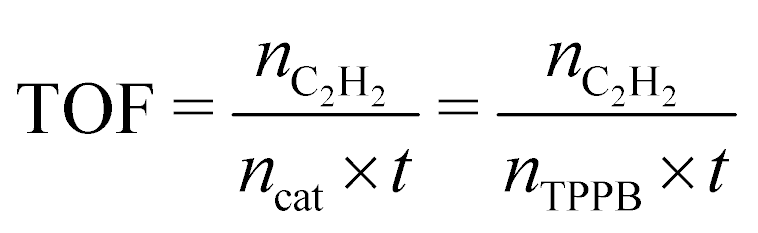Novel nonmetal catalyst of supported tetraphenylphosphonium bromide for acetylene hydrochlorination†
Received
10th October 2018
, Accepted 20th November 2018
First published on 20th November 2018
Abstract
Tetraphenylphosphonium bromide (TPPB) ionic liquid-supported catalysts were synthesized and evaluated for the acetylene hydrochlorination reaction for the development of highly efficient nonmetal catalysts as substitutes for the currently used industrial mercuric catalyst in the production of vinyl chloride (VCM). The optimal 15% TPPB/SAC catalyst exhibited favorable catalytic activity and stability, with the highest acetylene conversion of 97.1% and the selectivity for VCM above 99.5% under the conditions of 220 °C, an acetylene gas hourly space velocity (GHSV) = 30 h−1 and VHCl/VC2H2 = 1.15. Characterized by TPD, FTIR, XPS, etc., TPPB exhibits strong adsorption toward HCl but very weak adsorption toward C2H2 and VCM; in particular, the adsorbed HCl can change the conformational structure of TPPB. DFT calculations suggest that over the active catalytic site of TPPB, the activation energy of acetylene hydrochlorination is 21.15 kcal mol−1, which is much lower than that without catalyst (44.29 kcal mol−1). During the reaction, the H–Cl bond is preferentially activated through accepting the electrons transferred from the anion of TPPB, and then the C2H2 is activated to complete the addition reaction of H and Cl. Such unique preferential activation toward the H–Cl bond as well as the weak adsorption to the product VCM promotes the catalytic activity and the stability of the supported TPPB catalysts. The amount of carbon deposition on the 15% TPPB/SAC catalyst is as low as 2.99%, even after 300 h of reaction. The high activity and stability of the 15% TPPB/SAC catalyst indicate great promise for its application as a nonmetal catalyst for acetylene hydrochlorination.
Introduction
Vinyl chloride monomer (VCM) is a significant synthetic material for the industrial production of polyvinyl chloride (PVC), and the hydrochlorination of acetylene is the main synthetic method for preparing VCM in coal-rich regions.1–3 The current industrial catalyst for acetylene hydrochlorination is HgCl2-supported carbon, which can lead to serious environmental pollution because of the toxicity and volatility of mercuric chloride. To protect human health and the environment from the harmful effects of mercury compounds,4 it is necessary to develop environmentally benign catalysts as substitutes for the HgCl2 catalyst in acetylene hydrochlorination.
Hutchings and other researchers have found that many metal catalysts such as Au,5–8 Ru,9–12 Pd (ref. 13 and 14) and Cu (ref. 15 and 16) can be used as effective non-mercury catalysts for acetylene hydrochlorination. The gold-based catalyst has been evaluated on the pilot-plant scale. Hutchings and co-workers supported the Na3Au(S2O3)2 on carbon to obtain a Au-based catalyst for acetylene hydrochlorination; the 1.6 t catalyst has been evaluated for more than 4500 h online.3 Wei et al. utilized the Au with thiocyanate (–SCN) to synthesize the 0.25 wt% Au-based catalyst, the catalyst displayed favourable performance under 3000 h online testing with the acetylene conversion of 99% and the selectively to VCM of 99%.17 Luo et al. prepared the 0.2 wt% Au/Cu/TCCA catalyst with 6800 h of pilot-trial evaluation.18 However, metal catalysts are generally restricted due to cost and metal reserves and are susceptible to the impurities of sulfur and arsenic in the industrial feed of the acetylene hydrochlorination process. In contrast, nonmetallic catalysts are insensitive to these impurities, stimulating the R&D of efficient nonmetal catalysts for acetylene hydrochlorination.
At present, the reported nonmetallic catalysts for acetylene hydrochlorination mainly include N-doped carbon materials,19–21 carbon nitride materials,22–25 and B–N modified carbon materials.26 For example, Wei and co-workers reported that nitrogen-doped carbon nanotubes (N-CNTs) can promote covalent interactions between acetylene and N-CNT; the highest acetylene conversion was 7.2% under the conditions of T = 180 °C and GHSV(C2H2) = 180 h−1.20 Bao et al. synthesized the N-doped SiC@N–C nonmetallic catalyst, which exhibited the acetylene conversion of 80% under the conditions of GHSV(C2H2) = 30 h−1 and temperature of 200 °C.25 Li et al. also found that the N-doped nonmetal catalyst can enhance the adsorption of acetylene.27 Recently, Bao and co-workers synthesized the porous boron nitride (p-BN) material, displaying favorable catalytic performance for acetylene hydrochlorination, with the highest acetylene conversion of 99% and the selectively to VCM of 99% under the conditions of T = 280 °C and GHSV(C2H2) = 44 h−1, they concluded that the superior catalytic performance was due to the abundant defects and edge sites of the p-BN material, which enhanced the adsorption of acetylene.28 These results suggest a promising pathway to improve the activity of nonmetal catalysts for acetylene hydrochlorination via modulating the adsorption capacity of acetylene, since the carbon-based nonmetallic catalysts are still faced with relatively low activity and poor stability for acetylene hydrochlorination.29
Ionic liquids (ILs) possess unique physicochemical properties involving high solubility, high thermal stability, and negligible volatility, and have been applied in many fields including catalysis, adsorption and organic synthesis.30–32 On the other hand, some ILs exhibit favorable solubility for hydrogen chloride.33,34 Li and coworkers reported that imidazole chloride ILs showed high solubility for HCl, with the solubility being related to the size of the cation in ILs.35 Utilizing the unique properties of ILs, some metallic catalysts have been studied for acetylene hydrochlorination, including the nanoparticles of Pd, Au and Pt synthesized in the presence of anionic surfactant carboxylate ionic liquids (ASC-ILs),36 the Pd NPs@IL catalyst,37 the supported Au–IL complex,38 the Au–Cu–IL/AC catalyst,39 and the supported Ru@IL/AC catalyst, etc.12 It is suggested that these ILs can improve the catalytic activity of metallic species for acetylene hydrochlorination. In order to modulate the adsorption capacity of the reactants on carbon-based nonmetal catalysts, the effect of the preferential adsorption of ILs toward hydrogen chloride on the catalytic activity for acetylene hydrochlorination reaction should be studied.
Tetraphenylphosphonium bromide (TPPB) is not a commonly classified ionic liquid, having a high melting point of 295–300 °C. However, previous work has indicated that adding TPPB can enhance the adsorption HCl on ruthenium-based catalysts.12 In this article, we synthesized the supported TPPB on spherical activated carbon as a nonmetallic catalyst for acetylene hydrochlorination. The optimal 15% TPPB/SAC catalyst exhibits excellent catalytic activity and stability under the conditions of 220 °C, GHSV = 30 h−1 and VHCl/VC2H2 = 1.15. Combining DFT calculations with characterizations of TPD, FTIR, XPS, etc., the interactions between TPPB and the reactants were studied to disclose the catalytic mechanism on the catalyst TPPB/SAC. The high activity and stability of the 15% TPPB/SAC catalyst indicate that it is a promising nonmetal catalyst for acetylene hydrochlorination.
Experimental
Materials
Spherical activated carbon (SAC) was purchased from Shanghai Carbosino Material Co. Ltd. Tetraphenylphosphonium bromide was purchased from Shanghai D&B Biological Science and Technology Co. Ltd. The C2H2 (99.99%) and HCl (99.99%) gases were purchased from Tianjin Dong Xiang Gas Co. Ltd.
Catalyst preparation
A series of TPPB/SAC catalysts was prepared by an incipient wetness impregnation method with SAC as the support and TPPB as the active species. As a representative, TPPB (0.25 g) was dispersed in 8.0 mL ethanol solution, followed by the addition of the support, SAC (5.00 g), under ultrasonic mixing for 30 min. The obtained mixture was incubated at 60 °C for 6 h and then desiccated at 120 °C for 12 h, to prepare the supported catalyst, named 5% TPPB/SAC. Similarly, the catalysts 10% TPPB/SAC, 15% TPPB/SAC and 20% TPPB/SAC were prepared, respectively adopting the TPPB amounts of 0.50 g, 0.75 g and 1.00 g.
Catalyst characterization
The structural properties of the catalysts were investigated by nitrogen adsorption/desorption isotherms at −196 °C using the Quantachrome Autosorb-iQ. Temperature-programmed decomposition (TPD) profiles were recorded to determine the adsorption and desorption properties of the reactants and the product using Quantachrome Instruments AMI-90. The carbon deposition of catalysts and the decomposition temperature of TPPB were determined by thermogravimetric analysis (TGA, METTLER TOLEDO TGA/DSC 2). The catalysts were measured in the temperature range of 35–900 °C at a heating rate of 10 °C min−1 under an air atmosphere. Transmission electron microscopy (TEM) was conducted on a JEM-2010FEF instrument to detect the morphologies of the catalysts. The analysis of the elemental compositions on the surface of the catalysts was performed by X-ray photoelectron spectroscopy (XPS) on the Thermo ESCALAB 250Xi instrument. The functional groups of the catalysts were analyzed by Fourier transform infrared spectroscopy (FTIR) with a Bruker Vertex70 FT-IR spectrophotometer.
Catalyst tests
The catalytic performances of samples for acetylene hydrochlorination were investigated in the fixed-bed micro-reactor (i.d. 10 mm). The temperature controller (Chao Yang Automation Instrument factory) was used to control the reaction temperature. Before the reaction, the reaction system containing the catalyst (5 mL) was purged by nitrogen to remove the impurities of water and air. Hydrogen chloride was put into the reactor first to activate the catalyst, then acetylene was fed through the heated micro-reactor. The gas flow in the reaction was controlled by the mass flow controller. The reaction temperature was 180, 200, 220 or 240 °C, and the GHSV (C2H2) value was 50 or 30 h−1. The reaction effluents passed through the NaOH solution to absorb the excess hydrogen chloride gas, and then the effluent sample was analyzed by gas chromatography (GC-2014C).
Simulation details
All DFT calculations were performed by the Gaussian09 program package.40 No symmetry constraints were imported during the geometry optimization. The Becke-3-Lee-Yang-Parr (B3LYP)41 with the 6-311G** basis set were used for all atoms. Geometry optimization, frequency calculation and intrinsic reaction coordinate (IRC)42–44 were performed on the same level of calculation. All stationary points were calculated as minima (no imaginary frequencies) or transition states (one imaginary frequency) via a Hessian calculation. The sum of the electronic and zero-point energies was used as the configuration energy. The activation energy was calculated as the Gibbs free energy difference between the intermediate state and transition state at 220 °C and 1 atm. Atomic charges were calculated by the Hirshfeld charge. The adsorption energies (Eads) of the reactants or the product on TPPB are defined by eqn (1).| | | Eads = Eadsorption state − Emolecule − ETPPB | (1) |
where Emolecule denotes the energy of free HCl, C2H2 or the product VCM, respectively.
Results and discussion
Catalytic performance of TPPB/SAC catalysts
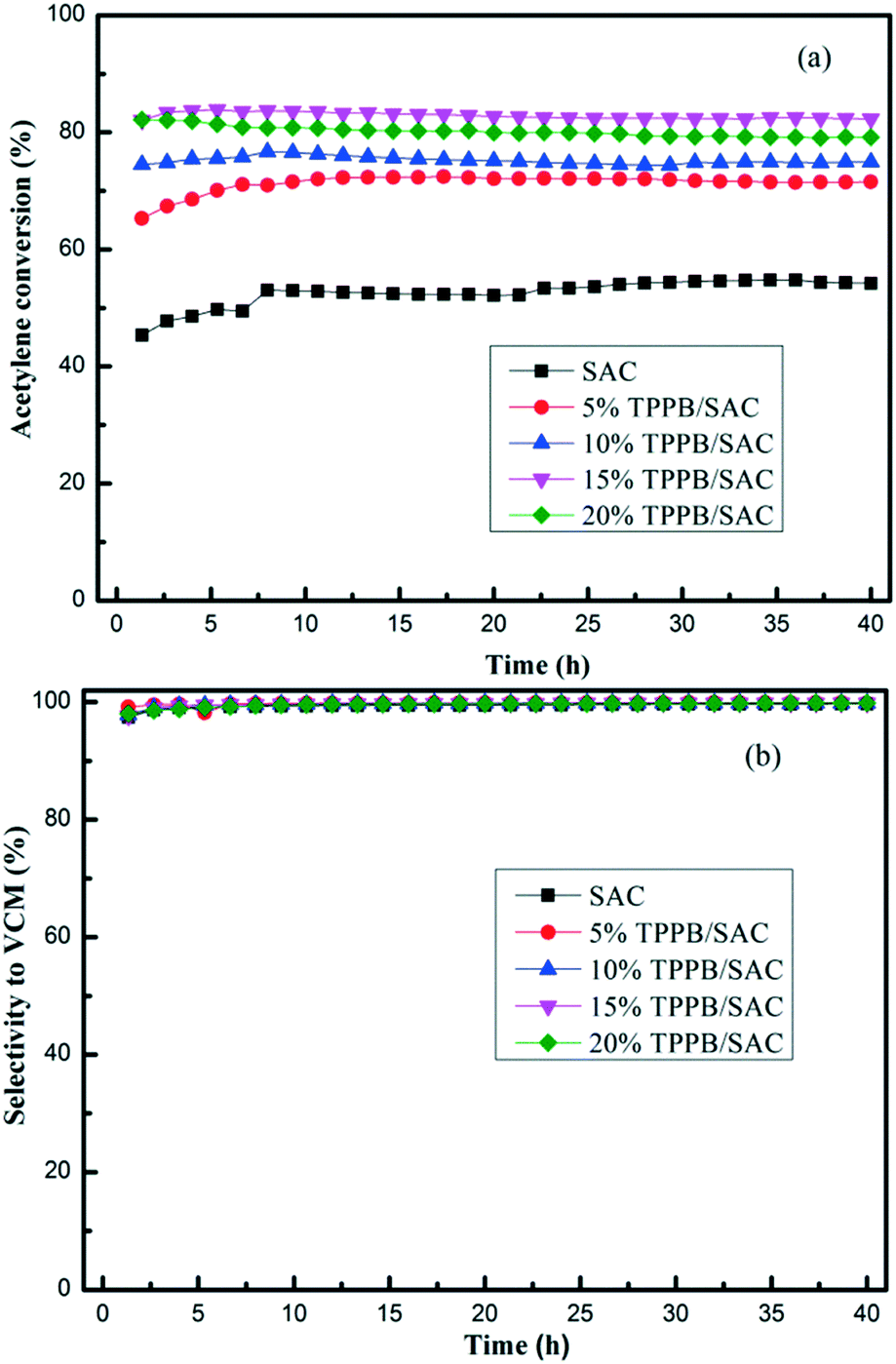
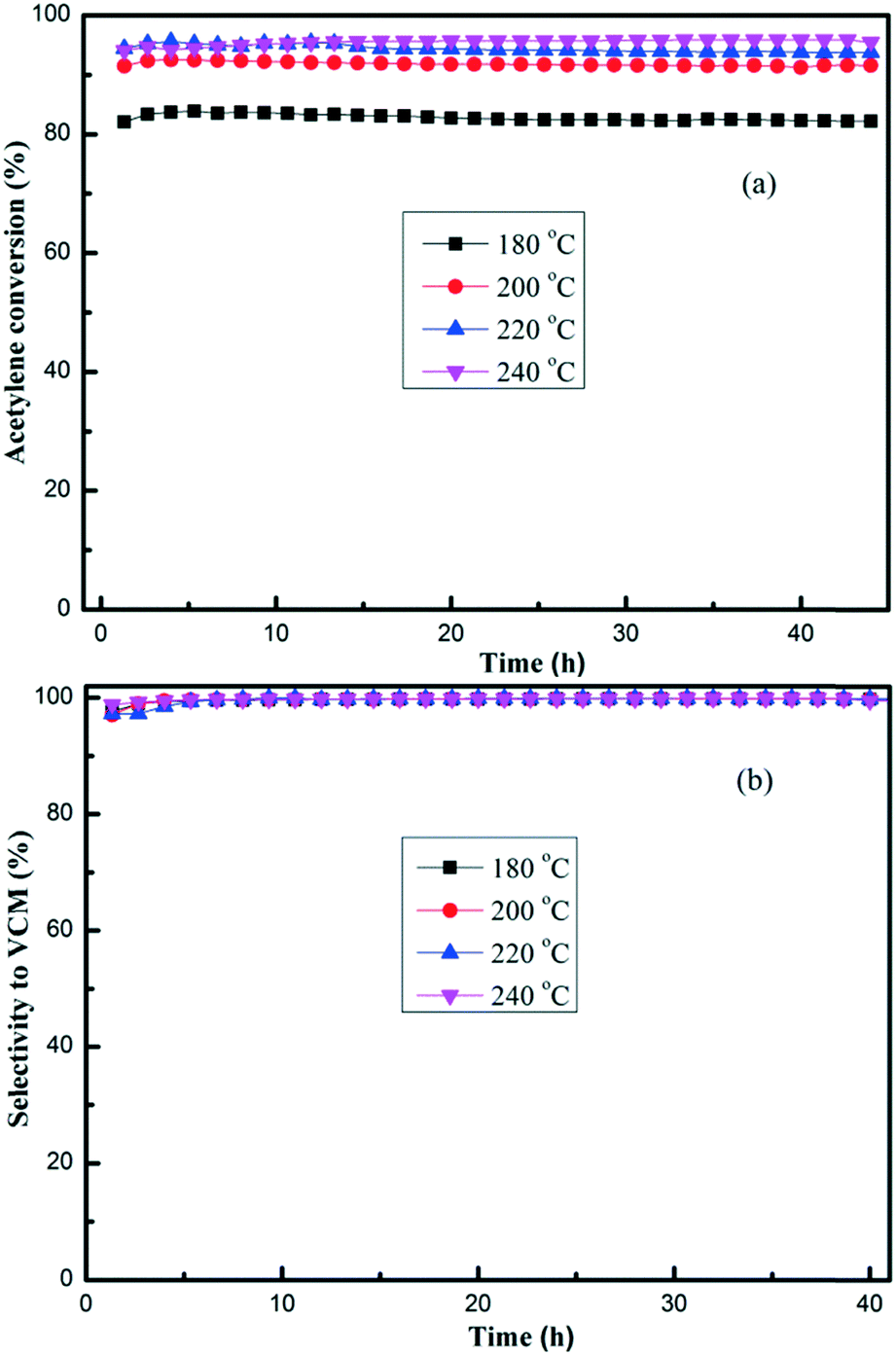
TPPB/SAC catalysts with different TPPB loading amounts were evaluated in the fixed-bed reactor for acetylene hydrochlorination under the conditions of 180 °C and the GHSV (C2H2) of 50 h−1. As shown in Fig. 1, the highest acetylene conversion was achieved over 15% TPPB/SAC (83.7%), followed by 20% TPPB/SAC (81.6%), 10% TPPB/SAC (76.7%) and 5% TPPB/SAC (72.4%), while over the pure support SAC, the acetylene conversion was about 53.2%. For all these catalysts, the selectivity for VCM was greater than 99.5%. The 15% TPPB/SAC catalyst displayed optimal catalytic performance for acetylene hydrochlorination due to the following: 1) when the mass percentage of TPPB was higher than 15%, the excess TPPB resulted in the blocking of micropores or the formation of thick layers of TPPB over the catalyst surface, and consequently increased the diffusion resistance of the reactants and the product during the reaction. Thus, over the 20% TPPB/SAC catalyst, the acetylene conversion was lower. 2) As the mass percentage of TPPB increased to 15%, there appeared more active sites on the catalyst surface, resulting in the increase in the catalytic activity. Thus, the highest acetylene conversion was achieved over the 15% TPPB/SAC. To further optimize the catalytic performance of the 15% TPPB/SAC catalyst, it was evaluated under different reaction temperatures in the range of 180–240 °C. As shown in Fig. 2, the highest acetylene conversion increased with the temperature, being 95.9% at 240 °C, 95.5% at 220 °C, 92.6% at 200 °C and 83.7% at 180 °C. Taking into account the energy consumption during the reaction, it was indicated that the optimal temperature over 15% TPPB/SAC was 220 °C for acetylene hydrochlorination.
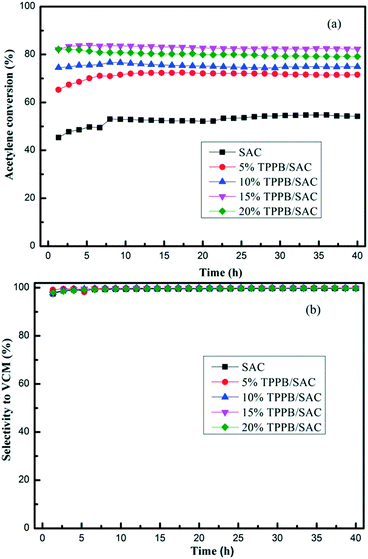 |
| | Fig. 1 Conversion of acetylene (a) and selectivity to VCM (b) over different catalysts including 5% TPPB/SAC, 10% TPPB/SAC, 15% TPPB/SAC and 20% TPPB/SAC. Reaction conditions: Temperature = 180 °C, GHSV (C2H2) = 50 h−1, VHCl/VC2H2 = 1.15. | |
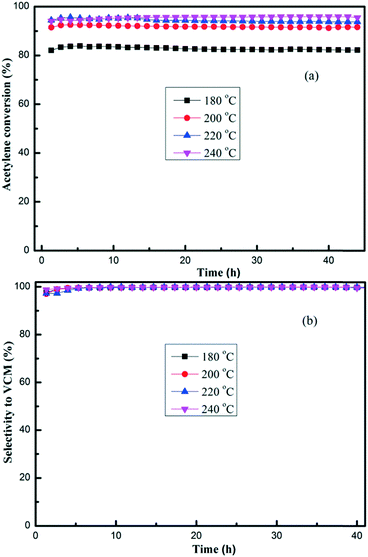 |
| | Fig. 2 Conversion of acetylene (a) and the selectivity for VCM (b) over the 15% TPPB/SAC catalyst under different temperatures in the range of 180–240 °C, with of GHSV (C2H2) = 50 h−1 and VHCl/VC2H2 = 1.15. | |
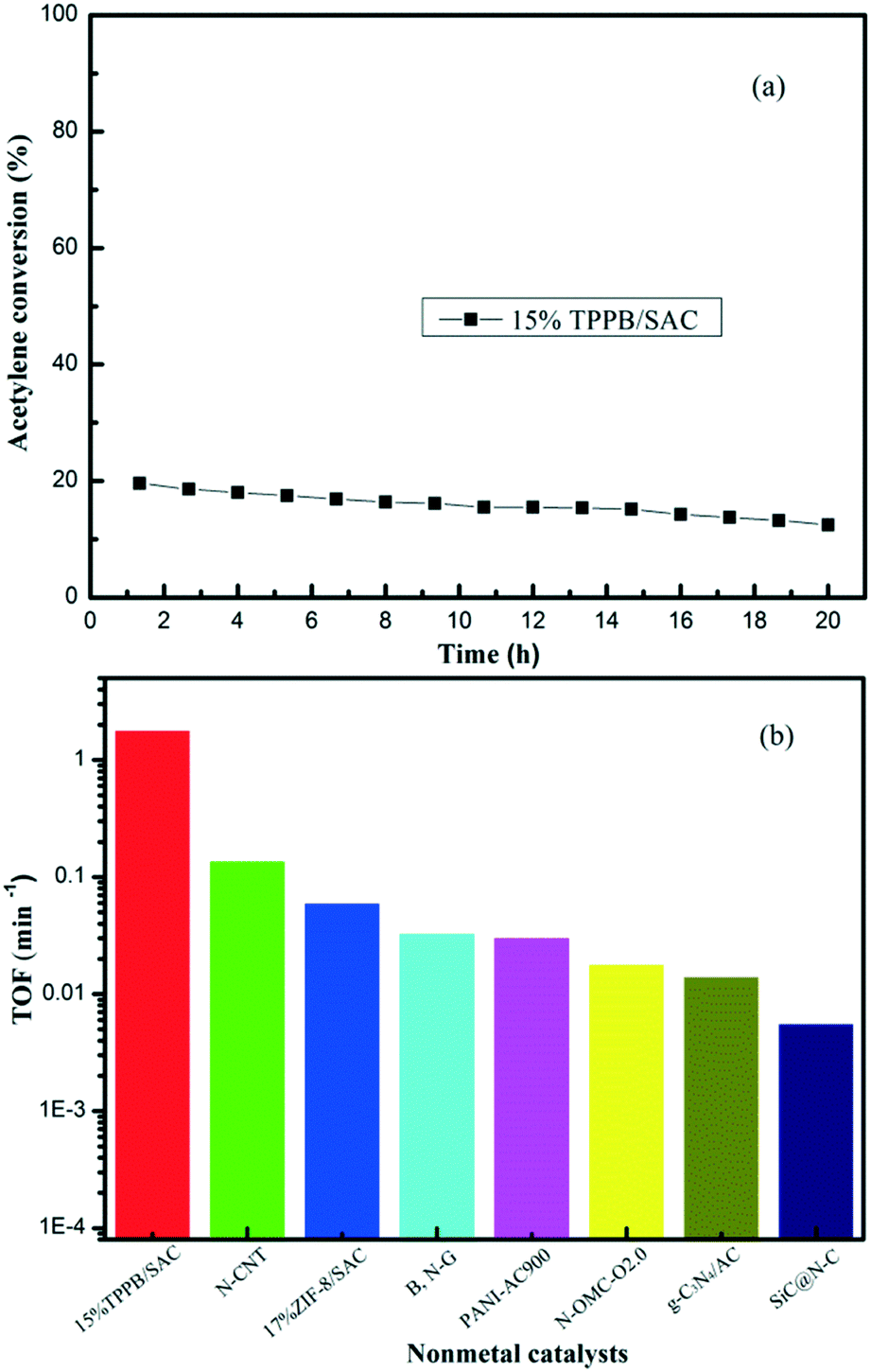
The turnover frequency (TOF) value was determined to evaluate the capacity for transforming acetylene into VCM over the 15% TPPB/SAC catalyst, under the reaction conditions of 220 °C, GHSV (C2H2) = 2300 h−1 and VHCl/VC2H2 = 1.15, as shown in Fig. 3a. The initial conversion of acetylene was 19.6%, which is valid for calculating the TOF.
| | 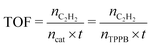 | (2) |
where
nC2H2,
nTPPB and
t represent the initial molar amount of acetylene, the molar amount of TPPB in the catalyst, and the reaction time, respectively.
 |
| | Fig. 3 (a) Conversion of acetylene over the 15% TPPB/SAC catalyst. Reaction conditions: Temperature = 220 °C, GHSV (C2H2) = 2300 h−1, VHCl/VC2H2 = 1.15. (b) The TOFs for different nonmetal catalysts: 15% TPPB/SAC, N-CNT, 17% ZIF-8/SAC, B, N-G, PANI-AC900, N-OMC-O2.0, g-C3N4/AC and SiC@N–C. | |
The TOF value of the 15% TPPB/SAC catalyst in acetylene hydrochlorination is 1.80 min−1. Previously, the TOFs of N-CNT,20 SiC@N–C,26 g-C3N4/AC,26 N-OMC-O2.0,45 PANI-AC900,46 B, N-G26 and 17% ZIF-8/SAC24 metal-free catalysts were reported to be 2.30 × 10−3 s−1, 5.63 × 10−3 min−1, 1.42 × 10−2 min−1, 3.00 × 10−4 s−1, 3.06 × 10−2 min−1, 3.32 × 10−2 min−1 and 6.04 × 10−2 min−1, respectively, as compared in Fig. 3b. The results indicate that the TOF value of the 15% TPPB/SAC metal-free catalyst is significantly higher than the previously reported metal-free catalysts, suggesting that the addition of TPPB can significantly enhance the catalytic performance for acetylene hydrochlorination.
Physicochemical properties of the 15% TPPB/SAC catalyst
Fig. 4 shows the TEM and elemental mapping images of the 15% TPPB/SAC catalyst. The elements of carbon, oxygen, bromine and phosphorus appeared in the 15% TPPB/SAC catalyst. The bromine and phosphorus are typical elements from TPPB, which are uniformly distributed on the surface of SAC, indicating the good dispersion of TPPB on the SAC support. Additionally, XPS was performed to analyze the chemical element on the surface of 15% TPPB/SAC catalyst, as listed in Table 1. Compared with the SAC support, the elements of bromine and phosphorus appeared in the 15% TPPB/SAC catalyst due to the supported TPPB. The atom percentages of bromine and phosphorus are, respectively, 0.32 at% and 0.40 at%, approximate to the atomic ratio of bromine and phosphorus in the TPPB molecule. The textural parameters of SAC, the fresh and spent TPPB/SAC catalysts were measured by the N2 adsorption–desorption experiments. The specific surface area, pore volume and average pore size of samples are listed in Table 2. The specific surface area and the pore volume of the support SAC are 1323 m2 g−1 and 0.62 cm3 g−1, respectively. The specific surface area decreased upon loading TPPB, equal to 1137 m2 g−1 for 5% TPPB/SAC, 1032 m2 g−1 for 10% TPPB/SAC, 873 m2 g−1 for 15% TPPB/SAC and 663 m2 g−1 for 20% TPPB/SAC. The total pore volume of TPPB/SAC catalysts decreased from 0.57 cm3 g−1 to 0.38 cm3 g−1, when the mass percent of TPPB increased from 5% to 20%, attributed to the partial blocking of the micropores on the support SAC by TPPB.
 |
| | Fig. 4 TEM image of the 15% TPPB/SAC catalyst, together with the elemental mapping images of C (red), O (green), Br (yellow) and P (purple). | |
Table 1 Surface element content of samples determined by XPS
| Samples |
Surface atomic composition of samples (atom.%) |
| C 1s |
O 1s |
P 2p |
Br 3d |
| SAC |
95.06 |
4.94 |
|
|
| 15% TPPB/SAC |
96.00 |
3.28 |
0.40 |
0.32 |
Table 2 Texture parameters of samples
| Samples |
S
BET (m2 g−1) |
V (cm3 g−1) |
D (nm) |
| Fresh |
Spent |
Fresh |
Spent |
Fresh |
Spent |
|
S
BET: surface area; V: total pore volume; D: average pore diameter.
|
| SAC |
1323 |
1258 |
0.62 |
0.57 |
1.89 |
1.82 |
| 5% TPPB/SAC |
1137 |
1026 |
0.57 |
0.55 |
2.06 |
2.05 |
| 10% TPPB/SAC |
1032 |
977 |
0.51 |
0.49 |
1.99 |
2.01 |
| 15% TPPB/SAC |
873 |
862 |
0.46 |
0.47 |
2.13 |
2.16 |
| 20% TPPB/SAC |
663 |
646 |
0.38 |
0.39 |
2.20 |
2.42 |
The average pore size of the TPPB/SAC catalysts increased with increasing the amount of TPPB in the TPPB/SAC catalysts, which was also attributed to the micropores of SAC being filled by the TPPB. For the spent 5% TPPB/SAC, 10% TPPB/SAC, 15% TPPB/SAC and 20% TPPB/SAC catalysts that experienced 40 h of reaction, the specific surface areas decreased by approximately 9.7%, 5.3%, 1.2% and 2.5%, respectively, as compared with the fresh individual counterparts. The pore volumes of the spent TPPB/SAC catalysts showed no obvious changes, as compared with the fresh catalysts. Generally, the reduction of specific surface area in the spent catalyst is mainly caused by the carbon deposition on the surface during acetylene hydrochlorination. However, such small variations do not affect the catalytic activities over TPPB/SAC for acetylene hydrochlorination, as reflected by the stable acetylene conversion in Fig. 1.
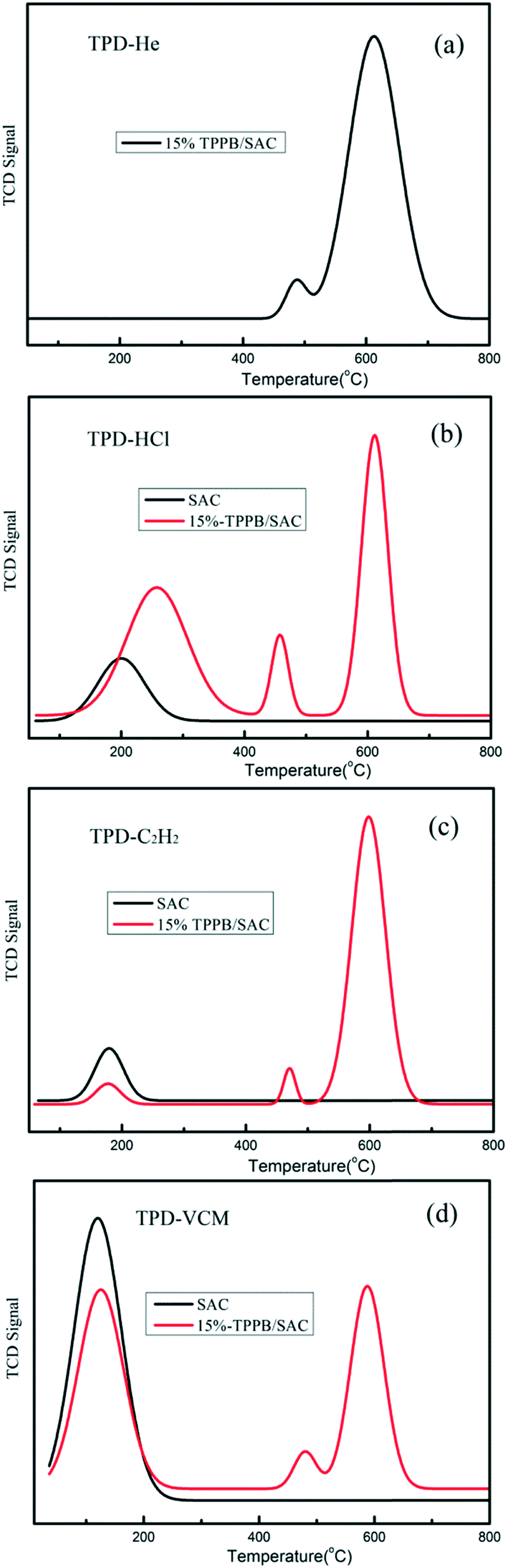
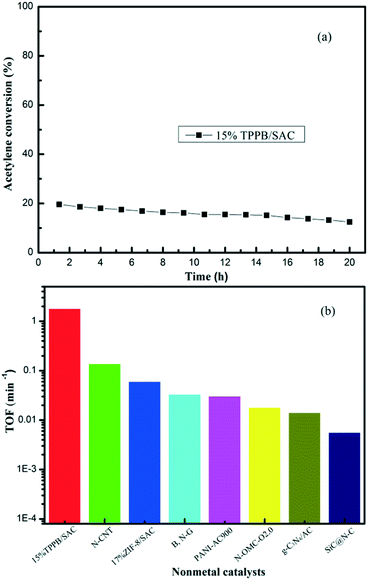
The adsorption properties of the reactants and the product on the optimal 15% TPPB/SAC catalyst were examined by the TPD experiments. In Fig. 5a, for the inert gas helium, the TPD profile shows two desorption peaks located at 488 and 613 °C respectively, representing the decomposition of TPPB in 15% TPPB/SAC catalyst.12 For the reactant HCl, the TPD profile on the 15% TPPB/SAC catalyst showed a peak at 258 °C (Fig. 5b), whereas on the pure support SAC, the peak was located at 200 °C. Moreover, the desorption peak area of the 15% TPPB/SAC catalyst was obviously higher than that of the support SAC. It is known that the peak location in TPD profiles indicates the adsorption strength and the peak area reflects the adsorption capacity. Hence, the 15% TPPB/SAC catalyst showed stronger adsorption toward HCl, as compared with the support SAC. In the case of another reactant, C2H2, the desorption peak in the TPD profiles is located at 178 °C for either 15% TPPB/SAC or SAC, while the peak area on the 15% TPPB/SAC is smaller than that on the SAC (Fig. 5c), suggesting the weaker adsorption toward C2H2 on the 15% TPPB/SAC catalyst. For the product VCM, the location of the desorption peak of VCM is located at 123 °C for both 15% TPPB/SAC and SAC. The TPD profiles indicate that the desorption area of VCM on the 15% TPPB/SAC catalyst is smaller than that on the support SAC (Fig. 5d). This illustrates that the adsorption of HCl is superior while the desorption of VCM is fast on the 15% TPPB/SAC catalyst, which is promising for obtaining high catalytic stability for acetylene hydrochlorination.
 |
| | Fig. 5 TPD profiles of HCl, C2H2 and VCM on the fresh 15% TPPB/SAC catalyst versus the support SAC: (a) TPD-He, (b) TPD-HCl, (c) TPD-C2H2 and (d) TPD-VCM. | |
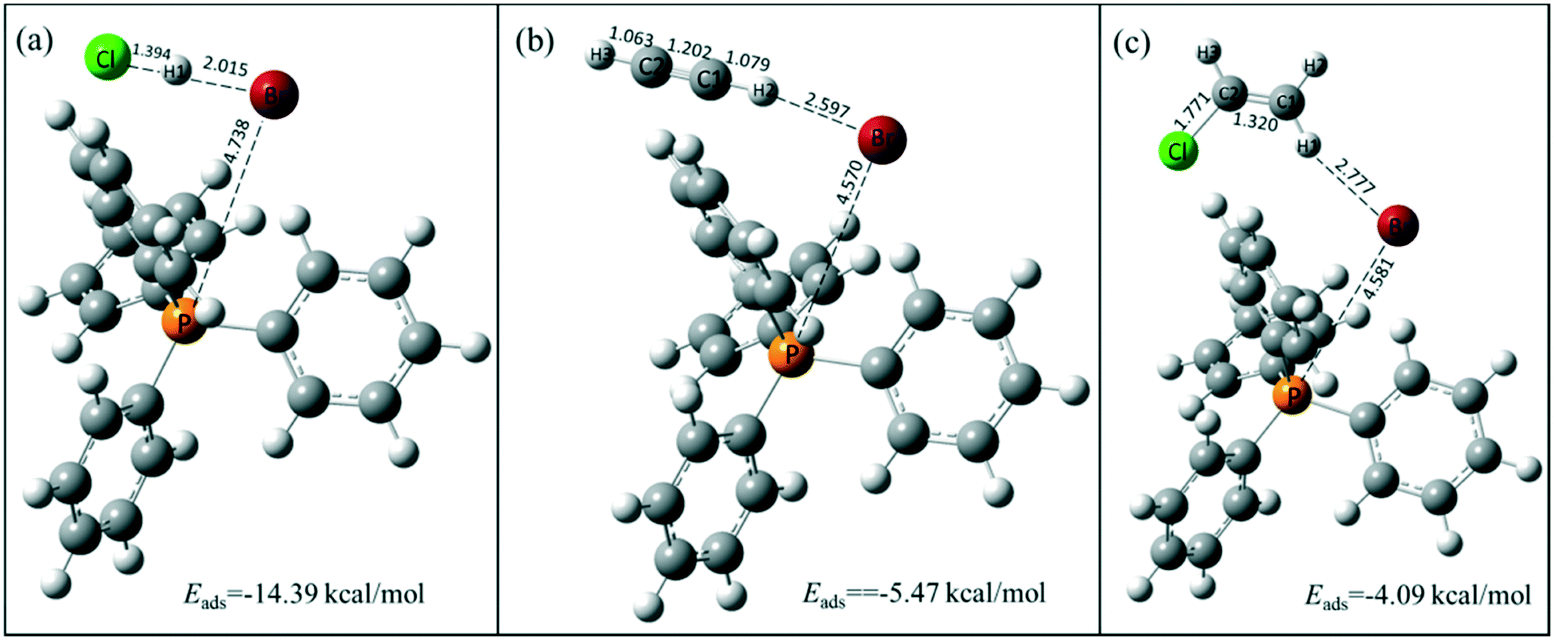
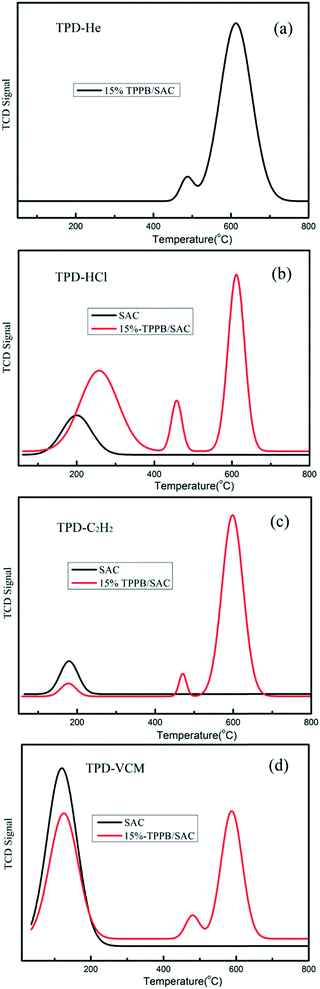
The active species of TPPB facilitates the adsorption of HCl and the desorption of product VCM, which was further demonstrated by the DFT calculation results. As shown in Fig. 6, the adsorption energies of HCl, C2H2 and VCM on TPPB are −14.39, −5.47 and −4.09 kcal mol−1, respectively, suggesting that the adsorption of HCl on TPPB is much easier than that of C2H2 and VCM. When HCl is adsorbed on TPPB (shown in Fig. 6a), the H1 atom of HCl interacts with the bromine anion of TPPB, resulting in the H–Cl bond being elongated from 1.287 Å (in the gas phase) to 1.394 Å. In the case of C2H2 and VCM adsorbed on TPPB (Fig. 6b and c), there are weak interactions between TPPB and C2H2 or VCM. In addition, we adopted the reduced density gradient (RDG) method to further analyze the interactions between TPPB and the reactants. As shown in Fig. 7, the results indicate that the Br anions of TPPB have distinct hydrogen bonds (blue region) with H in HCl, besides the van der Waals forces (green region) between HCl and TPPB. On the other hand, the interaction between TPPB and C2H2 is much weaker than that with HCl. Thus, TPPB can effectively activate the reactant of HCl to facilitate the occurrence of acetylene hydrochlorination.
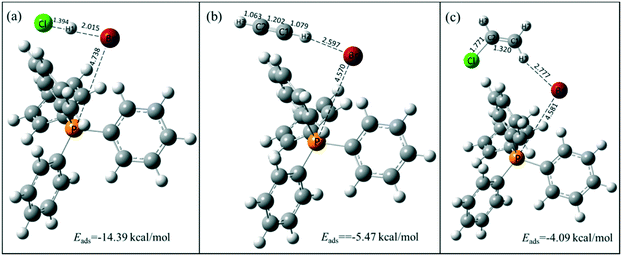 |
| | Fig. 6 The stable structures of HCl (a), C2H2 (b), and VCM (c) adsorption on TPPB. Carbon, hydrogen, bromine, phosphorus and chlorine atoms are depicted in gray, white, red, orange and green, respectively. The selected bond lengths are in Å. | |
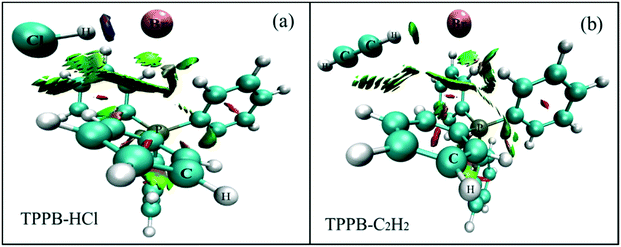 |
| | Fig. 7 The stable structures of HCl and C2H2 adsorption on TPPB using RDG method: (a) TPPB-HCl and (b) TPPB-C2H2. The red area indicates steric hindrance, the green area indicates van der Waals' forces, and the blue area indicates strong hydrogen bond. | |
As listed in Table 3, the Hirshfeld charge analysis shows that both the cation and the anion of TPPB donate electrons to the reactants and the product, but the electrons transferred from the anion of TPPB to the reactant HCl is twice that to the reactant C2H2. The sum of transferred electrons from TPPB to HCl is 0.224e, while 0.076e and 0.044e, respectively, are transferred to C2H2 and C2H3Cl. Moreover, the charge variations of individual atoms of the reactants and the product are listed in Table S1.† It is illustrated that most of the electrons are transferred from the anion of TPPB to the Cl atom of HCl, resulting in the H–Cl bond polarity increasing to 0.446, which is significantly higher than that in free HCl (0.316). Such unique preferential activation toward the H–Cl bond, as well as the weak adsorption to the product VCM, promotes the catalytic activity and the stability of the supported TPPB catalysts.
Table 3 The Hirshfeld charge variations in TPPB before and after adsorption
| Adsorbate |
The charge of the anion in TPPB (e) |
The charge of the cation in TPPB (e) |
| Before adsorption |
After adsorption |
The amount of electrons lost |
Before adsorption |
After adsorption |
The amount of electrons lost |
| HCl |
−0.668 |
−0.510 |
0.158 |
0.668 |
0.734 |
0.066 |
| C2H2 |
−0.668 |
−0.610 |
0.048 |
0.668 |
0.696 |
0.028 |
| C2H3Cl |
−0.668 |
−0.634 |
0.034 |
0.668 |
0.678 |
0.010 |
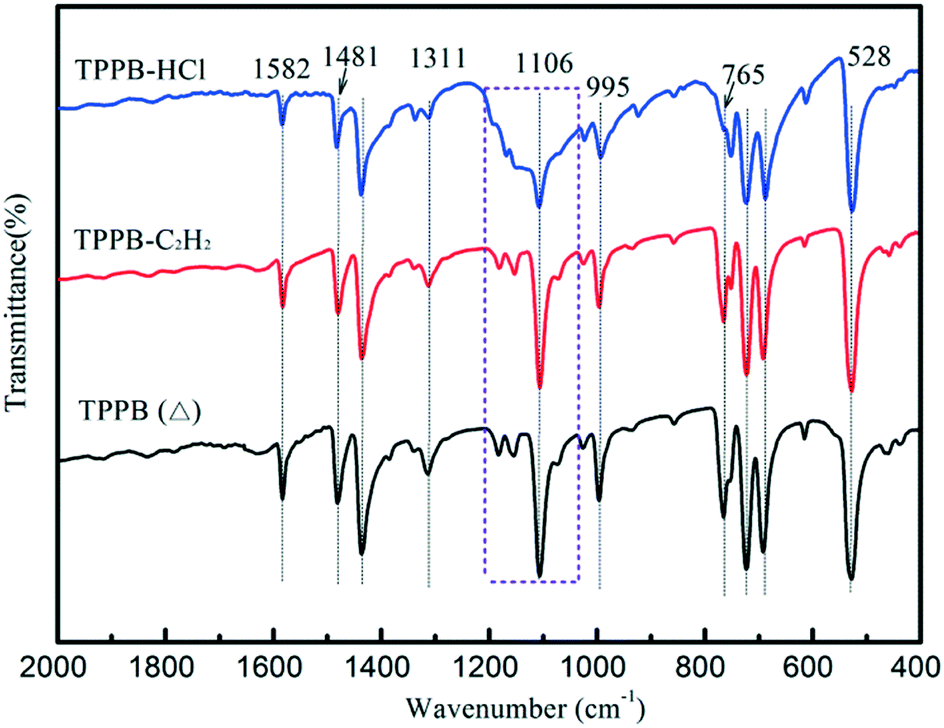
On the other hand, FTIR spectra were obtained to detect the variation in the structural conformation of TPPB caused by the adsorption of reactants. Fig. 8 shows the FTIR spectra of the TPPB (Δ), TPPB-C2H2 and TPPB-HCl samples, where TPPB (Δ) indicates the pure TPPB sample being heated at 220 °C for 1 h under an air atmosphere, TPPB-C2H2 is the TPPB sample that experienced heating at 220 °C for 1 h under the environment of C2H2, and TPPB-HCl is the TPPB sample that experienced heating at 220 °C for 1 h under the atmosphere of HCl. Generally, the peaks at 692 cm−1, 723 cm−1 and 765 cm−1 correspond to the out-of-plane deformation vibration of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H in aromatic rings; the peaks at 995 cm−1, 1154 cm−1, 1181 cm−1 and 1311 cm−1 are attributed to the in-plane deformation vibration of C–H, while the peaks at 1433 cm−1, 1481 cm−1 and 1582 cm−1 are due to the aromatic CC stretching vibration.47,48 For the bonds between aromatic carbon and phosphorus, the peak at 1106 cm−1 reflects the stretching vibration, whereas the peak at 528 cm−1 is assigned to the bending vibration. As seen in Fig. 8, TPPB-HCl has a special broad band at around 1106 cm−1, besides the weakening peaks at 1481 and 765 cm−1, illustrating that the adsorption of HCl results in variations in the conformation of TPPB, in particular, the interactions with HCl affect the covalent bonding between aromatic carbon and phosphorus in TPPB, which was confirmed by the change in the dihedral angles in the phenyl group (Table S2†).
C–H in aromatic rings; the peaks at 995 cm−1, 1154 cm−1, 1181 cm−1 and 1311 cm−1 are attributed to the in-plane deformation vibration of C–H, while the peaks at 1433 cm−1, 1481 cm−1 and 1582 cm−1 are due to the aromatic CC stretching vibration.47,48 For the bonds between aromatic carbon and phosphorus, the peak at 1106 cm−1 reflects the stretching vibration, whereas the peak at 528 cm−1 is assigned to the bending vibration. As seen in Fig. 8, TPPB-HCl has a special broad band at around 1106 cm−1, besides the weakening peaks at 1481 and 765 cm−1, illustrating that the adsorption of HCl results in variations in the conformation of TPPB, in particular, the interactions with HCl affect the covalent bonding between aromatic carbon and phosphorus in TPPB, which was confirmed by the change in the dihedral angles in the phenyl group (Table S2†).
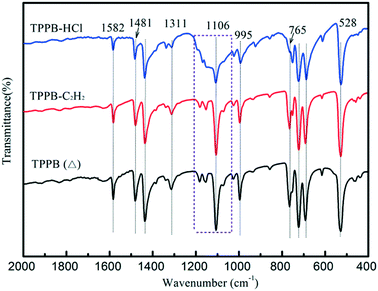 |
| | Fig. 8 FTIR spectra of TPPB (Δ), TPPB-C2H2 and TPPB-HCl samples. | |
Fig. S1† displays the stable structures of TPPB, TPPB-C2H2 and TPPB-HCl, indicating that the H atom in HCl interacts with the Br anion in TPPB, transferring the electrons to the H and Cl atoms in HCl. The Cl (47) atom in HCl is close to two H atoms (36 and 37) in the aromatic ring of TPPB (Fig. S1c†); the distances between Cl (47) and two H atoms (36 and 37) are 2.967 Å and 3.302 Å, respectively. The charges of the two H atoms (36 and 37) were increased to 0.039e and 0.053e from 0.044e and 0.056e (in TPPB), respectively, suggesting that the Cl (47) atom in HCl interacts with the aromatic ring of TPPB to alter the vibrational structure of phenyl in TPPB. These results confirm that TPPB has strong adsorption toward HCl and the adsorbed HCl can change the conformational structure of TPPB.
Catalytic mechanism of TPPB in acetylene hydrochlorination
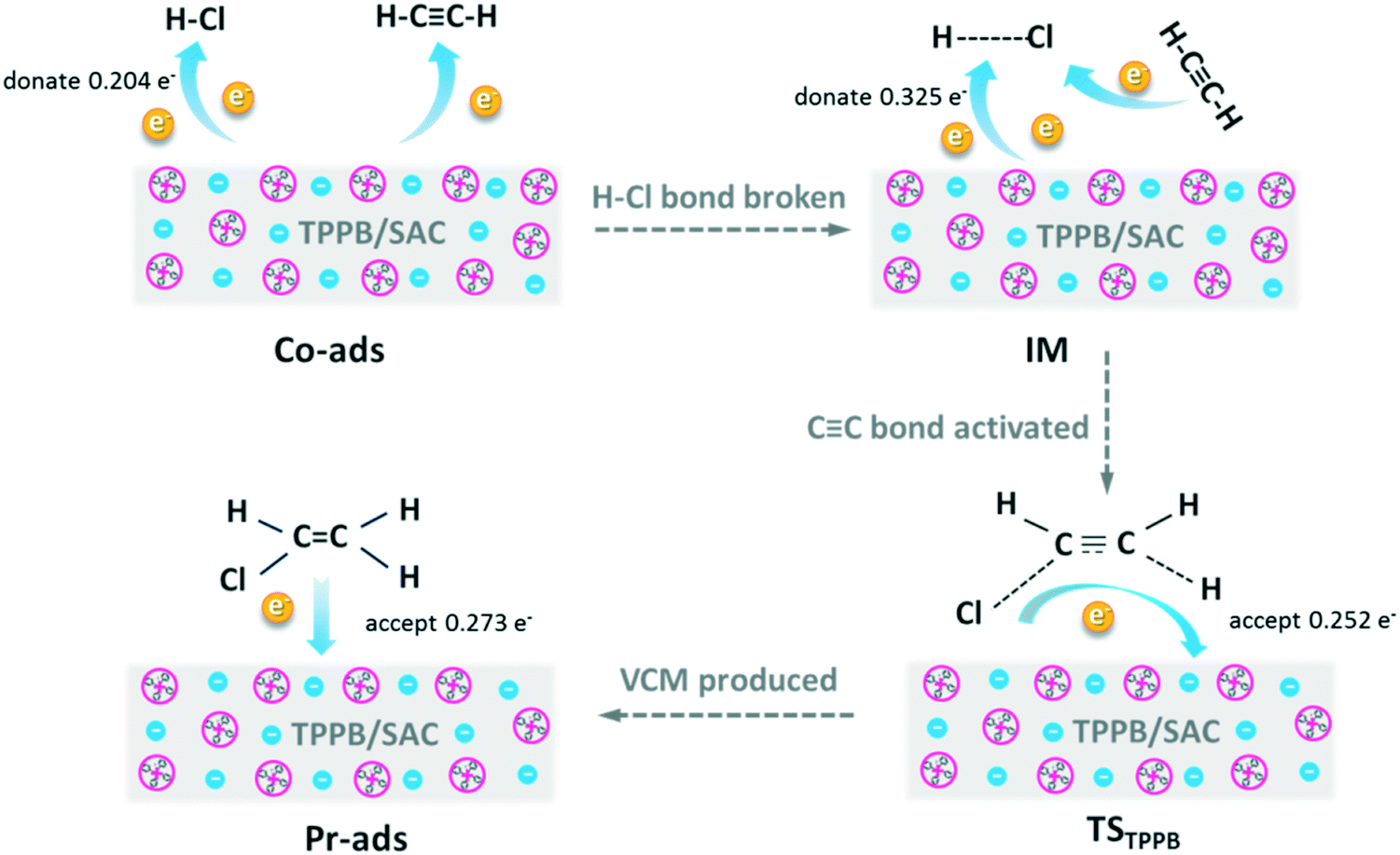
We used DFT calculations to understand the mechanistic pathway over the TPPB catalyst. Fig. 9 displays the reaction pathway over the TPPB catalyst, including the HCl and C2H2 co-adsorption on TPPB (Co-ads), the intermediate (IM), the transition state (TSTPPB), the production of VCM on TPPB (Pr-ads) and the desorption of VCM (Pr), as well as the details of electron transfer (Table S3†). In the Co-ads configuration, the distance between H1 and Br is 2.054 Å, which is slightly longer than the H–Br bond length of free HBr (1.427 Å). The cation and anion of TPPB donate electrons to HCl and C2H2, while the total number of electrons transferred from TPPB to HCl (0.204e) is much more than that to C2H2 (0.060e), indicating that the TPPB can preferentially activate HCl. In the IM configuration, the H1 atom in HCl moves toward the Br anion to form the H1–Br bond, with the bond length of 1.450 Å being close to that of the free HBr molecule (1.427 Å). Meanwhile, the distance between H1 and Cl is as great as 4.865 Å, suggesting the breakage of the H1–Cl bond. Compared with the Co-ads configuration, the chlorine atom in IM gets more electrons from the anion of TPPB and C2H2 (0.348e). The C1![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C2 bond polarity increases from 0.039 to 0.047. In the configuration of TSTPPB, the C1C2 bond polarity is greatly increased to 0.162, corresponding to the charge of −0.071e for C1 and 0.059e for C2. The distance between H1 and Br is stretched to 1.855 Å, and H1 atom attacks the C1 atom in C2H2. Meanwhile, some electrons on the chlorine atom and C2H2 are transferred back to TPPB, mainly on the Br anion. The scheme of the electron transfer process during the reaction is shown in Fig. 10. The transition state has one imaginary frequency (−276.95 cm−1), which is related to the stretching vibration of the H1 atom between the C1 and Br atoms and the bending vibration of the H3 atom. To confirm the rationality of this transition state, IRC was performed and the results are shown in Fig. S2.† The calculated reactant via IRC is the intermediate state and the calculated product via IRC is the C2H3Cl adsorption structure. Therefore, the transition state is reasonable and the reaction pathway is logical. Once the C2H3Cl molecule is generated, the desorption energy is only 4.09 kcal mol−1, which is conducive to the desorption of VCM from the surface of the catalyst, and inhibits the formation of carbon deposition by the polymerization of VCM. In contrast, Fig. S3† shows the reaction pathway in the absence of catalyst, including the reactant (Rea), transition state (TS) and product (Pr), with the energy barrier as high as 42.70 kcal mol−1. In the presence of TPPB catalyst, the acetylene hydrochlorination reaction is performed in two steps, with the first step involving activating the reactant of HCl by the anion of TPPB (with the energy barrier of 14.08 kcal mol−1), and the second step being the addition of H and Cl atoms to the activated C2H2 (with the energy barrier of 14.29 kcal mol−1). Through the two-step reaction pathway in the presence of TPPB, the energy barrier of acetylene hydrochlorination is significantly reduced. Converting to Gibbs free energy, the reaction activation energy was calculated as 21.15 kcal mol−1 over the TPPB catalyst, and 44.29 kcal mol−1 in the absence of a catalyst.
C2 bond polarity increases from 0.039 to 0.047. In the configuration of TSTPPB, the C1C2 bond polarity is greatly increased to 0.162, corresponding to the charge of −0.071e for C1 and 0.059e for C2. The distance between H1 and Br is stretched to 1.855 Å, and H1 atom attacks the C1 atom in C2H2. Meanwhile, some electrons on the chlorine atom and C2H2 are transferred back to TPPB, mainly on the Br anion. The scheme of the electron transfer process during the reaction is shown in Fig. 10. The transition state has one imaginary frequency (−276.95 cm−1), which is related to the stretching vibration of the H1 atom between the C1 and Br atoms and the bending vibration of the H3 atom. To confirm the rationality of this transition state, IRC was performed and the results are shown in Fig. S2.† The calculated reactant via IRC is the intermediate state and the calculated product via IRC is the C2H3Cl adsorption structure. Therefore, the transition state is reasonable and the reaction pathway is logical. Once the C2H3Cl molecule is generated, the desorption energy is only 4.09 kcal mol−1, which is conducive to the desorption of VCM from the surface of the catalyst, and inhibits the formation of carbon deposition by the polymerization of VCM. In contrast, Fig. S3† shows the reaction pathway in the absence of catalyst, including the reactant (Rea), transition state (TS) and product (Pr), with the energy barrier as high as 42.70 kcal mol−1. In the presence of TPPB catalyst, the acetylene hydrochlorination reaction is performed in two steps, with the first step involving activating the reactant of HCl by the anion of TPPB (with the energy barrier of 14.08 kcal mol−1), and the second step being the addition of H and Cl atoms to the activated C2H2 (with the energy barrier of 14.29 kcal mol−1). Through the two-step reaction pathway in the presence of TPPB, the energy barrier of acetylene hydrochlorination is significantly reduced. Converting to Gibbs free energy, the reaction activation energy was calculated as 21.15 kcal mol−1 over the TPPB catalyst, and 44.29 kcal mol−1 in the absence of a catalyst.
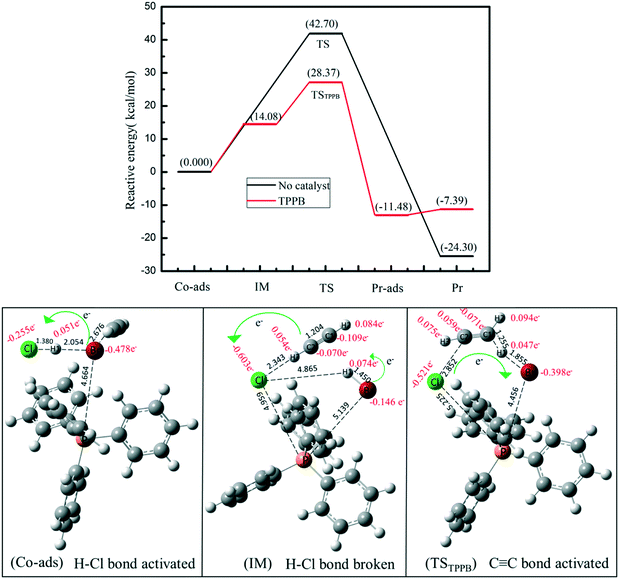 |
| | Fig. 9 Reaction energy diagram and the geometries of the substances involved in the reaction pathway: Co-adsorbed reactants (Co-ads), intermediate (IM), transition state (TS), product-adsorbed (Pr-ads) and product (Pr). Carbon, hydrogen, bromine, phosphorus and chlorine atoms are depicted in gray, white, red, orange and green, respectively. | |
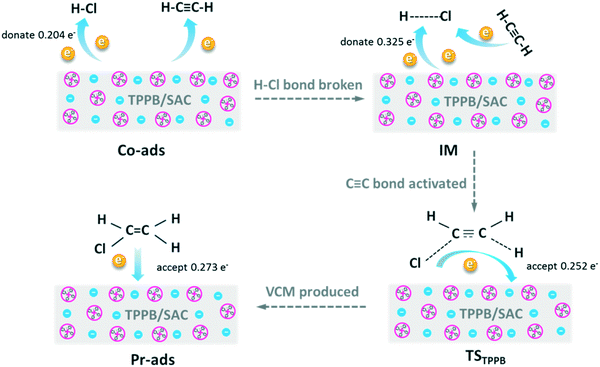 |
| | Fig. 10 The schematic diagram of electron transfer processes in view of the substances disclosed in the reaction pathway of Fig. 9. | |
Previously, Wei and his coworkers adopted the N-doped carbon nanotubes as the nonmetallic catalyst for acetylene hydrochlorination and found that N-CNTs could enhance the interaction between the highest occupied molecular orbital (HOMO) in NCNT and the lowest unoccupied molecular orbital (LUMO) in C2H2, suggesting that the electrons were transferred from NCNT to C2H2.20 In our previous studies, the Ru@IL/AC catalysts were applied in acetylene hydrochlorination, and the results indicated that the electrons prefer to transfer from the HOMO of ILs to the LUMO of HCl.12 Herein, in the presence of TPPB catalyst, the H–Cl bond is first activated via accepting electrons from the Br anion of TPPB, then the charge variations in both carbon atoms of C2H2 result in the increase in the C1C2 bond polarity and promote the completion of the addition reaction of H and Cl atoms to C2H2 to produce VCM.
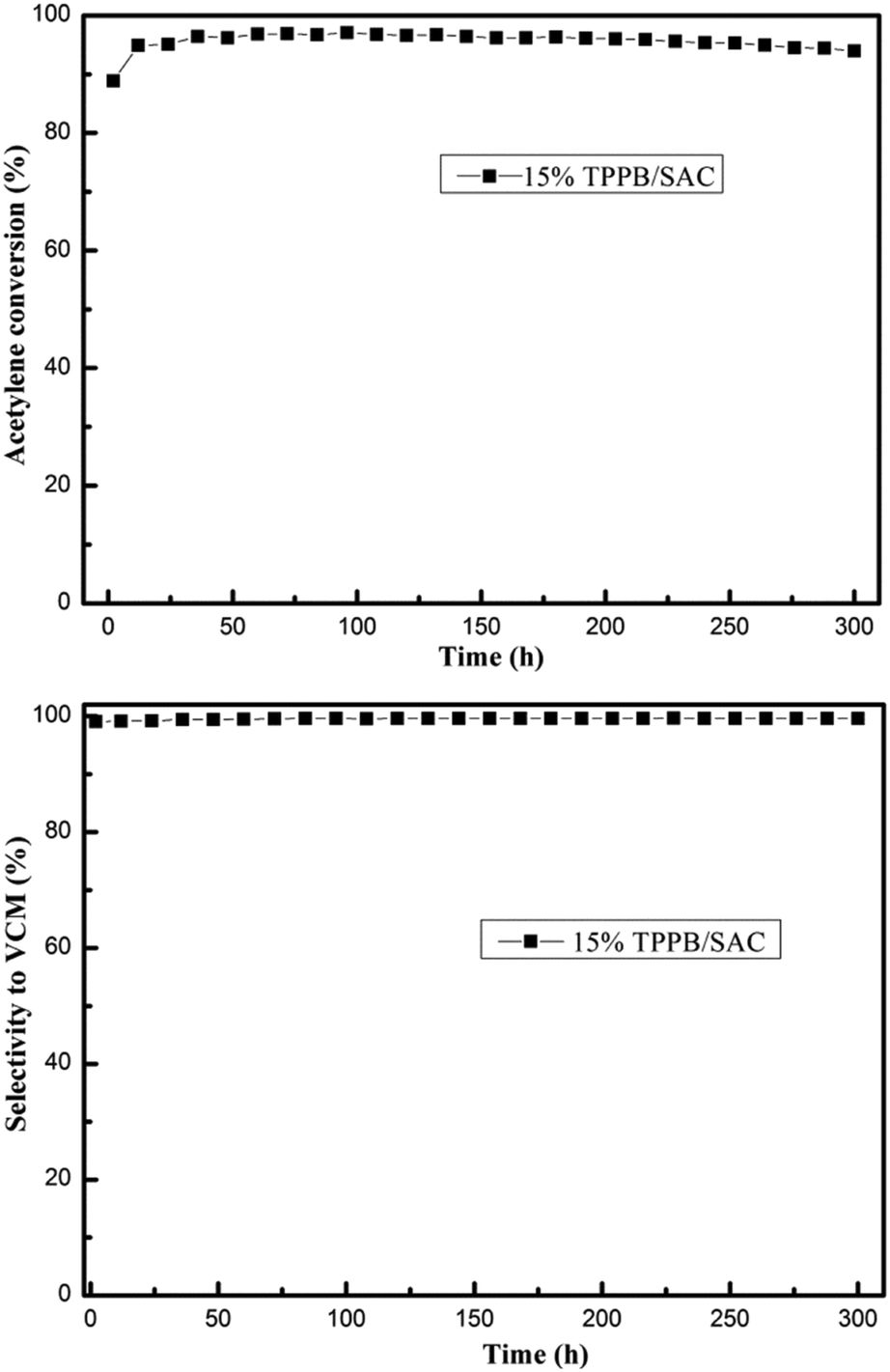
The stability evaluation of the 15% TPPB/SAC catalyst
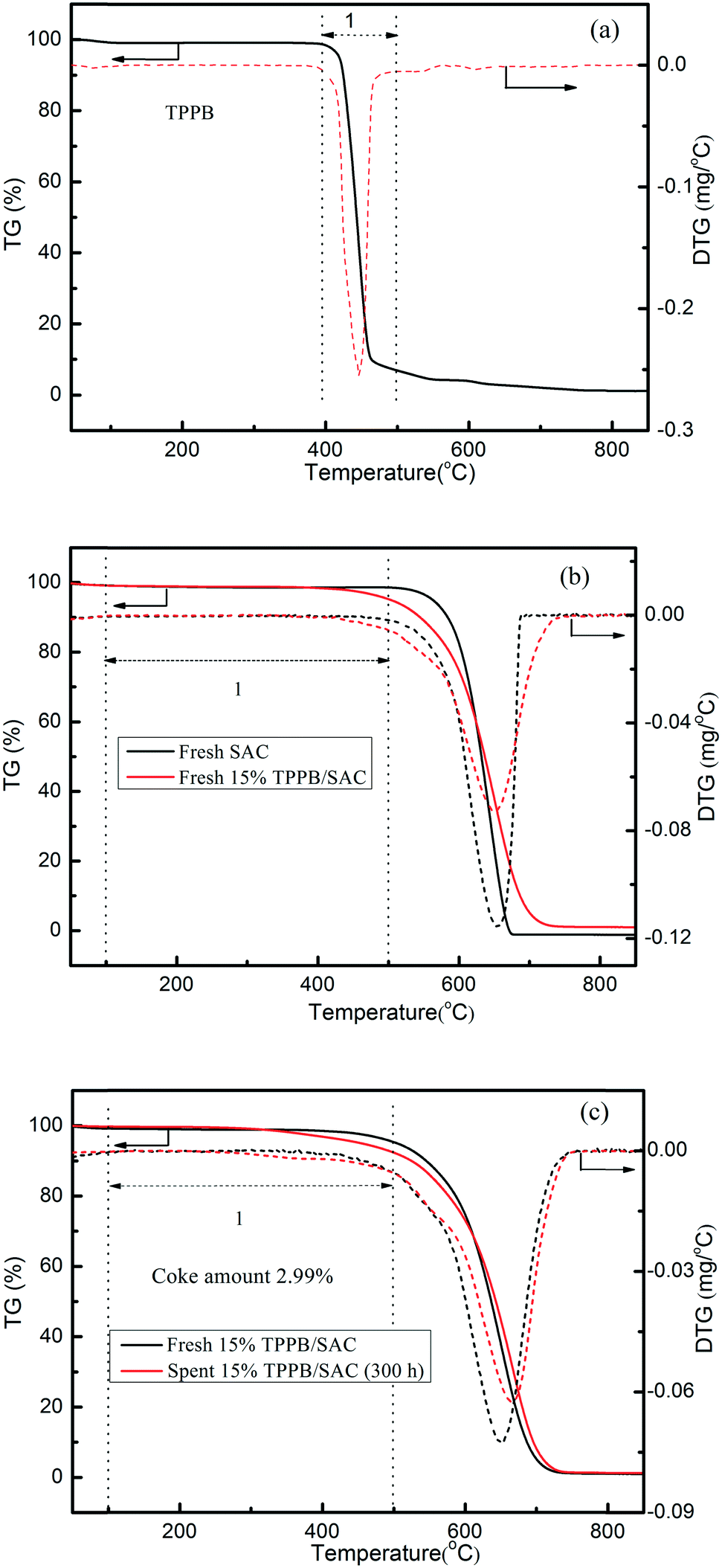
Previous literature indicated that most of the nonmetal catalysts, including PANI-AC900,3 N-OMC-700,4 g-C3N4/AC,5 B, N-GO,6 Z4M1 (ref. 22) and ZIF-8/SAC,24 showed poor catalytic stability for acetylene hydrochlorination. The stability evaluation of the 15% TPPB/SAC catalyst was performed under the conditions of 220 °C and GHSV (C2H2) of 30 h−1, approximating to the industrial space velocity. As shown in Fig. 11, the 15% TPPB/SAC catalyst exhibits superior catalytic stability. The highest acetylene conversion was 97.1%, and the selectivity for VCM was approximately 99.5%. With time, the acetylene conversion slightly decreased to 94.0% within 300 h. TGA profiles were performed to measure the amount of carbon deposition on the spent TPPB/SAC catalyst. For the unsupported pure TPPB, the decomposition occurred in the temperature range of 400–500 °C in an air atmosphere (Fig. 12a). For the fresh 15% TPPB/SAC, the mass loss was only 3.79% in the temperature range of 100–500 °C, while the quick mass loss at temperatures higher than 500 °C is associated with the burning of the carbon support in the air (Fig. 12b). It is suggested that the interactions between TPPB and the support retard the decomposition of TPPB. For the spent catalyst, 15% TPPB/SAC, after 40 h of reaction, no carbon deposition was detected (Fig. S4†), and after 300 h of reaction, the amount of carbon deposition was calculated as 2.99% (Fig. 12c).
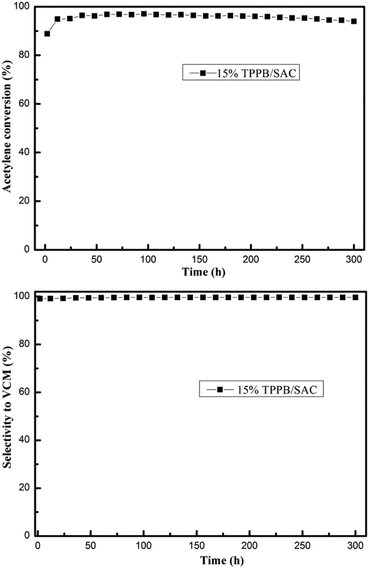 |
| | Fig. 11 Catalytic performance evaluation of the 15% TPPB/SAC catalyst. Reaction conditions: Temperature = 220 °C, GHSV (C2H2) = 30 h−1 and VHCl/VC2H2 = 1.15. | |
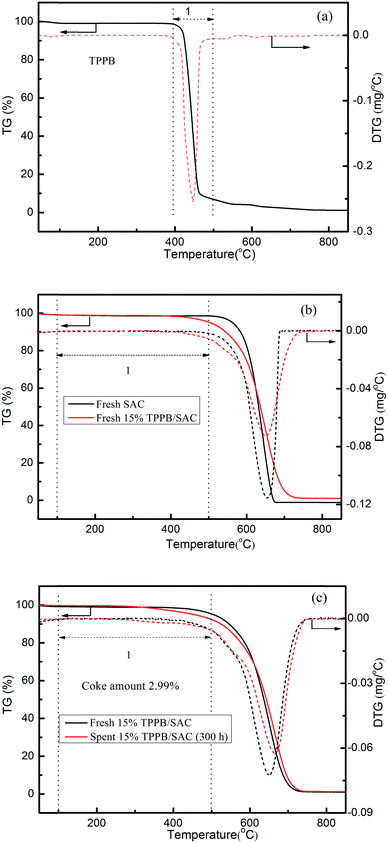 |
| | Fig. 12 TGA curves of the pure TPPB (a), the fresh SAC and fresh 15% TPPB/SAC catalyst (b), the fresh and spent 15% TPPB/SAC catalysts (c) (after 300 h reaction, under the reaction conditions of 220 °C, GHSV (C2H2) = 30 h−1 and VHCl/VC2H2 = 1.15) in air atmosphere. | |
Therefore, due to the low carbon deposition, the 15% TPPB/SAC catalyst exhibits superior stability for acetylene hydrochlorination. A combination of TPD profiles, DFT calculations and TGA indicated that the preferential activation toward H–Cl bonds and the weak adsorption of C2H2 and VCM resulted in the favorable catalytic activity of TPPB/SAC and also the low carbon deposition during the stability evaluation reaction process.
Conclusions
We adopted TPPB IL as the active species using the simple incipient wetness impregnation method to prepare the supported TPPB/SAC catalysts for acetylene hydrochlorination. The highest acetylene conversion over the optimal 15% TPPB/SAC metal-free catalyst was 97.1%, and the selectivity for VCM was above 99.5% under the conditions of 220 °C, GHSV = 30 h−1 and VHCl/VC2H2 = 1.15. Through catalyst characterization and the DFT calculation results, it was confirmed that the addition of TPPB can efficiently facilitate the adsorption and activation of HCl, with weak adsorption to C2H2 and VCM. The anion bromide of TPPB donates electrons to the chlorine atom of HCl, resulting in the H–Cl bond polarity increasing from 0.316 (free HCl) to 0.446. Compared with the reaction in the absence of the catalyst, the reaction activation energy with TPPB as the metal-free catalyst decreased from 44.29 kcal mol−1 (without catalyst) to 21.15 kcal mol−1, meaning that the TPPB active species reduced the reaction barrier by the adsorption and activation of HCl. Furthermore, the weak adsorption to C2H2 and VCM inhibited carbon deposition during the reaction; the amount of carbon deposition was calculated as 2.99% in the spent 15% TPPB/SAC catalyst after 300 h of reaction, suggesting that the 15% TPPB/SAC could be a promising metal-free catalyst for acetylene hydrochlorination.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Natural Science Foundation of China (21776215) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT_15R46).
References
- J. Zhang, N. Liu, W. Li and B. Dai, Front. Chem. Sci. Eng., 2011, 5, 514–520 CrossRef CAS.
- M. Zhu, Q. Wang, K. Chen, Y. Wang, C. Huang, H. Dai, F. Yu, L. Kang and B. Dai, ACS Catal., 2015, 5, 5306–5316 CrossRef CAS.
- P. Johnston, N. Carthey and G. J. Hutchings, J. Am. Chem. Soc., 2015, 137, 14548–14557 CrossRef CAS PubMed.
- T. K. Mackey, J. T. Contreras and B. A. Liang, Sci. Total Environ., 2014, 472, 125–129 CrossRef CAS PubMed.
- M. Conte, A. F. Carley, C. Heirene, D. J. Willock, P. Johnston, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 250, 231–239 CrossRef CAS.
- H. Zhang, B. Dai, X. Wang, L. Xu and M. Zhu, J. Ind. Eng. Chem., 2012, 18, 49–54 CrossRef CAS.
- J. Zhang, Z. He, W. Li and Y. Han, RSC Adv., 2012, 2, 4814–4821 RSC.
- X. Li, M. Zhu and B. Dai, Appl. Catal., B, 2013, 142–143, 234–240 CrossRef CAS.
- Y. Pu, J. Zhang, L. Yu, Y. Jin and W. Li, Appl. Catal., A, 2014, 488, 28–36 CrossRef CAS.
- H. Zhang, W. Li, Y. Jin, W. Sheng, M. Hu, X. Wang and J. Zhang, Appl. Catal., B, 2016, 189, 56–64 CrossRef CAS.
- J. Zhang, W. Sheng, C. Guo and W. Li, RSC Adv., 2013, 3, 21062–21068 RSC.
- S. Shang, W. Zhao, Y. Wang, X. Li, J. Zhang, Y. Han and W. Li, ACS Catal., 2017, 7, 3510–3520 CrossRef CAS.
- Q. Song, S. Wang, B. Shen and J. Zhao, Pet. Sci. Technol., 2010, 28, 1825–1833 CrossRef CAS.
- L. Wang, F. Wang and J. Wang, New J. Chem., 2016, 40, 3019–3023 RSC.
- H. Li, F. Wang, W. Cai, J. Zhang and X. Zhang, Catal. Sci. Technol., 2015, 5, 5174–5184 RSC.
- K. Zhou, J. Si, J. Jia, J. Huang, J. Zhou, G. Luo and F. Wei, RSC Adv., 2014, 4, 7766–7769 RSC.
- K. Zhou, J. Jia, C. Li, H. Xu, J. Zhou, G. Luo and F. Wei, Green Chem., 2015, 17, 356–364 RSC.
- Xu H, K. Zhou, J. Si, C. Lib and G. Luo, Catal. Sci. Technol., 2016, 6, 1357–1366 RSC.
- X. Li, Y. Wang, L. Kang, M. Zhu and B. Dai, J. Catal., 2014, 311, 288–294 CrossRef CAS.
- K. Zhou, B. Li, Q. Zhang, J. Huang, G. Tian, J. Jia, M. Zhao, G. Luo, D. Su and F. Wei, ChemSusChem, 2014, 7, 723–728 CrossRef CAS PubMed.
- X. Wang, B. Dai, Y. Wang and F. Yu, ChemCatChem, 2014, 6, 2339–2344 CrossRef CAS.
- X. Li, J. Zhang and W. Li, J. Ind. Eng. Chem., 2016, 44, 146–154 CrossRef CAS.
- S. Chao, F. Zou, F. Wan, X. Dong, Y. Wang, Y. Wang, Q. Guan, G. Wang and W. Li, Sci. Rep., 2017, 7, 39789–39795 CrossRef PubMed.
- X. Li, J. Zhang, Y. Han, M. Zhu, S. Shang and W. Li, J. Mater. Sci., 2018, 53, 4913–4926 CrossRef CAS.
- X. Li, X. Pan, L. Yu, P. Ren, X. Wu, L. Sun, F. Jiao and X. Bao, Nat. Commun., 2014, 5, 3688–3695 CrossRef CAS.
- B. Dai, K. Chen, Y. Wang, L. Kang and M. Zhu, ACS Catal., 2015, 5, 2541–2547 CrossRef CAS.
- T. Zhang, J. Zhao, J. Xu, J. Xu, X. Di and X. Li, Chin. J. Chem. Eng., 2016, 24, 484–490 CrossRef CAS.
- P. Li, H. Li, X. Pan, K. Tie, T. Cui, M. Ding and X. Bao, ACS Catal., 2017, 7, 8572–8577 CrossRef CAS.
- X. Li, P. Li, X. Pan, H. Ma and X. Bao, Appl. Catal., B, 2017, 210, 116–120 CrossRef CAS.
- M. J. Schneider, M. Lijewski, R. Woelfel, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2013, 52, 6996–6999 CrossRef CAS PubMed.
- W. Zhang, Q. Wang, H. Wu, P. Wu and M. He, Green Chem., 2014, 16, 4767–4774 RSC.
- N. Sahiner, A. O. Yasar and N. Aktas, Renewable Energy, 2017, 101, 1005–1012 CrossRef CAS.
- F. Pöhlmann, L. Schilder, W. Korth and A. Jess, ChemPlusChem, 2013, 78, 570–577 CrossRef.
- K. Murugappan and D. S. Silvester, Phys. Chem. Chem. Phys., 2016, 18, 2488–2494 RSC.
- R. He, B. Long, Y. Lu, H. Meng and C. Li, J. Chem. Eng. Data, 2012, 57, 2936–2941 CrossRef CAS.
- J. Hu, Q. Yang, L. Yang, Z. Zhang, B. Su, Z. Bao, Q. Ren, H. Xing and S. Dai, ACS Catal., 2015, 5, 6724–6731 CrossRef CAS.
- L. Yang, Q. Yang, J. Hu, Z. Bao, B. Su, Z. Zhang, Q. Ren and H. Xing, AIChE J., 2018, 64, 2536–2544 CrossRef CAS.
- J. Zhao, S. Gu, X. Xu, T. Zhang, Y. Yu, X. Di, J. Ni, Z. Pan and X. Li, Catal. Sci. Technol., 2016, 6, 3263–3270 RSC.
- J. Zhao, Y. Yu, X. Xu, S. Di, B. Wang, H. Xu, J. Ni, L. Guo, Z. Pan and X. Li, Appl. Catal., B, 2017, 206, 175–183 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. Stephens and F. J. Devlin, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- Y. Yang, G. Lan, X. Wang and Y. Li, Chin. J. Catal., 2016, 37, 1242–1248 CrossRef CAS.
- C. Zhang, L. Kang, M. Zhu and B. Dai, RSC Adv., 2015, 5, 7461–7468 RSC.
- G. Wang, X. Liu, Y. Ren, C. Zhang and X. Tao, Crystals, 2017, 7, 154–160 CrossRef.
- B. Haddad, A. Paolone, D. Villemin, M. Taqiyeddine, E-h. Belarbi, S. Bresson, M. Rahmouni, N. R. Dhumal, H. J. Kim and J. Kiefer, J. Mol. Struct., 2017, 1146, 203–212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02103a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Yao
Nian
ab,
Yao
Nian