect to suppress the electrolyte decomposition of 5 V class batteries composed of LiNi0.5Mn1.5O4 (LNMO) cathodes and carbonate-based electrolyte. Our strategy is to scavenge the free radicals during the charging process at the cathode interface by adopting a renewable biomass lignin binder with numerous phenol groups, which can significantly suppress the free radical chain reaction and subsequently generate a compatible multi-dimensional interphase between the electrode and electrolyte. The lignin based electrode exhibited a capacity retention of 94.1% after 1000 cycles, which is significantly higher than that of its PVDF counterpart (46.2%). This work represents a milestone contribution to the strategy for resolving the interfacial issue of high voltage cathode materials, initiating a big step in boosting 5 V batteries.
Broader context5 V high voltage cathodes have attracted more and more interest since the development of lithium-ion batteries with high energy density has become strongly desired. However, the commonly used carbonate-based electrolyte is susceptible to oxidative decomposition in the presence of high voltage cathodes. The electrolyte decomposition on the cathode is usually caused by the continuous generation of free radicals, which is often overlooked in the study of lithium-ion batteries. Lignin is one of the most promising binder materials for lithium-ion batteries owing to its outstanding adhesion capability and abundant reserves in nature. The phenolic groups in the backbone structure of lignin could scavenge free radicals and terminate the free radical chain reaction which helps to suppress the oxidative decomposition of the electrolyte. In contrast, conventionally used binders such as PVDF don’t possess the capability of scavenging free radicals. Therefore, an electrode with a lignin binder could effectively suppress the electrolyte decomposition caused by free radicals, protect the cathode/electrolyte interface, and ultimately exhibit outstanding electrochemical properties. It is worth noting that the LiNi0.5Mn1.5O4/Li batteries with a lignin binder encapsulated in the cathode could achieve the most stable cycling performance reported in the literature to date. |
Introduction
The electrochemical performance of high voltage lithium ion batteries (LIBs) is significantly influenced by the interfacial compatibility of the electrodes with electrolytes composed of solvents and Li salts.1 However, state-of-the-art carbonate based electrolytes are prone to oxidative decomposition when the cathode voltage exceeds 4.3 V.2–9 This is because carbonate based electrolytes are catalyzed to decompose during charging at the cathode/electrolyte interface, and thereby produce undesirable byproducts, leading to low coulombic efficiency and poor cycling performance.10,11 More importantly, the parasitic reactions between the carbonate based electrolyte and the fully-charged electrode proceed continuously during the entire cell life-span, resulting in the rapid capacity fading of LIBs.4,12It is widely accepted that free radical reactions are the decomposition culprits for carbonate based electrolytes in LIBs.13–16 At the interface of the cathode, once the carbonate solvent loses electrons during charging process,13,14 alkyl free radicals, supposed as the intermediate species of the carbonate solvent decomposition reaction, will generate gradually and react simultaneously with the neighbouring solvent molecules, leading to the polymerization of the solvent and the formation of a cathode/electrolyte interphase (CEI) in the presence of lithium salt.17,18 In most cases, the formation of an unstable CEI is achieved at the expense of the massive decomposition of the electrolytes19–26 despite surface modification27–30 and the introduction of additives.31–35 According to the aforementioned free radical reaction mechanism, it can be inferred that suppressing or even avoiding the generation and propagation of alkyl free radicals will be vital to suppressing the decomposition of the electrolyte and maintaining the interfacial compatibility in the electrolyte at the early stages. In this regard, there is great probability that scavenging the free radicals is an effective strategy to significantly stabilize the cathode/electrolyte interface and protect the electrolyte solvent from side reactions, and subsequently extending the battery life-span.
To scavenge the free radicals efficiently, it is better to embattle the radical scavenger at the cathode/electrolyte interface. A binder, which maintains the electrode integrity by encapsulating and binding cathode particles, is a preferential choice to function as the radical scavenger at the multi-dimensional cathode interface. To achieve excellent performance for high voltage LIBs, an ideal cathode/binder system36–38 should possess the attractive characteristics of an efficient radical scavenging capability as well as strong adhesion with the active material, fast Li-ion diffusion, and high resistance to electrochemical oxidation. It is also noted that the as-expected binders should be economical and abundant for large scale application. However, commercially available polyvinylidene fluoride (PVDF) binder fails to fulfil the aforementioned requirements, especially the properties of scavenging free radicals and suppressing the electrolyte decomposition.39–41 Enlighted by the free radical scavenging properties of phenolic architectures,42–44 biomass-based lignin with predominant phenolic groups could serve as a promising candidate, which can potentially capture free radicals efficiently.45 Herein, lignin is presented as the free radical scavenger based binder in a high voltage cell composed of a LNMO cathode and an EC-based electrolyte. The free radical scavenger method adopted here can greatly improve the performance of high voltage batteries; a capacity of 110.8 mA h g−1 is delivered after 1000 cycles at a 1C rate, which is significantly higher than that of PVDF based electrodes (44.2 mA h g−1). To the best of our knowledge, this is the first report of utilizing the multi-functional binder of lignin to solve the issue of an incompatible interface at the high voltage cathode with a free radical scavenger-based concept. We believe the free radical scavenger strategy can be expanded to boost the development of 5 V class batteries.39
Experimental section
Sample collection
LiNi0.5Mn1.5O4 microparticles (Xingneng new material), graphite nanoparticles and acetylene black were used after drying in an oven at 120 °C for 12 h. Lignin powder with a molecular weight of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Solarbio) and PVDF (5130, Sigma-Aldrich Inc.) were purchased and used without any further treatment. The lignin binder is water-soluble, and the concentration of the lignin solution was 10 wt% by dispersing a certain amount of lignin in alkaline water. As for the PVDF binder, N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich, anhydrous, 99.5%) was used as the solvent and the concentration was about 6 wt%. For the preparation of the working electrodes, the active materials, conductive agent and binders were thoroughly mixed in the ratio of 8:1:1 (wt%). Then the obtained homogeneous black slurry was cast onto aluminum foil (or copper foil) and dried at 60 °C in air for 2 h, and then further dried in a vacuum oven at 60 °C and 120 °C for the lignin electrode and PVDF electrodes, respectively.
000 (Solarbio) and PVDF (5130, Sigma-Aldrich Inc.) were purchased and used without any further treatment. The lignin binder is water-soluble, and the concentration of the lignin solution was 10 wt% by dispersing a certain amount of lignin in alkaline water. As for the PVDF binder, N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich, anhydrous, 99.5%) was used as the solvent and the concentration was about 6 wt%. For the preparation of the working electrodes, the active materials, conductive agent and binders were thoroughly mixed in the ratio of 8:1:1 (wt%). Then the obtained homogeneous black slurry was cast onto aluminum foil (or copper foil) and dried at 60 °C in air for 2 h, and then further dried in a vacuum oven at 60 °C and 120 °C for the lignin electrode and PVDF electrodes, respectively.
