
Electrochemically chopped WS2 quantum dots as an efficient and stable electrocatalyst for water reduction†
S.
Anantharaj‡
 ab,
Manila O.
Valappil‡
ab,
K.
Karthick
ab,
Vijayamohanan K.
Pillai
ab,
Subbiah
Alwarappan
*ab and
Subrata
Kundu
*ab
ab,
Manila O.
Valappil‡
ab,
K.
Karthick
ab,
Vijayamohanan K.
Pillai
ab,
Subbiah
Alwarappan
*ab and
Subrata
Kundu
*ab
aAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad – 201 002, Uttar Pradesh, India. E-mail: salwarap@gmail.com; kundu.subrata@gmail.com; skundu@cecri.res.in
bCSIR-Central Electrochemical Research Institute (CECRI), Karaikudi-630003, Tamil Nadu, India
First published on 22nd November 2018
Abstract
Advancements in the search for efficient H2 evolving electrocatalysts by engineering the structure and electronic properties of bulk materials at the nano regime have had a constructive impact on realizing better performance with ultralow catalyst loading. In this work, we investigate the electrochemical hydrogen evolution catalysis of WS2 quantum dots (QDs) synthesized electrochemically. WS2 QDs were evaluated for a hydrogen evolution reaction (HER) in 0.5 M H2SO4 as a binder-less electrocatalyst, which necessitated a low HER onset of 190 mV and reached −200 mA cm−2 at 473 mV. In contrast, the bulk WS2 exhibited a poor performance, which required 780 mV to drive −50 mA cm−2. The electrochemical disintegration of bulk WS2 into QDs is the key behind such activation.
Introduction
Hydrogen generated via facile electrochemical water splitting has been identified as a clean fuel to meet future energy demands in order to address the depletion of available fossil fuels.1 Energy and cost efficient H2 production from water demands extensive studies on the design and development of electrocatalysts to catalyze the half-cell reactions of water splitting, namely HERs and oxygen evolution reactions (OERs).2,3 Between these, an OER is considered important as it is kinetically more complex than a HER. At the same time, finding an efficient and non-precious electrocatalyst to initiate the HER to replace the expensive Pt is also very essential to ensure that the H2 produced is economically affordable.2–4 Many materials from the 3d transition metals and metals from Group IIIB and Group IVB are being discovered frequently with highly improved HER activities for both acidic and alkaline water electrolysis.2–4 These materials attracted greater attention in the latter half of the preceding decade due to the rise of chalcogenides, phosphides, carbides, nitrides and borides of metals such as Ni, Co, Fe, Mn, Mo, W, Ta and V as various nanostructured materials.2–4 The use of nanomaterials in water splitting has become desirable as it provides solutions for various problems encountered in earlier water electrolysis for H2 production with conventional larger and planar electrodes.5 The application of nanomaterials in the electrocatalysis of a HER and an OER has many advantages, like enhanced activity due to highly increased active sites as a consequence of a reduction in size, availability of defects and edges with high energy for further enhancement of electrocatalytic activity, realizing maximum activity with a minimum amount of material, etc.2–4 In parallel to the field of non-precious metals based HER electrocatalysis, there has also been some significant work done on metal-free and metal – non-metal based HER electrocatalysts.6–11Among the various non-precious metal chalcogenides, the disulphides of W are one of the frequently studied systems in the literature due to their intriguing layered structure and electrocatalytic HER activities comparable to those of Pt with lower additional overpotential.12 In many studies, it was implied that a reduction in the size of layered bulk WS2 sheets increased the HER activity. As a result, WS2 had been reported in various morphologies at the nanoscale with enhanced activities. Some of the intriguing morphologies of WS2 nanomaterials are nanosheets,13–21 dots,22,23 nanoflakes,24,25 nanoribbons,26–30 nanoplatelets31 and nanorods.32,33 In addition, the same WS2 has also been combined with synergistically enhancing materials like graphene and reduced graphene oxide which showed a further increased performance in HERs.13,15,20,32 Additionally, the role of metal dopants, namely Ni and Co, in WS2 has also been investigated for HERs.34,35 WS2 had such an excellent HER activity that it was applied to photocatalytic and photoelectrocatalytic HER with photo-harvesting materials like Si, TiO2 and CdS.14,19,32,36–38 Other than WS2, carbides, nitrides, phosphides and selenides of W were also reported for HERs.33,36,39–41 This particular observation leads to further work with WS2via tailoring its morphological features and electronic properties at nanoscale. Nanomaterials of WS2 reported in the literature utilized the top-down approach of tailoring bulk WS2 into the desired structure chemically, where the use of salts and surfactants was unavoidable which made those methods very cumbersome and the resultant nanomaterials were almost always anisotropic.19,24,25,29,42,43 Moreover, HER studies on WS2 QDs of sub-nanometer size are not extensively reported, though WS2 QDs have the potential to beat all other nanostructured WS2 catalysts due to the advantages of smaller size and a large number of catalytically active edges while retaining a layered structure.
The motivation of the present work lies in the recently reported one-step electrochemical synthesis of highly mono-dispersed WS2 QDs of size smaller than 5 nm using an applied bias of 2 V for 24 h in propylene carbonate containing LiClO4.44 The same was screened for HER in an acid medium with no binders for electrode fabrication as propylene carbonate itself possessed good adherence to the substrate electrode. The observed HER activity of the WS2 QDs is far better than the bulk WS2 sheets, or other nanostructured WS2 materials and is comparable to Pt/C.
