
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterplay of nucleophilic catalysis with proton transfer in the nitrile reductase QueF from Escherichia coli†
Jihye
Jung
ab,
Jan
Braun
a,
Tibor
Czabany
ab and
Bernd
Nidetzky
 *ab
*ab
aInstitute of Biotechnology and Biochemical Engineering, Graz University of Technology, NAWI Graz, Petersgasse 12/1, A-8010 Graz, Austria. E-mail: bernd.nidetzky@tugraz.at
bAustrian Centre of Industrial Biotechnology, Petersgasse 14, A-8010 Graz, Austria
First published on 14th January 2019
Abstract
Enzymatic transformations of the nitrile group are important in biology as well as in synthetic chemistry. The enzyme QueF catalyses the conversion of 7-cyano-7-deazaguanine (preQ0) to 7-aminomethyl-7-deazaguanine (preQ1), a unique approach towards biological four-electron reduction of a nitrile to an amine. The catalytic reaction involves a QueF–preQ0 thioimidate adduct that is converted to preQ1 in two NADPH dependent reduction steps via an imine intermediate. The QueF active site comprises a cysteine nucleophile flanked by an aspartic acid and additionally contains a histidine. Here, we used mutagenesis of E. coli QueF (C190A, C190S, D197A, D197H, and H229A) to study the functional interplay between these enzyme residues in covalent catalysis. Substitution of Cys190 or Asp197 annihilates preQ0 covalent binding and largely disrupts the nitrile-to-amine reductase activity. The H229A variant readily forms the thioimidate adduct and is 24-fold less active for preQ0 reduction than wild-type ecQueF (kcat = 7.2 min−1). Using isothermal titration calorimetry, we show that the non-covalent step of preQ0 binding involves proton uptake mediated by Asp197 with His229 as the likely protonated group. Catalytic proton transfer from the Cys190 thiol via Asp197 to the nitrile nitrogen promotes the covalent intermediate. We suggest that protonated (charged) His229 facilitates the polarization of the substrate nitrile for nucleophilic attack on carbon by Cys190, and through proton relay via Asp197, it could provide the proton for re-protonating Cys190 during the formation of the imine intermediate.
Introduction
Enzymatic transformations of the nitrile group are important biologically, for example, in secondary and xenobiotic metabolisms of plants and microorganisms.1–3 They also have significant applications in synthetic chemistry wherein nitriles often represent important intermediates.4,5 The nitrile group is converted by hydrolytic enzymes to an amide or to a carboxylic acid6–9 and by reductive enzymes to an amine.10–13 Despite the different reactivities, nitrile-converting enzymes share covalent catalysis from an active-site nucleophile as a common feature of their mechanisms. Nitrilase8,9,14–16 and nitrile reductase17–20 both utilize a cysteine to form a thioimidate adduct between the enzyme and nitrile substrate (Scheme 1A). The covalent catalysis in each enzyme requires assistance from catalytic proton transfer. The enzyme nucleophile is activated by deprotonation. Conversion of nitrile to the covalent intermediate and further on to the product necessitates acid–base catalysis, as shown in Scheme 1A. Nitrile reductase and nitrilase both have a candidate acid–base residue (Asp or Glu) positioned close to their cysteine nucleophile8,9,11,16,18,21 but the precise role of the Asp/Glu remains to be elucidated. Moreover, the critical interplay between nucleophilic and general acid–base catalysis is not well understood in both enzymes. In this study, therefore, we sought to clarify the involvement of catalytic proton transfer in the build-up and degradation of the covalent thioimidate intermediate during enzymatic nitrile reduction.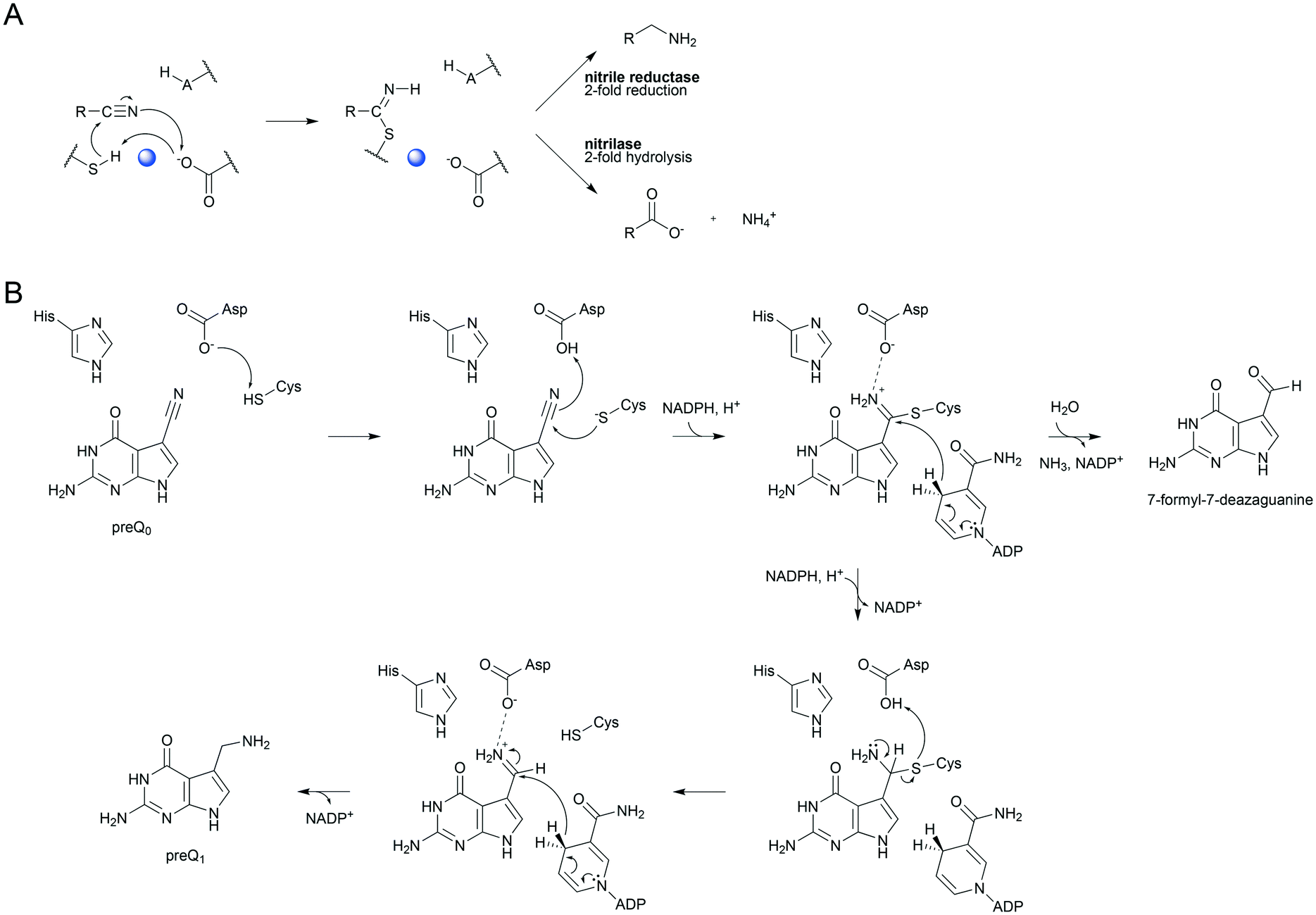 | ||
| Scheme 1 Mechanistic analogies of nitrile reductase and nitrilase (A) and the proposed mechanism of nitrile reduction by ecQueF (B). (A) Catalytic conversion of the nitrile substrate via a common covalent thioimidate enzyme intermediate is shown. Base-catalyzed activation of the cysteine nucleophile is water-mediated in the nitrilase. The water is shown in blue. Reduction and hydrolysis of the thioimidate both involve general acid–base catalysis. (B) A detailed mechanism of preQ0 reduction is shown. Interception of the imine intermediate by water results in the formation of 7-formyl-7-deazaguanine. This off-reaction is not significant in wildtype ecQueF but occurs in certain variants of the enzyme.27 | ||
The biological principle of nitrile reduction is embodied in the enzyme QueF.12,22–26 The natural reaction of QueF is conversion of 7-cyano-7-deazaguanine (preQ0) to 7-aminomethyl-7-deazaguanine (preQ1) by NADPH (Scheme 1B).17–19,21,27,28 QueF is a bacterial enzyme from the biosynthetic pathway for the modified nucleoside queuosine (Q).12,22,29 Inserted into tRNA as preQ1 and further converted to Q, queuosine modulates the codon–anticodon binding efficiency for decoding NAC/U codons to Asn, Asp, His, and Tyr.22,29,30 The proposed catalytic mechanism of QueF is summarized in Scheme 1B. The core characteristics of this mechanism are well supported by enzyme crystal structures and biochemical evidence.17,18,21,27,28 Therefore, the thioimidate adduct is formed from a non-covalent QueF–preQ0 complex that secludes the nitrile substrate completely from the solvent (Schemes 1B and 2).17–20 The covalent intermediate is converted to preQ1 in two NADPH dependent reduction steps via an imine. The imine is sequestered effectively in the QueF active site to prevent its decomposition.17,27 NADPH binds very tightly to the enzyme–imine complex.17 The enzyme thus ensures that every preQ0 converted is reduced fully to preQ1.
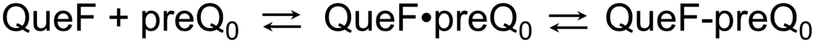 | ||
| Scheme 2 Two-step mechanism of preQ0 binding to QueF. QueF·preQ0, noncovalent complex; QueF–preQ0, covalent thioimidate adduct. In wildtype ecQueF, the conversion of QueF·preQ0 to QueF–preQ0 is irreversible within limits of detection.17 | ||
QueF structures reveal that the cysteine catalytic nucleophile is flanked by an aspartic acid. These two residues form the immediate catalytic center (Fig. 1A and B).18,21,31 A histidine residue is additionally present. In the enzyme from E. coli studied here (ecQueF), the relevant residues are Cys190, Asp197 and His229 and this amino acid numbering is used throughout.11,12,17,27 QM/MM computational analysis of the reaction path of QueF (from Vibrio cholerae, vcQueF) suggested involvement of Asp197 in each catalytic step (Scheme 1B).28 In thioimidate formation, Asp197 would deprotonate Cys190 to subsequently protonate the nitrile nitrogen atom of the preQ0 substrate. In the first and second steps of reduction by NADPH, respectively, Asp197 would stabilize positive charge development on the reactive nitrogen of the protonated thioimidate and imine intermediate. Additionally, upon decomposition of covalent thiohemiaminal to non-covalently bound imine, Asp197 would re-protonate Cys190. In the proposed mechanism,28 His229 had no immediate role and was excluded from involvement in proton transfer. However, the computational study started from the non-covalent complex between the enzyme and preQ0, not from the free enzyme, and it did not include macromolecular dynamics in the calculations. It also did not evaluate whether variation in the protonation states of the active-site residues affects the catalytic reaction. Crystal structures of thioimidate enzyme–preQ0 adducts of vcQueF and QueF from Bacillus subtilis (bsQueF) both reveal a hydrogen bond network connecting the thioimidate N10 via Asp197 Oδ2 and an intermediary water to His229 Nδ1, as shown in Fig. 1A and B.18,21 This suggests the possibility of a proton relay involving His229. We have shown in preliminary experiments that preQ0 binding by ecQueF was accompanied by net proton uptake from solution.17 This finding carries immediate implications for the enzymatic mechanism, but is unaccounted for in the computational reaction path of QueF. In this study, therefore, we used mutagenesis of active-site residues in ecQueF (C190A, C190S, D197A, D197H, and H229A) to study their functional interplay in covalent catalysis for nitrile reduction. We present evidence from kinetic and ligand binding studies that suggests a refined QueF mechanism. In particular, we show that the non-covalent step of preQ0 binding involves proton uptake from His229 via Asp197. Catalytic proton transfer from the Cys190 thiol via Asp197 to the nitrile nitrogen drives the formation of the covalent intermediate. We also show that His229 has an auxiliary role in ecQueF catalysis. The positive charge on His229 could facilitate the polarization of the substrate nitrile for nucleophilic attack on carbon by Cys190. Through proton relay via Asp197, His229 could provide the proton for re-protonation of the cysteine during the formation of the imine intermediate.
 | ||
| Fig. 1 Crystallographic evidence for a possible proton relay involving the conserved Asp and His residues in QueF active sites. (A and B) The covalent thioimidate adducts between preQ0 and the enzymes are shown for vcQueF (A, R262L variant, PDB code: 3S19) and bsQueF (B, wildtype, PDB code: 4F8B). Water molecules are shown as red balls. The full proton relay involving the substrate, Asp, water and His is established. (C) The apo-enzyme of vcQueF (C194A variant, PDB code: 3RJ4) is shown. (D and E) Noncovalent complexes of vcQueF with guanine (D, C194A variant, PDB code: 3BP1) and bsQueF with preQ0 (E, C55A variant, PDB code: 4FGC) are shown. In panel E, a proton relay involving Asp, water and His is suggested. (F) The covalent preQ0–vcQueF complex with NADPH (R262L variant, PDB code: 3UXJ) is shown. The full proton relay may be functional. In panels A–F, active-site residues are indicated and numbered. Note: the ecQueF residues corresponding to the vcQueF residues are Glu89, Cys190, Asp197, Phe228, His229, and Glu230. Elements of the secondary structure are indicated. In bsQueF (panels B and E) the prime is used to indicate secondary structure elements from another protein subunit. Crystal structures of vcQueF and bsQueF are from ref. 18 and 21 and from unpublished data in the database. | ||
Experimental
Chemicals
NADPH (purity >98%) and NADP+ (purity >97%) were from Carl Roth (Karlsruhe, Germany). Materials were of the highest purity available from Carl Roth and Sigma-Aldrich (St. Louis, MO, USA). preQ0 and 7-formyl-7-deazaguanine were synthesized as described previously.10,17Site-directed mutagenesis
Mutagenesis leading to site-directed substitution of Cys190 by Ser (C190S) and Asp197 by Ala (D197A) or His (D197H) was performed according to a standard two-stage PCR protocol.27,32 A pEHISTEV vector including the ecQueF gene (pEHISTEV:EcNRedWT) was used as the template. The oligonucleotide primers are shown with the mismatched bases underlined.C190S forward
5′-CTGCTGAAATCAAAC![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) CTGATCACCCATCAACC-3′
CTGATCACCCATCAACC-3′
C190S reverse
5′-GGTTGATGGGTGATCAG![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) GTTTGATTTCAGC-3′
GTTTGATTTCAGC-3′
D197A forward
5′-CCTGATCACCCATCAACCATGGGGTTCGCTCC-3′
D197A reverse
5′-GGAGCGAACCCCATGGTTGATGGGTGATCAGG-3′
D197H forward
5′-CCTGATCACCCATCAACCATGGGGTTCGCTCC-3′
D197H reverse
5′-GGAGCGAACCCCATGGTTGATGGGTGATCAGG-3′
Mutagenesis to substitute Cys190 by Ala (C190A) and His229 by Ala (H229A) was reported in an earlier study.11,17 All mutations were verified by gene sequencing.
Enzyme preparation
The ecQueF variants were obtained as N-terminally His-tagged proteins using expression in E. coli BL21-DE3 as described previously.17,27 All the enzymes were purified by immobilized metal ion affinity chromatography and gel filtration. The enzyme purity (≥99%) was confirmed by SDS-PAGE. The HisTrap affinity column (GE Healthcare, Buckinghamshire, UK) was regenerated fully after each use. The PD10-desalting columns (GE Healthcare) were always freshly used. Contamination with the protein carried over from previous purification runs was thus ruled out rigorously. The protein concentration was measured with a Pierce BCA protein assay kit (Thermo Fisher Scientific, Germering, Germany). Enzyme stock solutions (0.4–0.8 mM) were stored at −20 °C and used up within 3 weeks.Study of preQ0 binding by ecQueF variants
For isothermal titration calorimetry (ITC), a VP-ITC micro calorimeter from Microcal (Malvern Instruments Ltd., Malvern, UK) was used at 25 °C. The enzyme was gel-filtered twice to phosphate buffer (100 mM Na2HPO4–NaH2PO4, pH 7.5, additionally containing 50 mM KCl) using illustra NAP 5 columns (GE Healthcare). A DMSO concentration of maximally 2% (v/v) was used in both the enzyme and the preQ0 solution. Experiments were conducted and data evaluation done as described previously.17,27 The enzyme molar concentration was based on the protein concentration assuming a functional ecQueF homodimer with a molecular mass of 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 740 Da (C190S variant), 71684 Da (D197A variant), 71816 Da (D197H variant), or 71640 Da (H229A variant).
740 Da (C190S variant), 71684 Da (D197A variant), 71816 Da (D197H variant), or 71640 Da (H229A variant).
Proton uptake or release in conjunction with ligand binding was determined by ITC experiments done in different buffers featuring different ionization enthalpies (ΔHion). The principle of the method is that the change in ligand binding enthalpy (ΔH) is determined in dependence of ΔHion. The slope of the linear relationship between ΔH and ΔHion indicates the number of protons involved. A positive slope indicates proton uptake; a negative slope means proton release. Besides the phosphate buffer described above, HEPES and Tris buffers (each 100 mM, pH 7.5, additionally containing 50 mM KCl) were used, as reported previously.17 The number of protons taken up or released was calculated using the equation, nH+ = (ΔHbuffer 1 − ΔHbuffer 2)/(ΔHion, buffer 1 − ΔHion, buffer 2), where nH+ is the number of protons involved in the binding.33,34 The ΔHion of phosphate, HEPES, and Tris at pH 7.5 and 25 °C was obtained from the literature as 3.60, 20.40, and 47.45 kJ mol−1, respectively.35 The values are averages from three independent sets of experiments.
The covalent thioimidate adduct of ecQueF was detected from its characteristic absorbance with maximum absorption at around 380 nm.17 Absorbance titrations were carried out with a Beckman DU 800 spectrophotometer (Beckman Coulter, Pasadena, CA, USA) as described previously.17,27 preQ0 was titrated to the enzyme solution in the absence of NADPH.
Quenching of the intrinsic Trp fluorescence is a useful reporter of preQ0 binding, as shown previously for bsQueF and ecQueF.17,18 Fluorescence titrations were performed using a fluorescence spectrophotometer F-4500 (Hitachi, Ltd., Tokyo, Japan). Emission spectra were recorded in the range 300–500 nm at 1200 nm min−1 with an excitation wavelength of 280 nm. preQ0 was titrated to the enzyme solution in the absence of NADPH. The quenching yield was determined as the ratio (F0 − FS)/F0, where F0 and FS are the protein fluorescence intensities in the absence and presence of the substrate recorded at the same wavelength of emission.
Protein mass analysis
Samples were prepared in Tris-HCl buffer (100 mM, pH 7.5), additionally containing 50 mM KCl and 1.15 mM tris(2-carboxyethyl)phosphine, as described previously.17 The enzyme and preQ0 concentration was 130 μM and 500 μM, respectively. After the desalting process using Amicon Ultra 0.5 mL centrifugal filters (Merck Millipore, Burlington, MA, USA), a final protein concentration of 30 pmol μL−1 was obtained in water containing 5% acetonitrile and 0.1% trifluoroacetic acid. The samples were separated on a capillary HPLC system (Dionex Ultimate 3000, Thermo Fisher Scientific) and analysed in a maXis II electron transfer dissociation mass spectrometer (Bruker, Bremen, Germany) as described in our earlier study of wildtype ecQueF.17 The captive spray source in positive mode with a mass range of 250–3000 m/z was used. The obtained protein mass spectra (e.g., D197H variant after incubation with preQ0, see Fig. S1 in the ESI†) were deconvoluted by data analysis software, using the MaxEnt2 algorithm.Enzymatic reactions of ecQueF variants
HPLC analytics
The products of preQ0 reduction were analysed at 30 °C using an Agilent 1200 HPLC system (Santa Clara, CA, USA) equipped with a 5 μm SeQuant ZIC-HILIC column (200 Å, 250 × 2.1 mm; Merck, Billerica, MA, USA) and the corresponding guard column (20 × 2.1 mm; Merck), and a UV detector (λ = 254, 262 and 340 nm), as described previously.27Results and discussion
Covalent thioimidate formation
The thioimidate adduct of ecQueF with preQ0 is detectable by absorbance with maximum absorption at 380 nm (ε = 10.02 ± 0.14 mM−1 cm−1).17 In assessing ecQueF variants (D197A, D197H, and H229A) for thioimidate formation, the incompetent C190A variant served as a negative control.11,17 In absorbance titrations wherein preQ0 was used in up to 5-fold molar excess over the enzyme and the wavelength range 300–500 nm was scanned for the absorbance change, no thioimidate adduct was measured for the D197A and D197H variants. The limit of thioimidate detection was ∼0.2% of the enzyme concentration used (50 μM). The H229A variant formed the thioimidate adduct similar to the wildtype enzyme. The C190S variant could potentially form a covalent imidate involving serine as the enzyme nucleophile. From absorbance titrations, no evidence for such an intermediate was obtained.Protein mass analysis confirmed the absorbance data. In wildtype ecQueF, the covalent adduct (35906.0 ± 1.7 Da) was detected besides the unliganded enzyme (35732.1 ± 0.5 Da).17 Only the unliganded enzyme was detected in samples of C190A (35700 ± 2 Da), C190S (35716 ± 2 Da) and D197A variants (35689 ± 2 Da) incubated with preQ0 in 4-fold molar excess over the enzyme subunit present. For the D197H variant, as shown in Fig. 2, mass peaks corresponding to unliganded (35754 ± 1 Da) and preQ0-bound enzymes (35929 ± 1 Da) were detected. In the absence of the thioimidate adduct as demonstrated in absorbance titrations, the observable enzyme complex likely involved preQ0 tightly but noncovalently bound.
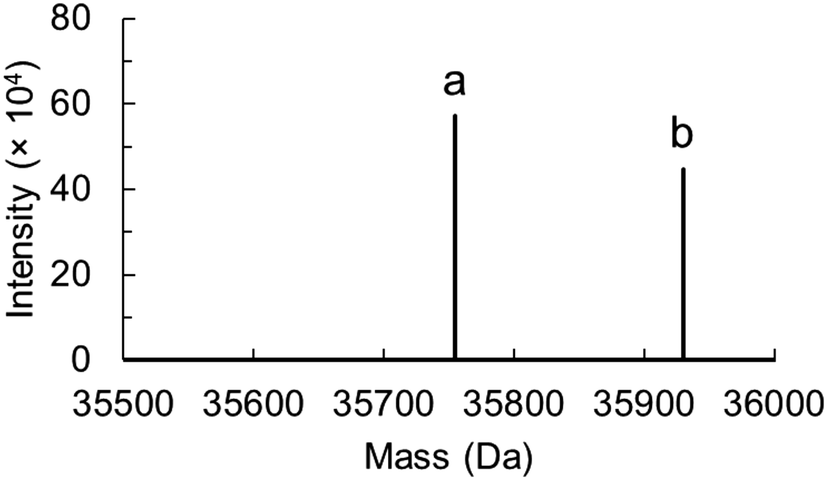 | ||
| Fig. 2 Protein mass analysis for the D197H variant of ecQueF after incubation with preQ0 is shown. Deconvoluted data was obtained from the protein mass spectrum (see Fig. S1 in the ESI†). The sample is shown to contain both the preQ0-free monomeric protein (a, 35754 ± 1 Da) and preQ0-bound monomeric protein (b, 35929 ± 1 Da). | ||
Noncovalent binding of preQ0
This was analysed with fluorescence titration. Binding of preQ0 is traceable by quenching of protein tryptophan fluorescence.17,18 We have shown in an earlier study comparing the wildtype and C190A enzyme that fluorescence quenching arises from the noncovalent step of preQ0 binding (Scheme 2).17 Results for the different ecQueF variants are shown in Fig. 3. The degree of fluorescence quenching at saturating preQ0 was similar in all the enzymes (85–90%). Dissociation constants (Kd) calculated from the data are summarized in Table 1. Among the enzymes not capable of thioimidate formation, the D197H variant showed the highest affinity for preQ0 binding. Its 0.55 μM Kd was on the same order of magnitude as the ∼0.1 μM Kd of the H229A variant which binds preQ0 covalently. The D197A variant showed a 25-fold loss of binding affinity as compared to the D197H variant. The Kd of the wildtype enzyme is extremely low (≤3 nM), reflecting nearly irreversible binding of preQ0 as a covalent adduct with the enzyme.17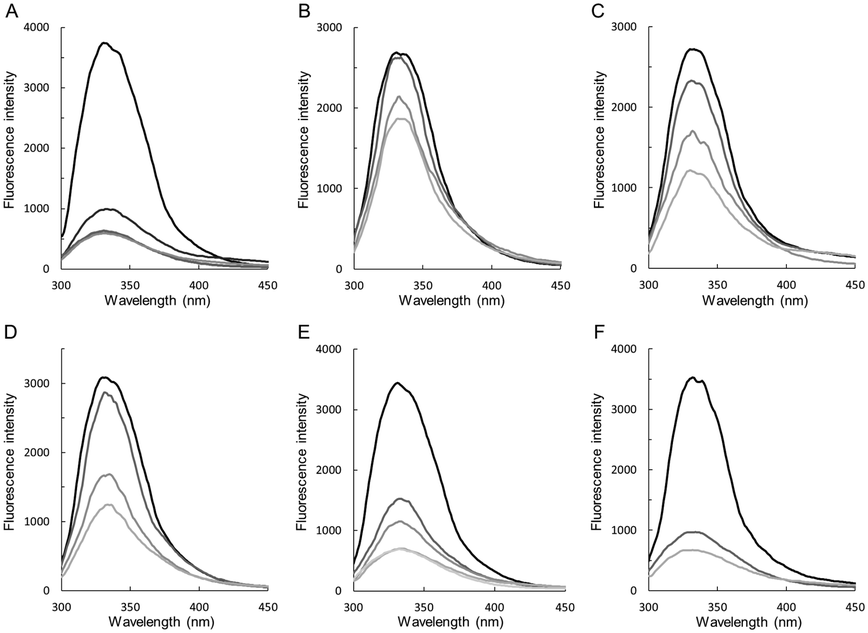 | ||
| Fig. 3 Fluorescence titration analysis of preQ0 binding to wildtype ecQueF and variants thereof. The enzymes used are wildtype ecQueF (A), C190A (B), C190S (C), D197A (D), D197H (E), and H229A variant (F). The enzyme concentration was 3 μM. Protein fluorescence was quenched upon addition of preQ0 (top to bottom: no preQ0; 1.5, 3, 7.5, 15, and 30 μM preQ0). The Kd values obtained from the data are summarized in Table 1. | ||
| K d (μM) | K d app (μM) | ΔHapp (kJ mol−1) | −TΔSapp (kJ mol−1) | ΔGapp (kJ mol−1) | n H+ | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| a Dissociation constants from fluorescence titrations (Fig. 3). b NADPH binding to the noncovalent complex of C190A with preQ0. c NADPH binding to the noncovalent complex D197H with preQ0. | ||||||||
| Wildtype | ≤0.003 | Phosphate | 0.039 | −80.3 | 37.9 | −42.4 | 0.78 ± 0.09 | 17 |
| HEPES | 0.034 | −65.7 | 22.9 | −42.8 | ||||
| Tris | 0.054 | −47.7 | 5.1 | −41.6 | ||||
| C190A | 46.5 | Phosphate | 5.5 (3.6)b | −44.7 (−31.3)b | 14.6 (0.07)b | −30.1 (−31.2)b | 0.46 ± 0.27 | 17 |
| HEPES | 4.26 | −42.3 | 11.5 | −30.8 | ||||
| Tris | 4.05 | −23.2 | −7.7 | −30.9 | ||||
| C190S | 15.3 | Phosphate | 5.3 | −38.9 | 8.7 | −30.2 | 0.31 ± 0.16 | This study |
| HEPES | 4.6 | −36.4 | 5.8 | −30.6 | ||||
| Tris | 3.7 | −23.8 | −7.3 | −31.1 | ||||
| D197A | 13.7 | Phosphate | 3.5 | −44.3 | 13.0 | −31.3 | ≈0 | This study |
| HEPES | 3.8 | −50 | 18.9 | −31.1 | ||||
| Tris | 3 | −42.7 | 11 | −31.7 | ||||
| D197H | 0.55 | Phosphate | 1.3 (1.0)c | −43.9 (−27.0)c | 10.1 (−7.5)c | −33.8 (−34.4)c | 0.61 ± 0.05 | This study |
| HEPES | 1.6 | −33.1 | −0.1 | −33.2 | ||||
| Tris | 1.9 | −17.5 | −15.3 | −32.8 | ||||
| H229A | ≤0.1 | Phosphate | 0.054 | −99.0 | 57.4 | −41.6 | 1.40 ± 0.37 | This study |
| HEPES | 0.127 | −68.9 | 29.4 | −39.5 | ||||
| Tris | 0.159 | −40.2 | 1.2 | −39.0 | ||||
Binding of preQ0 analysed with isothermal titration calorimetry
preQ0 binding to ecQueF is a strongly exergonic process that can be monitored well with ITC.17,27 Of note, a substantial amount (∼50%) of the total heat released during preQ0 binding is due to noncovalent complex formation (Scheme 2).17 ITC data of preQ0 binding to the ecQueF variants in 100 mM phosphate buffer (pH 7.5) are shown in Fig. 4. | ||
| Fig. 4 ITC analysis of preQ0 and NADPH binding to ecQueF variants at 25 °C is shown. Phosphate buffer (100 mM Na2HPO4–NaH2PO4, pH 7.5) containing 50 mM KCl was used. (A–D) C190S (A, 39 μM), D197A (B, 60 μM), D197H (C, 39 μM), and H229A (D, 8 μM) were used. The preQ0 solution (800 μM for C190S, D197A, and D197H; 250 μM for H229A) was titrated into the enzyme solution. The DMSO concentration did not exceed 2% in both the enzyme and the substrate solution. The c values (c = [dimeric protein]/Kd) obtained from the experiments were 7 for C190S, 17 for D197A, 30 for D197H, and 148 for H229A. (E) NADPH (1 mM) was titrated to the noncovalent preQ0–D197H complex (59 μM). The c value obtained from the experiment was 59. Results of fits of the data are summarised in Table 1. | ||
The corresponding thermodynamic parameters are summarized in Table 1. For the ecQueF variants unable to form the thioimidate adduct with preQ0, the Gibbs free energy of binding (ΔG) was consistently lower (ΔΔG = +8–12 kJ mol−1) than the corresponding ΔG for the wildtype enzyme. The ΔΔG for the enzyme variants involved a large decrease in the enthalpy of binding (ΔΔH = +36–41 kJ mol−1), thus rendering preQ0 binding less favorable. However, it also involved significant compensation from the entropy term (−TΔS), which decreased in the variants as compared to the wildtype enzyme. The H229A variant showed a ΔG of binding consistent with that for wildtype ecQueF. However, the ΔH was more negative and the −TΔS more positive than in the case of the wildtype enzyme.
The kinetic study of wildtype ecQueF has shown that NADPH binds to both the covalent and the noncovalent complex of the enzyme with preQ0.17 Binding of NADPH to the preQ0 complex of the C190A variant was previously studied by ITC (Table 1).17 The 3.6 μM Kd of NADPH binding to this complex was comparable to the corresponding Kd in the wildtype enzyme.17 Thermodynamic parameters of NADPH binding to the preQ0 complex of the D197H variant were determined (Fig. 4E and Table 1). They are highly similar to the corresponding parameters of the C190A variant. Substitution of Asp197 with a histidine, therefore, does not interfere with binding of NADPH.
Proton uptake during preQ0 reduction
The enzymatic reaction, preQ0 + 2NADPH + 2H+ → preQ1 + 2 NADP+, implies the uptake of two protons for each nitrile substrate converted into the amine product. Assuming a protonated amine in preQ1 at pH 7.5, a third proton would additionally be taken up in the reaction. Time-resolved analysis of proton consumption during preQ0 reduction by wildtype QueF is shown in Fig. 5. The ratio of steady-state rates of proton uptake and NADPH oxidation was 1.5 (± 0.1), consistent with a proton/preQ1 stoichiometry of 3. | ||
| Fig. 5 Proton uptake (dashed lines) and NADP+ formation (solid lines) during preQ0 reduction by wildtype ecQueF are shown. Reaction conditions: 3.6 μM enzyme; 100 μM NADPH; 20, 30 and 40 μM preQ0 (bottom to top, purple, green, and black). A Tris buffer (0.5 mM, pH 7.51) additionally containing 150 mM KCl and 34 μM phenol red was used. | ||
Proton uptake during preQ0 binding
The ITC study in buffers (phosphate, Tris, HEPES) differing in ionization enthalpy (ΔHion) was used to determine proton uptake/release during preQ0 binding.17 The change of binding enthalpy ΔH upon variation in ΔHion is measured and the number of protons exchanged (nH+) is determined from the slope (positive: proton uptake; negative: proton release) of the linear dependence of ΔH on ΔHion.33,34 Results for ecQueF variants are summarized in Table 1 along with previously reported data for the wildtype enzyme and C190A variant. The D197A variant stood out among all other enzymes in that it did not take up protons in conjunction with preQ0 binding (nH+ ≈ 0). Interestingly, the D197H variant recovered proton uptake to a level almost analogous to that seen with the wildtype enzyme. The two variants of Cys190 showed reduced proton uptake as compared to the wildtype enzyme. In the H229A variant, the preQ0 binding involved proton uptake larger than that in the wildtype enzyme.Enzymatic reduction of preQ0
Conversion of preQ0 into preQ1 was measured directly using HPLC.17,27 As shown recently for certain ecQueF variants, 7-formyl-7-deazaguanine is released during a preQ0 reduction wherein the imine intermediate can hydrolyse due to its incomplete sequestration from the solvent in the enzyme (Scheme 1B).27 Therefore, reactions were also analysed for 7-formyl-7-deazaguanine.Under assay conditions routinely used with the wildtype enzyme17 that involve measurement of NADPH consumption by absorbance at 340 nm, only the H229A variant showed activity. The kcat of the variant was 0.30 (± 0.06) min−1, that is, 24-fold lower than the kcat of the wildtype enzyme (7.2 ± 0.1 min−1).17 Initial rates of the H229A variant showed saturation at low concentrations (≤ 1 μM) of both preQ0 and NADPH. Determination of Km was therefore not pursued. Using 4R-D-NADPH as a coenzyme, the kinetic isotope effect of 2.2 (± 0.2) on the kcat of the H229A variant was determined. This is similar to the kinetic isotope effect of 2.4 on the kcat of the wildtype enzyme.17 When incubated for longer times (96 h) at high enzyme concentration (50 μM), a low level of nitrile reductase activity was confirmed for ecQueF Cys190 and Asp197 variants.
The loss of activity compared to the wildtype enzyme was substantial, about 104-fold for the Cys190 variants (C190A: 0.21 × 10−3 min−1; C190S: 0.25 × 10−3 min−1) and 105-fold for the Asp197 variants (D197A: 0.04 × 10−3 min−1; D197H: 0.10 × 10−3 min−1). Residual activity in these variants is interesting for it implies a reduction of the nitrile group that proceeds in the absence of a covalent ecQueF–preQ0 intermediate. Careful control was therefore necessary to ascertain this activity beyond doubt.
First of all, a close balance between preQ0 consumption and preQ1 formation was demonstrated in all the reactions. 7-Formyl-7-deazaguanine was not released. Non-enzymatic conversion of preQ0 by NADPH was not detected, indicating that catalysis from the ecQueF variants was required for the reaction.
Secondly, to ensure that the activity could not arise from an endogenous QueF contaminating the recombinant enzyme preparations used, we applied the E. coli cell extract to preQ0 conversion. One cell extract was from the expression of the D197A variant, which was the least active among the enzymes analysed here. The other was from an E. coli strain harbouring the empty plasmid vector and treated identically as the positive control. Whereas the cell-extract containing D197A variant showed preQ1 formation, the negative control was completely inactive. We also considered that proteins were purified by affinity via the His tag, and that the His tag was present in the recombinant but absent in the native enzyme.
Therefore, upon applying a purification procedure selective for the His-tagged target protein, it was inconceivable that a contaminating activity not detectable in the cell extract could become enriched to above detection limit in the course of purification.
Finally, interference from translational error, resulting in amino acid mis-incorporation to yield an active enzyme species, was unlikely due to the particular triplet codon changes used for site-directed mutagenesis. All codon changes involved substitutions of the first or second codon base (C190S, TGC → GC; C190A, TGC → C; D197H, GAT → AT, D197A, GAT → G; H229A, CAC → C). Translational misreading is however known to occur chiefly at the third base.38–40 In summary, therefore, these results reinforced the suggestion that Cys190 and Asp197 variants of ecQueF retained a low level of nitrile reductase activity that was intrinsic to their respective active sites.
The pH dependence of preQ0 reduction by the D197A variant was analysed by comparing reaction rates at pH 6.0, 7.5 and 9.0. The residual enzyme activity at pH 6.0 and pH 9.0, compared to the maximum activity at pH 7.5, was 19% and 24%, respectively. In a previous study,24 the wildtype enzyme was shown to exhibit a similar pH dependence of activity in the pH range 6.0–9.0. Therefore, these results suggest that Asp197 is not responsible for the pH dependence of activity of ecQueF.
Catalytic proton transfer coupled to covalent catalysis in ecQueF
QueF crystal structures capture the elementary steps of preQ0 binding.18,21 Upon noncovalent complex formation, the substrate is anchored tightly between the N-terminal ends of two helices (α2 and α5, in vcQueF; α1′ and α2, in bsQueF), as shown in Fig. 1D and E. The reactive nitrile group so becomes oriented towards the active-site loop comprising Cys190 and Asp197. The side chains of Asp197 and His229 adopt relatively flexible conformations that enable a dynamic, water-mediated interaction between the two residues (Fig. 6A). Upon covalent complex formation, the substrate nitrogen atom develops a hydrogen bond with Asp197, which in turn remains linked via a water molecule to His229 (Fig. 1A, B, and F). Biochemical evidence from the current study assigns proton relay function to this active-site network of hydrogen bonds.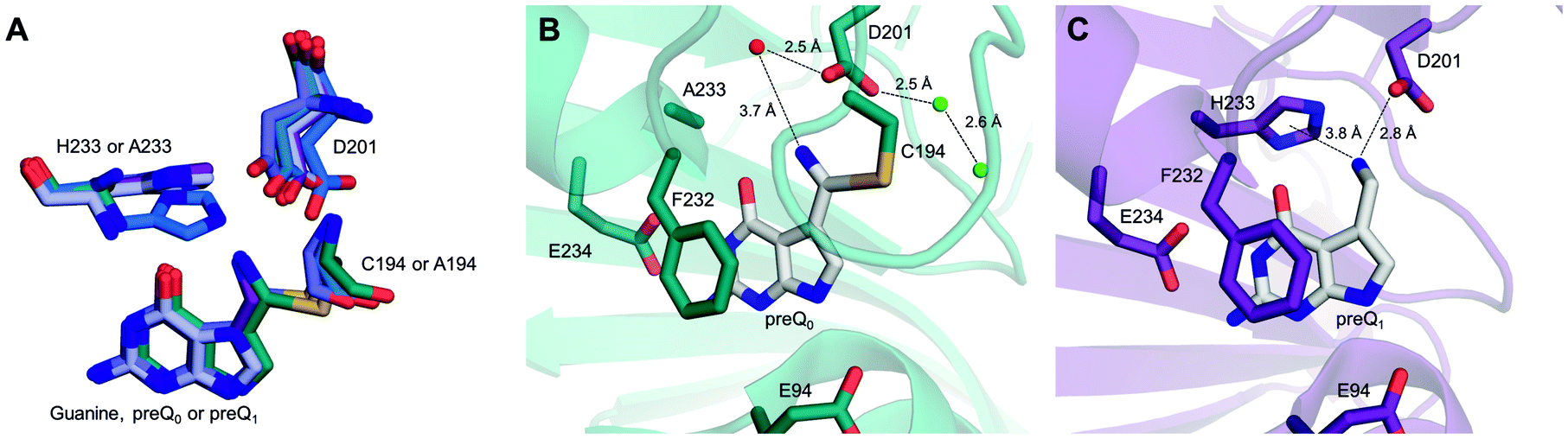 | ||
| Fig. 6 Conformational flexibility of His and Asp residues in the vcQueF active site and binding of the preQ1 product to the enzyme. (A) A structural overlay of vcQueF structures, used in Fig. 1 and 6, is shown: apo-enzyme (blue, C194A variant), holo-enzyme (deep blue, preQ0–R262L variant; light blue, guanine–C194A variant; teal blue, preQ0–H233A; violet, preQ1–C194A variant) and ternary complex structures (purple blue, preQ0–R262L variant–NADPH). (B) The covalent thioimidate complex of the vcQueF H233A variant with preQ0 (PDB code: 4GHM, teal blue) is shown. The structure reveals that Asp201 interacts with water molecules both inside (red sphere) and outside (green sphere) the active site. This could establish a proton relay despite the absence of His. (C) The complex of vcQueF with preQ1 (C194A variant, PDB code: 3RZP, violet) is shown. Crystal structures of vcQueF are from ref. 21 and from unpublished data in the database. | ||
The mechanism in Scheme 3 is proposed. Asp197 is central for proton transfer into and within the ecQueF active site. When preQ0 binds, Asp197 picks up a proton from water to protonate His229. The relative proton amount taken up by the enzyme (∼0.8 protons per active site) reflects the protonation state of the histidine at the pH of 7.5 used. Replacement of Asp197 by an alanine, whose side chain is incompetent in the proton transfer function considered, disrupts completely the proton uptake in conjunction with preQ0 binding. Replacement by a histidine, by contrast, restores the proton uptake to a level (∼0.6 protons per active site) comparable to that of the wildtype enzyme. The extra positive charge developed on the protonated His229 might assist in catalysis to covalent thioimidate formation. It could do so by promoting charge separation in the reactive nitrile group. Nucleophilic attack on carbon and proton transfer to nitrogen would thus be facilitated. Proton uptake by the H229A variant during preQ0 binding was larger than it was in the wildtype enzyme. This probably reflects the protonation of a water molecule in the H229A active site (Scheme 4). Indeed, the covalent complex structure of the His233A variant of Vibrio cholerae QueF reveals a candidate water molecule hydrogen-bonded to the active-site aspartate (Fig. 6B).21 Interestingly, as demonstrated by proton uptake measurements for the C190A and C190S variants of ecQueF, the catalytic cysteine is not essential for proton uptake during preQ0 binding.
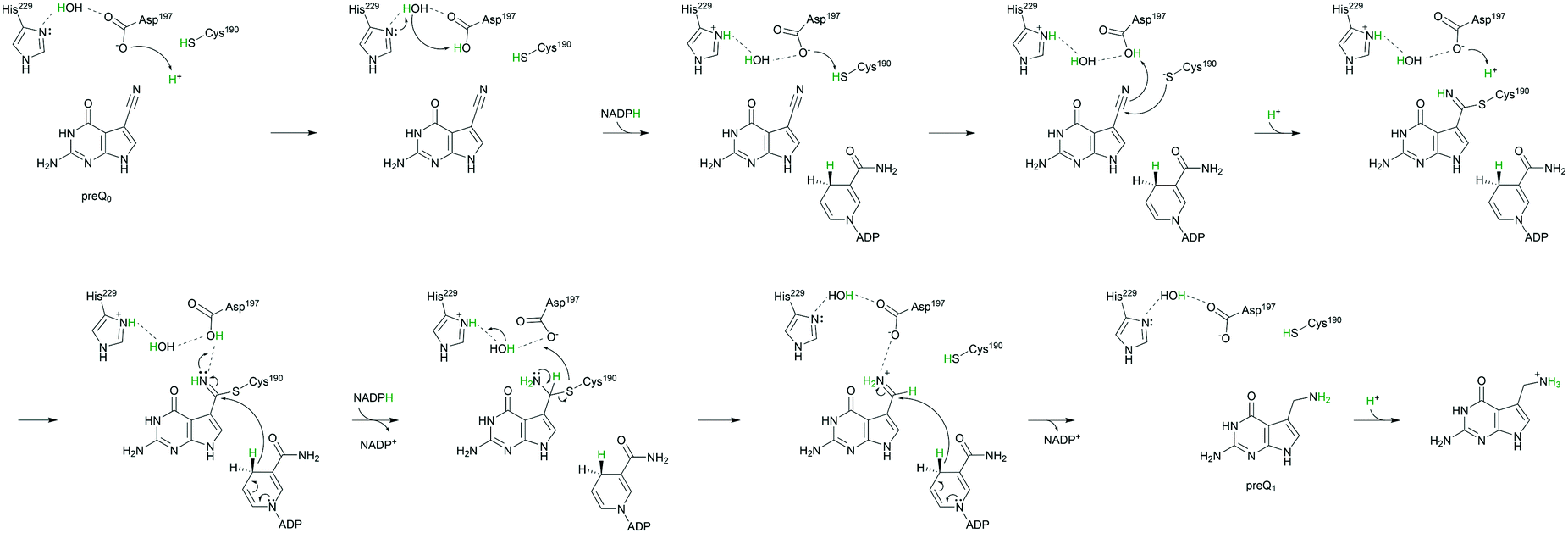 | ||
| Scheme 3 Proposed enzymatic mechanism of reduction of preQ0 to preQ1 showing the proton transfers involved. Amino acid numbering of ecQueF is used. The dashed lines indicate hydrogen bonds. Protons and the NADPH hydride involved in the reaction are marked green. | ||
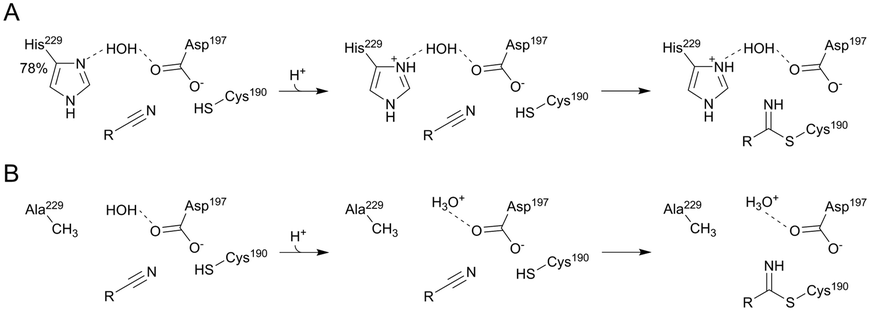 | ||
| Scheme 4 Proton uptake upon covalent thioimidate formation in the ecQueF wildtype (A) and H229A variant (B) is shown at pH 7.5. The wildtype enzyme takes up about 0.78 protons due to the pKa of His229 while the H229A variant takes up 1 proton by water. Amino acid numbering of ecQueF is used. The dashed line indicates hydrogen bonds. | ||
The absolute requirement for Asp197 in covalent thioimidate formation is explained by a twofold role of this residue in catalytic proton transfer. First, Asp197 activates through deprotonation of Cys190 for function as the catalytic nucleophile. Second, Asp197 promotes the covalent thioimidate through catalytic proton transfer to the nitrile nitrogen atom. The nucleophilic attack on carbon probably occurs in concert with the Asp197-mediated proton transfer events, as shown in Scheme 3. Evidence in support of this notion is that the D197A and D197H variants were both completely inactive to form the covalent thioimidate even under conditions (pH ≥ 9.0) in which a cysteine would be expected to become (partially) deprotonated anyway.
As shown in Scheme 3, conversion of the covalent thioimidate to preQ1 involves partial protonation–deprotonation, hence positive charge development, at the reactive nitrogen atom of the substrate. Proton relay via Asp197-water-His229 would help delocalizing the charge and could thus facilitate the hydride reduction from NADPH. Upon breakdown of covalent thiohemiaminal to noncovalently bound imine, Cys190 would become re-protonated via Asp197 in a reversion of the initializing proton transfer event. According to the mechanism proposed (Scheme 3), the preQ1 product would feature an unprotonated amino group and proton uptake by preQ1 probably takes place in solution. The notion is supported by the structure of V. cholerae QueF in complex with preQ1 (Fig. 6C). The structure shows the amino group of the product oriented towards the imidazole ring of His229. The orientation is suggestive of a cation–π interaction between the two groups, assuming the amino group to be protonated. The positioning of preQ1 in the enzyme complex structure thus appears to be unproductive catalytically. Of note, the preQ1 is not detectably oxidized by NADP+ in the presence of ecQueF within a pH range (7.5–9.0) accessible to a stable enzyme.17 The amino group pKa of preQ1 is predicted to be 8.39 (ChemSpider database). It may thus be difficult for the enzyme to bind preQ1 in the presumably active, unprotonated form. From the evidence presented, an auxiliary role of His229 in each catalytic step of the QueF reaction is suggested (Scheme 3). This is consistent with the kcat of the H229A variant being about 24-fold lower than the kcat of the wildtype enzyme.
QueF and nitrilase appear to utilize a highly similar mechanism to promote the covalent thioimidate intermediate (Scheme 1A).9,14,41 In the nitrilase, a triad of cysteine/glutamic acid/lysine constitutes the active-site apparatus.8,16,41 Like Asp197 in QueF, the glutamic acid is central for catalytic nucleophile activation and for acid–base catalysis. The role adopted by the positively charged lysine in nitrilase might be similar to that proposed for the protonated His229 in QueF (Scheme 3).
Enzymatic nitrile reduction in the absence of covalent catalysis
The conversion of preQ0 to preQ1 by the C190A variant implies a nitrile reduction by NADPH that proceeds in the absence of a covalent enzyme intermediate. The Asp197 variants and the C190S variant both could form covalent intermediates with preQ0 in principle, but no evidence in support of such intermediates was found in these enzymes. There is ample precedence for hydride reduction of nitriles to amines in synthetic organic chemistry.42–44 Borane reagents are often used as hydride sources/donors.43 The chemical conversions involve a twofold reduction at the nitrile carbon via the imine intermediate.42,44 The catalytic reactions of the ecQueF variants might proceed analogously, using NADPH as the hydride donor (Scheme 5A). From its one-electron redox potential,45–47 NADPH is able to reduce a nitrile group directly. An alternative possibility, shown in Scheme 5B, is that the reaction proceeds via base-catalysed attack of water on the nitrile group. Such conversion of the nitrile to iminol (amide) is well studied chemically.48 The resulting iminol could then undergo two-fold reduction to the amine as shown. Note that our LC-MS analysis of the conversion of preQ0 excludes the release of an amide (the iminol tautomer) or a carboxylic acid product in the enzymatic reactions. In both scenarios of Scheme 5, the enzyme might provide some catalytic facilitation from a general acid or base. Cys190 and Asp197 are candidates in the corresponding enzyme variants, but no clear assignment is possible on the evidence presented. Our results furthermore show that besides having the ability to position the nitrile substrate for the initial reaction (reduction or hydration–reduction; Scheme 5), the enzyme variants retain imine intermediate sequestration as a characteristic feature of the catalytic function of the ecQueF active site.27 Therefore, each preQ0 reduced by the variants, however slowly, makes it through to the preQ1 product.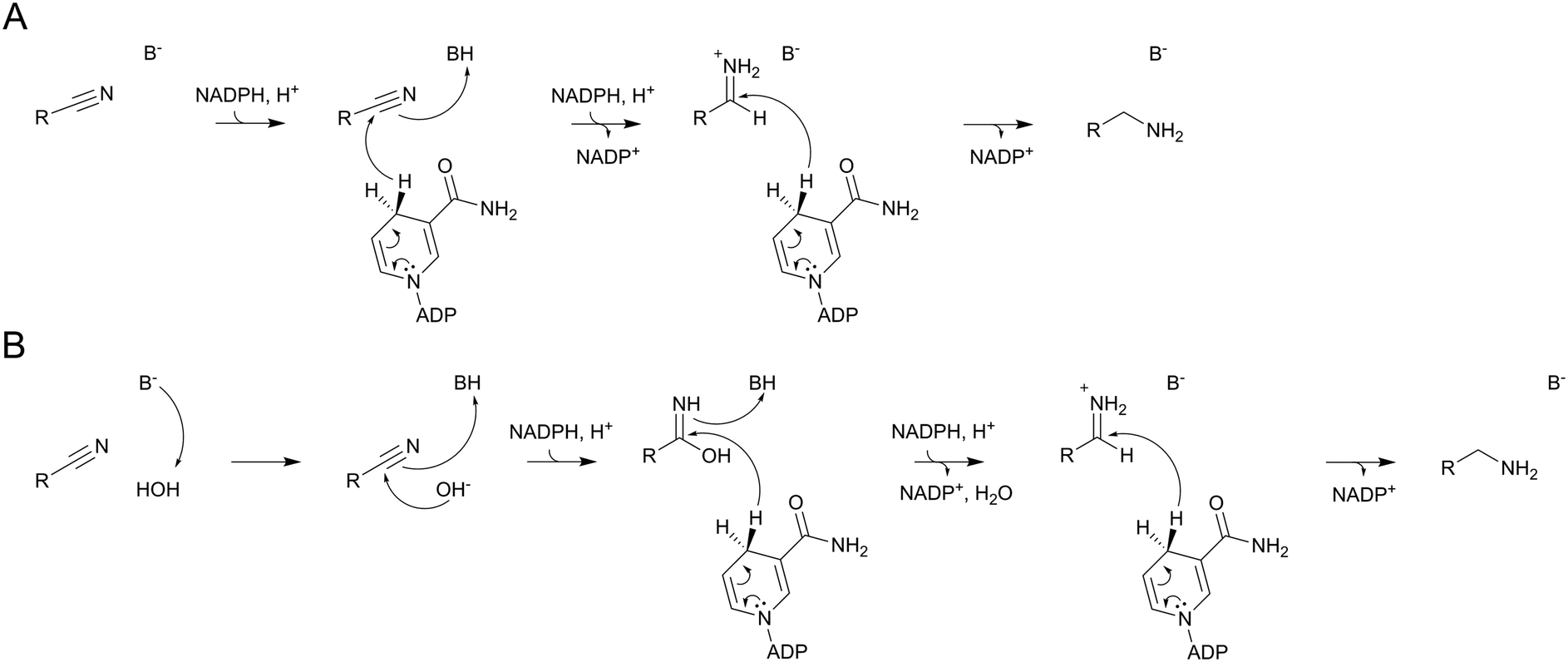 | ||
| Scheme 5 Possible mechanisms of enzymatic nitrile reduction by NADPH in variants of ecQueF unable to form a covalent thioimidate enzyme intermediate. (A) Direct hydride reduction of nitrile. (B) Hydration nitrile followed by reduction. Both reactions may involve facilitation from an unassigned enzyme acid (BH) or base (B−). | ||
Conclusions
We show in this study that proton relay through an active-site network of hydrogen bonds is central for ecQueF to promote nitrile reduction to amine via a covalent thioimidate intermediate. Asp197 is the key residue to manage the interplay between nucleophilic catalysis by Cys190 and the catalytic proton transfers. His229 has an auxiliary role. Deepened insight into the catalytic mechanism of ecQueF was thus obtained. The results have relevance in advancing mechanistic understanding of biological transformations of the nitrile group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Federal Ministry of Economy, Family and Youth (BMWFJ), the Federal Ministry for Transport, Innovation and Technology (BMVIT), the Styrian Business Promotion Agency SFG, the Vienna Business Agency, the Standortagentur Tirol, the Government of Lower Austria, and Styrian Provincial Government Economic Affairs and Innovation through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG. We thank Prof. Norbert Klempier and Dr. Birgit Wilding (Institute of Organic Chemistry, Graz University of Technology) for providing preQ0, and Prof. Ruth Birner-Grünberger and B. Darnhofer (Diagnostic and Research Institute of Pathology & Omics Center Graz, Medical University of Graz) for protein-mass analysis.References
- A. J. M. Howden, C. Jill Harrison and G. M. Preston, Plant J., 2009, 57, 243–253 CrossRef CAS PubMed.
- A. J. M. Howden and G. M. Preston, Microb. Biotechnol., 2009, 2, 441–451 CrossRef CAS PubMed.
- V. M. Luque-Almagro, P. Cabello, L. P. Sáez, A. Olaya-Abril, C. Moreno-Vivián and M. D. Roldán, Appl. Microbiol. Biotechnol., 2018, 102, 1067–1074 CrossRef CAS PubMed.
- P. W. Ramteke, N. G. Maurice, B. Joseph and B. J. Wadher, Biotechnol. Appl. Biochem., 2013, 60, 459–481 CrossRef CAS PubMed.
- J.-S. Gong, J.-S. Shi, Z.-M. Lu, H. Li, Z.-M. Zhou and Z.-H. Xu, Crit. Rev. Biotechnol., 2017, 37, 69–81 CrossRef CAS PubMed.
- C. Brenner, Curr. Opin. Struct. Biol., 2002, 12, 775–782 CrossRef CAS PubMed.
- J.-S. Gong, Z.-M. Lu, H. Li, J.-S. Shi, Z.-M. Zhou and Z.-H. Xu, Microb. Cell Fact., 2012, 11, 142 CrossRef CAS PubMed.
- L. Zhang, B. Yin, C. Wang, S. Jiang, H. Wang, Y. A. Yuan and D. Wei, J. Struct. Biol., 2014, 188, 93–101 CrossRef CAS PubMed.
- S. Jiang, L. Zhang, Z. Yao, B. Gao, H. Wang, X. Mao and D. Wei, Catal. Sci. Technol., 2017, 7, 1122–1128 RSC.
- B. Wilding, M. Winkler, B. Petschacher, R. Kratzer, A. Glieder and N. Klempier, Adv. Synth. Catal., 2012, 354, 2191–2198 CrossRef CAS.
- B. Wilding, M. Winkler, B. Petschacher, R. Kratzer, S. Egger, G. Steinkellner, A. Lyskowski, B. Nidetzky, K. Gruber and N. Klempier, Chem. – Eur. J., 2013, 19, 7007–7012 CrossRef CAS PubMed.
- S. G. Van Lanen, J. S. Reader, M. A. Swairjo, V. de Crécy-Lagard, B. Lee and D. Iwata-Reuyl, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4264–4269 CrossRef CAS PubMed.
- L. Yang, S. L. Koh, P. W. Sutton and Z.-X. Liang, Catal. Sci. Technol., 2014, 4, 2871 RSC.
- B. C. M. Fernandes, C. Mateo, C. Kiziak, A. Chmura, J. Wacker, F. van Rantwijk, A. Stolz and R. A. Sheldon, Adv. Synth. Catal., 2006, 348, 2597–2603 CrossRef CAS.
- H. C. Pace, S. C. Hodawadekar, A. Draganescu, J. Huang, P. Bieganowski, Y. Pekarsky, C. M. Croce and C. Brenner, Curr. Biol., 2000, 10, 907–917 CrossRef CAS PubMed.
- J. E. Raczynska, C. E. Vorgias, G. Antranikian and W. Rypniewski, J. Struct. Biol., 2011, 173, 294–302 CrossRef CAS PubMed.
- J. Jung, T. Czabany, B. Wilding, N. Klempier and B. Nidetzky, J. Biol. Chem., 2016, 291, 25411–25426 CrossRef CAS PubMed.
- V. M. Chikwana, B. Stec, B. W. K. Lee, V. de Crécy-Lagard, D. Iwata-Reuyl and M. A. Swairjo, J. Biol. Chem., 2012, 287, 30560–30570 CrossRef CAS PubMed.
- B. W. K. Lee, S. G. Van Lanen and D. Iwata-Reuyl, Biochemistry, 2007, 46, 12844–12854 CrossRef CAS PubMed.
- L. Gjonaj, M. Pinkse, E. Fernández-Fueyo, F. Hollmann and U. Hanefeld, Catal. Sci. Technol., 2016, 6, 7391–7397 RSC.
- Y. Kim, M. Zhou, S. Moy, J. Morales, M. A. Cunningham and A. Joachimiak, J. Mol. Biol., 2010, 404, 127–137 CrossRef CAS PubMed.
- D. Iwata-Reuyl, Bioorg. Chem., 2003, 31, 24–43 CrossRef CAS PubMed.
- P. Domínguez de María, ChemCatChem, 2011, 3, 1683–1685 CrossRef.
- K. Moeller, G.-S. Nguyen, F. Hollmann and U. Hanefeld, Enzyme Microb. Technol., 2013, 52, 129–133 CrossRef CAS PubMed.
- M. Li, Z. Zhou, Z.-J. Zhang, H.-L. Yu and J.-H. Xu, J. Mol. Catal. B: Enzym., 2016, 131, 47–54 CrossRef CAS.
- Z. Zhou, M. Li, J.-H. Xu and Z.-J. Zhang, ChemBioChem, 2018, 19, 521–526 CrossRef CAS PubMed.
- J. Jung and B. Nidetzky, J. Biol. Chem., 2018, 293, 3720–3733 CrossRef CAS PubMed.
- A. J. M. Ribeiro, L. Yang, M. J. Ramos, P. A. Fernandes, Z.-X. Liang and H. Hirao, ACS Catal., 2015, 5, 3740–3751 CrossRef CAS.
- B. El Yacoubi, M. Bailly and V. de Crécy-Lagard, Annu. Rev. Genet., 2012, 46, 69–95 CrossRef CAS PubMed.
- F. Harada and S. Nishimura, Biochemistry, 1972, 11, 301–308 CrossRef CAS PubMed.
- A. Mohammad, A. Bon Ramos, B. W. K. Lee, S. W. Cohen, M. Kiani, D. Iwata-Reuyl, B. Stec and M. Swairjo, Biomolecules, 2017, 7, 30 CrossRef PubMed.
- W. Wang and B. A. Malcolm, BioTechniques, 1999, 26, 680–682 CrossRef CAS PubMed.
- A. G. Kozlov and T. M. Lohman, Biochemistry, 1999, 38, 7388–7397 CrossRef CAS PubMed.
- S. Leavitt and E. Freire, Curr. Opin. Struct. Biol., 2001, 11, 560–566 CrossRef CAS PubMed.
- R. N. Goldberg, N. Kishore and R. M. Lennen, J. Phys. Chem. Ref. Data, 1999, 31, 231–370 CrossRef.
- S. L. Pival, M. Klimacek and B. Nidetzky, Biochem. J., 2009, 421, 43–49 CrossRef CAS PubMed.
- M. Klimacek, M. Brunsteiner and B. Nidetzky, J. Biol. Chem., 2012, 287, 6655–6667 CrossRef CAS PubMed.
- J. M. Ogle and V. Ramakrishnan, Annu. Rev. Biochem., 2005, 74, 129–177 CrossRef CAS PubMed.
- J. M. Ogle, D. E. Brodersen, W. M. Clemons Jr., M. J. Tarry, A. P. Carter and V. Ramakrishnan, Science, 2001, 292, 897–902 CrossRef CAS PubMed.
- M. Johansson, J. Zhang and M. Ehrenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 131–136 CrossRef CAS PubMed.
- L. Martínková and V. Křen, Curr. Opin. Chem. Biol., 2010, 14, 130–137 CrossRef PubMed.
- D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2015, 357, 883–900 CrossRef CAS.
- D. Haddenham, L. Pasumansky, J. DeSoto, S. Eagon and B. Singaram, J. Org. Chem., 2009, 74, 1964–1970 CrossRef CAS PubMed.
- C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge, K. Junge and M. Beller, Nat. Commun., 2014, 5, 4111 CrossRef CAS PubMed.
- K. Burton and T. H. Wilson, Biochem. J., 1953, 54, 86–94 CrossRef CAS PubMed.
- A. W. Munro, M. A. Noble, L. Robledo, S. N. Daff and S. K. Chapman, Biochemistry, 2001, 40, 1956–1963 CrossRef CAS PubMed.
- T. E. DeCoursey, D. Morgan and V. V. Cherny, Nature, 2003, 422, 531–534 CrossRef CAS PubMed.
- T. E. Schmid, A. Gómez-Herrera, O. Songis, D. Sneddon, A. Révolte, F. Nahra and C. S. J. Cazin, Catal. Sci. Technol., 2015, 5, 2865–2868 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Raw data of protein mass analysis. See DOI: 10.1039/c8cy02331j |
| This journal is © The Royal Society of Chemistry 2019 |