Electron spin resonance spectroscopy (ESR) tests
ESR tests of the PVDF and lignin based electrodes were conducted with a Bruker E500 electron-spin-resonance system with a frequency of 9.49 GHz. The work principle of ESR lies in the transverse relaxation induced by spin–spin dipolar interactions. It outperforms FTIR, X-ray photoelectron spectroscopy (XPS) and nuclear magnetic resonance (NMR) in sensitivity for detecting radicals containing unpaired electrons. Initially, the excitation energy of microwave power (<10−4 eV) is very low, which does not disturb the system in activated reaction processes. Secondly, ESR can detect unpaired electron signals up to 109 owing to its pretty high sensitivity. Moreover, ESR is specifically used in detecting unpaired electrons in the target system and for investigating radical species-involved trace reactions. Hence, ESR was selected to investigate and compare the free radical generation in the reaction between the fully-charged cathode and the electrolyte solvent after a certain amount of storage time. The preparation of ESR measurement samples is depicted as follows: a fully-charged LNMO electrode with 3 mm width was immersed in electrolyte solution for 1 h (40 μL, 1 M LiPF6 in EC/DMC (1:1, v/v)), and the samples were sealed into a quartz-synthesized ESR tube (id 5 mm). The preparation process was carried out in an argon atmosphere in a glovebox and the humidity and oxygen content were strictly maintained.
Sample characterization
Peeling tests of the two electrodes were conducted using a universal test(ing) machine (MTS, E43). The electrode matrixes were cut into rectangular shapes with 2 cm width and 7 cm length. The active material side was adhered to a wood bar using 3M double sided adhesive tape, and the Al foil side was adhered using traditional 3M tape. The value of the peeling strength could be output directly to a computer. Scanning electron microscopy (SEM, Hitachi S-4800 at 3 kV) and transmission electron microscopy (TEM) studies were conducted to observe the surface morphology of the lignin and PVDF based electrodes before and after cycling (JEM2100PLUS). X-ray diffraction (XRD, Ultima IV) tests were used to compare the structure evolution of the two electrodes. Moreover, the Fourier Transform Infrared spectrum (FT-IR, Bruker VERTEX 70) was used to investigate the chemical structures of lignin. The surface element composition was characterized using X-ray photoelectron spectroscopy (XPS, ESCALab220i-XL) and an Ultraviolet spectrophotometer (UV) was used to verify the lignin scavenging effect on free radicals. Coupled plasma-mass spectrometry (ICP-MS, Optima 8300) measurements were also utilized to compare the concentrations of dissolved manganese ions from the lignin and PVDF based electrodes. High performance liquid chromatography/mass spectrometry (HPLC-MS) spectra and gas chromatography-mass spectrometry (GC-MS) studies were conducted to investigate the solution decomposition byproducts with the two kinds of electrodes. In this section, fully-charged electrodes were soaked in electrolyte solvent, then the pure EC/DMC was taken as a fair comparison. Gel permeation chromatography liquid chromatography (GPC-LC) was utilized using a Breeze 2 HPLC system. The electrochemical decomposition of EC is sensitive to the Li content in the cathode in the fully-charged state, and the ring-opening reaction of EC becomes very easy. Therefore, we investigated the electrolyte solvent decomposition behavior in fully-charged batteries in the following work. Digital photographs (photos) were also used to observe the color changes of 1,1-diphenyl-2-picrylhydrazyl (DPPH) after the addition of lignin and other natural materials with similar structures. Thermogravimetric analysis (TG) was used to characterize the thermostability of the lignin polymer and the electrolyte solution.Electrochemical tests
2032 coin cells were assembled to investigate the extensive electrochemical properties of the different binders. The half-cell using lithium foil as the counter electrode and the full-cell using graphite as the counter electrode were both assembled in a glove box full of Ar, and the oxygen and water contents were strictly controlled below 0.1 ppm. The electrolyte of 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1:1, v/v) was used. Electrochemical impedance spectroscopy (EIS) measurements in the frequency range from 100 mHz to 7 MHz were carried out to compare the impedance and polarization evaluations of the two electrode system before and after cycling. Three-electrode polypropylene cells were used for electrolysis.
Galvanostatically charging/discharging tests were carried out on a Land battery test system (Land CT2001A, Wuhan Land Electronic Co. Ltd, China) in the voltage range between 3.5 V and 5.0 V (vs. Li+/Li). The loading density of the electrode materials was 1.5–1.8 mg cm−2.
Results and discussion
Investigation of the free radical scavenging ability of lignin
Lignin is able to exhibit considerable binding capability and robust peeling strength in electrodes, which mainly originates from the strong hydrogen bonding endowed by abundant –OH groups in the backbone (Fig. S1, ESI†).46 Furthermore, since lignin is water-soluble, the processing of the lignin based electrode is environmentally benign and sustainable. Therefore, it is expected that lignin has huge potential as an advanced multi-functional binder that can ensure the integrity of the electrode matrix (Fig. S2, ESI†) and simultaneously provide extraordinary free radical scavenging capability for LIBs.ESR is specialized in detecting unpaired electrons in the target system through investigating radical species-involved trace reactions. Hence, to verify the radical scavenging effect of the lignin binder, ESR was employed to investigate the free radicals generated between the fully-charged LNMO cathode and the electrolyte solvent (1 M LiPF6 in EC/DMC).13,19,47,48 In the symmetrical absorption spectrum of the PVDF based electrode in Fig. 1a, a hyperfine coupling tensor with a g value of 2.002 strongly confirms the existence of organic radical species.49 In stark contrast, no ESR peaks can be observed in the lignin based electrode, demonstrating the absence of free radicals and the efficient radical scavenging ability of the lignin binder.
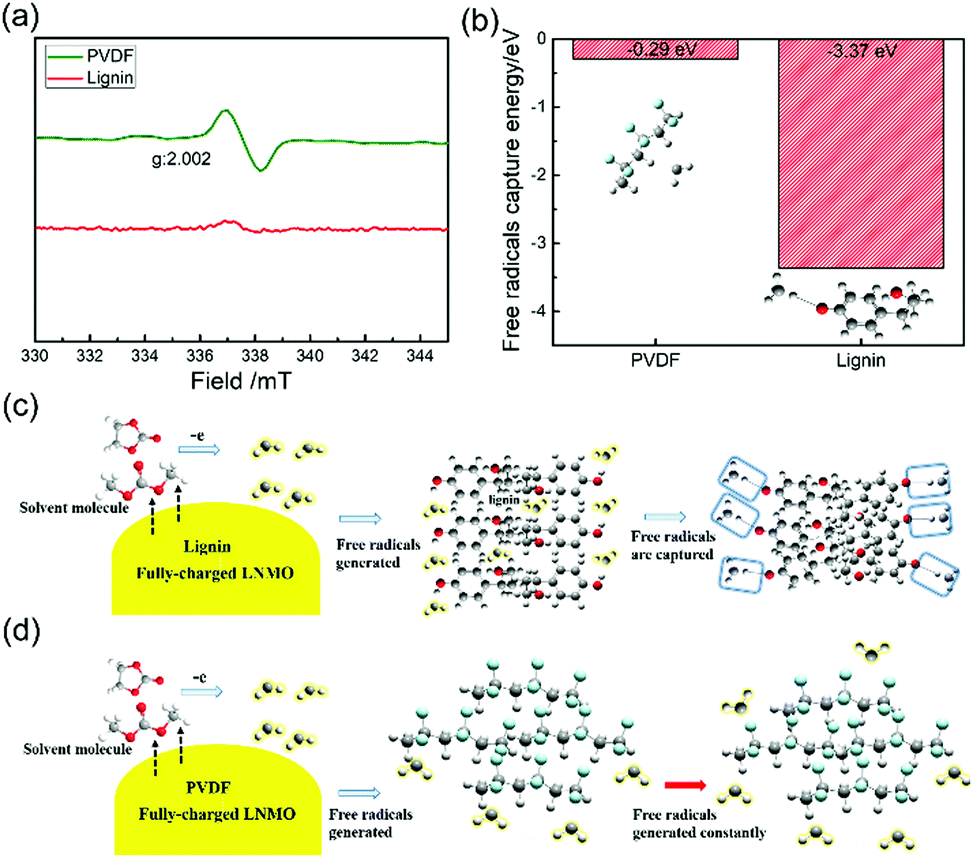 | ||
| Fig. 1 (a) ESR signals for electrolyte solvent from LNMO-half cells at a charged state with lignin and PVDF binders subjected to 5.0 V. (b) Potential energy diagram for the reaction between free radicals and the lignin/PVDF binder. Diagrammatic illustration of the free radical scavenging ability of (c) lignin and (d) PVDF. | ||
First principles simulations were utilized to investigate the trapping energies between the binder and alkyl free radicals, as shown in Fig. 1b, based on density functional theory (DFT). The DFT-B3LYP/6-311G level calculations were carried out using the Gaussian 09 program. The trapping energy of free radicals on lignin (−3.37 eV) is much stronger compared with that of PVDF (−0.29 eV), further demonstrating the effective free radical scavenging ability of the lignin binder. Ultraviolet (UV) spectroscopy was also investigated to indicate the free radical scavenging role of lignin by adding it into 1,1-diphenyl-2-picrylhydrazyl (DPPH), and the value of the absorbance profiles of the mixed solution decreases remarkably compared to pure DPPH, as shown in Fig. S3 (ESI†). This capability can also be observed in other natural molecules with similar structures to lignin (Fig. S4, ESI†). Based on the ESR spectra and DFT calculations as well as the UV-Vis results, we carefully propose the radical capturing process as shown in Fig. 1c. It is deduced that once free radicals generate at the LNMO/electrolyte interface, the lignin binder captures and devitalizes them effectively. The PVDF binder can’t display the capability of trapping free radicals, as depicted in Fig. 1d as a fair comparison.
Characterization of electrolyte decomposition
To investigate the free radical scavenger role of lignin in the suppression of electrolyte decomposition, gas chromatography-mass spectrometry (GC-MS) was conducted on the EC/DMC solvents before and after soaking the lignin and PVDF based fully-charged electrodes. The results are compared in Fig. S5 and Table S1 (ESI†). New species were detected in the solvent after adding the lignin and PVDF based electrodes, respectively, and many more kinds of by-product with higher amounts were generated with the PVDF based electrode, which results from oxidative decomposition of the EC/DMC solvent induced by free radical reactions.Detailed results from high performance liquid chromatography-mass spectrometry (HPLC-MS) shown in Fig. 2 further confirm the protective role of the lignin binder in alleviating the electrolyte solvent decomposition. The five peaks for the pristine EC/DMC sample correspond to EC/DMC species and the related polymerization products (Fig. 2a). Cation radicals would appear after the removal of one electron from EC/DMC molecules during the charging process.50–53 Then ring opening can occur, accompanied by a reaction attacking a neighbouring EC/DMC molecule, finally leading to the formation of various polycarbonates (Fig. S6, ESI†). A new characteristic peak appeared at 231.0 in the HPLC-MS of the PVDF based electrode soaked solvent, which was assigned to an oxidation product with a formula of C7H12O7. A likely structure of this compound may be CH3OCH2CH2OCOOCH2OCOOCH3. This structure may be regarded as the reaction product of a recombination of one EC molecule and two DMC molecules. Another HPLC peak of the oxidized solution at 261.0 corresponds to a compound with the formula C8H14O8, which may be assigned to the following structure: CH3OCOOCH2OCH2CH2OCH2OCOOCH3. This compound is supposed to be originated from the recombination reaction of two DMC molecules with one EC molecule (Fig. 2b).18 In sharp contrast, there is no possibility to detect these new peaks in the lignin based electrode soaked solvent (Fig. 2c) or in pristine EC/DMC, suggesting no severe decomposition reactions occur. This result indicates that a series of complicated solvent oxidation decomposition reactions occur in the PVDF based electrode in the presence of the fully charged LNMO cathode with PVDF binder. The possible chain reaction process is depicted in Fig. 2d.15,18 When the solvents lose electrons during the charging process, alkyl free radicals are generated, then the free radical chain reactions and the decomposition of the electrolyte solvent proceed continuously, leading to the formation of unfavourable decomposition products. It is deduced that the lignin binder could scavenge the free radicals at the beginning of the charging process, suppress the chain reaction of the radical cations and prevent the solvent from severe decomposition. Moreover, the generation, propagation and transfer reactions of free radicals were characterized in three-electrode polypropylene cells with LNMO as the cathode and Li foil as the reference electrode and counter electrode. The decreased UV absorbance value of DPPH suggested the generation of free radicals after mixing with the decomposed electrolyte (Fig. S7, ESI†). Gel permeation chromatography liquid chromatography (GPC-LC) was used to analyse the molecular weight of the decomposed electrolyte. The average molecular weight of the decomposed electrolyte increases as the electrolysis time increases (Fig. S8, ESI†). Furthermore, the TG profile of the decomposed electrolyte after electrolyzing for 24 h exhibits a lower weight loss of 56.9% than that of the sample electrolyzed for 5 min (67.4%). These results suggest the propagation of free radicals. In Fig. S9 (ESI†), a new peak corresponding to the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O appears in the FTIR spectrum of recycled lignin from the lignin/electrolyte after electrolyzing, which indicates the transfer of carbonic esters originated from the free radicals to lignin.
O appears in the FTIR spectrum of recycled lignin from the lignin/electrolyte after electrolyzing, which indicates the transfer of carbonic esters originated from the free radicals to lignin.
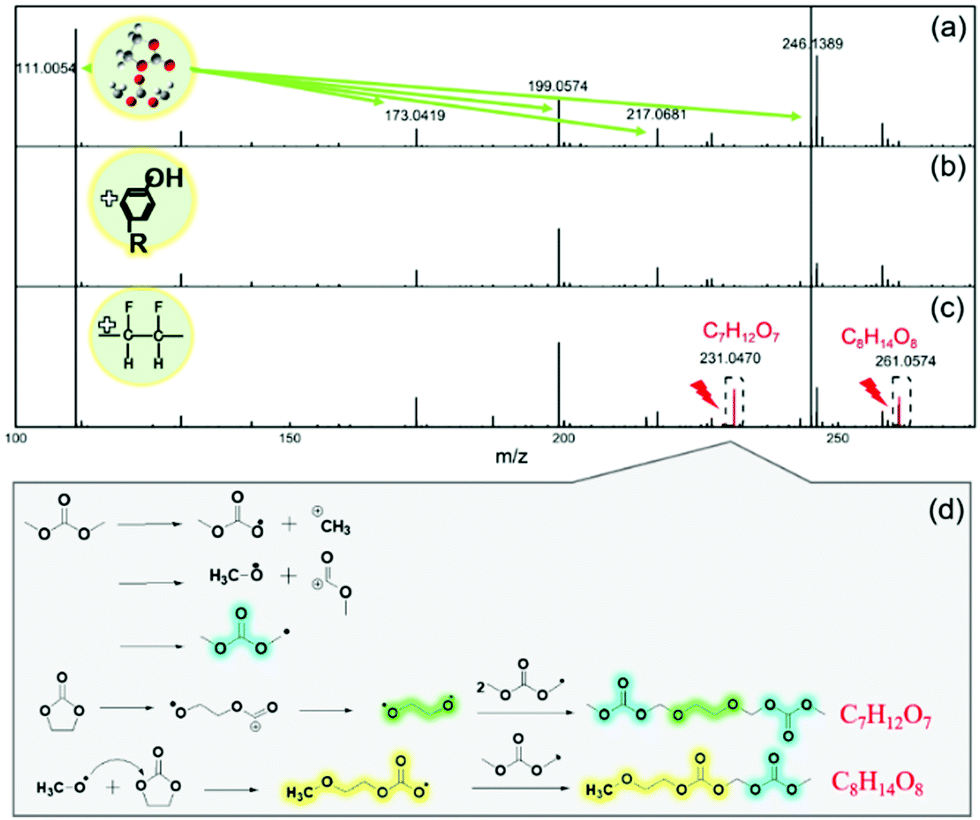 | ||
| Fig. 2 HPLC-MS chromatograms showing the composition of pure EC/DMC solution (a), EC/DMC solution in the presence of fully-charged (b) lignin based electrode and (c) PVDF based electrode. (d) Schematic illustration for the possible decomposition reactions of the electrolyte solvent.18,53 | ||
Electrochemical performance
Half-cell configurations were utilized to evaluate the electrochemical performance of the lignin and PVDF based electrodes in the voltage range between 3.5 V and 5.0 V at a current rate of 1C (150 mA g−1), as shown in Fig. 3a. For the PVDF based electrode, a discharge capacity lower than 100 mA h g−1 with an average coulombic efficiency (CE) of 99.0% was delivered after 1000 cycles (capacity retention: 46.2%). In sharp contrast, the lignin based electrode delivers a high capacity of 110.8 mA h g−1 after cycling with an average CE of 99.5% (capacity retention: 94.1%). In Fig. 3b, the long and flat plateau at 4.7 V represents the redox reaction involving Ni2+/3+/4+, and the plateau near 4.0 V can be attributed to the Mn3+/4+ couple.54 Compared to the PVDF based electrode, the lignin based electrode displays less capacity fading and polarization. In the LNMO/graphite full-cells shown in Fig. S10 (ESI†), the lignin full-cell delivers a higher capacity of 98.9 mA h g−1 after 100 cycles than that of its PVDF counterpart at a 1C rate (60 mA h g−1). Furthermore, the lignin based electrode shows better rate performance than the PVDF counterpart (Fig. S11 and S12, ESI†). Considering its extraordinary cyclability and rate capability, lignin is suggested to be a promising candidate for high performance binder of high voltage lithium batteries.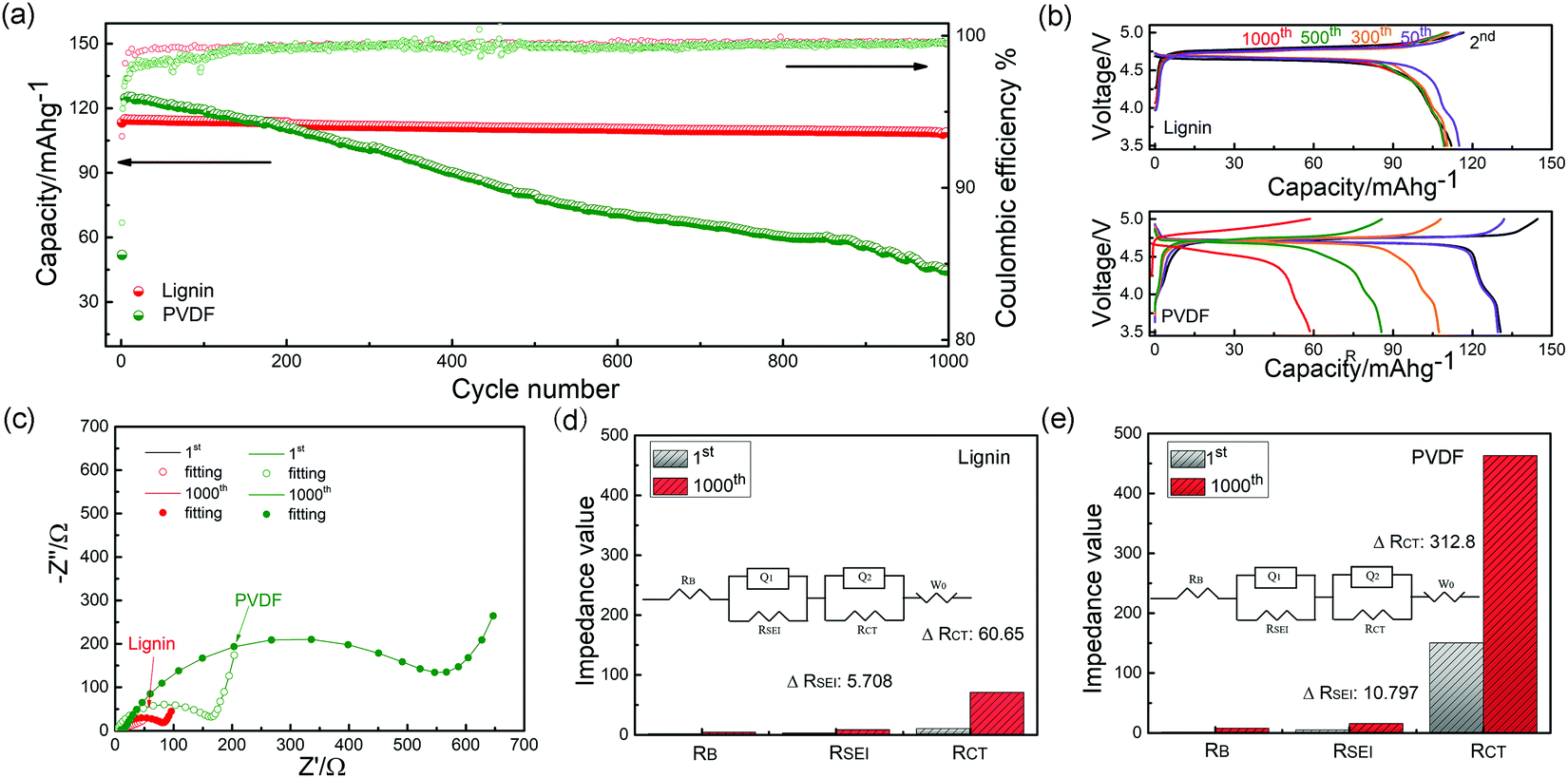 | ||
| Fig. 3 (a) Charge/discharge capacities and coulombic efficiencies of LNMO half-cells with lignin and PVDF binders at 1C (3.5–5.0 V vs. Li+/Li), and (b) The charge/discharge profiles of lignin and PVDF half-cells over 1000 cycles at a rate of 1C. (c) Impedance spectra of fully-discharged lignin and PVDF based electrodes after long-term cycling. Impedance analysis of cycled (d) lignin and (e) PVDF based electrodes. | ||
The interface impedance and charge transfer impedance seem closely related to the increased polarization and capacity fading during cycling. Hence, the electrochemical impedance spectra evolution with the elongated cycles was tested, as shown in Fig. 3c. The details of the impedance fitting data are shown in Fig. 3d and e, where the corresponding impedance for every component is calculated and compared. RB relates to the bulk impedance, RSEI stands for the interphase impedance, and RCT corresponds to the electron transfer impedance. After 1000 cycles, the ΔRSEI and ΔRCT for the lignin half-cell are only 5.4 Ω and 60.65 Ω, compared with 10.7 Ω and 312.8 Ω for its PVDF counterpart, respectively. The significant high impedance of the PVDF based electrode results from severe electrolyte decomposition since many of the decomposition species covered on the surface of electrode are poor electron conductors.27 In sharp contrast, the electrolyte decomposition in the lignin based electrode is suppressed owing to the free radical scavenging capability of the lignin binder and a minor impedance change can be observed. Moreover, a diffusion coefficient of 9.45 × 10−12 cm−2 s−1 for the lignin based electrode was calculated, as shown in Fig. S13 (ESI†), which is 1.5 times that of the PVDF counterpart (6.0 × 10−12 cm−2 s−1), indicating that the diffusion of lithium ions in the interface and bulk of the lignin based electrode is more facile.55 The morphology of the two electrodes after cycling is shown in Fig. S14 (ESI†). The PVDF based electrode shows severe electrode cracking, resulting in increased internal resistance and deteriorated capacity decay. On the contrary, the lignin based electrode is still intact with uniform surface, demonstrating the better binding capability of the lignin binder. The same conclusion can be drawn from the peeling test after cycling, as shown in Fig. S15 (ESI†).
To further explore the positive influence of these two binders on the electrode/electrolyte interface in high voltage batteries, the surface morphology and composition of the cycled LNMO electrode was characterized using TEM and XPS, respectively. As shown in Fig. 4a, a thin compact and homogeneous CEI layer of about 5 nm thickness was observed on the surface of the LNMO particles using the lignin binder, which may be generated from the decomposition of the electrolyte since the pristine LNMO particle demonstrates a smooth edge, as shown in Fig. S16 (ESI†).56 Meanwhile, the PVDF based LNMO particle displays an irregular and uncompact CEI layer, suggesting the favorable role of lignin in forming a uniform CEI layer, which could protect the solvent from further decomposition.57 The composition of the CEI was investigated using XPS. The O1s and F1s spectra of both the PVDF and lignin based electrodes in a pristine state and after 1000 cycles are displayed in Fig. 4c–f, along with the specific peak assignments on the C1s spectra provided in Fig. S17 (ESI†) (comprehensive peak assignments are recorded in Tables S2–S4, ESI†). In the O1s spectrum of LNMO powder, the lattice O can be observed at 529.8 eV and the peak centred at 531.7 eV is assigned to Li2CO3, which already exists from the material fabrication procedure.7 In the pristine state, the lignin based electrode presents a dramatic decrease in the lattice O peak, indicating that the lignin binder is covered on the surface of LNMO particles. Moreover, higher C–O peak intensity can be seen in the lignin based electrode due to the initial presence of C–O groups in lignin. After 1000 cycles, the lattice O can’t be observed, indicating that a compact CEI is generated and covered on the surface of LNMO. The relative ratio of the peak centred at 531.7 eV corresponding to ROCO2Li, Li2CO3, OC–O, and CO is higher in the PVDF based electrode, which may ascribed to the continuous decomposition of solvent caused by free radicals (Fig. S5, ESI†).49,50,58 In Fig. 4d, the F1s spectra of the pristine PVDF based electrode reveals the existence of PVDF binder. After long cycling, the relative ratio of the peak centred at 686 eV assigned to LixPOyFz, LixPFz and LiF, F–Ni/Mn is relatively higher in the PVDF based electrode, which may result from the decomposition of LiPF6 and the PVDF binder.59–61 Both the O1s spectra and F1s spectra indicate more severe electrolyte decomposition in the interface of the PVDF based electrode.