Experimental section
Electrochemical disintegration of bulk WS2 into WS2 QDs
The disintegration of bulk WS2 sheets into highly mono-dispersed WS2 QDs was performed using a three-electrode electrochemical cell. A Pt mesh counter electrode and a Pt wire quasi-reversible reference electrode were used along with a WS2 pellet as working electrode. The WS2 pellet was made with 1 g of bulk WS2 sheets by KBr hydraulic pressure that had a diameter of 13 mm and was about 2.5 mm thick. The electrochemical disintegration was done in a deoxygenated propylene carbonate electrolyte containing LiClO4 (0.1 wt%) at an applied potential of 2.0 V for 24 h, as we reported earlier. The colorless electrolyte solution turned yellow which was the indication of the formation of WS2 QDs. Then, the electrolyte was purified to exclude the residual ions, followed by the collection of the resultant WS2 QDs in propylene carbonate as a colloidal solution. The calculated molar concentration of WS2 in the resultant colloidal solution was 0.012 M.Catalyst ink preparation and electrode fabrication methods
For comparison, 3 mg of bulk WS2 sheets and Pt/C 20 wt% catalysts were mixed in separate vials by sonication with a 1 mL solution of water, isopropyl alcohol and a 5% Nafion ionomers suspension taken in the volume ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:0.5 for 20 min. About 68.5 μL of the resultant catalyst ink was drop casted on the CFP substrate electrode and dried overnight at ambient conditions and in the dark before the electrochemical studies. The corresponding mass loading of these catalysts was 0.205 mg cm−2. To fabricate the WS2 QDs/CFP electrode, the same volume of 68.5 μL of the colloidal solution of the WS2 QDs in propylene carbonate was used. This corresponded to a loading of 0.0132 mg cm−2, as per the calculated molar concentration, which is 15.47 times lower than the loadings of both the bulk WS2 and Pt/C catalysts. This particular electrode took a slightly longer time (18 h) to dry before it could be used as a working electrode.
:2.5:0.5 for 20 min. About 68.5 μL of the resultant catalyst ink was drop casted on the CFP substrate electrode and dried overnight at ambient conditions and in the dark before the electrochemical studies. The corresponding mass loading of these catalysts was 0.205 mg cm−2. To fabricate the WS2 QDs/CFP electrode, the same volume of 68.5 μL of the colloidal solution of the WS2 QDs in propylene carbonate was used. This corresponded to a loading of 0.0132 mg cm−2, as per the calculated molar concentration, which is 15.47 times lower than the loadings of both the bulk WS2 and Pt/C catalysts. This particular electrode took a slightly longer time (18 h) to dry before it could be used as a working electrode.
Results and discussion
Material characterizations
The X-ray diffraction (XRD) pattern (Fig. 1a) of the WS2 QDs synthesized by electrochemical disintegration at 2 V shows distinct peaks for the planes of (002), (004), (100) and (106), as per the reference ICDD card number of 08-0237, which confirmed that the synthesized WS2 QDs had retained a layered structure.19,43–45 Other cubical ((10n), n = 1, 3, 4 and 5) planes that are usually observed with bulk WS2 are absent with the WS2 QDs, except (106), which further indicated that the bulk WS2 sheets were rigorously disintegrated into QDs, mainly with a layered 2D structure which is highly anticipated for a better HER performance. Moreover, a shift of 2.1° at the (002) plane with the WS2 QDs indicates that the interlayer distance has also been increased as a consequence of the electrochemical disintegration. Such an increase in the interlayer distance is always advantageous in HER electrocatalysis as it leads to an increased electrochemically accessible surface area (ECSA). The morphology of the synthesized QDs was observed by transmission electron microscopy (TEM) and the corresponding TEM micrograph (Fig. 1b) shows the clear presence of WS2 QDs that are distinguishably separated from one another. The measured size (Fig. 1c) of the WS2 QDs was mainly in the range of 2 to 5 nm and the average diameter of the synthesized WS2 QDs is 3.5 ± 1 nm. This particular observation supports our claim of the formation of the WS2 QDs by the chosen method of electrochemical disintegration. The TEM micrograph obtained at a high magnification shows clear lattice planes, as can be seen in Fig. 1d. The measured d spacings were 0.2 nm and 0.27 nm which correspond to the miller indices of (006) and (100), respectively. This is in excellent agreement with the XRD analysis which confirms that the synthesized WS2 QDs are crystalline. The electron diffraction (ED) pattern of these WS2 QDs exhibits a typical dot plus ring feature indicating the presence of small and larger crystalline particles, which is in accordance with the microscopic and XRD analyses. Three predominant rings (highlighted) observed in the ED pattern were calibrated to assign the corresponding diffraction planes according to the literature where it was found that all three rings were of mainly (00n) planes in which n is 4, 6 and 10. This is also in agreement with the same ICDD card number of 08-0237. The combined microscopic and diffraction analyses thus confirm that the bulk WS2 sheets were successfully disintegrated into WS2 QDs of mainly particles smaller than 5 nm with some larger sheets of size 11 to 12 nm as counterparts. | ||
| Fig. 1 XRD pattern of the synthesized WS2 QDs (a), TEM micrograph showing the WS2 QDs (b), particle size distribution histogram (c) of the WS2 QDs, HRTEM micrograph (d) showing clear lattice planes with assigned miller indices and the ED pattern (e) of the WS2 QDs. | ||
The elemental composition of the synthesized WS2 QDs was analyzed by energy dispersive X-ray analysis (EDS) (Fig. S1 in ESI†) which indicates the presence of W and S for various shells such as W M, W L, S L, and S K. It also indicates the presence of some C and O peaks that are probably from the adsorbed O and from the residual solvent (propylene carbonate) that could have resided on the fabricated thin film during the analysis. The results of this EDS analysis confirmed that the synthesized WS2 QDs are free from other impurities, including Cl and Li, and are ready for their use in HER studies. Nonetheless, as the synthesized WS2 materials are QDs, their optical properties were also examined with UV-visible and photoluminescence (PL) spectroscopic techniques. Fig. S2a and b in ESI† show the UV-vis spectrum of bulk WS2 and the synthesized WS2 QDs in propylene carbonate. The UV-visible spectrum of bulk WS2 powder shows three broad characteristic peaks A, B and C at 620 nm, 530 nm and 460 nm, respectively. The peaks A and B correspond to the excitonic absorption due to the direct bandgap transitions at the K point of the Brillouin zone. The absorption located at around 460 nm is attributed to the optical transition from the valence band to the conduction band. However, the synthesized WS2 QDs showed a hump at 350 nm which is clear cut evidence for the formation of WS2 QDs. This is also in agreement with earlier findings.43–45 The spectrum of the bulk WS2 sheets had another broad and distinct peak around 620 nm which is absent the in case of WS2 dots and QDs.43–45 Since fluorescence is an intrinsic property of QDs, the PL characteristics of the electro-synthesized WS2 QDs were also analyzed. Fig. S3 in ESI† is the PL spectrum of the WS2 QDs in propylene carbonate, acquired by excitation with monochromatic radiation of a light with 350 nm, which was chosen because of a distinct peak observed in the UV-vis spectroscopic analysis for the same QDs. From the PL spectrum, it was observed that the WS2 QDs had shown an excitation wavelength dependent photoluminescence, which matches with earlier findings.43–45 Both UV-vis and PL spectral analyses have also ascertained the formation of WS2 QDs by the electrochemical disintegration of bulk WS2 sheets.
X-ray photoelectron spectroscopy (XPS) measurements were carried out to confirm the elemental composition. The deconvoluted XP spectra of W 4f, S 2p, C 1s and O 1s states are presented in Fig. 2a–d, respectively. The W 4f spectrum exhibits a doublet due to spin–orbit splitting, as can be seen in Fig. 2a. The first peak, located at 33.05 eV, is due to the 4f7/2 sublevel and the second peak, located at 35.41 eV, is due to the 4f5/2 sublevel. The occurrence of these peaks at these binding energies, with a separation of 2.36 eV, is clear cut evidence for the existence of W4+ in the synthesized sample, which is also in agreement with earlier reports of WS2.19,43–47 Interestingly, no sign of W6+ is observed here. In addition, one more peak with a relatively poor intensity was observed at 39 eV and assigned to the 5p3/2 state of W in the 4+ oxidation state. This further understates the tendencies of forming higher oxidation state oxysulfides of WS2. The observed peak features of W 4f and W 5p3/2 also resonate with earlier reports of WS2.19,43–47 Similarly, the XP spectrum of the S 2p state (Fig. 2b) upon deconvolution yielded two distinct peaks which are also due to the known spin-orbit coupling effect. The observed peaks are located at the binding energy values of 162.35 and 164.11 eV. The occurrence of these peaks at those binding energy values agrees well with the S in the 2− oxidation state.19,43–47 In addition, though there are C and O, there were no indications of C–S or S–O functionalities. This observation is also in agreement with our earlier report.44 The high resolution XPS spectrum of the C 1s state, provided in Fig. 2c, revealed the presence of more than one type of C. A careful deconvolution of the C 1s spectrum showed four distinct peaks located at 284.8, 285.3, 286.3 and 288.9 eV, respectively, for the presence of C–C, C–H, C–O–C and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionalities. All of these functional groups are present in the propylene carbonate solvent. Likewise, the high-resolution XP spectrum of the O 1s state (Fig. 2d), acquired for the same, also revealed similar results. The acquired raw spectrum for the O 1s state was deconvoluted into two distinct peaks located at 531.8 and 532.7 eV. These peaks correspond to the presence of O–CO and C–O–C functionalities, respectively.44 These two functional groups are present in the propylene carbonate used as the solvent for the electrochemical disintegration of bulk WS2 sheets into WS2 QDs. Overall, the XPS analysis indicated that the formed QDs are only WS2, and not any higher oxidation state sulphide of W, along with some residual solvent that still resided on the fabricated thin film sample.
O functionalities. All of these functional groups are present in the propylene carbonate solvent. Likewise, the high-resolution XP spectrum of the O 1s state (Fig. 2d), acquired for the same, also revealed similar results. The acquired raw spectrum for the O 1s state was deconvoluted into two distinct peaks located at 531.8 and 532.7 eV. These peaks correspond to the presence of O–CO and C–O–C functionalities, respectively.44 These two functional groups are present in the propylene carbonate used as the solvent for the electrochemical disintegration of bulk WS2 sheets into WS2 QDs. Overall, the XPS analysis indicated that the formed QDs are only WS2, and not any higher oxidation state sulphide of W, along with some residual solvent that still resided on the fabricated thin film sample.
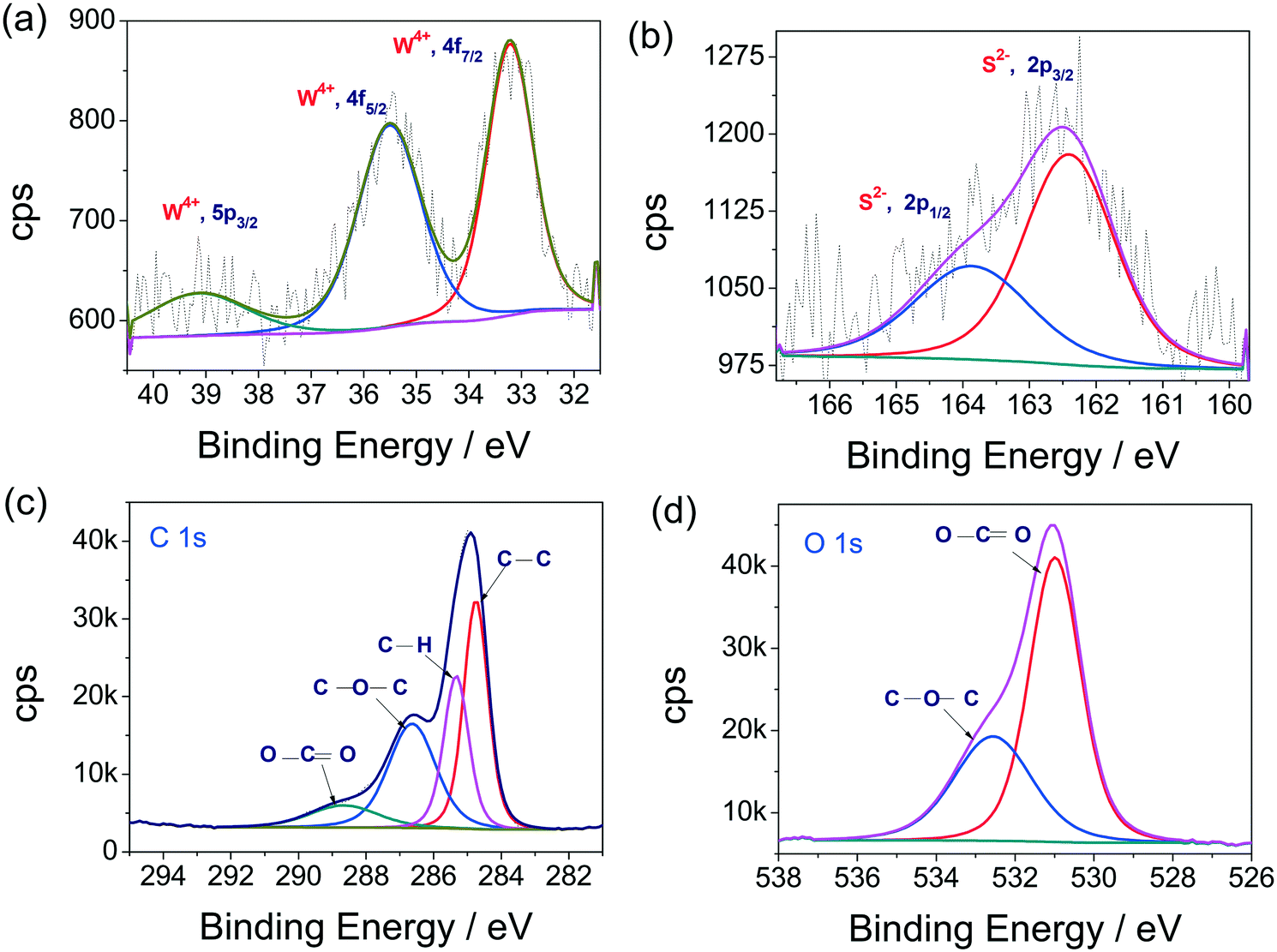 | ||
| Fig. 2 (a–d) XPS high resolution spectra of the W 4f, S 2p, C 1s and O 1s states, respectively, obtained with WS2 QDs thin films. | ||
Electrochemical characterizations
To examine the electrocatalytic HER activities of WS2 QDs, bulk WS2 and Pt/C 20 wt. catalysts were modified over carbon fiber paper (CFP) substrate electrodes 0.5 cm in width, 5 cm in length on only one side and an area of 1 cm−2. The remaining area of the substrate electrode was masked by insulating sticky tape, leaving some part of the substrate electrode on the other end which was destined for making an electrical connection with the electrochemical workstation. All electrochemical characterizations were done with a conventional three-electrode electrochemical cell containing Ar purged 0.5 M H2SO4 where an Hg/Hg2SO4 reference electrode and graphite counter electrodes were used. As a primary evaluation parameter, linear sweep voltammograms (LSVs) for WS2 QDs/CFP, WS2 bulk/CFP and Pt/C/CFP were recorded at a slower scan rate of 5 mV s−1. The same were corrected for the iR drop and the potential scales were converted to reversible hydrogen electrode (RHE), following earlier reports.48–50 The resultant iR compensated LSVs are given as Fig. 3a. The LSV profiles exhibit an improved HER activity of WS2 QDs which increased several fold compared to that of bulk WS2. This is clearly due to the reduction in size and the retention of a HER favoring layered structure having (00n) planes with more edges and corners. Moreover, it is also perceivable that the activity of the WS2 QDs is comparable to the state-of-the-art Pt at a higher overpotential region with a low overpotential difference. Specifically, the WS2 QDs required 190 mV to start the H2 evolution from that of Pt/C whereas the bulk WS2 required 310 mV for the same. The measured difference in the overpotentials at the current density of −50 mA cm−2 between WS2 QDs/CFP and WS2 bulk/CFP is 430 mV which is very high, indicating the advantage of disintegrating the bulk WS2 sheets into QDs for energy efficient HER electrocatalysis. Moreover, at the current density of −200 mA cm−2, the difference in the overpotential between Pt/C/CFP and WS2 QDs/CFP is just 338 mV. Interestingly, the activity observed with the WS2 QDs is 15.47-fold lower for catalyst loading than both bulk WS2 and Pt/C catalysts. These observations indicate that the electrochemical disintegration of bulk WS2 not only enhanced the activity but also reduced the catalyst loading. The reproducibility of the LSV features was confirmed by repeated measurements and the plot of j against overpotential (Fig. S4 in ESI†) also showed minimum error at each data point. | ||
| Fig. 3 (a) LSVs of CFP, WS2 bulk/CFP, WS2 QDs/CFP and Pt/C/CFP interfaces acquired at 5 mV s−1 in 0.5 M H2SO4. (b) Corresponding Tafel plots of the same. (c) Nyquist plots of CFP, WS2 bulk/CFP, WS2 QDs/CFP and Pt/C/CFP interfaces at onset overpotentials. (d) Plot of scan rate vs. double layer charging current density difference for CFP, WS2 bulk/CFP and WS2 QDs/CFP interfaces. | ||
The corresponding Tafel plots of bare CFP, WS2 QDs/CFP, WS2 bulk/CFP and Pt/C/CFP (Fig. 3b) showed that the Pt/C/CFP is the only interface that followed the Tafel mechanism by having the lowest Tafel slope of 33 mV dec−1. The WS2 QDs have shown a Volmer–Heyrovsky mechanism for HER by having 90 mV dec−1 as the Tafel slope.48 The relatively higher Tafel slopes observed for bulk WS2 and bare CFP indicated that HER kinetics are poor on those interfaces. In addition, the measured exchange current densities (j0) for CFP, WS2 bulk/CFP, WS2 QDs/CFP and Pt/C/CFP were 5.50 × 10−6, 1.58 × 10−4, 3.23 × 10−4 and 2.047 × 10−3 A cm−2, respectively. This is in exact agreement with the observed HER activities of all four interfaces studied here and implies that the j0 of WS2 was doubled as a consequence of forming QDs from bulk WS2 sheets. The electrochemical properties of the studied interfaces were examined by electrochemical impedance (EI) analysis at the onset overpotentials in the range of 100 kHz to 0.1 Hz. The corresponding Nyquist plots (Fig. 3c) show the charge transfer resistances (Rct) for WS2 QDs/CFP, WS2 bulk/CFP and Pt/C/CFP as 142, 1220 and 4.9 ohms, respectively, which agree well with HER activity order and imply that the formation of WS2 QDs from bulk WS2 enhances charge transfer. Further, the activity trend is correlated to the ECSA measured for WS2 QDs/CFP, WS2 bulk/CFP and only CFP by the widely accepted double layer capacitance (Cdl) method. Fig. 3d is the plot of scan rate against Δj (the difference in the anodic and cathodic double layer charging current, see ESI† for sample cyclic voltammograms (CVs) recorded for WS2 QDs (Fig. S5 in ESI†)). The plot shows a several hundred fold increase in the electrochemical surface area (ECSA) of the WS2 QDs which is a complementing factor in addition to the reduced Rct values. In addition to the activity parameters, the stability of WS2 QDs/CFP was also examined by high sweep rate (200 mV s−1) cycling using cyclic voltammetry and chronoamperometry (CA) for more than 66 k s at an iR uncorrected overpotential of 265 mV, respectively. The j–t response of the WS2 QDs (Fig. 4a) showed that there was a significant increase in the activity of the WS2 QDs as the reaction progressed with time. Such an increase in activity is observed earlier with various other metal chalcogenides and is usually attributed to negative polarization at the interface, unmasking of additional active sites and reduction of surface oxidized species. We believe that the first two factors are the probable ones in this case of activation. The post CA LSV (Fig. 4b) is also in agreement with such activation during CA. LSVs acquired before and after cycling for 1000 times at a high sweep rate (Fig. 4c) showed almost a slight decrease in activity, which could be due to the leaching of electrocatalysts from the surface of the substrate electrode. Nyquist plots obtained after such harsh stability studies (Fig. S6 in ESI†) showed no change in Rct at the WS2 QDs/CFP interface which demonstrated the robustness of the synthesized WS2 QDs. In addition, we have also checked the role of pH on the HER activity of the WS2 QDs' interface by taking 0.5, 0.05, 0.005, 0.0005 and 0.00005 M H2SO4 solutions as electrolytes (Fig. S7 in ESI†). As expected, we have observed a drastic decay in activity with an increase in pH.51 This study has revealed that for better HER kinetics and activity, the solution pH must be 0. Moreover, the observed HER activity with our method is superior to all other WS2 reports as shown in Table S1 in ESI.†
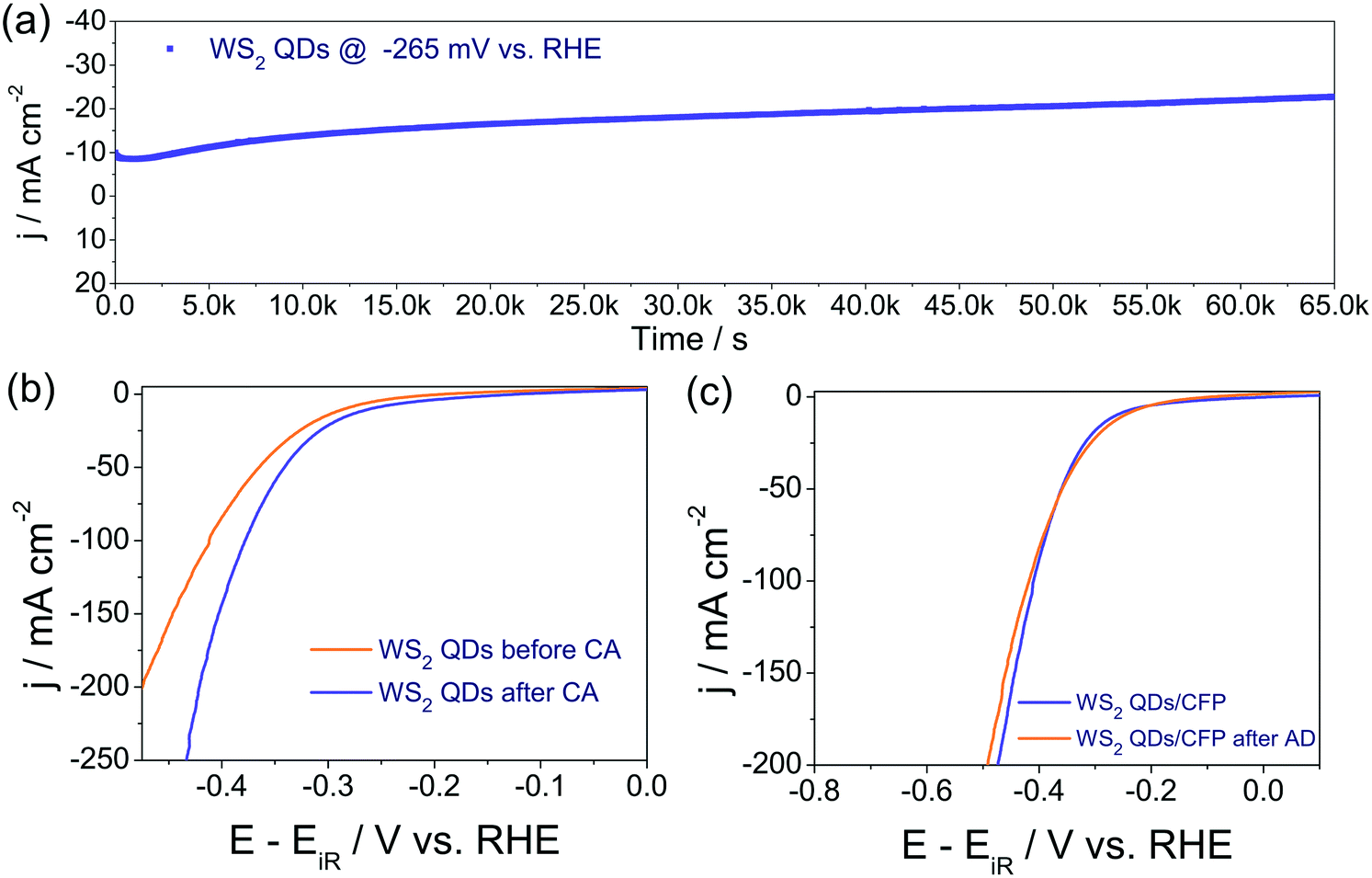 | ||
| Fig. 4 (a) Response of WS2 QDs in chronoamperometric analysis, which was carried out in 0.5 M H2SO4 at the overpotential of 265 mV (b) pre- and post-CA LSVs showing significant activation in the HER activity of the same after CA study. (c) LSVs of WS2 QDs recorded before and after 1000 cycles of rapid CV cycling at 200 mV s−1. | ||
Post-HER material characterizations
It is important now to show the morphological and chemical robustness of the studied WS2 QDs after prolonged HER studies. XRD, TEM and XPS analyses have been done to show such results. For XRD analysis, the WS2 QDs/CFP electrode was taken directly after chronoamperometry studies. The obtained diffraction pattern is provided in Fig. S8 in ESI.† We can see three distinguished peaks corresponding to graphitic and graphenic carbons of the CFP substrate. Other than these carbon peaks, no other peaks for WS2 or the other phases are observed. This could be due to the very low amount of catalyst that we loaded on the substrate. Fig. 5a–c show the TEM micrographs of the WS2 QDs after the HER studies. For the TEM analysis, the catalyst was re-dispersed in propylene carbonate again to make TEM specimens. From Fig. 5a–c, we can see that the WS2 QDs have retained their morphological characteristics by having average sizes of less than 5 nm. This strongly proves the morphological robustness of our WS2 QDs and suggests that they could be an efficient and robust alternate to the noble metals based HER electrocatalysts. Moreover, the SAED pattern of the WS2 QDs was also acquired and calibrated as shown in Fig. 5d. From this, the existence of (004), (006) and (0010) basal planes can be seen. This is in exact agreement with the pre-HER SAED results and suggests that the structural characteristics of the WS2 QDs are retained after HER studies. Fig. 5e shows the particle size distribution histogram that suggests that the average size of the WS2 QDs was also retained at 3.5 nm, which is more proof for the structural robustness of the WS2 QDs in HER studies. | ||
| Fig. 5 (a–c) TEM micrographs of WS2 QDs after HER studies. (d) Corresponding SAED pattern. (e) Particle size distribution histogram showing the average particle size. | ||
Having confirmed the structural robustness, it is now time to analyze the chemical stability of the WS2 QDs after HER studies. To do this, a detailed XPS analysis was carried out after the HER studies, taking the WS2 QDs/CFP electrode directly. Fig. 6a–d show the high resolution XPS spectra of the W 4f, S 2p, C 1s and O 1s states, respectively. Fig. 6a shows a notable difference from that of Fig. 2a. The exposure to an acidic environment for a considerably longer period of time resulted in the oxidation of W4+ to W6+ in considerable proportion. However, most of the WS2 QDs retained their chemical nature. Similarly, the S 2p high resolution spectrum (Fig. 6b) had also shown the oxidation of S2− to S4+ to a notable extent. Both results suggest that the surface oxidation of W and S in an acidic environment, even under reductive atmosphere, is irresistible. Other XPS spectra, such as C 1s (Fig. 6c) and O 1s (Fig. 6d), had shown the presence of CC, C–C, C–O, CO, S–O and H2O adsorbed moisture in it, as expected. The overall post-HER material characterization studies have revealed that our WS2 QDs are morphologically robust and also chemically stable to a significant extent.
 | ||
| Fig. 6 (a–d) XPS high resolution spectra of W 4f, S 2p, C 1s and O 1s of WS2 QDs/CFP electrode after chronoamperometry in acid, respectively. | ||
Conclusion
In summary, we have demonstrated how efficiently a simple electrochemical disintegration in a non-aqueous solvent can be utilized to abnormally enhance the moderately active bulk WS2 into a high performance HER electrocatalyst upon transforming to WS2 QDs. A detailed electrochemical characterization indicated that the WS2 QDs are a far better HER electrocatalyst than bulk WS2, which required 430 mV lesser overpotential than the latter for −50 mA cm−2. Moreover, the WS2 QDs required just 338 mV extra than that of Pt/C to drive −200 mA cm−2, which is a significant number in HER electrocatalysis, with a lower Tafel slope of 90 mV dec−1. Furthermore, an excellent stability without any added binders is another advantage of making WS2 QDs in propylene carbonate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. and M. O. V. acknowledge CSIR, India for SRF award and K. K. acknowledges UGC, India for SRF award.References
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5, 1500985–1500997 CrossRef.
- H. Wendt and G. Imarisio, J. Appl. Electrochem., 1988, 18, 1–14 CrossRef CAS.
- H. X. Zhong, Q. Zhang, J. Wang, X. B. Zhang, X. L. Wei, Z. J. Wu, K. Li, F. L. Meng, D. Bao and J. M. Yan, ACS Catal., 2018, 8, 3965–3970 CrossRef CAS.
- J. Wang, H. X. Zhong, Z. L. Wang, F. L. Meng and X. B. Zhang, ACS Nano, 2016, 10, 2342–2348 CrossRef CAS PubMed.
- K. Liu, H. Zhong, F. Meng, X. Zhang, J. Yan and Q. Jiang, Mater. Chem. Front., 2017, 1, 2155–2173 RSC.
- Z. L. Wang, X. F. Hao, Z. Jiang, X. P. Sun, D. Xu, J. Wang, H. X. Zhong, F. L. Meng and X. B. Zhang, J. Am. Chem. Soc., 2015, 137, 15070–15073 CrossRef CAS PubMed.
- K. H. Liu, H. X. Zhong, S. J. Li, Y. X. Duan, M. M. Shi, X. B. Zhang, J. M. Yan and Q. Jiang, Prog. Mater. Sci., 2018, 92, 64–111 CrossRef CAS.
- H.-X. Zhong, J. Wang, Q. Zhang, F. Meng, D. Bao, T. Liu, X.-Y. Yang, Z.-W. Chang, J.-M. Yan and X.-B. Zhang, Adv. Sustainable Syst., 2017, 1, 1700020–1700027 CrossRef.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- J. Yang, D. Voiry, S. J. Ahn, D. Kang, A. Y. Kim, M. Chhowalla and H. S. Shin, Angew. Chem., Int. Ed., 2013, 52, 13751–13754 CrossRef CAS PubMed.
- J. He, L. Chen, Z. Q. Yi, C. T. Au and S. F. Yin, Ind. Eng. Chem. Res., 2016, 55, 8327–8333 CrossRef CAS.
- T. A. Shifa, F. Wang, Z. Cheng, X. Zhan, Z. Wang, K. Liu, M. Safdar, L. Sun and J. He, Nanoscale, 2015, 7, 14760–14765 RSC.
- Y. S. Zhang, J. P. Shi, G. F. Han, M. J. Li, Q. Q. Ji, D. L. Ma, Y. Zhang, C. Li, X. Y. Lang, Y. F. Zhang and Z. F. Liu, Nano Res., 2015, 8, 2881–2890 CrossRef CAS.
- Z. Wu, B. Fang, A. Bonakdarpour, A. Sun, D. P. Wilkinson and D. Wang, Appl. Catal., B, 2012, 125, 59–66 CrossRef CAS.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef CAS PubMed.
- H. Zhou, F. Yu, J. Sun, R. He, Y. Wang, C. F. Guo, F. Wang, Y. Lan, Z. Ren and S. Chen, J. Mater. Chem. A, 2016, 4, 9472–9476 RSC.
- V. Štengl, J. Tolasz and D. Popelková, RSC Adv., 2015, 5, 89612–89620 RSC.
- X. Zhang, Z. Lai, Z. Liu, C. Tan, Y. Huang, B. Li, M. Zhao, L. Xie, W. Huang and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 5425–5428 CrossRef CAS PubMed.
- X. Zhao, X. Ma, J. Sun, D. Li and X. Yang, ACS Nano, 2016, 10, 2159–2166 CrossRef CAS PubMed.
- L. Cheng, W. Huang, Q. Gong, C. Liu, Z. Liu, Y. Li and H. Dai, Angew. Chem., Int. Ed., 2014, 53, 7860–7863 CrossRef CAS PubMed.
- C. L. Choi, J. Feng, Y. Li, J. Wu, A. Zak, R. Tenne and H. Dai, Nano Res., 2013, 6, 921–928 CrossRef CAS.
- M. J. Jaison, T. N. Narayanan, T. Prem Kumar and V. K. Pillai, J. Mater. Chem. A, 2015, 3, 18222–18228 RSC.
- M. J. Jaison, K. Vikram, T. N. Narayanan and V. K. Pillai, Appl. Phys. Lett., 2014, 104, 153111–153115 CrossRef.
- D. B. Shinde, J. Debgupta, A. Kushwaha, M. Aslam and V. K. Pillai, J. Am. Chem. Soc., 2011, 133, 4168–4171 CrossRef CAS PubMed.
- J. Lin, Z. Peng, G. Wang, D. Zakhidov, E. Larios, M. J. Yacaman and J. M. Tour, Adv. Energy Mater., 2014, 4, 1301875–1301881 CrossRef.
- F. Wang, J. Li, F. Wang, T. A. Shifa, Z. Cheng, Z. Wang, K. Xu, X. Zhan, Q. Wang, Y. Huang, C. Jiang and J. He, Adv. Funct. Mater., 2015, 25, 6077–6083 CrossRef CAS.
- W. F. Chen, J. M. Schneider, K. Sasaki, C. H. Wang, J. Schneider, S. Iyer, S. Iyer, Y. Zhu, J. T. Muckerman and E. Fujita, ChemSusChem, 2014, 7, 2414–2418 CrossRef CAS PubMed.
- Q. Xiang, F. Cheng and D. Lang, ChemSusChem, 2016, 9, 996–1002 CrossRef CAS PubMed.
- Z. Pu, Q. Liu, A. M. Asiri and X. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 21874–21879 CrossRef CAS PubMed.
- L. Yang, X. Wu, X. Zhu, C. He, M. Meng, Z. Gan and P. K. Chu, Appl. Surf. Sci., 2015, 341, 149–156 CrossRef CAS.
- P. D. Tran, S. Y. Chiam, P. P. Boix, Y. Ren, S. S. Pramana, J. Fize, V. Artero and J. Barber, Energy Environ. Sci., 2013, 6, 2452–2459 RSC.
- J. S. Jang, D. J. Ham, N. Lakshminarasimhan, W. Y. Choi and J. S. Lee, Appl. Catal., A, 2008, 346, 149–154 CrossRef CAS.
- X. Zong, J. Han, G. Ma, H. Yan, G. Wu and C. Li, J. Phys. Chem. C, 2011, 115, 12202–12208 CrossRef CAS.
- D. Jing and L. Guo, Catal. Commun., 2007, 8, 795–799 CrossRef CAS.
- X. Fan, H. Zhou and X. Guo, ACS Nano, 2015, 9, 5125–5134 CrossRef CAS PubMed.
- K. Xu, F. Wang, Z. Wang, X. Zhan, Q. Wang, Z. Cheng, M. Safdar and J. He, ACS Nano, 2014, 2, 8468–8476 CrossRef PubMed.
- H. Wang, D. Kong, P. Johanes, J. J. Cha, G. Zheng, K. Yan, N. Liu and Y. Cui, Nano Lett., 2013, 13, 3426–3433 CrossRef CAS PubMed.
- S. Yu, J. Kim, K. R. Yoon, J. W. Jung, J. Oh and I. D. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 28116–28121 CrossRef CAS PubMed.
- A. Bayat and E. Saievar-Iranizad, J. Lumin., 2017, 185, 236–240 CrossRef CAS.
- M. O. Valappil, A. Anil, M. Shaijumon, V. K. Pillai and S. Alwarappan, Chem. – Eur. J., 2017, 23, 9144–9148 CrossRef CAS PubMed.
- Y. Yan, C. Zhang, W. Gu, C. Ding, X. Li and Y. Xian, J. Phys. Chem. C, 2016, 120, 12170–12177 CrossRef CAS.
- J.-F. Lin, O. Pitkänen, J. Mäklin, R. Puskas, A. Kukovecz, A. Dombovari, G. Toth and K. Kordas, J. Mater. Chem. A, 2015, 3, 14609–14616 RSC.
- J. M. Woods, Y. Jung, Y. Xie, W. Liu, Y. Liu, H. Wang and J. J. Cha, ACS Nano, 2016, 10, 2004–2009 CrossRef CAS PubMed.
- S. Anantharaj, K. Karthick and S. Kundu, Materials Today Energy, 2017, 6, 1–26 CrossRef.
- S. Anantharaj, K. Karthick, M. Venkatesh, T. V. S. V. Simha, A. S. Salunke, L. Ma, H. Liang and S. Kundu, Nano Energy, 2017, 39, 30–43 CrossRef CAS.
- K. Karthick, S. Anantharaj, P. E. Karthik, B. Subramanian and S. Kundu, Inorg. Chem., 2017, 56, 6734–6745 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, E. K. Pitchiah and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of materials are given. Figures related to EDS, UV-vis, PL spectra and mentioned electrochemical data are available. See DOI: 10.1039/c8cy02168f |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |