
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of metal oxide semiconductors in light-driven organic transformations
Paola
Riente
 and
Timothy
Noël
*
and
Timothy
Noël
*
Micro Flow Chemistry and Synthetic Methodology, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, Het Kranenveld, Bldg 14 - Helix, 5600 MB, Eindhoven, The Netherlands. E-mail: t.noel@tue.nl
First published on 22nd August 2019
Abstract
The search for greener alternatives to perform organic reactions has become the order of the day in the chemistry community. In this regard, the use of heterogeneous photocatalysts has emerged as a powerful alternative to replace transition metal-based complexes and organic dyes to enable light-driven organic transformations. Within this realm, metal oxide semiconductors (MOS) have become increasingly popular due to their recyclability, availability, energy efficiency, photo- and chemical stability and generally low toxicity. Here, we cover the most relevant literature related to the use of MOS as photocatalysts to light induce organic reactions, including oxidations, C–C and C-heteroatom bond formations. We also discuss the mechanisms involved in these processes, as well as the hitherto best known MOS modifications able to enhance their photocatalytic performance.
 Paola Riente | Paola Riente received her B.Sc. degree (2001) in Industrial Pharmacy from the Federal Fluminense University (Brazil) and Ph.D. degree (Summa Cum Laude) in Organic Chemistry from Alicante University (Alicante-Spain) in 2009 under the supervision of Profs. Miguel Yus and Francisco Alonso. After that, she moved to the Pericàs group at ICIQ (Tarragona-Spain) to work, as a postdoctoral fellow, with magnetic nanoparticles-supported (organo)catalysts and semiconductor photocatalysis. Currently, she is a Marie Curie postdoctoral fellow in the Noël Research Group at the Eindhoven University of Technology (The Netherlands). Her research interests are mainly focused on the application of metal oxide semiconductors in photocatalysis. |
 Timothy Noël | Timothy Noël received in 2004 his MSc degree (Industrial Chemical Engineering) from the KaHo Sint-Lieven in Ghent. He then moved to Ghent University to obtain a PhD under the guidance of Professor Johan van der Eycken at the Laboratory for Organic and Bioorganic Synthesis (2005–2009). Next, he moved to Massachusetts Institute of Technology (MIT) as a Fulbright Postdoctoral Fellow with Professor Stephen L. Buchwald. He currently holds a position as an associate professor (UHD1) and he chairs the Micro Flow Chemistry & Synthetic Methodology group at Eindhoven University of Technology. His research interests are flow chemistry, synthetic methodology and chemical engineering. His research on photochemistry in microfluidic reactors was awarded the DECHEMA award 2017 and he is the editor in chief of Journal of Flow Chemistry. |
1. Introduction
Throughout history, humanity has been inspired by Nature to find elegant solutions for complex problems in their daily life. In the same way that natural photosynthesis drives chemical processes using solar light, photocatalysis has emerged as a Nature-inspired approach for harvesting and converting solar energy to enable challenging synthetic transformations.1 Defined by IUPAC Gold Book as ‘a change in the rate of a chemical reaction or its initiation under the action of ultraviolet, visible or infrared radiation in the presence of a substance—the photocatalyst—that absorbs light and is involved in the chemical transformation of the reaction partners’,2 the concept of photocatalysis may be traced back to Becquerel in 1839 with his demonstration of the photovoltaic effect.3 In this work, he observed the effect of sunlight irradiation over silver halogen electrodes to establish a potential difference and an electric current. The keyword photocatalysis was first mentioned in 1911 in a scientific report where ZnO was used as a photocatalyst to bleach dyes.4 Although this seminal work can be regarded as a breakthrough discovery, it was not given much attention until the late 1960s, when Honda and Fujishima carried out their pioneering work on the electrochemical photolysis of water at a semiconductor electrode.5 Following the work of Honda and Fujishima, heterogeneous photocatalysis based on metal oxide semiconductors (MOS) has gained enormous traction giving rise to a wide variety of applications, including water splitting, CO2 reductions, photovoltaic cells, and pollutant degradation.6 However, only in 1998, their potential as catalytic platforms in organic synthesis was demonstrated by Albini and coworkers. In their initial work, these researchers used TiO2 as a photocatalyst to induce C–C bond formation under solar irradiation.7 Another decade later, the scientific contributions of MacMillan,8 Yoon9 and Stephenson10 demonstrated the utility of homogeneous transition metal-based photocatalysts for synthetic organic chemistry. Their findings proved to be the spark that ignited the field resulting in the development of a massive amount of synthetic methodologies based on photocatalysis.11However, in a tireless effort to find greener alternatives for visible-light photoredox catalysis,12 the use of heterogeneous photocatalysts have received significant amounts of attention.13 More specifically, metal oxide based semiconductors (MOS) have emerged as promising candidates to substitute expensive and toxic transition metal-based complexes and organic dyes. MOS-based photocatalysts offer several advantages over these homogeneous variants, such as (i) high recyclability and reusability; (ii) high abundancy (most of them are earth-abundant) and thus low cost; (iii) low toxicity; (iv) high chemical stability and (v) tunability of electronic and optical properties.
This review focuses on the application of MOS as photocatalysts to drive synthetically useful transformations. In section 2, we introduce different MOS and highlight their unique electronic structure and properties, as well as, potential modifications to enhance their visible light absorption capacity. Section 3 focuses on the most applied wide and narrow bandgap MOS and their applications as photocatalysts in organic synthesis.
2. Background: metal oxide semiconductors (MOS)
Metal oxide semiconductors (MOS) are mainly characterized by a large band gap (>3.0 eV) (Scheme 1). They have been widely applied in the photodegradation of organic pollutants in water and air, energy conversion, biosensing, microelectronics, optoelectronics, and storage.14 Usually, most of the MOS are inexpensive, stable, safe, and earth-abundant. Also, due to their solid insoluble nature, chemical and photo-stability, they can be easily removed from the reaction media and be reused in subsequent catalytic processes. MOS also present great ability to form both oxidizing and reducing species under appropriate irradiation, being a promising greener alternative to the use of expensive and toxic transition metal-based complexes to promote important organic transformations.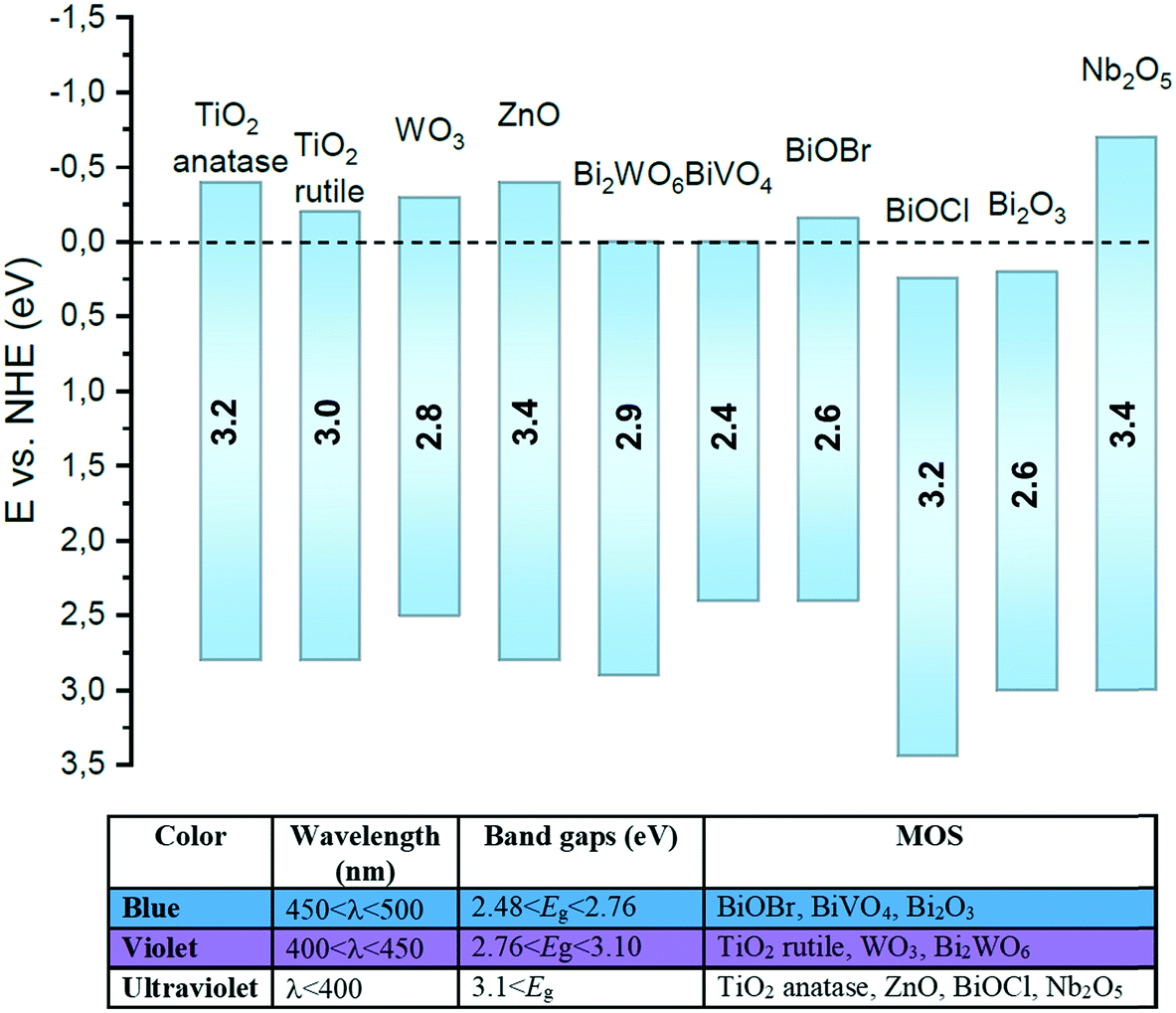 | ||
| Scheme 1 Representative diagram of the redox potentials of valence and conduction bands and bandgap energies estimated at pH 7 of a range of MOS presented in this review and relationship between band gap and wavelength. | ||
Typically, the electronic structure of MOS is composed of a valence band (VB, highest occupied energy band), a conduction band (CB, lowest unoccupied energy band) and a band gap (Eg, forbidden energy zone). If the energy of the incident photons matches or is higher than that of Eg, a charge separation takes place leading to an electron–hole (e−–h+) pair. Once these photogenerated electrons and holes are formed, they become usually trapped at the surface of the MOS which, by interfacial electron transfer (IFET), can reduce or oxidize substrates with appropriate redox potentials (Scheme 2a).
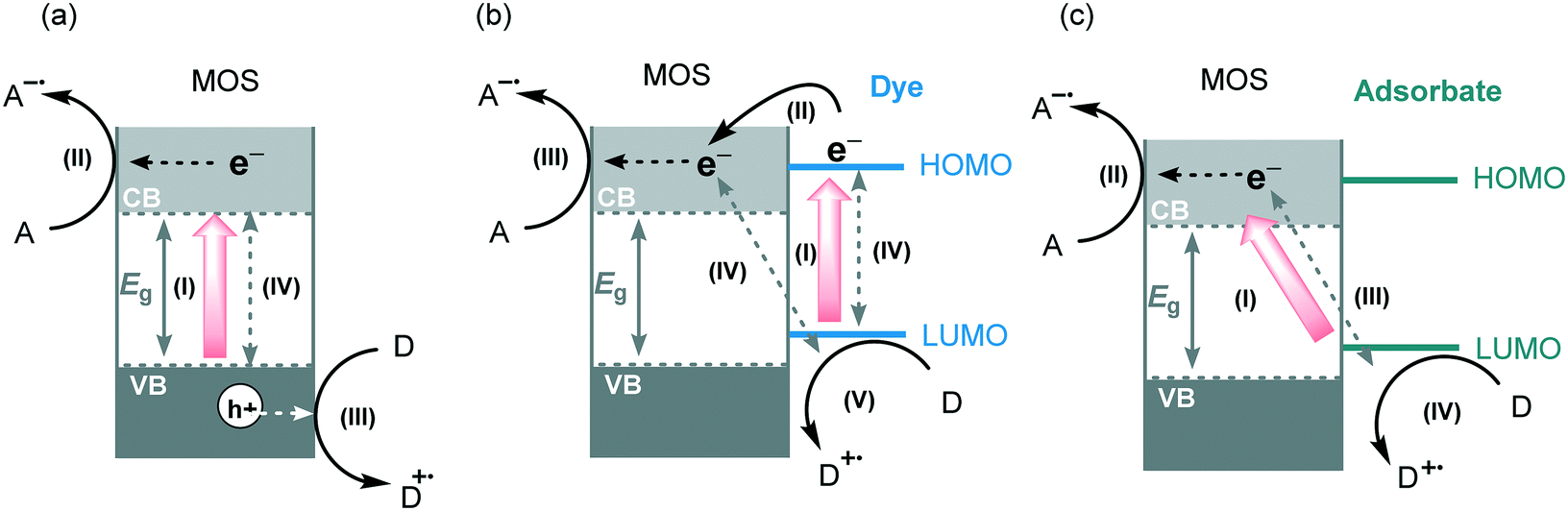 | ||
| Scheme 2 A representative illustration of MOS photocatalytic mechanisms. (a) General MOS photocatalytic mechanism: (I) photoexcitation and charge generation, (II) electron transfer to an acceptor, (III) oxidation of a donor (IV) charge recombination. (b) Dye photosensitization: (I) visible light photoexcitation, (II) electron transfer from the excited state of the dye to MOS CB, (III) electron transfer to an acceptor, (IV) charge recombination, (v) regeneration of the sensitizer by an electron donor. (c) LMCT photosensitization: (I) visible light induced LMCT transfer, (II) electron transfer to an acceptor, (III) charge recombination, (IV) regeneration of adsorbates by an electron donor or in its absence degradation of adsorbates into small molecules. A and D represent electron acceptor and the electron donor, respectively. | ||
Despite the positive aspects of the use of MOS in photoredox processes, their unique electronic structure could generate some drawbacks that could hamper their practical photocatalytic performance. Mainly, these drawbacks are associated with their wide band gaps, which may reduce their capacity to absorb visible light. Another remarkable drawback is the fast charge carrier recombination that can eventually reduce the number of electrons and holes available to carry out the photocatalytic process. Finally, photostability can become an important obstacle which can significantly limit the application of MOS.15 Other aspects which may negatively affect the photocatalytic performance of MOS are related to their atomic structure parameters, such as geometry, crystalline phases, surface defects, specific surface area and particle sizes.16
As previously stated, most of the MOS have large band gaps and are mainly photoexcited by UV irradiation to form the required electron–hole pairs which can subsequently trigger the photocatalytic processes. Additional drawbacks associated with the use of UV irradiation have to be taken into consideration as well. These high energy photons can directly activate chemical bonds of some organic molecules to form radical intermediates giving rise to non-selective reactions. Also, the use of UV light implies the use of more expensive light sources and specialized glassware. The most employed strategies to overcome these drawbacks and to enhance their visible light photocatalytic activity are based on tuning or doping of the surface of the MOS. Chemical doping with metals, non-metal based dopants or surface loading of the MOS with visible light sensitizers have been intensely studied.17 Amongst these alternatives, the photosensitization of MOS using dyes or ligand-to-metal-charge transfer (LMCT) constitute the most employed strategies to increase the visible light response of the MOS.
Pioneered by Grätzel and O'Regan, the dye-sensitization strategy is widely applied in solar cells, water splitting and the photodegradation of organic pollutants.18 This strategy is based on the attachment or adsorption of a dye or sensitizer (metal complex, metal-free organic dye, noble metals) onto the surface of the MOS. After visible light irradiation, the dye undergoes a HOMO–LUMO photoexcitation generating electron–hole pairs.19 Next, the photogenerated electrons are injected into the CB of the MOS forming conduction band electrons (e−CB). These electrons can be transferred to an electron acceptor and the oxidized dye can be regenerated by the presence of a sacrificial electron donor (Scheme 2b). In this process, the CB of the MOS mediates the electron transfer from the dye, or sensitizer, to the substrate extending indirectly the visible light photo-response of the wide band gap semiconductors. Apart from the positive aspects of the use of dye-sensitization, some fundamental requisites have to be taken into account in the selection of an appropriate dye: (i) the dye should possess a broad range absorption in the visible region (ii) its LUMO must be more negative than the CB of the semiconductor, and its HOMO more positive than the sacrificial electron donor redox potential; (iii) the dye should display high photostability towards long-term light irradiation and (iv) the dye should exhibit extended excited state lifetimes. Despite ruthenium-based polypyridyl complexes and other metal complexes being the most used dyes for sensitization of MOS,20 there is a recent trend to use more eco-friendly and low-cost metal-free organic dyes.21
Ligand-to-metal-charge transfer (LMCT) is another method to enable a visible light induced charge transfer. This type of sensitization is less studied than dye-sensitization but has great potential for visible light photocatalytic applications.22 LMCT is extensively used for degradation of pollutants adsorbed onto the surface of TiO2.23 Unlike dye-sensitization, the compounds adsorbed onto the surface of the MOS do not absorb visible light and the excited state of the adsorbates is not involved in the process. The electron is photoexcited directly from the ground state of the adsorbate to the MOS CB. The oxidized adsorbate can be degraded in small molecules or be regenerated by recombination with the photoexcited electrons or by the presence of an electron donor in the reaction medium (Scheme 2c). The formation of CT complexes on MOS is generally characterized by the appearance of a visible light absorption band which is not observed with either the adsorbate or the MOS alone. A wide range of organic and inorganic compounds with an appropriate HOMO level can become suitable LMCT sensitizers. For instance, electron-rich adsorbates containing hydroxyl and carboxyl linkers, such as catechol, EDTA (ethylenediaminetetraacetic acid), ascorbic acid and salicylic acid can be LMCT sensitizers. Also, substrates containing heteroatoms (X = S, N or O) may adsorb onto the surface of the MOS by weak interaction and can subsequently inject electrons into the CB of MOS giving rise to a weak light absorbance and activation of the substrates (Scheme 3).
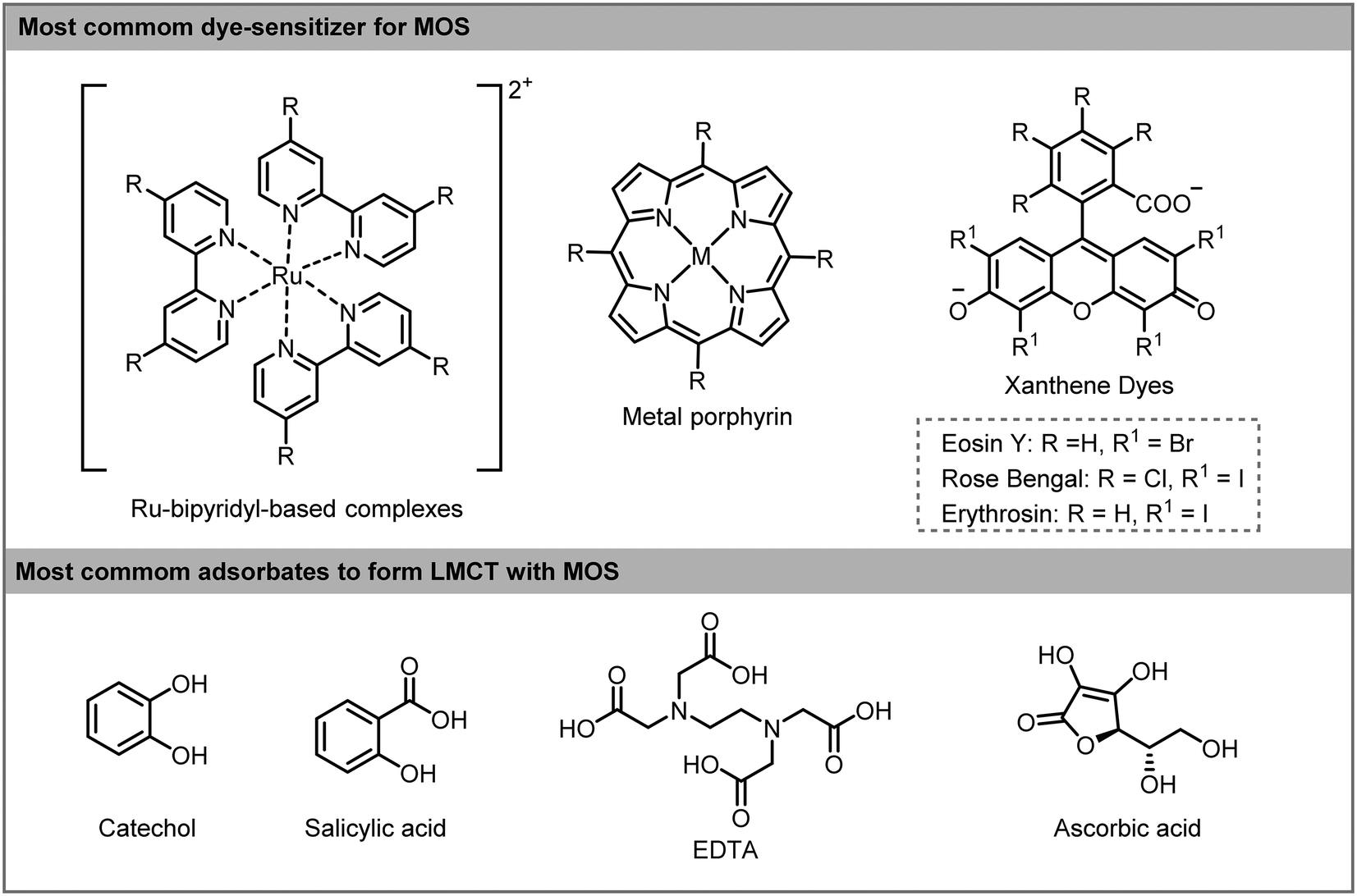 | ||
| Scheme 3 Most common dyes (top) and adsorbates (bottom) used in sensitizer-MOS for photocatalytic applications. | ||
In both strategies, the efficiency of the photosensitization process relies on several parameters, including (i) the way that the foreign compounds are connected to the semiconductor (physically or chemically bonded), (ii) the nature of the anchored groups, (iii) the distance between the compound and the MOS surface and (iv) the electronic nature of functional groups on the anchored compound.
3. Application of MOS as photocatalysts in organic transformations
Despite TiO2 being by far the most used MOS due to its low toxicity, low cost, high photoreactivity and -stability, a variety of different MOS have emerged as potential alternatives to TiO2. Particularly, ZnO and other narrow bandgap semiconductors, such as bismuth-based oxides, have received quite some attention. In this section, we will highlight selected examples of the most used MOS capable of driving specific organic transformations either under UV or visible light irradiation. Moreover, we will also detail the different strategies to increase the visible-light response of wide bandgap MOS.3.1. TiO2
The photosensitization of TiO2 dates back to the early 20th century.24 However, the interest in TiO2 as a photocatalyst only started to grow after the appearance of the paper by Fujishima and Honda in 1972.5 Since then, TiO2 has been intensely studied triggering numerous applications in the photocatalysis field.25 Among these applications, its use as photocatalyst to promote organic transformations specifically has emerged only in the last 20 years as a great alternative to replace the use of toxic and expensive transition metal complexes. Mainly this augmented interest can be attributed to its low cost, low toxicity, photo-stability and -reactivity and abundance. However, its large band gap (3.2 eV for anatase and brookite, 3.0 eV for rutile) together with a fast charge recombination limited its application potential in photocatalytic processes. In this respect, we direct the reader to a number of interesting reviews, detailing the tuning and/or doping of the TiO2 surface to enhance its visible light absorption and/or reduce charge recombination.26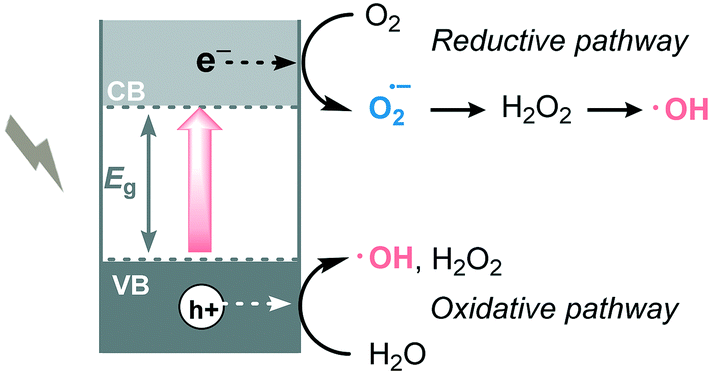 | ||
| Scheme 4 Generation of reactive oxygen species in MOS photocatalysis. | ||
In 2008, Zhao and co-workers reported the selective aerobic oxidation of alcohols in benzotrifluoride (BFT) using alizarin red (AR)-sensitized TiO2 under visible light irradiation (λ > 450 nm).31 The nitroxyl radical TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) was used as co-catalyst and acted as a sacrificial electron donor to prevent the degradation of the dye. The photocatalytic system composed by AR/TiO2/TEMPO/O2 successfully promoted the oxidation of a variety of functionalized benzylic alcohols to the corresponding aldehydes with good to excellent conversions (23 to 100%) and high selectivities (up to 99%). Aliphatic and cyclic secondary alcohols were also oxidized under the optimized photocatalytic conditions to the corresponding carbonyl compounds albeit in low conversion. For the proposed mechanism, the authors suggested that the excited state of the dye injects electrons in the CB of the TiO2 furnishing the dye radical. This radical species promotes the oxidation of TEMPO which selectively oxidizes the alcohols to the corresponding carbonyl products by a two-electron-transfer mechanism. In turn, the reduction of oxygen leads to the formation of O2˙− species (that further reacts to form H2O and H2O2) which could promote the regeneration of the dye (Scheme 5).
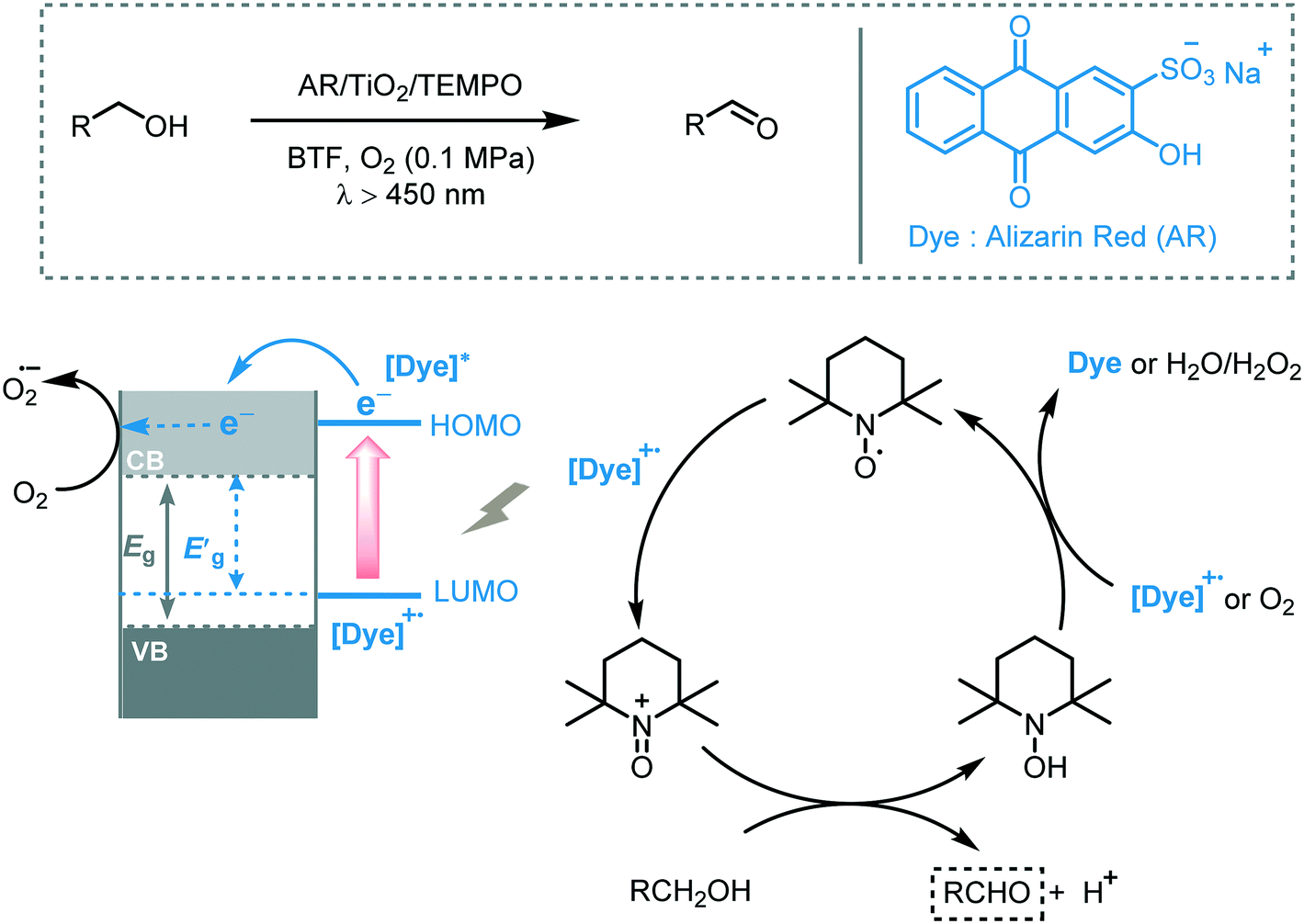 | ||
| Scheme 5 The proposed mechanism for the oxidation of alcohols photocatalyzed by AR/TiO2/TEMPO under visible light irradiation. | ||
In a similar approach, Lang and co-workers recently reported an exhaustive study of the use of eosin Y as a sensitizer for TiO2.32 In this work, the eosin Y-TiO2/TEMPO system was very effective to oxidize alcohols in acetonitrile, such as benzylic alcohols in short reaction times (1 to 4 hours) under visible light irradiation (green light, λ ≈ 530 nm). However, moderate to poor conversions were obtained when the system was applied to the oxidation of allylic, aliphatic and secondary alcohols.
The formation of charge transfer complexes between colorless adsorbates and MOS to extend their visible light response through LMCT is also a widely used approach to promote the oxidation of alcohols under visible light irradiation. For instance, Higashimoto and co-workers reported the photooxidation of alcohols into the corresponding carbonyl compounds using unmodified TiO2, under both visible and UV light irradiation.33 In both cases, the developed method showed excellent efficiency and selectivity in the photooxidation of benzylic alcohols (up to 99% in yields and selectivities) using acetonitrile as solvent. To explain the photoreactivity of the system under visible light irradiation, they proposed that the interaction of the starting benzylic alcohols with the surface of the semiconductor enables visible light absorption mediated through LMCT.
Zhao et al. carried out successfully the selective oxidation of amines to imines using air as oxidant and TiO2 as the photocatalyst under UV irradiation (500 W Xe lamp, cutoff filter λ < 350 nm).34 A range of benzylamines, including heterocyclic benzyl amines, were converted into the desired imines generally with high conversions (44 to 100%) and selectivities (up to 94%). Next, they proposed the use of visible light to induce this oxidation.35 They observed that the benzylic amines adsorbed onto the surface of the TiO2 could extend the light absorption into the visible region by LMCT. The underlying idea was to test whether the photooxidation could be carried out under visible light irradiation. Remarkably, a great variety of imines derivatives, ranging from primary and secondary benzylamines, were prepared in high yields and selectivities using a 300 W Xe lamp with a cutoff filter λ < 420 nm. However, the photooxidation of disubstituted benzylamines yields the formation of a mixture of aldehydes, symmetric and unsymmetric imines. The proposed mechanism comprises the activation of the CT complex by visible light irradiation to generate electron–hole pairs. The holes locate on the adsorbed amine and promote its oxidation to furnish a carbon-center radical. The corresponding electrons are localized in the CB of the TiO2 and form Ti3+ species. The interaction between the carbon-centered radical, Ti3+ and oxygen lead to the formation of an oxygen bridged intermediate which undergoes cleavage of both C–N and O–O bonds to yield the aldehyde intermediate. The condensation of the latter intermediate with the free amine yields the desired imine. Next, TiO2 is regenerated from the complex with the assistance of H+, thus completing the catalytic cycle (Scheme 6).
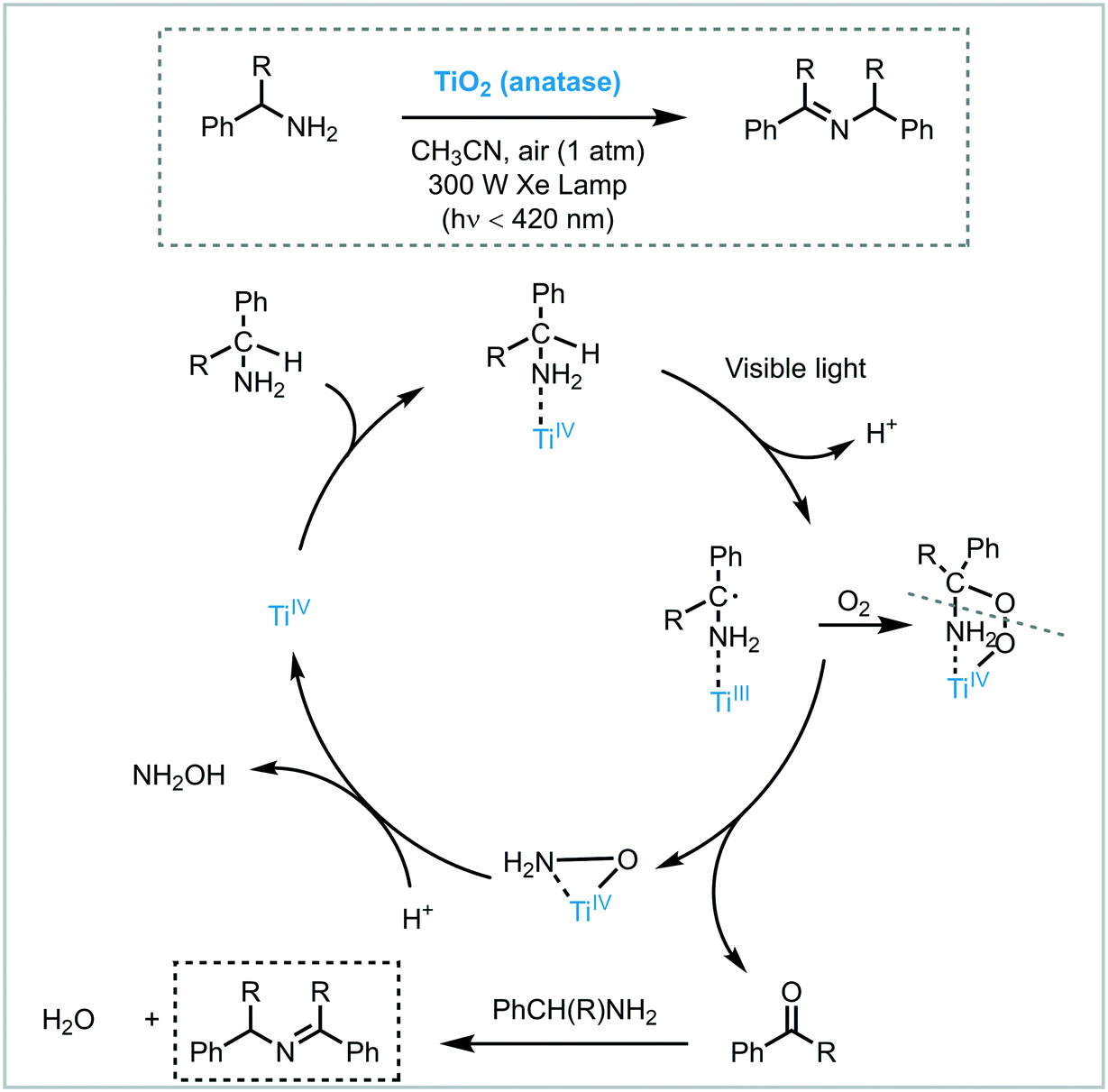 | ||
| Scheme 6 Proposed mechanism for the oxidation of amines to imines photocatalyzed by TiO2 under visible light irradiation. | ||
Li et al. anticipated that the photocatalytic activity of TiO2 may be strongly affected by its different crystalline phases. For this study, they prepared mixed-phase TiO2 nanowires with different ratios of TiO2(B)/anatase.36 The best photocatalytic performance was demonstrated with a mixed-phase TiO2(B)/anatase TiO2 (TW-550, 65%/35%, respectively) in the oxidation of benzylic amines (46 to 95% in conversions) under visible light irradiation. Also, the results obtained using this mixed-phase photocatalyst showed higher activity in comparison with bare TiO2 or pure TiO2 anatase. The authors suggested that the higher photocatalytic activity of TW-550, compared to other mixed phase TiO2 compositions, is mostly due to the optimal surface-phase junctions between TiO2(B) and anatase which can efficiently promote the interfacial charge separation by their staggered band gap energy. Moreover, the good adsorption ability of the TiO2(B) in comparison with anatase TiO2, allows enhanced adsorption of the amines onto their surface favoring the key charge transfer event (Scheme 7). As mentioned in the latter report, the crystalline phase of the semiconductor not only can affect their optical properties but also its surface structural arrangements which can lead to different surface reactivity hindering the selectivity of the reaction.37 A good example of this effect is described in the sub-section Miscellaneous transformations.
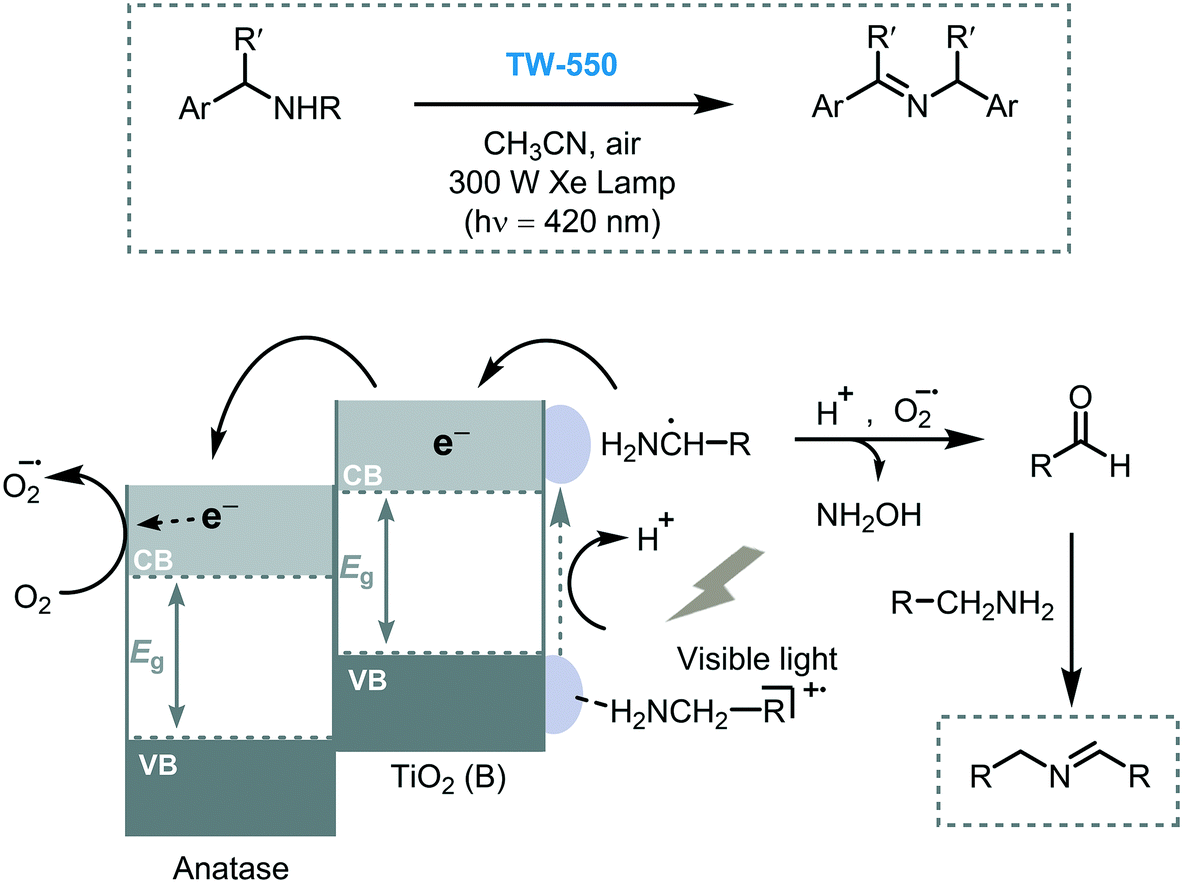 | ||
| Scheme 7 Proposed mechanism for the oxidation of benzylic amines into imines on mixed-phase TiO2(B)/anatase TiO2 (TW-550) under visible light irradiation. The staggered gap at the interface is clearly observed in the scheme. | ||
Similar to dye sensitization, another strategy to overcome intrinsic drawbacks associated with the use of wide bandgaps semiconductors photocatalysts comprises the deposition of noble metal plasmonic nanoparticles (PNP), such as Au and Ag, onto their surfaces to form so-called plasmonic photocatalytic composites. These systems exhibit efficient visible light absorption over a wide wavelength range and also reduces the carrier recombination rate.38 In these composites, the plasmon activation of the PNP onto the surface of the semiconductor by visible light generates positive charges on the PNP and e−CB on the nearby semiconductor. Shiraishi et al. described the application of the plasmonic photocatalyst Pt/TiO2 (Degussa P25) for the visible light aerobic photooxidation of aniline to nitrosobenzene.39 Higher yields and selectivities (∼90%) were observed compared to other photocatalytic systems, which contain either P25, Au2/P25 or Pd/P25 and Pt/anatase or rutile. The authors proposed that the Lewis base sites located on the Pt nanoparticles promote the reductive deprotonation of aniline to form the corresponding anion. This further reacts with hydroperoxides species, which are formed through photogenerated electrons in the CB of TiO2, furnishing the targeted nitrosobenzene and water. They also claimed that the photocatalytic activity of the composite was strongly dependent on the amount of Pt loaded onto the TiO2, the temperature and the particle size.
The photooxidation power of TiO2 was also demonstrated in other organic oxidations. For instance, Cong and co-workers reported visible light-mediated selective aerobic oxidation of benzyl ethers to benzoates using an eosin Y-sensitized TiO2 (P25).40 The photocatalytic system showed high efficiency and allowed to obtain the corresponding benzoates in high yields (up to 92%). The transformation displayed also high functional and protect groups tolerance, including alkyl bromide, alkyl nitrile, pyridyl, silyl ether and acetal among others and could be used to modify natural products, such as serine, threonine and monosaccharide derivatives. Based on kinetic studies and EPR (electron paramagnetic resonance) experiments, the authors suggested that the reaction mechanism goes through a single electron transfer (SET)-mediated oxidation of benzyl ethers. The homolytic cleavage of the O–O bond of the pre-formed peroxide intermediate (A) by TiO2 photocatalysis, could result in the formation of radical species. Subsequently, these species would react with acetone (solvent) or (B) during the radical chain propagation, including a series of hydrogen atom transfer processes (HAT), to yield the corresponding benzoates (Scheme 8).
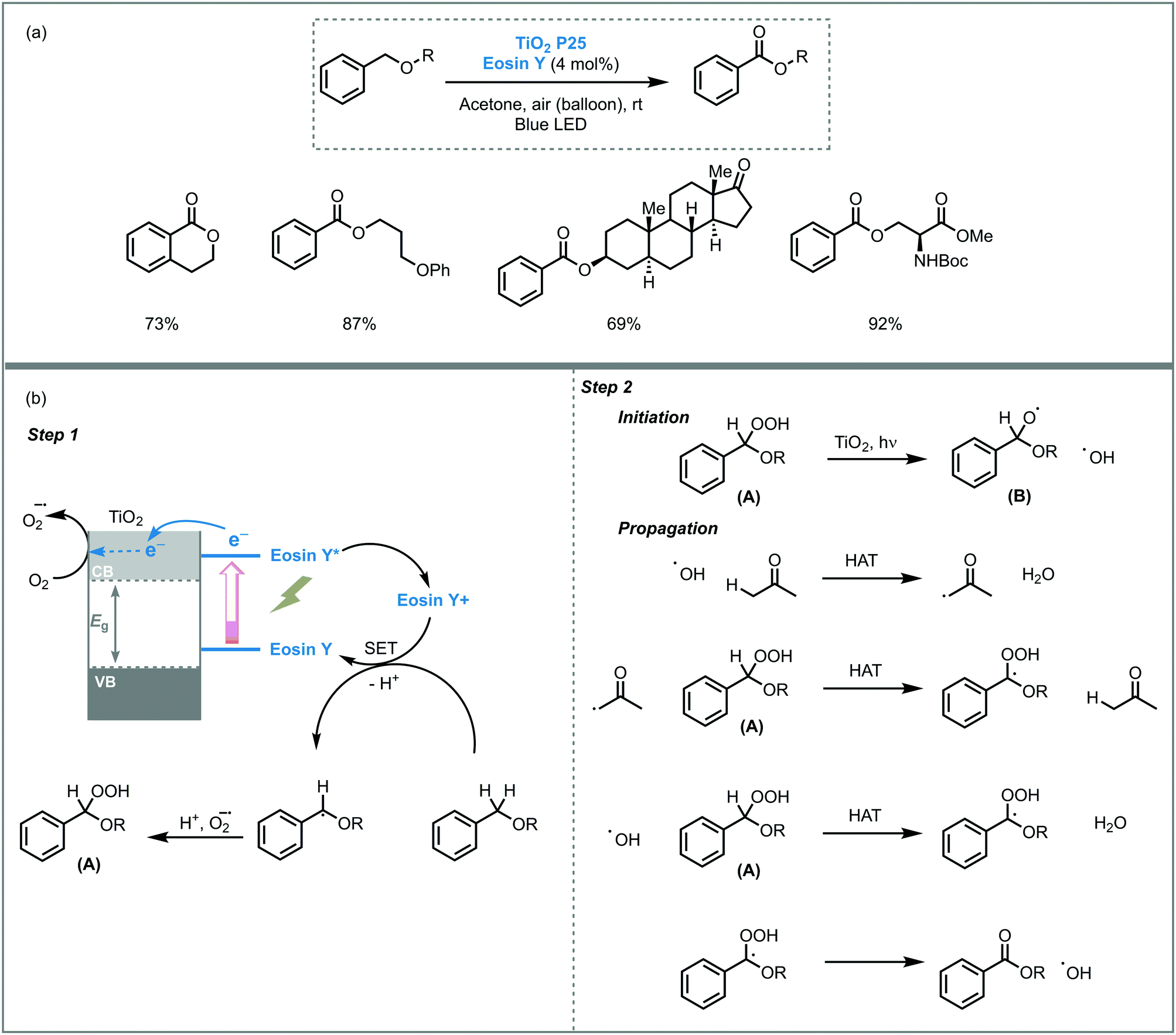 | ||
| Scheme 8 Oxidation of benzyl ethers photocatalyzed by eosin Y-sensitised TiO2. (a) Selected scope; (b) proposed mechanism. | ||
Phenol is an important chemical compound widely applied in the chemical and pharmaceutical industry. There are several methods for the synthesis of phenol starting from benzene, such as cumene–phenol process, thermal catalysis, oxidation with H2O2 and Fenton process. However, most of these methods require harsh reaction conditions in combination with toxic reagents. More recently, the application of semiconductor photocatalysis has emerged as a greener alternative for the synthesis of phenol. The mechanism for the photooxidation of aromatics into phenols involves the activation of the unsaturated Csp2–H bonds by hydroxyl radicals or holes to form radical cationic benzene species. Next, these radical cations are attacked by active oxygen species leading to the desired phenol. For instance, Di Paola et al. promoted the hydroxylation of various aromatic compounds using TiO2 under UV irradiation (125 W Hg lamp, λ ≈ 360 nm).41 Interestingly, benzene derivatives bearing electron donor groups favor the oxidation of ortho- and para-positions, while electron withdrawing groups result in unselective reactions furnishing all possible isomers.
Huang et al. reported the synthesis and use of metal plasmonic photocatalysts (M@TiO2, M = Au, Pt, Ag) to promote the visible light induced photooxidation of benzene to phenol in water (300 W Xe arc lamp with UV cutoff filter, λ ≥ 400 nm).42 The best performance was achieved using Au@TiO2, obtaining phenol in good yield (63%) and selectivity (91%). They suggested that the photoexcitation of the plasmonic photocatalyst induces an electron transfer from the plasmonic metal to the semiconductor by surface plasmon resonance (SPR) effects, leading to oxygen reduction and an electron deficient Au. The electron-depleted Au subsequently adsorbs phenoxy anions (present in the reaction medium) and provoke their oxidation to form free phenoxy radicals. These radical species are able to oxidize benzene to phenol regenerating the anion. The key step in this photocatalytic process is the oxidation of the adsorbed phenoxy anion, which is strongly influenced by the electronegativity of the metal (Au > Pt > Ag) (Scheme 9).
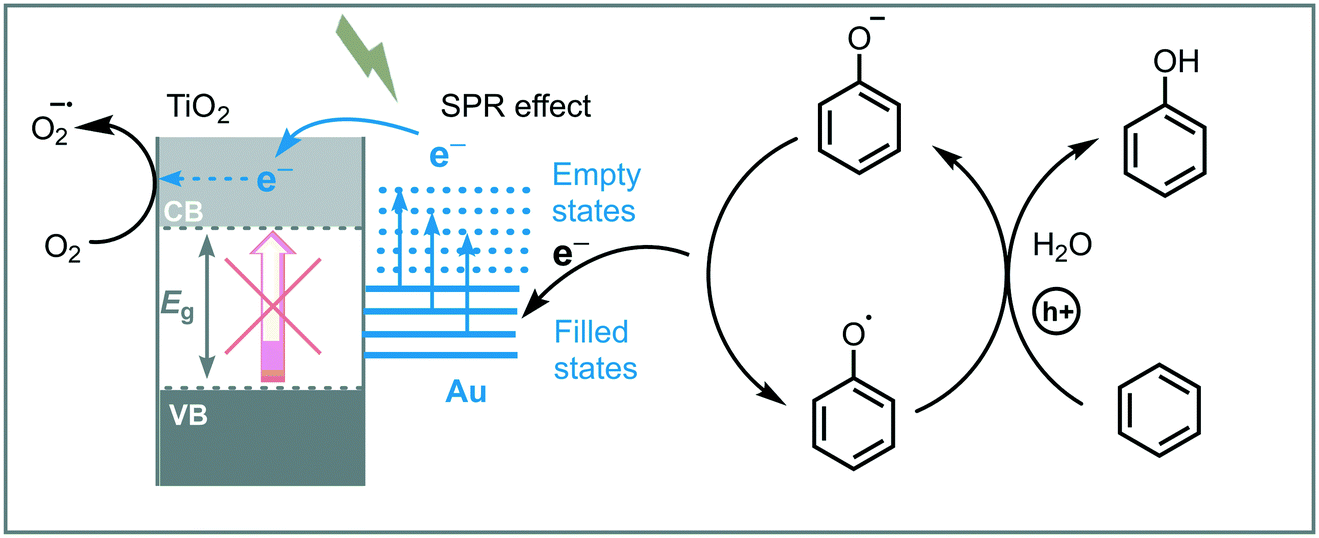 | ||
| Scheme 9 Proposed mechanism for the photooxidation of benzene into phenol photocatalyzed by Au@TiO2 under visible light. | ||
It is well known that carbon allotropes, such as C60, carbon nanotubes (CNT) and graphene (GR) may improve the photocatalytic performance of MOS due to their excellent electron conductivity, which may increase the lifetime of the photoexcited electron–hole pairs by charge separation.43 In an interesting example, Shiraishi et al. developed a photocatalytic system containing reduced graphene oxide (rGO) and TiO2 to carry out the photooxidation of cyclohexane into cyclohexanone using UV irradiation (2 kW Xe lamp, λ > 300 nm).44 The system displayed higher yields and selectivities (>80%) in comparison to the use of bare TiO2 (ca. 60%). They proposed that the enhancement of the selectivity is due to efficient charge separation promoted by rGO, which traps the e−CB of TiO2 after being photoexcited, favoring the selective formation of the cyclohexanone. This event avoids the formation of single electron reduced species (e.g. O2˙−), via the reduction of oxygen by the e−CB in TiO2. These species are known to be involved in the photodecomposition of cyclohexanone. Similar studies also describe the use of hybridized rGO/TiO2 to promote photooxidations of alcohols45 and reduction of nitrosobenzene.46
The oxidation of organic sulfur compounds is a cornerstone in organosulfur chemistry.47 Especially sulfoxides have received increasing amounts of attention due to their presence in biologically active compounds. The oxidation of sulfides using oxygen is the most straightforward approach to obtain these important moieties. The oxidation of sulfides into sulfoxides using light to activate oxygen was achieved by Chen and co-workers. Based on their experience in the synergetic photocatalytic oxidation of sulfides and the oxidative formylation of amines using TiO2,48 they explored the synergy between triethylamine (Et3N) and TiO2 for the oxidation of sulfides.49 With a system comprising Et3N/TiO2/O2, they successfully achieved the photooxidation of several thioanisole derivatives in moderate to excellent conversions (33 to 86%) and selectivities (79 to 98%). The presence of a strong electron withdrawing groups, such as –NO2 group, resulted in lower conversions. They suggested that the presence of the –NO2 negatively affected the Et3N stability. However, when they increased the amount of Et3N (from 0.03 mmol to 0.1 mmol), the desired sulfoxide was obtained in higher conversion (33 to 62%). Based on control experiments and DFT calculations, the authors could unravel the role of Et3N and CH3OH in the photocatalytic process. They proposed that the interaction between Et3N and TiO2 favors the LMCT and results in an increased visible light response. After irradiation, the electrons in the HOMO of the adsorbed amine are excited and injected into the CB of the TiO2. The hole located on the tertiary amine acts as a redox mediator and favors the oxidation of thioanisole to form the corresponding sulfide cationic radical, thus regenerating the amine. Subsequent oxygen atom transfer to the sulfide restores the TiO2 electronic state and after further electron and proton transfer (from the methanol), the targeted sulfoxide is formed (Scheme 10).
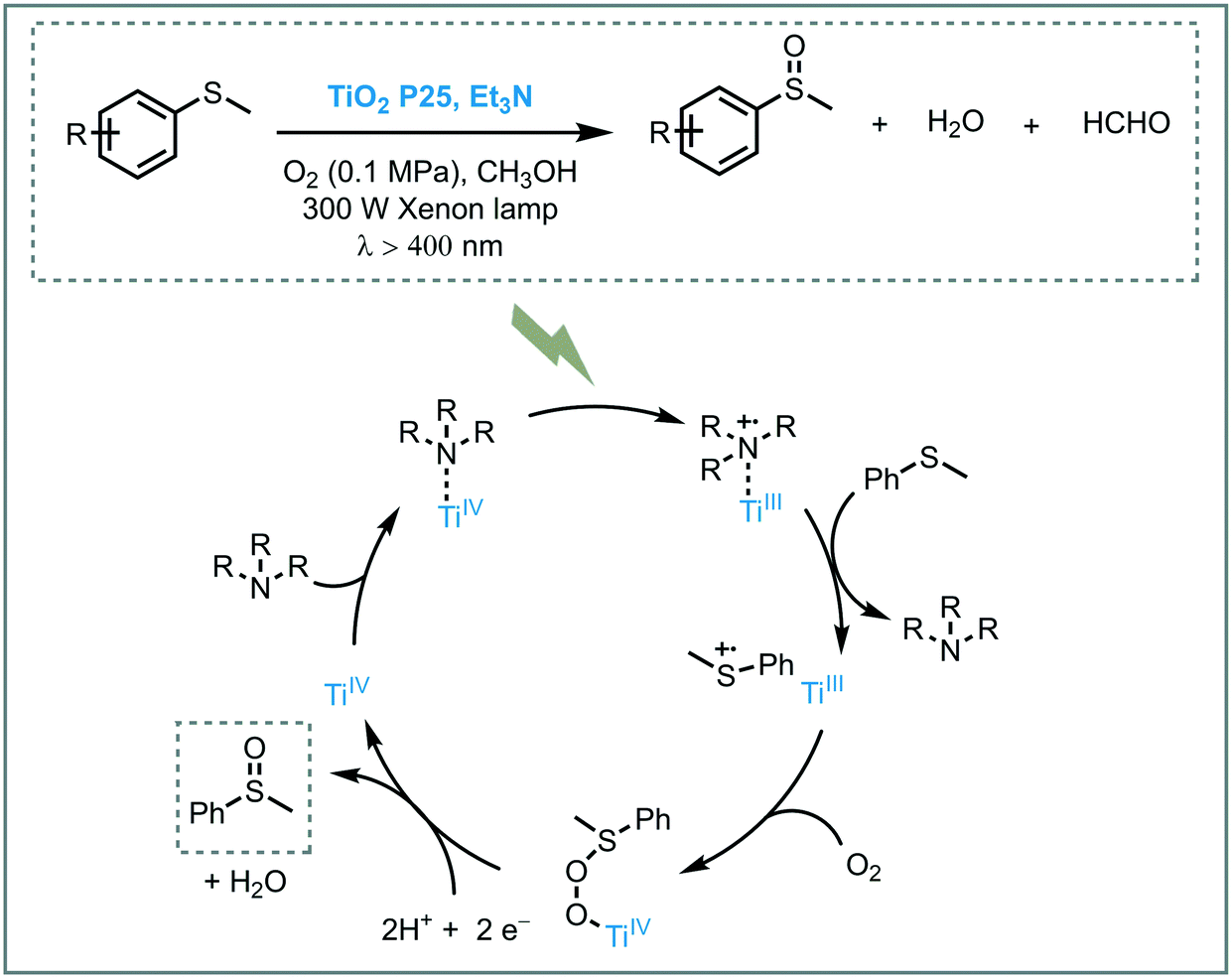 | ||
| Scheme 10 Proposed mechanism for the aerobic oxidation of thioanisole photocatalyzed by TiO2 under visible light irradiation and a tertiary amine as the redox mediator. | ||
Rezaeifard and co-workers reported that Co–ascorbic acid complex-sensitized–TiO2 (TiO2–AA–Co) is able to induce a variety of organic transformations when subjected to visible light irradiation. The photocatalyst was prepared through incorporation of Co(acac)2 into ascorbic acid-surface-modified-TiO2 nanoparticles. Diffuse reflectance spectroscopy (DRS) data revealed a synergetic effect between Co–ascorbic acid and TiO2, resulting in an enhanced visible light response due to a decrease of the MOS band gap (∼0.4 eV). The nanohybrid photocatalyst was successfully applied to promote the aerobic oxidation of benzylic alcohols, benzylic hydrocarbons, and olefins in the presence of N-hydroxyphthalimide (NPHI) as a co-catalyst.50 More recently, they expanded the reaction scope of this catalytic system to promote a cross-dehydrogenative-coupling (CDC) between aldehydes and N-hydroxyimides. The photocatalyst showed excellent recyclability and retained a good stability and activity after several cycles (Scheme 11).
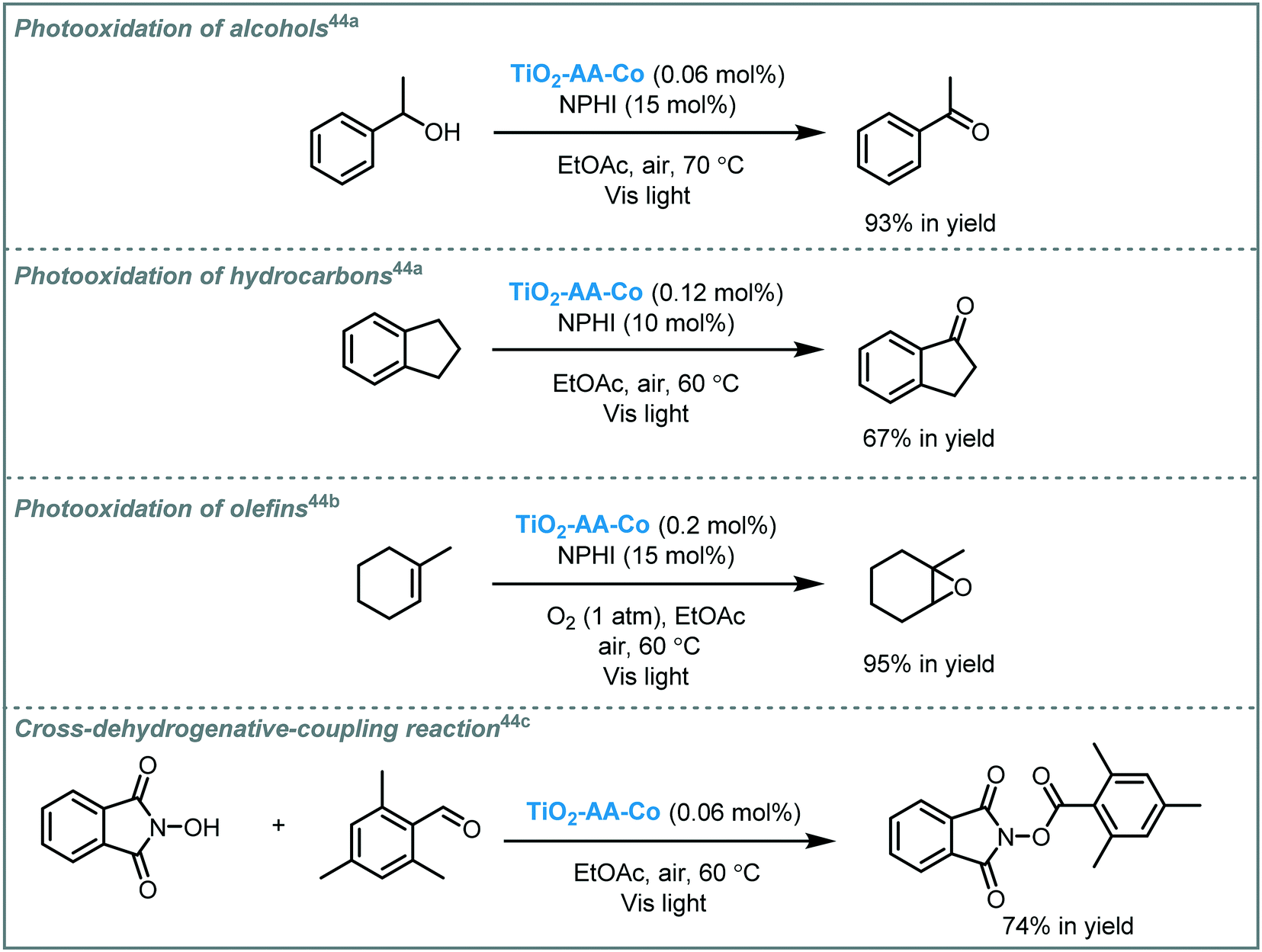 | ||
| Scheme 11 Selected examples for the aerobic oxidation and CDC photocatalyzed by TiO2–AA–Co under visible light. | ||
Scaiano and co-workers reported the use of Pd@TiO2 to enable a photochemical Ullmann reaction between two aryl iodides using both visible and UV irradiation.51 They claimed that, by subjecting Pd@TiO2 to two-color light sources, enhanced selectivity and yields were obtained for the corresponding cross-coupled biaryl compound. According to the authors, upon UV irradiation, the photoexcited TiO2 facilitated the formation of THF radical species (I), which are key for the photocatalytic process.52 This radical species subsequently transfers an electron to the aryl iodide bearing electron withdrawing groups to form a short-lived aryl radical (II), which generates the oxidative addition complex with palladium. Next, the aryl iodide bearing electron donor groups associates with the surface of the Pd or TiO2. Through visible light induced photoreductive elimination, the desired cross-coupled product is formed (Scheme 12). Various aryl iodides could be cross-coupled using this method with yields up to 94%.
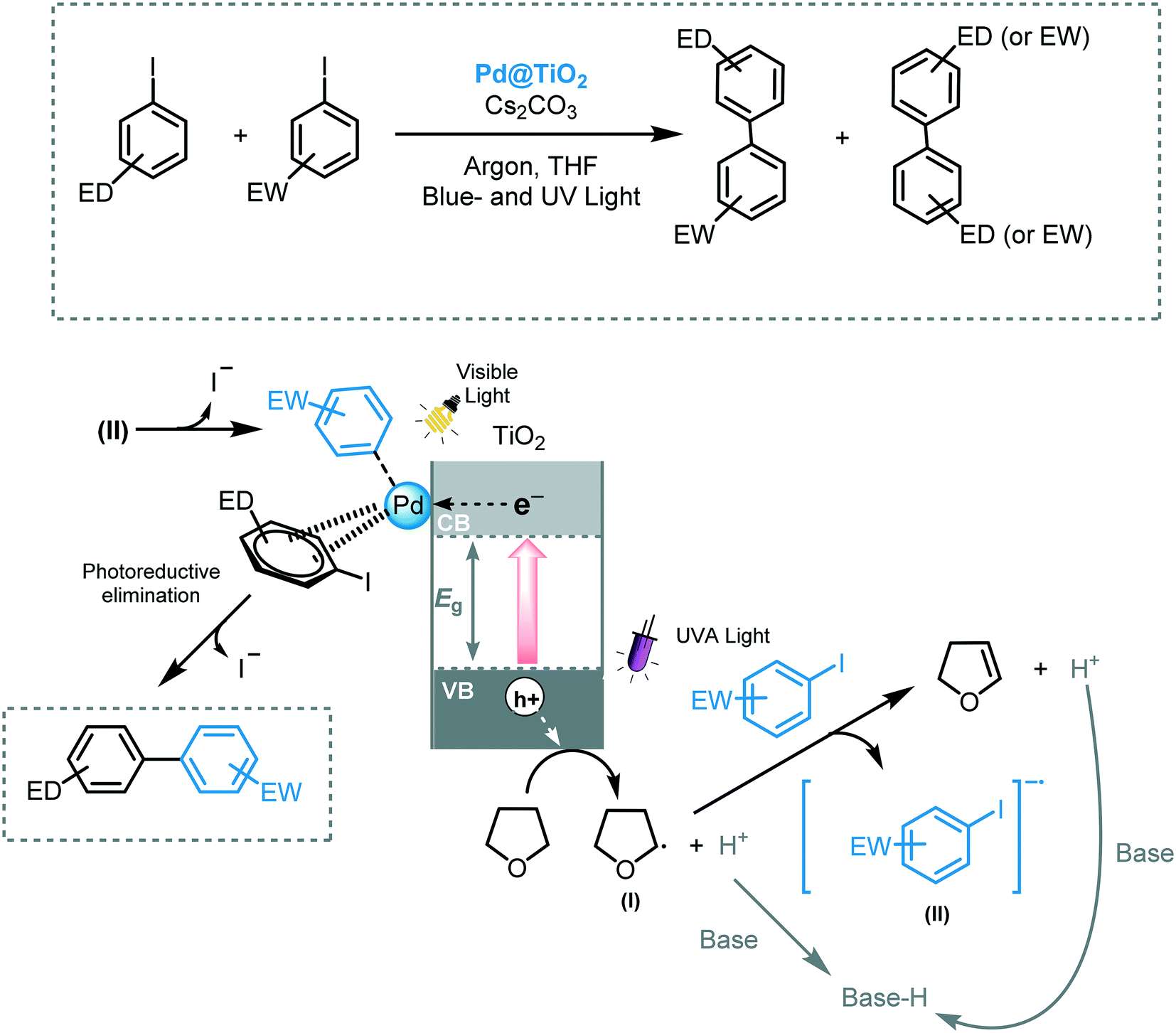 | ||
| Scheme 12 Dual light enables to induce Ullmann reaction photocatalyzed by Pd@TiO2. | ||
Yoshida et al. described the cross-coupling between tetrahydrofuran and a variety of alkanes via UV-induced C–H bond activation using Pt/TiO2 as a photocatalyst (xenon lamp, λ ≥ 350 nm).53 They proposed that the Pt nanoparticles have a dual role in this process: (i) reducing the recombination of the photogenerated electron–hole pairs and; (ii) acting as metal catalyst to activate the Csp3–H bond of the THF or alkane molecules. Next, the same authors reported the cross-coupling between benzene and cyclohexane using metal modified-TiO2 (metal = Rh, Pt, Au, Pd, Ag, Ni, Co) under UV and visible light irradiation (xenon lamp with a long pass filter, λ > 350 or 400 nm).54 Pd modified-TiO2 exhibited higher selectivity for the formation of phenylcyclohexane than other metals when UV irradiation was used. A further enhancement of the selectivity towards the desired cross-coupled product was observed when the system was irradiated with visible light (λ > 400 nm). The authors disclosed that the high selectivity observed for the Pd/TiO2 catalytic system could be attributed to the LMCT excitation, established through π-interaction between the benzene complex and the TiO2 surface. Based on these observations, they proposed that under visible light irradiation the CT complex favors the formation of a benzene radical cation which can selectively activate cyclohexane to yield the cyclohexyl radical. Next, the cyclohexyl radical attacks the benzene molecule through addition–elimination and yields the desired cross-coupled product. Conversely, under UV irradiation, the formation of cyclohexyl and benzene radicals is caused by the holes on the VB of TiO2 and the coupling between these radicals decreases the selectivity of the reaction (Scheme 13).
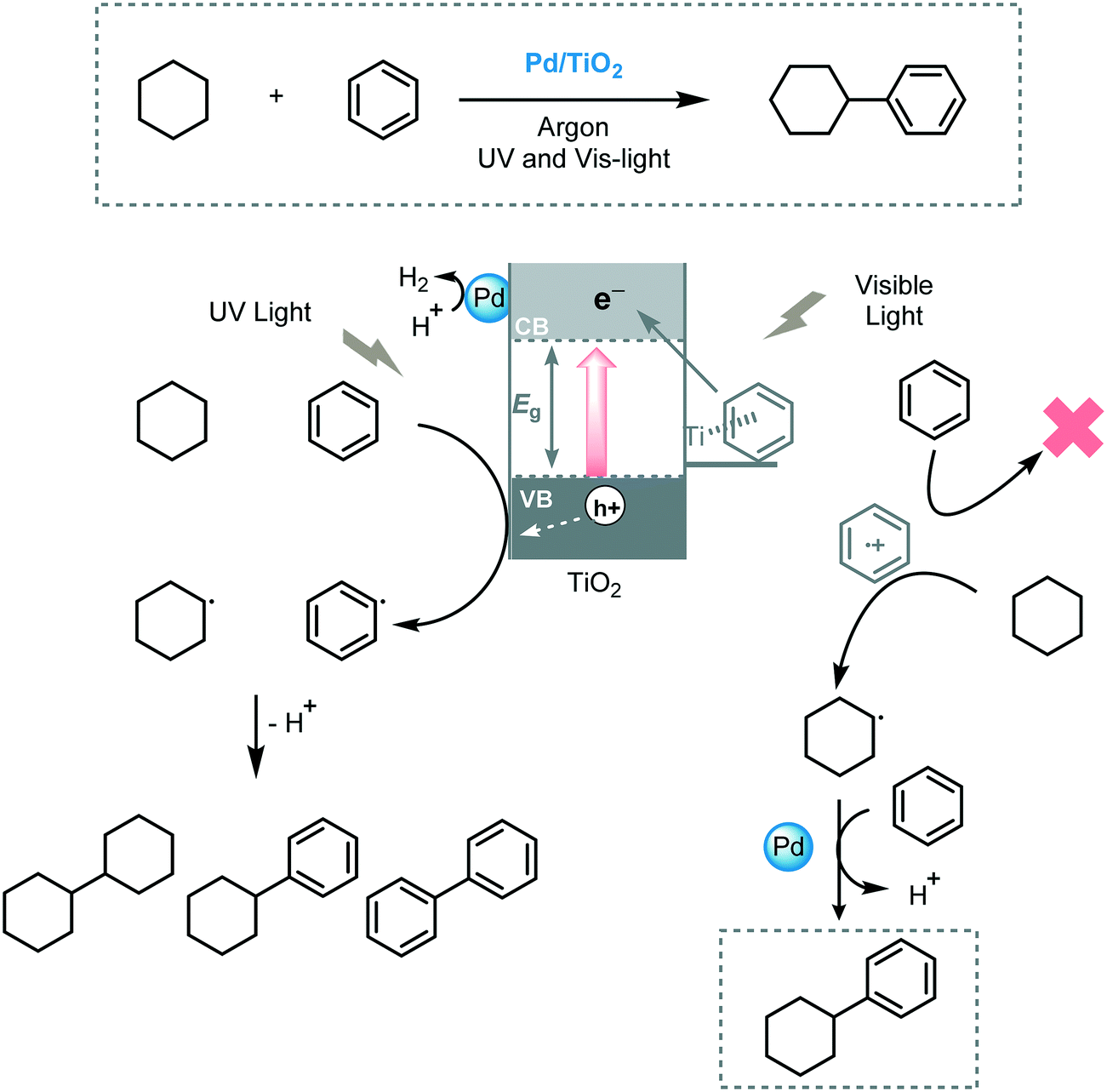 | ||
| Scheme 13 Proposed mechanism for the benzene/cyclohexane cross-coupling reaction photocatalyzed by Pd/TiO2. | ||
The light-induced radical decarboxylation of carboxylic acids is another valuable approach to obtain carbon-centered radicals. Inspired by the photo-Kolbe reaction,55 Walton and co-workers reported the decarboxylation of carboxylic acids by photolysis using TiO2 (P25 Degussa) under UVA irradiation (15 W Cleo tubes, λ ≈ 350 nm), to obtain a range of C-C coupled products.56 The photocatalytic protocol was successfully applied for the photolysis of a variety of carboxylic acids furnishing the corresponding homocoupled products in moderate to good yields (38–83%) and high selectivity. In this case, Pt (0.1%) was used to enhance the photocatalytic activity of the TiO2. They also extended the scope of the decarboxylation reaction to promote the alkylation of electron-deficient alkenes using TiO2 (P25). In all cases, the formation of moderate amounts of the corresponding reduced alkenes was observed. Notably, the reaction between phenoxyacetic acid and maleic anhydride furnished the desired alkylated product along with dihydrofurochromenedione derivatives (C). The authors claimed that the appearance of these adducts might be the result of an intramolecular closure onto the phenyl ring followed by re-aromatization (Scheme 14). Nevertheless, the selectivity of the reaction could be tuned, in favor of the alkyl- or cycloadducts, using platinized-TiO2 or by changing the reaction concentration. Taking into account these observations, they expanded the protocol for intramolecular reactions with acceptor-functionalized aryloxyacetic acids. However, the cycloadducts were obtained in low yields (up to 58%). The proposed mechanism involves the oxidation of the carboxylic acid by the photogenerated holes on the VB of the TiO2 furnishing the corresponding radical cation (I). Deprotonation of the radical species followed by decarboxylation leads to an alkyl radical (II) that is added across the double bond of the alkene. Electron transfer from TiO2 and subsequent protonation afforded the alkylated alkene (Scheme 14).
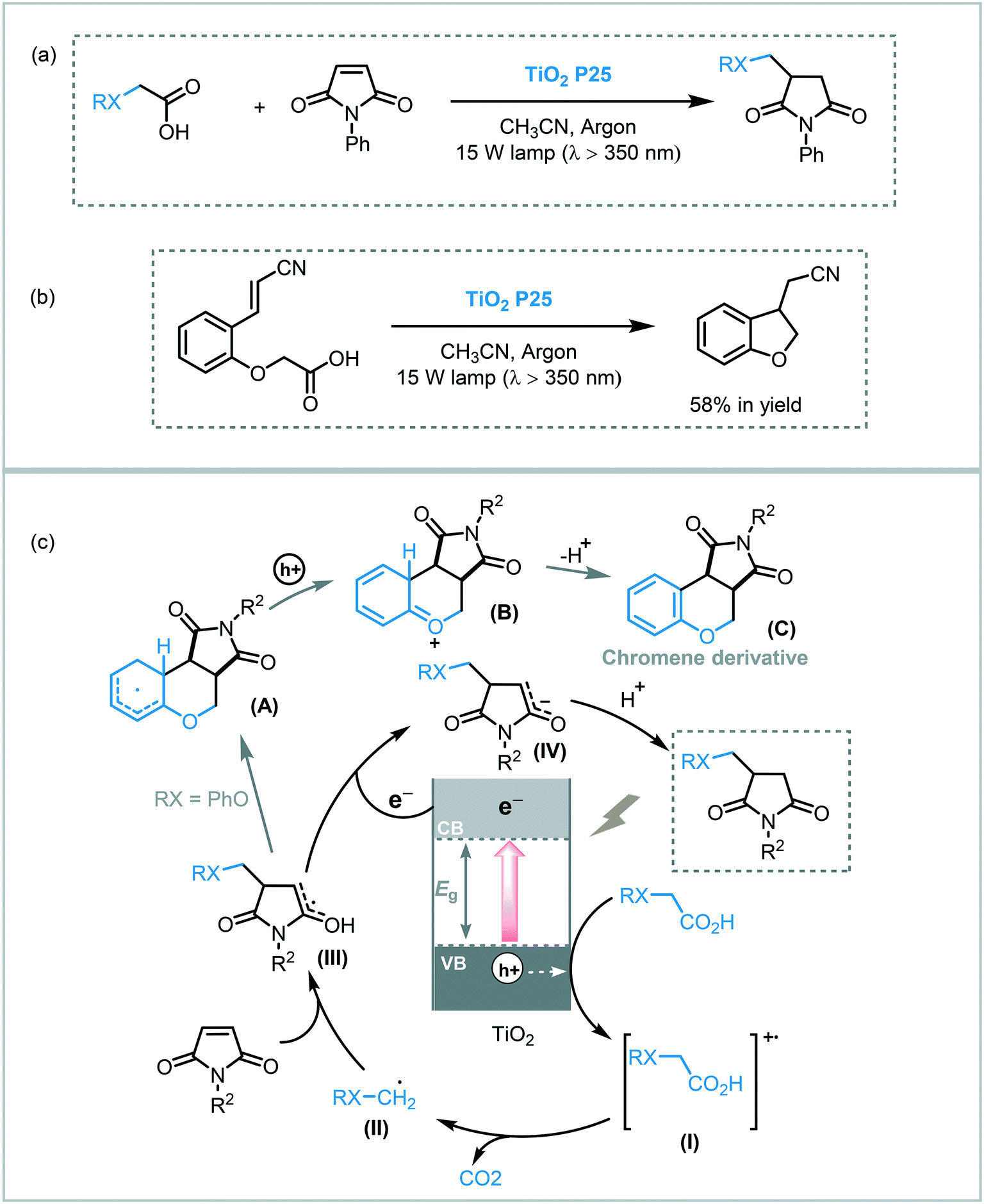 | ||
| Scheme 14 Alkylation of alkenes with carboxylic acids promoted by TiO2. (a) Alkylation of N-phenylmaleimide with carboxylic acids; (b) addition–cyclization reaction; (c) proposed mechanism. | ||
Cong and Ren promoted the decarboxylative alkylation of the benzylic C–H bond of aryltetrahydroisoquinolines in 2,2,2-trifluoroethanol (TFE) using erythrosine B sensitized-TiO2 as photocatalyst and N-acyloxy-phthalimide as alkylating agent under visible light irradiation.57 A range of aryltetrahydroisoquinolines were successfully subjected to the photocatalytic protocol furnishing the corresponding alkylated products in moderate to excellent yields (44 to 93%). It was suggested that the formation of the desired products could be attributed to the radical–radical coupling between the alkyl radical and the benzylic radical generated by the electrons in the CB of TiO2 and the photogenerated holes on the dye, respectively (Scheme 15).
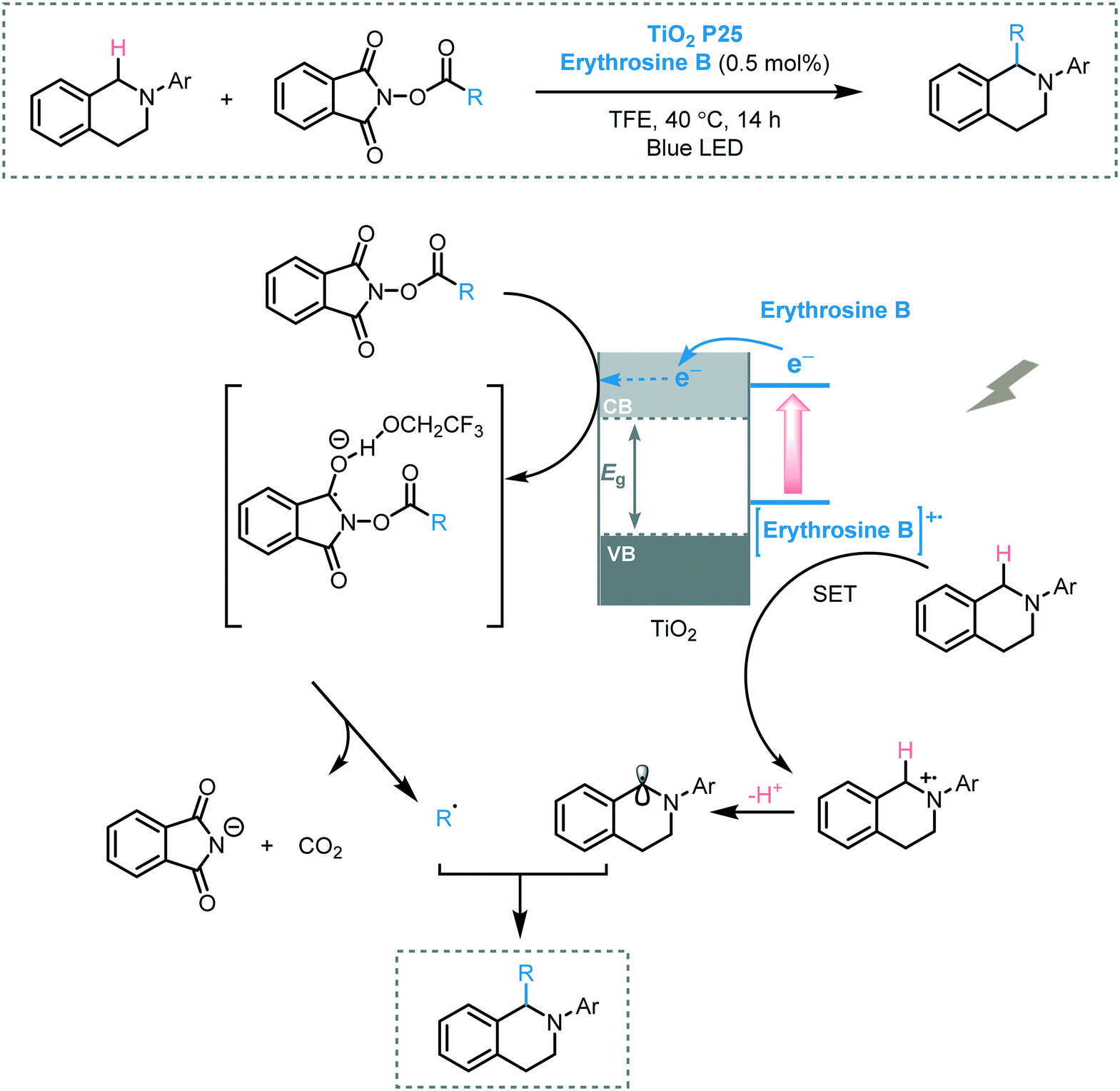 | ||
| Scheme 15 Proposed mechanism for the decarboxylative alkylation of C–H bond photocatalyzed by erythromycin B-sensitized TiO2. | ||
Arylation of heteroarenes is another useful transformation to establish C–C bond formation and to access valuable heterobiaryl structures.58 The most efficient methods to forge aryl–heteroaryl bonds relay on the direct arylation of heteroarenes through C–H bond activation using transition metals in the presence of ligands and an aryl source such as aryl halides, aryl boronic acids or aryl diazonium salts.59 Nevertheless, photocatalysis offers an eco-friendly alternative for the use of transition metals. In this context, Rueping et al. reported the use of commercially available TiO2 (P25) as a photocatalyst to carry on the C–H arylation of heteroarenes under visible light irradiation (11 W household lamp).60 The reaction of a variety of heteroarenes, including furans, thiophenes, pyridines, could be established using aryl diazonium salts as coupling partners. The corresponding heterobiaryl compounds could be obtained in good to excellent yields (53–94%). In addition, the photocatalyst could be recycled and reused for 5 cycles with a negligible decrease in reactivity. Based on spectroscopic experiments, they proposed that the formation of the TiO2-azoether (I) is responsible for the visible light response of the photocatalytic system. Thus, after irradiation, the excited state of the modified-TiO2 induces the formation of the corresponding aryl radical species (III) through SET. The aryl radical subsequently reacts with the heteroarene to yield a biaryl radical species (IV). The oxidation of the biaryl radical (IV) results in the formation of the carbocation ion (V) and is followed by proton removal and rearomatization, yielding the corresponding heterobiaryl compound (Scheme 16).
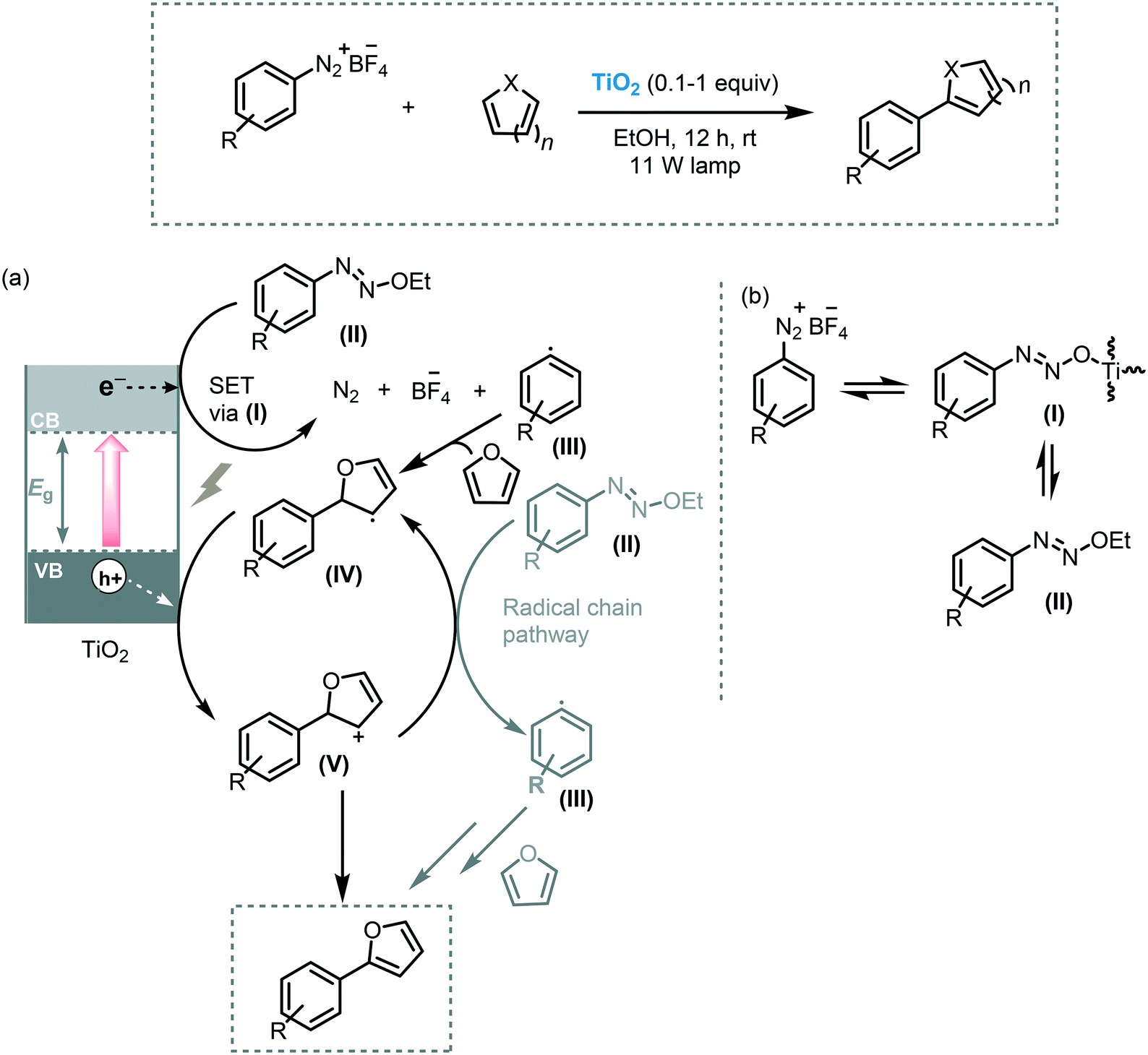 | ||
| Scheme 16 α-Arylation of heteroarenes photocatalyzed by TiO2 under visible light. (a) Proposed mechanism and (b) formation of TiO2-diazoether. | ||
Wang and co-workers prepared a ternary system composed of a polyaniline (PANI)–graphitic-carbon nitride (g-C3N4) and TiO2 composite (PANI–g-C3N4–TiO2). The photocatalytic activity of this composite was demonstrated for the arylation of terminal alkynes, using aryl diazonium salts as the aryl source, under visible light irradiation (14 W CFL).61 Although the reaction worked smoothly with a variety of functionalized aryl diazonium salts and phenylacetylene derivatives leading to the corresponding α-chloro aryl ketones in good yields (up to 78%), the reaction failed when electron-rich aryl diazonium salts were employed. The difference in potentials of the band edges of the three components forms the basis of the operative mechanism and can explain these observations. After irradiation, the photogenerated electrons in PANI sequentially decay into the g-C3N4 CB first, and then into the TiO2 CB, so gradually reducing their energy. The electrons in the TiO2 CB are able to reduce the aryl diazonium salts to form the corresponding aryl radical species. This aryl radical adds subsequently to the alkyne system. Simultaneously, the holes on the TiO2 migrate back to the VB of the g-C3N4 and to the π-orbital of PANI, promoting the oxidation of the radical VI to furnish the desired α-chloro aryl ketone. Therefore, the lack of reactivity for the electron-rich aryl diazonium salts can be explained by the low edge oxidation potential of the π-orbital of PANI, which is unable to oxidize intermediate (VI). Nevertheless, it cannot be excluded that the oxidation can be carried through the VB of the g-C3N4 instead of the VB of the PANI (Scheme 17). The same group extended the application potential of this photocatalytic ternary system PANI–g-C3N4–TiO2 to visible light induced C–H arylation of heteroarenes such as enol acetates or benzoquinones using in situ generated diazonium salts in aqueous conditions.62
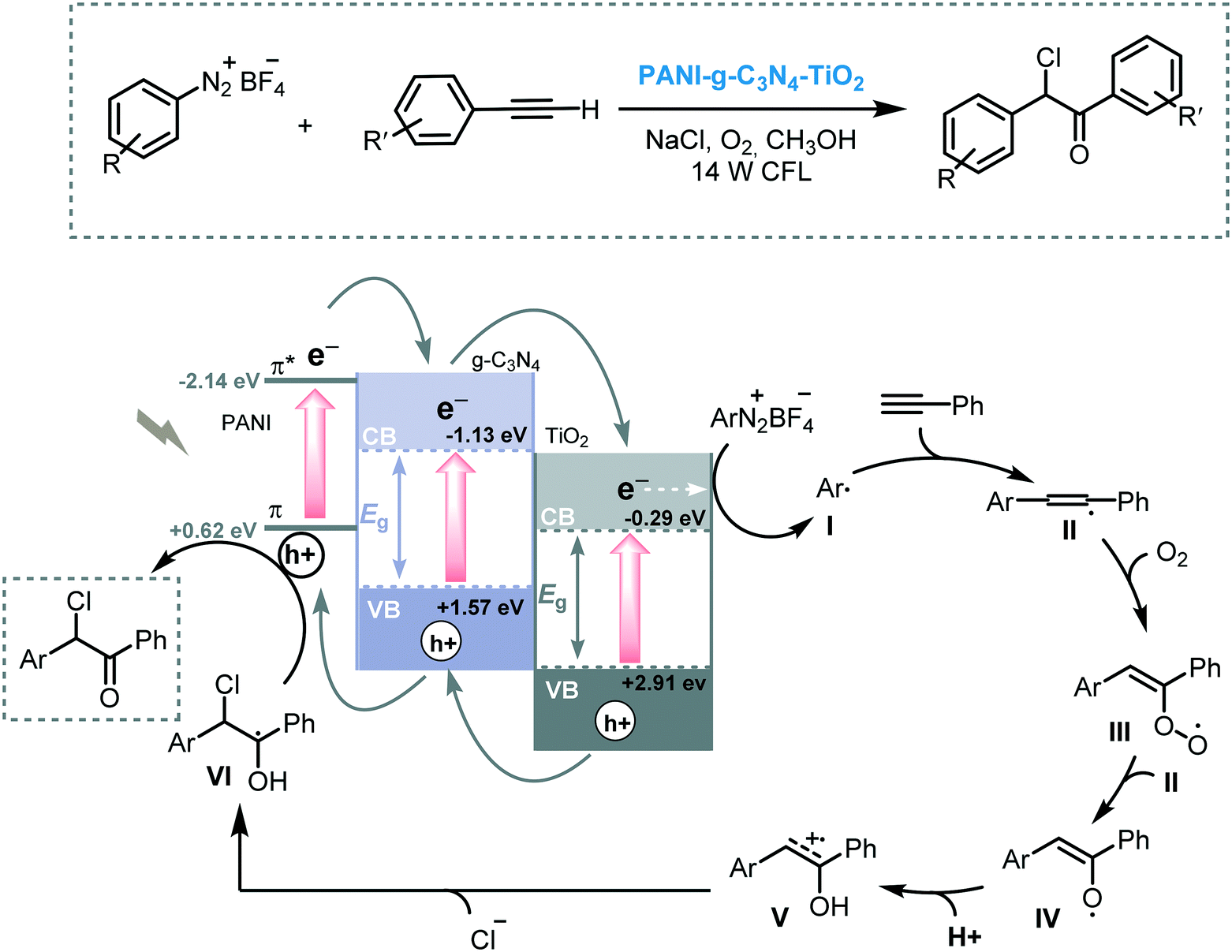 | ||
| Scheme 17 Proposed mechanism for the arylation of phenylacetylene derivatives photocatalyzed by PANI–g-C3N4–TiO2. | ||
The photocatalytic functionalization of C–H bonds adjacent to a nitrogen atom has been thoroughly studied in the last 10 years.63 The importance of this transformation resides in that valuable building blocks can be accessed in a single step using this approach. Shen and co-workers contributed to this field by demonstrating the application of MOS to promote the cyclization of tertiary anilines with maleimides through C–H bond activation under visible light irradiation (3 W blue LED).64 For that purpose, they decorated the surface of TiO2 with highly dispersed NiO particles, extending the light response of the MOS to the visible region and also reducing the hole–electron pair recombination. A strong correlation between the amount of NiO nanoparticles loaded onto the TiO2 surface and the photocatalytic activity of the system was observed. Starting from a wide range of tertiary anilines and maleimides, a large variety of tetrahydroquinolines was synthesized in good to excellent yields (45 to 93%) in relatively short reaction time (12 h). The TiO2/NiO photocatalyst could be recycled and reused for at least nine consecutive cycles without significant detrimental effect on its photocatalytic activity. In the proposed mechanism, a SET from the tertiary amine to the hole of the photoexcited photocatalyst occurs, after which a proton transfer of the cationic radical (I) to the superoxide anion happens, yielding the α-amino radical species (II). The addition of this species to the Michael acceptor is followed by an intramolecular cyclization leading to radical intermediate (IV). Subsequent oxidation of IV, by oxygen or photogenerated electrons of the photocatalyst, and deprotonation yields the target product (Scheme 18).
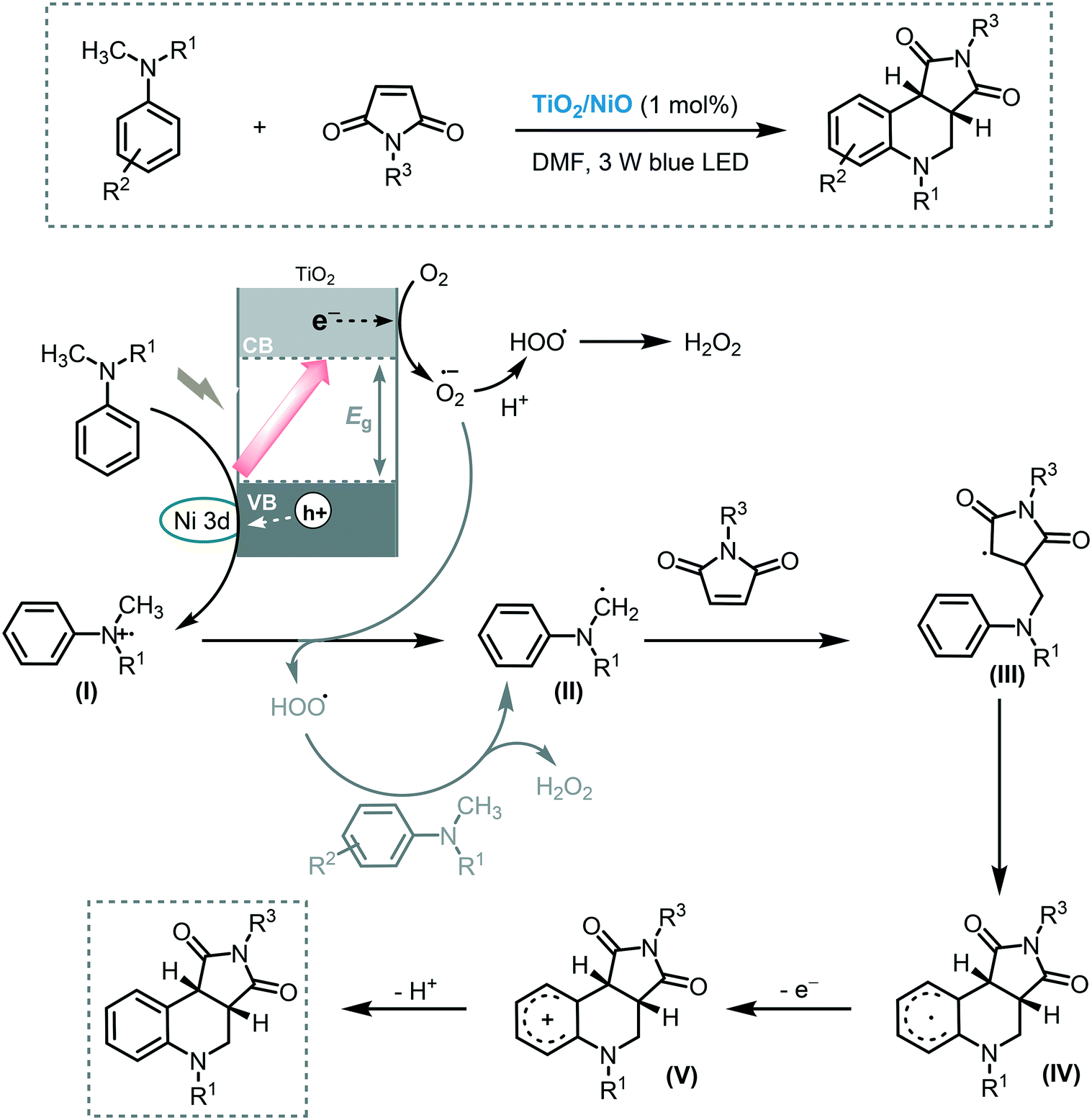 | ||
| Scheme 18 Proposed mechanism for the synthesis of α-chloro ketones using TiO2/NiO photocatalysis under visible light irradiation. | ||
The reaction of tertiary amines with cyanide sources affords α-amino nitriles which are versatile building blocks for the synthesis of nitrogen-containing compounds. Especially the Strecker reaction65 and transition metal catalyzed C–CN bond formation66 are the most straightforward methodologies to obtain α-amino nitriles. Using light to drive these transformations was first reported by Santamaria et al. in 199067 and, since then, various reports were published on the use of photoredox catalysis to promote the cyanation of amines to aminonitriles.68 One elegant example of the application of MOS to light-induce cyanation of amines was reported recently by Opatz and Tremel and co-workers.69 They anchored a 6,7-dihydroxy-2-methylisoquinolinium (DHMIQ) chromophore onto the surface of TiO2 nanoparticles (DHMIQ–TiO2) triggering a panchromatic sensitization of the photocatalyst which was able to promote the aerobic photocyanation of tertiary amines. With this catalyst, they were able to synthesize a variety of α-aminonitriles with excellent yields (up to 97%) starting from a range of tertiary amines using trimethylsilyl cyanide (TMSCN) and oxygen under visible light irradiation (λ = 462 nm). From a mechanistic point of view, they proposed that, after the irradiation of the photocatalytic system, the catechol-based ligand is excited and is able to inject an electron into the TiO2 CB. The reduction of O2 by the e−CB leads to the formation of the hyperoxide radical anion O2˙− that can displace CN− from TMSCN, yielding a TMS-peroxo species. These species can accept an H-atom from the amine radical cation to form an iminium ion which can be subsequently be trapped by the CN− furnishing the desired α-aminonitrile compound (Scheme 19).
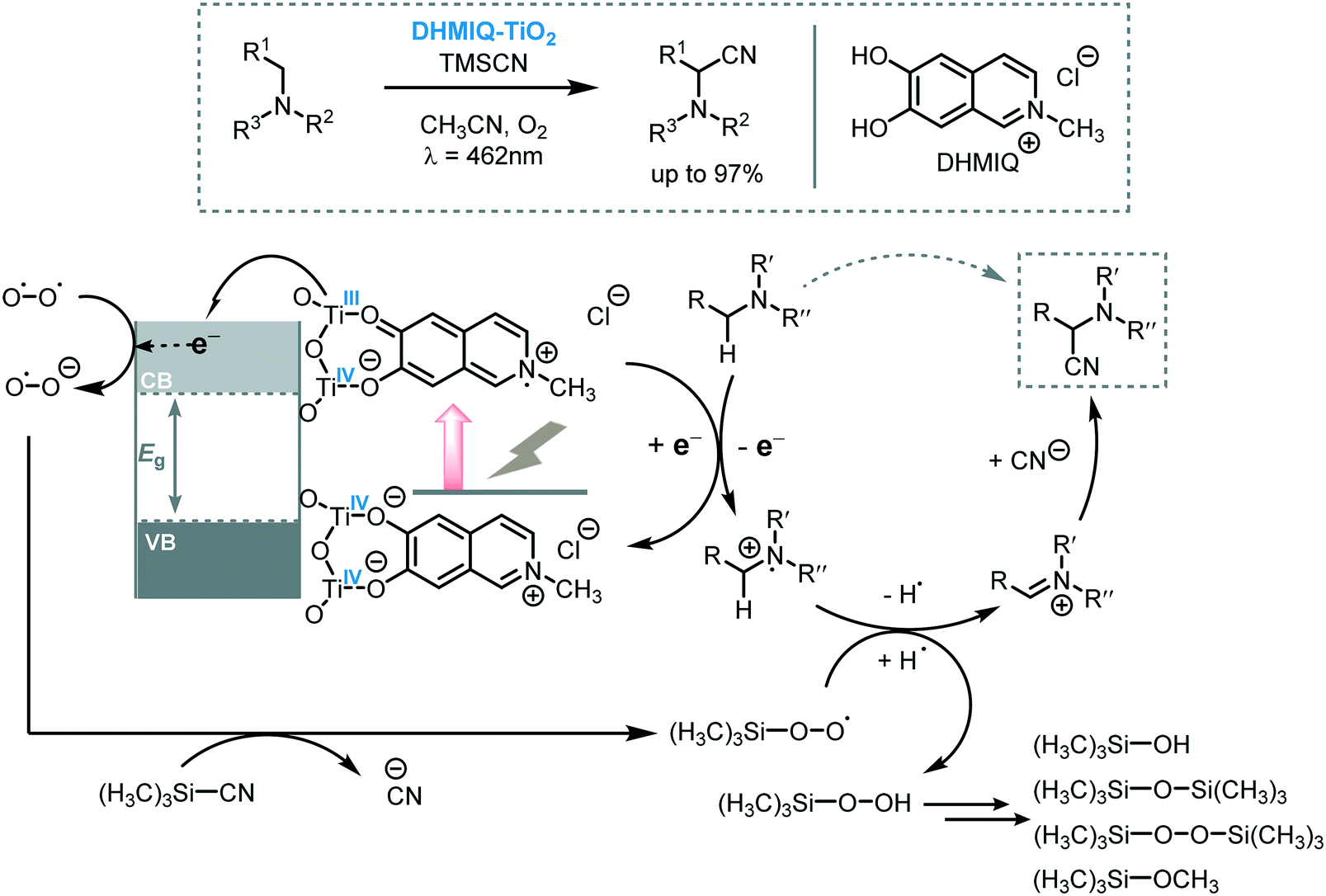 | ||
| Scheme 19 Proposed mechanism for the cyanation of amines photocatalyzed by DHMIQ–TiO2 under visible light. | ||
Scaiano et al. reported the use of Pt nanoparticles-decorated TiO2 as a photoredox catalyst to successfully promote the reductive dehalogenation–cyclization of aryl and alkyl iodides under UVA/visible light irradiation (solar simulator with 375 nm cut off filter).70 The desired products were obtained in good to excellent yields (up to 87%). Despite the fact that the catalyst could be reused for 4 cycles, poor recyclability was observed, which can be attributed to the agglomeration of the Pt nanoparticles. The approach was also extended to carry out intramolecular cyclization of bisenones. However, for this reaction, the corresponding products were obtained with moderate conversions and low selectivity. The authors surmised that TiO2 may present a dual role in this reaction: (i) photoinduced the electron transfer; and (ii) act as a weak Lewis acid, which could activate the enone favoring the formation of the radical anion upon electron reduction. Also, they claimed that the presence of Pt nanoparticles doping the TiO2 surface could enhance the catalytic activity of TiO2 and increase its visible light response (Scheme 20).
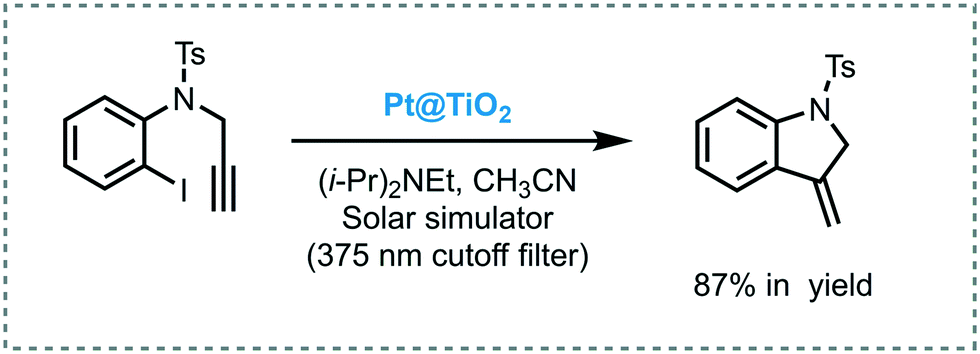 | ||
| Scheme 20 Selected example for the hydrodehalogenation and cyclization of iodine compound photocatalyzed by Pt@TiO2. | ||
More recently, the same research group reported the [4 + 2] cycloaddition of indoles and electron-rich dienes promoted by Pt nanoparticles supported on TiO2 under visible light irradiation (10 W LED, λ = 460 nm).71 The reaction worked successfully for a variety of indoles and cis-1,3-dienes providing the corresponding alkaloids in moderate to good yields (up to 72%) and stereoselectivities. The heterogeneous photocatalyst could be easily recuperated and reused for three cycles. However, a deleterious effect on the photoactivity of the catalyst was observed. The proposed mechanism starts with the adsorption of the indole nitrogen onto the surface of the MOS leading to a weakly visible light absorbance. Upon irradiation, the nitrogen of the indole injects electrons into the CB of the semiconductor, which are trapped by the Pt nanoparticles and subsequently quenched by either MeNO2 or O2, decreasing the probability of electron–hole recombination. Once the indole radical-cation is formed, it can undergo an [4 + 2] cycloaddition with 1,3-cyclohexadiene, which is followed by reduction of the as-formed tetrahydrocarbazole radical cation with an electron originating from the Pt@TiO2. The rapid acylation of the tetrahydrocarbazole prevents the overoxidation and cycloreversion of the reaction products (Scheme 21).
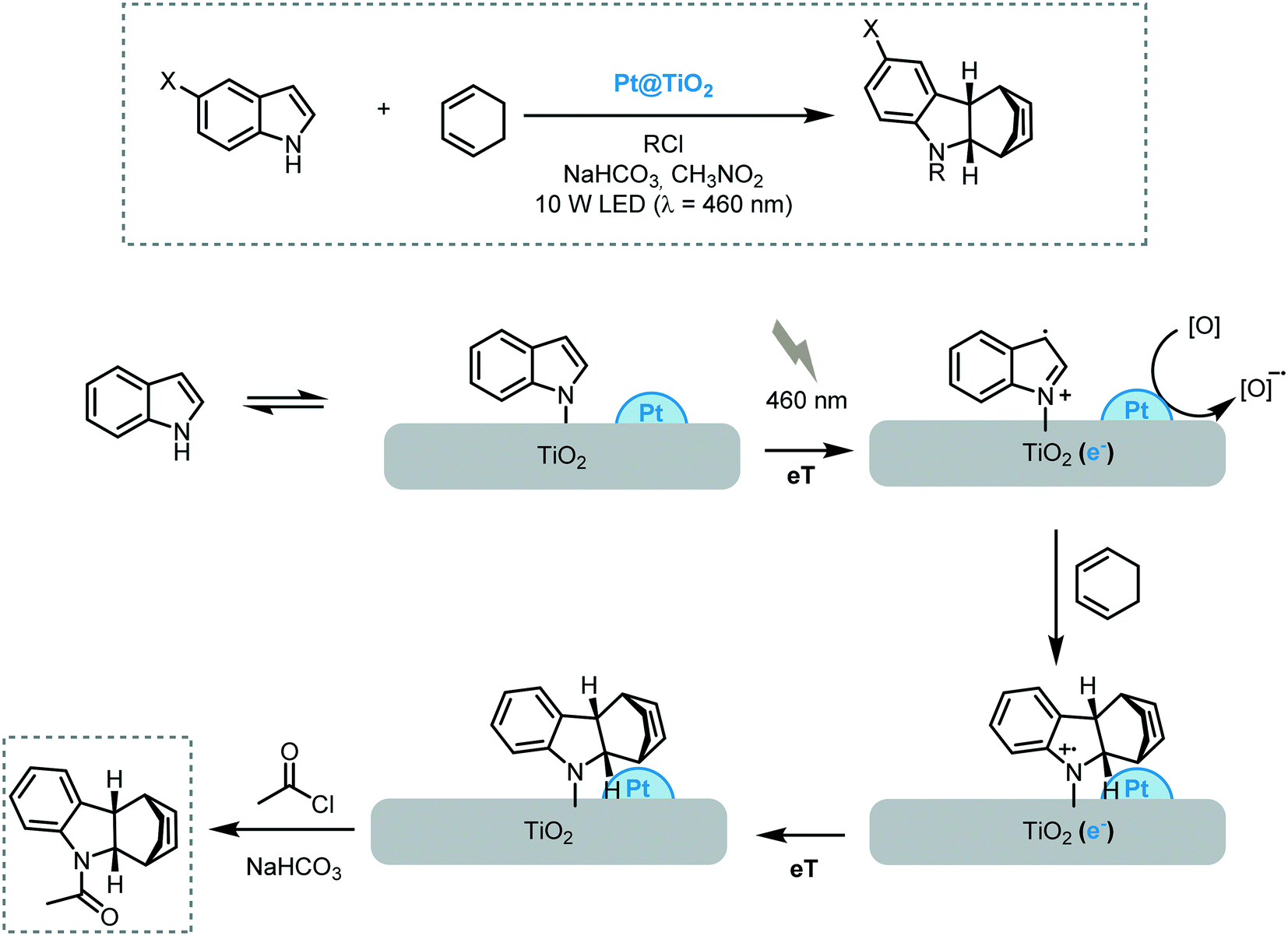 | ||
| Scheme 21 Proposed mechanism for the Diels–Alder reaction photocatalyzed by Pt@TiO2. | ||
Inspired by the last findings, Scaiano et al. reported the heterogeneous dual photoredox–Lewis acid catalysis.72 For this purpose, they supported samarium oxide nanoparticles onto nanostructured TiO2 and CeO2 semiconductors. In a first attempt, the efficiency of the photocatalytic system was studied in the reductive cyclization of chalcones under visible light irradiation (90 W LED, λ = 400 nm) using diisopropylethyl amine as an electron donor. The best yield was achieved with SmxOy@TiO2 for the formation of the corresponding cyclopentanol (70%). The authors proposed that the modest reactivity of the SmxOy@CeO2 (up to 40%) in this transformation can be attributed to the lower response in the visible light region compared to SmxOy@TiO2, which possesses a narrower bandgap. The narrower band gap observed for the latter nanocomposite is related to the presence of in-gap electronic states corresponding to the 4f manifold of Sm, with the filled states near the valence edge of TiO2, which reduces the band gap effectively. Based on these results, they investigated the scope of the reaction for functionalized chalcone derivatives and the recyclability of the photocatalytic system. The corresponding cyclic dimers could be obtained in modest to excellent yields (27 to 90%) and diastereoselectivities (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Only traces of [2 + 2] cycloaddition products and pinacol coupling, as by-products, were observed. In some cases, the yields were superior to the ones obtained with previously reported dual homogeneous photoredox catalysis. Using the same photocatalytic system, they also investigated the [2 + 2] photocycloaddition of (bis)enones using SmxOy@TiO2 as the photocatalyst and DABCO as a sacrificial electron donor. The desired cycloadduct products were obtained in modest to good yields (4 to 72%) and selectivities (10 to 81%). In both cases, the nanocomposite photocatalyst could be reused and recycled without apparent loss in reactivity. Regarding the mechanism, a SET from the photoexcited nanocomposite to the Lewis acid-activated substrate was suggested to yield the radical anion species. Thereafter, an intramolecular addition Michael and subsequent cyclobutanation lead to the samarium coordinate ketyl radical, which could release one electron back to the TiO2 nanocomposite yielding the cycloadduct (Scheme 22).
:1). Only traces of [2 + 2] cycloaddition products and pinacol coupling, as by-products, were observed. In some cases, the yields were superior to the ones obtained with previously reported dual homogeneous photoredox catalysis. Using the same photocatalytic system, they also investigated the [2 + 2] photocycloaddition of (bis)enones using SmxOy@TiO2 as the photocatalyst and DABCO as a sacrificial electron donor. The desired cycloadduct products were obtained in modest to good yields (4 to 72%) and selectivities (10 to 81%). In both cases, the nanocomposite photocatalyst could be reused and recycled without apparent loss in reactivity. Regarding the mechanism, a SET from the photoexcited nanocomposite to the Lewis acid-activated substrate was suggested to yield the radical anion species. Thereafter, an intramolecular addition Michael and subsequent cyclobutanation lead to the samarium coordinate ketyl radical, which could release one electron back to the TiO2 nanocomposite yielding the cycloadduct (Scheme 22).
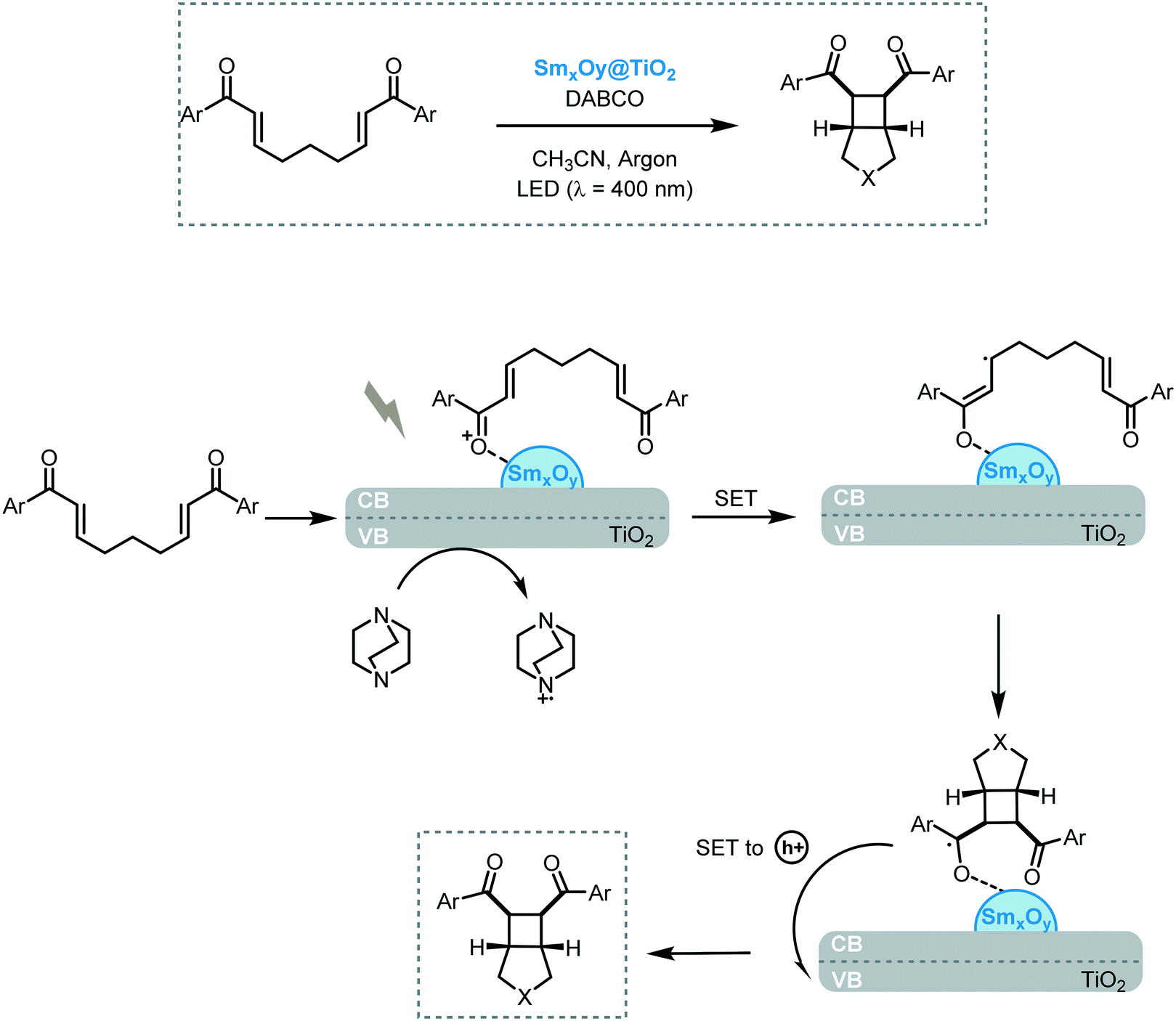 | ||
| Scheme 22 Proposed mechanism for the cycloaddition of (bis)enones photocatalyzed by SmxOy@TiO2 | ||
Finally, Okada and coworkers demonstrated that TiO2 can successfully photocatalyze radical cation Diels–Alder reactions and [2 + 2] cycloadditions using LiClO4 and a nitromethane electrolyte solution under visible light irradiation.73
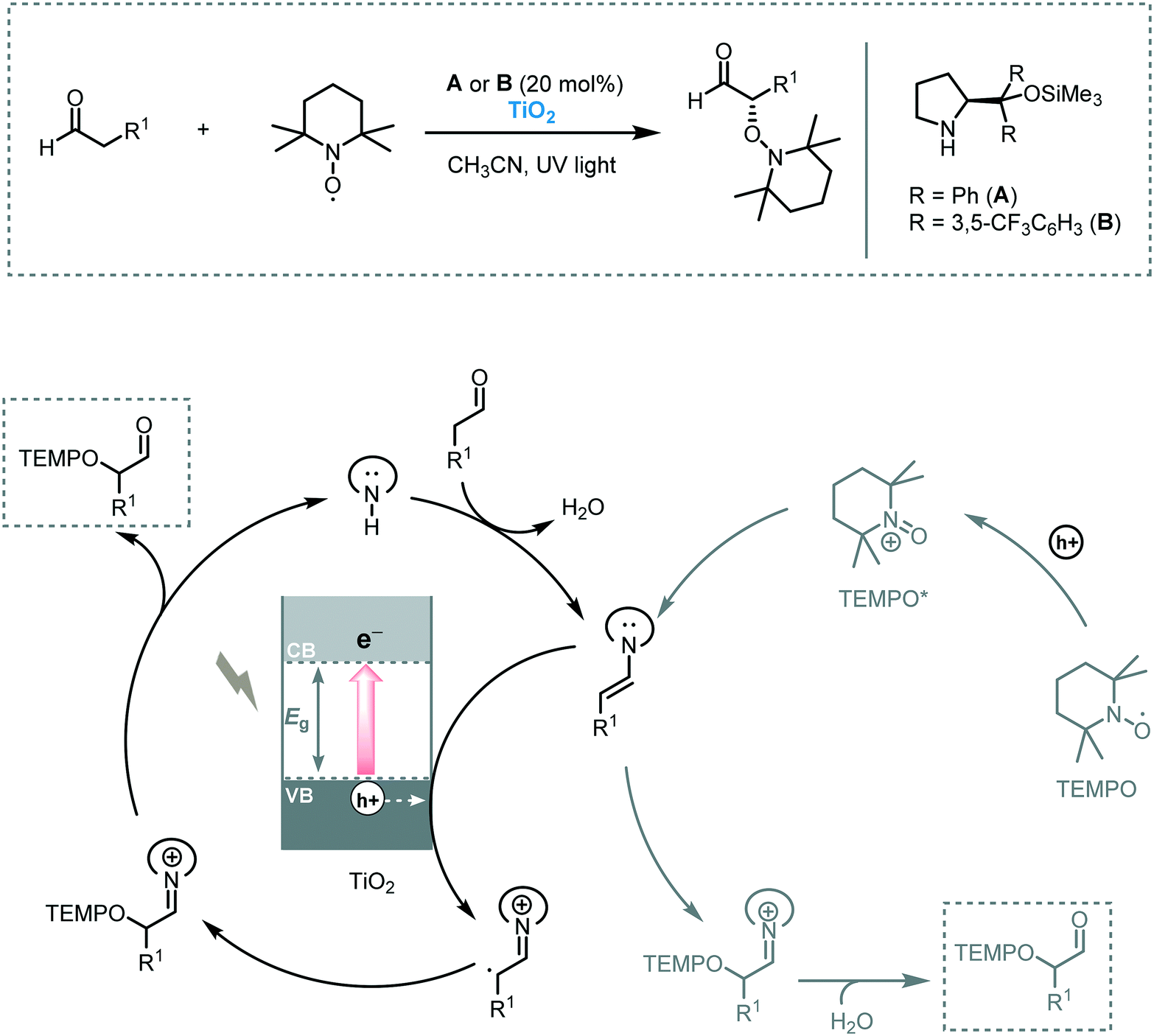 | ||
| Scheme 23 Proposed mechanism for the α-oxyamination of aldehydes with TEMPO photocatalyzed by TiO2. | ||
Organofluorides play a pivotal role in the pharmaceutical and agrochemical sector.77 Hence, the development of novel or improved synthetic approaches install fluorine into organic molecules is a contemporary research objective. Especially because conventional methods to incorporate fluorine typically suffer from low selectivities and poor functional group tolerance. In this regard, the conversion of carboxylic acids into the corresponding fluorinated-compounds by photoredox catalysis has emerged in the last years as a very mild way to obtain organofluorides.78 For instance, Hammond and co-workers promoted the C–F bond formation using TiO2 (P25), as a photocatalyst, by decarboxylative electrophilic fluorination of a wide range of aliphatic carboxylic acids using selectfluor as the fluorine source.79 In addition to the very high turnover frequencies (up to 1050 h−1), the photocatalyst could be recovered and reused during four cycles without a significant loss in catalytic performance. However, the use of more challenging primary aliphatic carboxylic acids, as starting materials, proved ineffective. Spectroscopic and mechanistic studies, suggested that two mechanisms could be involved in the photocatalytic process. First, the photogenerated holes could promote the oxidation of the carboxylate species, which are adsorbed onto the TiO2 surface, affording the formation of the carboxyl radical. After decarboxylation, the corresponding alkyl radical can be fluorinated by selectfluor. The selectfluor cationic species can be next trapped by free electrons generated after the photoexcitation of TiO2 yielding the more stable reduced species. In addition to the main photocatalytic cycle, another free-radical pathway process could be involved. In this case, the selectfluor radical cation could react with other species, such as carboxylate ions or adsorbed carboxylates leading to the alkyl radical and CO2 (Scheme 24). The authors also proposed that the base may have a dual role in the mechanism: it can deprotonate the carboxylic acid and can act as a sacrificial electron donor.
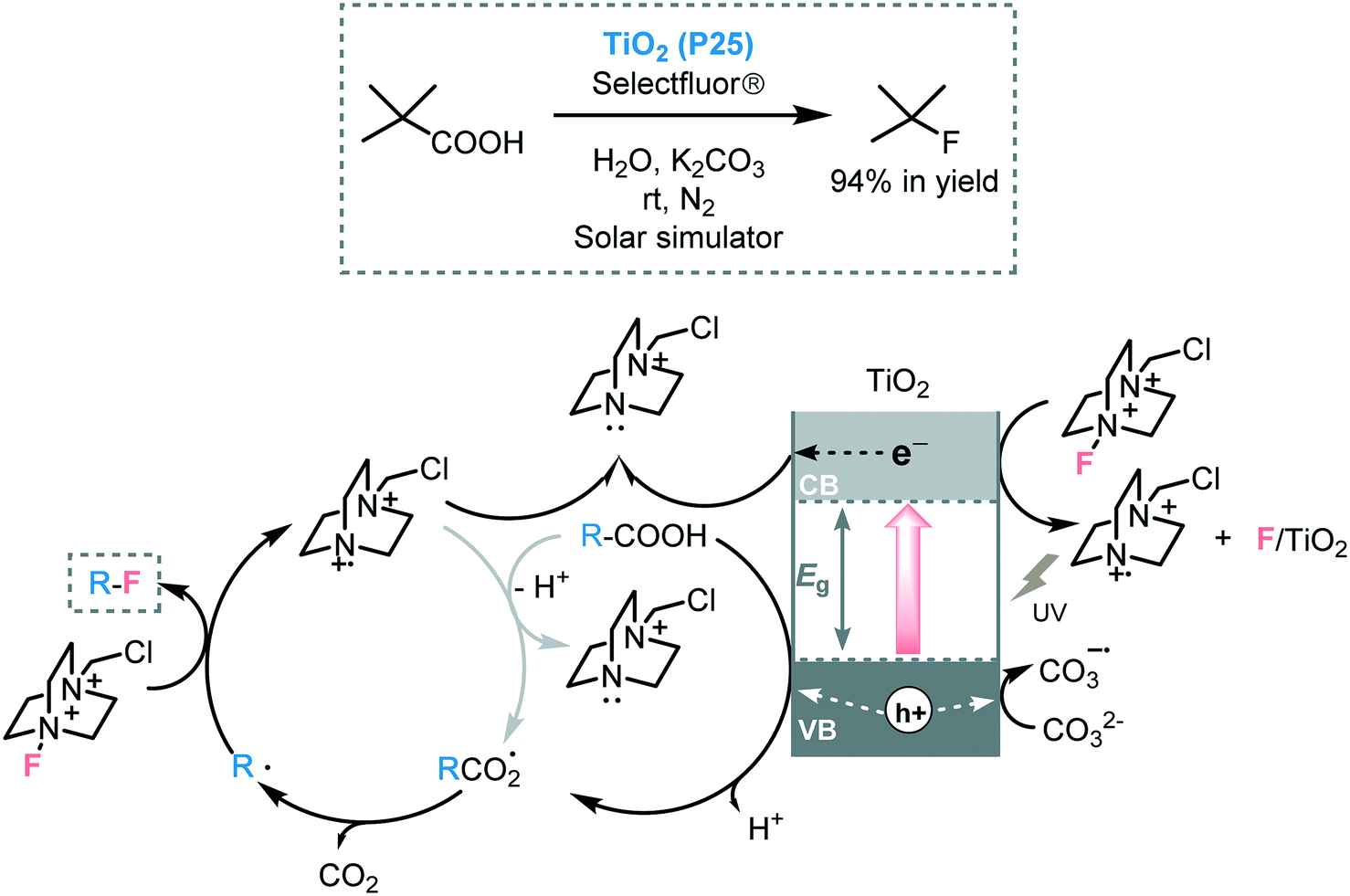 | ||
| Scheme 24 Selected example and the proposed mechanism for the decarboxylative fluorination of carboxylic acids photocatalyzed by TiO2. | ||
Sulfoxidation of hydrocarbons is a widespread and well-known method for the synthesis of alkyl sulfonic acids.80 Normally, this reaction requires the excitation of the SO2 by UV or the presence of radical initiators at high-temperature. The use of visible light is appealing as a desired green alternative for the synthesis of organosulfur compounds. For instance, Kisch et al. promoted the photosulfoxidation of alkanes using TiO2 under visible light irradiation (λ ≥ 400 nm).81 According to the authors, the visible light response of the photocatalyst is due to the formation of a charge-transfer (CT) complex between the surface of the TiO2 and sulfur dioxide generating a CT band in the visible region of the solar spectrum. The photocatalytic system composed by TiO2/O2/SO2 worked smoothly using n-heptane, cyclohexane and adamantine, as starting alkanes, showing high chemoselectivity for the formation of the alkyl sulfonic acids. Moreover, the photocatalyst could be reused after washing with methanol. Based on experimental evidence, the authors suggested that, after visible light irradiation of the TiO2–SO2 complex, electrons are transferred into the CB of the TiO2, through LMCT, promoting the oxygen reduction to furnish superoxides. The SO2 radical cation oxidizes the alkane to give the C-centered radical and a proton, which could induce a radical chain mechanism (Scheme 25).
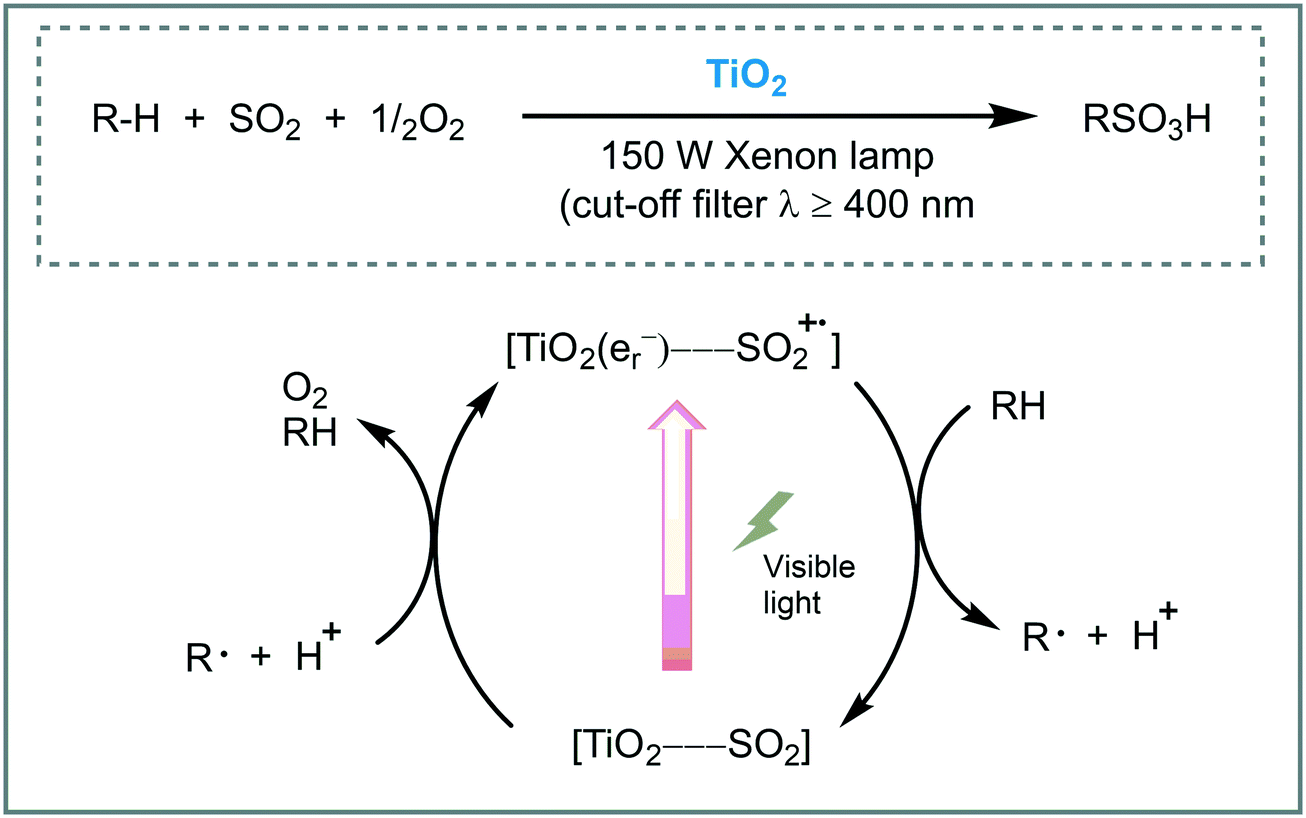 | ||
| Scheme 25 Proposed mechanism for the primary reaction steps for the alkane sulfoxidation photocatalyzed by TiO2. | ||
The classical thiol–ene reaction is an anti-Markovnikov radical addition of a thiyl radical to non-activated alkenes. Similar to sulfoxidation, the reaction is generally initiated by UV irradiation, heat or radical initiators. Yoon and Stephenson's groups pioneered the application of visible light photocatalysis to induce this transformation.82 Inspired by this work, Greaney and co-workers used TiO2 nanoparticles and visible light (20 W light) (Scheme 26).83 They were able to successfully click a variety of allyl and vinyl alkenes with thiols in acetonitrile using TiO2 (1 equiv. or 0.1 equiv.) under visible light irradiation. The authors also suggested that the intriguing visible light response of the TiO2 could be associated with the interactions between the substrates and the surface of the MOS.
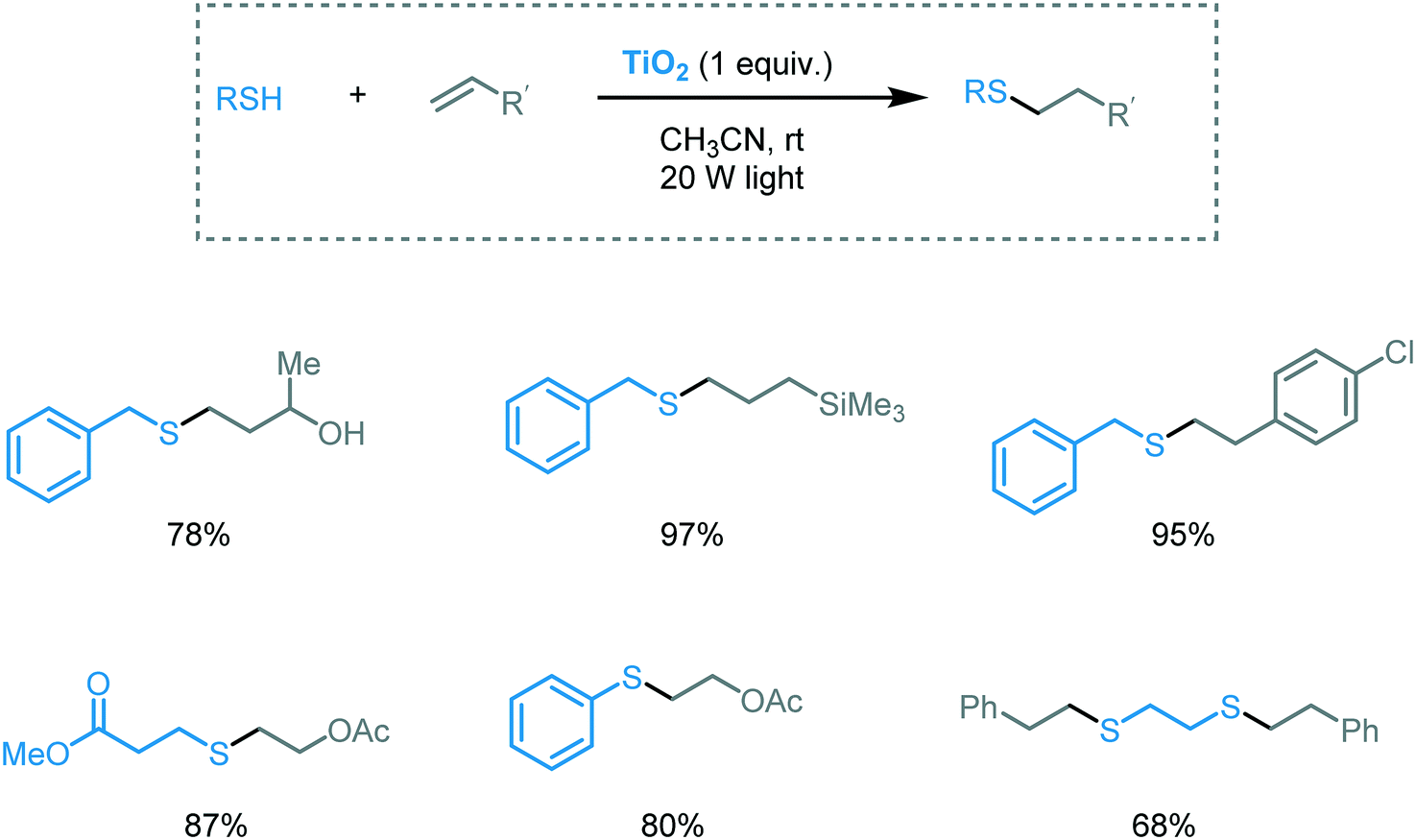 | ||
| Scheme 26 Selected scope for the thiol–ene reaction photocatalyzed by TiO2 under visible light irradiation. | ||
Photoredox-mediated Csp2–P bond formation can be regarded as a strategy for the phosphorylation of arenes using the narrow bandgap Cu2O/TiO2 composite under visible light irradiation.84 Starting from arylhydrazines and a range of trialkylphosphites, a variety of arylphosphonate derivatives was prepared in good to excellent yields (up to 94%). Moreover, the photocatalyst could be easily recovered and reused without significant loss in activity. The authors claimed that the synergetic combination of Cu2O with TiO2 may prevent the fast recombination of the photogenerated electron–hole pairs due to the electron transfer from Cu2O to the CB of TiO2. The mechanism involves an aryl radical species (I), formed after the reduction of arylhydrazine by the e−CB of TiO2, which reacts with trialkylphosphite furnishing the phosphoryl radical (II). Then, the desired product was generated after a displacement step generating an ethyl radical (III) (Scheme 27).
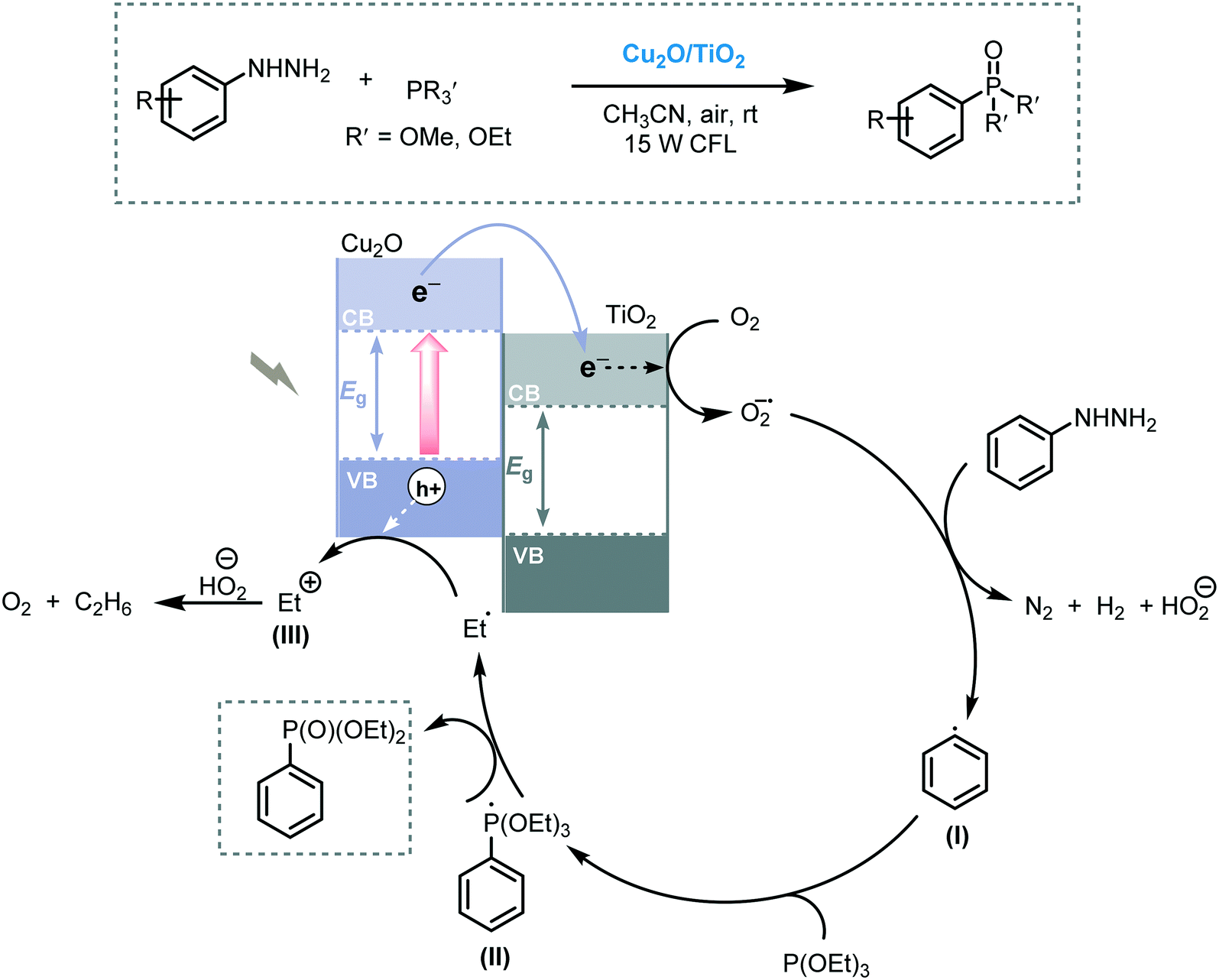 | ||
| Scheme 27 Proposed mechanism for the phosphorylation of arenes photocatalyzed by Cu2O/TiO2 under visible light irradiation. | ||
Naka and co-workers reported a mild approach to promoting the self-condensation of primary amines in cyclopentyl methyl ether (CPME) using Pd/TiO2, as a photocatalyst, under ultraviolet irradiation (300 W Xe lamp, λ = 365 nm).86 The photocatalytic self-condensation afforded a variety of secondary amines in moderate to excellent yields (52 to 96%). Moreover, the protocol was extended to the one-pot photocatalytic synthesis of Alverine, a drug used for irritable bowel syndrome (Scheme 28).
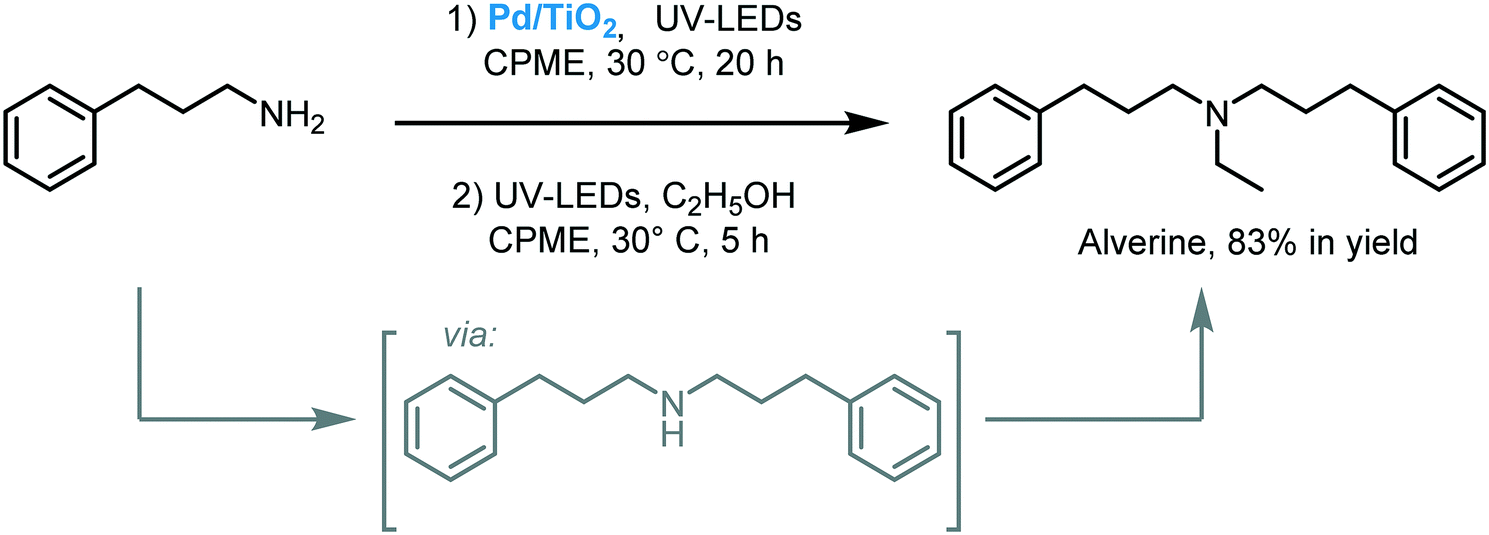 | ||
| Scheme 28 One-pot photocatalytic synthesis of Alverine. | ||
The use of CT complexes to enable the C–H functionalization of tertiary amines was reported by Rueping et al. using TiO2 and visible light (11 W lamp).87 The α-functionalization of N,N-dimethylaniline and its derivatives with isocyanides furnished the corresponding amino amides with moderate to good yields (40 to 82%). After separation by centrifugation, the semiconductor could be reused five times without deleterious effect on the reactivity and selectivity. The proposed mechanism goes through the photogeneration of electron–hole pairs which could reduce oxygen and oxidize the starting amine into iminium ion (II), respectively. Nucleophilic attack with isocyanide leads to the formation of the nitrilium ion (III). After tautomerization, the corresponding α-amino amido product is formed (Scheme 29).
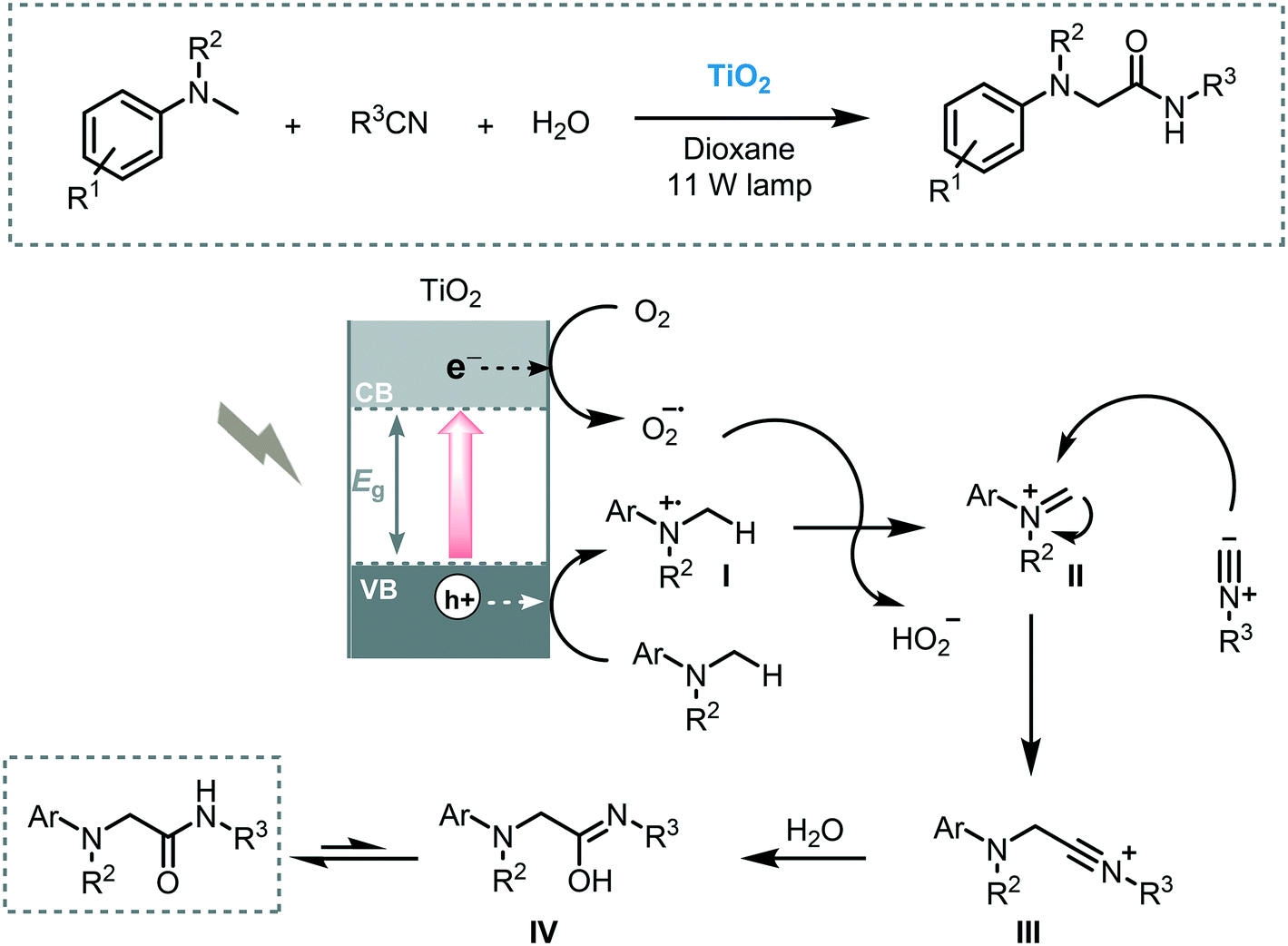 | ||
| Scheme 29 Proposed mechanism for the oxidative multi-component reaction photocatalyzed by TiO2 under visible light irradiation. | ||
Atom transfer radical addition (ATRA), also known as the Kharasch reaction, is an efficient reaction for the synthesis of difunctionalized alkanes (or alkenes) by the addition of haloalkanes across carbon–carbon double (or triple) bonds.88 Typically, the photoinduced ATRA reaction is carried out using transition metal complexes. However, Stephenson et al. developed a protocol to carry out the ATRA reaction using ruthenium and iridium-based photocatalysts in the presence of a Lewis acid.89 Next, other groups also made important advances to drive this photocatalytic reaction using Cu- or EDA-complexes and visible light irradiation.90 Cong and Mao combined a TiO2 semiconductor with hypervalent iodine(III) reagents, such as Togni I or PIDA ((diacetoxyiodo)benzene), as co-initiators to drive the ATRA reaction using visible light irradiation (blue LED).91 The reaction system worked smoothly using alkyl halides containing different functional groups and unactivated olefins (up to 95% in yields). Also, a more challenging ATRA donor could be used under the optimized conditions: chloroform was used as both alkyl halide partner and solvent leading to the corresponding hydrotrichloromethylation product with good yield (62%). The reaction takes place through reduction of the Togni reagent I by the photogenerated electrons in the TiO2 CB to yield the benzoate anion (II) and trifluoromethyl radical species (I). The latter is added to the alkene to generate the new alkyl radical (III), which reacts with the atom transfer reagent to form the byproduct (IV) and the alkyl radical (V). This radical (V) is finally involved in a radical chain pathway (Scheme 30).
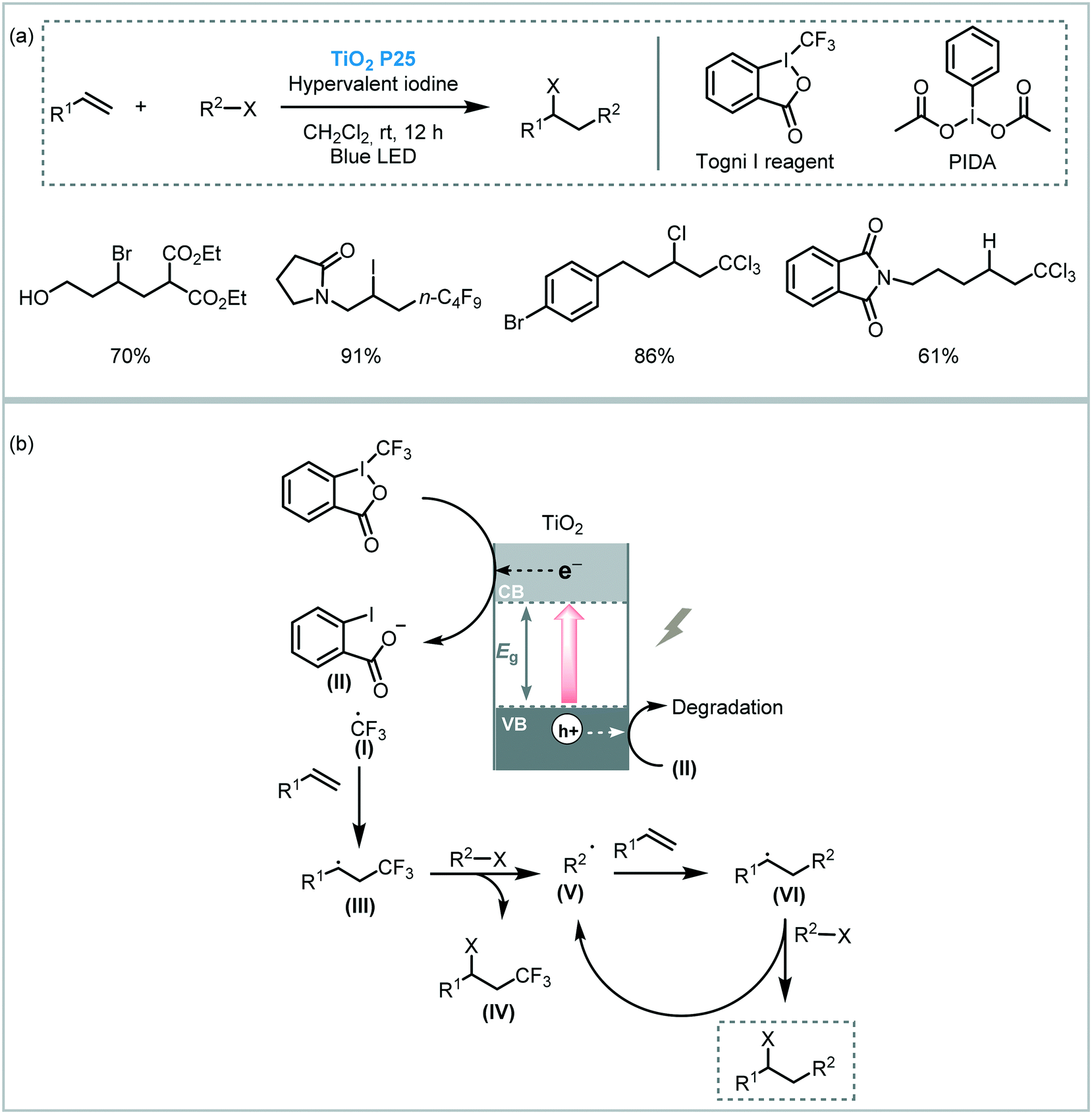 | ||
| Scheme 30 ATRA reaction photocatalyzed by TiO2 in the presence of additives. (a) Selected scope; (b) proposed mechanism. | ||
Bahnemann and co-workers reported a valuable study concerning the effect of the crystalline phase of the semiconductor in its surface properties.92 In this work, they investigated the selectivity of four different types of TiO2 (rutile, anatase, aeroxide P25 and mesoporous anatase) and its surface properties towards the photocatalytic conversion of nitroaromatic derivatives into nitrogen-containing compounds using ethanol as solvent and hole scavenger. In this study, they observed that the selectivity of the reaction was closely linked to the surface properties of the crystalline phase of the TiO2 used in the photocatalytic process. By measuring the acidic properties of the surface sites of the photocatalysts, they disclosed that this property had a strong influence on the selectivity of the reaction. For instance, the condensation of the amine with the aldehyde to form imine was favored on the Lewis acidic site of the anatase. On the contrary, the formation of the amines as a side product was more pronounced when the poor acidic rutile is employed. Interestingly, the formation of quinoline from meta-nitrotoluene, through the cyclization of the pre-formed imine, was favored by the Brønsted acidic sites on the surface of P25. Moreover, when the reaction was carried out with a mixture of anatase and rutile in the same ratio as P25 the products were obtained with similar selectivity obtained for the reaction with pure anatase (Scheme 31). Also, in this work, they study the effect of the platinization of the TiO2 photocatalysts against the N-alkylation and N,N-dialkylation reactions of anilines.
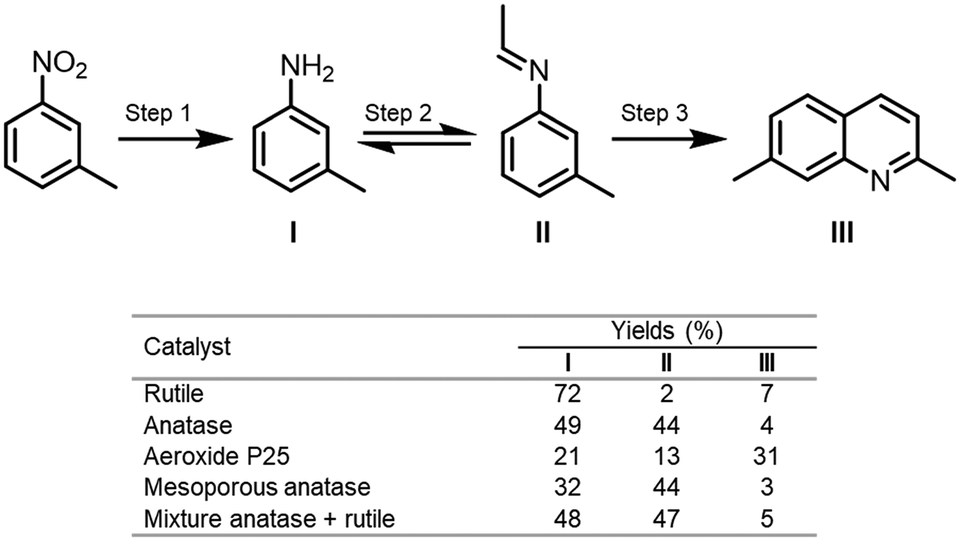 | ||
| Scheme 31 Results for the photocatalytic conversion of m-nitrotoluene in EtOH using TiO2 photocatalysts under UVA irradiation (λ > 320 nm). | ||
Continuous-flow technology has been embraced as an enabling technology for photochemistry.93 This is due to its intrinsic advantages compared to batch, such as shorter diffusion distances, larger surface-to-volume ratios, efficient mass transfer, improved irradiation, etc. However, the use of heterogeneous catalysts in flow has been challenging due to problems associated with microreactor clogging.94 Up to now, continuous processing technology has almost exclusively been applied to enable wastewater treatment and fuel processing using TiO2.95 Often, TiO2 is either deposited as a layer on the channel wall or packed in a fixed bed.
As an example, the C–H arylation of heteroarenes using a variety of aryl diazonium salts as starting materials was performed in a microstructured falling film reactor (FFMR) with TiO2 immobilized on the stainless steel microchannel walls (Scheme 32).96 The reaction medium was irradiated with royal blue light (λ ≈ 455 nm). Reusability of the semiconductor was demonstrated by carrying out a long term run over 3 hours. In this experiment, the arylation of pyridine with 4-trifluoromethylbenzene diazonium salt served as a benchmark example. The desired product was obtained with a constant conversion of 77% during the entire process. Notably, the combination of heterogeneous photocatalysis and microreactor technology showed a significant enhancement of the performance of the reaction in contrast to the batch synthesis. For justify that, they calculated the specific reactor performance for the batch system and the continuous flow reactor. They evaluated the performance using as a reaction model the synthesis of 2-(4-chlorophenyl)pyridine, starting from 4-chlorobenzenediazonium salt with pyridine. For the flow synthesis, the performance was estimated to be around 6000 higher than the used batch system.60
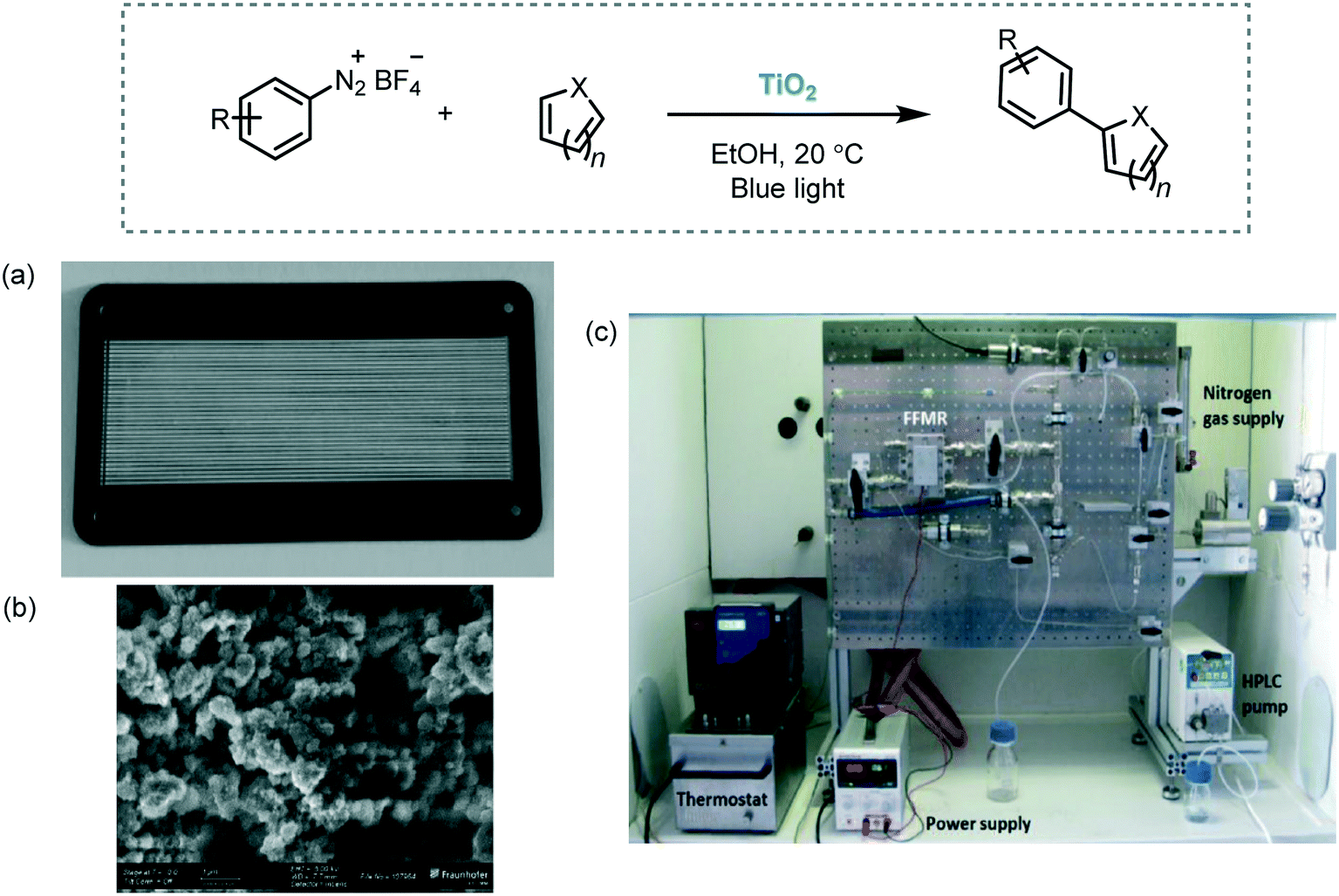 | ||
| Scheme 32 C–H arylation of heteroarenes photocatalyzed by TiO2 immobilized in a continuous flow reactor. (a) FFMR reaction plate with TiO2 immobilized in 32 microchannels; (b) SEM image of TiO2 surface in the microchannels; (c) set up of the reaction (Fig. 32a–c: reprinted from ref. 96 published by the Royal Society of Chemistry). | ||
Another example of flow photocatalysis involves the aerobic synthesis of disulfides using TiO2 as a photocatalyst (Scheme 33).97 The photocatalyst was loaded into a packed bed type-reactor together with glass beads and irradiated with visible light using white LEDs. Interestingly, the presence of TMEDA induced the agglomeration of TiO2 nanoparticles thus avoiding the leaching of the particles and subsequent clogging of the capillaries. The use of continuous-flow resulted in a substantial rate acceleration compared to batch (hours to minutes). Moreover, the photocatalyst could be reused and recycled in both batch (10 consecutive times) and flow (28 h) and the reactions could be carried out at full conversion without appreciable deactivation of the catalyst.
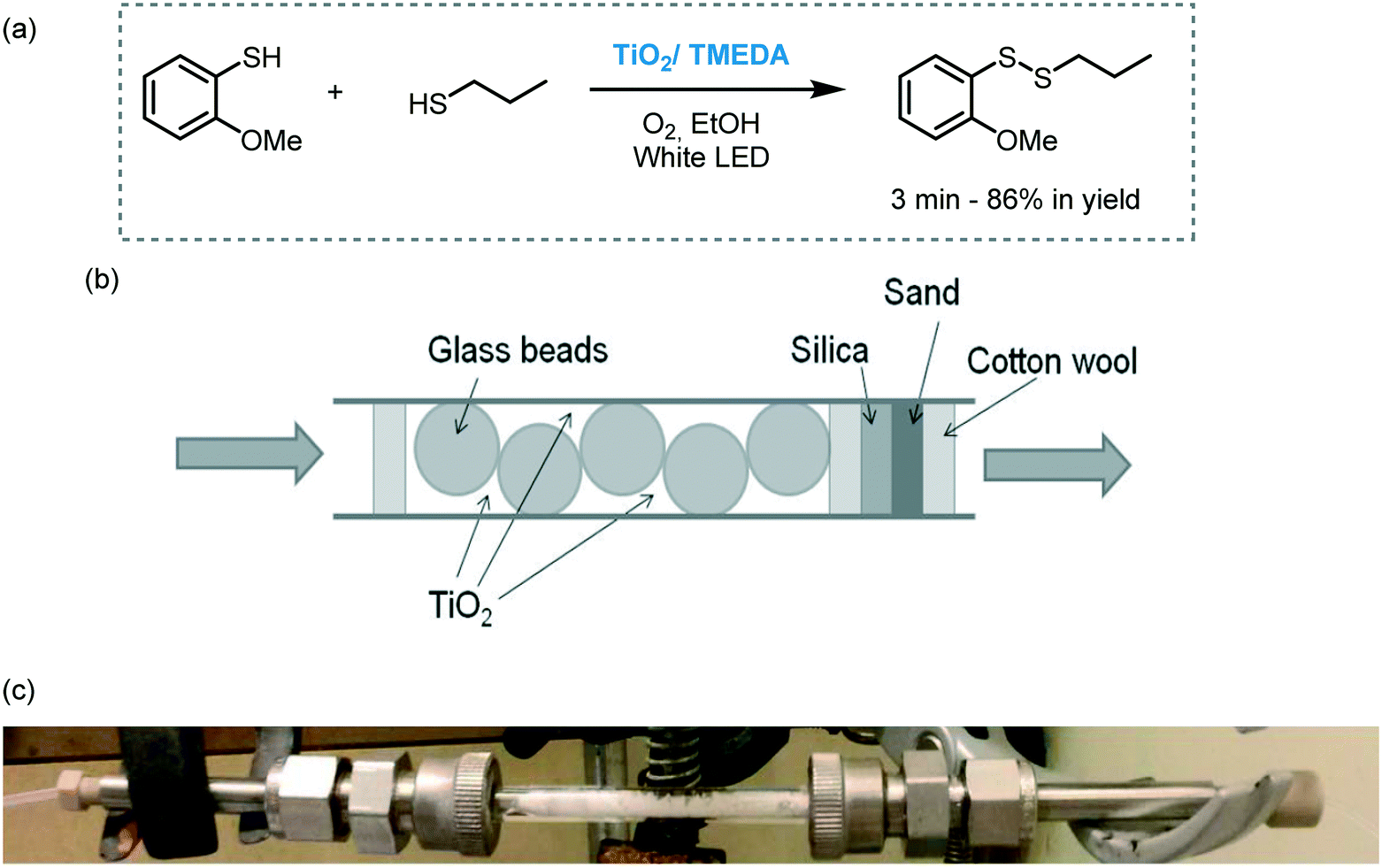 | ||
| Scheme 33 Synthesis of disulfides photocatalyzed by TiO2/TMEDA in flow under visible light irradiation. (a) Selected example; (b) schematic view of the packed bed reactor; (c) picture of the packed bed reactor (Fig. 33b and c: reprinted from ref. 97 Copyright (2016), with permission from John Wiley and Sons). | ||
The synthesis of L-pipecolinic acid from L-lysine was achieved by a photocatalytic process using TiO2/Pt nanoparticles supported on the channel walls in a Pyrex microreactor (Scheme 34).98 The reaction mixture was irradiated with a high-pressure mercury lamp with UV transmitting filter (110 mW cm−2) to obtain the desired product in 52 seconds of residence time (14% yield, 50% ee). Despite the similar yield and enantioselectivity compared to batch, the conversion rate in the microreactor was 70 times larger.
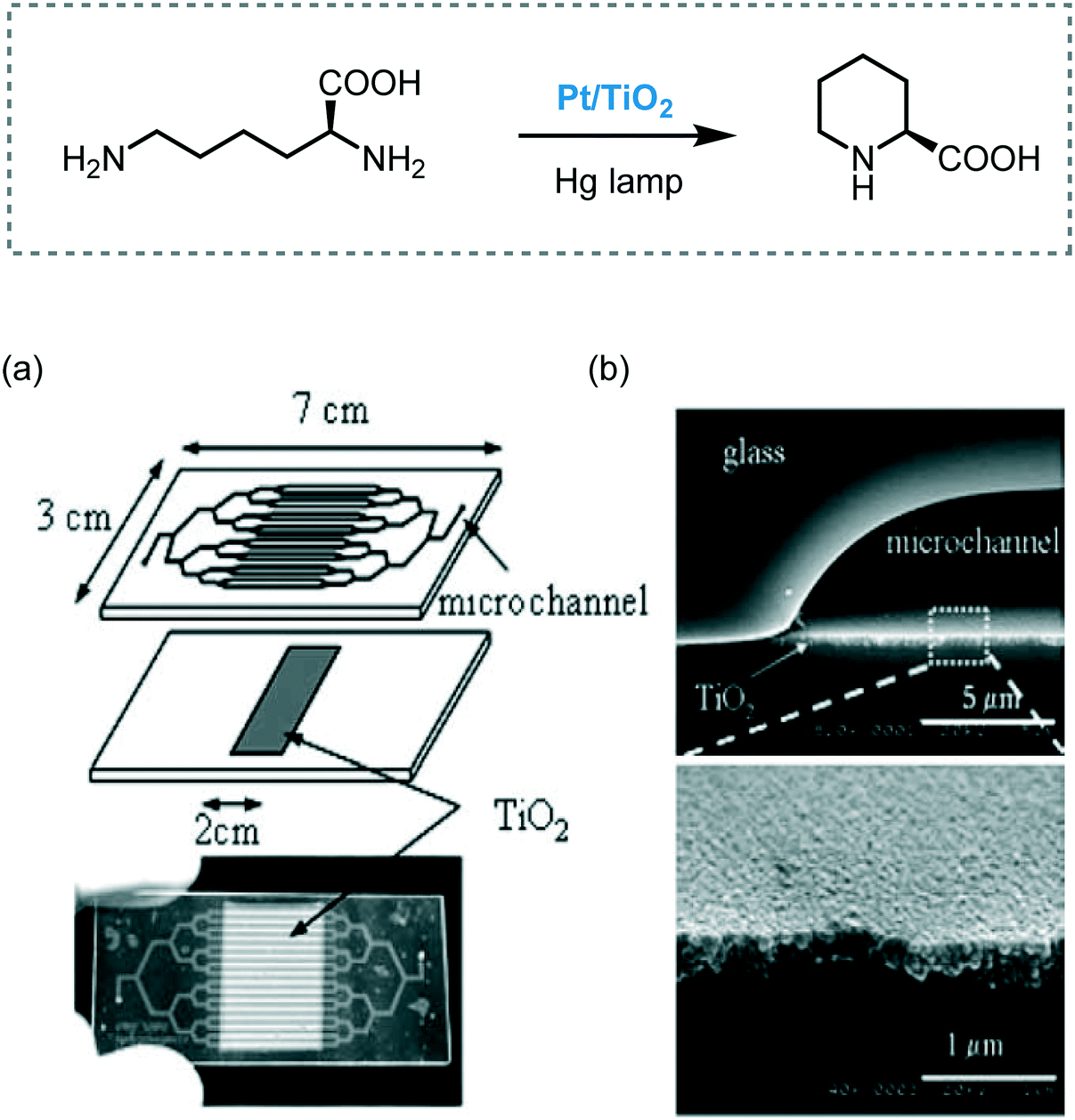 | ||
| Scheme 34 Synthesis of L-pipecolinic acid photocatalyzed by Pt/TiO2. (a) Pt/TiO2-modified microchannel chip and; (b) cross-sectional SEM images of the channel (Fig. 34a and b: reprinted from ref. 98 Copyright (2005), with permission from Elsevier). | ||
Kappe et al. developed a scalable continuous-flow process to prepare TiO2 nanocrystals (NCs). Monodisperse TiO2 NCs with a rod or spherical shape in various, tunable sizes were synthesized. Their photocatalytic activity was investigated in flow using as a model reaction the tandem addition–cyclization reaction of N,N-dimethylaniline and N,N-methylmaleimide under ultraviolet irradiation (Scheme 35).99 The best result was achieved using spherical TiO2 NCs (91% in yield) with 0.5 mL min−1 flow rate for 5 hours. It should be noted that TiO2 was used as a colloidal suspension.
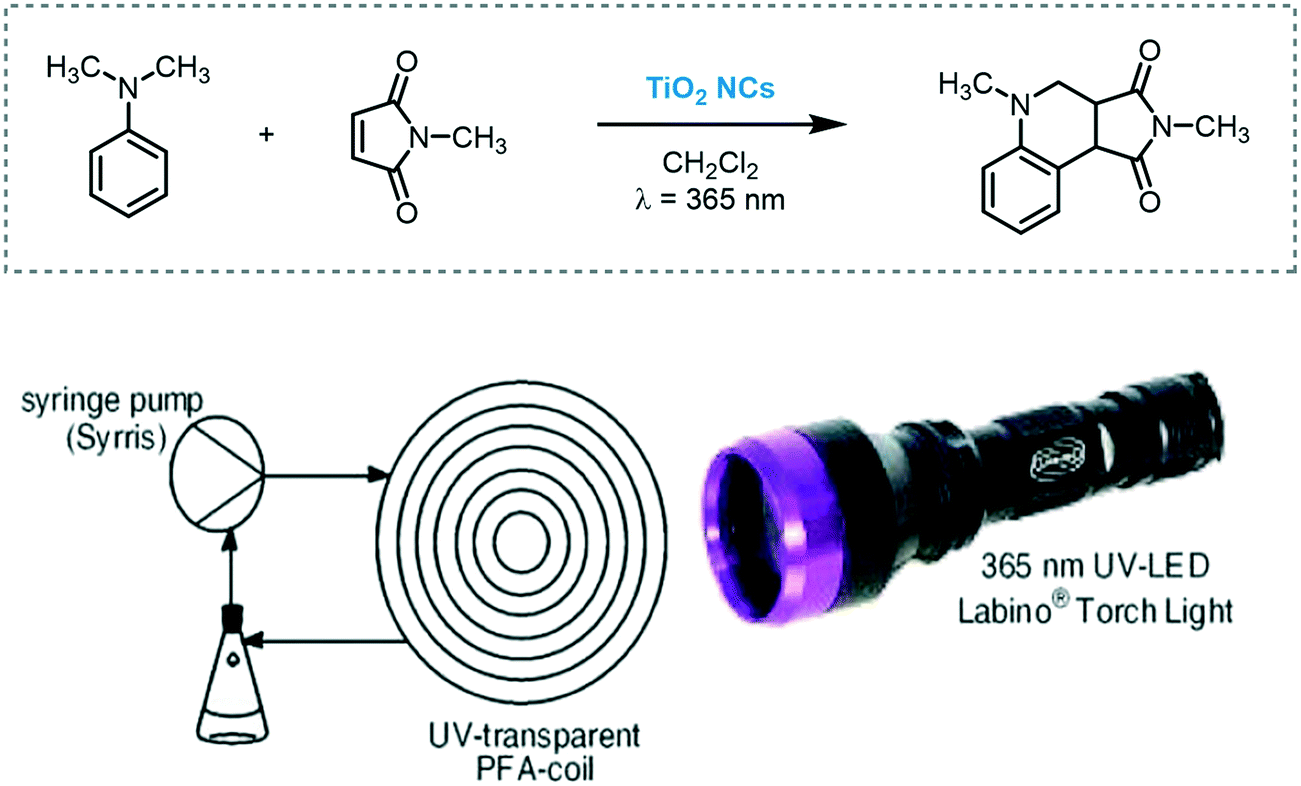 | ||
| Scheme 35 Tandem addition–cyclization reaction photocatalyzed by TiO2 NCs in a continuous-flow photoreactor setup (figure reprinted from ref. 99 Copyright (2013), with permission from Springer Nature). | ||
The transmission, as well as the uniform distribution of photons in the design of a photoreactor, are crucial factors to carry out an effective photocatalytic process.93b An interesting approach, based on internal illumination, was developed by Bloh and co-workers for the photo-reduction of nitrobenzene to aniline in isopropanol using TiO2.100 For that aim, they integrated the heterogeneous photocatalyst into light emitter units by embedded wireless light emitters (WLEs), previously coated with a cyclic olefin polymer, with a multilayer SiO2/TiO2 (Scheme 36). The photocatalytic spheres powered wirelessly by inductive coupling demonstrated high photonic efficiency (26%), reusability and maintain free mobility within the reaction media.
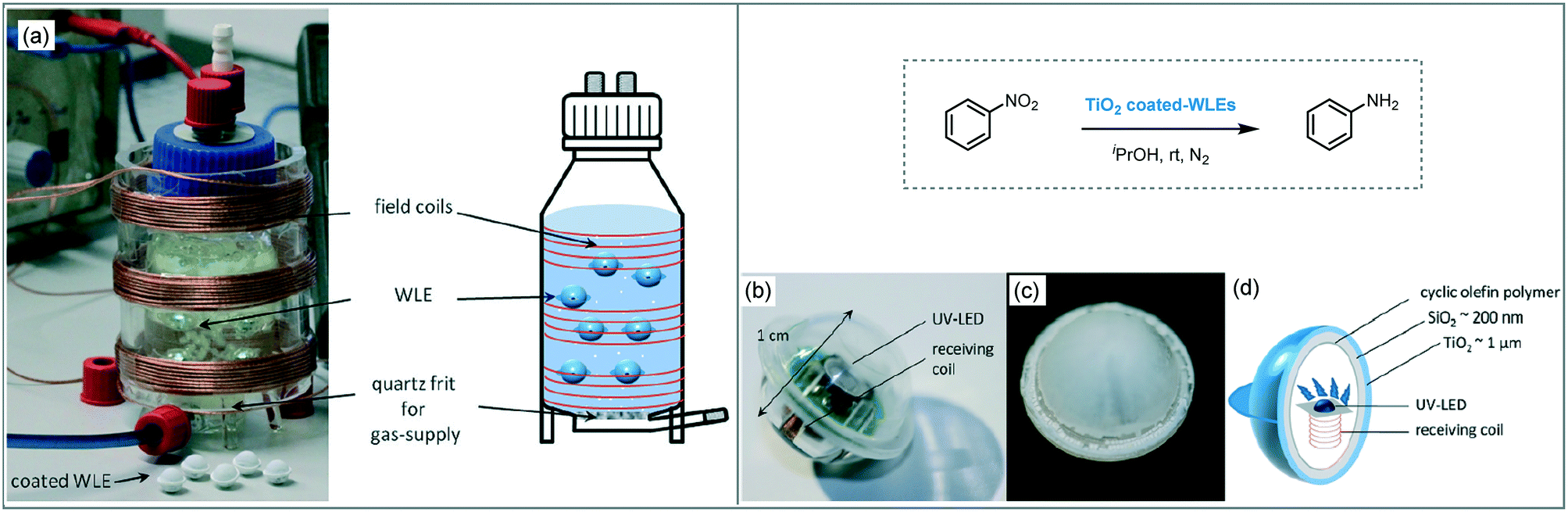 | ||
| Scheme 36 Photo-reduction of nitrobenzene using TiO2/SiO2 supported onto WLEs. (a) Photograph and schematic view of the modified photoreactor containing WLEs; (b) WLE; (c) coated-WLE and; (d) schematic view of WLE and coating. (Fig. 36a–d: reprinted from ref. 100 published by the Royal Society of Chemistry). | ||
Finally, the combination of enzymes and photocatalysts to perform photo-biocatalysis is a highly valuable approach for the sustainable synthesis of relevant organic compounds. The integration of photocatalysts, such as organic dyes, quantum dots or MOS into photo-biocatalysis has shown high potential when light-insensitive enzymes are present. In this regard, the widespread applications of TiO2 as a photocatalyst were also demonstrated in biocatalysis. Recently, very important contributions were reported in this topic and we encourage the readers to check them (Table 1).101
| Reaction type | Photocatalyst/modifications | Irradiation | Ref. |
|---|---|---|---|
| Oxidation of alcohols | Dye-sensitized-TiO2 | Vis | 31, 32 |
| TiO2/LMCT | Vis | 33 | |
| rGO/TiO2 | Vis | 45 | |
| Co–Ascorbic acid/TiO2 | Vis | 50a | |
| Oxidation of amines | TiO2 anatase | UV | 34 |
| TiO2/LMCT | Vis | 35 | |
| TiO2(B)/anatase | Vis | 36 | |
| Pt/TiO2 | Vis | 39 | |
| Oxidation of hydrocarbons | TiO2 | UV | 41 |
| M@TiO2 (M = Au, Pt, Ag) | Vis | 42 | |
| Co–Ascorbic acid/TiO2 | Vis | 50a,b | |
| rGO/TiO2 | UV | 44 | |
| Other oxidations | |||
| Oxidation of benzyl ether | Dye-sensitized-TiO2 | Vis | 40 |
| Oxidation of sulfides | TiO2-LMCT | Vis | 49 |
| C–C bond formation | |||
| Cross-coupling | TiO2 | Vis | 7, 60 |
| Pt/TiO2 | UV/vis | 53 | |
| Pd/TiO2 | UV/vis | 51 | |
| M@TiO2 (M = Rh, Pt, Au, Pd, Ag, Ni, Co) | UV/vis | 54 | |
| Decarboxylative alkylation | TiO2 | UVA | 56 |
| Dye-sensitized-TiO2 | Vis | 57 | |
| Arylation of unsaturated systems | TiO2 | Vis | 60, 96 |
| PANI–g-C3N4–TiO2 | Vis | 61, 62 | |
| Cyanation of amines | DHMIQ–TiO2 | Vis | 69 |
| α-Alkylation of aldehydes | TiO2 | Vis | 124 |
| [4 + 2] cycloaddition | Pt@TiO2 | Vis | 71 |
| TiO2/LiClO4/CH3NO2 | Vis | 73b | |
| [2 + 2] cycloaddition | SmxOy@TiO2 | Vis | 72 |
| TiO2/LiClO4/CH3NO2 | Vis | 73a | |
| Dehalogenation–cyclization | Pt@TiO2 | UVA/vis | 70 |
| C–O bond formation | |||
| α-Oxyamination of aldehydes | TiO2 | UV | 76 |
| C–S bond formation | |||
| Thiol–ene | TiO2 | Vis | 83 |
| Sulfoxidation of HCs | TiO2-LMCT | Vis | 81 |
| C–F bond formation | |||
| Decarboxylative fluorination | TiO2 | Solar simulator | 79 |
| C–P bond formation | |||
| Phosphorylation of arenes | Cu2O/TiO2 | Vis | 84 |
| Miscellaneous | |||
| ATRA | TiO2 | Vis | 91 |
| Self-condensation of amines | Pd/TiO2 | UV | 86 |
| CDC | Co–Ascorbic acid/TiO2 | Vis | 50c |
| TiO2 | Vis | 131 | |
| Cyclization of tertiary anilines with maleimides | TiO2/NiO | Vis | 64 |
| TiO2 | UV | 99 | |
| Synthesis of disulfides | TiO2-LMCT | Vis | 97 |
| Multicomponent reaction | TiO2 | Vis | 87 |
| Synthesis of L-pipecolinic acid | Pt/TiO2 | UV | 98 |
| Reduction of nitro aromatics | rGO/TiO2 | UV | 46 |
| TiO2 | UVA | 92 | |
| TiO2/WLEs | UVA | 100 | |
| CuAAc | CuOx/TiO2 | UVA | 148 |
| Biocatalysis | TiO2 | Vis | 101 |
3.2. Bismuth(III)-Based semiconductors
Although Bi is ranked as the 69th most abundant element in the earth's crust, a large amount of Bi is obtained each year as a side product in the refining other metals, such as copper, lead, tin, and tungsten. The estimated world reserves exceed 320000 tons. Therefore, bismuth and its derivatives are quite inexpensive.
Bismuth-Based semiconductors, such as BiO(X) (X = Cl, Br and I), Bi2MO6 (M = Cr, Mo, W), BiVO4 and Bi2O3, have been widely applied in several areas such as photocatalytic water splitting and photocatalytic decontamination of wastewater and air.102 More recently, Bi semiconductors have emerged as a very promising alternative to promote photocatalytic processes in organic synthesis, including oxidations, C–C and C–X bond formation, etc. The hybridization of 6s Bi and 2p O orbitals at the top of the valence band allows for a narrow band gap (generally Eg < 3.0 eV) and also helps to increase the mobility of the photogenerated charge carriers. Their excellent electronic properties, in combination with their low toxicity, makes them good candidates to carry out organic reactions driven by visible light.
While the application of Bi2O3 as a photocatalyst in organic synthesis is quite recent, valuable synthetic methodologies have been developed in recent years. Pericàs et al. were the pioneers in the use of Bi2O3 as a photocatalyst to light induce an organic transformation. Inspired by previous work on dual organo-photocatalysis,8 they disclosed the use of commercially available Bi2O3 bulk or Bi2S3 nanoparticles to promote the visible light-enabled asymmetric α-alkylation of aldehydes.104 Despite both semiconductors showing excellent activities at low concentrations, Bi2O3 was generally more active in the photocatalytic process. The authors suggested that this higher reactivity was due to the solubility of this MOS in the reaction medium. Moreover, when bromomalonate was used as an alkylated agent, the results obtained with the above mentioned bismuth-based semiconductors showed slightly higher enantioselectivity and were faster compared to the Ru-based homogeneous system.8 Analogous to the proposed mechanism for homogeneous photocatalysis, the photo-α-alkylation of aldehydes by MOS may involve the reduction of the α-bromocarbonyl compounds to generate the alkyl radicals (I). These radicals react with the chiral enamine leading to an α-amino radical (II) that delivers an electron to the hole of the MOS closing the catalytic cycle (Scheme 37). Based on the work of Melchiorre and co-workers,12b Pericàs et al. also proposed a secondary competitive cycle involving the formation of an electron–donor–acceptor (EDA) complex which does not include the participation of any external photocatalyst. However, based on control experiments, the authors claimed that the suggested semiconductor cycle should be much faster than the EDA cycle since the latter involves the coupling of two low-abundance radical species.
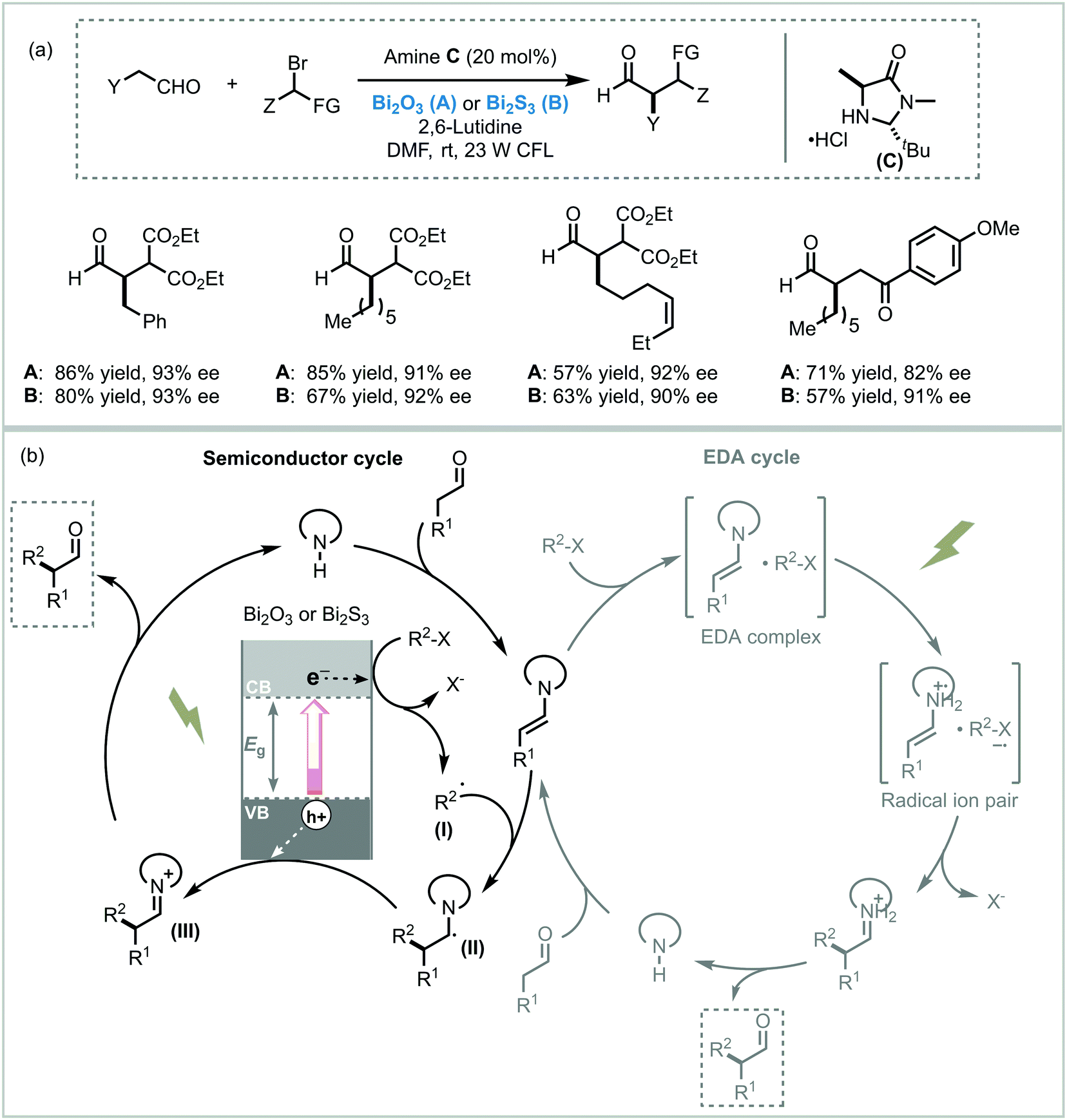 | ||
| Scheme 37 Asymmetric α-alkylation of aldehydes with α-bromocarbonyl compounds photocatalyzed by bismuth-based semiconductors under visible light irradiation. (a) Selected scope for the α-alkylation of aldehydes and; (b) proposed reaction mechanism. | ||
The versatility of bismuth oxide as a photocatalyst was also proven in reactions involving a C–X bond formation. For instance, Fadeyi et al. applied commercially available Bi2O3, as a photocatalyst, and bromotrichloromethane (BrCCl3), as a single electron acceptor, to promote the formation of the thiyl radical under visible light irradiation (25 W CFL).105 The corresponding thiol–ene adducts were obtained in high yields (up to 96%) and displayed the expected anti-Markovnikov regioselectivity. Moreover, they explored the application of this methodology to late-stage diversification of biomolecules and active pharmaceutical agents, such as S-acetyl protected precursor to NCH-31 (A, potent antitumor agent) and the cyclic tetrapeptide (B, potent histone deacetylase inhibitor) (Scheme 38). Similar to the mechanism proposed for TiO2,83 the authors suggested that the photogenerated electrons induce the reduction of the bromotrichloromethane leading to the corresponding trichloromethyl radical. This radical readily abstracts the thiol hydrogen atom producing the thiyl radical, which in turn initiates the radical-thiol process.
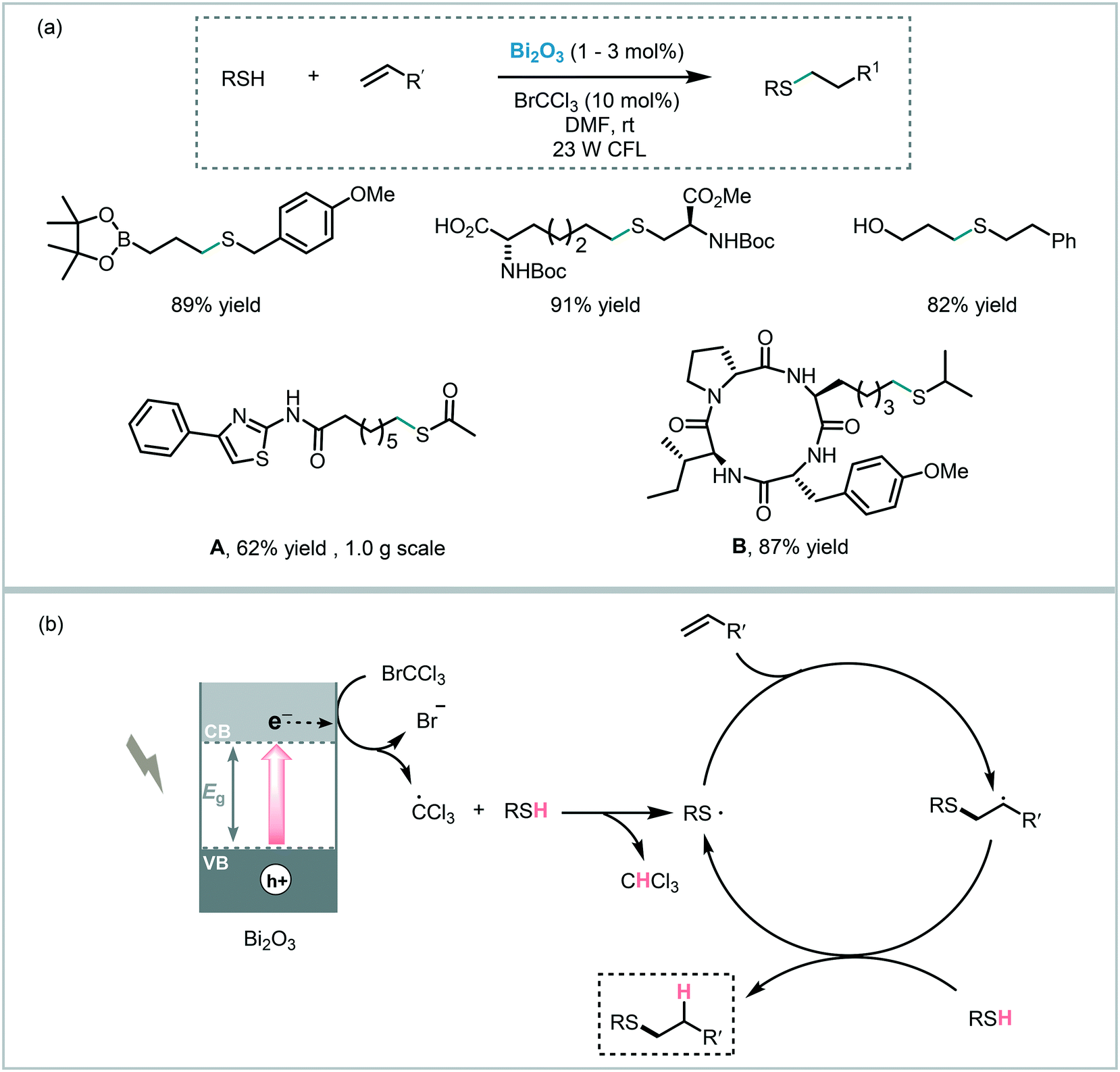 | ||
| Scheme 38 Thiol–ene reaction photoinitiated by Bi2O3. (a) Selected scope for the thiol–ene reaction and; (b) proposed reaction mechanism. | ||
Pericàs et al. tested the feasibility of Bi2O3 to induce the arylation of heteroarenes under visible light irradiation (23 W CFL).106 A variety of heteroarenes, including pyridine, thiophene, furan and N-protected pyrrole, were engaged in this transformation, furnishing the corresponding heterobiaryls in moderate to excellent yields (32 to 97%). A reaction mechanism involving the reduction of the diazonium salt by the photogenerated electrons in the conduction band of the Bi2O3 leading to the aryl radical (I) were suggested. This aryl radical (I) would then attack the heteroarene to afford radical (II). Next, radical (II) is oxidized by the photogenerated holes to yield the cation (III). In the last step, the deprotonation of the cation furnishes the target arylated product (Scheme 39). In a competitive pathway, a radical chain propagation mechanism could be also involved.
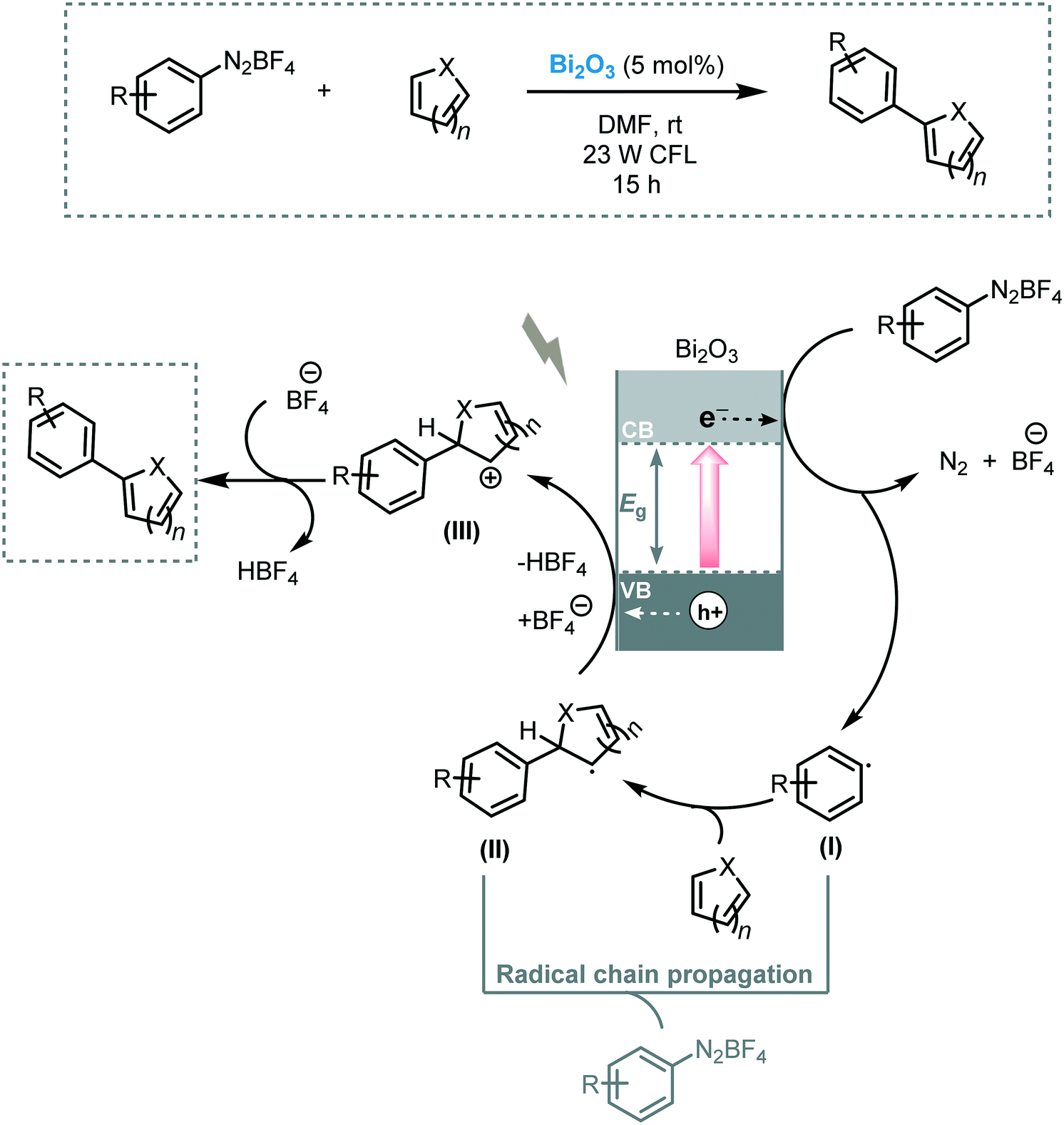 | ||
| Scheme 39 Proposed reaction mechanism for α-arylation of heteroarenes photocatalyzed by Bi2O3. | ||
Pericàs et al. also demonstrated that the commercially available Bi2O3 semiconductor could be an efficient photocatalyst in the ATRA of bromoalkenes across double bonds and triple bonds.107 A variety of organobromides and olefins were subjected to the reaction conditions yielding the desired ATRA products in good to excellent yields (up to 91%). The reaction worked particularly well when using dialkyl bromomalonate, as ATRA donor, and could be carried out using solar light. Mechanistically, the authors suggested that both radical-polar crossover and radical chain propagation routes could be involved in the photocatalytic process. Therefore, the photogenerated electron would reduce the organobromide compound forming the carbon-centered radical (I). This radical species is subsequently added to the double bond of the olefin giving rise to radical (II). In a radical-polar crossover pathway, radical (II) is oxidized by the holes of the valence band of Bi2O3 resulting in the formation of a carbocation (III). This carbocation ultimately reacts with bromide forming the target ATRA product. Alternatively, radical (II) could subtract a bromine atom from the starting material leading directly to the desired product and regenerating the radical (I) that continues the chain (Scheme 40).
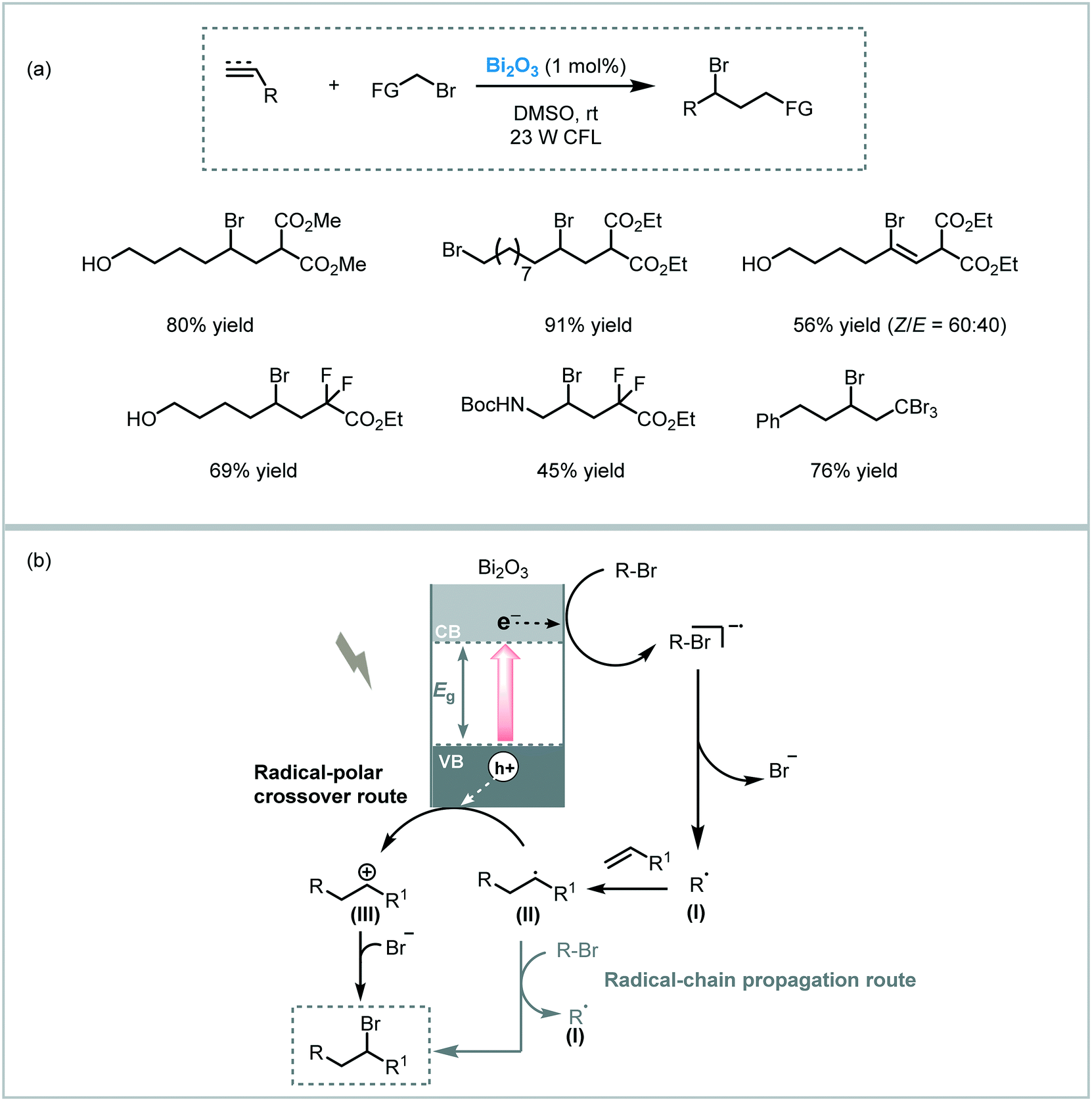 | ||
| Scheme 40 Bi2O3 photocatalyzed ATRA reaction. (a) Selected scope and; (b) proposed mechanism. | ||
Free radicals are well-known species able to induce a polymerization process. Typically the radical precursor is thermally decomposed to yield the free-radical initiators. However, the poor control over chain length makes this approach less efficient and selective for industrial applications. To overcome this drawback, atom-transfer radical polymerization (ATRP) or reversible addition fragmentation chain transfer (RAFT) have been developed to generate uniform polymer chain growth.108 Generally, these polymerization methodologies make use of stoichiometric amounts of transition metals and high reaction temperatures, hindering their application in industry. Recently, the introduction of light enabled new modes of reactivity, such as single electron transfer (PET) processes.109 Light activation is also an attractive alternative for the existing classical methods offering a low-cost, eco-friendly and energy-efficient approach to initiate polymerizations.
Müllner et al. demonstrated that Bi2O3 can photo-induce polymerizations (Scheme 41).110 The synthesis of di-block copolymers with different activated monomers and narrow molecular weight distribution was achieved using very mild conditions (room temperature, visible light irradiation). Interestingly, the polymerization could be carried out in a range of different solvents, including water. Moreover, the photocatalytic protocol displayed excellent control over degenerative chain transfer polymerizations, such as PET-MADIX and PET-RAFT. The catalyst could be easily recycled and reused for four cycles without a significant loss in catalytic performance.
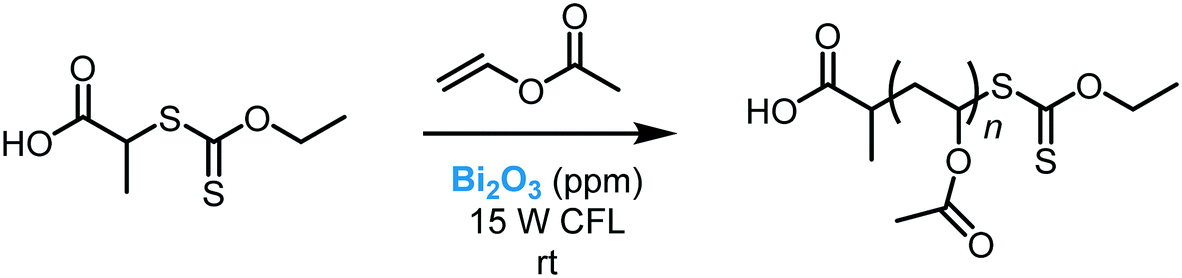 | ||
| Scheme 41 PET-MADIX polymerization of vinyl acetate photocatalyzed by Bi2O3 under visible light irradiation. | ||
One of the first applications of Bi2WO6 in photocatalysis was presented by Xu et al. in 2013.112 They reported a chemoselective aerobic oxidation of glycerol to dihydroxyacetone (DHA) in water using flower-like Bi2WO6 crystals irradiated with visible light (λ > 420 nm). The most optimal result yielded the target compound in 87% yield and in high selectivity (91%), with glyceraldehyde being the main byproduct. Moreover, the photocatalyst could be recycled and reused for six consecutive cycles without significant loss in reactivity. Based on control experiments, the high selectivity in the formation of DHA could be explained by the absence of non-selective ˙OH radical species in the photocatalytic process (the redox potential of the photogenerated holes in the valence band of Bi2WO6 are more negative than that of ˙OH/OH−). Therefore, the proposed mechanism relies on the direct oxidation of the adsorbed glycerol by the photogenerated positive holes on the Bi2WO6 valence band and subsequent formation of the corresponding intermediate. The reaction between the intermediate and oxygen, or activated oxygen (e.g. O2˙−), furnishes the desired DHA product (Scheme 42). Inspired by these results, they extended the protocol to promote the oxidation of benzylic alcohols also under visible light irradiation (λ = 420–760 nm).113
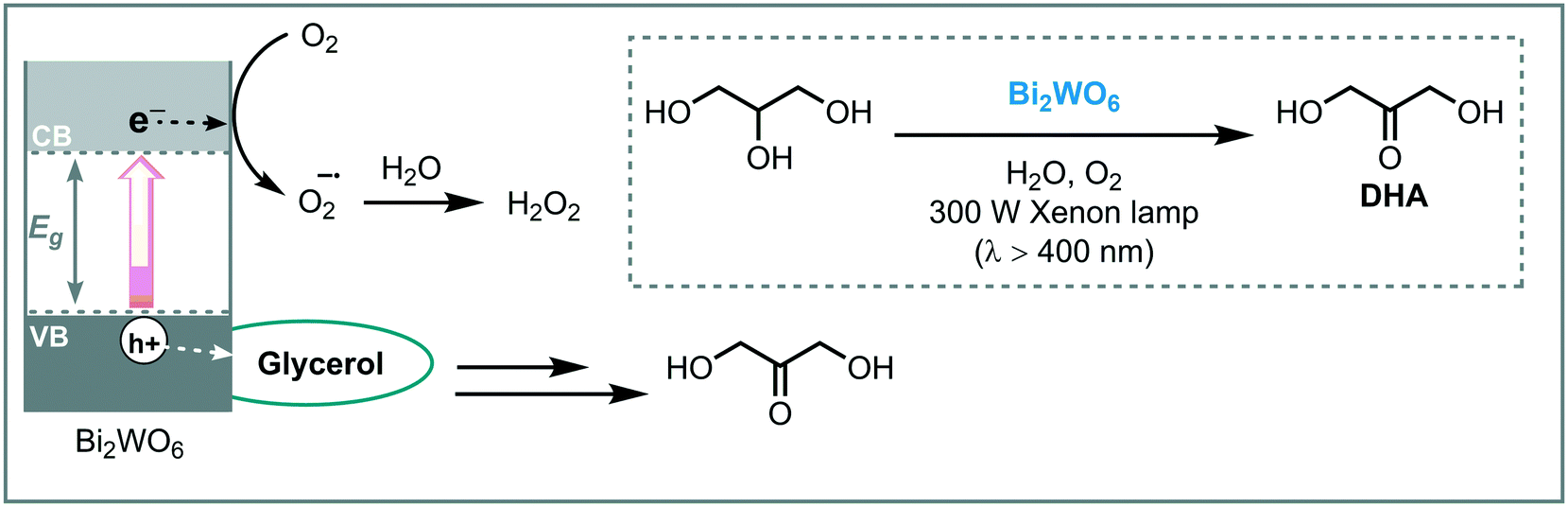 | ||
| Scheme 42 Proposed mechanism for the oxidation of glycerol to DHA photocatalyzed by Bi2WO6 under visible light. | ||
The synergetic effect between MOS and metal complexes can form highly efficient photocatalysts with a strong absorption in a broad spectral range. This type of photocatalysts has received special attention in the last years, especially in the photogeneration of H2 and reduction of CO2.116 As an example, Naya et al. demonstrated that monoclinic sheelite BiVO4 containing bis(acetylacetonate)copper(II) binuclear complexes (Cu(acac)2/ms-BiVO4) on the surface constitute an efficient electrocatalyst for the multi-electron reduction of O2 under visible-light irradiation, which shows high activity for the oxidation of amines, imines and alcohols.117 They observed that the addition of trimethylstearylammonium chloride (C18TAC) drastically enhanced the activity and selectivity of the Cu(acac)2/ms-BiVO4 towards the photooxidation of benzylalcohol analogs to the corresponding benzaldehydes. Although the reaction was highly dependent on the stirring speed, the authors claimed that a C18TAC monolayer was eventually growing on the ms-BiVO4 surface, thus forming an adsorption bilayer or admicelle. This bilayer allowed for a high concentration of substrates near the ms-BiVO4 provoking a good particle dispersity in water by electrostatic repulsion between surface charges.
Next, Li et al. prepared BiVO4 crystals selectively doped in the (010) and (110) facets using Pt as reduction cocatalyst and MnOx as oxidation cocatalyst.118 They demonstrated that the spatial separation of reduction and oxidation sites, avoided recombination and reversed reactions, improving conversions and selectivities for the oxidation benzyl alcohols (Scheme 43).
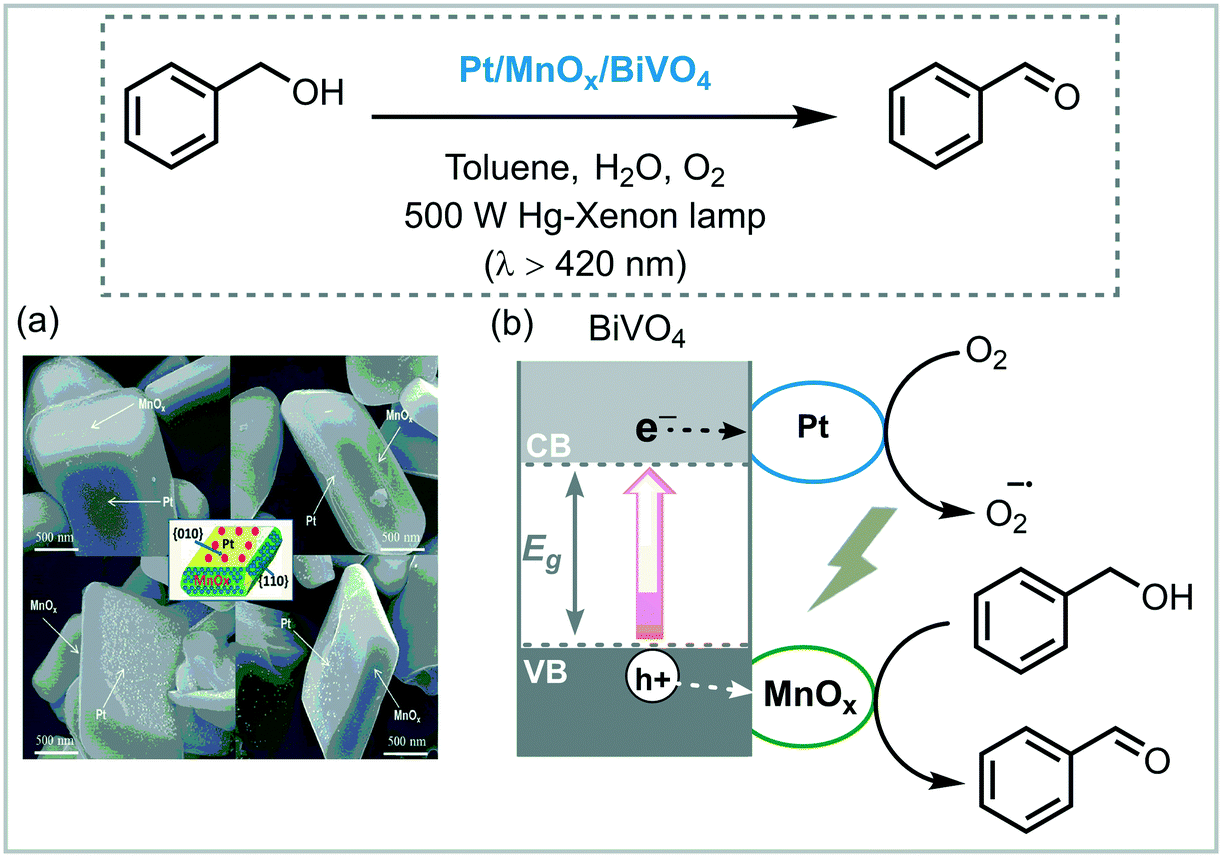 | ||
| Scheme 43 Oxidation of benzylic alcohol to benzaldehyde photocatalyzed by BiVO4 under visible light. (a) SEM images of BiVO4 crystals after selective deposition of Pt and MnOx on {010} and {110} facets and; (b) schematic mechanism (Fig. 43a: reprinted from ref. 118 Copyright (2018), with permission from the Royal Society of Chemistry). | ||
Similarly, Li et al. reported the application of BiVO4 to drive the oxidation of amines.119 BiVO4 crystals with more reactive (040) and (110) facets, using a hydrothermal procedure, were prepared. The photocatalytic activity of the crystals towards the oxidation of a variety of benzylic primary and secondary amines was investigated using visible light irradiation (xenon lamp, λ > 420 nm) and oxygen as the oxidant. The developed approach gave the corresponding target imines in high yields and selectivities (up to 99%), even when the sterically hindered N-t-butyl benzylamine was used (92% in yield and selectivity). BiVO4 could be separated from the reaction media by filtration and could be reused (seven cycles) without significant activity loss. Regarding the mechanism, it was suggested that the photooxidation with BiVO4 may have a different pathway than with TiO2. In TiO2 photooxidation catalysis, the formation of the amide, via the adsorbed amine, is a pre-requisite in the photocatalytic process. However, based on kinetic experiments, the photogenerated electrons in the CB of BiVO4 reduce dioxygen, forming activated oxygen species. At the same time, the amine is oxidized by the holes in the VB to form the carbocation radical intermediate. Removal of the protons yield the corresponding imine (Scheme 44).
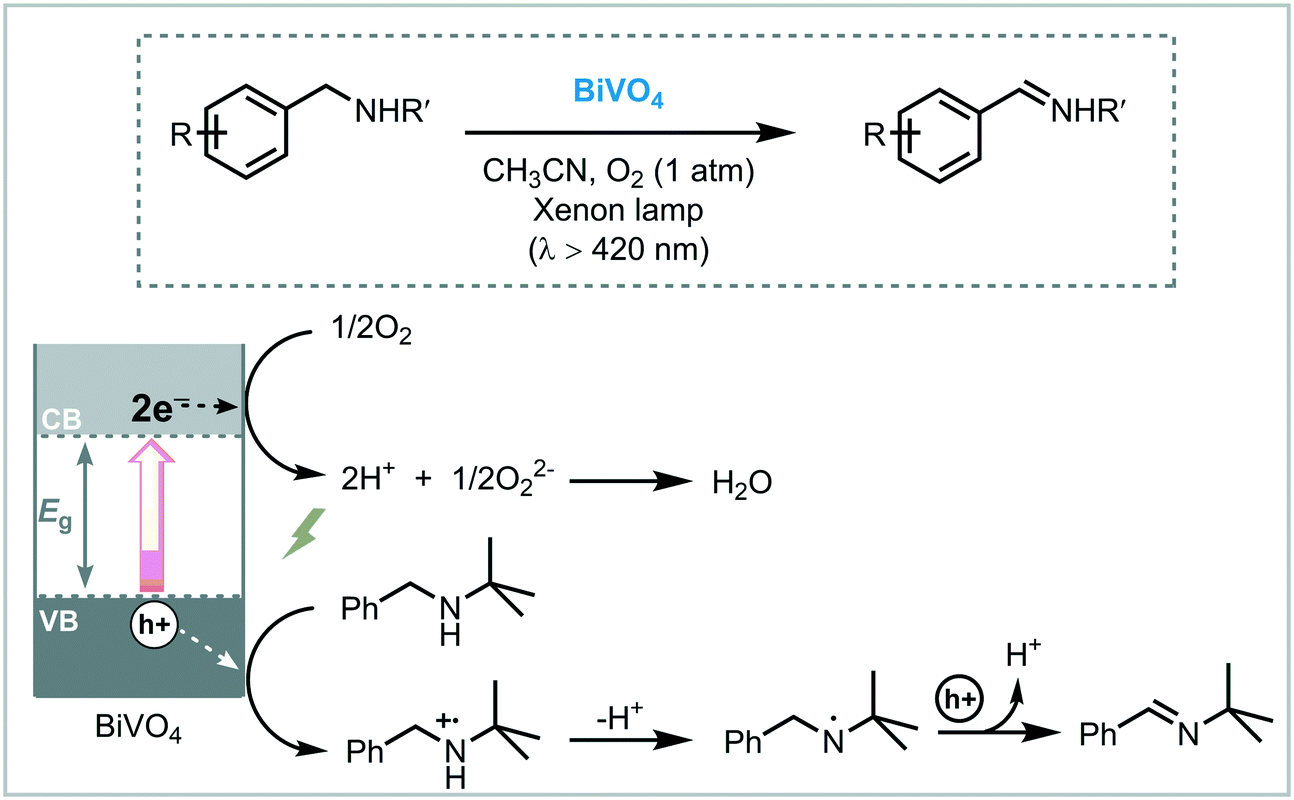 | ||
| Scheme 44 Proposed mechanism for the oxidation of amines into imines photocatalyzed by BiVO4 under visible light. | ||
Barlett et al. compared the reactivity of CuWO4 and BiVO4 powders in the photooxidation of benzylamine under visible light irradiation (blue LED, λmax = 460 nm).120 Despite the fact that both showed near quantitative yields and high selectivity for the formation of the N-benzylidenebenzylamine (98 to 99% in yields), it was observed that BiVO4 catalyzed the reaction at about twice the rate (0.34 h−1 g−1 for CuWO4 and 0.70 h−1 g−1 for BiVO4). They asserted that the differences in the electronic structures of the employed photocatalysts might explain the difference in CuWO4 and BiVO4 reaction rates. More specifically, BiVO4 showed a larger absorption coefficient due to a direct band transition, which is 0.2 eV greater than the indirect electronic bandgap.
One elegant example of the application of BiOCl as a photocatalyst for light-driven organic transformations was reported by Li et al. After using BiVO4 as the photocatalyst for the oxidation of amines,119 the authors exploited the potential of BiOCl to carry out the same transformation (Scheme 45). The importance of the structure of the MOS on its photocatalytic activity was shown using ultrathin graphene-like BiOCl nanosheets (BiOCl C-UTNSs).122 The unique layered structure enables the recombination of the photogenerated charge carriers and reduces their surface trapping. Moreover, this behavior explains the intriguing visible light response (in contrast to the bulk material which is ultraviolet responsive). This is due to the abundant presence of oxygen vacancies in the nanosheets, which create a new impurity band between the VB and CB thus efficiently reducing the energy gap.123
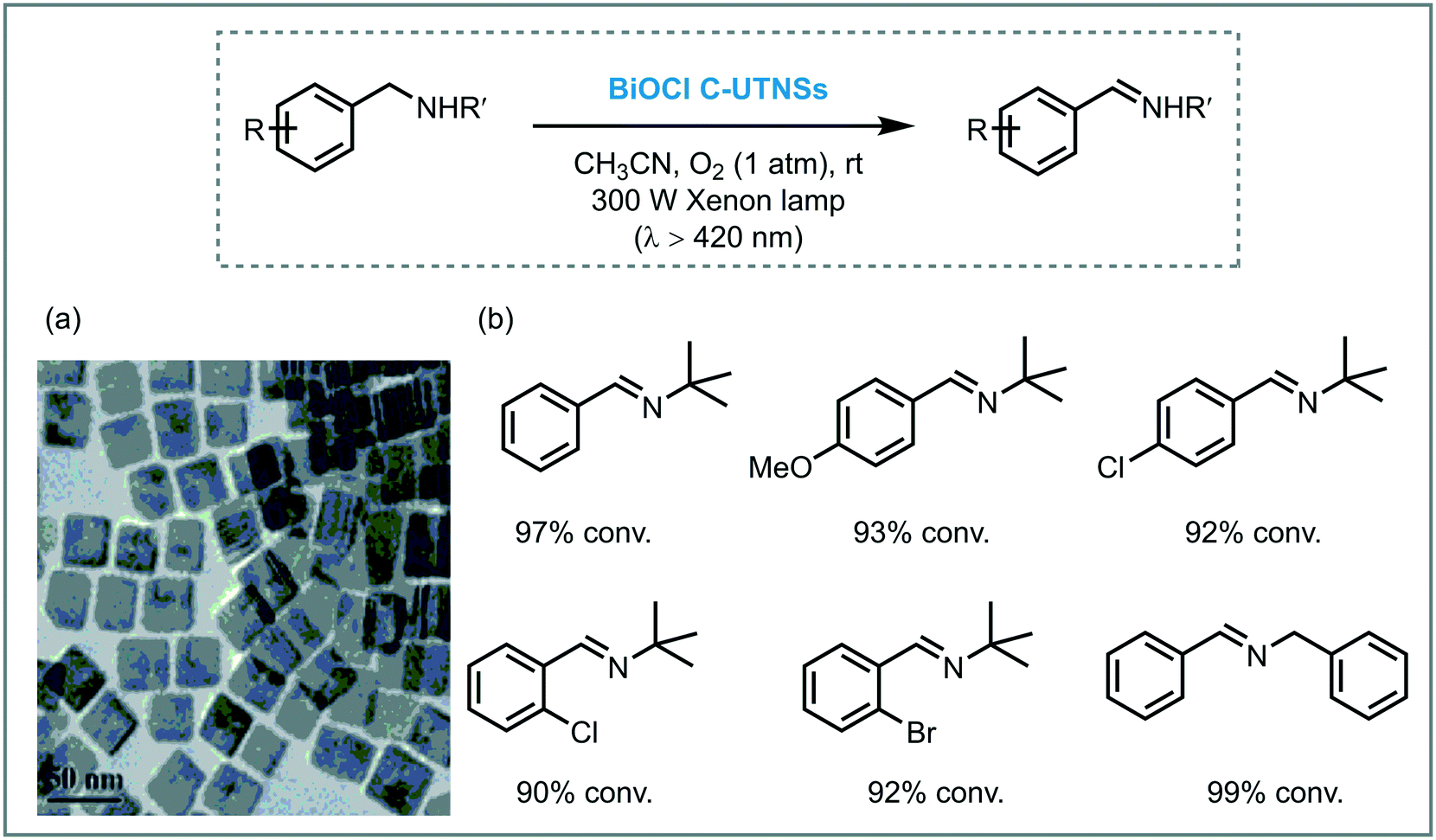 | ||
| Scheme 45 Oxidation of benzylamines to imines photocatalyzed by BiOCl-UTNSs under visible light. (a) TEM images of BiOCl-UTNSs and; (b) selected scope (Fig. 45a: reprinted from ref. 122 published by the Royal Society of Chemistry). | ||
Regarding oxybromides, König et al. applied perovskite-like semiconductors, PbBiO2Br, as the photocatalyst. This catalyst was combined with chiral imidazolidinone under visible light irradiation (high-power LED, 3 W, LUXEOM) to promote the asymmetric α-alkylation of aldehydes.124 The starting octanal was alkylated with organobromo derivatives leading to the corresponding chiral alkylated aldehydes in good yields and enantioselectivities (Scheme 46).
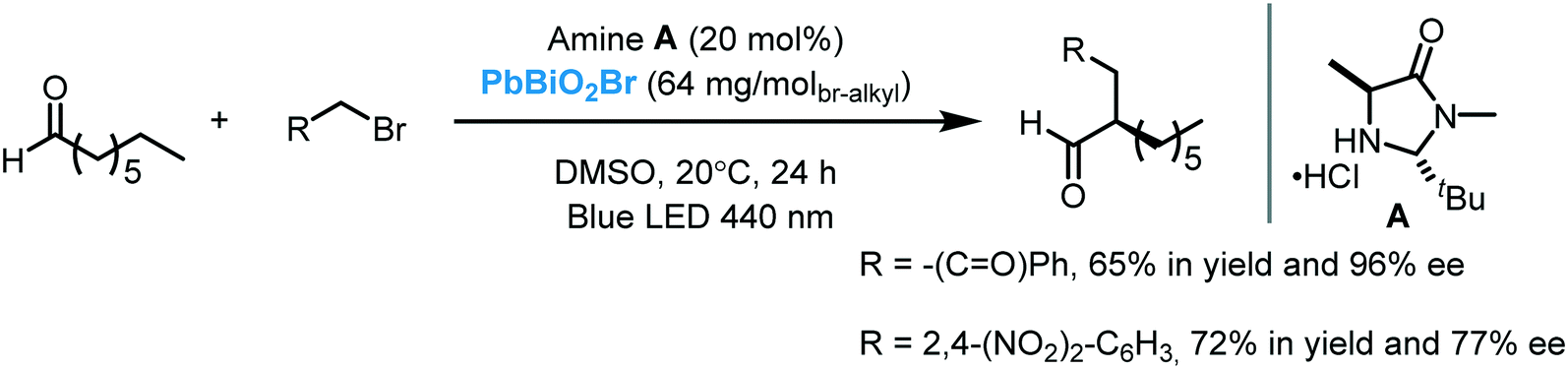 | ||
| Scheme 46 α-Alkylation of octanal with organobromides using PbBiO2Br. | ||
Su et al. described the selective photooxidation of primary alcohols to aldehydes in air using Bi24O31Br10(OH)δ as a photocatalyst.125 The photocatalyst was prepared using a microwave-assisted method with Bi(NO3)3·5H2O as a salt precursor in an alkaline aqueous solution. The microstructure analysis revealed that the material was highly porous which favors the diffusion of bulky alcohols to the active sites. Under visible light (LED, λ = 450 nm), a series of primary, secondary and aromatic alcohols were successfully oxidized to the corresponding aldehydes or ketones in excellent conversions and selectivities (up to 99%). Notably, the quantum efficiency for the photooxidation of isopropanol was reported to be high (71% and 55% at 410 and 450 nm, respectively) (Scheme 47). In this study, it was also observed that aromatic alcohols reacted faster than their aliphatic counterparts. This could be attributed to the presence of conjugated systems leading to a stronger adsorption onto the surface of the photocatalyst. Based on spectroscopic and spectrometric analysis, they proposed that the surface basic sites and active lattice oxygen (Ol) on the photocatalyst have a pivotal role in the dehydrogenation of the alcohols. This favors the oxidation process even in the absence of oxygen. Hence, Bi24O31Br10(OH)δ first promotes the dehydrogenation of the alcohol, leading to the absorption of hydrogen on the photocatalyst.126 Then, the Ol species of the photocatalyst is involved in the oxidation yielding the formation of water, aldehydes and oxygen vacancies. These vacancies can be restored in the presence of oxidants (O2 or benzoquinone) (Table 2).
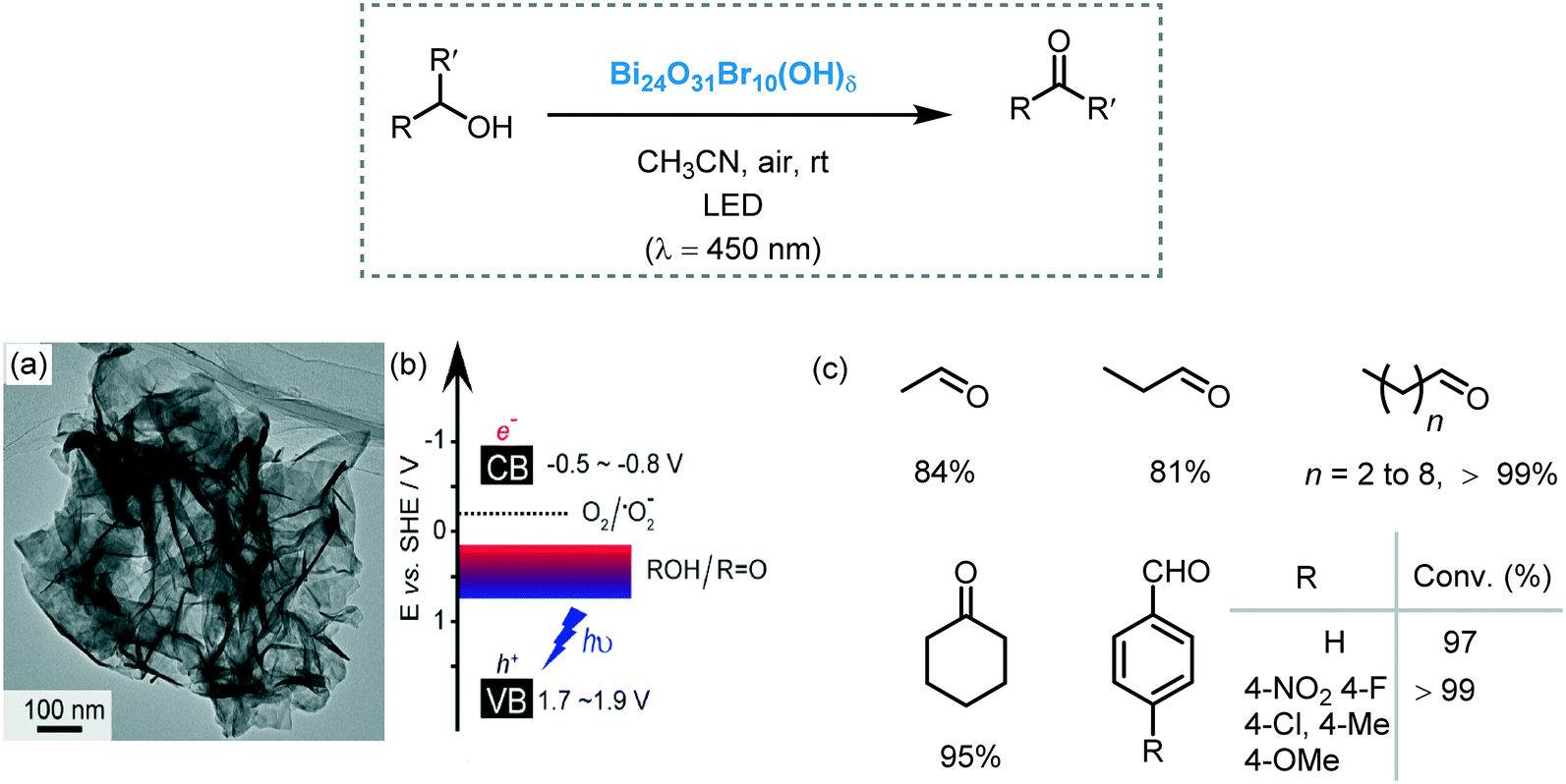 | ||
| Scheme 47 Oxidation of alcohols photocatalyzed by Bi24O31Br10(OH)δ under visible light irradiation. (a) TEM image and (b) band positions of the Bi24O31Br10(OH)δ sample. The redox potentials of O2 and isopropanol are also shown as references and; (c) scope for the reaction (Fig. 47a and b: reprinted from ref. 125 Copyright (2019), with permission from John Wiley and Sons). | ||
| Reaction type | Photocatalyst/modifications | Irradiation | Ref. |
|---|---|---|---|
| Oxidation of alcohols | Bi2WO6 | Vis | 112, 113 |
| Cu(acac)2/ms-BiVO4 | Vis | 117b | |
| Pt/MnOx/BiVO4 | Vis | 118 | |
| Bi24O31Br10(OH)δ | Vis | 125 | |
| Oxidation of amines | BiVO4 | Vis | 119, 120 |
| BiOCl | Vis | 122 | |
| Cu(acac)2/ms-BiVO4 | Vis | 117a | |
| Oxidation of imines | Cu(acac)2/ms-BiVO4 | Vis | 117a |
| C–C bond formation | |||
| Arylation of unsaturated systems | Bi2O3 | Vis | 106 |
| α-Alkylation of aldehydes | Bi2O3 | Vis | 104 |
| PbBiO2Br | Vis | 124 | |
| C–S bond formation | |||
| Thiol–ene | Bi2O3 | Vis | 105 |
| Miscellaneous | |||
| ATRA | Bi2O3 | Vis | 107 |
| Polymerization | Bi2O3 | Vis | 110 |
3.3. ZnO
ZnO is a white powder mostly obtained from metallurgical processes based on the roasting of zinc ore. However, in Nature, it can occur as a rare mineral named zincite. Due to its low toxicity and favorable medicinal properties, ZnO is widely used in domestic care products and food factory, such as baby cream, sunscreen, calamine lotion, oral care products, food additive, etc.127 As a semiconductor, it is widely applied in electronic and photonic devices and used for environmental photocatalysis.128 Despite its apparent advantages, the use of ZnO as a photocatalyst to drive organic transformations is less investigated. This is mainly due to the fact that ZnO has a low visible light response (Eg ≈ 3.4 eV, λ ≈ 380 nm) and also displays fast charge recombination, limiting the overall photocatalytic efficiency. However, similarly to TiO2, ZnO can be doped (e.g. sensitizers, metals, etc.) or tuned, allowing to extend its absorption from the UV to the visible light region and to decrease the charge recombination rate.129Inspired by the work of Zhao on the use of dyes to sensitize TiO2,31 Robinson et al. used ZnO for the photooxidation of organic molecules. The photocatalytic system was composed of an alizarin (Ar) sensitized-ZnO/AgNO3/TEMPO and was able to promote the aerobic oxidation of alcohols under visible light irradiation (λ > 450 nm).130 The versatility of the protocol was demonstrated in the oxidation of a variety of benzylic alcohols and cinnamyl alcohol leading the corresponding aldehydes in high yields (82–99%). Remarkably, the photooxidation of more challenging secondary alcohols such as 1-phenyl ethanol gave rise to acetophenone in good yield (67%) after 2 hours reaction time. However, the photocatalytic system proved to be sluggish in the photooxidation of cyclohexanol (12% yield). The authors claimed that the presence of AgNO3 had a dual rule in the photocatalytic process: (i) preventing the detachment of the dye and; (ii) increasing the power of the photooxidation system by its high electron acceptance capacity.
Another interesting example on the use of ZnO as a photocatalyst was reported by Rueping et al.131 The photooxidation of tertiary amines to form highly reactive iminium ions intermediates was possible with this photocatalyst. The iminium intermediates can be subsequently trapped with various nucleophiles leading to the formation of C–C and C-heteroatoms coupled products. This includes oxidative aza-Henry, Mannich, cyanation reactions as well as phosphonylation of amines. Although ZnO showed interesting photocatalytic activity toward cross dehydrogenative coupling reactions, lower conversions or even decomposition of the starting material were typically observed compared to TiO2. Nevertheless, ZnO showed higher photoactivity in the phosphonylation of tetrahydroisoquinolines yielding the amino-phosphonate adducts in moderate to excellent yields (40 to 96%) (Scheme 48). In addition, both ZnO and TiO2 could be recycled by simple centrifugation and could be reused in consecutive cycles without apparent loss in reactivity and selectivity.
 | ||
| Scheme 48 Phosphonylation of tetrahydroisoquinoline photocatalyzed by ZnO under visible light irradiation. | ||
More recently, Hosseini-Sarvari and Bazyar reported the photocatalytic activity of Pd nanoparticles-supported onto ZnO semiconductors (Pd/ZnO). This catalyst system could light promote cross-coupling reactions, such as Suzuki–Miyaura, Hiyama and Buchwald–Hartwig reactions as well as carbonylation of aryl halides using visible light irradiation (11 W lamp).132 Moreover, the photocatalyst showed good chemical and photostability. The authors surmised that the intriguing visible-light response of the nanocomposite could be ascribed to the SPR effect of the Pd nanoparticles due to the interaction of the conduction band with the incident photons on the surface of the ZnO. Also, in the proposed mechanism for the carbonylation, they suggested that Pd(0) nanoparticles could favor the oxidative addition (Scheme 49).
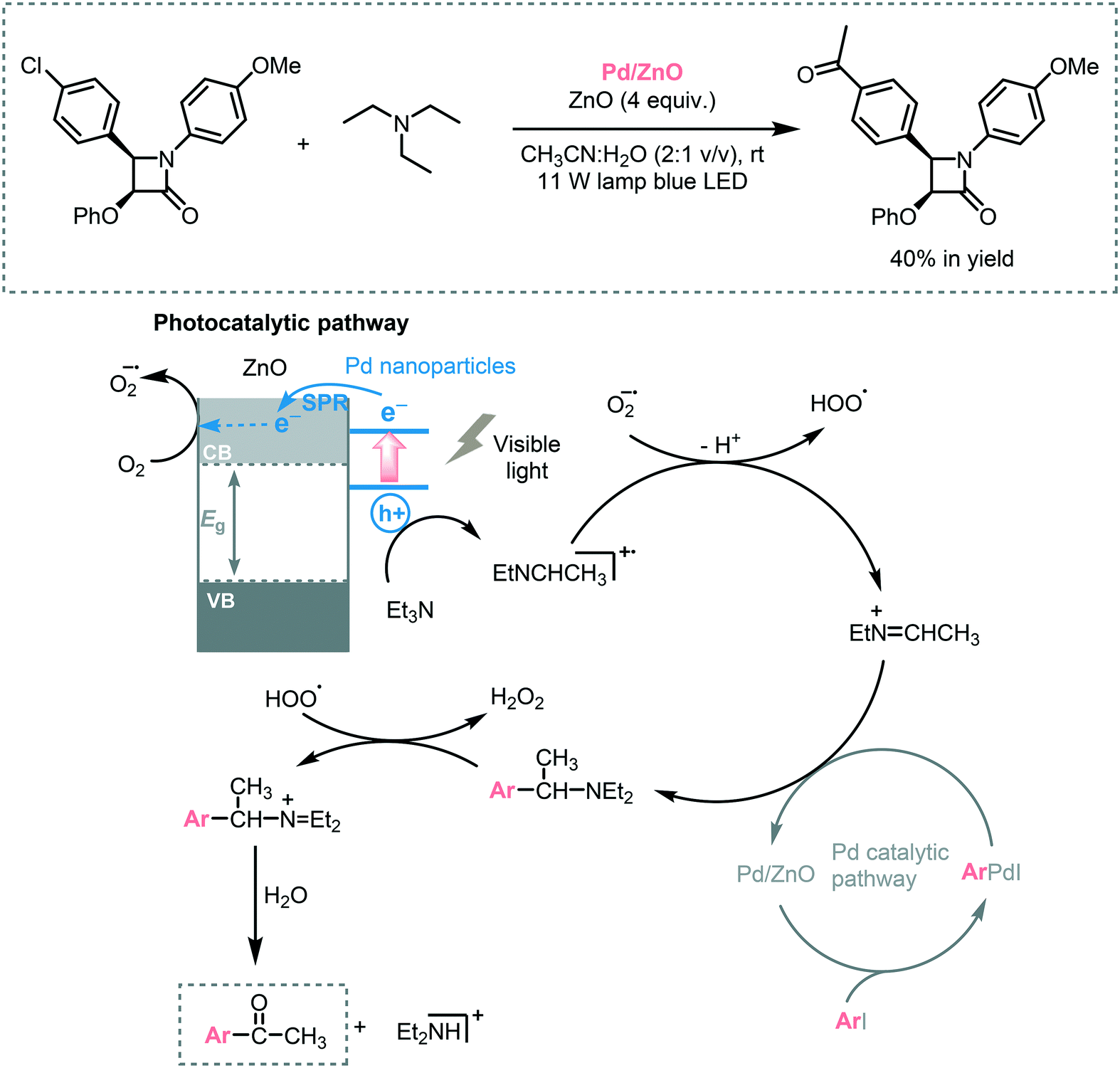 | ||
| Scheme 49 Selected example and the proposed mechanism for the carbonylation of aryl iodides using tertiary amines photocatalyzed by Pd/ZnO under visible light irradiation. | ||
Yagci and co-workers applied ZnO nanoparticles to the atom transfer radical polymerization (ATRP) of methyl acrylate (MMA).133 Two different systems, composed either of ZnO or Fe-doped ZnO/CuBr2/N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), were used to furnish the corresponding polymers under UV light (λ > 350 nm) using α-bromoisobutyrate as a radical initiator. Moreover, the growth of the polymer chains could be tuned by controlling the irradiation time. Regarding the mechanism, the authors suggested that the reduction of Cu(II)/PMDETA to Cu(I)/PMETA by e−CB of ZnO could activate the alkyl bromide initiator. Based on these results, they also reported the application of ZnO or Fe-doped ZnO, as photocatalysts, to promote the aerobic free-radical polymerization of vinyl monomers in both aqueous and organic media under UV irradiation (350 nm).134
The same authors described the application of homemade ZnO nanoparticles in the presence of CuCl2 and PMDETA to promote the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction in acetonitrile under visible light irradiation (λ = 400–500 nm).135 A range of aromatic and aliphatic azides and alkynes compounds were subjected to the photocatalytic protocol giving the corresponding adducts in moderate to excellent yields (up to 98%) (Scheme 50). Remarkably, in control experiments, both the absence of light or ZnO inhibited the reaction and only traces of the final product were observed.
 | ||
| Scheme 50 Selected scope for the visible-light-induced CuAAC photocatalyzed by ZnO under visible light irradiation. | ||
3.4. Nb2O5
Niobium pentoxide (Nb2O5) is another wide band gap semiconductor (Eg ≈ 3.4 eV) with a conduction band formed by empty Nb5+ 4d orbitals.136 Due to its remarkable acidic properties, its chemical and thermal stability and low toxicity, Nb2O5 is widely used as a solid acid catalyst to drive important organic transformations.137 Nb2O5, as a photocatalyst, is widely applied in gas sensing and energy storage as well as in degradation of organic pollutants.138 It can also be used as a dopant of other semiconductors to form heterojunctions (e.g. Nb2O5/TiO2, g-C3N4/Nb2O5, Nb2O5/Bi2WO6).139 These heterojunctions show a better photocatalytic activity than the isolated systems by decreasing the electron–hole recombination rate. Although the application of Nb2O5 to drive photocatalytic organic processes is not common, few examples have been already reported. As an example, Tanaka et al. contributed to demonstrate the feasibility of Nb2O5 to promote the photooxidation of alcohols using molecular oxygen and solvent-free conditions.140 They suggested that the photooxidation of alcohols over Nb2O5 follows a different reaction pathway than observed with TiO2. They proposed that the adsorption of the alcohol onto the Nb2O5 surface as an alcoholate species is the rate-determining step. Thus, this intermediate would transfer an electron to the conduction band of the MOS under appropriate irradiation, leading to its reduction to Nb4+ and to the formation of a hole on the alcoholate. Then, the alkenyl radical is dehydrogenated to form the carbonyl compound. Re-oxidation of Nb4+ to Nb5+ happens with molecular oxygen (Scheme 51). To further improve the photocatalytic activity of Nb2O5, they doped its surface with small amounts of copper. As a result, they could enhance significantly the conversions by increasing the desorption rate of the formed carbonyl compounds.141 Encouraged by the results, the authors also used Nb2O5 to improve the photocatalytic activity of TiO2 for the photooxidation of alcohols.142 The amount of Nb2O5 loaded onto the surface of TiO2 has a strong impact on the selectivity of the reaction and the photocatalytic activity of the composite. The enhancement of the selectivity can be attributed to the inhibition of ozonide ion (O3−) formation, by surface coverage of TiO2 with Nb2O5. This avoids further oxidation of the carbonyl compounds to carbon dioxide. | ||
| Scheme 51 Proposed mechanism for the oxidation of alcohols photocatalyzed by Nb2O5. | ||
Another remarkable example of the combination of Nb2O5 and TiO2 was reported by Li et al.143 The photocatalytic activity of the Nb2O5/TiO2 heterojunction was successfully demonstrated in the photocatalytic oxidation of primary and secondary aromatic alcohols as well as in the photocatalytic reforming of methanol. The conversions obtained with the heterojunction were higher than with pure TiO2 or Nb2O5. Regarding the mechanism, upon ultraviolet irradiation (200 W, 320–780 nm), electron–hole pairs are generated. Due to the difference of the valence band edges of the semiconductors (2.81 eV for TiO2 and 2.75 eV for Nb2O5), the photogenerated electrons in the conduction band of Nb2O5 can be transferred to the CB of TiO2. Simultaneously, the photogenerated holes can move from the TiO2 to the Nb2O5. Therefore, the heterojunction has a dual role: Nb2O5 acts as an oxidant and TiO2 as the reducing agent in the photocatalytic process (Scheme 52).
 | ||
| Scheme 52 Schematic diagram of Nb2O5/TiO2 heterojunction and relative CB and VB structure and charge-transfer of the photogenerated electrons and holes. | ||
Another photochemical transformation with Nb2O5 as a photocatalyst is the aerobic oxidation of amines to imines. A range of primary, secondary, cyclic and benzylic amine derivatives were converted into the corresponding imines generally with high conversions and selectivities under UV irradiation (λ > 300 nm).144 Although primary benzylamines were converted into the corresponding imines with high yields and selectivities, the oxidation of primary alkyl amines or more sterically hindered amines, such as N-phenyl- or N-tert-butyl benzylamines, led to the corresponding imines in moderated selectivity (61%) or in low conversions (up to 30%) (Scheme 53). The reaction was also carried out using visible light irradiation (λ > 390 nm), even though lower conversions were observed (up to 30%). A complete mechanistic study was reported using a combination of experimental techniques and theoretical calculations.145 Similar to the reaction pathways for the photooxidation of alcohols catalyzed by Nb2O5, the rate-determining step is the adsorption of the amine onto the MOS surface.
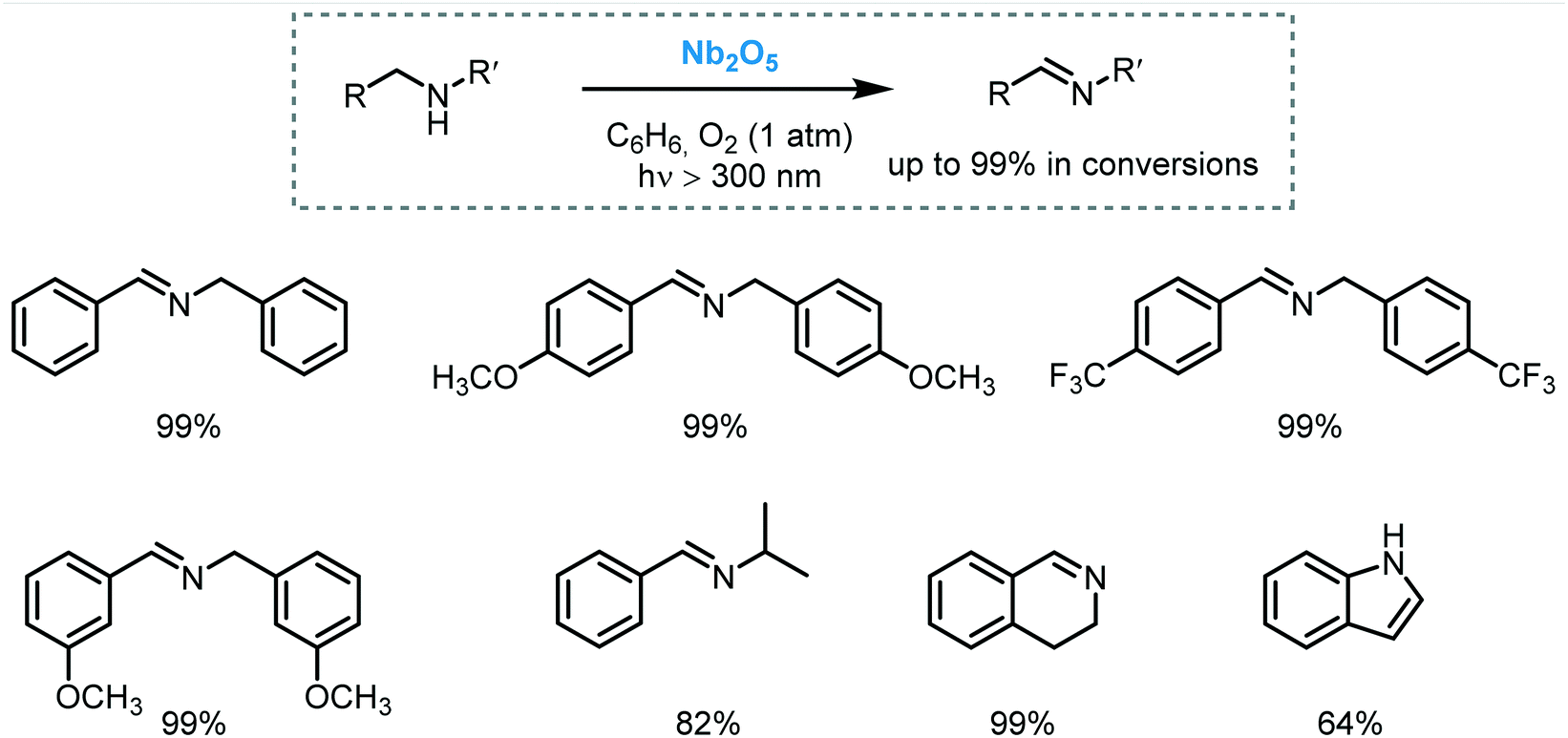 | ||
| Scheme 53 Selected scope for the oxidation of amines photocatalyzed by Nb2O5. | ||
Yoon et al. reported the formation of CT complexes through adsorption of polycyclic hydrocarbons onto the surface of TiO2.146 Inspired by these findings, Tanaka et al. described the photooxidation of aromatic hydrocarbons into carbonyl compounds using Nb2O5 under visible light irradiation (Hg–Xe lamp with cutoff filter, λ > 390 nm).147 A vacuum heat treatment of Nb2O5 enhanced the photocatalytic activity of the system by favoring the LMCT between the semiconductor surface and small aromatic hydrocarbons adsorbed onto its surface. Similar to other LMCT-based photocatalytic systems, the mechanism is based on the reduction of oxygen by the electrons injected in the CB of the Nb2O5. These reduced radical species are next able to dissociate the benzylic C–H bond to form the benzylic radical and ˙OOH. The oxidation of the benzylic radical, by O2, gives rise the benzylperoxy radicals which further decompose in benzaldehyde (Scheme 54).
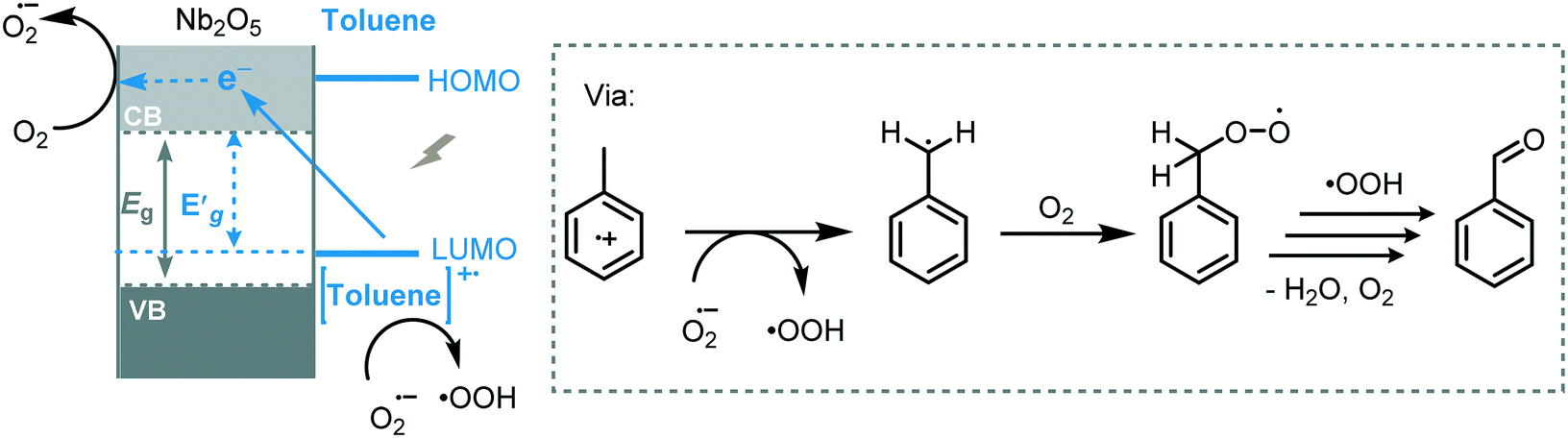 | ||
| Scheme 54 Proposed mechanism for the visible light photooxidation of aromatic hydrocarbons via LMCT on the surface of Nb2O5. | ||
Scaiano et al. reported the use of TiO2 and Nb2O5 semiconductors doped with CuOx nanostructures to carry out aerobic copper-catalyzed azide–alkyne cycloaddition reactions under UVA irradiation.148 The authors claimed that the CuO decorated onto the surface of the MOS trap the electrons, thus decreasing the electron–hole recombination and yielding Cu(I). They argued that the presence of an amine in the system is required to act as a hole scavenger hindering the oxidation of Cu(I). Both semiconductors showed excellent versatility with a great variety of substituted alkynes and azides leading to 1,4-disubstituted-1,2,3-triazole derivatives in moderate to excellent yields (up to 99%). Also, both catalysts showed good stability and could be reused in four consecutive runs.
3.5. WO3
Tungsten trioxide (WO3) is a stable and non-toxic semiconductor with a narrow band gap (Eg ≈ 2.8 eV), which can absorb light up to 480 nm. A large variety of applications of this semiconductor have been reported, including solar cells components, gas sensors and water splitting.149 For these reasons, it is an excellent candidate for applications in visible light photocatalysis. However, WO3 exhibits a relatively positive conduction band (e.g., +0.5 V vs. NHE for WO3) that limits its capacity to reduce O2, thus hindering its application in the oxidation of organic compounds by light. Moreover, it displays a rapid recombination of the photogenerated charges, limiting further its application in photocatalysis. The improvement of its photocatalytic activity can be achieved by loading its surface with metals.150 This allows to construct catalytic materials which are able to promote the multi-electron reduction of O2.Abe et al. reported the oxidation of benzene to phenol by using Pt loaded onto WO3, as a visible light photocatalyst.151 The authors observed that the system composed by Pt/WO3 showed higher selectivity for the phenol production (74% of selectivity) when compared with platinized or bare TiO2. It was hypothesized that the high selectivity is due to the presence of the platinum co-catalyst, which favors the two-electron reduction of O2.152 This results in the formation of mainly H2O2 and thus avoids the formation of stronger oxidizing radical species, such as ˙O2− and ˙HO2. The formed H2O2 can be rapidly decomposed in water and oxygen, consequently, having an insignificant effect on the oxidation pathway (Scheme 55). Moreover, since benzene and phenol show less affinity with the WO3 surface than with the TiO2 surface, the photogenerated holes on WO3 mainly react with H2O to form ˙OH radical species, which subsequently react with benzene to produce selectively phenol.
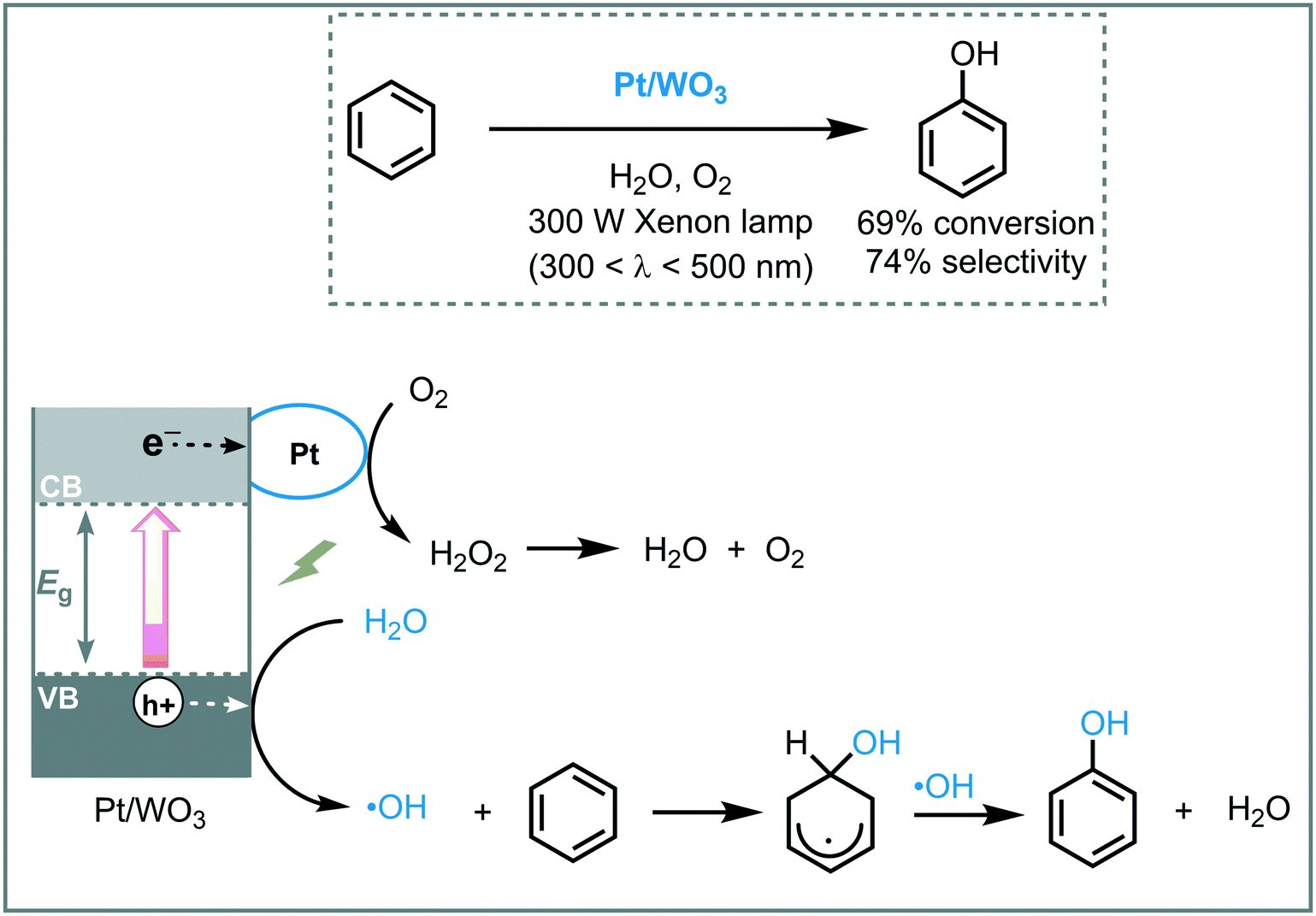 | ||
| Scheme 55 Proposed mechanism for phenol production from benzene photocatalyzed by Pt/WO3. | ||
Next, Abe et al. expanded the approach for the photooxidation of 2-propanol to acetone using WO3 loaded with palladium oxide (PdOx/WO3).153 This photocatalytic system demonstrated higher selectivity under UV-visible light irradiation (300 W Xe lamp, 300 < λ < 500 nm) compared to other photocatalysts, such as Pt/WO3, Pd/TiO2 or Pt/TiO2. Similar to the case of Pt-loaded WO3, the role of the PdOx on the photocatalytic composite was to promote the highly selective two-electron reduction of O2 to form H2O2. Next, it reacts with the holes generated in PdOx/WO3 system, suppressing the overoxidation of the formed acetone. Moreover, the higher selectivity observed in PdOx/WO3 seems to be related to the photogenerated holes in WO3. Due to the low affinity of the WO3 surface with the starting material and acetone, the photogenerated holes engage preferentially with water molecules generating ˙OH radical species that react specifically with alcohols to produce acetone (Scheme 56).
 | ||
| Scheme 56 Proposed mechanism for the oxidation of 2-propanol into acetone photocatalyzed by PdOx/WO3. | ||
Wang et al. developed an efficient photocatalytic system for the oxidation of benzyl alcohol to benzaldehydes replacing the use of noble metals as a doping agent by fluor anion. The as-prepared fluorinated-mesoporous WO3 (Fm-WO3) showed good conversion (>57%) and excellent selectivity (>99%) towards the photooxidation of benzyl alcohol to benzaldehyde. The effect of fluorination in the MOS included the enhancement of the adsorption of the benzyl alcohol on Fm-WO3 surface through H–F bond formation triggering an efficient photocatalytic process. Moreover, its large surface area, furnish more active sites for O2 absorption which benefit efficient multi-electron reduction of O2 to H2O2 and, therefore, its photocatalytic activity.154
The creation of defect states in MOS, such as oxygen vacancies, by defect engineering, can have a significant impact on its electronic structure, ionic/electronic conductivity and catalytic activity. Regarding the creation of oxygen vacancies, such defects can modify the bulk material changing the interactions of the catalyst with the substrates enhancing the performance of the MOS.155 For instance, Xiong and co-workers demonstrated that the creation of oxygen vacancies in WO3 nanosheets led to numerous coordinatively unsaturated sites on its surface for oxygen activation.156 As a proof of concept, the as-formed MOS was used as a photocatalyst to promote the aerobic coupling of amines into imines under visible light. The photocatalytic system showed high efficiency, enhancing the kinetic rate six times compared to defect-deficient WO3. Moreover, its high stability was demonstrated in recycling-reuse experiments. They asserted that the oxygen chemisorption, favored at the defect sites, yielded the formation of superoxide radicals in a chemisorbed state 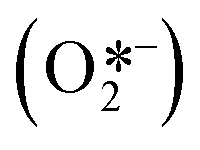 . In a subsequent step, these radical species could react with the amines adsorbed at the neighboring sites (Table 3).
. In a subsequent step, these radical species could react with the amines adsorbed at the neighboring sites (Table 3).
| Reaction type | Photocatalyst/modifications | Irradiation | Ref. |
|---|---|---|---|
| Oxidation of alcohols | Dye-sensitized-ZnO | Vis | 130 |
| Nb2O5/TiO2 | UV | 142, 143 | |
| Nb2O5 | UV | 140 | |
| Cu/Nb2O5 | UV | 141 | |
| PdOx/WO3 | Vis | 153 | |
| Fm-WO3 | Vis | 154 | |
| Oxidation of amines | Nb2O5 | UV/vis | 144, 145 |
| Oxidation of hydrocarbons | Nb2O5/LMCT | Vis | 147 |
| Pt/WO3 | Vis | 152 | |
| C–C bond formation | |||
| Cross-coupling | Pd/ZnO | Vis | 132a,b |
| C–N bond formation | |||
| Cross-coupling | Pd/ZnO | Vis | 132a |
| C–P bond formation | |||
| Phosphonylation | ZnO | Vis | 131 |
| Miscellaneous | |||
| CuAAC | ZnO | Vis | 135 |
| CuOx/Nb2O5 | Vis | 148 | |
| CDC | ZnO | Vis | 131 |
| Polymerization | ZnO or Fe/ZnO | UV | 133, 134 |
| Condensation of amines | WO3 | Vis | 156 |
4. Outlook
In recent years, the application of heterogeneous metal oxide semiconductors (MOS) in photocatalytic organic synthesis has gained considerable traction as a viable alternative for the use of homogeneous transition metal-based complexes and organic dyes. The most attractive features of MOS photocatalysts are their heterogeneous nature, abundance, low cost, low toxicity, and unique and tunable electronic and optical properties. Moreover, most MOS catalysts display also a high chemical and thermal stability, which enables their use even under aggressive reaction conditions.Despite those apparent advantages, there are still some considerable challenges which hamper their use in all potential photocatalytic transformations. These hurdles for further implementation of MOS include (i) the – sometimes – low chemical selectivity, which is mostly associated with the photogeneration of high-energetic holes in wide bandgap semiconductors; (ii) the limited capacity of visible light absorption due to their wide bandgap; (iii) the charge-carrier and fast charge recombination which may also hinder their photon-efficiency and; (iv) the non-ideal band edge potentials. Considerable research efforts should be devoted to these specific aspects to increase the application potential of MOS photocatalysis in organic synthesis. Given the interdisciplinarity of the application of MOS photocatalysis, collaborative efforts between, amongst others, organic chemists, catalysis experts and material scientists will be required to solve these important challenges.
Concerning the band edge potentials, MOS possess valence band edges with sufficiently high potentials to oxidize most of the organic molecules. However, the energy of their conduction bands is frequently lower than the HOMO of these molecules. The latter aspect may hamper MOS-based photocatalytic processes involving reduction pathways.
Finally, recent advances in continuous-flow processes have allowed the use of heterogeneous reagents and catalysts, even in microchannels. Despite this progress, the application of MOS in flow is still rare. However, the combination of an improved irradiation of the MOS photocatalyst in flow together with increased mass transfer characteristics should allow for more selective and faster photochemical processes. Hence, we do expect that more flow applications will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Dutch Science Foundation (NWO) for a VIDI grant for T. N. (SensPhotoFlow, No. 14150). P. R. received financial support from EU through the MSCA Individual Fellowship program (MOSPhotocat, Grant No. 793677).References
- For reviews on solar photochemistry, see: (a) M. Oelgemoeller, Chem. Rev., 2016, 116, 9664–9682 CrossRef PubMed; (b) D. Cambié and T. Noël, Top. Curr. Chem., 2018, 376, 45 CrossRef PubMed.
- PAC, 2007, 79, 293, Glossary of terms used in photochemistry, IUPAC Recommendations, 3rd edn, 2006, p. 384 Search PubMed.
- (a) A. E. Becquerel, Comptes Rendus de L'Academie des Sciences, 1839, 9, 561–567 Search PubMed; (b) A. E. Becquerel, Comptes Rendus de L'Academie des Sciences, 1839, 9, 145–149 Search PubMed.
- (a) L. Bruner and J. Kozak, Z. Elektrochem. Angew. Phys. Chem., 1911, 17, 354–360 CAS; (b) A. Eibner, Chem.-Ztg., 1911, 35, 753–788 CAS; (c) A. Eibner, Chem.-Ztg., 1911, 35, 774–776 Search PubMed; (d) A. Eibner, Chem.-Ztg., 1911, 35, 786–788 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- For reviews on applications of MOS, see: (a) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC; (b) S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed; (c) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed; (d) C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- L. Cermenati, C. Richter and A. Albini, Chem. Commun., 1998, 805–806 RSC.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- For selected reviews on photoredox catalysis, see: (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (b) C.-S. Wang, P. H. Dixneu and J.-F. Soulé, Chem. Rev., 2018, 16, 7532–7585 CrossRef PubMed; (c) C. Bottecchia and T. Noël, Chem. – Eur. J., 2019, 25, 26–42 CrossRef CAS PubMed.
- (a) K. Fidaly, C. Ceballos, A. Falguières, M. S.-I. Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293–1297 RSC; (b) E. Arceo, I. D. Jurberg, A. A. Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (c) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed.
- For reviews, see: (a) A. M. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef; (b) D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391–5411 RSC; (c) J. Chen, J. Cen, X. Xu and X. Li, Catal. Sci. Technol., 2016, 6, 349–362 RSC; (d) C. Huang, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem. – Eur. J., 2018, 24, 11530–11534 CrossRef CAS PubMed; (e) H. Hao and X. Lang, ChemCatChem, 2019, 11, 1378–1393 CrossRef CAS.
- For reviews, see: (a) M. R. Hoffmann, S. Martin, W. Choi and D. W. Bahnemannl, Chem. Rev., 1995, 96, 69–96 CrossRef; (b) X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2016, 15, 383–396 CrossRef CAS PubMed; (c) Y. S. Rim, H. Chen, B. Zhu, S.-H. Bae, S. Zhu, P. J. Li, I. C. Wang and Y. Yang, Adv. Mater. Interfaces, 2017, 4, 1700020 CrossRef.
- For review, see: B. Weng, M.-Y. Qi, C. Han, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2019, 9, 4642–4687 CrossRef CAS.
- For selected examples, see: (a) F. Amano, A. Yamakata, K. Nogami, M. Osawa and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 17650–17651 CrossRef CAS PubMed; (b) A. Mclaren, T. Valdes-Solis, G. Li and S. C. Tsang, J. Am. Chem. Soc., 2009, 131, 12540–12541 CrossRef CAS PubMed; (c) F. Amano, E. Ishinaga and A. Yamakata, J. Phys. Chem. C, 2013, 117, 22584–22590 CrossRef CAS; (d) X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2014, 4, 4596 CrossRef PubMed.
- For review, see: (a) C. Gao, J. Wang, H. Xu and Y. Xiong, Chem. Soc. Rev., 2017, 46, 2799–2823 RSC; (b) C. Xu, P. R. Anusuyadevi, C. Aymonier, R. Luque and S. Marre, Chem. Soc. Rev., 2019, 48, 3868–3902 RSC.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- For reviews, see: (a) M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef; (b) X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC.
- For reviews, see: (a) N. A. Anderson and T. Lian, Coord. Chem. Rev., 2004, 248, 1231–1246 CrossRef CAS; (b) M. Watanabe, Sci. Technol. Adv. Mater., 2017, 18, 705–723 CrossRef CAS PubMed.
- For review, see: A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- For review, see: G. Zhang, G. Kim and W. Choi, Energy Environ. Sci., 2014, 7, 954–966 RSC.
- For selected examples, see: (a) W. Zhao, W. Ma, C. Chen, J. Zhao and Z. Shuai, J. Am. Chem. Soc., 2004, 126, 4782–4783 CrossRef CAS PubMed; (b) Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS PubMed; (c) X. Zhang, X. Li and N. Deng, Ind. Eng. Chem. Res., 2012, 51, 704–709 CrossRef; (d) X. Xiao, W.-W. Zhu, Y.-B. Lei, Q.-Y. Liu, Q. Lia and W.-W. Li, RSC Adv., 2016, 6, 35449–35454 RSC.
- C. F. Goodeve and J. A. Kitchener, Trans. Faraday Soc., 1938, 34, 570–579 RSC.
- For reviews, see: (a) X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed; (b) U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS; (c) M. Nasr, C. Eid, R. Habchi, P. Miele and M. Bechelany, ChemSusChem, 2018, 11, 3023–3047 CrossRef CAS PubMed.
- For reviews, see: (a) R. Asahi, T. Morikawa, H. Irie and T. Ohwak, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS PubMed; (b) H. Xu, S. Ouyang, L. Liu, P. Reunchan, N. Umezawaace and J. Ye, J. Mater. Chem. A, 2014, 2, 12642–12661 RSC; (c) H. Park, H.-I. Kim, G.-H. Moon and W. Choi, Energy Environ. Sci., 2016, 9, 411–433 RSC; (d) M. Humayun, F. Raziq, A. Khan and W. Luo, Green Chem. Lett. Rev., 2018, 11, 86–102 CrossRef CAS; (e) H. Hao, L. Zhang, W. Wang and S. Zeng, Catal. Sci. Technol., 2018, 8, 1229–1250 RSC.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- For reviews, see: (a) M. C. De Rosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS; (b) C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS PubMed; (c) A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- (a) R. Konaka, E. Kasahara, W. C. Dunlap, Y. Yamamoto, K. C. Chien and M. Inoue, Free Radical Biol. Med., 1999, 27, 294–300 CrossRef CAS; (b) Y. Nosaka, T. Daimon, A. Y. Nosaka and Y. Murakami, Phys. Chem. Chem. Phys., 2004, 6, 2917–2918 RSC; (c) T. Daimon and Y. Nosaka, J. Phys. Chem. C, 2007, 111, 4420–4424 CrossRef CAS; (d) A. V. Demyanenko, A. S. Bogomolov, N. V. Dozmorov, A. I. Svyatova, A. P. Pyryaeva, V. G. Goldort, S. A. Kochubei and A. V. Baklanov, J. Phys. Chem. C, 2019, 123, 2175–2181 CrossRef CAS.
- For selected examples, see: (a) A. Jańczyk, E. Krakowska, G. Stochel and W. Macyk, J. Am. Chem. Soc., 2006, 128, 15574–15575 CrossRef PubMed; (b) M. Buchalska, M. Kobielusz, A. Matuszek, M. Pacia, S. Wojtyła and W. Macyk, ACS Catal., 2015, 5, 7424–7431 CrossRef CAS; (c) C. Cantau, T. Pigot, J.-C. Dupin and S. Lacombe, J. Photochem. Photobiol., A, 2010, 216, 201–208 CrossRef CAS; (d) H. Wang, D. Yong, S. Chen, S. Jiang, X. Zhang, W. Shao, Q. Zhang, W. Yan, B. Pan and Y. Xie, J. Am. Chem. Soc., 2018, 140, 1760–1766 CrossRef CAS PubMed.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS PubMed.
- Y. Zhang, Z. Wang and X. Lang, Catal. Sci. Technol., 2017, 7, 4955–4963 RSC.
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279–285 CrossRef CAS.
- X. Lang, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934–3937 CrossRef CAS PubMed.
- X. Lan, W. Ma, Y. Zhao, C. Chen, H. Ji and Z. Zhao, Chem. – Eur. J., 2012, 18, 2624–2631 CrossRef PubMed.
- J. Dai, J. Yang, X. Wang, L. Zhang and Y. Li, Appl. Surf. Sci., 2015, 349, 343–352 CrossRef CAS.
- (a) D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS; (b) E. Oi, M.-Y. Choo, H. V. Lee, H. C. Ong, S. B. A. Hamid and J. C. Juan, RSC Adv., 2016, 6, 108741–108754 RSC.
- For reviews, see: (a) P. Wang, B. Huang, Y. Daia and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC; (b) K. H. Leong, A. A. Aziz, L. C. Sim, P. Saravanan, M. Jang and D. Bahnemann, Beilstein J. Nanotechnol., 2018, 9, 628–648 CrossRef CAS PubMed.
- Y. Shiraishi, H. Sakamoto, K. Fujiwara, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 2418–2425 CrossRef CAS.
- L. Ren, M.-M. Yang, C.-H. Tung, L.-Z. Wu and H. Cong, ACS Catal., 2017, 7, 8134–8138 CrossRef CAS.
- G. Palmisano, M. Addamo, V. Augugliaro, T. Caronna, A. Di Paola, E. G. López, V. Loddo, G. Marcì, L. Palmisano and M. Schiavello, Catal. Today, 2007, 122, 118–127 CrossRef CAS.
- Z. Zheng, B. Huang, X. Qin, X. Zhang, Y. Dai and M.-H. Whangbo, J. Mater. Chem., 2011, 21, 9079–9087 RSC.
- W. Tu, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2013, 23, 4996–5008 CrossRef CAS.
- Y. Shiraishi, S. Shiota, H. Hirakawa, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2017, 7, 293–300 CrossRef CAS.
- (a) M.-Q. Yang, N. Zhang and Y.-J. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 1156–1164 CrossRef CAS PubMed; (b) L. Yuan, Q. Yu, Y. Zhang and Y.-J. Xu, RSC Adv., 2014, 4, 15264–15270 RSC.
- (a) C. Xu, Y. Yuan, R. Yuan and X. Fu, RSC Adv., 2013, 3, 18002–18008 RSC; (b) X. Pan, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, J. Phys. Chem. C, 2014, 118, 27325–27335 CrossRef CAS.
- For reviews, see: (a) A. Levai, ARKIVOC, 2003, 14, 14–30 Search PubMed; (b) I. Barnes, J. Hjorth and N. Mihalopoulos, Chem. Rev., 2006, 106, 940–975 CrossRef CAS; (c) H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- X. Lang, W. R. Leow, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 1075–1082 RSC.
- X. Lang, W. Hao, W. R. Leow, S. Li, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 5000–5005 RSC.
- (a) M. Jafarpour, F. Feizpour and A. Rezaeifard, RSC Adv., 2016, 6, 54649–54660 RSC; (b) M. Jafarpour, F. Feizpour and A. Rezaeifard, Synlett, 2017, 28, 235–238 CrossRef CAS; (c) M. Jafarpour, F. Feizpour and A. Rezaeifard, New J. Chem., 2018, 42, 807–811 RSC.
- N. Marina, A. B. Lanterna and J. C. Scaiano, ACS Catal., 2018, 8, 7593–7597 CrossRef CAS.
- A. Hainer, N. Marina, S. Rincon, P. Costa, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2019, 141, 4531–4535 CrossRef CAS PubMed.
- A. Tyagi, A. Yamamoto, T. Kato and H. Yoshida, Catal. Sci. Technol., 2017, 7, 2616–2623 RSC.
- A. Yamamoto, T. Ohara and H. Yoshida, Catal. Sci. Technol., 2018, 8, 2046–2050 RSC.
- B. Kraeutler, C. D. Jaeger and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 4903–4905 CrossRef CAS.
- (a) D. W. Manley, R. T. McBurney, P. Miller, R. F. Howe, S. Rhydderch and J. C. Walton, J. Am. Chem. Soc., 2012, 134, 13580 CrossRef CAS PubMed; (b) D. W. Manley, R. T. McBurney, P. Miller and J. C. Walton, J. Org. Chem., 2014, 79, 1386–1398 CrossRef CAS PubMed.
- L. Ren and H. Cong, Org. Lett., 2018, 20, 3225–3228 CrossRef CAS PubMed.
- For reviews, see: (a) M. M. Conn and J. Rebek Jr, Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS PubMed; (b) R. Capdeville, E. Buchdunger, J. Zimmermann and A. Matter, Nat. Rev. Drug Discovery, 2002, 1, 493–502 CrossRef CAS PubMed; (c) A. R. Murphy and J. M. J. Frèchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed; (d) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- For reviews, see: (a) F. Shibahara and T. Murai, Asian J. Org. Chem., 2013, 2, 624–636 CrossRef CAS; (b) H. Bonin, M. Sauthier and F.-X. Felpin, Adv. Synth. Catal., 2014, 356, 645–671 CrossRef CAS; (c) R. Rossi, M. Lessia, C. Manzinia, G. Marianettic and F. Bellina, Synthesis, 2016, 48, 3821–3862 CrossRef CAS; (d) P. Gao, Y.-R. Gu and X.-H. Duan, Synthesis, 2017, 49, 3407–3421 CrossRef CAS For a recent example, see: H. P. L. Gemoets, I. Kalvet, A. V. Nyuchev, N. Erdmann, V. Hessel, F. Schoenebeck and T. Noël, Chem. Sci., 2017, 8, 1046–1055 Search PubMed.
- J. Zoller, D. C. Fabry and M. Rueping, ACS Catal., 2015, 5, 3900–3904 CrossRef CAS.
- W. Liu, C. Wang and L. Wang, Ind. Eng. Chem. Res., 2017, 56, 6114–6123 CrossRef CAS.
- L. Wang, J. Shen, S. Yang, W. Liu, Q. Chen and M. He, Green Chem., 2018, 20, 1290–1296 RSC.
- For reviews, see: (a) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC; (b) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- J. Tang, G. Grampp, Y. Liu, B.-X. Wang, F.-F. Tao, L.-J. Wang, X.-Z. Liang, H.-Q. Xiao and Y.-M. Shen, J. Org. Chem., 2015, 80, 2724–2732 CrossRef CAS PubMed.
- For review, see: V. V. Kouznetsov and C. E. P. Galvis, Tetrahedron, 2018, 74, 773–810 CrossRef CAS.
- For review, see: Y. Ping, Q. Ding and Y. Peng, ACS Catal., 2016, 6, 5989–6005 CrossRef CAS.
- J. Santamaria, M. T. Raddachi and J. Rigaudy, Tetrahedron Lett., 1990, 314735–314738 Search PubMed.
- For selected examples, see: (a) S.-I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931–6933 CrossRef CAS PubMed; (b) M. Rueping, S. Zhua and R. M. Koenigsa, Chem. Commun., 2011, 47, 12709–12711 RSC; (c) Y. Pan, S. Wang, C. W. Kee, E. Dubuisson, Y. Yang, K. P. Loh and C.-H. Tan, Green Chem., 2011, 13, 3341–3344 RSC; (d) D. B. Ushakov, K. Gilmore, D. Kopetzki, D. T. McQuade and P. H. Seeberger, Angew. Chem., Int. Ed., 2014, 53, 557–561 CrossRef CAS PubMed.
- A. M. Nauth, E. Schechtel, R. Dören, W. Tremel and T. Opatz, J. Am. Chem. Soc., 2018, 140, 14169–14177 CrossRef CAS PubMed.
- C. D. McTiernan, S. P. Pitre, H. Ismaili and J. C. Scaiano, Adv. Synth. Catal., 2014, 356, 2819–2824 CrossRef CAS.
- S. P. Pitre, J. C. Scaiano and T. P. Yoon, ACS Catal., 2017, 7, 6440–6444 CrossRef CAS PubMed.
- G. K. Hodgson and J. C. Scaiano, ACS Catal., 2018, 8, 2914–2922 CrossRef CAS.
- (a) Y. Okada, N. Maeta, K. Nakayama and H. Kamiya, J. Org. Chem., 2018, 83, 4948–4962 CrossRef CAS PubMed; (b) K. Nakayama, N. Maeta, G. Horiguchi, H. Kamiya and Y. Okada, Org. Lett., 2019, 21, 2246–2250 CrossRef CAS.
- (a) M. P. Sibi and M. Hasagawa, J. Am. Chem. Soc., 2007, 129, 4124–4125 CrossRef CAS PubMed; (b) S. P. Simonovich, J. F. Van Humbeck and D. W. C. MacMillan, Chem. Sci., 2012, 3, 58–61 RSC.
- T. Koike and M. Akita, Chem. Lett., 2009, 38, 166–167 CrossRef CAS.
- X.-H. Ho, M.-J. Kang, S.-J. Kim, E. D. Park and H.-Y. Jang, Catal. Sci. Technol., 2011, 1, 923–926 RSC.
- For reviews, see: (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) A. Harsanyi and G. Sandford, Green Chem., 2015, 17, 2081–2086 RSC.
- For review, see: J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed.
- G. Tarantino and C. Hammond, ACS Catal., 2018, 8, 10321–10333 CrossRef CAS.
- C. Platz and K. Schimmelschimidt, Chem. Abstr., 1940, 38, 1249 Search PubMed.
- (a) F. Parrino, A. Ramakrishnam and H. Kisch, Angew. Chem., Int. Ed., 2008, 47, 7107–7109 CrossRef CAS PubMed; (b) F. Parrino, A. Ramakrishnam, C. Damn and H. Kisch, ChemPlusChem, 2012, 77, 713–720 CrossRef CAS.
- (a) E. L. Tyson, M. S. Ament and T. P. Yoon, J. Org. Chem., 2013, 78, 2046–2050 CrossRef CAS PubMed; (b) E. L. Tyson, Z. L. Niemeyer and T. P. Yoon, J. Org. Chem., 2014, 79, 1427–1436 CrossRef CAS PubMed; (c) M. H. Keylor, J. E. Park, C.-J. Wallentin and C. R. J. Stephenson, Tetrahedron, 2014, 70, 4264–4269 CrossRef CAS.
- V. T. Bhat, P. A. Duspara, S. Seo, N. S. B. A. Bakar and M. F. Greaney, Chem. Commun., 2015, 51, 4383–4385 RSC.
- M. H. Sarvari, F. Jafari, A. Mohajeri and N. Hassani, Catal. Sci. Technol., 2018, 8, 4044–4051 RSC.
- For selected examples: (a) A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162–1167 CrossRef CAS; (b) R. D. Patil and S. Adimurthy, Adv. Synth. Catal., 2011, 353, 1695–1700 CrossRef CAS; (c) W. He, L. Wang, C. Sun, K. Wu, S. He, J. Chen, P. Wu and Z. Yu, Chem. – Eur. J., 2011, 18, 13308–13317 CrossRef PubMed; (d) S. Zhao, C. Liu, Y. Guo, J.-C. Xiao and Q.-Y. Chen, J. Org. Chem., 2014, 79, 8926–8931 CrossRef CAS PubMed.
- L.-M. Wang, K. Kobayashi, M. Arisawa, S. Saito and H. Naka, Org. Lett., 2019, 21, 341–344 CrossRef CAS PubMed.
- C. Vila and M. Rueping, Green Chem., 2013, 15, 2056–2059 RSC.
- (a) M. S. Kharasch, E. V. Jensen and W. H. Urry, Science, 1945, 102, 128 CrossRef CAS PubMed; (b) M. S. Kharasch, P. S. Skell and P. Fisher, J. Am. Chem. Soc., 1948, 70, 1055–1059 CrossRef CAS.
- (a) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160–4163 CrossRef CAS PubMed; (b) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed.
- (a) M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – Eur. J., 2012, 18, 7336–7340 CrossRef CAS PubMed; (b) S. Paria, M. Pirtsch, V. Kais and O. Reiser, Synthesis, 2013, 45, 2689–2698 CrossRef CAS; (c) E. Arceo, E. Montroni and P. Melchiorre, Angew. Chem., Int. Ed., 2014, 53, 12064–12068 CrossRef CAS PubMed.
- L.-L. Mao and H. Cong, ChemSusChem, 2017, 10, 4461–4464 CrossRef CAS PubMed.
- A. Hakki, R. Dillert and D. W. Bahnemann, Phys. Chem. Chem. Phys., 2013, 15, 2992–3002 RSC.
- For reviews, see: (a) Y. Su, N. J. W. Straathof, V. Hessel and T. Noël, Chem. – Eur. J., 2014, 20, 10562–10589 CrossRef CAS PubMed; (b) D. Cambié, C. Bottechia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef; (c) T. Noël, J. Flow Chem., 2017, 7, 87–93 CrossRef; (d) J. Yue, Catal. Today, 2018, 308, 3–19 CrossRef CAS.
- For review, see: R. L. Hartman, Org. Process Res. Dev., 2012, 165, 870–887 CrossRef.
- For selected examples: (a) R. F. P. Nogueira and W. F. Jardim, Sol. Energy, 1996, 56, 471–477 CrossRef CAS; (b) J. Grzechulska and A. W. Morawski, Appl. Catal., B, 2003, 46, 415–419 CrossRef CAS; (c) M. A. Ferguson and J. G. Hering, Environ. Sci. Technol., 2006, 40, 4261–4267 CrossRef CAS PubMed; (d) M. Dilla, A. E. Becerikli, A. Jakubowski, R. Schlögl and S. Ristig, Photochem. Photobiol. Sci., 2019, 18, 314–318 RSC.
- D. C. Fabry, Y. A. Ho, R. Zapf, W. Tremel, M. Panthöfer, M. Rueping and T. H. Rehm, Green Chem., 2017, 19, 1911–1918 RSC.
- C. Bottechia, N. Erdmann, P. M. A. Tijssen, L.-G. Milroy, L. Brunsveld, V. Hessel and T. Noël, ChemSusChem, 2016, 9, 1781–1785 CrossRef PubMed.
- G. Takei, T. Kitamiro and H.-B. Kim, Catal. Commun., 2005, 6, 357 CrossRef CAS.
- M. Bagbanzadeh, T. N. Glasnov and C. O. Kappe, J. Flow Chem., 2013, 3, 109–113 CrossRef.
- B. O. Burek, A. Sutor, D. W. Bahnemann and J. Z. Bloh, Catal. Sci. Technol., 2017, 7, 4977–4983 RSC.
- For reviews, see: (a) L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist and W. Kroutil, ACS Catal., 2019, 9, 4115–4144 CrossRef CAS; (b) C. J. Seel and T. Gulder, ChemBioChem, 2019, 20, 1871–1897 CrossRef CAS PubMed.
- For review, see: X. Meng and Z. Zhang, J. Mol. Catal. A: Chem., 2016, 423, 533–549 CrossRef CAS.
- L. Zhang, W. Wang, J. Yang, Z. Chen, W. Zhang, L. Zhou and S. Liu, Appl. Catal., A, 2006, 308, 105–110 CrossRef CAS.
- P. Riente, A. M. Adams, J. Albero, E. Palomares and M. A. Pericàs, Angew. Chem., Int. Ed., 2014, 53, 9613–9616 CrossRef CAS PubMed.
- O. O. Fadeyi, J. J. Mousseau, Y. Feng, C. Allais, P. Nuhant, M. Z. Chen, B. Pierce and R. Robinson, Org. Lett., 2015, 17, 5756–5759 CrossRef CAS PubMed.
- L. Buglioni, P. Riente, E. Palomares and M. A. Pericàs, Eur. J. Org. Chem., 2017, 6986–6990 CrossRef CAS.
- P. Riente and M. A. Pericàs, ChemSusChem, 2015, 8, 1841–1844 CrossRef CAS PubMed.
- For review, see: (a) K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 CrossRef CAS PubMed; (b) C. Boyer, N. A. Corrigan, K. Jung, D. Nguyen, T.-K. Nguyen, N. N. M. Adnan, S. Oliver, S. Shanmugam and J. Yeow, Chem. Rev., 2016, 116, 1803–1949 CrossRef CAS PubMed.
- For a review on photo-induced polymerizations, see: (a) M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed; (b) Q. Michaudel, V. Kottisch and B. P. Fors, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 CrossRef CAS PubMed.
- K. Hakobyan, T. Gegenhuber, C. S. P. McErlean and M. Müllner, Angew. Chem., Int. Ed., 2019, 58, 1828–1832 CrossRef CAS PubMed.
- T. Saison, P. Gras, N. Chemin, C. Chanéac, O. Durupthy, V. Brezová, C. Colbeau-Justin and J.-P. Jolivet, J. Phys. Chem. C, 2013, 117, 22656–22666 CrossRef CAS.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, Chem. Sci., 2013, 4, 1820–1824 RSC.
- Y. Zhang and Y.-J. Xu, RSC Adv., 2014, 4, 2904–2910 RSC.
- A. Kudo, K. Ueda, H. Kato and I. Mikami, Catal. Lett., 1998, 53, 229–230 CrossRef CAS.
- For reviews, see: (a) H. L. Tan, R. Amal and Y. H. Ng, J. Mater. Chem. A, 2017, 5, 16498–16521 RSC; (b) B. Lamm, B. J. Trzésniewski, H. Döscher, W. A. Smith and M. Stefik, ACS Energy Lett., 2018, 3, 112–124 CrossRef CAS; (c) A. Malathi, J. Madhavan, M. Ashokkumar and P. Arunachalam, Appl. Catal., A, 2018, 555, 47–74 CrossRef CAS.
- For reviews, see: (a) M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS; (b) R. M. N. Yerga, M. C. Á. Galván, F. del Valle, J. A. V. de la Mano and J. L. G. Fierro, ChemSusChem, 2009, 2, 471–485 CrossRef PubMed; (c) S. R. Lingampalli, M. M. Ayyub and C. N. R. Rao, ACS Omega, 2017, 2, 2740–2748 CrossRef CAS PubMed.
- (a) S.-I. Naya, T. Niwa, R. Negishi, H. Kobayashi and H. Tada, Angew. Chem., Int. Ed., 2014, 53, 13894–13897 CrossRef CAS PubMed; (b) R. Negishi, S.-I. Naya and H. Tada, J. Phys. Chem. C, 2015, 119, 11771–11776 CrossRef CAS.
- X. Jin, R. Li, Y. Zhao, X. Liu, X. Wang, H. Jiao and J. Li, Catal. Sci. Technol., 2018, 8, 6173–6179 RSC.
- B. Yuan, R. Chong, B. Zhang, J. Li, Y. Liu and C. Li, Chem. Commun., 2014, 50, 15593–15596 RSC.
- A. D. Proctor, S. Panuganti and B. M. Barlett, Chem. Commun., 2018, 54, 1101–1104 RSC.
- (a) W. Zhang, Q. Zhang and F. Dong, Ind. Eng. Chem. Res., 2013, 52, 6740–6746 CrossRef CAS; (b) H. Li, J. Li, Z. Ai, F. Jia and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 122–138 CrossRef CAS PubMed.
- Y. Wu, B. Yuan, M. Li, W.-H. Zhang, Y. Liu and C. Li, Chem. Sci., 2015, 6, 1873–1878 RSC.
- X. Zhang, L. Zhao, C. Fan, Z. Liang and P. Han, Comput. Mater. Sci., 2012, 61, 180–184 CrossRef CAS.
- M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062–4066 CrossRef CAS PubMed.
- Y. Dai, P. Ren, Y. Li, D. Lv, Y. Shen, Y. Li, H. Niemantsverdriet, F. Besenbacher, H. Xiang, W. Hao, N. Lock, X. Wen, J. P. Lewis and R. Su, Angew. Chem., Int. Ed., 2019, 58, 6265–6270 CrossRef CAS PubMed.
- Y. Dai, C. Li, Y. Shen, S. Zhu, M. S. Hvid, L.-C. Wu, J. Skibsted, Y. Li, J. W. H. Niemantsverdriet, F. Besenbacher, N. Lock and R. Su, J. Am. Chem. Soc., 2018, 140, 16711–16719 CrossRef CAS PubMed.
- J. Liu, Y. Wang, J. Mab, Y. Peng and A. Wang, J. Alloys Compd., 2019, 783, 898–918 CrossRef CAS.
- For reviews, see: (a) C. Klingshirn, ChemPhysChem, 2007, 8, 782–803 CrossRef CAS PubMed; (b) X. Gu, C. Li, S. Yuan, M. Ma, Y. Qiang and J. Zhu, Nanotechnology, 2016, 27, 402001 CrossRef PubMed; (c) C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS.
- J. Wang, R. Chen, L. Xiang and S. Komarneni, Ceram. Int., 2018, 44, 7357–7377 CrossRef CAS.
- V. Jeena and R. S. Robinson, Chem. Commun., 2012, 48, 299–301 RSC.
- M. Rueping, J. Zoller, D. C. Fabry, K. Poscharny, R. M. Koenigs, T. E. Weirich and J. Mayer, Chem. – Eur. J., 2012, 18, 3478–3481 CrossRef CAS PubMed.
- (a) M. Hosseini-Sarvari and Z. Bazyar, ChemistrySelect, 2018, 3, 1898–1907 CrossRef CAS; (b) Z. Bazyar and M. Hosseini-Sarvari, Eur. J. Org. Chem., 2019, 2282–2288 CrossRef CAS.
- S. Dadashi-Silab, M. A. Tasdelen, A. M. Asiri, S. B. Khan and Y. Yagci, Macromol. Rapid Commun., 2014, 35, 454–459 CrossRef CAS PubMed.
- S. Dadashi-Silab, A. M. Asiri, S. B. Khan, K. A. Alamry and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1500–1507 CrossRef CAS.
- O. Yetiskin, S. Dadashi-Silab, S. B. Khan, A. M. Asiri and Y. Yagci, Asian J. Org. Chem., 2015, 4, 442–444 CrossRef CAS.
- I. Nowak and M. Ziolek, Chem. Rev., 1999, 99, 3603–3624 CrossRef CAS.
- S. M. A. Siddiki, M. N. Rashed, M. A. Ali, T. Toyao, P. Hirunsit, M. Ehara and K. Shimizu, ChemCatChem, 2018, 11, 383–396 CrossRef.
- For selected examples, see: (a) A. G. S. Prado, L. B. Bolzon, C. P. Pedroso, A. O. Moura and L. L. Costa, Appl. Catal., B, 2008, 82, 219–224 CrossRef CAS; (b) L. Yan, X. Rui, G. Chen, W. Xu, G. Zou and H. Luo, Nanoscale, 2016, 8, 8443–8465 RSC; (c) B. Boruah, R. Gupta, J. Modak and G. Madras, Nanoscale Adv., 2019, 1, 2748–2760 RSC.
- (a) J. Wu, J. Li, J. Liu, J. Baia and L. Yang, RSC Adv., 2017, 7, 51046–51054 RSC; (b) I. Khan, N. Baig and A. Qurashi, ACS Appl. Energy Mater., 2019, 2, 607–615 CrossRef CAS.
- T. Shishido, T. Miyatake, K. Teramura, Y. Hitomi, H. Yamashita and T. Tanaka, J. Phys. Chem. C, 2009, 113, 18713–18718 CrossRef CAS.
- S. Furukawa, A. Tamura, T. Shishido, K. Teramura and T. Tanaka, Appl. Catal., B, 2011, 110, 216–220 CrossRef CAS.
- S. Furukawa, T. Shishido, K. Teramura and T. Tanaka, ACS Catal., 2012, 2, 175–179 CrossRef CAS.
- J. Ya, G. Wu, N. Guan and L. Li, Appl. Catal., B, 2014, 152, 280–288 Search PubMed.
- S. Furukawa, Y. Ohno, T. Shishido, K. Teramura and T. Tanaka, ACS Catal., 2011, 1, 1150–1153 CrossRef CAS.
- S. Furukawa, Y. Ohno, T. Shishido, K. Teramura and T. Tanaka, J. Phys. Chem. C, 2013, 117, 442–450 CrossRef CAS.
- Y. S. Seo, C. Lee, K. H. Lee and K. B. Yoon, Angew. Chem., Int. Ed., 2005, 44, 910–913 CrossRef CAS PubMed.
- K. Tamai, K. Murakami, S. Hosokawa, H. Asakura, K. Teramura and T. Tanaka, J. Phys. Chem. C, 2017, 121, 22854–22861 CrossRef CAS.
- B. Wang, J. Durantini, J. Nie, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2016, 138, 13127–13130 CrossRef CAS PubMed.
- P. Dong, G. Hou, X. Xi, R. Shao and F. Dong, Environ. Sci.: Nano, 2017, 4, 539–557 RSC.
- For selected examples: (a) R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780–7781 CrossRef CAS; (b) J. Kim, C. W. Lee and W. Choi, Environ. Sci. Technol., 2010, 44, 6849–6854 CrossRef CAS PubMed; (c) S. Mohammadi, M. Sohrabi, A. N. Golikand and A. Fakhri, J. Photochem. Photobiol., B, 2016, 161, 217–221 CrossRef CAS PubMed.
- O. Tomita, R. Abe and B. Ohtani, Chem. Lett., 2011, 40, 1405–1407 CrossRef CAS.
- O. Tomita, B. Ohtani and R. Abe, Catal. Sci. Technol., 2014, 4, 3850–3860 RSC.
- O. Tomita, T. Otsubo, M. Higashi, B. Ohtani and R. Abe, ACS Catal., 2016, 6, 1134–1144 CrossRef CAS.
- Y. Su, Z. Han, L. Zhang, W. Wang, M. Wang, M. Duan, X. Li, Y. Zheng, Y. Wang and X. Lei, Appl. Catal., B, 2017, 217, 108–114 CrossRef CAS.
- For review, see: Y. Zhu, X. Liu, S. Jin, H. Chen, W. Lee, M. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875–5897 RSC.
- N. Zhang, X. Li, H. Ye, S. Chen, H. Ju, D. Liu, Y. Lin, W. Ye, C. Wang, Q. Xu, J. Zhu, L. Song, J. Jiang and Y. Xiong, J. Am. Chem. Soc., 2016, 138, 8928–8935 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |