
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium(II) complexes with thiosemicarbazones derived from pyrene as topoisomerase IB inhibitors†
Carolina G.
Oliveira
 abc,
Isolda
Romero-Canelón
cd,
Monize M.
Silva
e,
James P. C.
Coverdale
e,
Pedro Ivo S.
Maia
f,
Alzir A.
Batista
e,
Silvia
Castelli
g,
Alessandro
Desideri
*g,
Peter J.
Sadler
*c and
Victor M.
Deflon
*a
abc,
Isolda
Romero-Canelón
cd,
Monize M.
Silva
e,
James P. C.
Coverdale
e,
Pedro Ivo S.
Maia
f,
Alzir A.
Batista
e,
Silvia
Castelli
g,
Alessandro
Desideri
*g,
Peter J.
Sadler
*c and
Victor M.
Deflon
*a
aSão Carlos Institute of Chemistry, University of São Paulo, 13560-970, São Carlos, Brazil. E-mail: deflon@iqsc.usp.br
bInstitute of Chemistry, Federal University of Uberlândia, 38400-902, Uberlândia, Brazil
cDepartment of Chemistry, University of Warwick, CV4 7AL, Coventry, UK
dSchool of Pharmacy, University of Birmingham, B15 2TT, Birmingham, UK
eDepartment of Chemistry, Federal University of São Carlos, 13565-905, São Carlos, Brazil
fDepartment of Chemistry, Federal University of the Triângulo Mineiro, 38025-440, Uberaba, Brazil
gDepartment of Biology, University of Rome Tor Vergata, Via Della Ricerca Scientifica, 00133, Rome, Italy
First published on 30th October 2019
Abstract
New palladium complexes with thiosemicarbazonate ligands derived from pyrene exhibit potent antiproliferative activity against A2780 and cisplatin-resistant A2780Cis human ovarian cancer cells, which is dependent on substituent groups of the thiosemicarbazone ligands. Cellular accumulation and distribution studies confirmed that palladium enters the cell nucleus. DNA and topoisomerase IB studies show that one complex is a potent TopIB inhibitor, with selectivity for cancer versus normal cells.
Introduction
Thiosemicarbazones (TSCs) have been extensively studied due to their ability to chelate several metal ions1 and to suppress tumor growth by inhibiting ribonucleotide reductase (RR), a key enzyme required for DNA synthesis.2,3 Another well-known target for some TSCs derivatives is Topoisomerase (Top), responsible for DNA topology regulation during cell division.4 Recently, it was shown that the cytotoxicity of Dp44mT (di-2-pyridylketone-4,4-dimethyl-3-thiosemi-carbazone, Chart 1) against breast cancer cells is caused by poisoning topoisomerase IIa.5 Dp44mT is structurally similar to triapine (Chart 1), a compound which has undergone Phase 1 and 2 clinical trials.6,7 Both topoisomerase I (Top I) and topoisomerase II (Top II) have been established as effective molecular targets for many antitumor drugs. Several transition metal complexes have been shown to react with topoisomerase IB and act as poisons and/or catalytic inhibitors, since tumor cells present high levels of Top enzymes.8,9 Top IB is the cellular target of camptothecin (CPT), a compound which generated the analogues topotecan and irinotecan, currently in clinical use to treat ovarian and colorectal cancers.10,11 Besides, the functionalization of TSCs provides a strategy to improve their efficiency and to incorporate specific properties. A pyrene group present in a thiosemicarbazone compound might bring new characteristics to the ligand, like fluorescence,12 that would enable its use as a luminescent probe for tracking complexes inside cells. Furthermore, the intercalating properties of pyrene can be used to inhibit cellular DNA replication.13,14 Hence, thiosemicarbazone ligands containing a pyrene moiety (Chart 1) might be promising to achieve metal complexes with mechanisms of action different from cisplatin, making them potentially active against platinum-resistant cell lines. | ||
| Chart 1 Molecular structure of thiosemicarbazones Dp44mT, triapine and the ligands studied here. | ||
In addition to the potential use of thiosemicarbazones in cancer therapy, their derivatives with metal complexes are of considerable interest. New metal complexes might have increased efficacy and the possibility of overcoming resistance, which are the major limitations of the platinum anticancer drugs used in chemotherapy.15,16 In this regard, palladium complexes are natural analogues of platinum. However, palladium complexes usually exhibit high lability towards ligand exchange which limits their use.17 The use of thiosemicarbazone chelating ligands is a strategy to suppress this problem. Furthermore, the coordination of such ligands to palladium could give rise to compounds with a mechanism of action distinct from that of ‘cisplatin like’ complexes, since thiosemicarbazone derivatives complexes have shown different molecular targets.5,18,19
In this work, new palladium complexes derived from pyrene thiosemicarbazones were prepared, and designed to have sufficient stability against hydrolysis in solution together with a ‘non-cisplatin like’ mechanism of action against cancer cells, to overcome the acquired resistance of platinum-based drugs. In view of such potential, the compounds have been studied against A2780 and cisplatin-resistant A2780Cis cancer cell lines and also towards non cancer cells (MRC5). Furthermore, the complexes, cellular accumulation and distribution, interaction with DNA and potential activity as TopIB inhibitors were evaluated to explore their mechanism of action.
Results and discussion
Synthesis and characterization of the compounds
The ligands HPrR (R = ethyl and cyclohexyl) were synthesized via condensation reactions of the corresponding thiosemicarbazide derivative with 1-pyrenecarboxaldehyde (more details in ESI†). The synthesis of the complexes [PdCl(PPh3)(PrCh)] (1) and [PdCl(PPh3)(PrEt)] (2) consisted on 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reactions between [PdCl2(MeCN)2] and the appropriate HPrR ligand, with final addition of PPh3. 1H and 13C NMR results support the proposed structures for all ligands and complexes prepared. The palladium complexes were further studied by 31P NMR. The purity of these compounds was confirmed by elemental analyses and HPLC studies. The high resolution electrospray ionization mass spectrometry (ESI-MS) spectra showed peaks for [M + Na]+ for the free ligands and [M − Cl]+ for the palladium complexes (m/z 752.1485 for 1 and m/z 698.1019 for 2), indicating that the Pd–Cl bond breaks under the experimental conditions used.
:1 reactions between [PdCl2(MeCN)2] and the appropriate HPrR ligand, with final addition of PPh3. 1H and 13C NMR results support the proposed structures for all ligands and complexes prepared. The palladium complexes were further studied by 31P NMR. The purity of these compounds was confirmed by elemental analyses and HPLC studies. The high resolution electrospray ionization mass spectrometry (ESI-MS) spectra showed peaks for [M + Na]+ for the free ligands and [M − Cl]+ for the palladium complexes (m/z 752.1485 for 1 and m/z 698.1019 for 2), indicating that the Pd–Cl bond breaks under the experimental conditions used.
The absorption and emission spectra of the free ligands and complexes were recorded. The ligands HPrCh and HPrEt absorb in the region 382–407 nm and around 282 nm. These bands are most probably due to π–π* electronic transitions associated to the polycyclic aromatic fragment of the ligands.20 The electronic spectra of complexes 1 and 2 are similar, displaying strong absorptions in the 280–410 nm range. These bands may be attributed to a combination of intraligand and ligand-to-metal charge transfer (LMCT) transitions.21
The room temperature emission spectroscopic properties of the thiosemicarbazone ligands were investigated using an excitation wavelength of 415 nm. The emission spectra obtained for both free ligands show two emission bands, at 429 and 448 nm, while complexes 1 and 2 display only one broad band at 540 nm. The fluorescence intensity for both complexes 1 and 2 is clearly less than that of the respective free ligands, evidence that the fluorescence is quenched in the metal complexes.22
X-ray crystal structure analyses were determined for the ligands HPrCh and HPrEt (ESI, Fig. S1†) and complexes 1 and 2. Fig. 1 shows the molecular structure of complex 1, while that of complex 2 is in Fig. S2.† The molecular structures of both the complexes are very similar and show that the metal center is coordinated by the thiosemicarbazone ligands in monoanionic bidentate mode through the nitrogen and sulfur atoms, forming a 5-membered ring. The PPh3 phosphorus atom and the chlorido ligand occupy the remaining coordination sites, forming a distorted square-planar environment around the palladium(II) atom. The bond distances Pd–N(1), Pd–S(1), Pd–Cl(1) and Pd–P(1) for both complexes 1 and 2 are comparable with those of other thiosemicarbazone derivatives reported in literature.23–25 Upon complexation with the metal ion, an elongation of the S(1)–C(2) bond length is observed, in accordance with the coordination via the sulfur atom in a thioenolate form. The N(2)–C(2) bond lengths in 1 [1.294(3) Å] and in 2 [1.302(3) Å] are also consistent with the deprotonation of the thiosemicarbazone ligands after coordination, as observed for other thiosemicarbazone derivatives.26,27 Selected bond lengths and angles are listed in Tables S1 and S2.†
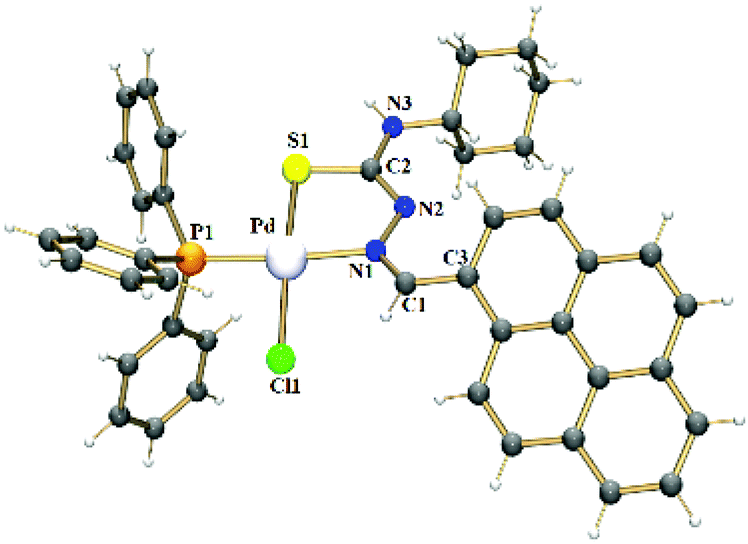 | ||
| Fig. 1 Crystal structure of the complex 1. | ||
The stability of complex 1 was monitored by 1H and 31P NMR in DMSO-d6:D2O (80%:20%) solution for a 24-hour period. The spectra remained unchanged during the assay time, showing that the compound does not hydrolyze under such conditions (Fig. S3†).
Antiproliferative activity of the compounds
Biological studies were carried out on A2780 human ovarian carcinoma cells for both ligands and complexes. The complexes were further evaluated against A2780Cis (human ovarian carcinoma cisplatin-resistant cells). The results are shown in Table 1. The free ligands were inactive towards the A2780 cell line, with IC50 values >50 μM. In contrast, the complexes are highly potent. Changing the R substituent group in the thiosemicarbazone ligand affects the antiproliferative activity of the complex, as observed for complexes 1 (cyclohexyl derivative, IC50 = 7.59 μM) and 2 (R = ethyl, IC50 = 0.74 μM). The experiments using cisplatin-resistant A2780Cis cell line showed that the potency of complex 1 is increased when compared to that observed for A2780, with an IC50 value of 3.16 ± 0.02 μM. In contrast, complex 2 loses its potency in A2780 cells resistant to cisplatin, with the IC50 value increasing to 35.8 ± 0.8 μM. Cisplatin (CDDP) cross-resistance factors are shown in Fig. 2A (values in Table 1). The bars are expressed as the ratio between the IC50 values in the resistant cell line divided by the IC50 values for the parental cell line. These results suggest that complex 1 may have a different mechanism of action compared to CDDP, since this complex overcomes CDDP resistance and increases potency in the A2780Cis cells, with a cross-resistance factor of 0.41 ± 0.01 μM. On the other hand, the resistance factor for complex 2 (48 ± 0.6) is very high compared to complex 1 and cisplatin, being less potent in A2780cis. Additionally, we investigated activity of the complexes towards non-cancerous cell line MRC-5 human embryonal lung fibroblasts (Table 1). The complexes exhibit high IC50 values for MRC5 human fibroblasts, showing low toxicity against healthy normal cells. Both palladium(II) complexes are nontoxic towards the healthy cells and present relatively high activity, affording high SF values for A2780 cells as well as for A2780Cis. In the case of A2780, complex 2 is the most selective (SFA2780 > 135). On the other hand, considering the selectivity for the cisplatin resistant cell line, complex 1 presents the higher selectivity (SFA2780Cis > 31.6). In both cell lines, the complexes are more selective than cisplatin, with SF = 9.08 ± 0.3, Table 1. Therefore, complex 1 appears to be a promising candidate to circumvent cisplatin resistance in ovarian cancer cells.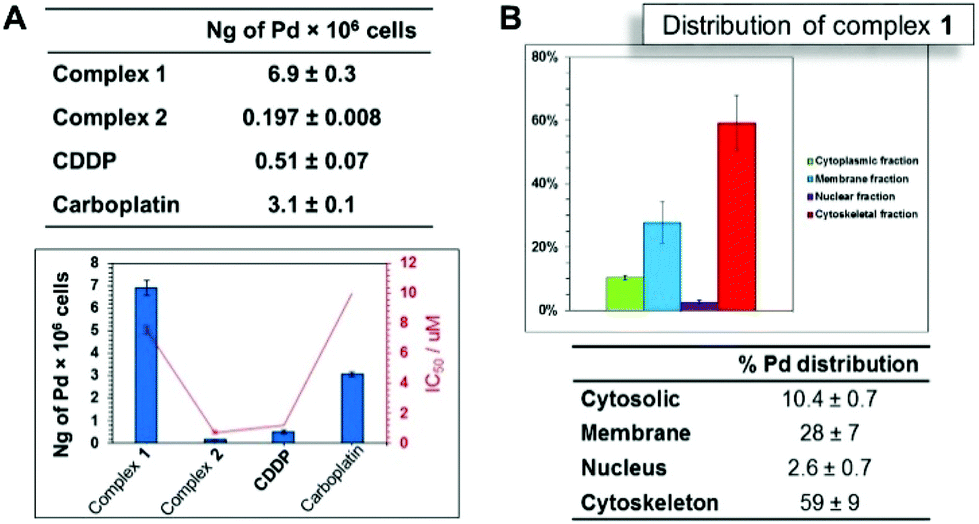 | ||
| Fig. 2 (A) Cellular accumulation of palladium (ng Pd per 106 cells) (up) uptake results and correlation of IC50 values (μM) for complexes 1 and 2 (down). (B) Distribution of Pd for complex 1 in A2780 ovarian cells. Concentration used was IC50, pre-incubation time in drug free medium was 24 h and drug exposure time was 24 h. | ||
| A2780 (μM) | A2780cis (μM) | RF | MRC5 (μM) | SFA2780 | SFA2780Cis | |
|---|---|---|---|---|---|---|
| HPrEt | >50 | nd | nd | nd | nd | nd |
| HPrCh | >50 | nd | nd | nd | nd | nd |
| 1 | 7.6 ± 0.3 | 3.16 ± 0.02 | 0.41 ± 0.01 | >100 | >13 | >31.6 |
| 2 | 0.74 ± 0.09 | 35.8 ± 0.8 | 48 ± 6 | >100 | >135 | >2.8 |
| CDDP | 1.20 ± 0.03 | 12.3 ± 04 | 10 ± 2 | 10.9 ± 0.3 | 9.08 ± 0.3 | 0.88 |
| CARB | 10 ± 1 | nd | nd | nd | nd | nd |
Cellular accumulation and distribution in A2780 cell line
The cellular accumulation of the palladium complexes, cisplatin and carboplatin was determined in A2780 cells, in order to correlate with the IC50 values in this cell line. The structural modification of changing a cyclohexyl group to an ethyl group affected significantly the cellular uptake. The results indicate that the accumulation for complex 1 (R = cyclohexyl) is higher than that of complex 2 (R = ethyl), with almost 7 ng of Pd per 106 cells (Fig. 2A) inside the A2780 cell line. These data are in accordance with the available amount of palladium for cellular uptake for complexes 1 and 2, which are 10 ng and 1.1 ng of palladium, respectively. Thus, 70% of complex 1 enters the cells, while only 18% of complex 2 is able to cross the cellular membrane. However, the results show that there is no correlation between cellular accumulation and the antiproliferative activities, since the complex 1 is the most lipophilic and presented the higher uptake but is less cytotoxic compared with complex 2.We also investigated cell uptake by fluorescent imaging of A2780 cells treated with complex 1. It was possible to observe transport of the complex was into the interior of the cells, through a weak fluorescent green signal, when the compound was excited at 514 nm. The cell distribution of complex 1 was determined. Fig. 2B shows the distribution of Pd and its percentage in four cellular fractions, the cytoplasm, membrane, nucleus and cytoskeletal fraction. It is notable that the highest amount is found in the cytoskeletal. The amount of palladium accumulated in the nuclear fraction is high (ca. 3%), compared to the amount of platinum from cisplatin in the nuclear portion <1%.28 Considering the amount of palladium in the nucleus, DNA binding studies were performed, since DNA is generally known to be the main target of cisplatin.
DNA interaction
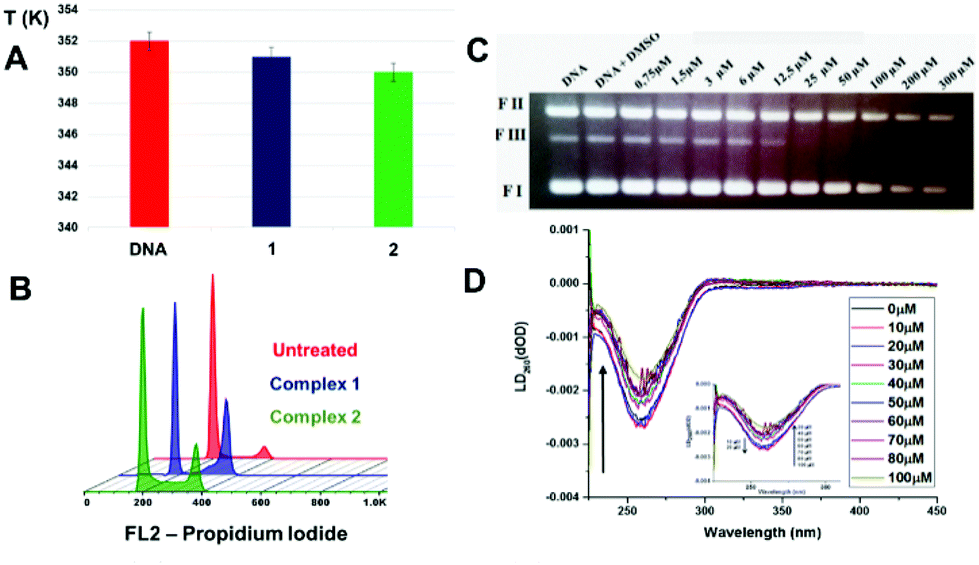 | ||
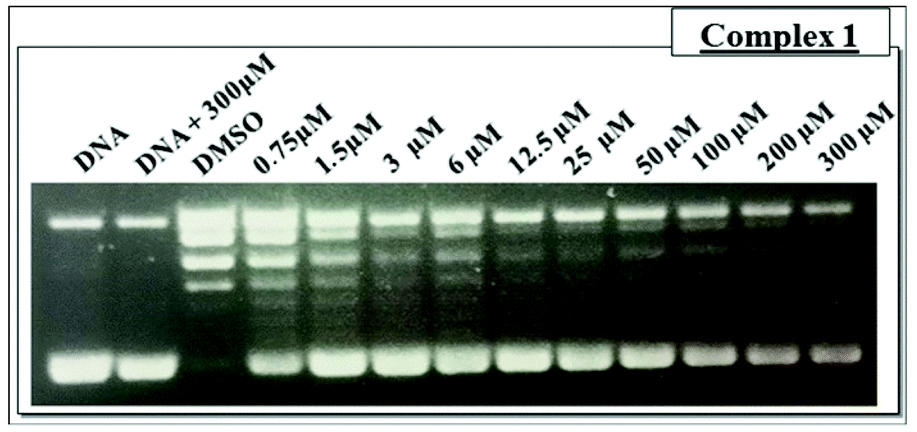
| Fig. 3 (A) Comparison of the Tm (K) for CT-DNA when incubated with palladium complexes 1 and 2 (B) FL2-H histograms obtained by flow cytometry for cell cycle analysis of control untreated cells; cells treated using 7.5 μM of complex 1; cells treated using 0.7 μM of complex 2; (C) ethidium bromide stained agarose gel (0.5 g ml−1) of pBlue Script KSII(+) plasmid DNA (0.25 μg μL−1) in the presence of increased concentration of the complexes after 30 min of incubation at 37 °C: lane 1, plasmid DNA control; lane 2, plasmid DNA + DMSO control, lane 3–10, plasmid DNA + complexes 1 at 0.75, 1.5, 3, 6, 12.5, 25, 50, 100, 200 and 300 μM. FI: supercoiled DNA, FII: opened circular, FIII: linear DNA; (D) linear dichroism (LD) spectra of 100 μM DNA with increasing concentration of complex 1. For all experiments, drug exposure time was 24 h, cells were treated with RNAse and stained using PI. | ||
 | ||
| Fig. 4 Relaxation of negative supercoiled plasmid DNA by TopIB in presence of increasing concentrations of complex 1. Lane 1, no protein added; lane 2, control reaction with DNA and the maximum compound concentration (300 μM); lane 3, positive control reaction with DNA and enzyme in absence of the compound. The reaction products were resolved in an agarose gel and visualized with ethidium bromide. SC, supercoiled plasmid DNA. | ||
An analysis of the cleavage and religation kinetics was carried out in order to better understand the modality of topoisomerase I inhibition by complex 1. The cleavage assay was carried out using the CL14/CP25 suicide substrate, radiolabelled at the 5′ end of the CL14. The enzyme is able to cleave this substrate but it is not able to carry on the religation step because the cleaved dinucleotide is too short to be religated.35 The PAGE analysis of the reaction products, as a function of time, is shown in Fig. S5† (upper panel), where the band corresponding to the cleaved DNA fragment is indicated as “cleavage”. It is interesting that the substrate is cleaved in a fast and efficient way either in the absence or in the presence of complex 1 at 25 μM concentration, i.e. a concentration that produce full relaxation inhibition (Fig. 4). The complete inability of complex 1 to inhibit the cleavage reaction is confirmed by plotting the percentage of the cleavage product as a function of time in presence and absence of the compound (Fig. S4,† lower panel).
The religation reaction was carried out by incubating, for 30 min, the oligonucleotide CL14/CP25 substrate with the TopIB enzyme, allowing the enzyme substrate complex to produce the cleaved complex (Fig. 5). After the cleavage has occurred, the R11 complementary strand is added, to start the religation process. At different times, aliquots were removed, treated with SDS, trypsinized and analyzed by PAGE electrophoresis (Fig. 5, upper pannel). The percentage of religation, plotted in Fig. 5 (lower panel), shows that in the presence of 25 μM complex 1 the inhibitory effect is > 60%. This result is very interesting since it clearly shows that complex 1 acts as a poison since it is able to inhibit the DNA religation but not the cleavage.
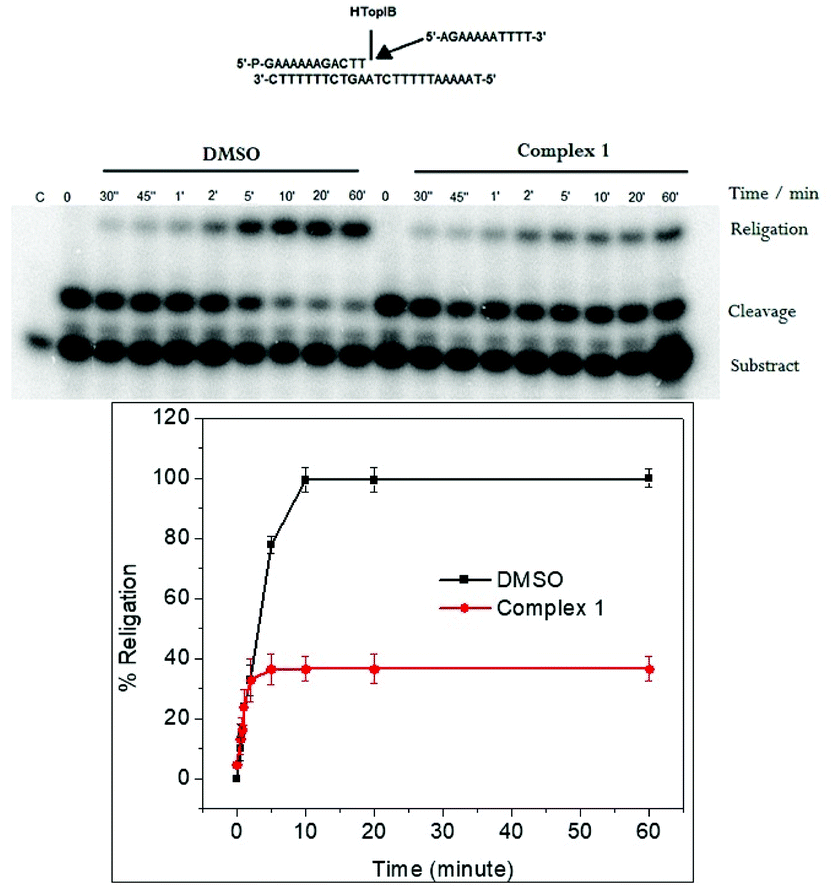 | ||
| Fig. 5 Upper panel: Gel analysis of the religation kinetics with the CL14/CP25 substrate for TopIB in DMSO (lanes 2–9) and in presence of 25 μM of complex 1 (lanes 10–18). Lower panel: The graphic shows the percentage of the religation product plotted at different times for topoisomerase IB. The data reported are the average ± SD of three independent experiments. | ||
Conclusions
In conclusion, we have described the synthesis and characterization of stable new palladium(II) complexes with thiosemicarbazonate ligands derivatized with pyrene. The in vitro biological data show promising low micromolar antiproliferative activity against A2780 and also A2780cis resistance cell lines. Complexes 1 and 2 were relatively less-toxic towards normal cells, but complex 2, in contrast to complex 1, was cross-resistant with the anticancer drug cisplatin I in ovarian cancer cell lines. Cell cycle studies in A2780 ovarian cells showed that complexes 1 and 2 exhibit cytotoxicity by causing G2/M and S phase arrest. Investigations of the mechanism of action, show that TopIB is a plausible target for complex 1. Interestingly the compound inhibits the religation reaction without inhibiting the DNA cleavage. TopIB can be inhibited by several compounds and the inhibitors are divided into two classes depending on the different ways they interact with the enzyme.36–38 Poisons inhibit the enzyme at the level of the religation stabilizing the DNA-TopI cleavage complex and catalytic inhibitors prevent the DNA binding or inhibit the cleavage reaction.39–41 Some of them are able to inhibit both the cleavage and religation reaction.10,42 The poisons, such as camptothecin (CPT), are the most interesting inhibitors, in fact CPT derivatives such as topotecan and irinotecan are in clinical use.43,44 The here studied complex 1 behaves as a poison making it an interesting candidate to chemotherapy. Moreover, since resistance is one of the major challenges to be overcome in the use of chemotherapy for cancer treatment, palladium complexes which are active against resistant A2780cis cell lines are interesting as alternative antitumor agents. Therefore, the complexes studied here might be considered promising lead candidates for cancer therapy.Experimental section
General methods and materials
Orange crystals were produced via slow evaporation of the mother solution of complex 1 and mixture of solvents CH2Cl2:MeOH (1:1) of complex 2 at room temperature. Data collection was performed at 296 K by applying Mo-Kα radiation (λ = 71.073 pm) using a Bruker Kappa APEX II Duo diffractometer (Bruker®, Karlsruhe, Germany). The multi-scan method was applied for absorption correction. The structures were solved with SHELXS9747 software using direct methods, and all non-hydrogen atoms were refined with anisotropic displacement parameters in SHELXL97.48 The hydrogen atoms were calculated at idealized positions using the riding model option of SHELXL2014.48 Table S3† presents more detailed information about the structure determinations. The structural data have been deposited in The Cambridge Crystallographic Data Centre with accession numbers of CCDC 1866632–1866635† for compounds HPrCh, HPrEt, 1 and 2, respectively.
Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) analyses were carried out on a PerkinElmer Optima 5300 DV series ICP-OES. The water (18.2 MΩ) used for analysis was doubly deionized (DDW) using a Millipore Milli-Q water purification system and a USF Elga UHQ water deionizer. ICP standards for platinum (1001 ± 2 μg ml−1, Fluka) and palladium (1015 μg mL−1, Aldrich) were diluted with 3.6% HNO3 DDW to freshly prepare calibrants at concentrations of 50–700 ppb, which were spiked with NaCl to match the salt content of the samples being analyzed. All Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) analyses were carried out on an Agilent Technologies 7500 series ICP-MS instrument. ICP standards were diluted with 3.6% HNO3 DDW to prepare freshly calibrants at concentrations of 0.1–1000 ppb. The ICP-MS instrument was set to detect Pd and Pt with typical detection limits of ca. 2 ppt using no-gas mode, with an internal calibration standard of Er (50 pp).
:MeOH (1:1).
Data for [PdCl(PPh
3
)(PrCh)] (1) (788.6 g mol−1). Color: orange. Yield: 40 mg (76%). M.P.: decomposes 278–280 °C. IR (νmax/cm−1): 3394 ν(N–H), 1583, 1527 ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) + ν(CC), 1095 ν(P–C), 746 ν(CS). 1H NMR (CD2Cl2): δ 1.05–1.28 (m, 6H,
N) + ν(CC), 1095 ν(P–C), 746 ν(CS). 1H NMR (CD2Cl2): δ 1.05–1.28 (m, 6H, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) –H), 1.65–1.69 (m, 2H, Ch–H), 2.00–2.03 (m, 2H, Ch–H), 3.48–3.55 (m, 1H,
–H), 1.65–1.69 (m, 2H, Ch–H), 2.00–2.03 (m, 2H, Ch–H), 3.48–3.55 (m, 1H, ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –Ch), 4.64 (s, 1H,
–Ch), 4.64 (s, 1H, ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) –Ch), 7.44–7.53 (m, 9H, PPh3–H), 7.80–7.5 (m, 6H, PPh3–H), 7.99–8.22 (m, 6H, Pr–H), 8.62 (d, 1H, 3J = 8 Hz, Pr–H), 9.15 (d, 3J = 8 Hz, 1H, Pr–H), 9.77 (d, 3J = 4 Hz, 1H, Pr–H). 31P NMR (CDCl3): δ 27.53. 13C NMR (APT, CD2Cl2): δ 24.92 (CH2), 25.63 (CH2), 32.89 (CH2), 55.75 (CH), 123.16 (CH), 123.91 (CH), 124.42 (C), 124.51 (C), 125.27 (C), 126.01 (CH), 126.05 (CH), 126.20 (CH), 127.26 (CH), 128.20 (CH), 128.34 (CH), 128.77 (CH), 128.94 (CH), 129.14 (CH), 129.91 (C), 130.56 (C), 130.63 (C), 131.11 (C), 131.13 (CH), 131.17 (CH), 131.24 (C), 132.73 (C), 134.54 (CH), 134.69 (CH), 150.51 (CH). Anal. Calcd for C42H38ClN3PPdS·H2O: C, 62.53; H, 4.87; N, 5.21%. Found: C, 62.23; H, 4.69; N, 5.11%. IV (νmax/cm−1): 3394 ν(N–H), 1583, 1517 ν(CN) + ν(CC), 1095 ν(P–C), 746 ν(C–S). UV–Vis, CH2Cl2 solution concentration: 9.8 × 10−5 M [λmax (ε, L mol−1 cm−1)]: 400.50 nm (41430), 284.00 nm (32448). MS (ESI+): m/z for C42H38ClN3PPdS [M − Cl]+: calcd 752.1490, found 752.1485. HPLC: Rt = 40.5 min (at 254 nm).
–Ch), 7.44–7.53 (m, 9H, PPh3–H), 7.80–7.5 (m, 6H, PPh3–H), 7.99–8.22 (m, 6H, Pr–H), 8.62 (d, 1H, 3J = 8 Hz, Pr–H), 9.15 (d, 3J = 8 Hz, 1H, Pr–H), 9.77 (d, 3J = 4 Hz, 1H, Pr–H). 31P NMR (CDCl3): δ 27.53. 13C NMR (APT, CD2Cl2): δ 24.92 (CH2), 25.63 (CH2), 32.89 (CH2), 55.75 (CH), 123.16 (CH), 123.91 (CH), 124.42 (C), 124.51 (C), 125.27 (C), 126.01 (CH), 126.05 (CH), 126.20 (CH), 127.26 (CH), 128.20 (CH), 128.34 (CH), 128.77 (CH), 128.94 (CH), 129.14 (CH), 129.91 (C), 130.56 (C), 130.63 (C), 131.11 (C), 131.13 (CH), 131.17 (CH), 131.24 (C), 132.73 (C), 134.54 (CH), 134.69 (CH), 150.51 (CH). Anal. Calcd for C42H38ClN3PPdS·H2O: C, 62.53; H, 4.87; N, 5.21%. Found: C, 62.23; H, 4.69; N, 5.11%. IV (νmax/cm−1): 3394 ν(N–H), 1583, 1517 ν(CN) + ν(CC), 1095 ν(P–C), 746 ν(C–S). UV–Vis, CH2Cl2 solution concentration: 9.8 × 10−5 M [λmax (ε, L mol−1 cm−1)]: 400.50 nm (41430), 284.00 nm (32448). MS (ESI+): m/z for C42H38ClN3PPdS [M − Cl]+: calcd 752.1490, found 752.1485. HPLC: Rt = 40.5 min (at 254 nm).
Data for [PdCl(PPh
3
)(PrEt)] (2) (734.59 g mol−1). Color: orange. Yield: 38 mg (52%). M.P.: 215–217 °C. IR (νmax/cm−1): 3356 ν(N–H), 1584, 1527 ν(CN) + ν(CC), 1091 ν(P–C), 756 ν(CS). 1H RMN (CD2Cl2): δ 1.13 (t, 3J = 6.0 Hz, 3H, –CH23), 3.31 (dq, 3J1 = 6.0 Hz, 3J2 = 3 Hz, 2H, –NH2CH3), 4.82 (s, 1H, –Et), 7.47–7.57 (m, 9H, PPh3–H), 7.78–7.84 (m, 6H, PPh3–H), 8.03–8.27 (m, 8H, Pr–H), 8.56 (d, 3J = 9 Hz, 1H, Pr–H), 9.18 (d, 3J = 9 Hz, 1H, Pr–H), 9.72 (d, 1H, 3J = 6 Hz, 1H, Pr–H). 31P NMR (CD2Cl2): δ 27.56. 13C NMR (APT, CD2Cl2): δ 14.45 (CH3), 41.41 (CH2), 123.17 (CH), 124.02 (CH), 124.41 (C), 125.34 (C), 126.02 (CH), 126.05 (CH), 126.20 (CH), 127.26 (CH), 128.20 (CH), 128.35 (CH), 128.75 (CH), 128.95 (CH), 129.11 (CH), 129.88 (C), 130.56 (C), 130.61 (C), 131.11 (C), 131.15 (CH), 131.18 (CH), 132.72 (C), 134.54 (CH), 134.68 (CH), 150.94 (CH). Anal. Calcd for C38H31ClN3PPdS: C, 62.13; H, 4.25; N, 5.72%. Found: C, 61.51; H, 4.18; N, 5.64%. UV–Vis, CH2Cl2 solution concentration: 5.1 × 10−5 M [λmax (ε, L mol−1 cm−1)]: 408.00 nm (15128), 393.50 nm (15326), 286.50 (13702). MS (ESI+): m/z for C38H31ClN3PPdS [M − Cl]+: calcd 698.1019, found 698.1019. HPLC: Rt = 38.4 min (at 254 nm).
:1) (v/v) following serial dilutions in RPMI-1640. The drug exposure period was 24 h. After this, supernatants were removed by suction and each well was washed with PBS. The cells were allowed to recover for a further 72 h in drug-free medium at 310 K. The SRB assay was used to determine cell viability.49 Absorbance measurements of the solubilized dye (on a BioRad iMark microplate reader using a 470 nm filter) allowed the determination of viable treated cells compared to untreated controls. IC50 values were determined as duplicates of triplicates in two independent sets of experiments and their standard deviations were calculated.
Metal accumulation in cancer cells
:1 Pd(II):CT-DNA (45 μM of both complex and CT-DNA). The value of the melting temperature (Tm) as the temperature when 50% of the present double-stranded CT-DNA converts into single-stranded CT-DNA was determined as the corresponding maximum on the first-derivative profile of the melting curves. Complexes were incubated for 24 hours with 5% DMSO.
:100 in SC-uracil plus 2% raffinose, cells were induced with 2% galactose for 6 h. After enzyme extraction, the extracts were loaded into ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich) column, already equilibrated according to manufactures’ instructions. The FLAG-fusion topoisomerase IB was eluted by competition with five column volumes of a solution containing 200 μg ml−1 FLAG peptide in 50 mM Tris-HCl, 150 mM KCl pH 7.4. Glycerol was added into each collected fraction up to a final concentration of 40%. All the fractions were stored at −20 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (grant no. EP/F034210/1 to PJS) and Wellcome Trust (grant no 107691/Z/15/Z to PJS and IRC) for support. The authors thank CNPq (Grant 141845/2013-9 and 201134/2015-2), CAPES (finance code 001), FAPESP (Grant 2009/54011-8) and the Minas Chemical Network (RQ-MG) supported by FAPEMIG (Project: CEX-RED-00010-14).References
- C. G. Oliveira, P. I. da S. Maia, P. C. Souza, F. R. Pavan, C. Q. F. Leite, R. B. Viana, A. A. Batista, O. R. Nascimento and V. M. Deflon, J. Inorg. Biochem., 2014, 132, 21 CrossRef CAS PubMed.
- E. O. Lopes, C. G. Oliveira, P. B. Silva, C. E. Eismann, A. C. Suárez, A. A. Menegário, C. Q. F. Leite, V. M. Deflon and F. R. Pavan, Int. J. Mol. Sci., 2016, 17, 1 Search PubMed.
- Z. G. Jiang, M. S. Lebowitz and H. A. Ghanbari, CNS Drug Rev., 2006, 12, 77 CrossRef CAS.
- T. K. Li and L. F. Liu, Annu. Rev. Pharmacol. Toxicol., 2001, 41, 53 CrossRef CAS.
- J. C. Yalowich, X. Wuc, R. Zhang, R. Kanagasabai, M. Hornbaker and B. B. Hasinoff, Biochem. Pharmacol., 2012, 84, 52 CrossRef CAS PubMed.
- C. A. Kunos and T. M. Sherertz, Front. Oncol., 2014, 4, 1 Search PubMed.
- C. A. Kunos, Oncol. Hematol. Rev., 2012, 8, 55 Search PubMed.
- F. Bacher, E. A. Enyedy, N. V. Nagy, A. Rockenbauer, G. M. Bogná, R. Trondl, M. S. Novak, E. Klapproth, T. Kiss and V. B. Arion, Inorg. Chem., 2013, 52, 8895 CrossRef CAS PubMed.
- J. L. Nitiss, Nat. Rev., 2009, 9, 327 CAS.
- S. Castelli, P. Katkar, O. Vassallo, M. Falconi, S. Linder and A. Desideri, Anticancer Agents Med. Chem., 2013, 13, 356 CrossRef CAS PubMed.
- J. Dancey and E. A. Eisenhauer, Br. J. Cancer, 1996, 74, 327 CrossRef CAS PubMed.
- S. Lim, K. A. Price, S. F. Chong, B. M. Paterson, A. Caragounis, K. J. Barnham, P. J. Crouch, J. M. Peach, J. R. Dilworth, A. R. White and P. S. Donnelly, J. Biol. Inorg. Chem., 2010, 15, 225 CrossRef CAS PubMed.
- A. Coleman and M. T. Pryce, Inorg. Chem., 2008, 47, 10980 CrossRef CAS PubMed.
- R. R. Avirah and G. B. Schuster, J. Photochem. Photobiol., 2013, 89, 332 CrossRef CAS PubMed.
- G. McGowan, S. Parsons and P. J. Sadler, Inorg. Chem., 2005, 44, 7459 CrossRef CAS PubMed.
- A. A. Ammar, R. Raveendran, D. Gibson, T. Nassar and S. A. Benita, J. Med. Chem., 2016, 59, 9035 CrossRef PubMed.
- J. S. Casas, E. E. Castellano, J. Ellena, M. S. García-Tasende, M. L. Pérez-Parallé, A. Sánchez, Á. Sánchez-González, J. Sordo and A. Touceda, J. Inorg. Biochem., 2008, 102, 33 CrossRef CAS PubMed.
- A. Sîrbu, O. Palamarciuc, M. V. Babak, J. M. Lim, K. Ohui, E. A. Enyedy, S. Shova, D. Darvasiová, P. Rapta, W. H. Ang and V. B. Arion, Dalton Trans., 2017, 46, 3833 RSC.
- B. M. Zeglis, V. Divilov and J. S. Lewis, J. Med. Chem., 2011, 54(7), 2391 CrossRef CAS PubMed.
- R. N. Prabhu and S. Pal, J. Chem. Sci., 2015, 127, 589 CrossRef CAS.
- P. I. S. Maia, A. Graminha, F. R. Pavan, C. Q. F. Leite, A. A. Batista, D. F. Back, E. S. Lang, J. Ellena, S. de S. Lemos, H. S. S. Araujo and V. M. Deflon, J. Braz. Chem. Soc., 2010, 21, 1177 CrossRef CAS.
- J. E. Expósito, M. Álvarez-Paíno, G. Aullón, J. A. Miguel and P. Espinet, Dalton Trans., 2015, 44, 16164 RSC.
- P. Paul, P. Sengupta and S. Bhattacharya, J. Organomet. Chem., 2013, 724, 281 CrossRef CAS.
- J. Ruiz, M. D. Villa, N. Cutillas, G. López, C. Haro, D. Bautista, V. Moreno and L. Valencia, Inorg. Chem., 2008, 47, 4490 CrossRef CAS PubMed.
- J. S. Casas, E. E. Castellano, J. Ellena, M. S. García-Tasende, M. L. Pérez-Parallé, A. Sánchez, A. Sánchez-González, J. Sordo and A. Touceda, J. Inorg. Biochem., 2008, 102, 33 CrossRef CAS PubMed.
- S. Mandal, V. Kundi, D. K. Seth, K. Srikanth and P. Gupta, Polyhedron, 2014, 80, 290 CrossRef CAS.
- C. G. Oliveira, P. I. S. Maia, M. Miyata, F. R. Pavan, C. Q. F. Leite, E. T. Almeida and V. M. Deflon, J. Braz. Chem. Soc., 2014, 25, 1848 CAS.
- B. T. Benedetti, E. J. Peterson, P. Kabolizadeh, A. Martínez, R. Kipping and N. P. Farrell, Mol. Pharmaceutics, 2011, 8, 940 CrossRef CAS PubMed.
- L. H. Hurley and F. L. Boyd, Trends Pharmacol. Sci., 1988, 9, 402 CrossRef CAS PubMed.
- A. Rodger, Sci. Prog., 2008, 91, 377 CrossRef.
- S. Betanzos-Lara, N. P. Chmel, M. T. Zimmerman, L. R. Barrón-Sosa, C. Garino, L. Salassa, A. Rodger, J. L. Brumaghim, I. Gracia-Moraa and N. Barba-Behrens, Dalton Trans., 2015, 44, 3673 RSC.
- M. A. Bjornsti, P. Benedetti, G. A. Vigilanti and J. C. Wang, Cancer Res., 1989, 49, 6318 CAS.
- B. Zhivotovsky and G. Kroemer, Nat. Rev. Mol. Cell Biol., 2004, 5, 752 CrossRef CAS PubMed.
- N. Raja, N. Devika, G. Gupta, V. L. Nayak, A. Kamal, N. Nagesh and B. Therrien, J. Organomet. Chem., 2015, 794, 104 CrossRef CAS.
- J. Q. Svejstrup, K. Christiansen, I. I. Gromova, A. H. Andersen and O. Westergaard, J. Mol. Biol., 1991, 222, 669 CrossRef CAS PubMed.
- J. A. Holden, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1(1), 1 CrossRef CAS PubMed.
- Y. Pommier, ACS Chem. Biol., 2013, 8(1), 82 CrossRef CAS PubMed.
- S. Castelli, A. Coletta, I. D'Annessa, P. Fiorani, C. Tesauro and A. Desideri, Biol. Chem., 2012, 393(11), 1327 CAS.
- D. Strumberg, Y. Pommier, K. Paull, M. Jayaraman, P. Nagafuji and M. Cushman, J. Med. Chem., 1999, 42, 446 CrossRef CAS PubMed.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17(5), 421 CrossRef CAS PubMed.
- L. F. Liu, S. D. Desai, T. K. Li, Y. Mao, M. Sun and S. P. Sim, Ann. N. Y. Acad. Sci., 2000, 922, 1 CrossRef CAS PubMed.
- S. Castelli, S. Vieira, I. D'Annessa, P. Katkar, L. Musso, S. Dallavalle and A. Desideri, Arch. Biochem. Biophys., 2013, 530, 7 CrossRef CAS PubMed.
- S. Castelli, A. Campagna, O. Vassallo, C. Tesauro, P. Fiorani, P. Tagliatesta, F. Oteri, M. Falconi, H. K. Majumder and A. Desideri, Arch. Biochem. Biophys., 2009, 486(2), 103 CrossRef CAS PubMed.
- H. Ulukan and P. W. Swaan, Drugs, 2002, 62, 2039 CrossRef CAS PubMed.
- T. S. Lobana, P. Kumari, G. Hundal, R. J. Butcher, A. Castineiras and T. Akitsu, Inorg. Chim. Acta, 2013, 394, 605 CrossRef CAS.
- M. S. Inkpen, A. J. P. White, T. Albrecht and N. J. Long, Chem. Commun., 2013, 49, 5663 RSC.
- G. A. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. A. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112 CrossRef CAS PubMed.
- H. Hong-Liang, L. Zheng-Zheng, L. Zhen-Hua, Y. Jun-Hua and L. Yun-Jun, Eur. J. Med. Chem., 2011, 46, 3282 CrossRef PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1866632–1866635. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt02570g |
| This journal is © The Royal Society of Chemistry 2019 |