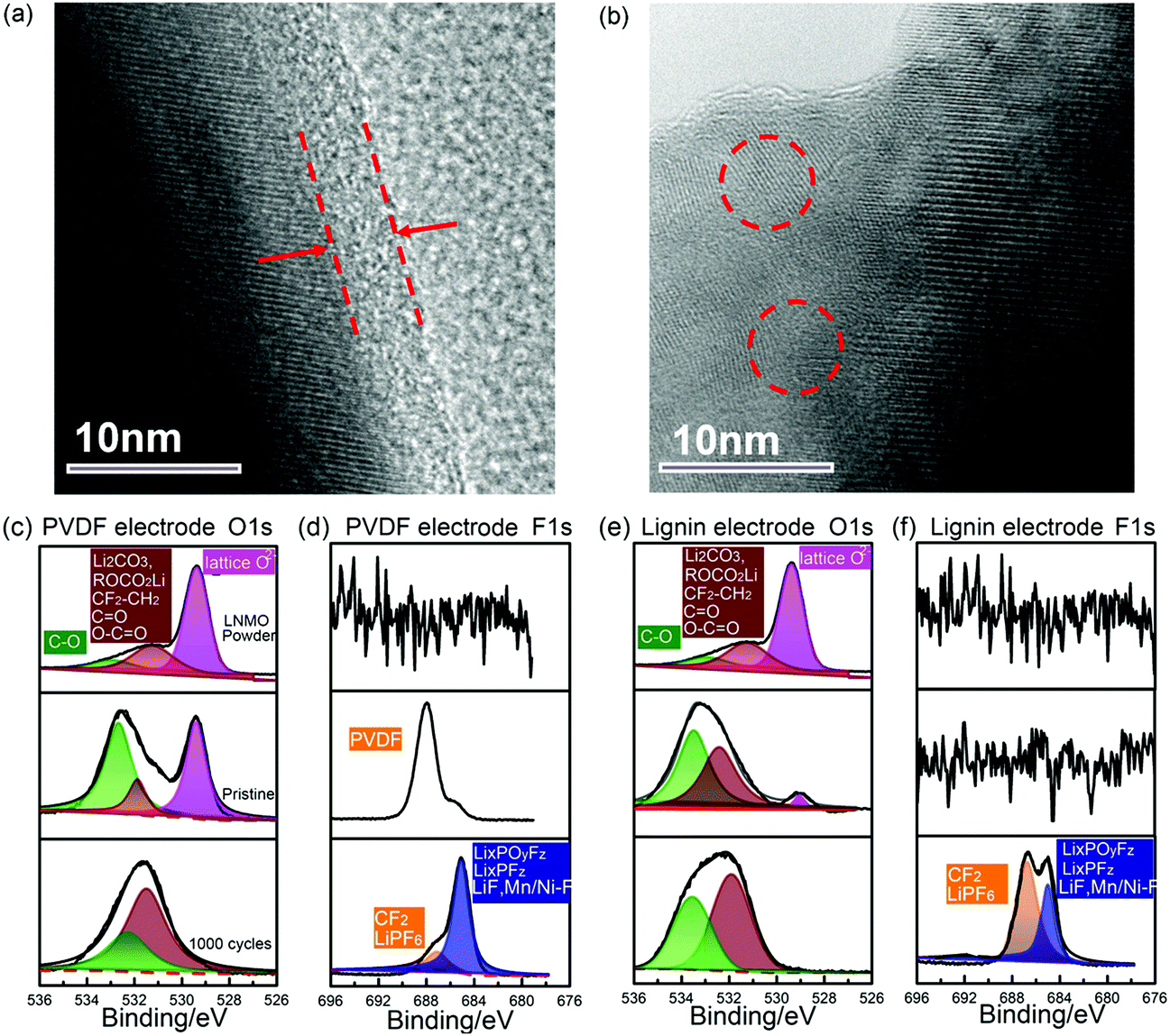 | ||
| Fig. 4 Typical TEM images of (a) the lignin based electrode and (b) the PVDF based electrode after cycling. Surface composition evolution of the PVDF and lignin based electrodes investigated using high-resolution XPS measurements upon long cycling. The (c) O1s and (d) F1s spectra of the PVDF based electrodes in a pristine state and after 1000 cycles. The (e) O1s and (f) F1s spectra of the lignin based electrodes in a pristine state and after 1000 cycles. The pure LNMO powder is taken as a fair comparison. The spectra were calibrated versus the C1s peak of hydrocarbon species at 284.8 eV. | ||
The formation of compatible electrode/electrolyte interfaces
Although the oxidative decomposition of the electrolyte and the dissolution of Mn are usually considered as two isolated issues,62 the PVDF based electrode with severe interfacial electrolyte decomposition shows serious Mn dissolution and structure evolution.63–65 This can be evidenced from the ICP-MS test results shown in Fig. S18 (ESI†) and the XRD pattern exhibited in Fig. S19 (ESI†), from which more dissolved Mn ions are detected and new phases belonging to Li0.08Mn1.72O3.4(OH)0.6 and Li0.2Mn2O4 are generated. The migrated transition metal ions will deposit on the anode and decrease the stability of the anodic interface.66–70 Typical SEM images of lithium foils obtained from the lignin and PVDF half-cells are shown in Fig. S20 (ESI†). Lithium foil obtained from the PVDF based half-cell displays an uneven surface morphology with lots of fractures. The related EDS spectra demonstrate the existence of Mn and Ni elements, which can’t be observed in the lignin counterpart, indicating more transition metal deposited on the lithium foil of the PVDF half-cell. Thereafter, the XPS spectra of lithium foil from the lignin and PVDF half-cells were investigated, as shown in Fig. S21 (ESI†), which demonstrates that higher amounts of species containing Mn or Ni elements are generated on the surface of the lithium foil in the PVDF half-cell. The influence of lignin on the cathode/electrolyte and anode/electrolyte interfaces is depicted in Fig. 5a and b. With abundant phenol structure groups in the backbone structure, lignin scavenges the active free radicals, impedes the free radical chain reaction, and suppresses the severe electrolyte solvent decomposition. This specific structure is favorable for maintaining the integrity of the original cathode material structure, mitigating the dissolution of transition metals, contributing to the formation of stable cathode/electrolyte and anode/electrolyte interfaces, and ultimately improving the cycling performance of the batteries.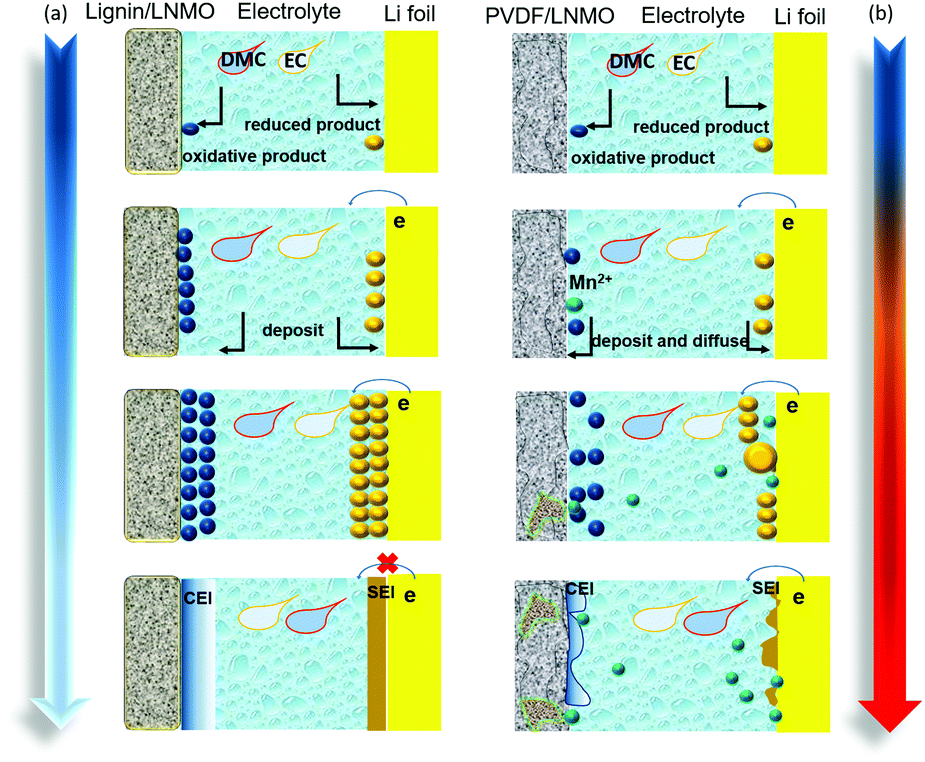 | ||
| Fig. 5 Schematic illustrations of CEI film formation processes based on (a) lignin binder, and (b) PVDF binder. | ||
Conclusion
In this paper, we have demonstrated that a biomass-based lignin binder can address the issues of 5 V class LiMn1.5Ni0.5O4 cathodes. It was deduced that biomass lignin performed as a free radical scavenger to stabilize the electrolyte solvent by continuously capturing the free radicals and constructing favourable multi-dimensional interfaces throughout the battery. Accordingly, a superior cycling life than that of the PVDF counterpart can be achieved. A capacity value of 110.8 mA h g−1 with an overall coulombic efficiency of 99.5% is still delivered after 1000 cycles. Also, the biomass-based lignin binder allows for environmentally benign processes by utilizing water as the solvent in electrode fabrication. The aforementioned advantages would rank lignin as a promising candidate for upcoming high energy density LIBs. It is expected that this strategy can be expanded to other energy storage devices such as Na+, Mg2+ and Zn2+ based ion batteries to achieve superior cycling performances.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation for Distinguished Young Scholars of China (Grant No. 51625204), National Natural Science Foundation of China (Grant No. 51502319), Qingdao Science and Technology Program (Grant No. 17-1-1-26-jch), The Key Research Program of the Chinese Academy of Sciences (Grant No. KFZD-SW-414), and “135” Projects Fund of CAS-QIBEBT Director Innovation Foundation, the Key Scientific and Technological Innovation Project of Shandong (Grant No. 2017CXZC0505).Notes and references
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
- C. Huang, S. Zhuang and F. Tu, J. Electrochem. Soc., 2013, 160, A376–A382 CrossRef CAS.
- D. Lu, M. Xu, L. Zhou, A. Garsuch and B. L. Lucht, J. Electrochem. Soc., 2013, 160, A3138–A3143 CrossRef CAS.
- J. Vetter, P. Novák, M. R. Wagner, C. Veit, K. C. Möller, J. O. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, J. Power Sources, 2005, 147, 269–281 CrossRef CAS.
- R. Dedryvère, D. Foix, S. Franger, S. Patoux, L. Daniel and D. Gonbeau, J. Phys. Chem. C, 2010, 114, 10999–11008 CrossRef.
- L. Suo, Y. S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- J. Cabana, B. J. Kwon and L. Hu, Acc. Chem. Res., 2018, 51, 299–308 CrossRef CAS PubMed.
- L. L. Wang, B. B. Chen, J. Ma, G. L. Cui and L. Q. Chen, Chem. Soc. Rev., 2018, 47, 6505–6602 RSC.
- J. Ma, P. Hu, G. L. Cui and L. Q. Chen, Chem. Mater., 2016, 28, 3578–3606 CrossRef CAS.
- B. Aktekin, M. J. Lacey, T. Nordh, R. Younesi, C. Tengstedt, W. Zipprich, D. Brandell and K. Edström1, J. Phys. Chem. C, 2018, 122, 11234–11248 CrossRef CAS.
- D. Aurbach, B. Markovsky, Y. Talyossef, G. Salitra, H.-J. Kim and S. Choi, J. Power Sources, 2006, 162, 780–789 CrossRef CAS.
- L. Schafzahl, H. Ehmann, M. Kriechbaum, J. Sattelkow, T. Ganner, H. Plank, M. Wilkening and S. A. Freunberger, Chem. Mater., 2018, 30, 3338–3345 CrossRef CAS.
- S. Matsuta, Y. Kato, T. Ota, H. Kurokawa, S. Yoshimura and S. Fujitani, J. Electrochem. Soc., 2001, 148, A7–A10 CrossRef CAS.
- E. Endo, M. Ata, K. Tanaka and K. Sekai, J. Electrochem. Soc., 1998, 145, 3757–3764 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. Abraham, J. Phys. Chem. C, 2013, 117, 19255–19269 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- M. Moshkovich, M. Cagocaru, H. E. Gottlieb and D. Aurbach, J. Electroanal. Chem., 2001, 497, 84–96 CrossRef CAS.
- D. Aurbach, B. Markovsky, A. Shechter, Y. Ein-Eli and H. Cohen, J. Electrochem. Soc., 1996, 143, 3809–3820 CrossRef CAS.
- A. Schechter, D. Aurbach and H. Cohen, Langmuir, 1999, 15, 3334–3342 CrossRef CAS.
- D. Aurbach and H. Gottlieb, Electrochim. Acta, 1989, 34, 141–156 CrossRef CAS.
- T. Eriksson, A. M. Andersson, A. G. Bishop, C. Gejke, T. Gustafsson and J. O. Thomas, J. Electrochem. Soc., 2002, 149, A69–A78 CrossRef CAS.
- M. Kunduraci and G. Amatucci, J. Electrochem. Soc., 2006, 153, A1345–A1352 CrossRef CAS.
- Z. Zhang, L. Hu, H. Wu, W. Weng, M. Koh, P. C. Redfern, L. A. Curtiss and K. Amine, Energy Environ. Sci., 2013, 6, 1806–1810 RSC.
- T. Sasaki, T. Abe, Y. Iriyama, M. Inaba and Z. Ogumi, J. Power Sources, 2005, 150, 208–215 CrossRef CAS.
- T. Sasaki, S.-K. Jeong, T. Abe, Y. Iriyama, M. Inaba and Z. Ogumi, J. Electrochem. Soc., 2005, 152, A1963–A1968 CrossRef CAS.
- J. Liu and A. Manthiram, Chem. Mater., 2009, 21, 1695–1707 CrossRef CAS.
- J.-H. Cho, J.-H. Park, M.-H. Lee, H.-K. Song and S.-Y. Lee, Energy Environ. Sci., 2012, 5, 7124–7131 RSC.
- J.-H. Kim, N. P. W. Pieczonka, P. Lu, Z. Liu, R. Qiao, W. Yang, M. M. Tessema, Y.-K. Sun and B. R. Powell, Adv. Mater. Interfaces, 2015, 2, 1500109 CrossRef.
- G. D. Wu, P. J. Li, C. X. Zhu, Y. Lei, H. X. Zhao, T. T. Li, H. W. Yue, B. P. Dou, Y. H. Gao and X. G. Yang, Electrochim. Acta, 2017, 246, 720–729 CrossRef CAS.
- A. von Cresce and K. Xu, J. Electrochem. Soc., 2011, 158, A337–A342 CrossRef CAS.
- Z. Qin, B. Hong, B. Duan, S. Hong, Y. Chen, Y. Lai and J. Feng, Electrochim. Acta, 2018, 276, 412–416 CrossRef CAS.
- Y. Xu, J. Liu, L. Zhou, L. Zeng and Z. Yang, J. Electroanal. Chem., 2017, 791, 109–116 CrossRef CAS.
- H. Rong, M. Xu, L. Xing and W. Li, J. Power Sources, 2014, 261, 148–155 CrossRef CAS.
- Y. Li, G. M. Veith, K. L. Browning, J. Chen, D. K. Hensley, M. P. Paranthaman, S. Dai and X.-G. Sun, Nano Energy, 2017, 40, 9–19 CrossRef CAS.
- M. Wu, X. Xiao, N. Vukmirovic, S. Xun, P. K. Das, X. Song, P. Olalde-Velasco, D. Wang, A. Z. Weber, L. W. Wang, V. S. Battaglia, W. Yang and G. Liu, J. Am. Chem. Soc., 2013, 135, 12048–12056 CrossRef CAS PubMed.
- S. J. Park, H. Zhao, G. Ai, C. Wang, X. Song, N. Yuca, V. S. Battaglia, W. Yang and G. Liu, J. Am. Chem. Soc., 2015, 137, 2565–2571 CrossRef CAS PubMed.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- Y. Tang, J. Deng, W. Li, O. I. Malyi, Y. Zhang, X. Zhou, S. Pan, J. Wei, Y. Cai, Z. Chen and X. Chen, Adv. Mater., 2017, 1701828 CrossRef PubMed.
- T. Zhang, J. T. Li, J. Liu, Y. P. Deng, Z. G. Wu, Z. W. Yin, D. Guo, L. Huang and S. G. Sun, Chem. Commun., 2016, 52, 4683–4686 RSC.
- H. Wang, V. Sencadas, G. Gao, H. Gao, A. Du, H. Liu and Z. Guo, Nano Energy, 2016, 26, 722–728 CrossRef CAS.
- I. Vulic, G. Vitarelli and J. M. Zenner, Polym. Degrad. Stab., 2002, 78, 27–34 CrossRef CAS.
- Y. Ohkatsu, T. Matsuura and M. Yamato, Polym. Degrad. Stab., 2003, 81, 151–156 CrossRef CAS.
- D. Villano, M. S. Fernandez-Pachon, M. L. Moya, A. M. Troncoso and M. C. Garcia-Parrilla, Talanta, 2007, 71, 230–235 CrossRef CAS PubMed.
- J. M. Rosas, R. l. Berenguer, M. a. J. Valero-Romero, J. RodrÃguez-Mirasoland and T. S. Cordero, Front. Mater., 2014, 1, 29 Search PubMed.
- N. P. W. Pieczonka, V. Borgel, B. Ziv, N. Leifer, V. Dargel, D. Aurbach, J.-H. Kim, Z. Liu, X. Huang, S. A. Krachkovskiy, G. R. Goward, I. Halalay, B. R. Powell and A. Manthiram, Adv. Energy Mater., 2015, 5, 1501008 CrossRef.
- D. Aurbach, Y. Ein-Eli, O. Chusid, Y. Carmeli, M. Babai and H. Yamin, J. Electrochem. Soc., 1994, 141, 603–611 CrossRef CAS.
- D. Aurbach, A. Zaban, Y. Gofer, Y. E. Ely, I. Weissman, O. Chusid and O. Abramson, J. Power Sources, 1995, 54, 76–84 CrossRef CAS.
- K. Kanamura, S. Toriyama, S. Shiraishi and Z. I. Takehara, J. Electrochem. Soc., 1995, 142, 1383–1389 CrossRef CAS.
- L. D. Xing, W. S. Li, C. Y. Wang, F. L. Gu, M. Q. Xu, C. L. Tan and J. Yi, J. Phys. Chem. B, 2009, 113, 16596–16602 CrossRef CAS PubMed.
- O. Borodin, W. Behl and T. R. Jow, J. Phys. Chem. C, 2013, 117, 8661–8682 CrossRef CAS.
- P. Novák and R. Imhof, J. Electrochem. Soc., 1999, 146, 1702–1706 CrossRef.
- X. R. Zhang, J. K. Pugh and P. N. Ross, J. Electroanal. Chem., 2001, 148, E183–E188 CAS.
- N. S. Norberg, S. F. Lux and R. Kostecki, Electrochem. Commun., 2013, 34, 29–32 CrossRef CAS.
- S. L. Chou, J. Z. Wang, H. K. Liu and S. X. Dou, J. Phys. Chem. C, 2011, 115, 16220–16227 CrossRef CAS.
- Y.-H. Lee, J. Min, K. Lee, S. Kim, S. H. Park and J. W. Choi, Adv. Energy Mater., 2017, 7, 1602147 CrossRef.
- A. Jarry, S. Gottis, Y. S. Yu, J. Roque-Rosell, C. Kim, J. Cabana, J. Kerr and R. Kostecki, J. Am. Chem. Soc., 2015, 137, 3533–3539 CrossRef CAS PubMed.
- N.-S. Choi, J.-G. Han, S.-Y. Ha, I. Park and C.-K. Back, RSC Adv., 2015, 5, 2732–2748 RSC.
- J. H. Kim, N. P. Pieczonka and L. Yang, Chem. Phys. Chem., 2014, 15, 1940–1954 CrossRef CAS PubMed.
- T. Yoon, S. Park, J. Mun, J. H. Ryu, W. Choi, Y.-S. Kang, J.-H. Park and S. M. Oh, J. Power Sources, 2012, 215, 312–316 CrossRef CAS.
- L. Fransson, K. Eriksson, K. Edström, T. Gustafsson and J. O. Thomas, J. Power Sources, 2001, 101, 1–9 CrossRef CAS.
- P. Ye, H. Dong, Y. L. Xun, C. J. Zhao and D. Liu, Appl. Surf. Sci., 2018, 428, 469–477 CrossRef CAS.
- L. Zhao, E. Chénard, Ö. Ö. Çapraz, N. R. Sottos and S. R. White, J. Electrochem. Soc., 2018, 165, A345–A348 CrossRef CAS.
- A. Bhandari and J. Bhattacharya, J. Electrochem. Soc., 2017, 164, A106–A127 CrossRef CAS.
- R. Qiao, Y. Wang, P. Olalde-Velasco, H. Li, Y.-S. Hu and W. Yang, J. Power Sources, 2015, 273, 1120–1126 CrossRef CAS.
- D. H. Jang, Y. J. Shin and S. M. Oh, J. Electrochem. Soc., 1996, 143, 2204–2211 CrossRef CAS.
- K. Kanamura, S. Toriyama, S. Shiraish and Z.-I. Takehara, J. Electrochem. Soc., 1995, 142, 1383–1389 CrossRef CAS.
- N. P. W. Pieczonka, Z. Liu, P. Lu, K. L. Olson, J. Moote, B. R. Powell and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 15947–15957 CrossRef CAS.
- G. Pistoia, A. Antonini, A. Rosati and D. Zane, Electrochim. Acta, 1996, 41, 2683–3689 CrossRef CAS.
- B. Xiao, J. Liu, Q. Sun, B. Wang, M. N. Banis, D. Zhao, Z. Wang, R. Li, X. Cui, T. K. Sham and X. Sun, Adv. Sci., 2015, 2, 1500022 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ee02555j |
| ‡ Yue Ma and Kai Chen contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |