
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitching the substrate preference of fungal aryl-alcohol oxidase: towards stereoselective oxidation of secondary benzyl alcohols†
Ana
Serrano‡
a,
Ferran
Sancho‡
b,
Javier
Viña-González
c,
Juan
Carro
a,
Miguel
Alcalde
c,
Victor
Guallar
bd and
Angel T.
Martínez
 *a
*a
aCentro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, E-28040 Madrid, Spain. E-mail: atmartinez@cib.csic.es
bBarcelona Supercomputing Center, Jordi Girona 31, E-08034 Barcelona, Spain
cDepartment of Biocatalysis, Institute of Catalysis, CSIC, Cantoblanco, E-28049, Madrid, Spain
dICREA, Passeig Lluís Companys 23, E-08010, Barcelona, Spain
First published on 22nd January 2019
Abstract
Oxidation of primary alcohols by aryl-alcohol oxidase (AAO), a flavoenzyme that provides H2O2 to fungal peroxidases for lignin degradation in nature, is achieved by concerted hydroxyl proton transfer and stereoselective hydride abstraction from the pro-R benzylic position. In racemic secondary alcohols, the R-hydrogen abstraction would result in the selective oxidation of the S-enantiomer to the corresponding ketone. This stereoselectivity of AAO may be exploited for enzymatic deracemization of chiral mixtures and isolation of R-enantiomers of industrial interest by switching the enzyme activity from primary to secondary alcohols. A combination of computational simulations and mutagenesis has been used to produce AAO variants with increased activity on secondary alcohols, using the already available F501A variant of Pleurotus eryngii AAO as a starting point. Adaptive-PELE simulations for the diffusion of (S)-1-(p-methoxyphenyl)-ethanol in this variant allowed Ile500 to be identified as one of the key residues with a higher number of contacts with the substrate during its transition from the solvent to the active site. Substitution of Ile500 produced more efficient variants for the oxidation of several secondary alcohols, and the I500M/F501W double variant was able to fully oxidize (after 75 min) with high selectivity (ee >99%) the S-enantiomer of the model secondary aryl-alcohol (±)-1-(p-methoxyphenyl)-ethanol, while the R-enantiomer remained unreacted.
Introduction
There is an increasing interest in the production of pure enantiomers of a variety of chemical compounds for preparation of drugs and fine chemicals.1 The majority of chiral molecules of industrial interest are obtained by kinetic resolution from racemic mixtures in which two enantiomers react at different rates with a chiral catalyst resulting in a sample enantio-enriched with the less reactive isomer.2 For this purpose, the use of biological systems (microorganisms and enzymes) provides an alternative to chemical reagents due to the regio- and enantio-selectivity of many biocatalysts and their mild reaction conditions.3–6For oxidation of secondary alcohols in an enantioselective manner, both dehydrogenases and oxidases have been used.3,4,7 Due to the intrinsic asymmetry of these enzymes, their action on secondary alcohols often results in kinetic resolution with selectivity and enantiomeric excesses (ee) depending on the characteristics of their active sites. Several NAD(P)H-dependent alcohol dehydrogenases have been described for deracemization of secondary alcohols.8,9 However their use implies the need for auxiliary enzymes and stoichiometric amounts of reductants to recycle the NAD(P)H co-substrate. Thus, oxidases, which only need molecular oxygen as an oxidizing agent, are an interesting option for the stereoselective oxidation of secondary alcohols.6 Polyvinyl-alcohol oxidase is described as a secondary alcohol oxidase, although no information on its eventual selectivity is available.10 Other oxidases have been reported to oxidize secondary alcohols,4,6 including cholesterol oxidase,11 glycolate oxidase12 and alditol oxidase.13 Moreover, due to the importance of stereoselective oxidation of these substrates, several studies that widen the specificity of other oxidases on secondary alcohols have been reported in the last few years.14,15
One potential candidate for oxidation of secondary alcohols is fungal aryl-alcohol oxidase from Pleurotus eryngii (AAO, E.C 1.1.3.7), whose biotechnological potential has been demonstrated.16,17 This flavooxidase catalyzes the oxidation of a range of primary alcohols conjugated to an aromatic group (mainly phenyl but also naphthalenyl and furanyl) or even to an aliphatic-polyunsaturated system.18 The AAO catalytic mechanism consists of proton transfer from the hydroxyl group to a catalytic base, His502, taking place in a concerted (but asynchronous) way with hydride abstraction from the benzylic position by the oxidized flavin.19,20 Due to its active-site architecture and the concerted nature of the hydride and proton transfers, hydride abstraction in AAO is stereoselective, taking place only from the pro-R position.21 This stereoselectivity could be exploited for deracemization since it is maintained when secondary benzyl alcohols (with chiral centers) are assayed as substrates, although the AAO activity on these compounds is almost residual due to its narrow active site. In fact, it has been reported that the widening of the active site by substitution of the bulky Phe501 in variant F501A increases the enzyme activity on secondary alcohols, and improves its stereoselectivity.21
In this work, we took advantage of computational simulations to design AAO variants with increased activity on secondary alcohols. These variants were kinetically characterized to evaluate their oxidation efficiency (on primary and secondary benzyl alcohols) and their selectivity for deracemization reactions (Scheme 1) was assessed. A switch in the substrate pattern of the variants was observed and rationalized at the atomic level.
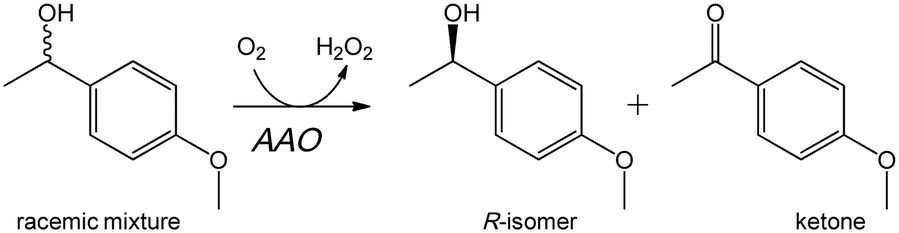 | ||
| Scheme 1 Stereoselective oxidation of a racemic mixture of model secondary benzyl alcohol (1-[p-methoxyphenyl]-ethanol) by AAO leading to kinetic resolution. | ||
Material and methods
Chemicals
p-Methoxybenzyl alcohol, (±)1-(p-methoxyphenyl)-ethanol (racemic mixture), (R)-1-(p-fluorophenyl)-ethanol, (S)-1-(p-fluorophenyl)-ethanol, (±)1-phenylpropanol (racemic mixture), 2-phenyl-2-propanol and horseradish peroxidase (HRP) were purchased from Sigma-Aldrich. (±)2-Methyl-1-phenyl-1-propanol (racemic mixture) was purchased from Alfa Aesar. AmplexRed® was obtained from Invitrogen.Directed mutagenesis, enzyme production and purification
Wild-type recombinant (hereinafter native) AAO from P. eryngii was obtained by expressing the mature AAO cDNA (GenBank AF064069) in Escherichia coli followed by in vitro activation and purification, as previously described.22 Variants were produced by site-directed mutagenesis using the following synthetic primers (bold substituted nucleotides in underlined mutated triplets are indicated below): 5′-GGG TCT AGC TCT GTT CAC![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/b_char_0054_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ATG GTC ATG ATG CG-3′ for Y92F, 5′-GAC AAC GCC AAC ACG
ATG GTC ATG ATG CG-3′ for Y92F, 5′-GAC AAC GCC AAC ACG ![[G with combining low line]](https://www.rsc.org/images/entities/b_char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) TTC CAC CCA GTT GG-3′ for I500A, 5′-GAC AAC GCC AAC ACG
TTC CAC CCA GTT GG-3′ for I500A, 5′-GAC AAC GCC AAC ACG ![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) TTC CAC CCA GTT GG-3′ for I500M, 5′-GAC AAC GCC AAC ACG TTC CAC CCA GTT GG-3′ for I500W, 5′-C AAC GCC AAC ACG ATT CAC CCA GTT GGA ACG-3′ for F501A and 5′-GAC AAC GCC AAC ACG ATT CAC CCA GTT GG-3′ for F501W, using the plasmid containing the native AAO sequence as template. The double mutations were obtained using the I500A plasmid as a template and the primers 5′-C CTA TCC GAC CAT TTG CTT CCT GCT GCC TTC TTC G-3′ for L315A/I500A and 5′-GCT CAT TGG GAG ACC TTT TCC AAC CAA TGG-3′ for I391A/I500A and the F501W plasmid as a template and the primer 5′-GAC AAC GCC AAC ACG CAC CCA GTT GG-3′ for I500M/F501W including both mutations. Mutations were confirmed by gene sequencing, the variants were purified to electrophoretic homogeneity following the same protocol as for the native protein,22 and their electronic absorption spectra were recorded. Enzymes were quantified with a Cary-4000 spectrophotometer using extinction coefficients (Table S1†) taken from the literature or calculated in the present work by heat denaturation,23 using ε450 = 11
TTC CAC CCA GTT GG-3′ for I500M, 5′-GAC AAC GCC AAC ACG TTC CAC CCA GTT GG-3′ for I500W, 5′-C AAC GCC AAC ACG ATT CAC CCA GTT GGA ACG-3′ for F501A and 5′-GAC AAC GCC AAC ACG ATT CAC CCA GTT GG-3′ for F501W, using the plasmid containing the native AAO sequence as template. The double mutations were obtained using the I500A plasmid as a template and the primers 5′-C CTA TCC GAC CAT TTG CTT CCT GCT GCC TTC TTC G-3′ for L315A/I500A and 5′-GCT CAT TGG GAG ACC TTT TCC AAC CAA TGG-3′ for I391A/I500A and the F501W plasmid as a template and the primer 5′-GAC AAC GCC AAC ACG CAC CCA GTT GG-3′ for I500M/F501W including both mutations. Mutations were confirmed by gene sequencing, the variants were purified to electrophoretic homogeneity following the same protocol as for the native protein,22 and their electronic absorption spectra were recorded. Enzymes were quantified with a Cary-4000 spectrophotometer using extinction coefficients (Table S1†) taken from the literature or calculated in the present work by heat denaturation,23 using ε450 = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 M−1 cm−1 for the free FAD.24
300 M−1 cm−1 for the free FAD.24
Steady-state kinetics for alcohol oxidation
The kinetic parameters for oxidation of primary p-methoxybenzyl alcohol were calculated by following spectrophotometrically the oxidation initial rate (1 min reaction) of the alcohol to the corresponding aldehyde using the difference molar absorptivity (Δε285 = 16950 M−1 cm−1).25
The kinetic parameters for oxidation of the secondary alcohol (±)1-(p-methoxy-phenyl)-ethanol were calculated by monitoring the production of H2O2 in the HRP-coupled assay with AmplexRed (Δε563 = 52000 M−1 cm−1) as an alternative to the spectrophotometric estimation of the ketone product.
Measurements were performed at 25 °C in air-saturated (0.256 mM O2 concentration)26 50 mM phosphate, pH 6.0. Kinetic parameters were determined by fitting the initial reaction rates at different alcohol concentrations to the Michaelis–Menten equation (eqn (1)):
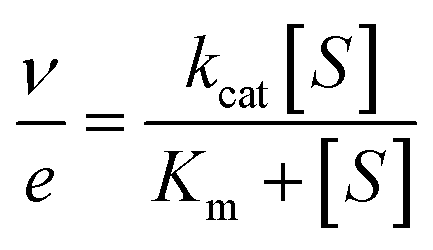 | (1) |
In the case of (±)1-(p-methoxy-phenyl)-ethanol, the enantiomer concentration was considered to be 50% of the racemic mixture concentration, and an apparent turnover number (appkcat) was estimated in the presence of both enantiomers.
Analysis of stereoselective reactions
The time course of mid-term (up to 75 min) and long-term (up to 24 or 72 h) reactions of native AAO and variants with (±)1-(p-methoxyphenyl)-ethanol, (±)1-phenyl-1-propanol and (±)2-methyl-1-phenyl propanol was followed at 25 °C in air saturated 50 mM phosphate, pH 6.0, under continuous shaking. The samples were taken at different times and, after addition of an internal standard (Table S2†), were liquid–liquid extracted with hexane, and analyzed by chiral HPLC.The enantiomers were separated in a Chiralcel IB column (4.6 × 250 mm, 5 μm; Daicel Chemical Industries, Ltd.) using a pre-column of the same material, and 98:2 (v/v) n-hexane:isopropanol (98:1 for 2-methyl-1-phenyl propanol) as a mobile phase (at 1 mL min−1 and 25 °C). Detection was performed at 206 nm (see spectra of the different compounds in Fig. S1†). The retention times for the corresponding R and S enantiomers (Table S2†) were obtained from the racemic standards assuming the published elution order.21 Quantification was performed using internal standards and calibration curves (Fig. S2†).
The ee was calculated using eqn (2):
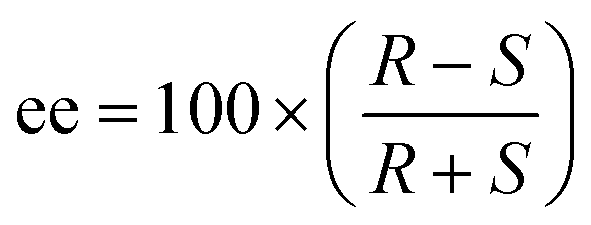 | (2) |
Enantiomeric ratios (E-values)27 were calculated from eqn (3):
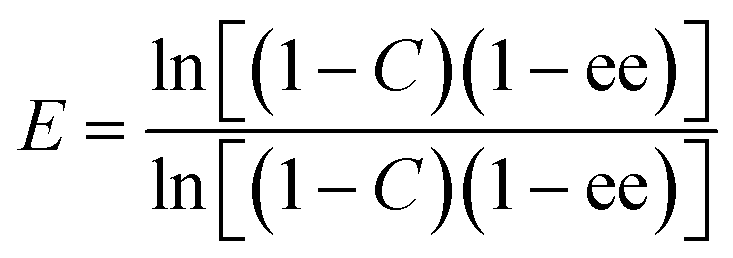 | (3) |
The oxidation of (R)- and (S)-1-(p-fluorophenyl)-ethanol to p-fluoroacetophenone by native AAO and variants was evaluated in air-saturated 50 mM phosphate, pH 6.0, at 25 °C, after long-term incubations (up to 48 h). The amount of p-fluoroacetophenone formed was calculated using the difference in the molar absorbance coefficients of the alcohol and the ketone at 248 nm (εketone − εalcohol, Δε248 = 12606 M−1 cm−1) (Fig. S3†).
Computational analysis
The new adaptive-PELE (protein energy landscape exploration) software28 was used to study (R)- and (S)-1-(p-methoxyphenyl)-ethanol diffusion and binding on native AAO and four variants. The adaptive protocol improves sampling in PELE by running multiple short simulations (epochs) where the initial conditions in each of them are selected through a reward function aiming at sampling non-visited areas. Briefly, PELE uses a Monte Carlo (MC) procedure including protein structure prediction algorithms and a ligand rotamer library for sampling enhancement. Each MC iteration includes three main steps: 1) ligand and protein (backbone) perturbation; 2) side chain sampling; 3) overall minimization. For this study, ligand perturbation involved the [0.5–1.5] translation and [0.05–0.1] rotation ranges, in Å and rad, respectively. Backbone flexibility was allowed using the lowest 6 modes in an anisotropic network model,29 while all side chains within 6 Å of the ligand were predicted on each step. The ligand was allowed to move enforcing (its center of mass) a 5 Å radius sphere center on the FAD N5 coordinates. Each simulation involved 192 trajectories with 40 epochs and 20 MC PELE iterations per epoch. To improve the sampling towards the FAD cofactor, we used an epsilon value of 0.1, meaning that 10% of the processors started each epoch from the best ligand-FAD distance previously sampled. More details and examples on running PELE can be found elsewhere.30PELE uses an all atom OPLS2005 force field,31 with an implicit generalized born model. All ligands and FAD charges, however, were extracted from quantum mechanical (QM) calculations. The FAD cofactor (in its quinone state) was optimized with mixed quantum mechanics/molecular mechanics (QM/MM) calculations at the M06(6-31G*)/OPLS2005 level of theory using Qsite.32 The initial model was derived from the 3FIM crystal structure, solvated with an 8 Å layer of water molecules and prepared with Maestros's protein wizard.33 Only the FAD, which does not present any covalent interaction with the rest of the protein, was included in the QM region. Ligands were optimized at the same QM level of theory with an implicit PBF solvent using Jaguar.34 Ligands were then parameterized in accordance to OPLS2005, keeping the electrostatic QM charges and a rotamer library was built with Macromodel.33
Results and discussion
Although the activity of AAO on secondary alcohols is nearly residual, a variant with increased activity was obtained by substituting the bulky Phe501 by an alanine.21 Taking this variant as a starting point, we combined computational simulations and site-directed mutagenesis to obtain new variants with higher activity on secondary alcohols of interest in deracemization reactions.Computational approach for AAO engineering
To identify mutations along the access path to the AAO active site (Fig. 1A) that could improve the secondary alcohol access and binding, PELE simulations of (S)-1-(p-methoxyphenyl)-ethanol diffusion in the F501A variant (molecular structure from in silico mutation of PDB 3FIM) were carried out. The goal was to identify residues at the active-site access channel potentially-involved in the substrate diffusion and oxidation. The strategy included exploring the energy profiles for the entrance of this secondary alcohol and determining which amino acids can be limiting the ligand's access or constraining it in an incorrect orientation for oxidation at the active site.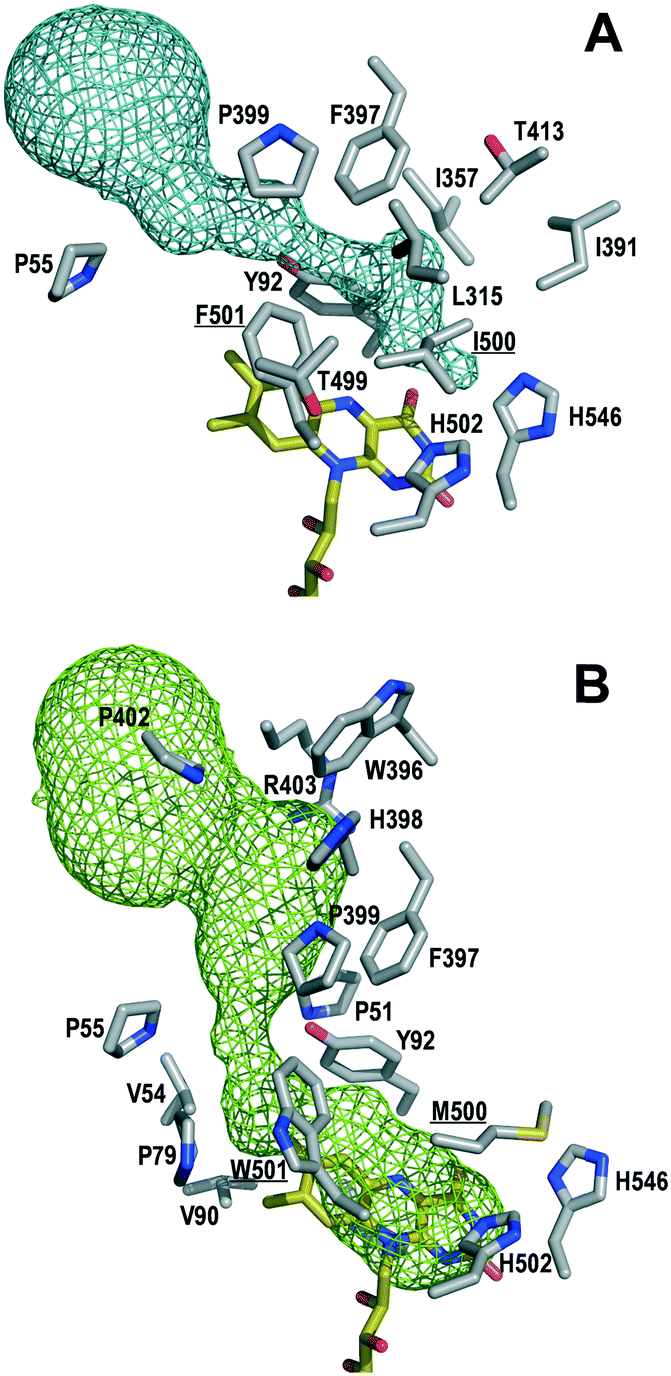 | ||
| Fig. 1 Detail of the access channel and active site cavity (around flavin N5) in native AAO (A) and its regioselective I500M/F501W variant (B), including FAD (yellow-C CPK-colored sticks) and residues at less than 4 Å from the channel (side chains as gray-C CPK-colored sticks; mutated residues underlined). | ||
During the ligand transition from the solvent to the active site, several residues showed significantly higher number of interaction contacts with the alcohol (Fig. 2) being the main direct obstacles that the substrate has to bypass. Among them, Tyr92 and Phe397 together with Phe501 in native AAO constitute a hydrophobic bottleneck (Fig. 1A) for the access of substrates, being also involved in their stabilization at the active site.35–38 According to these computational data, residues with contact numbers higher than 35000—such as Pro79, Tyr92, Leu315, Ile391, Phe397, Pro399 and Ile500, together with previously mutated Ala501, all of them located at less than 4 Å of the active-site channel—were selected for site-directed mutagenesis. In vitro folding of the mutated proteins was performed after replacing the above residues with alanines, and alternative mutations were introduced in several cases.
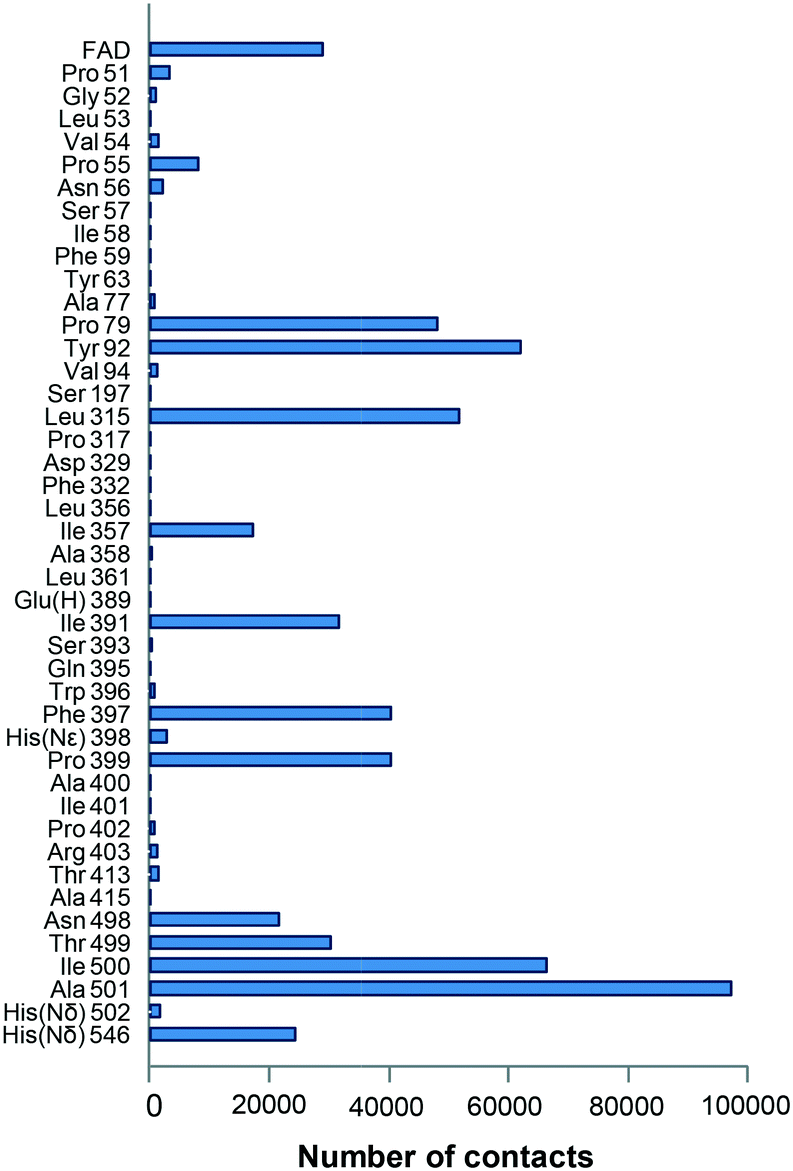 | ||
| Fig. 2 Residues (up to a total of 43 plus the FAD cofactor) interacting with the model secondary alcohol (S)-1-(p-methoxyphenyl)-ethanol during PELE simulations (on the F501A variant). Interactions were selected considering contact distances below 2.7 Å between the ligand and enzyme atoms. See Fig. 1 for the location of the main residues. Glu(H) indicates a protonated glutamate, and His(Nε) and His(Nδ) indicate histidines protonated in Nε and Nδ, respectively. | ||
Proper incorporation of the FAD cofactor was shown by the presence of typical bands I and II in their electronic absorption spectra (with only slight displacements due to mutations in the flavin environment; Table S1†). The ability of these variants to oxidize secondary alcohols was tested by incubating them with (±)1-(p-methoxyphenyl)-ethanol, as a model chiral substrate, and analyzing the resulting ketone and the remaining R and/or S substrate enantiomers (Fig. S2†). Quantification of both isomers by chiral HPLC after 24 h of reaction indicated that, with the exception of I500W, the variants oxidized (S)-1-(p-methoxyphenyl)-ethanol to the corresponding ketone with different rates, without any activity on (R)-1-(p-methoxyphenyl)-ethanol. I500A showed 15-times higher activity than the native enzyme, with 50% conversion of the racemic substrate in the first 4 h of reaction (Fig. 3 and Table S3†) due to the almost total oxidation of the S-enantiomer.
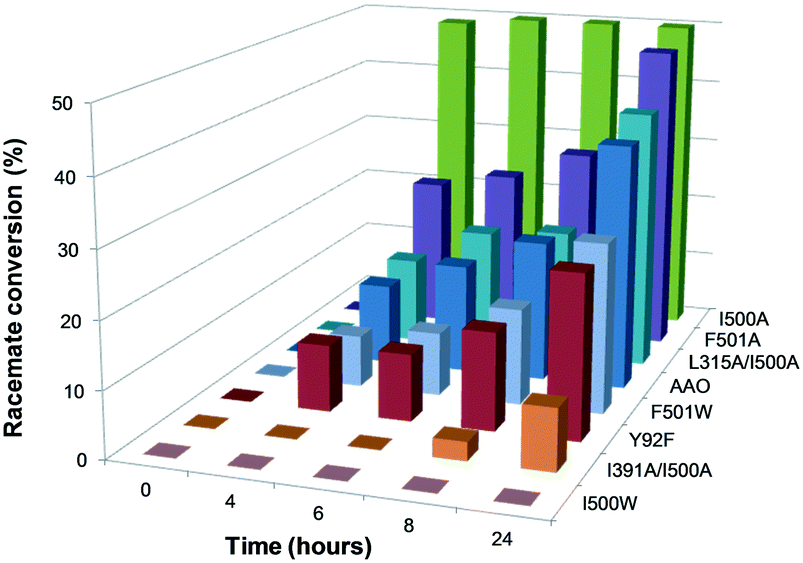 | ||
| Fig. 3 Conversion of (±)1-(p-methoxyphenyl)-ethanol during 24 h reaction with native AAO and seven variants. The reactions between the alcohol (2.5 mM racemic mixture) and the enzyme (5 μM) were performed in 50 mM phosphate, pH 6.0, at 25 °C, and analyzed by HPLC. | ||
Rationalizing the effect of the I500A mutation
PELE simulations for (S)-1-(p-methoxyphenyl)-ethanol diffusion and positioning at the active site of the I500A variant, compared to native AAO, contributed to explaining its higher activity on secondary alcohols mentioned above.In the case of native AAO, PELE diffusion leads to two main locations of the alcohol in the active site, in which its hydroxyl group is located near the catalytic His502 at an adequate distance (∼2.5 Å) for the proton abstraction to be produced (Fig. 4A). These two structures differ in the position of the benzylic hydrogen (in the R position) with regard to the flavin (hydrogen–FAD distances of 2.5 Å and 5.3 Å) only the first one being compatible with hydride transfer to flavin N5 and ketone formation.
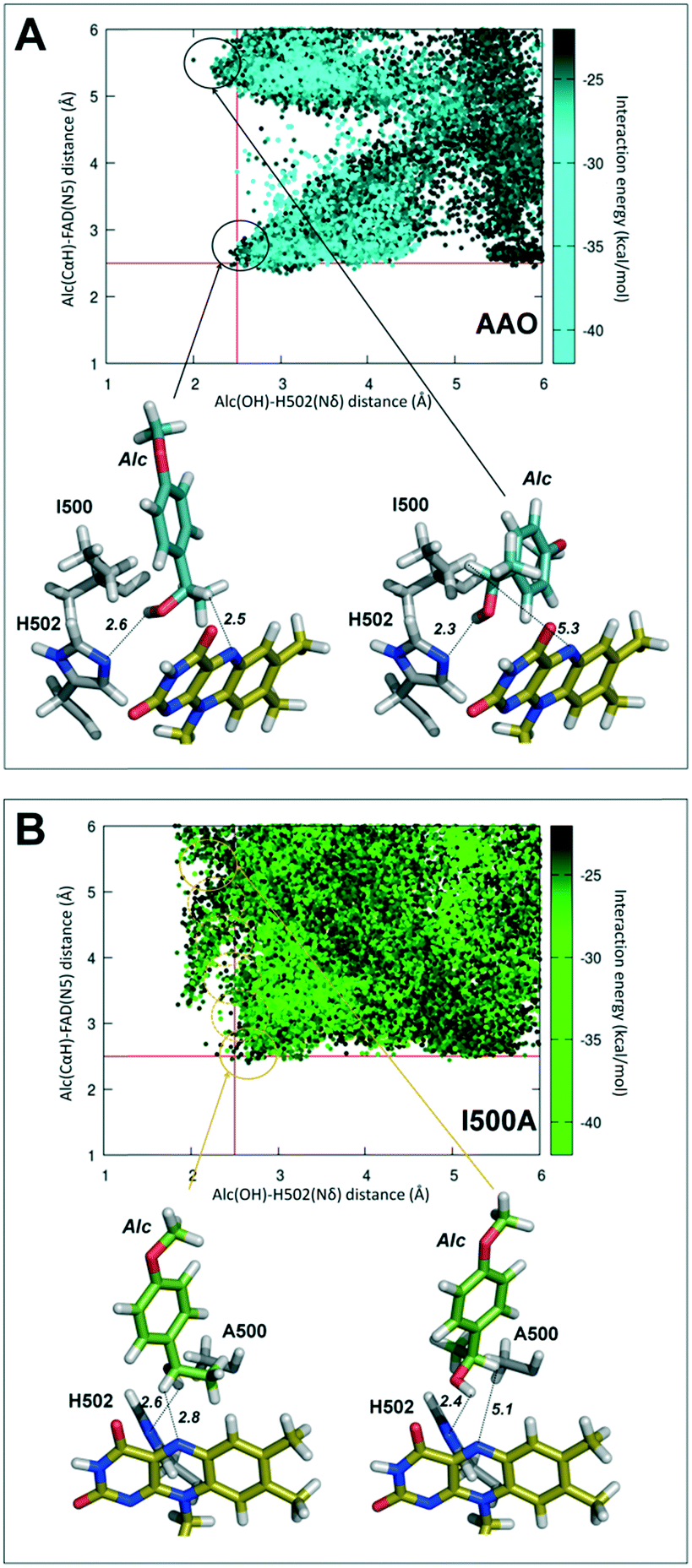 | ||
| Fig. 4 PELE diffusion of (S)-1-(p-methoxyphenyl)-ethanol (Alc) on native AAO (A) and its I500A variant (B) including: i) plot of the PELE results (top) with each position represented as a function of the distances between the hydroxyl hydrogen and the Nδ of His502 (axis-x) and the benzylic hydrogen (Cα) and the flavin N5 (axis-y); and ii) two selected substrate poses (bottom) for each simulation, including the substrate, flavin ring, catalytic His502 and the residue at the mutated position. Each point is colored according to the interaction energy scale. | ||
By contrast, alcohol diffusion in I500A showed that in this variant the ligand is able to move more freely in the catalytic site (due to its increased size) with a range of positions (circles in Fig. 4B, top) at 2.0–2.5 Å from His502 and progressively shorter distances of the FAD, to finally attain a position compatible with catalysis (Fig. 4B, bottom, left).
Combinatorial saturation mutagenesis of AAO expressed in Saccharomyces cerevisiae,39 taking the above results into account, yielded the I500M/F501W variant with a noticeable increase in the oxidation of 1-(p-methoxyphenyl)-ethanol.40
Alcohol oxidation by I500 and Phe501 variants
Therefore, to get insights into the AAO stereoselective oxidation of secondary benzyl alcohols, two additional variants (I500M and I500M/F501W) were expressed in E. coli, in vitro activated and purified to homogeneity, showing correct folding and FAD incorporation (with only slight displacements in bands I and II due to mutations in the flavin environment; Table S1†).We first evaluated the effect of four selected single and double substitutions of Ile500 and Phe501 on the AAO activity towards its preferred substrate, p-methoxybenzyl alcohol, by measuring the release of anisaldehyde (Table 1, top). Aldehyde estimation has been confirmed to provide similar kinetic constants to those obtained when the equimolecular H2O2 release is followed.36 The variants showed lower turnover numbers (kcat) but increased affinity, as shown by lower Km values, with regard to the native enzyme. Thus, the I500M/F501W and I500M mutations resulted in 1.5-, and 2.5-fold more efficient oxidation of the primary alcohol, respectively.
| p-Methoxybenzyl alcohol | |||
|---|---|---|---|
| k cat (s−1) | K m (μM) | k cat/Km (mM−1 s−1) | |
| AAO | 142 ± 5 | 27 ± 4 | 5230 ± 620 |
| F501A | 3.1 ± 0.03 | 12.3 ± 0.6 | 251 ± 12 |
| I500A | 1.5 ± 0.1 | 1.3 ± 0.1 | 1140 ± 45 |
| I500M | 5.3 ± 0.3 | 0.4 ± 0.1 | 13100 ± 200 |
| I500M/F501W | 3.3 ± 0.1 | 0.4 ± 0.03 | 7930 ± 640 |
| (±)1-(p-Methoxyphenyl)-ethanol | |||
|---|---|---|---|
| app k cat (s−1)b | K m (mM)c | app k cat/Km (mM−1 s−1) | |
| a Determined at 25 °C in air-saturated (0.256 mM O2) 50 mM phosphate, pH 6.0. b Apparent kcat estimated in the racemic mixture. c Referring to the S-enantiomer, representing 50% of the racemic mixture. | |||
| AAO | 0.18 ± 0.002 | 25.0 ± 0.6 | 0.0072 ± 0.0002 |
| F501A | 0.05 ± 0.003 | 10.0 ± 1.5 | 0.0051 ± 0.0008 |
| I500A | 0.22 ± 0.01 | 2.9 ± 0.3 | 0.079 ± 0.009 |
| I500M | 0.42 ± 0.01 | 1.4 ± 0.1 | 0.30 ± 0.02 |
| I500M/F501W | 2.2 ± 0.04 | 3.1 ± 0.2 | 0.71 ± 0.05 |
Then, kinetic measurements of (±)1-(p-methoxyphenyl)-ethanol oxidation were followed by H2O2 release (Table 1, bottom). Increased affinity for the secondary alcohol was reflected in the lower Km values that the variants showed. As a consequence, I500A, I500M and I500M/F501W present 11-, 42- and 97-fold higher catalytic efficiencies (kcat/Km) than native AAO, respectively, in agreement with the results from 24 h experiments (Table S3†). Comparison of the action of the variants on secondary and primary alcohols was based on their turnover (kcat) values, since in deracemization reactions in an industrial context substrates are used under saturated conditions and the rates are therefore independent of the Km value.41 Interestingly, the substitution of Ile500 produced an increase of the activity on the model secondary alcohol at the expense of reducing its activity on the primary p-methoxybenzyl alcohol, as shown by kinetic comparison (up to 500-fold higher secondary/primary ratio for I500M/F501W) (Table 2). Therefore, the more active a variant is on secondary alcohols, the less active it is on primary alcohols, revealing an interesting switch in the substrate preference of AAO. The higher activity of the double variant agrees with a broader active site (Fig. 1B) enabling entering and adequate positioning of the bulkier secondary alcohol.
| Secondary (sec; s−1) | Primary (pri; s−1) | Relative sec/pri ratio | |
|---|---|---|---|
| AAO | 0.18 | 142.0 | 1 |
| F501A | 0.05 | 3.1 | 13 |
| I500A | 0.22 | 1.5 | 116 |
| I500M | 0.42 | 5.3 | 63 |
| I500M/F501W | 2.2 | 3.3 | 526 |
Stereoselective oxidation of (±)1-(p-methoxyphenyl)-ethanol
The oxidation of the model secondary aryl alcohol by native AAO and its variants was monitored in time-course reactions using chiral HPLC. Reactions were performed at 25 °C under continuous shaking and aliquots were taken every 15 min for 2 h (Fig. S4†). As shown in Fig. 5, only 3% conversion (with 2% ee) was found after 2 h incubation with native AAO, and the results were only slightly improved with the F501A variant.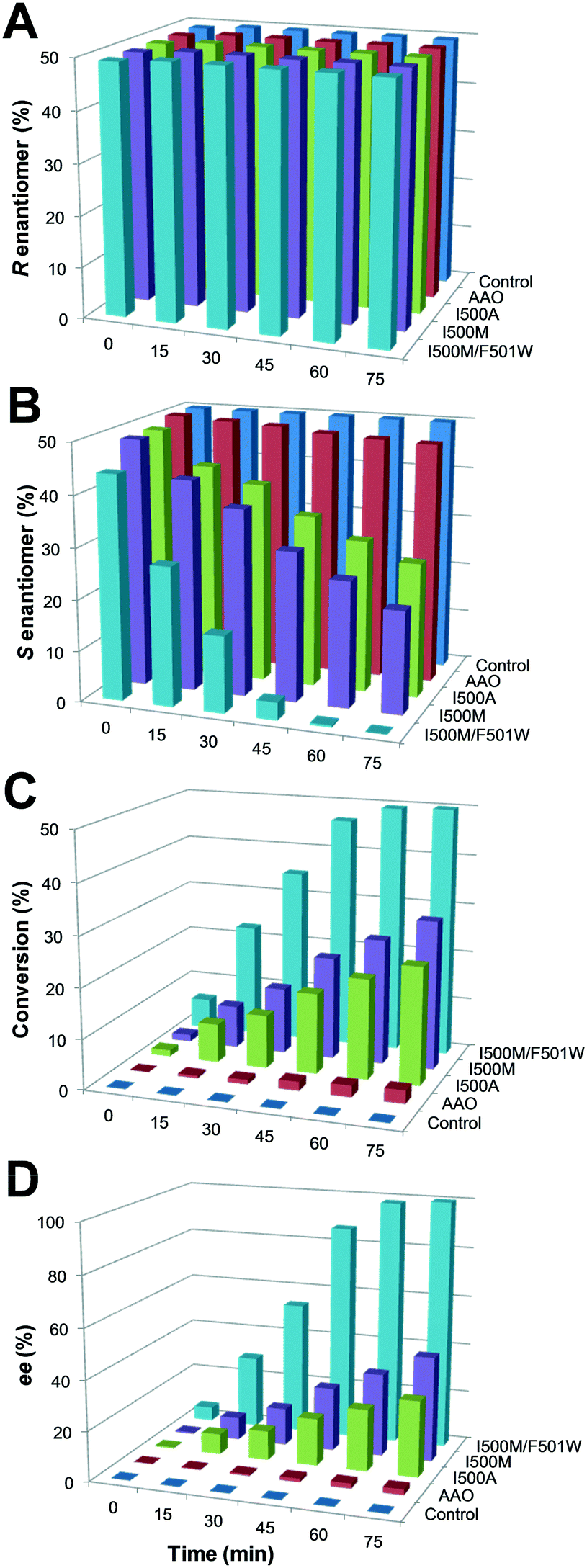 | ||
| Fig. 5 (±)1-(p-Methoxyphenyl)-ethanol oxidation to the corresponding ketone for 75 min by three selected AAO variants compared with the native enzyme (and control without enzyme): A) remaining R-enantiomer; B) remaining S-enantiomer; C) conversion yield (racemic mixture); and D) R-isomer ee. Reactions between alcohol (2.5 mM racemic mixture) and enzyme (2.5 μM) were performed in 50 mM phosphate, pH 6.0, at 25 °C, and the remaining substrates and product were analyzed by chiral HPLC. | ||
However, other variants were considerably more active on the secondary alcohol, with up to 18- and 66-fold higher conversion rates in the cases of I500M and I500M/F501W, respectively (Fig. 5B and C). The substitution of Ile500 by either methionine or alanine considerably increases both conversion and ee with regard to native AAO. After 75 min, the conversion reached 24% and 30% for I500A and I500M (with ee of 30% and 42%), respectively. These values increased up to 50% conversion of the total racemic mixture (with ee 100%) for I500M/F501W, indicating a high enantioselectivity of this double variant (Fig. 5C and D, 6A and Table 3).
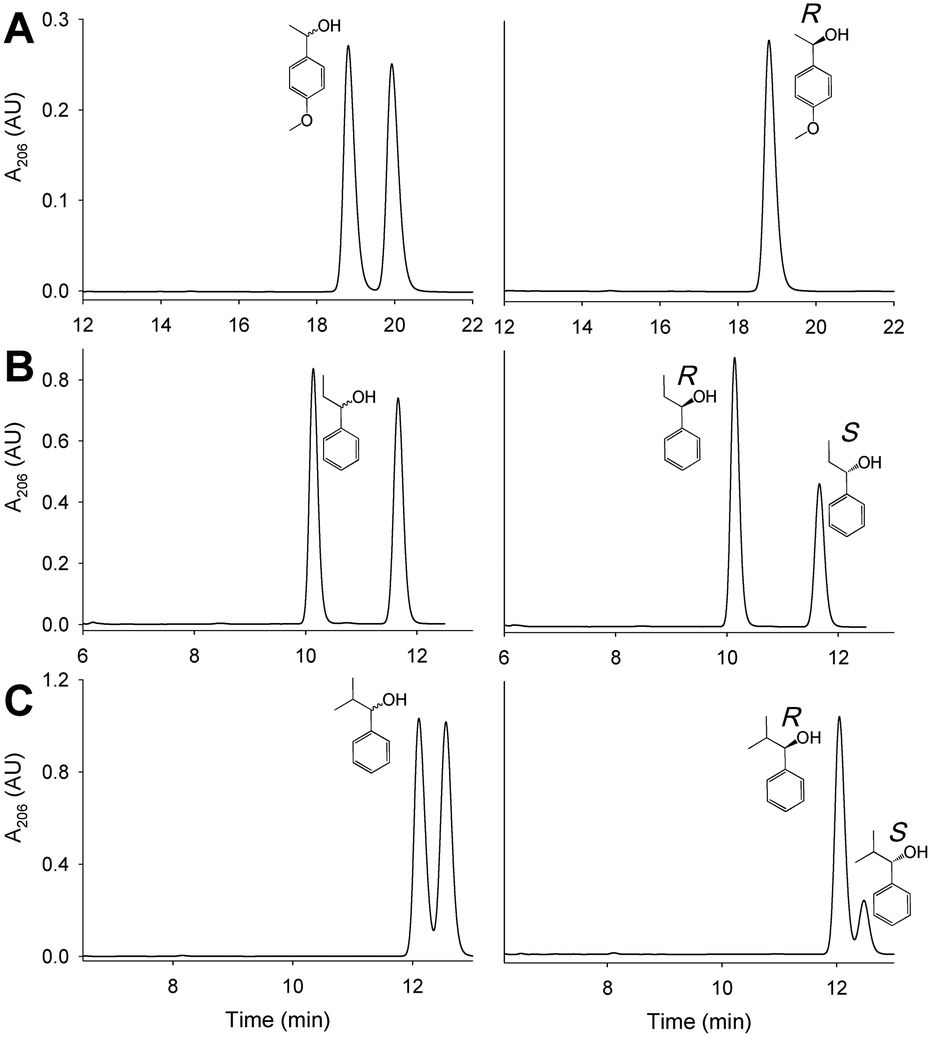 | ||
| Fig. 6 Kinetic resolution of (±)1-(p-methoxyphenyl)-ethanol (A), (±)1-phenyl-1-propanol (B) and (±)1-phenyl-2-methyl-1-propanol (C) by the I500M/F501W variant: chromatograms before (left) and after incubation with the enzyme (for 1 h in A and 72 h in B and C) resulting in R-enriched samples (right). | ||
| Conversionb (%) | ee (%) | E-Value | |
|---|---|---|---|
| a Mid-term (75 min) reaction of 2.5 mM alcohol and 2.5 μM enzyme in air-saturated 50 mM phosphate, pH 6.0, at 25 °C. b Referring to the racemic mixture. c —, too low E-values (<15) for practical purposes. | |||
| AAO | 3 | 2 | —c |
| F501A | 4 | 5 | — |
| I500A | 24 | 30 | 204 |
| I500M | 30 | 42 | 352 |
| I500M/F501W | 50 | 100 | 89000 |
Reaction with other secondary alcohols
To extend the deracemization potential of AAO, revealed by the (±)1-(p-methoxyphenyl)-ethanol reactions, to other secondary benzyl alcohols, the I500A, I500M and I500M/F501W variants (10 μM) were incubated with (±)1-phenyl-1-propanol and (±)1-phenyl-2-methylpropanol (2.5 mM) up to 72 h.Chiral HPLC analyses indicated that I500M and I500M/F501W were able to oxidize the two alcohols to their corresponding ketones (Fig. 6B and C) although with low conversion yields (4% and 22% of the above racemic compounds, respectively, by I500M; and 13% and 31%, respectively, by I500M/F501W) leading to an R-enantiomer ee up to 62% in the case of I500M/F501W (Table 4). These results suggest that the active site in these two variants has been sufficiently enlarged (as illustrated in Fig. 1B for I500M/F501W) to accommodate not only the methyl group of the model secondary alcohol but also larger groups, such as the ethyl and isopropyl groups of the two other secondary alcohols assayed.
| 1-Phenyl-1-propanol | 1-Phenyl-2-methylpropanol | |||
|---|---|---|---|---|
| Conversionb (%) | ee (%) | Conversionb (%) | ee (%) | |
| a Long-term (72 h) reaction of 2.5 mM alcohol and 10 μM enzyme in air-saturated 50 mM phosphate, pH 6.0, at 25 °C. b Referring to the racemic mixture. | ||||
| AAO | 1 | 2 | 0 | 0 |
| I500A | 1 | 2 | 0 | 0 |
| I500M | 4 | 9 | 22 | 39 |
| I500M/F501 W | 13 | 26 | 31 | 62 |
The enantioselectivity of the Ile500 variants was confirmed by incubating them with pure R- and S-enantiomers of 1-(p-fluorophenyl)-ethanol. After 48 h of reaction, the formation of p-fluoroacetophenone (up to 30%) was shown by difference spectra when (S)-1-(p-fluorophenyl)-ethanol was treated with the single and double Ile500 variants (Fig. 7A). By contrast, hardly any reaction was observed for (R)-1-(p-fluorophenyl)-ethanol (Fig. 7B) confirming the enantioselectivity shown using racemic mixtures.
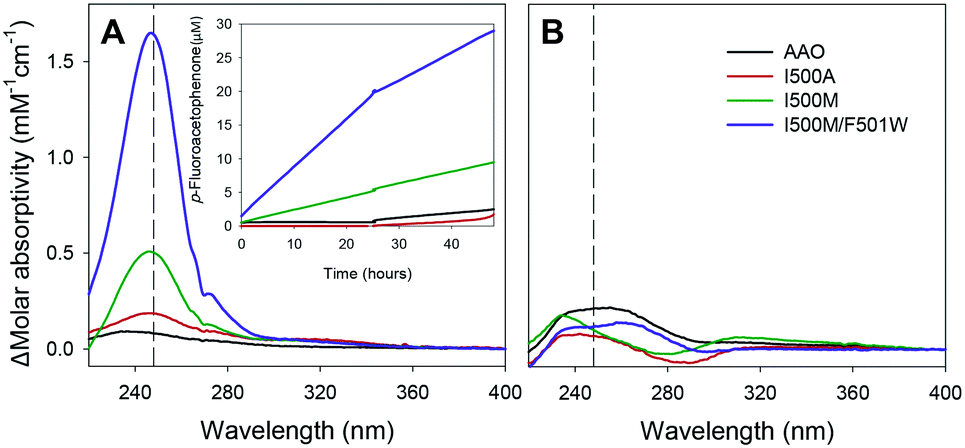 | ||
| Fig. 7 Molar absorptivity difference spectra (final – initial) after 48 h of reaction of (S)-1-(p-fluorophenyl)-ethanol (A) and (R)-1-(p-fluorophenyl)-ethanol (B) with native AAO and three variants. The inset shows the p-fluoroacetophenone production. Reactions between alcohol (100 μM) and enzyme (∼0.2 μM) were performed in 50 mM phosphate, pH 6.0, at 25 °C. | ||
Stereoselectivity explained by R and S simulations
The AAO selectivity for the S-enantiomers, during secondary alcohol oxidation, is consistent with the catalytic mechanism reported for the oxidation of p-methoxybenzyl alcohol, in which the hydrogen in the pro-R position is selectively abstracted by the flavin.21 To further investigate the stereoselectivity in secondary (±)1-(p-methoxyphenyl)-ethanol oxidation, the energy profiles of the diffusion of both enantiomers, from the solvent to the active site of AAO and the I500A, I500M, F501A and I500M/F501W variants, were analyzed in PELE simulations.According to these simulations, the S-enantiomer is capable of reaching the active site of the selected variants with good binding energies and adequate distances from both the FAD and the catalytic His502 for hydroxyl oxidation (Fig. 8, left). This means that the oxidation of the S-enantiomer will be more favorable for these variants than for the native enzyme, being consistent with the experimental kinetic parameters (Table 1). Moreover, the catalytic efficiency (kcat/Km) values – which cluster into three main groups: native AAO and F501A (low efficiency), I500A (intermediate efficiency) and I500M and I500M/F501W (high efficiency) (Table 1) – qualitatively correlate with the catalytic population observed for the S-enantiomer (Fig. 8, left).
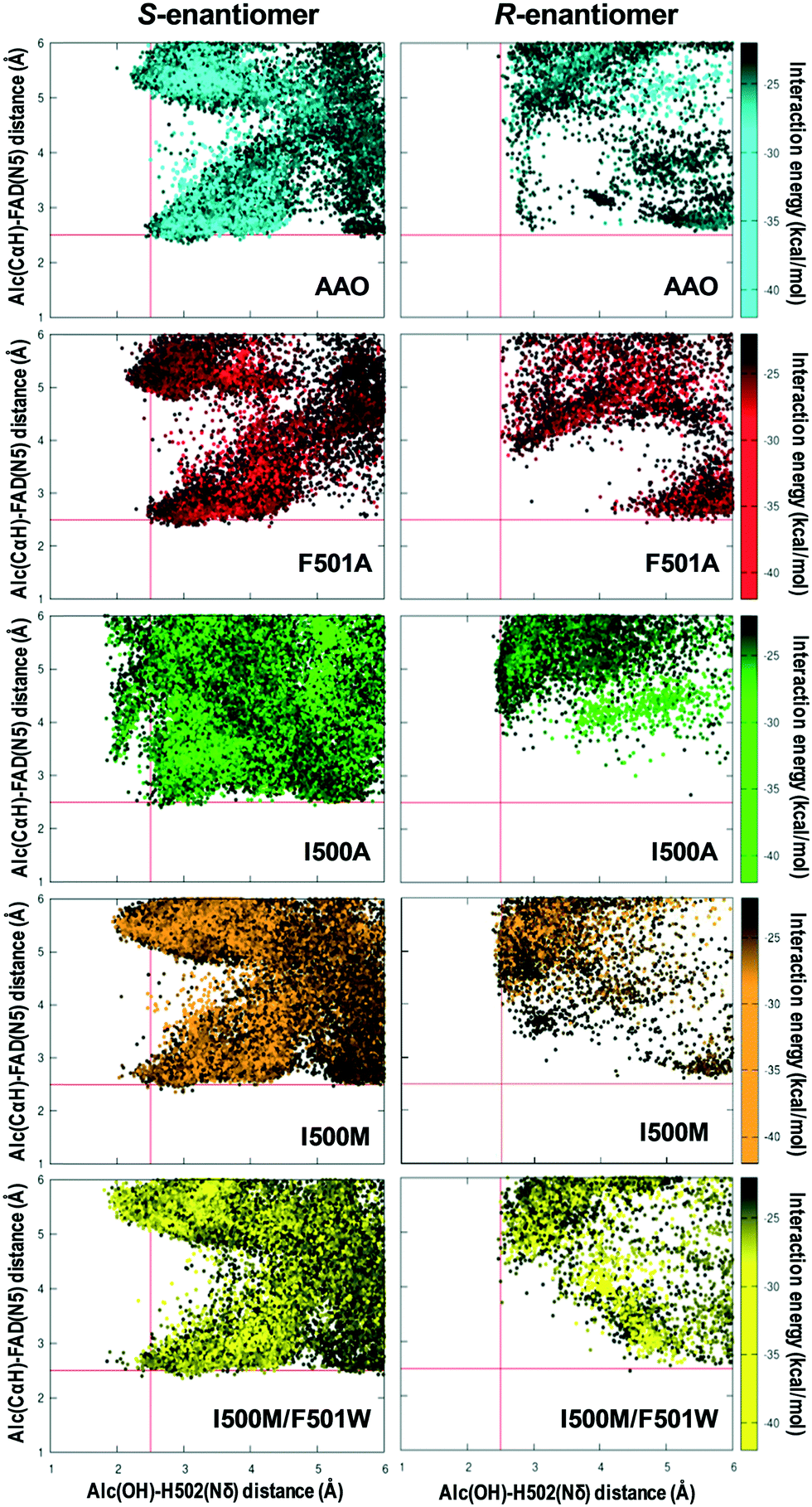 | ||
| Fig. 8 PELE diffusion of (S)-1-(p-methoxyphenyl)-ethanol (left) and (R)-1-(p-methoxyphenyl)-ethanol (right) on AAO (blue) and its F501A (red), I500A (green), I500M (orange) and I500M/F501W (yellow) variants. Each ligand position is represented by the distances from the hydroxyl hydrogen to the Nδ of His502 (axis-x) and from the benzylic hydrogen (Cα) to the flavin N5 (axis-y) and colored according to the interaction energy scale. | ||
In contrast to the above calculations for the S-enantiomer, when simulations were performed with (R)-1-(p-methoxyphenyl)-ethanol in the native enzyme and variants (Fig. 8, right), the alcohol did not reach the active site at distances that would allow the simultaneous abstraction of the proton from the alcohol and the hydride transfer (which in this case should be from the S position) in agreement with the experimental data (Fig. 5A).
Conclusions
The ability to oxidize secondary alcohols has been introduced in AAO by site-directed mutagenesis guided by computational simulations with the adaptive PELE software. The I500M/F501W double variant appears as a biocatalyst of biotechnological interest since it produces enantiomerically-enriched secondary alcohols (up to ee >99%), only at the expense of molecular oxygen, during the kinetic resolution of racemic mixtures. According to the S stereo-preference of AAO, the R-isomers remain unreacted, together with the ketone formed during oxidation of the S-isomers (Scheme 1). This stereoselectivity, which agrees with the binding energy at catalytically-relevant positions (near the active-site catalytic base and the flavin cofactor) in the computational diffusion of the R- and S-isomers, can be exploited for deracemization of chiral secondary alcohols. In summary, we show how computational simulations can guide protein engineering to switch the oxidase preference on primary benzyl alcohols towards enantioselective oxidation of secondary alcohols. Analysis of the resulting variants reveals that the mutations introduced facilitate the entrance and accommodation of bulkier secondary alcohols at the active site of AAO.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the INDOX (KBBE-2013-7-613549) EU project and by the BIO2017-86559-R (GenoBioref), CTQ2016-79138-R and BIO2016-79106-R projects of the Spanish Ministry of Economy, Industry and Competitiveness, cofinanced by FEDER funds. Pedro Merino (University of Zaragoza, Spain) is acknowledged for his suggestions on chiral HPLC analyses.References
- R. N. Patel, Coord. Chem. Rev., 2008, 252, 659–701 CrossRef CAS.
- S. Servi, D. Tessaro and G. Pedrocchi-Fantoni, Coord. Chem. Rev., 2008, 252, 715–726 CrossRef CAS.
- T. Matsuda, R. Yamanaka and K. Nakamura, Tetrahedron: Asymmetry, 2009, 20, 513–557 CrossRef CAS.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Adv. Synth. Catal., 2004, 346, 125–142 CrossRef CAS.
- R. N. Patel, Biomolecules, 2013, 3, 741–777 CrossRef PubMed.
- N. J. Turner, Chem. Rev., 2011, 111, 4073–4087 CrossRef CAS PubMed.
- J. Liu, S. Wu and Z. Li, Curr. Opin. Chem. Biol., 2018, 43, 77–86 CrossRef CAS PubMed.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieile, J. G. Zeikus and R. S. Phillips, J. Org. Chem., 2007, 72, 30–34 CrossRef CAS PubMed.
- C. V. Voss, C. C. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 741–745 CrossRef CAS PubMed.
- Y. Kawagoshi and M. Fujita, World J. Microbiol. Biotechnol., 1997, 13, 273–277 CrossRef CAS.
- S. Dieth, D. Tritsch and J. F. Biellmann, Tetrahedron Lett., 1995, 36, 2243–2246 CrossRef CAS.
- W. Adam, M. Lazarus, B. Boss, C. R. Saha-Möller, H. U. Humpf and P. Schreier, J. Org. Chem., 1997, 62, 7841–7843 CrossRef CAS.
- E. W. van Hellemond, L. Vermote, W. Koolen, T. Sonke, E. Zandvoort, D. P. Heuts, D. B. Janssen and M. W. Fraaije, Adv. Synth. Catal., 2009, 351, 1523–1530 CrossRef CAS.
- F. Escalettes and N. J. Turner, ChemBioChem, 2008, 9, 857–860 CrossRef CAS PubMed.
- W. P. Dijkman, C. Binda, M. W. Fraaije and A. Mattevi, ACS Catal., 2015, 5, 1833–1839 CrossRef CAS.
- J. Carro, P. Ferreira, L. Rodríguez, A. Prieto, A. Serrano, B. Balcells, A. Ardá, J. Jiménez-Barbero, A. Gutiérrez, R. Ullrich, M. Hofrichter and A. T. Martínez, FEBS J., 2015, 282, 3218–3229 CrossRef CAS PubMed.
- J. Carro, E. Fernández-Fueyo, M. Alcalde, A. T. Martínez, P. Ferreira, R. Ullrich and M. Hofrichter, Spain Pat., P201730805, 2017 Search PubMed.
- F. Guillén, A. T. Martínez and M. J. Martínez, Eur. J. Biochem., 1992, 209, 603–611 CrossRef.
- P. Ferreira, A. Hernández-Ortega, B. Herguedas, A. T. Martínez and M. Medina, J. Biol. Chem., 2009, 284, 24840–24847 CrossRef CAS PubMed.
- A. Hernández-Ortega, K. Borrelli, P. Ferreira, M. Medina, A. T. Martínez and V. Guallar, Biochem. J., 2011, 436, 341–350 CrossRef PubMed.
- A. Hernández-Ortega, P. Ferreira, P. Merino, M. Medina, V. Guallar and A. T. Martínez, ChemBioChem, 2012, 13, 427–435 CrossRef PubMed.
- F. J. Ruiz-Dueñas, P. Ferreira, M. J. Martínez and A. T. Martínez, Protein Expression Purif., 2006, 45, 191–199 CrossRef PubMed.
- A. Aliverti, B. Curti and M. A. Vanoni, Methods Mol. Biol., 1999, 131, 9–23 CAS.
- R. M. C. Dawson, D. C. Elliot, W. H. Elliot and K. M. Jones, Data for biochemical research, Oxford Science Publications, Oxford, 1986 Search PubMed.
- P. Ferreira, M. Medina, F. Guillén, M. J. Martínez, W. J. H. van Berkel and A. T. Martínez, Biochem. J., 2005, 389, 731–738 CrossRef CAS PubMed.
- S. A. Rounds, F. D. Wilde and G. F. Ritz, U. S. Geological Survey Techniques of WaterResources Investigations, book 9, chap. A6, sec. 6.2 (http://water.usgs.gov/owq/FieldManual/Chapter6/6.2_v3.0.pdf), 2013 Search PubMed.
- K. Faber and W. Kroutil, http://biocatalysis.uni-graz.at/enantio/DataFiles/Selectivity-Help.pdf, 2012.
- D. Lecina, J. F. Gilabert and V. Guallar, Sci. Rep., 2017, 7, 8466 CrossRef PubMed.
- A. R. Atilgan, S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin and I. Bahar, Biophys. J., 2001, 80, 505–515 CrossRef CAS PubMed.
- J. F. Gilabert, D. Lecina, J. Estrada and V. Guallar, in Biomolecular Simulations in Drug Discovery, ed. F. L. Gervasio and V. Spiwok, Wiley, 2018, pp. 87–96 Search PubMed.
- J. L. Banks, H. S. Beard, Y. Cao, A. E. Cho, W. Damm, R. Farid, A. K. Felts, T. A. Halgren, D. T. Mainz, J. R. Maple, R. Murphy, D. M. Philipp, M. P. Repasky, L. Y. Zhang, B. J. Berne, R. A. Friesner, E. Gallicchio and R. M. Levy, J. Comput. Chem., 2005, 26, 1752–1780 CrossRef CAS PubMed.
- R. B. Murphy, D. M. Philipp and R. A. Friesner, J. Comput. Chem., 2000, 21, 1442–1457 CrossRef CAS.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS.
- I. S. Fernández, F. J. Ruiz-Dueñas, E. Santillana, P. Ferreira, M. J. Martínez, A. T. Martínez and A. Romero, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 1196–1205 CrossRef PubMed.
- J. Carro, P. Amengual-Rigo, F. Sancho, M. Medina, V. Guallar, P. Ferreira and A. T. Martínez, Sci. Rep., 2018, 8, 8121 CrossRef PubMed.
- J. Carro, M. Martínez-Júlvez, M. Medina, A. T. Martínez and P. Ferreira, Phys. Chem. Chem. Phys., 2017, 19, 28666–28675 RSC.
- A. Hernández-Ortega, F. Lucas, P. Ferreira, M. Medina, V. Guallar and A. T. Martínez, J. Biol. Chem., 2011, 286, 41105–41114 CrossRef PubMed.
- J. Viña-González, D. González-Pérez, P. Ferreira, A. T. Martínez and M. Alcalde, Appl. Environ. Microbiol., 2015, 81, 6451–6462 CrossRef PubMed.
- J. Viña-González, D. Jiménez-Lalana, A. Serrano, A. T. Martínez and M. Alcalde, Abs. XL Congr. SEBBM, Barcelona, 2017, vol. P10-20, p. 90 Search PubMed.
- R. J. Fox and M. D. Clay, Trends Biotechnol., 2009, 27, 137–140 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02447b |
| ‡ These two authors equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2019 |