
Applications of 2D MXenes in energy conversion and storage systems
Jinbo
Pang†
 ab,
Rafael G.
Mendes†
ac,
Alicja
Bachmatiuk
acd,
Liang
Zhao
c,
Huy Q.
Ta
c,
Thomas
Gemming
a,
Hong
Liu
*be,
Zhongfan
Liu
*cf and
Mark H.
Rummeli
*acd
ab,
Rafael G.
Mendes†
ac,
Alicja
Bachmatiuk
acd,
Liang
Zhao
c,
Huy Q.
Ta
c,
Thomas
Gemming
a,
Hong
Liu
*be,
Zhongfan
Liu
*cf and
Mark H.
Rummeli
*acd
aThe Leibniz Institute for Solid State and Materials Research Dresden (IFW Dresden), Helmholtzstr. 20, Dresden, D-01069, Germany
bInstitute for Advanced Interdisciplinary Research (iAIR), University of Jinan, Jinan 250022, China. E-mail: ifc_liuh@ujn.edu.cn
cSoochow Institute for Energy and Materials InnovationS (SIEMIS), Optoelectronics and Energy & Collaborative Innovation Center of Suzhou Nano Science and Technology, and Key Laboratory of Advanced Carbon Materials and Wearable Energy Technologies of Jiangsu Province, School of Energy, Soochow University, Suzhou, 215006, China. E-mail: mhr1967@yahoo.com
dCentre of Polymer and Carbon Materials, Polish Academy of Sciences (CMPW PAN), ul. M. Curie-Sklodowskiej 34, Zabrze, PL-41-819, Poland
eState Key Laboratory of Crystal Materials, Center of Bio & Micro/Nano Functional Materials, Shandong University, 27 Shandanan Road, Jinan, Shandong 250100, China. E-mail: hongliu@sdu.edu.cn
fCenter for Nanochemistry (CNC), Beijing Science and Engineering Center for Nanocarbons, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China. E-mail: zfliu@pku.edu.cn
First published on 2nd November 2018
Abstract
Transition metal carbides and nitrides (MXenes), a family of two-dimensional (2D) inorganic compounds, are materials composed of a few atomic layers of transition metal carbides, nitrides, or carbonitrides. Ti3C2, the first 2D layered MXene, was isolated in 2011. This material, which is a layered bulk material analogous to graphite, was derived from its 3D phase, Ti3AlC2 MAX. Since then, material scientists have either determined or predicted the stable phases of >200 different MXenes based on combinations of various transition metals such as Ti, Mo, V, Cr, and their alloys with C and N. Extensive experimental and theoretical studies have shown their exciting potential for energy conversion and electrochemical storage. To this end, we comprehensively summarize the current advances in MXene research. We begin by reviewing the structure types and morphologies and their fabrication routes. The review then discusses the mechanical, electrical, optical, and electrochemical properties of MXenes. The focus then turns to their exciting potential in energy storage and conversion. Energy storage applications include electrodes in rechargeable lithium- and sodium-ion batteries, lithium–sulfur batteries, and supercapacitors. In terms of energy conversion, photocatalytic fuel production, such as hydrogen evolution from water splitting, and carbon dioxide reduction are presented. The potential of MXenes for the photocatalytic degradation of organic pollutants in water, such as dye waste, is also addressed, along with their promise as catalysts for ammonium synthesis from nitrogen. Finally, their application potential is summarized.
 Jinbo Pang | Jinbo Pang (H-Index 12) joined the Institute for Advanced Interdisciplinary Research (iAIR), University of Jinan, Shandong, China in 2018 as an assistant professor. He received his MEng from the Institute of Photoelectronics, Nankai University, China, in 2011 and his PhD in Material Science from Dresden University of Technology (TU Dresden), Germany in 2017. He completed a Postdoctoral Fellowship at the Leibniz Institute for Solid State and Materials Research Dresden (IFW Dresden) in 2017. His research focuses on the synthesis, electronics, and electrochemical energy applications of graphene and other novel 2D materials as well as their characterization with electron microscopy. |
 Top left: Rafael G. Mendes, top right: Alicja Bachmatiuk, bottom left: Liang Zhao, bottom middle: Huy Q. Ta and bottom right: Thomas Gemming | Rafael G. Mendes (H-Index 14) is currently working as a postdoctoral fellow at IFW Dresden. He obtained a MSc. in Nanobiophysics (2010) and a PhD in Physics from TU Dresden (2015). Alicja Bachmatiuk (H-Index 32) currently heads the Graphene Group at the Polish Academy of Sciences and manages a double Cs corrected TEM at the Polish Center for Technology Development, PORT (former Wroclaw Research Centre EIT+), Poland. She is a guest professor at SIEMIS. She obtained her PhD at Szczecin University and joined IFW Dresden with a Humboldt postdoctoral fellowship. Liang Zhao (H-Index 5) is a PhD candidate at SIEMIS. Huy Q. Ta (H-Index 12) is currently working as a postdoctoral fellow at IFW-Dresden. He received his MSc. at Sungkyunkwan University and his PhD from the Polish Academy of Sciences (2017). Thomas Gemming (H-Index 44) is currently the Acting Director of the Institute for Complex Materials (IKM) at IFW Dresden and is Head of the Department of Micro- and Nanostructures. He obtained his MSc from the University of Karlsruhe (TH) 1994 and his PhD from the University of Stuttgart (1998); he joined the MPI for Metal Research Stuttgart as a staff scientist. He has been the Executive Secretary of the German Society for Electron Microscopy since 2006. |
 Hong Liu | Hong Liu (H-Index 56) is a professor at the State Key Laboratory of Crystal Materials, Shandong University. He also acts as the director of the Institute for Advanced Interdisciplinary Research (iAIR), University of Jinan. He received his PhD degree in 2001 from Shandong University (China). His current research is mainly focused on the chemical processing of nanomaterials for energy-related applications, including photocatalysis, tissue engineering (especially the interaction between stem cells and the nanostrucutures of biomaterials) and nonlinear optical crystals. |
 Zhongfan Liu | Zhongfan Liu (H-Index 88) received his PhD from the University of Tokyo (1990) and performed postdoctoral work at the Institute for Molecular Science (IMS) in Japan (1991 to 1993). He has been a Full Professor at Peking University since 1993 and a Changjiang Chair Professor since 1999. He was elected as a member of the Chinese Academy of Sciences (2011). His research interest focuses on carbon nanomaterials and novel 2D atomic crystals for energy applications. He was elected a Fellow of the RSC and IOP. He also serves as an editor or advisory board member for 10 journals, including Advanced Materials. |
 Mark H. Rummeli | Mark H. Rummeli (H-Index 50) heads the electron microscopy laboratories at Soochow Institute for Energy and Materials InnovationS (SIEMIS), Soochow University, where he is a full professor and director of the Characterization Center. He heads a gas sensor laboratory at the Polish Academy of Sciences, Zabzre. He earned his PhD from London Metropolitan University. His research focuses on the growth mechanisms of nanostructures and their functionalization. He is a Fellow of the RSC and IMMM. He has contributed to over 300 journal papers, including Science and Nature Nanotechnology. He serves as an editor or international advisory board member for several journals. |
1 Introduction
The exfoliation of graphene and other layered materials has driven a tremendous amount of research in 2D materials for more than a decade, which is attributed to their extraordinary electrochemical and optoelectronic properties. The incorporation of 2D layered materials has enhanced the performance of conventional devices, e.g. rechargeable lithium-ion batteries, fuel production, photodetectors, solar steam production, and photothermal therapies. Among 2D materials, graphene, MoS2, phosphorene, and their derivatives represent the most intensively and successfully investigated materials. The potential of MXenes can only be completely explored by fully understanding their fundamentals, ranging from their synthesis and properties to their complex applications. To date, few scientific questions have been answered and many more remain to be investigated, especially concerning technical issues for mass production and process integration. MXenes, a family of 2D layered structures (Fig. 1), remain insufficiently studied and are still in the early stages of understanding since their discovery in 2011.1 This may be due to the limited synthesis methods for their production, which for a long time required the use of hazardous hydrofluoric acid.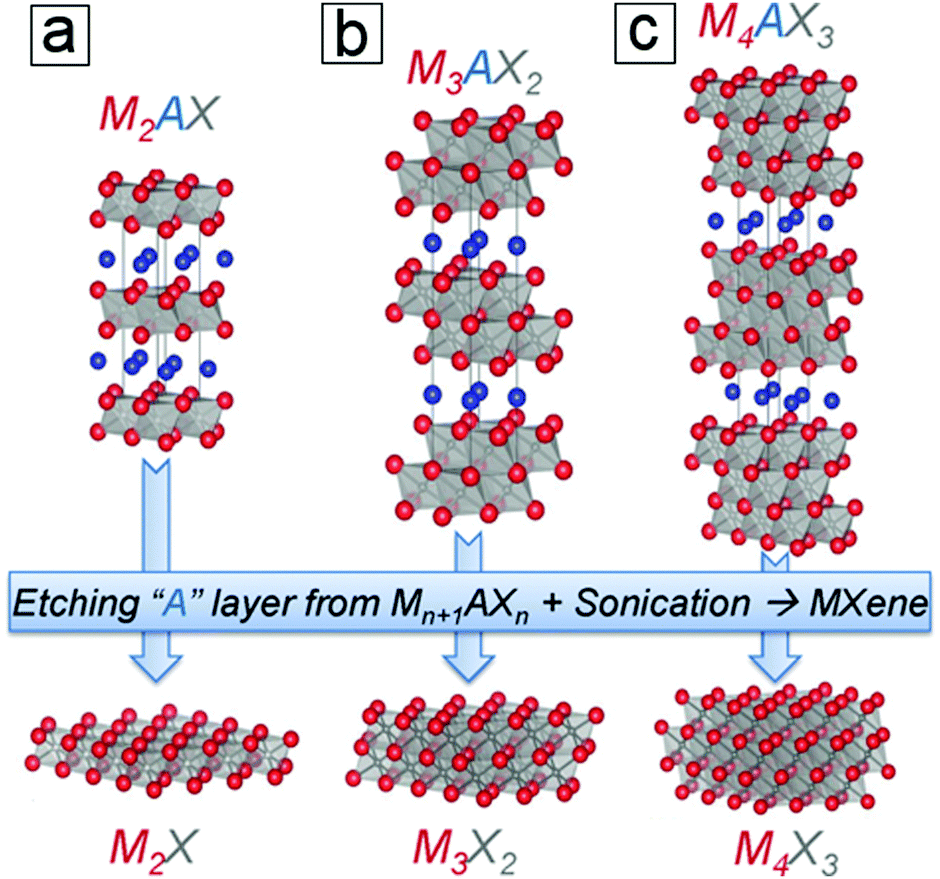 | ||
| Fig. 1 Structures of MAX 211, 312, and 413 phases and their derived pristine MXene monosheets. M is a transition metal, e.g., Ti; A is an element from group 13 or 14, e.g., Al. X is C, N, or their combination. From ref. 3. Copyright © [2013] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
However, the alternative LiF salt precursor, first developed in 2014, has elevated research on MXenes to a new era;2 since this discovery, numerous exciting applications have emerged. MXenes show great promise for application in electrodes of rechargeable batteries, supercapacitors, photocatalysts, catalysts, transparent conducting films, electromagnetic interference shielding and sensors, adsorption agents for crude oil and heavy metals, and flexible high-strength composites. Among these, electrochemical energy storage and energy conversion are of great interest for providing clean and renewable energy strategies to address energy shortage issues. Therefore, there is a great need to update the recent progress and milestones of MXene electrochemistry and energy applications.
Herein, we summarize recent advances in MXene energy storage and energy conversion. Initially, we introduce the fundamentals of MXenes, such as their discovery, family types, properties, and synthesis. Subsequently, we emphasize the latest energy-related progress in both unconventional device configurations and proof of concept applications, e.g. rechargeable batteries, supercapacitors, photocatalysts, catalysts, electrocatalysts, photovoltaic devices, solar thermal usage, and thermoelectric power generation. To finalize, we provide a perspective on future opportunities and possibilities for rising star MXene materials.
1.1 MXenes in brief
The term MXenes refers to 2D layered materials derived from transition metal carbides, nitrides, or carbonitrides,3 such as Ti3C2. Unlike graphene and phosphorene, which respectively have graphite and black phosphorus natural 3D bulk precursors, MXenes do not have a straightforward 3D precursor in nature. Instead, MXene multilayer flakes are usually produced by the selective removal of the A layers in MAX phases, e.g., the removal of Al from Ti3AlC2. Thus, upon delamination using intercalation agents such as DMSO and LiF, few-layer MXene nanosheets can be readily created. MXenes were first experimentally used in lithium-ion batteries for energy storage, and research rapidly expanded to the prediction of new MXene family members with the assistance of theoretical computational chemistry.To understand the structures of MXenes, one must recall their origins, from the MAX phase to the isolated 2D layer, as discussed in the next sections.
1.2 MAX phases
MAX and Mn+1AXn phases refer to hexagonal layered transition metal carbides and nitrides.4–6 The variable n can be assigned three different integers, n = 1, 2, and 3, while the MAX phases are classified as three types, 211, 312, and 413 structures. M represents an early transition metal element, such as Ti; A represents an element from group 13 or 14 (formerly groups 3A and 4A), such as Al or Si; and X refers to C, N, or their blends. For example, the simplest crystal structure of Ti2AlC comprises an MAX phase with periodic stacking of four atom layers, Ti–C–Ti–Al, along its cross section (Fig. 1a). The Ti bilayers consist of two layers of close-packed atoms. The C monolayer is composed of C atoms occupying the octahedral sites between two Ti atomic layers, while the Al monolayer interleaves two periodic units of the Ti–C–Ti trilayer. The Ti6C octahedral structure is analogous to rock salt, while the Al atoms occupy the centers of trigonal prisms.The slight difference between the MAX 211, 312, and 413 phases lies in the number of Ti layers between each Al layer, i.e. 2, 3, and 4 Ti layers, respectively. Moreover, the number of C layers is always one layer less than the number of Ti layers (Fig. 1b and c). The experimentally synthesized MAX phases comprise >60 family members,7 most of which are in the 211 phase, followed by the 312 phase and finally the 413 phase. The discovery of more MAX members is ongoing, especially for 413 phase members with single M elements or binary metal alloys (M′xM′′y) that replace single elemental M layers (Section 1.4). The M layers comprise one element from Ti, Mo, V, Cr, Zr, Hf, Nb, and Ta. The A layers consist of an element from Al, Si, P, S, Ga, Ge, As, Cd, In, Sn, Tl, and Pb, and the X layers comprise C, N, or both. MAX phases have attractive thermal, mechanical, and electrical properties. They are ideal for application in high temperature ceramics and other “extreme condition” materials,7 including gas burner nozzles for aircraft engines and molten Pb containers in nuclear power plants. However, MAX phases themselves have limited potential in lithium-ion batteries. In contrast to MAX phases, 2D MXenes are in high demand for energy storage applications. This is due to their large specific areas for providing redox sites as well as their suitable interlayer spacings, which allow ion intercalation and deintercalation.
1.3 MXene discovery
The first MXene, Ti3C2, was isolated from MAX powders by soaking Ti3AlC2 phase in hydrofluoric acid solution.1 Subsequently, many MXene family members have been synthesized using selective etching approaches.3,8–12 As interest in these materials grew,13–21 theoretical chemists predicted a number of new MXenes.22–26In general, the MX-bound layers are more stable than the A layers, which exhibit relatively weak binding to the M–X layer. Therefore, selective reactions of A layer atoms with foreign species and continual out-diffusion are the preferred strategies to create M–X-layered structures (Fig. 2).
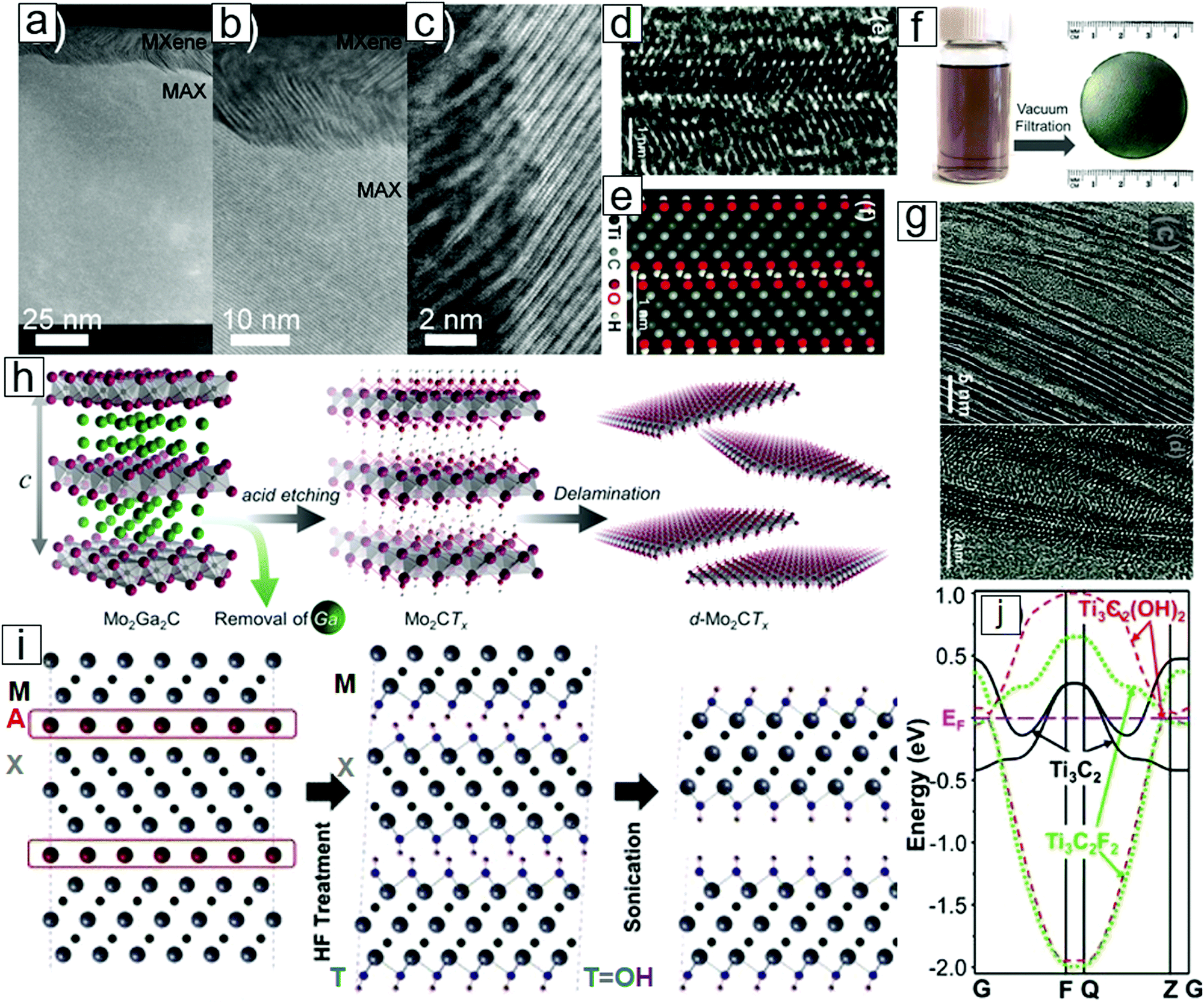 | ||
| Fig. 2 Structures and schematic of MXene formation from MAX phases with selective etching. Transmission electron microscopy (TEM) micrographs of the (a) MAX phase thin film, partly etched at the top to produce a very thin MXene; (b) MXene attached to the MAX phase, where the A layer has been selectively removed; and (c) high resolution micrograph of the MAX–MXene interface. (d) Atomic resolution TEM micrograph illustrating an MXene with surface OH functional groups. (e) Atomic configuration in the simulation image. (f) Photographs of MXene dispersion and MXene paper obtained from filtering MXene dispersions. (g) TEM micrographs displaying the delamination of MXene sheets. (h) Scheme of MXene formation from acid-assisted A removal in MAX. A further delamination step thins the MXene into nanosheets with few atomic layers. (i) Cross-sectional view of layered MXene formation. (j) Band structures of monolayer MXene with OH and F surface terminal groups and pristine MXene. The surface chemistry leads to a difference between metals and semiconductors. (a–c) Reprinted with permission from ref. 27. Copyright 2013 IOP Publishing and (d, e, g, i and j) from ref. 1. Copyright © [2011] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. (f and h) From ref. 28. Copyright © [2015] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
Interestingly, HF is the etchant of choice for the preparation of MXenes. Early work revealed that MAX phases are inert in the presence of common acids (HCl, H2SO4, and HNO3), common NaOH base solutions, and common salts (NaCl and Na2SO4). Neither molten metals nor molten salts have enabled MAX phase exfoliation for MXene formation. Eventually, the immersion of MAX phases in dilute HF solution succeeded in selective removal of the A layers and led to the formation of M–X multilayer flakes. Upon delamination with the assistance of intercalation agents, 2D-layered Ti3C2 and Ti2C nanosheets were synthesized.1,29,30
Synthetic Ti3C2 nanosheets are a few atomic layers thick and can be converted into conical scrolls.1,31,32 These nanosheets are analogous to graphene33–35 and other 2D materials.36–39 Because of the removal of the A layer in the MAX phases, the family of 2D metal carbides and nitrides derived from MAX phase materials was termed MXenes.
MXene nanosheets frequently contain surface functional groups such as O, F, and OH (Fig. 2i). These functional groups lead to the formation of semiconducting functionalized MXenes [Mn+1XnTx; e.g., Ti3C2(OH)2], while pristine MXenes [Mn+1Xn; e.g., Ti3C2] are metallic (Fig. 2j).
1.4 Binary transition metal MXenes
MXenes mostly refer to compounds based on single metal carbides, such as Ti3C2 and Ti4C3. However, compounds with binary metals as M layers also exist and are thermodynamically stable. Both M′FM′′C2 and M′2M′′2C3 phases have been successfully synthesized,22 where M′ and M′′ are two different metals (Fig. 3a). Two examples are Mo2TiC2 and Mo2Ti2C3 (Fig. 3b). These binary MXenes display similar crystal structures, atomic configurations, and morphologies to the layered structures (Fig. 3c and d).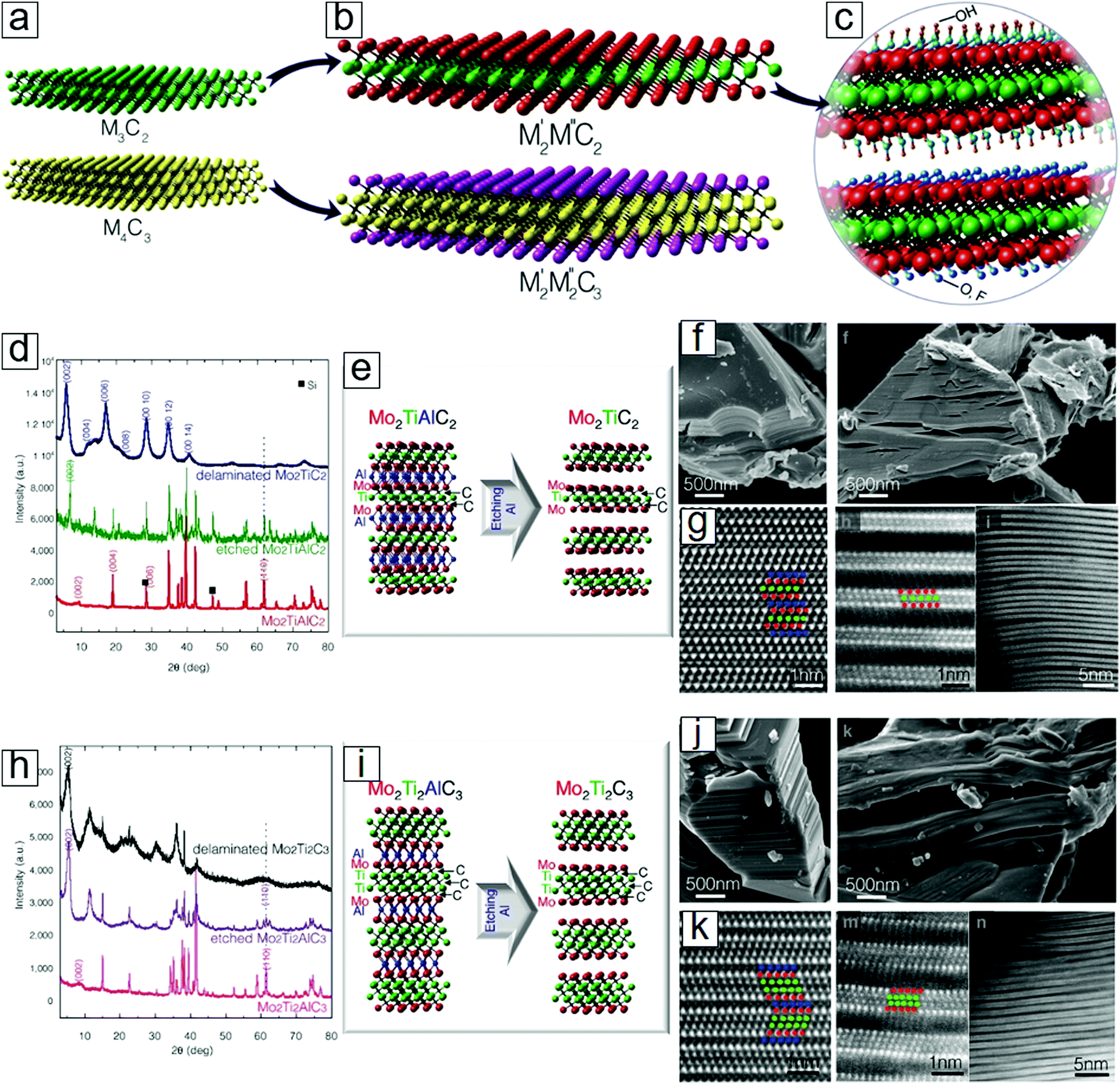 | ||
| Fig. 3 Double transition metal MXenes. (a) Schematics of conventional single transition metal MXenes. (b) Schematics of double transition metal MXenes M′2M′′C2 and M′2M′′2C3. M′ and M′′ are different elements: Ti, V, Nb, Ta, Cr, or Mo. (c) Functional groups over the MXene surfaces: OH, O, or F. (d) XRD patterns of MXenes M′2M′′C2 related species. (e) Atomic configuration of the selective etching step for producing MXenes M′2M′′C2. (f) SEM images of MXenes M′2M′′C2 related species. (g) HRTEM images of the M′2M′′C2 related samples. (h) XRD patterns of MXenes M′2M′′2C2 related species. (i) Atomic configuration of the selective etching step for producing MXenes M′2M′′2C2. (j) SEM images of MXenes M′2M′′2C2 species. (k) HRTEM images of the M′2M′′2C2 samples. Here, M′=Mo, M′′=Ti. Reprinted with permission from ref. 22. Copyright 2015 American Chemical Society. | ||
The discovery of surface termination groups has led to the prediction of 256 more MXene members through different combinations of the metal element M′, M′′ pairs C and N with the functional groups O, F, or OH.40 Moreover, the compositional ratio can be tuned to afford the binary transition metal couples V1−xMox, Nb1−xMox, Ta1−xMox, Ti1−xMox, Ti1−xNbx, Ti1−xTax, Ti1−xVx, and Nb1−xVx. The MXene family may consist of millions of members, as suggested in a study that considered the variation in the atomic composition ratios of two alloyed metals with different synthetic temperatures.23
2 Properties
2.1 Atomic structures
The MXene monolayer sheet has a hexagonal lattice with sixfold symmetry. Analogous to graphene, it has a rhombus unit cell from the top view, which is indicated by the dashed lines in Fig. 4a (upper panel). From the side view, the MXene monolayer consists of trilayer sheets where two M transition metal layers sandwich one X layer.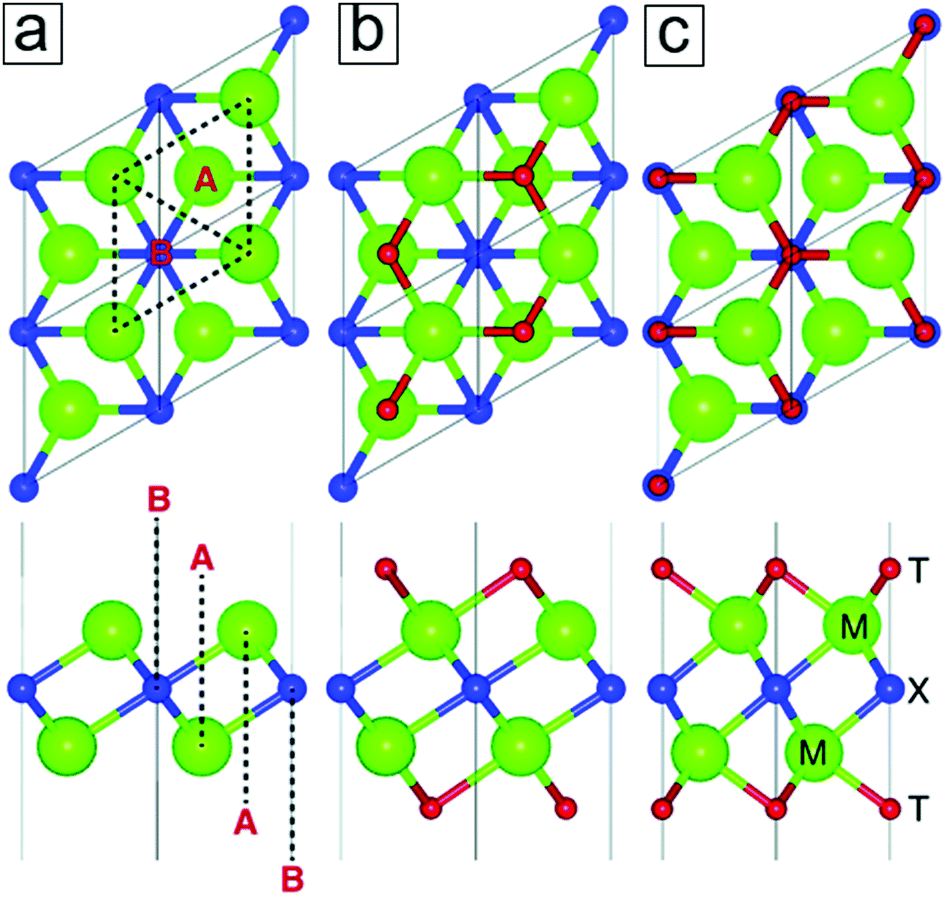 | ||
| Fig. 4 Crystal structures of two-dimensional (2D) M2X MXenes. (a) The Ti2C MXene model comprises a rhombus unit cell (dashed line) with hexagonal symmetry (top view). It consists of trilayer configurations where the C atomic layer is sandwiched between two Ti atomic layers (bottom panel). (b and c) MXene models with two different surface termination group configurations. Reprinted with permission from ref. 41. Copyright 2017 The Royal Society of Chemistry. | ||
As determined by computational methods, the transition metals in MXenes are configured by six chemical bonds with the closest X atoms. This is due to the typical six-coordination observed in transition metal ions and leads to the formation of M2XF2, M2XO2, and M2X(OH)2. When taking the functional groups into account, the hollow sites of the M2X surfaces can be classified as two types: A and B (Fig. 4a, lower panel). In type A, there are no X atoms between the M layers and the hollow sites, while type B sites have X atoms under them. Two energetically stable configurations for the attachment of the terminal groups are therefore demonstrated. One configuration allows two functional groups to locate over a hollow site, A (Fig. 4b), while the other enables two functional groups to reside over a hollow site in the B configuration.
Functionalized MXenes are thermodynamically stable due to the negative formation energies required for the functionalization reactions. Density functional theory (DFT) revealed the formation of strong interactions between the transition metal atoms and specific surface groups.42 Indeed, the surfaces of fully functionalized MXenes are thermodynamically more favored than those of partially functionalized MXenes. Eventually, the MXene surfaces can be fully functionalized at the same chemical potential.42,43
2.2 Electronic band structures
Ideal pristine MXenes are metallic and, hence, are analogous to MAX phases. With functional termination groups over their surfaces, some MXenes can acquire semiconducting natures. Their band gaps match the solar spectrum, thereby providing opportunities for band gap alignment in heterostructures for optoelectronics. Later, several semiconducting MXenes with suitable band gaps will be introduced.Depending on their spin orbital coupling, MXenes can be one of two types, topologically trivial42 or non-trivial.44–46 Moreover, based on their electrical conductance, MXenes can be divided into metallic, semi-metallic, and semiconducting types.
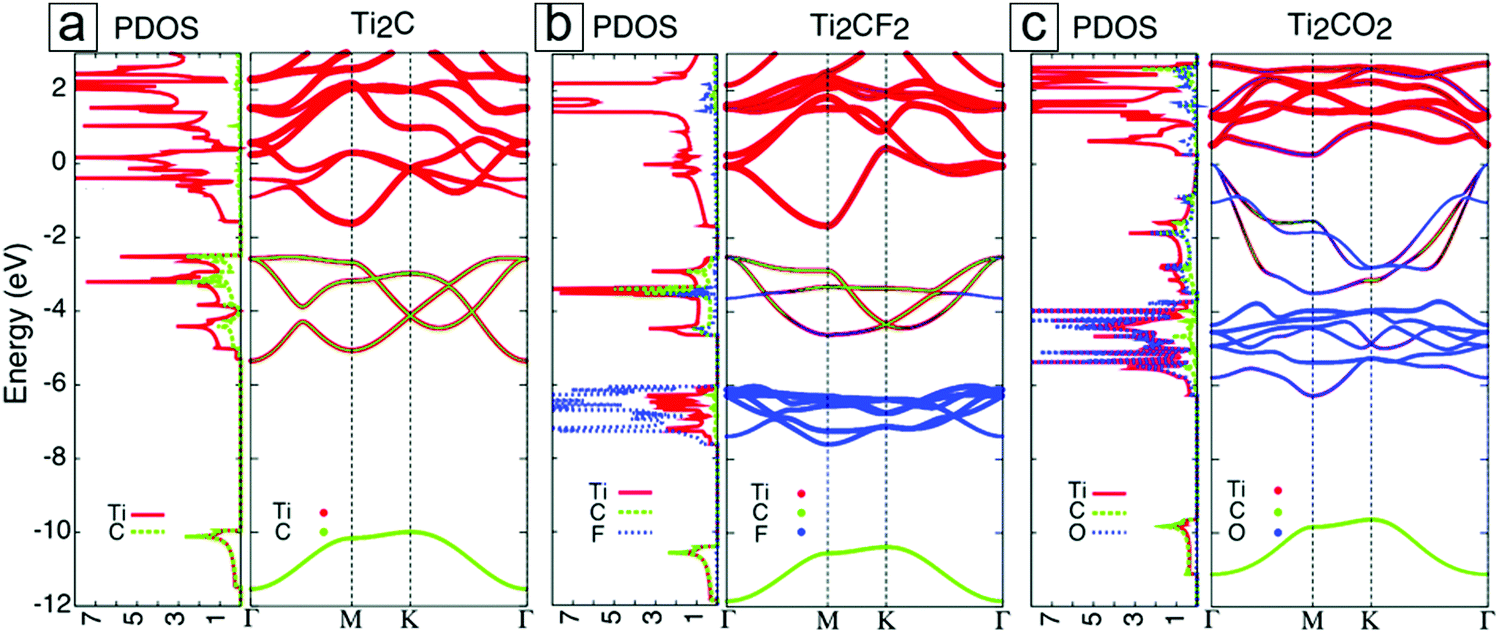 | ||
| Fig. 5 Densities of states and electronic band structures for MXenes with different terminal groups: (a) Ti2C, (b) Ti2CF2, and (c) Ti2CO2. These results were first published in ref. 42. The Fermi energy is set at zero energy. Reprinted with permission from ref. 41. Copyright 2017 The Royal Society of Chemistry. | ||
For most MXenes, the p bands in X occur beneath the d bands, with a small band gap in between them. In the presence of surface groups, new types of bands are formed below the Fermi energy. These are attributed to the hybridization of the d orbitals of M with the p orbitals of F and O. In F-functionalized MXenes, e.g. Ti2CF2, the Fermi energy is still located at the d band of the M layers; thus, they remain metallic (Fig. 5b). However, in O-functionalized MXenes, e.g. Ti2CO2, the d band is lifted above the Fermi level, and they become semiconducting (Fig. 5c).
It is also possible to tune the band gaps of these compounds through strain49,50 and external electric fields.51,52 For example, the MXene Sc2CO2 undergoes a direct band gap transition from a pristine indirect band gap with a 2% strain.49 This band gap modulation also applies to Ti2CO2, Zr2CO2, and Hf2CO2 within a 14% strain.50 With an external electric field of 4 V nm−1, a direct band gap from Γ to Γ forms in Sc2CO2, which undergoes an indirect band gap transition from K to Γ without an external electric field.52 With a further 2 V nm−1 increase in the electric field, the band gap increases to ≤183 meV.52 Theoretical studies on band gap modulation suggest that MXenes can provide suitable platforms for valley electronics.53–60
2.3 Morphologies
Analogous to exfoliated graphene, MXene exhibits typical flake features when drop-coated onto a flat substrate. These morphologies are presented through scanning electron microscopy (SEM) and atomic force microscopy (AFM) micrographs (Fig. 6a and e). The typical height for a monolayer sheet is 2.7 nm. On the other hand, a folded structure displays an interlayer distance of 1.5 nm, which is larger than that of graphene.2,61,62 The sheet–sheet spaces between MXene sheets have been experimentally measured as 1 to 1.5 nm;63–67 these values are larger than those observed for both graphene and phosphorene. This large interlayer distance accounts for the extraordinary performance of MXenes when they are applied as active materials in sodium-ion batteries (Section 4). | ||
| Fig. 6 Morphologies and chemical bonding environments of the MXene Ti3C2 formed with LiF selective etching. (a) Scanning electron microscopy (SEM) micrographs of MXene flakes. (b) Transmission electron microscopy (TEM) micrograph of an MXene flake over a Lacey carbon grid. (c) Magnified TEM micrograph of MXene. Inset is a selected area electron diffraction (SAED) pattern displaying sixfold reflexes. (d) High resolution TEM micrograph illustrating the hexagonal symmetry in the lattice structures. (e) Atomic force microscopy (AFM) micrograph displaying the few-layer nature of an MXene flake. (f) X-ray photoelectron spectroscopy (XPS) spectra of MXenes with C–Ti–T bonding; T is F or OH. From ref. 40. Copyright © [2016] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
The TEM micrographs reveal significantly low mass contrast compared to the supporting carbon, which indicates the thin nature of the few-atom layer (Fig. 6b and c). Moreover, high resolution transmission electron microscopy (TEM) illustrates the hexagonal lattice structures in the axis zone along the c direction. Selected area electron diffraction (SAED) patterns confirm the sixfold reflexes of the lattice (inset in Fig. 6c). The chemical bond information (Fig. 6f) is discussed in the subsequent section.
2.4 Chemical bond environments
Analysis of the local chemical environment of a material is important to determine its chemical composition and elemental stoichiometric ratio. Two typical characterization tools are X-ray photoelectron spectroscopy (XPS) and electron energy loss spectroscopy (EELS). In addition, the spectral shift generated in nuclear magnetic resonance (NMR) spectroscopy depends on the local magnetic field that is supplied by the electron spin from molecular orbital hybridization between the chemical bonds. Thus, NMR spectroscopy is also included in this section for local chemical environment examination.XPS is often adopted for 2D materials to probe the synthetic reaction mechanisms and the extent of chemical reaction,68–75i.e., partial or full conversion of the reactants. For example, graphene formation over Ni has demonstrated that Ni3C forms as an intermediate phase and eventually produces graphene upon decomposition during cooling.76–79 Another example is MoS2 synthesis from MoO3 sulfurization, where XPS can be used to determine whether the oxidation phase disappears, thereby revealing whether full conversion to MoS2 has occurred.80–84 Moreover, XPS can reveal the degradation status of phosphorene in air and the humidity dependence of this process.
Synthesis of MXenes by selective etching is highly dependent on XPS to provide useful information; for example, whether the remaining Ti–C bonds and newly emerging Ti–F, Ti–O, and Ti–OH functional groups interact.85–88 In more detail, the as-obtained MXenes revealed the presence of Ti–C and Ti–O bonds in the Ti 2p spectrum as well as C–Ti and C–C bonds in the C 1s spectrum (Fig. 7a and b). Because the surface functional groups determine the electronic band structure, diagnosis of the bond information can reveal material property changes which can occur in an intended manner. Indeed, functional grafting of phenylsulfonic acid groups (SO3H)89 can improve MXene dispersion in water and provide more redox reaction sites for electrochemical reactions to occur. This is also of significance for determining foreign species introduced with other etching agents, e.g., LiF/HCl in immersion etching and HCl in electrochemical etching.
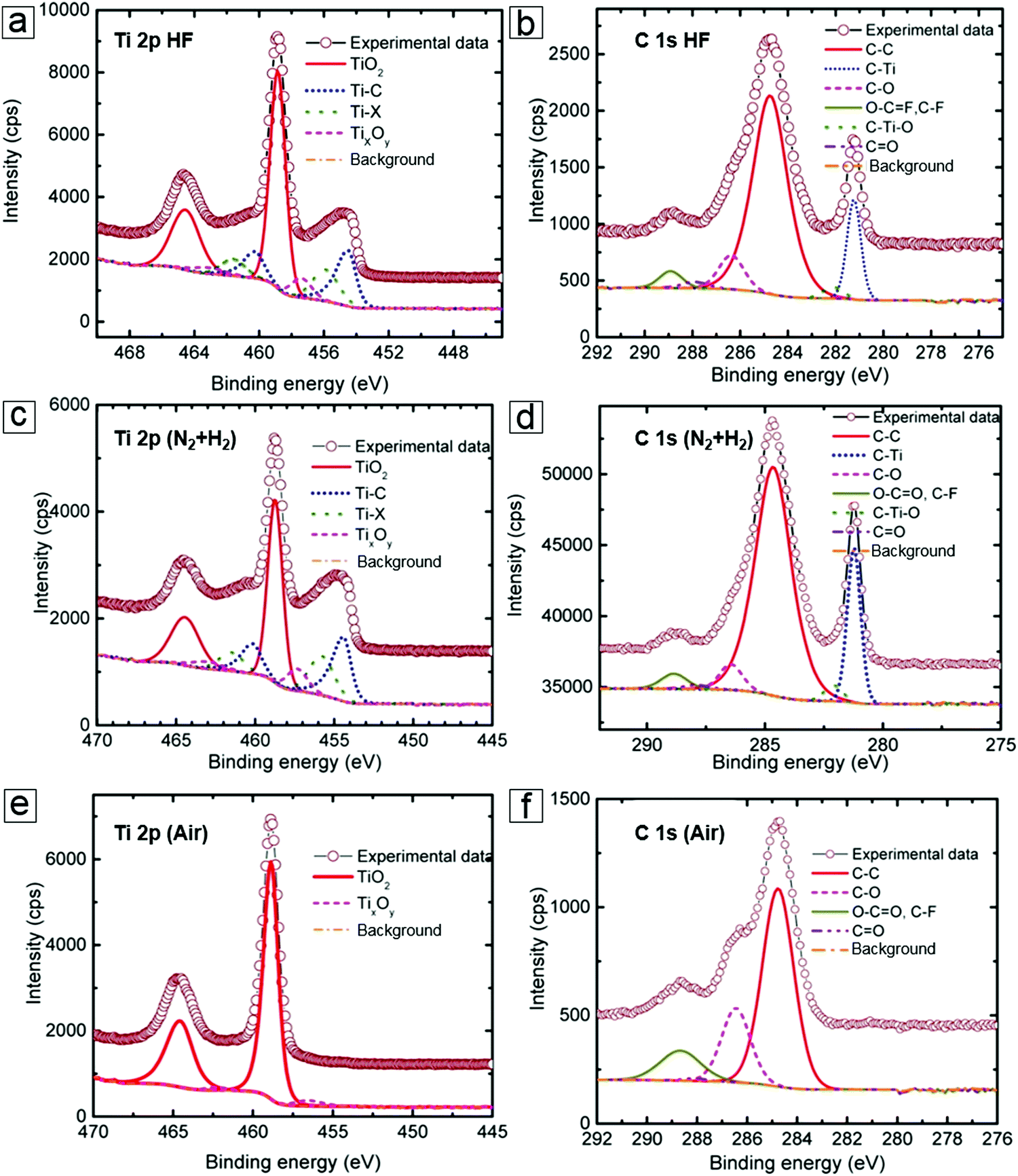 | ||
| Fig. 7 X-ray photoelectron spectroscopy (XPS) spectra of MXenes. (a and b) Ti 2p and C 1s peaks of the as-prepared MXene Ti2CTx from the HF etching MAX method. (c and d) Ti 2p and C 1s peaks of the H2-annealed MXene samples. (e and f) Ti 2p and C 1s peaks of the air-annealed MXene samples. Reprinted with permission from ref. 15. Copyright 2015 American Chemical Society. | ||
The oxidation or reduction of MXenes can occur through post-treatment, such as annealing under oxygen or hydrogen atmosphere. For example, the Ti–C bond peaks are enhanced in XPS compared to the peaks of the pristine material after hydrogen annealing (Fig. 7c and d). In contrast, annealing in air induces complete oxidation to yield TiO2. Therefore, the Ti–C signal in the spectrum disappears (Fig. 7e and f).
XPS typically offers low spatial resolution. Thus, when combined with scanning transmission electron microscopy (STEM), EELS with a sub-nm electron beam probe is an important core level spectroscopic technique that provides high spatial resolution data.90–92 Because EELS93–95 can provide precise chemical bond information at the nanometer scale, the spectral information can describe the bonding environment at the atomic level, e.g., at the Ti vacancy and atomic levels in the MXene monolayer sheets. The representative EELS spectra for the C-K, Ti M2,3, F-K, and O-K edges of MXenes are presented in Fig. 8. This allows comparison between the selective etching methods using different etching media (HF and LiF/HCl).96
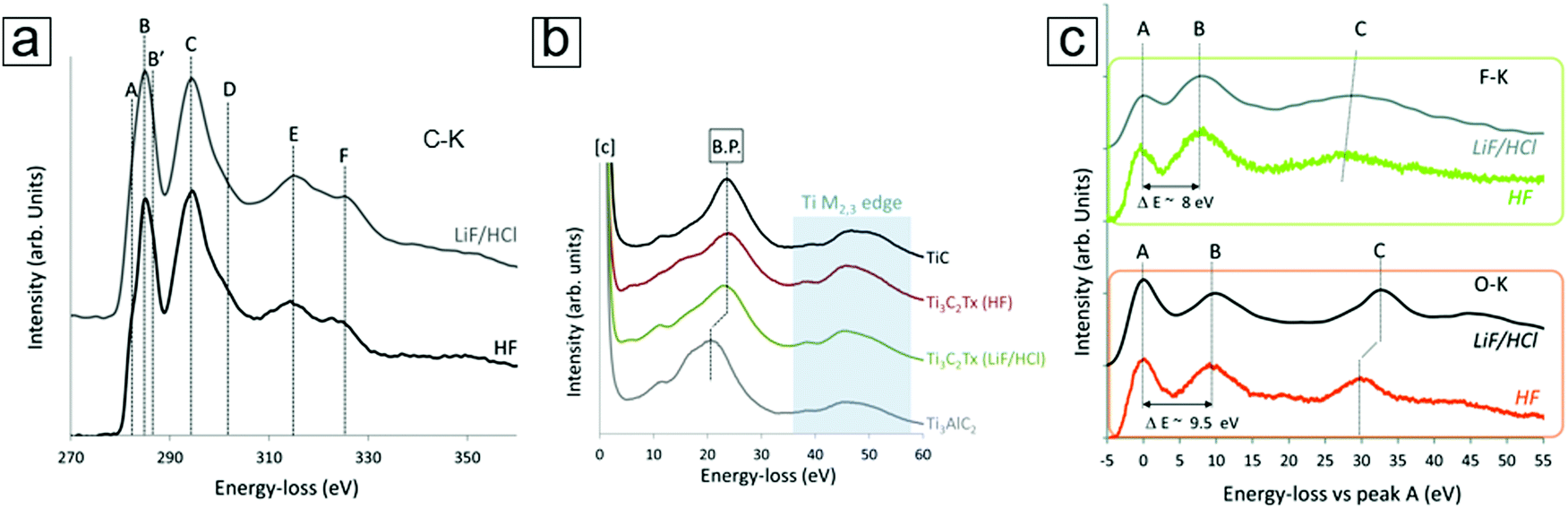 | ||
| Fig. 8 Electron energy loss spectroscopy (EELS) curves of the MXene Ti3C2Tx. (a) C-K edge data of MXenes exfoliated with HF and LiF/HCl selective etching. The four main peaks are labelled C, D, E, and F. (b) Ti M2,3 edge peaks of the MXenes, TiC, and MAX phases. Exfoliation causes a shift in the bulk plasmon (B.P. in the figure panel). (c) F-K and O-K edge peaks of MXenes with characteristic peaks at ∼535 eV and 685 eV, respectively. Both spectra use relative values by setting their first peak (A) to zero. Reprinted with permission from ref. 96. Copyright 2016 The Royal Society of Chemistry. | ||
EELS also opens an avenue for understanding single-atom chemical reaction chemistry97–99 when accompanied by other imaging and spectral techniques, e.g., STEM100–103 and energy dispersive X-ray spectroscopy (EDX).104,105 This combined approach allows the determination of Ti adatoms and vacancies in MXene monolayer sheets. This approach can reveal the dynamics of individual atoms, such as individual Cr106 and Fe107 atoms.
NMR provides information on the absorption and emission of electromagnetic radiation by atomic nuclei in a molecule under magnetic conditions. The resonance frequency energy depends on the intrinsic magnetic properties of the atomic isotope, which are determined by the chemical bonds. The most commonly studied nuclei are 1H, 7F, and 13C. Thus, NMR spectroscopy employs the specific quantum mechanical magnetic properties of a nucleus and provides rich information on the chemical environment of a complicated molecule. NMR spectroscopy can determine MXene information for materials derived from both HF and LiF etching methods. For example, NMR spectroscopy demonstrated the presence of residual water in an MXene material even after annealing at 200 °C (Fig. 9a and b). Additionally, NMR spectroscopy can reveal information on the surface functionalization of MXenes, viz. whether HF etching to produce an MXene results in more F surface terminal groups than LiF etching (Fig. 9d). With complementary 1H and 7F spectral images, F and OH surface groups are also revealed (Fig. 9e and f).
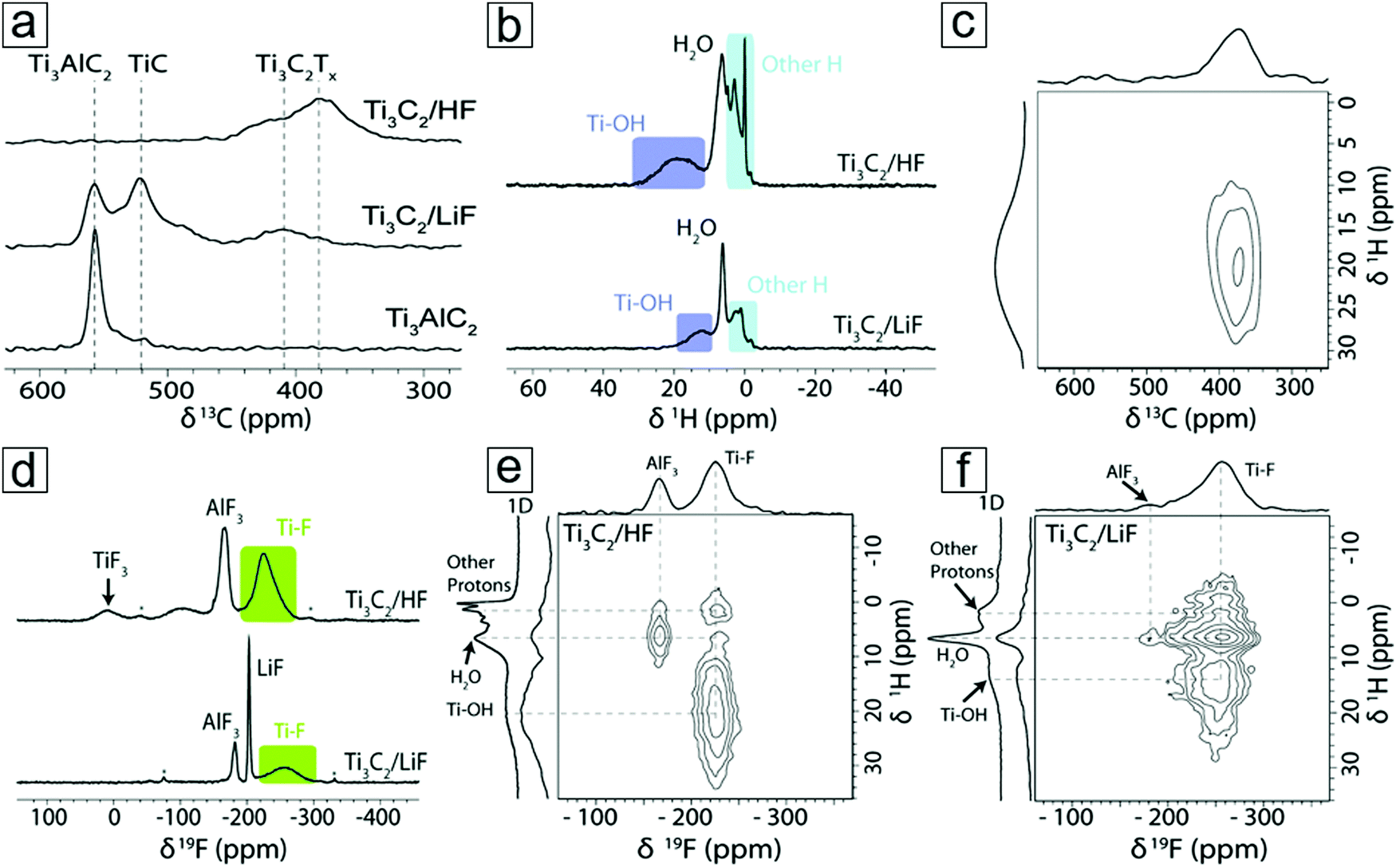 | ||
| Fig. 9 Nuclear magnetic resonance (NMR) spectra of the MXene Ti3C2. (a) 13C NMR spectra of MXene and MAX captured at 50 kHz magic-angle spinning in a Carr–Purcell–Meiboom–Gill setup. The echoes are summed to display the line shapes. (b) 1H NMR spectra of Ti3C2 MXenes from HF and LiF–HCl etching synthesis. (c) 1H–13C two-dimensional (2D) NMR spectrum of HF etching-derived Ti3C2 MXene. (d) 19F NMR spectra of MXenes. (e and f) 1H–19F 2D NMR spectra of MXenes obtained from HF and LiF etching methods, respectively. Reprinted with permission from ref. 108. Copyright 2016 The Royal Society of Chemistry. | ||
NMR spectroscopy can also determine the chemical environments of alkali metal nuclei during electrochemical reactions. With NMR spectroscopy, one can even observe Na evolution from a freestanding electrolyte to intercalation in the MXene interlayer spacing (Section 4.2).
2.5 Vibrational modes
The vibrational modes depend on the covalent bonds as well as the interlayer interactions in the MXene structures. Thus, they can be used to determine the quality and functional groups of the synthetic layered material. Raman and infrared (IR) spectroscopy are two frequently used methods to reveal the vibrational modes of molecular nanostructures. Indeed, Raman spectroscopy has been widely adopted to determine the layer number and quality of graphene with the relative ratio of the 2D/G bands and the intensity of the D band.109–113 Layer number information can be extracted through multi-peak fitting in 2D mode. These layer-dependent vibrational modes can also be seen in the vibrational spectra of phosphorene114–117 and molybdenum disulfide.118–125 Analogous to MoS2, MXenes exhibit A1g and Eg modes, which respectively correspond to in-plane and out-of-plane modes.126,127 A typical Raman spectrum for an as-synthesized MXene sheet is presented in Fig. 10a and b. Depending on the M, X, and T selection, the Eg modes can reside at 138, 278, 437, or 622 cm−1 for the in-plane vibrations, while the A1g modes reside at 218, 514, 684, or 3734 cm−1 as the out-of-the plane vibrations.128 | ||
| Fig. 10 Vibrational modes of MXene monosheets. (a) Atomic bond vibrations for different characteristic wavenumbers in Raman and infrared (IR) active modes. (b) A typical Raman spectrum of the MXene Ti3C2. The inset is an optical micrograph of an MXene flake. (c) Fourier-transform IR spectra of an MXene and an MXene after surface modifications with foreign diazonium salt groups. (a and b) Reprinted with permission from ref. 128. Copyright 2015 The Royal Society of Chemistry. (c) Reprinted with permission from ref. 89. Copyright 2016 Elsevier. | ||
Unfortunately, Raman spectroscopy is relatively insensitive to surface functional groups on 2D-layered materials. For example, reduced graphene oxide (rGO) displays a peak at ∼1350 cm−1 (D mode) that can vary with surface functionalization levels.129–132 However, a lack of information inhibits the identification of the types of surface functional groups present. IR spectroscopy is more sensitive to functional groups; thus, it plays an important complementary role to Raman spectroscopy. IR spectroscopy uses IR light to excite molecules to yield information on covalent bonding through the different vibrational modes, e.g., the –OH and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups on the surface of graphene oxides produced by the Hummers method can be identified.133–137 IR spectroscopy is well suited for the examination of MXenes because of the rich functional groups introduced to their surfaces during production. The IR light excites molecular vibrations that can be divided into Eu and Au modes. The Eu modes occur at 244, 275, 435, and 637 cm−1, while the A2u modes are observed at 348, 498, 577, and 3732 cm−1. Indeed, typical peaks for the functional groups of MXenes include the O–H hydroxyl stretching mode at 3500 cm−1, the CO stretching mode at 1700 cm−1, and the C–H deformation mode at 1460 cm−1.138–142
O groups on the surface of graphene oxides produced by the Hummers method can be identified.133–137 IR spectroscopy is well suited for the examination of MXenes because of the rich functional groups introduced to their surfaces during production. The IR light excites molecular vibrations that can be divided into Eu and Au modes. The Eu modes occur at 244, 275, 435, and 637 cm−1, while the A2u modes are observed at 348, 498, 577, and 3732 cm−1. Indeed, typical peaks for the functional groups of MXenes include the O–H hydroxyl stretching mode at 3500 cm−1, the CO stretching mode at 1700 cm−1, and the C–H deformation mode at 1460 cm−1.138–142
Beyond these intrinsic functional groups, foreign species can be introduced via post modification. For example, the intentional introduction of SO3H can be identified at ∼1020 cm−1 and 1160 cm−1 (Fig. 10c). Thus, IR spectroscopy allows the examination of newly tailored functional groups beyond the conventional O, F, and OH surface terminations. IR spectroscopy can also be applied to recognize the C–Cl stretching modes in a range of 600 to 800 cm−1 when HCl is the sole medium in the electrochemical etching process for MXene production.143–145
2.6 Optical absorption and photoluminescence spectroscopy
Optical absorption and photoluminescence spectroscopic techniques have been used to sort chiral and diameter-dependent single-walled carbon nanotubes and to determine the sizes and thicknesses of exfoliated nanosheets of 2D materials, including chemically exfoliated graphene, MoS2, and phosphorene. This allows homogeneity control during the preparation of a monodispersed suspension of colloids with nanoflakes or quantum dots used in ink for printed electronics, biological imaging agents, and photothermal therapy. The same types of applications can be achieved with MXenes, e.g. MXene quantum dots synthesized by hydrothermal treatment.146TEM micrographs have confirmed the formation of MXene nanodiscs, termed MXene quantum dots. Fig. 11 presents the absorption (black solid line) and photoluminescence (blue solid line) spectra for the three types of MXene quantum dots, with different sizes according to their synthetic temperatures. Photoluminescence excitation spectra are also provided; these are consistent with the absorption spectra due to the resonant excitation and transitions that reflect the optical band gaps. In contrast, photoluminescence is a non-resonant excitation and emission depends on the size and density of the states to provide fingerprints for the different-sized quantum dots.
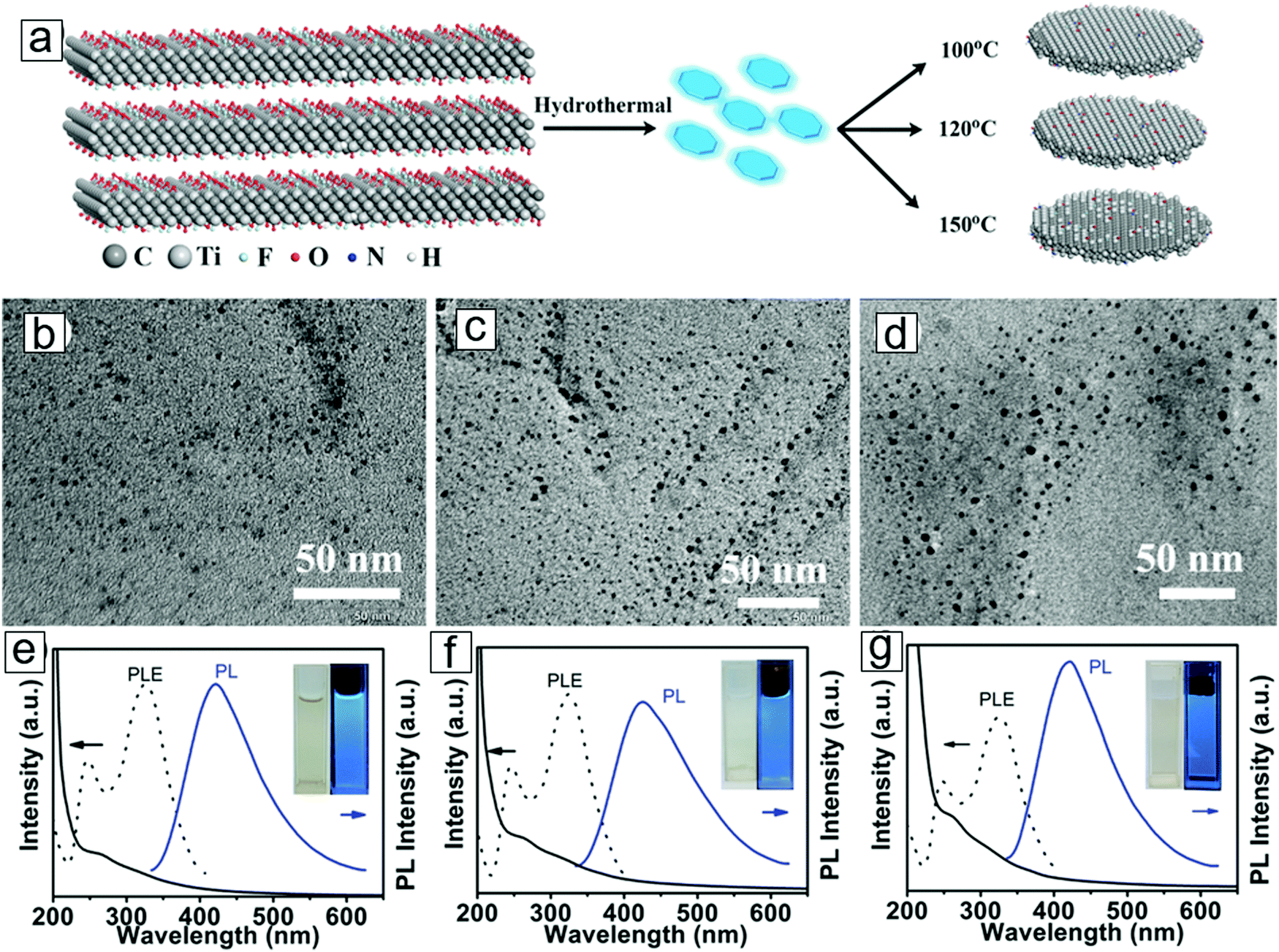 | ||
| Fig. 11 Synthesis of MXene quantum dots. (a) Reaction paths displaying MXene derivation of MXene quantum dots in a hydrothermal method in ammonia solution. Higher temperatures lead to larger average quantum dot sizes. (b–d) Transmission electron microscopy (TEM) micrographs for MXene quantum dots formed by hydrothermal treatments at 100 °C, 120 °C, and 150 °C, respectively. (e–g) Optical absorption (black solid line), photoluminescence excitation (black dashes), and photoluminescence (blue solid line) spectra for MXene quantum dots synthesized from the three respective temperatures. Insets are photographs for the quantum dot dispersions. From ref. 146. Copyright © [2016] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
2.7 Optoelectronic properties
MXene thin films can be produced from the layer-by-layer deposition of MXene dispersions. These films exhibit outstanding optical transmittance (>80%) at a thickness of 15 nm. MXene thin films created by this method exhibit the lowest optical absorption over reduced graphene and sputtered MXene. This is essential for enhancing transparent conductor performance and allowing greater illumination of active materials, such as displays and photovoltaic cells.The transmittance versus electrical resistance curves show high significance in transparent conducting electrodes. About 85% optical transmittance is observed at 550 nm with a sheet resistance of 10 kΩ sq−1 (Fig. 12). For transparent conductors,147–150 70% transmittance is required for an electrical resistance <1 kΩ sq−1. The variation of resistance with bending radius has shown a minimal radius of 3.8 mm with only a 15% change in resistance. This suggests long cycling performance. After 10 cycles, the resistance observed in the third cycle is retained.
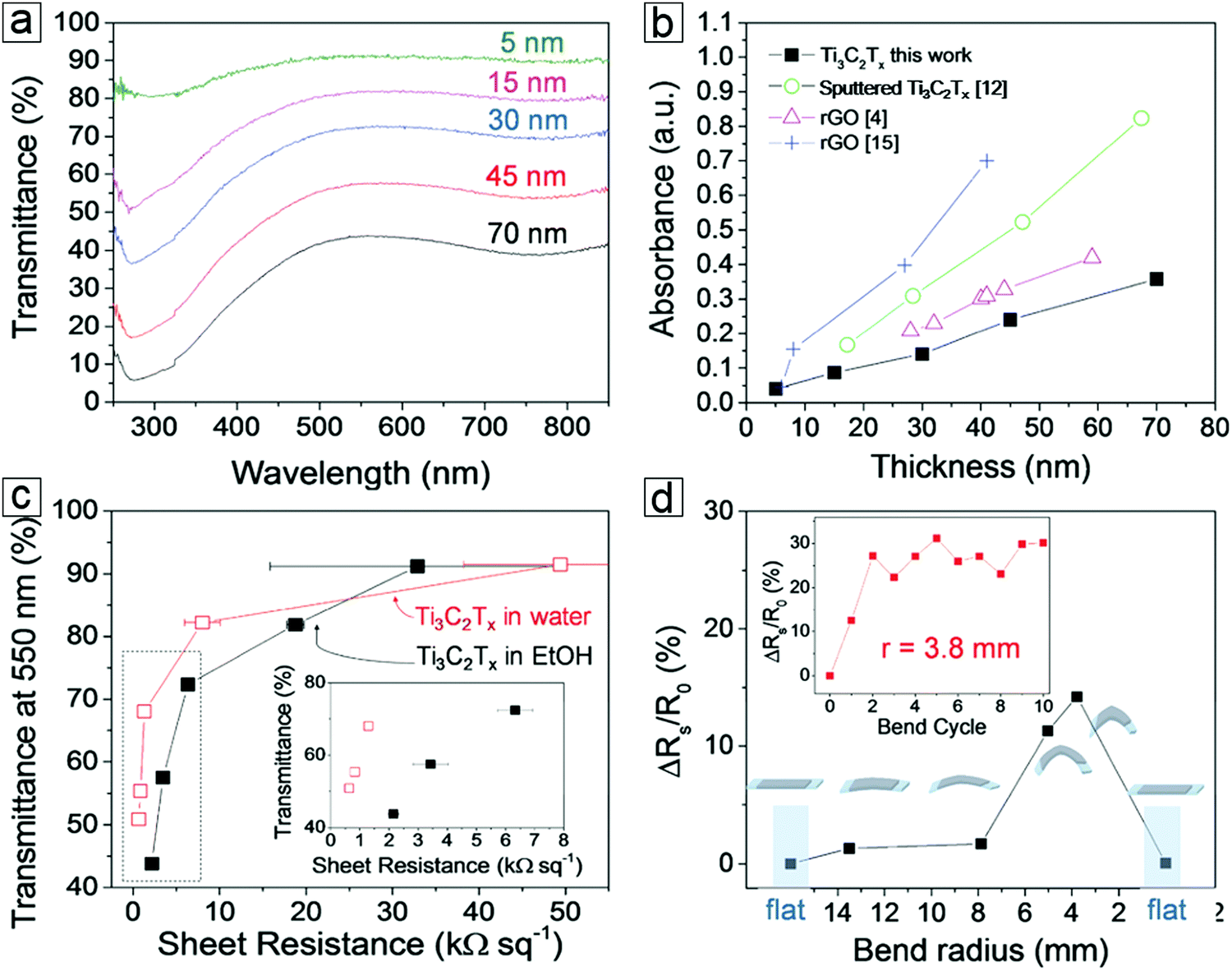 | ||
| Fig. 12 Flexible transparent conducting properties of MXene Ti3C2Tx films. (a) Thickness-dependent transmittance spectra of MXenes. (b) Thickness-dependent absorbance spectra of MXene and reduced graphene oxides (rGO). (c) Transmittance–sheet resistance curves of MXene films displaying transparent conducting performance. The inset is the low sheet resistance region. Thin films formed from spray deposition of a Ti3C2Tx water dispersion (red unfilled squares) and ethanol dispersion (solid black squares). (d) Variation of MXene film sheet resistance over a polyester substrate in a bending test. Inset displays the long cycling stability after 10 bending cycles. From ref. 151. Copyright © [2016] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
2.8 Mechanical properties
Computational methods were employed to study the mechanical properties of MXenes according to thickness, surface functionalization, and composite formation.152 The Young's moduli of the pristine MXenes were determined from the slopes of the strain–stress curves. The values were 597, 502, and 534 GPa for Ti2C, Ti3C2, and Ti4C3, respectively, revealing that the thinnest Ti2C 2D monolayer sheet possesses the highest Young's modulus. The high tensile stress and bond breakage in Ti2C and Ti3C2 initiated at the outer surface of the layers at the point with highest local stress and propagated towards the center of the carbon layers. However, for Ti4C3, breakage began in the center of the layer and expanded until a full fracture led to small fragment formation.153The functional MXene surface exhibited a different response to tensile stress than that of the pristine MXene. Indeed, functionalized Ti2CO2 can sustain large strain with uniaxial and biaxial tensions that exceed or are comparable to those observed for graphene and other 2D materials.154 Conversely, the pristine version is fragile to tensile strain due to the ready collapse of the surface Ti layers. Surface functional groups can suppress this collapse and therefore enhance mechanical flexibility.155
MXene/PVA composite films display high flexibility and can sustain 5000 times their own weight (Fig. 13). The micrographs of MXene-layered structures and the blending of MXene nanosheets with polymers are illustrated in Fig. 13. A 40% MXene mass ratio can provide a Young's modulus exceeding that observed for PVA polymers.156
 | ||
Fig. 13 MXene polymer composites and their mechanical properties. (a and b) Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) micrographs of as-synthesized MXene flakes. (c) Synthetic protocols for MXene films and MXene/polymer composite films. (d) Stress–strain curves of Ti3C2Tx/PVA composite films depending on the Ti3C2Tx weight ratio. (e) Photograph of a pristine MXene film supporting 4000 times its own weight. (f) Photograph of a 90 wt% MXene/PVA composite film supporting 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times its own weight. Reprinted with permission from ref. 156. Copyright 2014 National Academy of Sciences. 000 times its own weight. Reprinted with permission from ref. 156. Copyright 2014 National Academy of Sciences. | ||
3 Synthetic methods
3.1 Top-down from MAX powders to MXene dispersions
| Ti3AlC2(s) + 3HF(aq) = Ti3C2(s) + AlF3(aq) + 3/2H2(g) | (1) |
| Ti3C2(s) + 2HF(aq) = Ti3C2F2(s) + H2(g) | (2) |
| Ti3C2(s) + 2H2O(aq) = Ti3C2(OH)2(s) + H2(g) | (3) |
The as-prepared MXene samples consist of multilayer flakes that stack together with weak interlayer interactions. Ultrasonication of the resulting products leads to exfoliation of MXene nanosheets with thicknesses of 11 ± 3 nm as determined by the Sherrer formula.157 This thickness along the c axis corresponds to 10 layers of Ti2C2(OH)2.
| LiF(aq) + HCl(aq) = HF(aq) + LiCl(aq) | (4) |
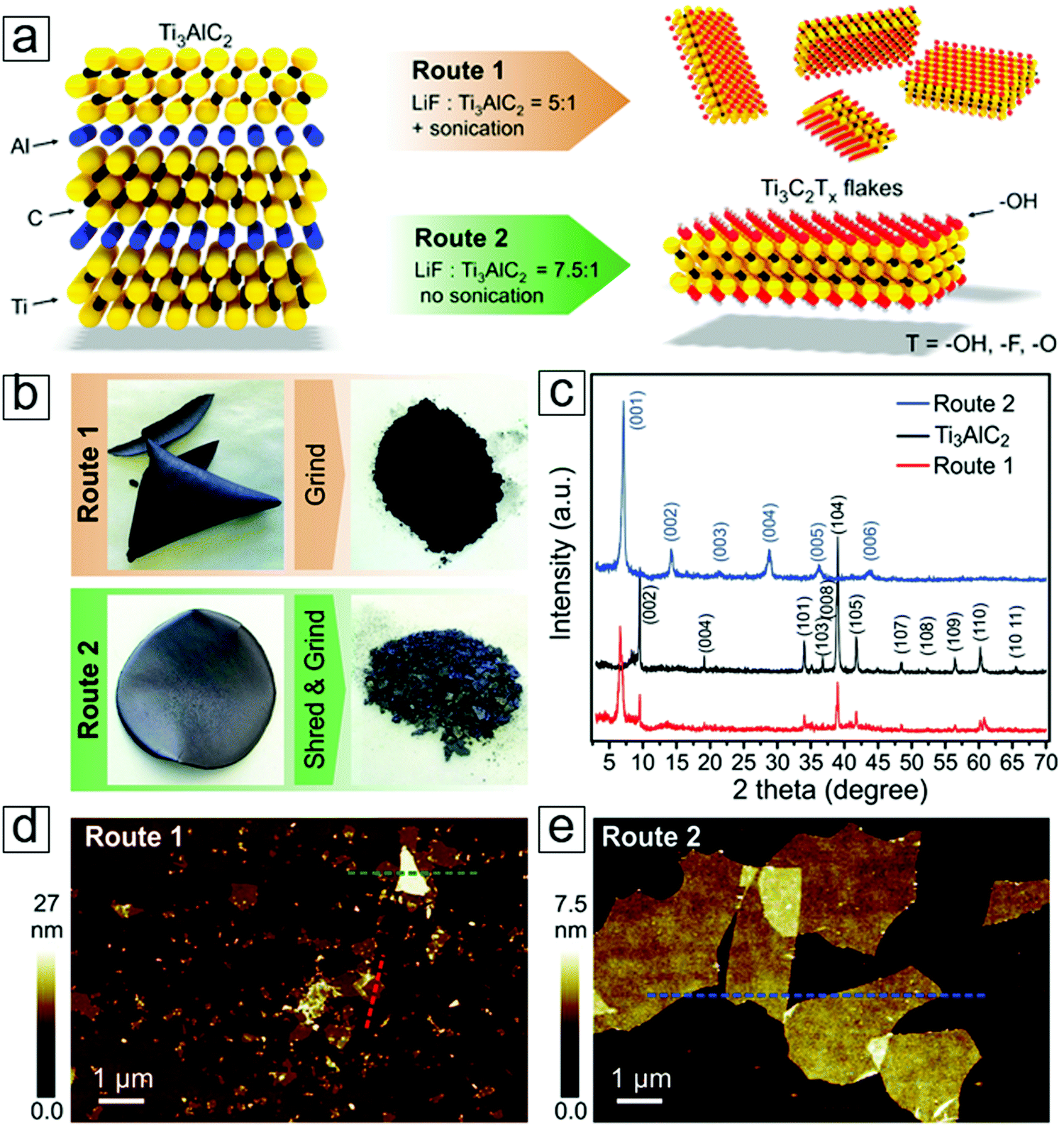 | ||
| Fig. 14 MXene Ti3C2 exfoliated with LiF etching methods. (a) Schematic of exfoliation reactions. (b) Photographs of an MXene paper formed via filtering an MXene dispersion on a polyvinylidene difluoride membrane. (c) X-ray diffraction (XRD) curves of the MAX and MXene phases. (d) Atomic force microscopy (AFM) micrographs of the MXene flakes. From ref. 40. Copyright © [2016] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
Li ions can intercalate between these MXene sheets, and studies have shown high volumetric capacitance in their electrochemistry performance.2 Moreover, this mild fabrication method leads to a larger planar size with fewer atomic defects than those observed for MXene sheets extracted with pure HF etching. This also applies to fluoric salts in the presence of common acids, e.g., HCl and H2SO4. Indeed, anions such as Na, Ka, Cs, Mg, and Ca lead to intercalation of the cations, applicable to one-step anion intercalation techniques for anode materials in rechargeable secondary ion batteries.
| Ti3AlC2(s) + 3NH4HF2(aq) = Ti3C2(s) + (NH4)3AlF6(aq) + 3/2H2(g) | (5) |
| Ti3C2(s) + aNH4HF2(aq) + bH2O(aq) = (NH3)c(NH4)dTi3C2(OH)xFy(s) + zH2(g) | (6) |
After selective etching and ammonia species intercalation, MXene thin films become thicker than the initial MAX thin films (60 nm). This feature is advantageous in MXene thin film production because the morphology of the initial film does not break.
Moreover, the MXenes produced from NH4HF2 etching methods display more homogeneity in the interlayer spacing of the layered structures than the HF-produced material. In addition, the c lattice parameters (2.5 nm) are 25% larger than those of HF-derived MXene thin films.164 This is advantageous in the intercalation of large ions, such as Na and K, for electrochemical energy storage applications.
Selective etching steps frequently lead to the production of MXene multilayer flakes. However, their limited specific area suppresses their potential in electrochemical storage applications. The larger interlayer space is also important because delamination becomes more efficient for isolating MXene thin nanosheets with one- or few-atom-layer thicknesses.
Organic molecules are gradually being developed to isolate thin nanosheets of MXenes such as Ti3CN, V2C, and Nb2C. They include amine n-butylamine172 and organic bases such as tetrabutylammonium hydroxide TBAOH173 and choline hydroxide.173,174 The immersion of MXene multilayer flakes in these solvents leads to swelling of the MXene flakes; upon shaking by hand or by mild sonication, MXene colloids form through delamination. Further centrifugation can produce uniform-sized enriched MXene nanosheets.173
This MXene dispersion yields a stable colloid of delaminated few-layer MXene sheets, which is negatively charged with surface exposed C atoms as well as F and OH groups.42,66,175,176 The negatively charged MXenes hold great promise, with the bridging assistance of positively charged rGO,177,178 to couple negatively charged components such as MnO2.177,179–181 This can be achieved through a simple hydrothermal approach182 and is useful for enhancing electrochemical performance.
These MXene nanosheets are so well-dispersed that they are ideal filler materials for enhancing the mechanical and electrical properties of composite materials. However, MXene nanosheets are stored in colloids or suspensions, thereby requiring a further deposition step, e.g., spray deposition, to form a thin film over a flat substrate such as glass for optoelectronic applications. To simplify the fabrication protocol, MAX epitaxial thin film deposition followed by an etching step will render a readily prepared MXene thin film of homogeneous (high) quality over a large area.
3.2 Bottom-up strategies from Ti, Al, and C atoms to form MXene thin films
The direct bottom-up preparation of an MXene thin film directly involves two steps, namely MAX thin film epitaxy with compositional atoms and subsequent MXene thin film formation by the removal of A layers. This route has been shown to provide readily usable transparent conducting films over transparent sapphire glasses. A brief description of the synthesis protocol follows.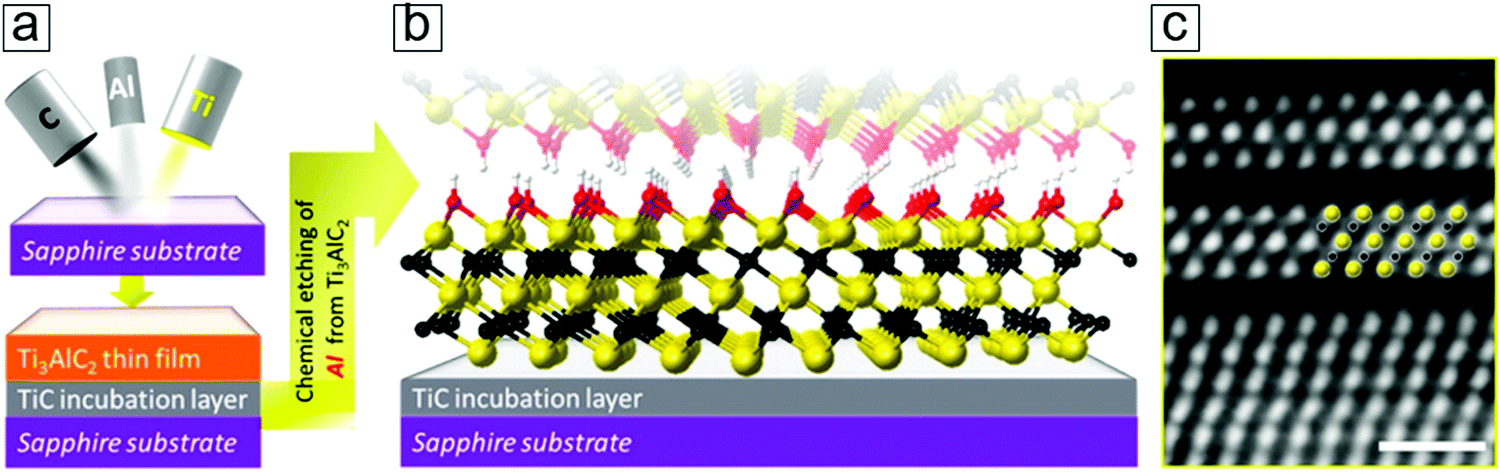 | ||
| Fig. 15 Epitaxy method for MXene thin film synthesis. (a) Schematic of magnet sputtering for depositing the Ti3AlC2 phase over a supporting substrate. (b) Atomic configuration of the MXene Ti3C2 thin film after selectively removing the Al layers. The colors indicating Ti, C, O, and H are yellow, black, red, and white, respectively. (c) Scanning transmission electron microscopy (STEM) micrographs of the initial two layers of MXene. Reprinted with permission from ref. 164. Copyright 2014 American Chemical Society. | ||
The starting Ti3C2 sheets, counted from the TiC incubation layer, exhibit an unambiguous ordered structure (Fig. 15c). This provides solid evidence of the synthesis of monolayer MXene nanosheets by gaseous vacuum deposition methods, such as chemical vapor deposition and atomic layer deposition. These findings may lead to advances in the synthetic optimization of MXene monolayer film epitaxy. This provides clear evidence that the synthesis of monolayer MXene remains an open field that is of fundamental importance in providing a platform to investigate the chemistry, physics, and electronics of genuine monolayer MXene sheets.
Another example of an MXene epitaxial thin film is Mo2CTx, with a starting MAX thin film comprising Mo2GaC phase as a hexagonal ternary laminated carbide.183–185 In this case, two layers of Ga are stacked between the simple hexagonal configurations present between the Mo2C layers. The selective removal of Ga with hydrofluoric acid leads to the formation of Mo2CTx.186 Furthermore, Mo-containing binary-metal MXenes such as Mo2TiC2Tx and Mo2Ti2C3Tx have been demonstrated (Section 1.4).
After introducing the types, properties, and synthetic methods of MXenes, we now turn to their energy-related applications.
Global energy consumption is rising, and significant effort is being devoted towards improving energy storage, conversion, and direct utilization. Herein, we first discuss strategies for clean energy production, including photocatalytic fuel production and photovoltaic devices. We then consider avenues of energy storage in the form of batteries and supercapacitors with MXene materials.
The gradual depletion of fossil fuels has contributed to increasing interest in renewable energy sources. These include solar and wind energy sources, which already contribute to total electricity production. However, most renewable electricity is not fully utilized because the peak daytime output does not match the night-time electricity consumption maxima. Therefore, smart grid strategies that employ enormous numbers of energy storage devices as electricity reservoirs are being developed to automatically adjust electricity delivery according to real-time consumption.
Herein, we concentrate on certain significant results to highlight the latest advances in the application of MXenes to electrochemical energy systems, e.g., supercapacitors, rechargeable batteries, photocatalysts, and electrocatalysts. Conventional chemical reactions covering catalytic NH3 synthesis and photodegradation of organic macromolecule pollutants are also introduced. In addition, solar thermal applications and solar steam productions are reviewed. The use of MXenes for heating purposes in biomedicine, including photothermal therapies, is also briefly discussed. Finally, other energy conversion applications, such as photovoltaics and thermoelectronic power generation, are introduced.
4 Electrochemical energy storage
Most energy conversion and storage systems implement electrochemical reactions that have employed carbon-based materials (graphite) for centuries.191–193 Recent advances in sp2-hybridized carbon materials, e.g., fullerenes,194–196 carbon nanotubes,197–200 and graphene,201–204 have enhanced electrochemical performance due to their large specific surface areas, high electrical conductance, and extraordinary mechanical strength. Other 2D materials, such as TMDCs205–208 and black phosphorene,209–212 also exhibit exciting energy storage potential, thereby attracting extensive research interest.213–218 Additionally, following breakthroughs in mild synthetic methods (Section 3) for MXenes,16,219–221 these materials also exhibit high intrinsic specific areas, electrical conductance, proton exchange, and mechanical strength.The most representative energy storage systems are supercapacitors and secondary batteries. Supercapacitors have large power densities and fast charge–discharge rates; these have enabled mobile applications, such as power sources for the electric automotive industry.222–228 The demand for carbon emission suppression has triggered the current interest in hybrid-powered automobiles, which are occupying an increasing share of the market. This has greatly stimulated research interest in batteries with low cost and long cycle life. Lithium-ion batteries are one of the most successful secondary batteries because they have large specific capacities, which is ideal for compact volume use.229–232 Lithium batteries have been commercially integrated in portable electronics, such as smartphones and tablets, which require high capacity and low weight to satisfy the rising energy consumption of large displays in smartphones.
We first examine the performance of MXene electrodes in supercapacitors.
4.1 Supercapacitors
Supercapacitors fall in the category of electrical energy storage systems that store electricity by forming double charge layers at the dual electrode/electrolyte interfaces. Their power densities are superior to those of conventional dielectric capacitors and other electrochemical energy storage systems. In addition, their fast charging/discharging rates and long cycle life facilitate their use in vehicles and backup power stations.Based on variations in the actual charge storage mechanism, supercapacitors can be divided into three types, namely electrical double layer capacitors (EDLCs),233–237 pseudocapacitors,238–244 and asymmetrical supercapacitors, i.e., one electrode for the EDLC and a pseudocapacitive electrode on the other side.245,246 Recently, a hybrid battery capacitor247,248 with two symmetric (or identical) electrodes has emerged. Both electrodes consist of battery cathode materials or asymmetric electrodes, viz. one electrode for EDLC storage and the other electrode for the redox reaction (lithium-ion storage).
For EDLCs with electrodes made from carbon-related materials, the electrical energy is stored through rapid ion adsorption at their numerous pores through a physical electrosorption mechanism.
Pseudocapacitors involve faradaic charge transfer at, or close to, the surface of the electrodes. Here, transition metal oxides, hydroxides, or conducting polymers provide active electrochemical redox sites.
Hybrid capacitors mix both first and second capacitor mechanisms. The total capacity is the sum of the double layer capacitance for physical ion adsorption and the pseudocapacitance for electrochemical redox reactions. Hybrid capacitors also refer to asymmetrical capacitors with one porous material electrode for fast ion adsorption and another electrode composed of a pseudocapacitive redox transition metal oxide.
Schematics of all three types of supercapacitors are presented in Fig. 16. The anode, cathode, and electrolyte are the three main components of a capacitor. The equation E = 1/2CV2 determines the energy stored in a supercapacitor, where C is the capacitance and V is the working voltage.
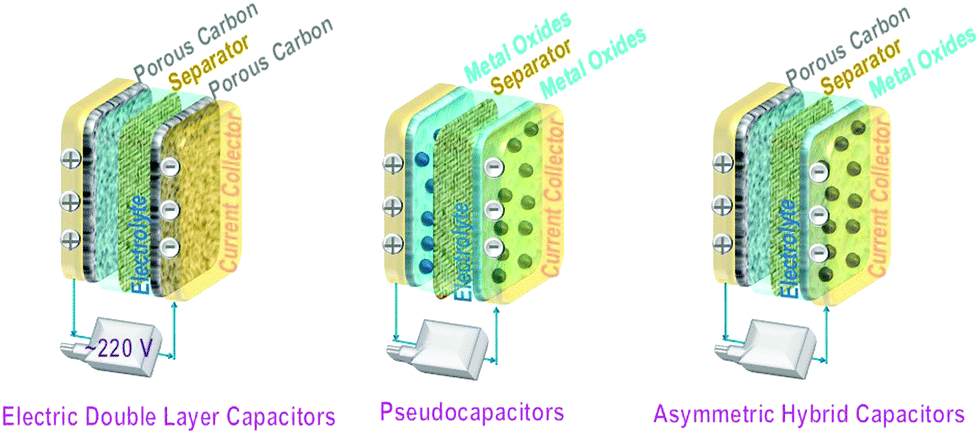 | ||
| Fig. 16 Schematics of the three types of supercapacitors. (left) Electric double layer capacitors (EDLCs) using porous carbon as both symmetric electrodes. (middle) Pseudocapacitors employing transition metal oxide-related materials, which promote electrochemical reactions, as symmetric electrodes. (right) Asymmetric hybrid capacitors using a porous carbon cathode and a transition metal oxide anode (battery type). | ||
The capacitance of EDLCs is proportional to the specific surface areas of their electrodes. Examples include porous carbon, mesoporous carbon, carbon aerogel, and graphene. Commercial supercapacitors with carbon electrodes249,250 have a specific gravimetric capacitance of 80 to 200 F g−1, volumetric capacitance of 35 to 112 F cm−3, energy density of 3.5 to 4.5 W h kg−1, and power density of 80 to 120 W kg−1. The representative capacitance of graphene thin films displays a specific gravimetric capacitance of 78 F g−1,251,252 a volumetric capacitance of 198 to 736 F cm−3,253,254 an energy density of 62.8 to 136 W h kg−1,255,256 and a power density of 7.2 to 500 kW kg−1.257–259 MXenes exhibit superior performance in supercapacitors, especially in the electrodes.220,260,261 The typical performance of these supercapacitors is listed in Table 1.
| MXene type | Capacitance (F g−1) @ rate (mV s−1) | Rate capability (%) | Rate range (mV s−1) | Cycle number | Capacitance retention (%) | Ref. |
|---|---|---|---|---|---|---|
| Notes: PANI, polyaniline; rGO, reduced graphene oxide; GO, graphene oxide; SWCNT, single-walled carbon nanotube; PEDOT:PSS, poly(3,4-ethylenedioxythiophene):polystyrene sulfonate; d-Ti3C2, delaminated Ti3C2; PFD, polyfluorene derivatives; CTAB, cetyltrimethylammonium bromide; PPy, polypyrrole; PVDF, polyvinylidene difluoride; DMSO, dimethyl sulfoxide; PVA, polyvinyl alcohol. | ||||||
| 1. Pseudocapacitors with metal oxides and hydroxides | ||||||
| NixAly(OH)2/Ti3C2 | 1061 @ 1 A g−1 | 44.7 | 1 to 10 A g−1 | 4000 | 70 | 140 |
| MnOx/Ti3C2 | 602 F cm−3 @ 2 | 39 | 2 to 200 | 10000 |
89.8 | 262 |
| LiMn2O4/Ti3C2Tx Li-ion capacitor | 243 @ 100 | 62.1 | 100 to 5000 | 500 | 88.2 | 263 |
| Ar-annealed MnO2/Ti3C2Tx | 212 @ 1 A g−1 | — | — | 10000 |
88 | 264 |
| MoO3/Ti3C2Tx | 151 @ 2 | 66.2 | 2 to 100 | 8000 | 93.7 | 265 |
| TiO2/Ti3C2 | 143 @ 5 | 82 | 5 to 200 | 6000 | 92 | 266 |
| SnO2/Ti3C2 | 125.63 @ 1 A g−1 | 79.8 | 1 to 10 A g−1 | 8000 | 82 | 267 |
| ZnO/Ti3C2 | 120 @ 2 | 75 | 2 to 100 | 10000 |
85 | 268 |
| 2. Pseudocapacitors with conducting polymers | ||||||
| H2SO4/PEDOT:PSS/Mo1.33C | 1310 F cm−3 @ 2 | 45 | 2 to 100 | 10000 |
90 | 269 |
| PVA–KOH/Ti3C2Tx | 530 F cm−3 @ 2 | 58.5 | 2 to 100 | 10000 |
83.7 | 156 |
| PPy/Ti3C2 | 416 @ 5 | 60.1 | 5 to 100 | 25000 |
92 | 270 |
| PPy/Ti3C2 | 406 F cm−3 | — | — | 20000 |
100 | 271 |
| PFDs/Ti3C2Tx | 380 @ 2 | 63.2 | 2 to 100 | 10000 |
98 | 272 |
| Ag/nylon fiber/Ti3C2Tx | 328 mF cm−2 @ 2 | 45.7 | 2 to 100 | 10000 |
96 | 273 |
| PANI/Ti3C2 | 164 @ 2 | 76.8 | 2 to 10 | 3000 | 96 | 274 |
| PVDF/Ti3C2 | 117 @ 2 | 68 | 2 to 50 | 10000 |
98 | 275 |
| PEDOT:PSS/Ti3C2Tx | 30.8 @ 2 | 15.5 | 2 to 200 | 10000 |
90 | 276 |
| 3. Pseudocapacitors with O and N functional groups | ||||||
| N-Doped d-Ti3C2 | 266.5 @ 5 | 79 | 5 to 200 | 2000 | 86.4 | 277 |
| GO/Ti3C2 fiber | 257 | — | — | 20000 |
100 | 278 |
| 4. Electrical double-layer capacitors with foreign electric enhancement | ||||||
| CNT/Ti2CTx paper | 515.3 @ 2 | — | — | 5000 | 95.3 | 279 |
| Ni foam/Ti3C2Tx films | 499 @ 2 | 70 | 2 to 100 | 10000 |
100 | 280 |
| Ni foam/Ti3C2 | 370 @ 2 | 31.6 | 2 to 100 | 10000 |
86.3 | 281 |
| Graphene/Ti3C2Tx fiber | 327.5 @ 10 | — | — | 3000 | 95 | 282 |
| rGO/Ti3C2Tx | 230 @ 2 | 72.6 | 2 to 100 | 1000 | 97 | 283 |
| Sandwiched Ti3C2Tx CNT/Ti3C2Tx paper | 150 @ 2 | 78 | 2 to 200 | 10000 |
100 | 284 |
| CNF/Ti3C2Tx | 123 @ 0.5 | 57.7 | 0.5 to 200 | 500 | 100 | 285 |
| CNT/Ti3C2 | 85 @ 2 | 75 | 2 to 100 | 1000 | 90 | 286 |
| rGO/Ti3C2Tx | 1040 F cm−3 @ 2 | 61 | 2 to 1000 | 20000 |
100 | 158 |
| CNT/Ti3C2Tx | 393 F cm−3 @ 5 | 80 | 5 to 100 | 10000 |
100 | 287 |
| SWCNT/Ti3C2Tx | 314 F cm−3 @ 2 | 65.3 | 2 to 100 | 1000 | 95 | 288 |
| rGO/Ti3C2Tx | 80 F cm−3 @ 10 | — | — | 10000 |
97 | 261 |
| Graphene/Ti3C2Tx | 33 F cm−3 @ 5 | 67 | 5 to 100 | 2500 | 82 | 289 |
| Carbon/Ti3C2Tx | 362 mF cm−2 | — | — | 1000 | 96 | 290 |
| rGO/Ti3C2Tx | 405 @ 1 A g−1 | — | — | 10000 |
100 | 163 |
| rGO/Ti3C2Tx | 154.3 @ 1 A g−1 | — | — | 6000 | 77.8 | 291 |
| 5. Electrical double-layer capacitors with fillers to expand the spacing between the MXene nanosheets | ||||||
| 90 nm Ti3C2 film | 450 @ 10 | 97.8 | 10 to 100 | — | — | 292 |
| 3 μm Ti3C2 hydrogel | 380 @ 2 | 52.6 | 2 to 100 | 10000 |
90 | 292 |
| CTAB–Sn(IV)/Ti3C2 Li ion capacitor | 268 @ 0.2 A g−1 | — | — | 4000 | 71.1 | 293 |
| 5 μm Ti3C2 clay | 250 @ 2 | 80 | 2 to 100 | 10000 |
100 | 2 |
| Electrophoretic deposited Ti3C2Tx | 140 @ 5 | 78.5 | 5 to 50 | 10000 |
100 | 294 |
| K-ion/Ti3C2 | 135 @ 5 | 83 | 5 to 200 | 5000 | 95 | 295 |
| Delaminated Ti3C2Tx | 134 @ 20 | 61.2 | 20 to 1000 | 5000 | 94 | 296 |
| KOH/Ti3C2Tx paper | 132 @ 2 | 60.6 | 2 to 100 | 10000 |
100 | 176 |
| 216 h HF treated Ti3C2 | 118 @ 5 | 91.5 | 5 to 100 | 5000 | 100 | 297 |
| Ti3C2Tx | 70 @ 20 | 97 | 20 to 100 | 1000 | 85 | 298 |
| Ti3C2Tx | 676 F cm−3 @ 20 | 91 | 20 to 100 | 20000 |
100 | 299 |
| DMSO delaminated Ti3C2 | 520 F cm−3 @ 2 | 42.3 | 2 to 100 | 10000 |
100 | 300 |
| 6. Electrical double-layer capacitors with pristine MXenes | ||||||
| 200 °C-annealed N-Ti3C2Tx | 192 @ 1 | 67 | 1 to 200 | 10000 |
92 | 88 |
| H2-annealed Ti2C | 51 @ 1 A g−1 | 86 | 1 to 40 A g−1 | 6000 | 93 | 15 |
| 7. Asymmetric supercapacitors | ||||||
| Ti3C2 aerogel | 87.1 @ 2 | 76.6 | 2 to 100 | 10000 |
95.1 | 301 |
| Ti3C2Tx paper | 295.4 F cm−3 @ 2 | 79.7 | 2 to 100 | 15000 |
94.4 | 302 |
| NiO/Ti3C2Tx | 60.7 mA h g−1@ 1 A g−1 | — | — | 5000 | 72.1 | 303 |
| 8. Microcapacitors | ||||||
| Ti3C2Tx solid micro capacitor | 357 F cm−3 @ 20 | 87.3 | 20 to 100 | 10000 |
100 | 304 |
| Ti3C2 clay on paper | 20 mF cm−2 @ 20 | 88 | 20 to 100 | 10000 |
92 | 305 |
| Ti3C2Tx micro pattern | 27.29 mF cm−2 | — | — | 5000 | 70 | 306 |
| Wire type Ti2C | 4.64 @ 0.1 | — | 5 to 100 | 1000 | 92.62 | 307 |
A progressive report on mesoporous MXene films has promoted their use in supercapacitors. MXene hydrogels292 comprising a 3 μm thick Ti3C2Tx thin film possess a capacitance of 380 F g−1 at a scan rate of 10 V s−1 and a volumetric capacitance of 1500 F cm−3 (Fig. 17). These values are comparable to those observed for commercial supercapacitor electrodes using RuO2.308–312
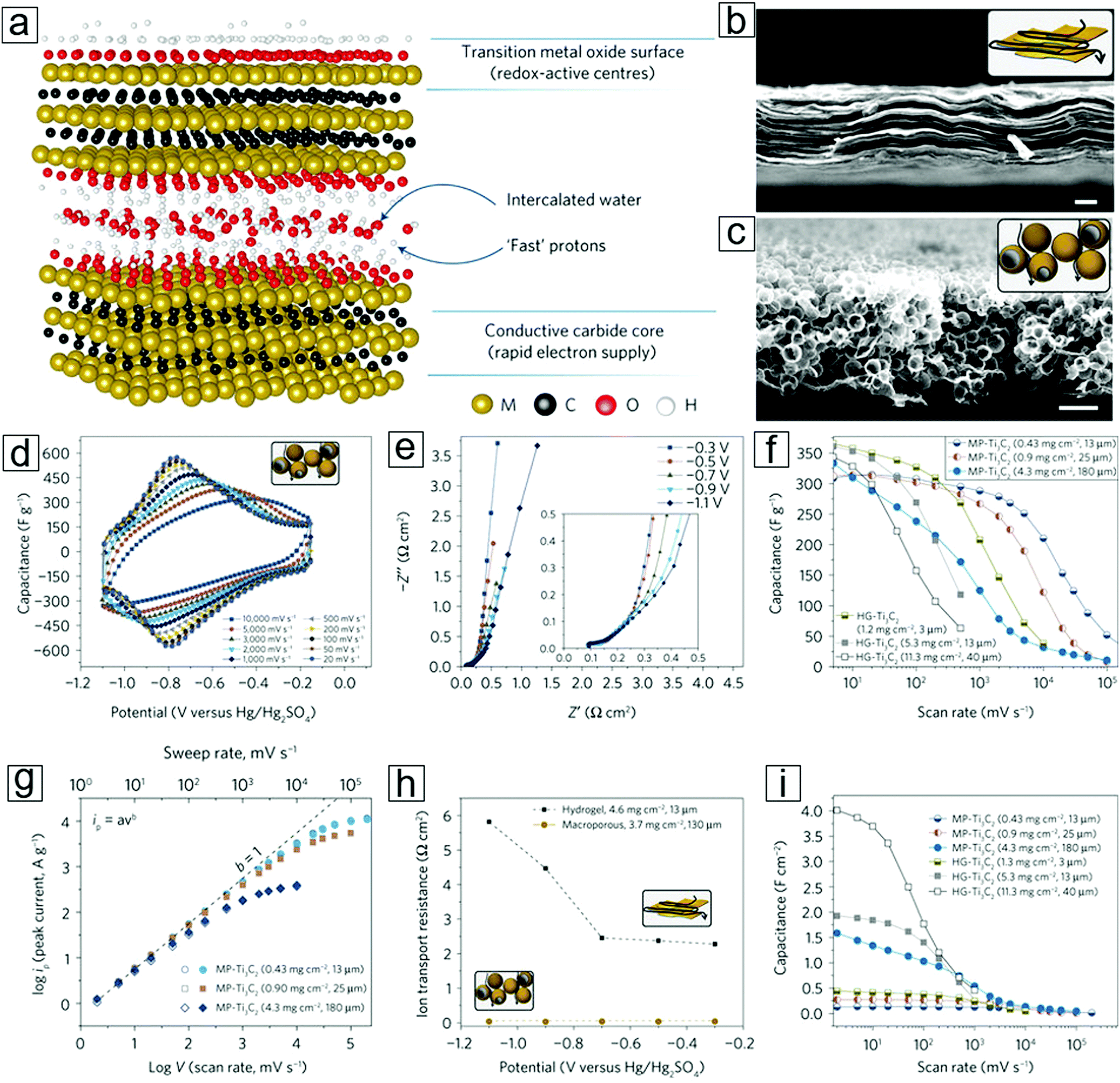 | ||
| Fig. 17 Supercapacitors with macroporous or planar MXene Ti3C2Tx as double-layer symmetrical electrodes. (a) Schematic of MXene-facilitated proton exchange with water intercalation for energy storage. (b) Schematic and scanning electron microscopy (SEM) image of the MXene hydrogel. (c) Schematic and SEM micrograph of the macroporous MXene synthesized over a template of porous poly(methyl methacrylate) (PMMA) spheres. (d) Cyclic voltammetry profiles of macroporous MXenes at different scan rates. (e) Electrochemical impedance spectroscopy information recorded at different potentials for a macroporous MXene film. (f) Rate performance of MXene films with different preparation methods and mass loadings. Gravimetric capacitance is used. HG represents the hydrogel method, while MP represents the macroporous MXene method. (g) Peak current versus voltage scan rate for macroporous films with different mass loadings; anodic current (open shapes) and cathodic current (filled shapes). (h) Ion transport resistances for both macroporous and hydrogel MXene electrodes. Data are extracted from panel (e). (i) Rate performance of MXene films with different areal capacitances. Reprinted by permission from Macmillan Publishers Ltd: [Nature Energy] ref. 292 copyright (2017). | ||
Double-layer supercapacitors have a limited theoretical capacitance that is solely determined by their physical properties, e.g., specific area. Their charging and discharging rates are suppressed by the large ionic resistance in the porous carbon structures. Pseudocapacitors employ a rapid surface redox storage mechanism that enables a larger power density than that afforded by double-layer capacitors.
The charge storage mechanism in Ti3C2Tx is generally recognized as an electrochemical pseudocapacitance process292 which depends on variations in the oxidation states of titanium [reaction (7)]:
| Ti3C2Ox(OH)yFz + δe− + δH+ = Ti3C2Ox−δ(OH)y+δFz | (7) |
To elucidate the large difference between the theoretical and experimental results, an ultrathin 90 nm MXene film was implemented to eliminate the influence of ionic transport resistance (Fig. 17). A high capacitance of 450 F g−1 was achieved using glassy carbon to minimize the ionic transport limitations, e.g., avoiding the water splitting that frequently occurs at the Pt or Au supporting electrodes. On increasing the MXene thickness to 5 μm, the capacitance decreased to 250 F g−1. Hence, the origin of the discrepancy between the theoretical and experimental values was attributed to the lower accessibility of the electrochemical active redox sites with thicker films in a planar electrode configuration. This is due to the large ionic resistance caused by the diffusion mechanisms. To overcome these diffusion obstacles, an MXene hydrogel was employed. Using this type of electrode facilitates the transport of electrolytes between the MXene sheets, eventually leading to a gravimetric capacitance of 380 F g−1 or a volumetric capacitance of 1500 F cm−3. This occurs because the bulky electrolyte ions can access the active redox sites. The potential window was enlarged to 1 V. However, the hydrogel electrodes displayed limited rate performance, viz., the gravimetric and volumetric capacitance rapidly decreased after charging at a rate of 700 mV s−1. Thus, the rate handling ability of the MXene electrode requires a breakthrough. Strategies involving macroporous electrode design and suppressed pore tortuosity have shown efficient enhancements in rate performance (Fig. 17a–c). Gogotsi, Simon et al.292 introduced homogeneous macroporosity in MXene films using polymeric spheres as templates. Gravimetric and volumetric capacitances of 210 F g−1 and 0.09 F cm−2, respectively, were achieved for ∼12 μm thick macroporous MXene films at 10 V s−1. This success has exceeded some of the milestone results.248,254,255
A 25 μm macroporous MXene thin film displayed gravimetric capacitances of 280 F g−1 at 1 V s−1 and 120 F g−1 at 10 V s−1 (Fig. 17f, h and i). Moreover, a 180 μm thick MXene macroporous film292 yielded a capacitance of 125 F g−1 at 1 V s−1. This macroporous electrode strategy is successful for Ti3C2Tx and can be applied to Mo2CTx. For example, a 30 μm thick macroporous Mo2CTx film shows a capacitance of 100 F g−1 at 10 V s−1. Therefore, this concept can be reasonably expanded to other experimentally discovered and theoretically predicted MXene types.7,313 In addition, this strategy can elevate areal capacitances, e.g. capacitances of 4 F cm−2 at 5 mV s−1 and 0.5 F cm−2 at 1 V s−1 for a 40 μm thick hydrogel are maintained for macroporous electrodes with a 43 mg cm−2 MXene mass loading.
A macroporous architecture can be used to improve ionic electrolyte transport between the MXene sheets. Thus, due to the enlargement of the layer spacing via the introduction of foreign species (e.g., graphene nanosheets), alkaline Na or K ions can be intercalated between the MXene sheets. Yan, Gogotsi et al.158 fabricated an rGO/MXene composite film via self-assembly of negatively charged Ti3C2 MXene and positively charged rGO in a layer-by-layer electrostatic fashion. The graphene nanosheets between the MXene layers act as supporting pillars, enabling fast electrolyte ion delivery in the enlarged sheet–sheet spaces. This is eventually reflected in high rate performance. In an electrochemical test with a three-electrode system, the MXene/rGO composite film exhibited a specific capacitance of 332 F g−1 and a volumetric capacitance of 1040 F cm−3 at 2 mV s−1. The retention ratio of the capacitance was 61% at 1 V s−1. The capacitance displayed extraordinary cycling performance and no significant decay after 20000 cycles. The supercapacitor with symmetric MXene/rGO electrodes exhibited a high volumetric energy density of 32.6 W h L−1 and a volumetric power of 744 kW L−1.
K-ion intercalation between the MXene sheets leads to an interlayer spacing of 0.77 nm, which is much larger than the initial 0.2 nm spacing in the OH/F terminated MXene sheets. The preparation protocol is schematically presented in Fig. 18a. X-ray diffraction (XRD) results revealed a downshift of 2θ for the (002) phases with K-ion intercalated MXenes (Fig. 18b), indicating that lattice expansion occurred in the c axis. The conventional OH/F-terminated MXene sheets have a layer–layer distance of 0.98 nm (Fig. 18c and d). K-ion intercalation followed by annealing at 400 °C in an Ar environment eliminates the surface functional groups, further increasing the void space to 1.26 nm (Fig. 18e and f).
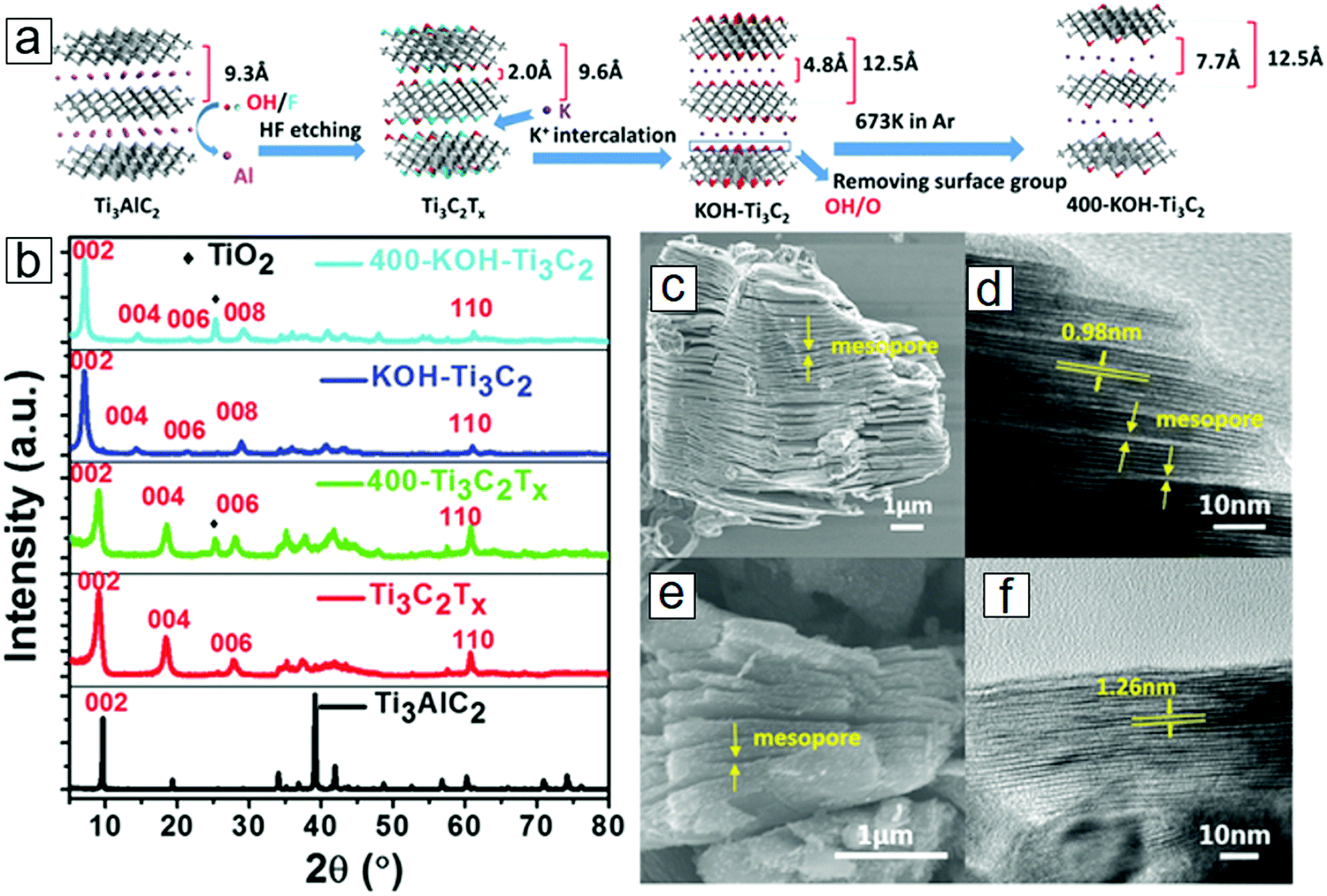 | ||
| Fig. 18 MXene Ti3C2 modified with K-ion intercalation. (a) Synthetic protocol of K-ion-modified MXene. After surface coating with KOH solutions and thermal annealing in Ar, the MXene was successfully intercalated with K ions. (b) X-ray diffraction (XRD) curves of K-ion-modified MXene, MXene, and MAX. MXene after annealing at 400 °C in Ar is also included. (c and d) Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) micrographs of conventional Ti3C2Tx. (e and f) SEM and TEM micrographs of 400 °C-annealed K-ion-modified Ti3C2. From ref. 314. Copyright © [2016] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
In the electrochemical evaluation of a three-electrode platform, a gravimetric capacitance of 517 F g−1 has been achieved for K-ion intercalated MXene electrodes.314 The increased number of accessible redox sites of the Ti atoms account for the enhanced intercalation-induced pseudocapacitance. The electrodes display high reversibility of the redox reactions during the charging/discharging process and exhibit extraordinary cycling performance with 99% capacitance retention after 10000 cycles. In the symmetric supercapacitor configuration, the operation potential increases to 1.6 V (Fig. 19a). The Ragone plots present a maximum energy density of 27.4 W h kg−1 at 1 A g−1 current density (Fig. 19b). The supercapacitor exhibits long cycling life, with 90.4% capacitance retention after 5000 cycles. This cation intercalation strategy indeed enhanced the pseudocapacitance of the electrodes.
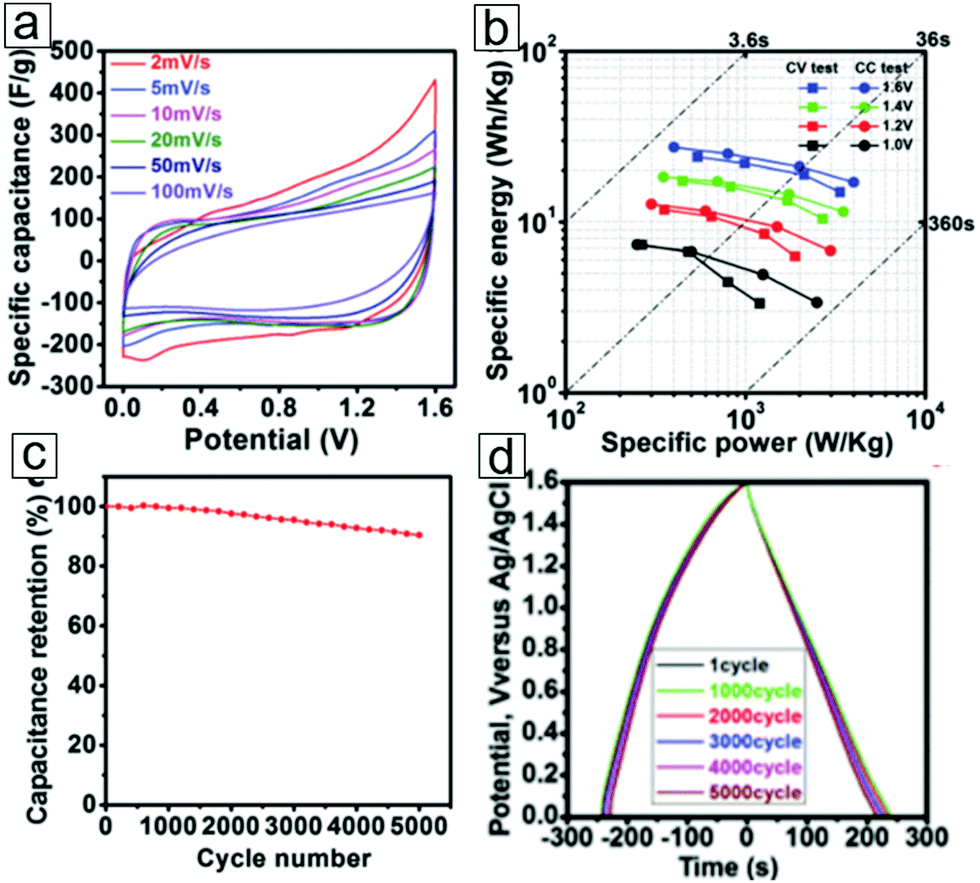 | ||
| Fig. 19 Electrochemical performance of K-ion-intercalated MXene Ti3C2 symmetric pseudocapacitors. (a) Cyclic voltammetry curves at various scan rates within a 1.6 V potential. (b) Energy density versus power density curves under various potentials in cyclic voltammetry measurements and galvanostatic charging/discharging tests. (c) Cycling life performance ≤5000 cycles at a current density of 1 A g−1. A mixed 1 M H2SO4/1 M Li2SO4 solution was used as the electrolyte. (d) Galvanostatic charging/discharging plots for K-ion-intercalated Ti3C2 symmetric supercapacitors after 1, 1000, 2000, 3000, 4000, and 5000 cycles. The curves exhibit steady discharging profiles. From ref. 314. Copyright © [2016] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
One can gain insight from theoretical predictions to explore future opportunities. The theoretical capacitances of MXene materials are listed in Table 2.
| MXene type | Capacitance (volumetric or gravimetric) | Ref. |
|---|---|---|
| Li6/Nb2C | 1982 F cm−3 | 315 |
| Li6/Nb2CF2 | 1406 F cm−3 | 315 |
| Li6/Nb2CO2 | 1617 F cm−3 | 315 |
| Bare Nb2C cathode | 1828 F g−1 | 316 |
| Nb3C2 | ca. 1600 F g−1 | 316 |
| Nb4C3 | ca. 1200 F g−1 | 316 |
| Nb3C2T2, Nb4C3T2 | 441 to 663 F g−1 | 316 |
| Graphene oxide | 189 to 191 F g−1 | 255, 317 and 318 |
The capacitance values for Nb2C, Nb3C2, and Nb4C3 are higher than those for MXenes with surface functional groups. This suggests that removal of functional surface groups, for example by annealing, can improve the capacitance.
4.2 Anodes in lithium-ion batteries
Rechargeable batteries represent another dominant energy storage device, featuring high energy capacity and work voltage, light weight, long cycle life, and good environmental stability. From early-stage lead–acid batteries dating back to 1859319,320 to the latest lithium-ion battery introduced in 1991,321–323 rechargeable battery technology has been advancing in parallel with the advent of novel materials. 2D MXenes have been employed in batteries as anodes since the second year of their discovery.11,29,30,171,176,324 Indeed, investigation of MXene as a useful material in batteries is continuously growing.325–329There are emerging trends in the architectures of rechargeable batteries: first, low cost, abundant elements such as Na are replacing Li to pave the way for sodium-, magnesium-, K-, and Ca-ion batteries.330–335 Second, by using sulfur as the high energy density cathode material to replace the LiCoO2 cathode, lithium–sodium–sulfur batteries are produced.336–338 In addition, Li–metal passivated with a protection layer has been revisited as an anode material by exploiting its high theoretical capacity potential.339–343 Third, metal–oxygen batteries,344–347 which enable chemical and electric energy conversion through the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR), possess light weights and high energy densities. These batteries use oxygen in air as a reactant to decrease their weight.348–353
The graphite anode/LiCoO2 spinel cathode system remains the most successful electrode couple in commercial lithium-ion batteries. For example, single lithium cells provide power for smartphones, tablets, and six-cell modules for laptops. Furthermore, battery packs with an energy density range of 10 to 30 kW h are employed to power electric vehicles.356 Large battery packs with an energy density of 1 MW h are targeted for renewable electrical energy storage. These will eventually level the load and integrate into the electrical grid. This transport power and renewable electricity storage requires more power density, lower cost, and high energy density. Because cobalt is an expensive noble metal and LiCoO2 has limited capacity357–360 (theoretical limit = 274 mA h g−1; experimental value = 140 mA h g−1), new materials with high abundance and low cost are being sought. Trends in new materials include LiMn2O4,361–363 LiMn1−xFexPO4,364–367 LiFePO4,368 and others.369–371
The commercial anode is based on Li-ion intercalation mechanisms. For example, in layered graphite, a theoretical capacity of 372 mA h g−1 and an experimental capacity of 330 mA h g−1 have been observed.372,373
The alloying mechanism applies to elements such as Si and Sn, which form reversible alloys with lithium. These bulk material anodes are at a disadvantage because of pulverization induced by large volume changes, e.g. a 300% volume expansion for Si during the charging and discharging process.374,375 The advent of nanotechnology has enabled new directions in anode development, such as the use of nanoparticles with higher specific areas. Various elements have been shown to have high specific capacities for lithium storage that exceed that of graphite; examples include Si (3579 mA h g−1),376–380 Ge (1624 mA h g−1),381–384 Sn (994 mA h g−1),374,385 SnO2 (790 mA h g−1),386–388 Fe2O3 (1005 mA h g−1),389 Fe3O4 (924 mA h g−1),390 NiO (717 mA h g−1),391–397 CoO (716 mA h g−1),398 other oxides,399 and of course Li (3860 mA h g−1).374 The high mechanical strength of 2D materials plays a special role in wrapping and protecting these materials from large volume variations, thereby decreasing pulverization.400–402 For example, graphene shells137,403 over Si and Sn nanoparticles can enhance their electrochemical performance and cycling life as anode materials.404 In addition, small dimensions provide short paths for electron and ion motion in ion-exchange anodes, which allows rapid diffusion of Li ions between the electrodes.
Nanosized materials are more active than their bulky counterparts toward lithium. When some transition metal (TM) oxides and fluorides react with Li ions electrochemically, nanocomposites of TM/Li2O and TM/LiF405 that can decompose and form reversibly are readily formed. These lithiation and delithiation reactions favor an electrochemical mechanism. In addition, new mechanisms can occur in certain transition metal oxides, i.e., RuO2 nanoparticles, as active anode materials. Here, a capacitive charge separation between the Li2O grains and metal nanoparticles occurs until the Li potential approaches that of pure Li.406 This allows excess lithium ions to be stored at the interface of the grain boundaries.406 Indeed, nanomaterials,407–411 especially 2D materials such as graphene and phosphorene, show great promise as anode materials in lithium- and sodium-ion batteries. 2D MXenes with larger interlayer spacings (0.7 to 1.1 nm) and extraordinary electron and ion conductivities have led to great success in batteries, with theoretical Li capacities ranging from 259 mA h g−1 to 1767 mA h g−1.412,413
We will first examine the battery related performance of individual Mn+1XnTx materials, i.e., standalone MXene nanosheets, without coupling with other anode materials or functionalization with foreign species. The electrochemical performance of MXene anodes in a pioneering lithium-ion battery was studied at the Simon and Gogotsi labs from 2012 to 2013.29,30,171
The MXene, Ti2CTx, presented an experimental gravimetric capacity of 160 mA h g−1 for Li intercalation, which is 1.5 times larger than that found for Ti3C2Tx (110 mA h g−1).29,171 Assuming an intercalation mechanism for Li storage, the two layers of Ti atoms (estimated as 2-mass) in Ti2C host one layer of Li-atom intercalation, while three layers of Ti (estimated as 3-mass) in Ti3C2 provide one layer of Li-atom accommodation. This is equivalent to one Li-atom layer in 2 masses of Ti2C, which is 1.5 times denser than one Li-atom layer in three masses of Ti3C2. This was confirmed by DFT calculations, which showed theoretical capacities of 383 mA h g−1 for Ti2CO2Li2 and 268 mA h g−1 for Ti3C2O2Li2. Hence, the Li storage mechanism in Ti2CTx involves an intercalation mechanism. In addition, the transition metal type affects the specific capacitance. For example, Ti2CTx presents a capacitance of 110 mA h g−1 at a rate of 1C, while Nb2CTx exhibits a capacity of 180 mA h g−1 at the same rate. The highest specific capacity of the tested MXenes11 was observed in V2CTx, which presented capacities of 280 mA h g−1 at a rate of 1C and 125 mA h g−1 at a rate of 10C.
Surface termination groups can also influence the electrochemical performance. Theoretical calculations show capacities of 320 mA h g−1 for bare MXene, 268 mA h g−1 for an O-terminated MXene, 130 mA h g−1 for the F-terminated MXene, and 67 mA h g−1 for the OH-terminated MXene.174,324,416 The oxygen surface configuration facilitates the highest capacity in all three types of surface terminated MXenes due to the bilayer Li atom formation at the MXene interlayer spacing.174,324 DFT theoretical calculations predict a reversible bilayer formation of Li atoms between the MXene Ti3C2O2 sheets, while in situ X-ray absorption spectroscopy experimentally proved the Li-ion charge mechanism. When the potential decreases continuously from 3.0 V to 0.5 V versus Li/Li+ during the charging/discharging process, the continuous change in the oxidation state of Ti is analogous to the potential variation. A further decrease in potential does not change the Ti edge energy but leads to the formation of a second Li-atom layer. The double layer capacity for Li-ion storage can also apply to other MXenes.24,417
Moreover, the specific capacity of MXenes can be greatly improved depending on their composite architectures with other materials. The performance and anode configurations of various MXene batteries are listed in Table 3.
| MXene type | Current rate (mA g−1) | Initial discharge capacity (mA h g−1) | Cycle number | Last capacity after cycling (mA h g−1) | Ref. |
|---|---|---|---|---|---|
| Notes: main voltage range used for the measurements = 0.01 to 3 V; the stepwise voltage scanning differs slightly according to the cyclic voltammetry equipment employed. For the current rate, the unit is mainly mA g−1; however, the unit C is also sometimes employed. Here 1 C equals a current rate with a value of the theoretical capacity. Abbreviations: CNT, carbon nanotube; CNF, carbon nanofiber; PVP, polyvinylpyrrolidone. | |||||
| PVP–Sn(IV)/Ti3C2 | 500 | 1626 | 200 | 544 | 418 |
| CNT/Ti3C2Tx | 500 | 1250 | 100 | 500 | 419 |
| MoS2/Ti3C2 | 20000 |
1210 | 3000 | 580 | 420 |
| SnO2/Ti3C2 | 300 | 1030 | 200 | 360 | 421 |
| SnO2/Ti3C2 | 500 | 916 | 50 | 843 | 422 |
| MoS2/Ti3C2 | 500 | 656 | 50 | 230 | 139 |
| CNT/Nb2CTx | 2.5C | 500 | 320 | 400 | 172 |
| H2O2–Ti2CTx | 500 | 429 | 1000 | 280 | 423 |
| Ti3C2 | 1C | 410 | 100 | 410 | 171 |
| MoS2/Ti3C2 | 1000 | 386.4 | 200 | 131.6 | 424 |
| CNF/Ti3C2 | 1000 | 320 | 295 | 320 | 425 |
| Ag/Ti3C2(OH)0.8F1.2 | 1000 | 310 | 5000 | 310 | 426 |
| Nb4C3Tx | 500 | 278 | 100 | 380 | 427 |
| V2C | 500 | 260 | 500 | 252 | 428 |
| V2C | 10C | 260 | 150 | 125 | 11 |
| Li4Ti5O12/Ti3C2Tx | 500 | 236 | 200 | 125 | 429 |
| Nb2O5/Nb4C3Tx | 250 | 235 | 400 | 208 | 430 |
| Ti2C | 0.04C | 225 | 120 | 80 | 29 |
| (V0.7Ti0.3)2CTx | 500 | 221 | 20 | 254 | 431 |
| CNT/Ti2C | 100 | 179.6 | 200 | 155.5 | 279 |
| Ti3CNTx | 500 | 174 | 1000 | 300 | 432 |
| Nb2C | 10C | 170 | 150 | 110 | 11 |
| Ti3C2 | 1C | 123.6 | 75 | 118.7 | 433 |
| Ti2C | 10C | 70 | 1000 | 60 | 30 |
| Carbon black/Nb2C | 30 | 16 | 50 | 6.7 | 434 |
| Carbon black/Ti3C2 | 30 | 15 | 50 | 5.9 | 434 |
The strategies for creating MXene composites with anode applications include the implementation of heterostructure blending with other nanostructures and interlayer spacing enlargement with foreign atoms or molecules. A hybrid of carbon nanotubes and MXene flakes as an anode material exhibited a specific capacity of 750 mA h g−1.419 TMDCs such as MoS2 can also form composites with MXenes to enhance the capacity of Li-ion batteries.
Another blending protocol for cetyltrimethylammonium bromide (CTAB)–Sn/MXene formation (Fig. 20) also presented highly promising electrochemical performance.
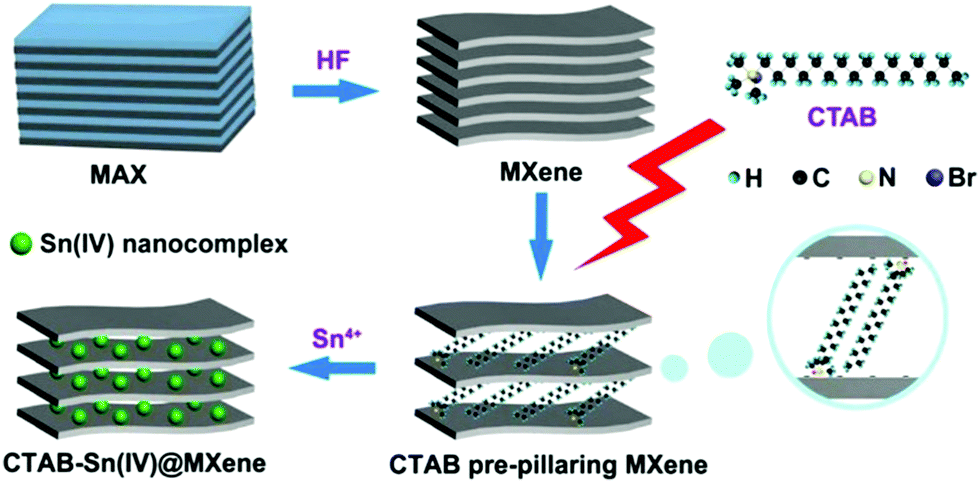 | ||
| Fig. 20 Schematic of Sn-ion modified MXene. Steps include selective etching of MAX, MXene interlayer spacing extension with cetyltrimethylammonium bromide (CTAB), and Sn-ion complex intercalation. Reprinted with permission from ref. 293. Copyright 2017 American Chemical Society. | ||
Cyclic voltammetry tests show reversible charging/discharging at different scan rates (Fig. 21a). The CTAB–Sn/Ti3C2 composite achieved a capacity of 800 mA h g−1 at a current density of 0.1 A g−1, which far exceeds the performance of Ti3C2 itself. This indicates that Sn nanoparticle/Ti3C2 composites function as well as CTAB/Ti3C2 composites (Fig. 21b).
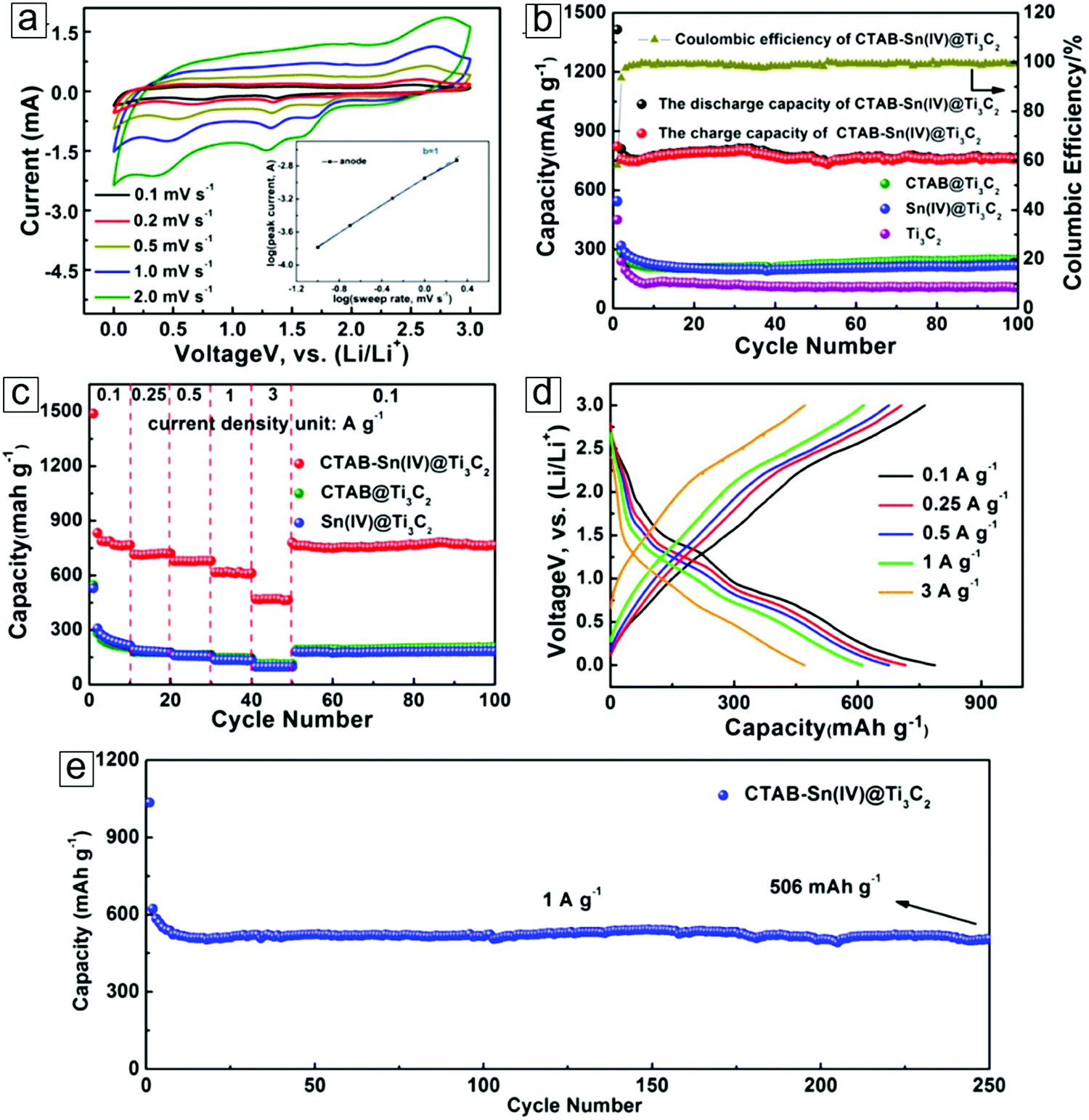 | ||
| Fig. 21 Electrochemical performance of Sn-modified MXene electrodes. (a) Capacitance–voltage (CV) curves at different scan rates. The inset is the b-slope determination of the peak currents extracted from the CV curves. (b) Cycling performance of four MXene samples at 0.1 A g−1. (c) Rate performance of these samples. (d) Discharge–charge curves of Sn-modified MXene Ti3C2 electrodes at various current densities. (e) Long cycling performance at a high current density of 1 A g−1. Reprinted with permission from ref. 293. Copyright 2017 American Chemical Society. | ||
The coulombic efficiency has a high retention ratio of 100% after 100 cycles. The CTAB–Sn/MXene architecture displays excellent rate performance and a capacity of 600 mA h g−1 at a current density of 1 A g−1. When a current density of 0.1 A g−1 was employed, the capacity recovered to 800 mA h g−1 (Fig. 21c).
The charge/discharge plots of the Sn-modified MXenes reveal high reversibility at various current densities (Fig. 21d). After >200 cycles, the electrode yields extraordinary cycling performance, remaining at 96% with 506 mA h g−1 at a current density of 1 A g−1.
TMDCs have presented extraordinary electrochemical performance for energy storage applications.435–438 MoS2 possesses a high specific capacity of 950 mA h g−1 at a rate of 0.1 A g−1;439 however, it has the disadvantage of low electrical conductance. Therefore, MoS2 composites with good electrically conductive materials are efficient strategies for enhancing Li storage close to its theoretical capacity. The electrochemical performance was compared following the blending of MoS2 with rGO, oxidized MXenes, and MXenes. The charging/discharging plots of the first three cycles display high reversibility of Li-ion intercalation and deintercalation (Fig. 22a). The MXene/MoS2 composite presents the lowest electrochemical impedance (Fig. 22b).
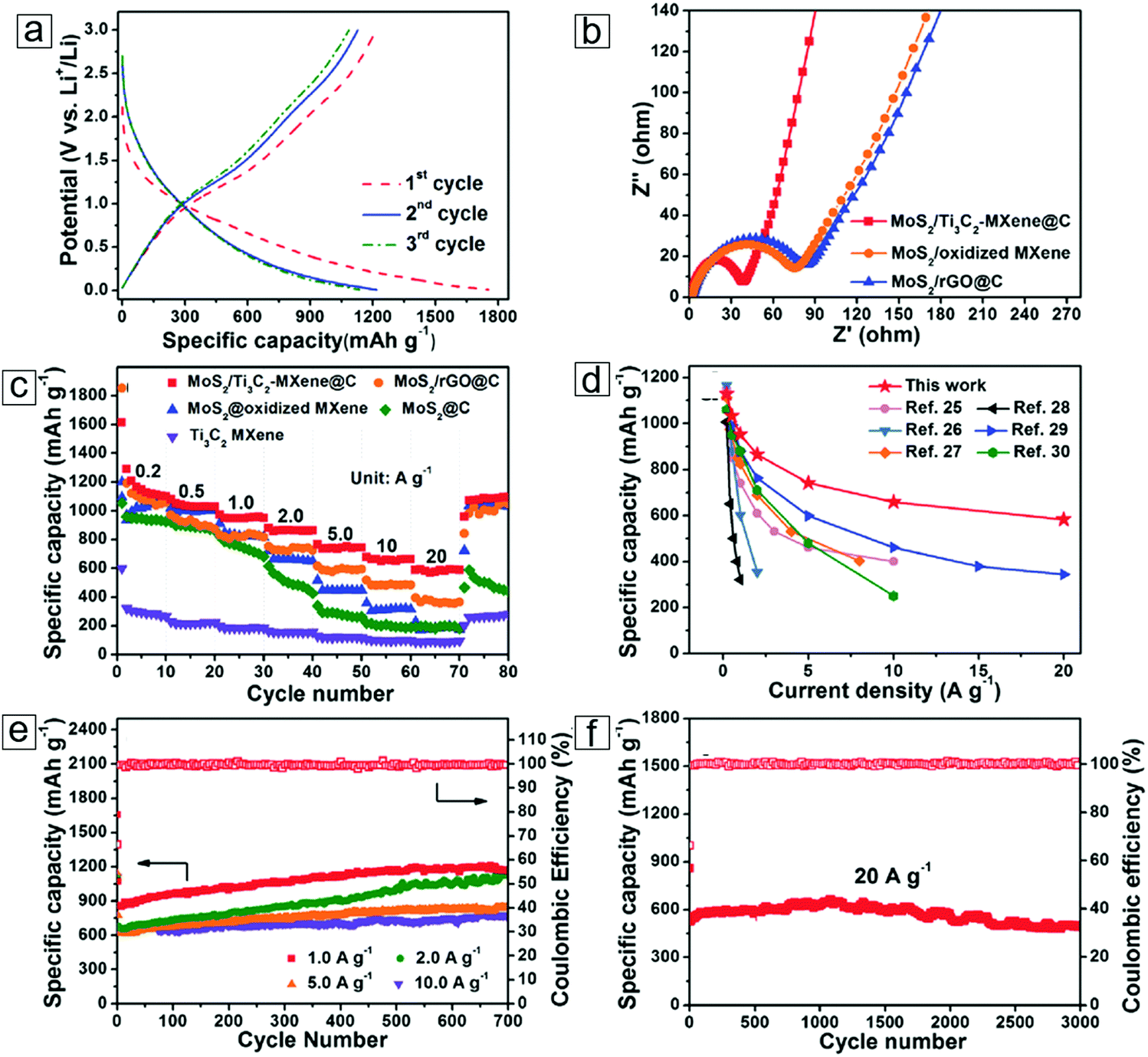 | ||
| Fig. 22 Electrochemical performance of MoS2–MXene@C composite electrodes. (a) Charge–discharge voltage profiles for the first three cycles at a current density of 0.2 A g−1. (b) Electrochemical impedance spectra of an MoS2–MXene composite and MoS2-other two-dimensional (2D) material composites. (c) High-rate specific capabilities of MoS2–MXene composite electrodes at a high current density. (d) Comparison of the capacity of this composite with those of other MoS2 hybrid composites. (e) Coulombic efficiencies and long-term cycling performance of MXene composite electrodes at high current densities. (f) Long cycle life (3000 cycles) of MoS2/MXene electrodes at a high current rate of 20 A g−1. From ref. 420. Copyright © [2016] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
When a MoS2/rGO composite was formed, the capacity was enhanced to 1100 mA h g−1, while a MoS2/MXene Ti3C2 composite presented an even larger capacity of 1200 mA h g−1 (Fig. 22c). These MoS2/MXene hybrids exceeded the other composites in rate performance, e.g., 600 mA h g−1 at a current density of 20 A g−1 (Fig. 22d). Extraordinary cycling performance with the capacity remaining steady at 1200 mA h g−1 after a 700-cycle test at 1 A g−1 was revealed. Additionally, the coulombic efficiency was maintained at 100%, possibly due to the mechanical strength of the system (Fig. 22e). Furthermore, the MoS2/MXene anode system exhibited excellent high-rate performance, e.g., 500 mA h g−1 at 20 A g−1, even after 3000 cycles (Fig. 22f).
This confirms that the extraordinary electrical conductance of MXenes can elevate the performance of MoS2-based anodes. The excellent conductance of MXenes can also be relevant to other anode materials that possess high capacity but relatively low conductance. These include nanoparticles of Si, Ge, Fe3O4, Co3O4, and MnO2 as well as 2D nanosheets of phosphorene, C3N4, and WS2.
The theoretical capacities of different types of MXenes have been determined by theoretical chemists. A few examples are listed in Table 4. The theoretical capacity of V2C for F- and OH-terminated MXenes (<200 mA h g−1) is much lower440 than the 940 mA h g−1 capacity observed for bare V2C. This large difference was attributed to side reactions and steric effects. The surface groups extruding in the interlayer spacing of the MXenes suppress the diffusion of Li atoms from edge adsorption (steric effect).324 On the other hand, the functional groups react with Li to form LiF and LiOH. These species tend to cause collapse of the MXene structures with the formation of the mesostable intermediates440 V2CF2Li and V2C(OH)2Li1.5; thus, they are not thermodynamically preferable. Hence, strategies for eliminating surface terminal groups in MXenes will help enhance their capacity in lithium-ion batteries.
| MXene type | Theoretical capacity (mA h g−1) | Ref. |
|---|---|---|
| Si2–Ti2C | 1767 | 412 |
| P2–Ti2C | 1264 | 412 |
| Li6–V2C | 1412 | 441 |
| Li6–Cr2C | 1386 | 441 |
| Li4–V2C | 941 | 440 |
| Li–V2C(OH)2 | 181 | 440 |
| Li0.5–V2CF2 | 116 | 440 |
| Mn2C | 879 | 442 |
| Li6 Mo2C | 789 | 443 |
| Li4–Mo2C | 526 | 443 |
| Nb2C | 542 | 444 |
| Mo2C | 526 | 445 |
| Ti2N | 484 | 446 |
| Ti3C2 | 448 | 447 |
| Ti3C2 | 447 | 447 |
| Sc2C, Ti2C | >400 | 416 |
| Ti3CN | 320 | 448 |
| Zr2C | 310 | 413 |
| V2CO2 | 276 | 449 |
| Ti3CNO2 | 269 | 448 |
| Ta2C | 264 | 450 |
| S2–Zr2C | 259 | 413 |
| Others | ||
| Si | 3579–4212 | 378 and 451–453 |
| Li | 3680 | 454 |
| P | 2596 | 455 and 456 |
| Ge | 1624 | 381 and 384 |
| Fe2O3 | 1005 | 457 |
| Sn | 994 | 374 |
| SnO2 | 790 | 458 |
| Fe3O4 | 924 | 459 |
| NiO | 717 | 460 |
| CoO | 716 | 398 and 461–463 |
| Graphite | 372 | 409 |
This will provide future opportunities for the preparation of MXene composites blended with high-capacity materials such as Si, Ge, and Sn.
4.3 Anodes in sodium-ion batteries
The sodium ion battery represents another important secondary ion battery; it adopts the inexpensive and abundant element Na as the shuttling ion for intercalation/deintercalation-based electrochemical energy storage. However, the current selection of materials for sodium ion energy storage has limited capability of satisfying electrochemical performance.MXenes have larger interlayer distances than graphite, which enables them to host the intercalation/deintercalation of different-sized ions, including Na, K, Mg, Ca, and Al. Indeed, a theoretical study has reported analogous capacities of Na and K ions in MXenes compared to that of Li ions.24 These calculated capacities were predicted with atomic models by counting one-metal-atom layer intercalation between two MXene sheets. However, a more detailed study revealed that a double layer of metal-atom formation is thermodynamically favorable between MXene interlayer spaces. This may enable a two-fold theoretical capacity.
Na-ion intercalated MXene nanosheets were also successfully employed in a sodium-ion hybrid capacitor. Sodium-ion hybrid capacitors possess the merits of high energy and power in one electrochemical storage device. In this system, Ti2CTx was implemented as the electrode material in a sodium-ion hybrid capacitor or the equivalent of a pseudocapacitive anode sodium-ion battery; viz., the anode in a sodium ion battery does not involve a Na intercalation mechanism but relies on an electrochemical redox reaction at the anode for electric energy storage. Unlike aqueous alkali metal solutions, in which the alkali metal ions spontaneously intercalate into the MXene interlayer spaces, Na ions from non-aqueous liquids do not intercalate into MXene sheets via simple immersion. Conversely, when directly immersing MXenes into a non-aqueous electrolyte from a NaPF6/ethylene carbonate/diethyl carbonate mixture, no sodium ion intercalation occurs. After the first cathodic scan, an irreversible current of <1.0 V versus Na/Na+ is found. This indicates that the intercalation and/or adsorption of Na ions into the MXene nanosheets is electrochemical and the MXene layers are pillared with Na+ ions (Fig. 23). After activation, the interlayer distance (1.01 nm) of the Na-pillared MXene sheets does not change with further charging/discharging.
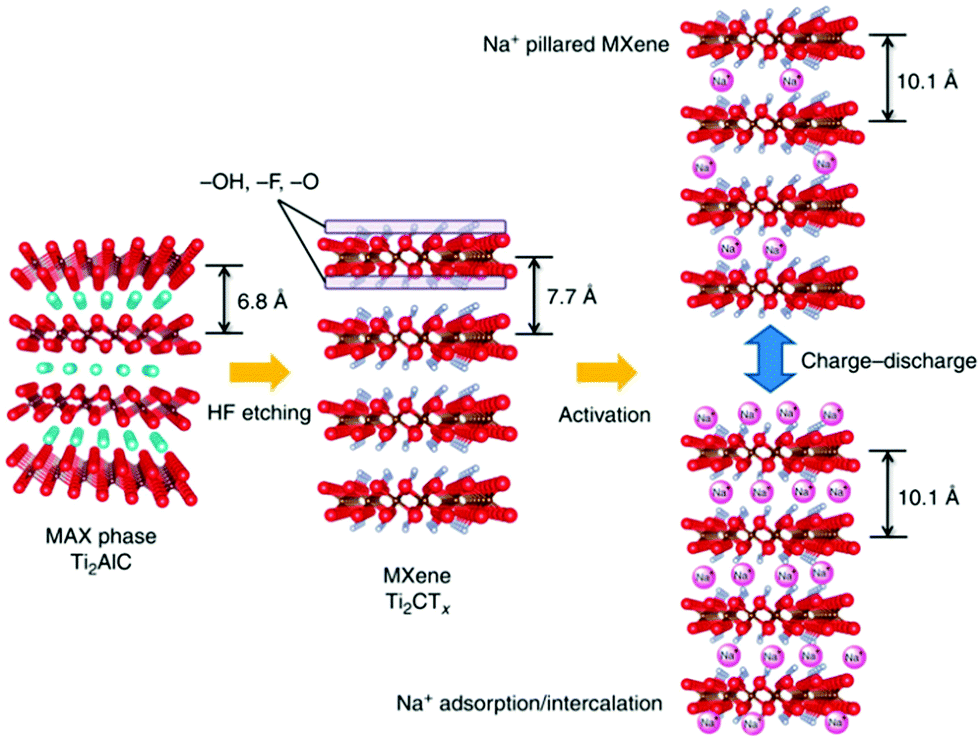 | ||
| Fig. 23 Schematic of Na-ion-pillared MXene Ti2CTx formation from electrochemical reactions. Left is the MAX phase; middle is the MXene. The right upper panel is the activation of MXene with initial absorption and intercalation of Na ions. The right bottom panel is the Na-ion-pillared MXene without changes in the layer distance after several charge/discharge cycles. Reprinted by permission from Macmillan Publishers Ltd: [Nature Communications] ref. 20 copyright (2015). | ||
A prototype full cell of this hybrid device consists of a Ti2CTx anode and a Na2Fe2(SO4)3 alluaudite cathode (Fig. 24a). In this cell, the Na2Fe2(SO4)3 cathode displays a specific capacitance of 100 mA h g−1 at an operation potential of 3.8 V versus Na/Na+. In the charging/discharging plots of the full cell, at an operating voltage of 2.4 V, reversible capacities of 133 and 107 mA h g−1 are achieved at 150 and 600 mA g−1, respectively (Fig. 24b). The coulombic efficiency and cycle performance display 99.7% efficiency when cycling at 600 mA g−1, while the capacity presents retention of 96% after 100 cycles (Fig. 24c).
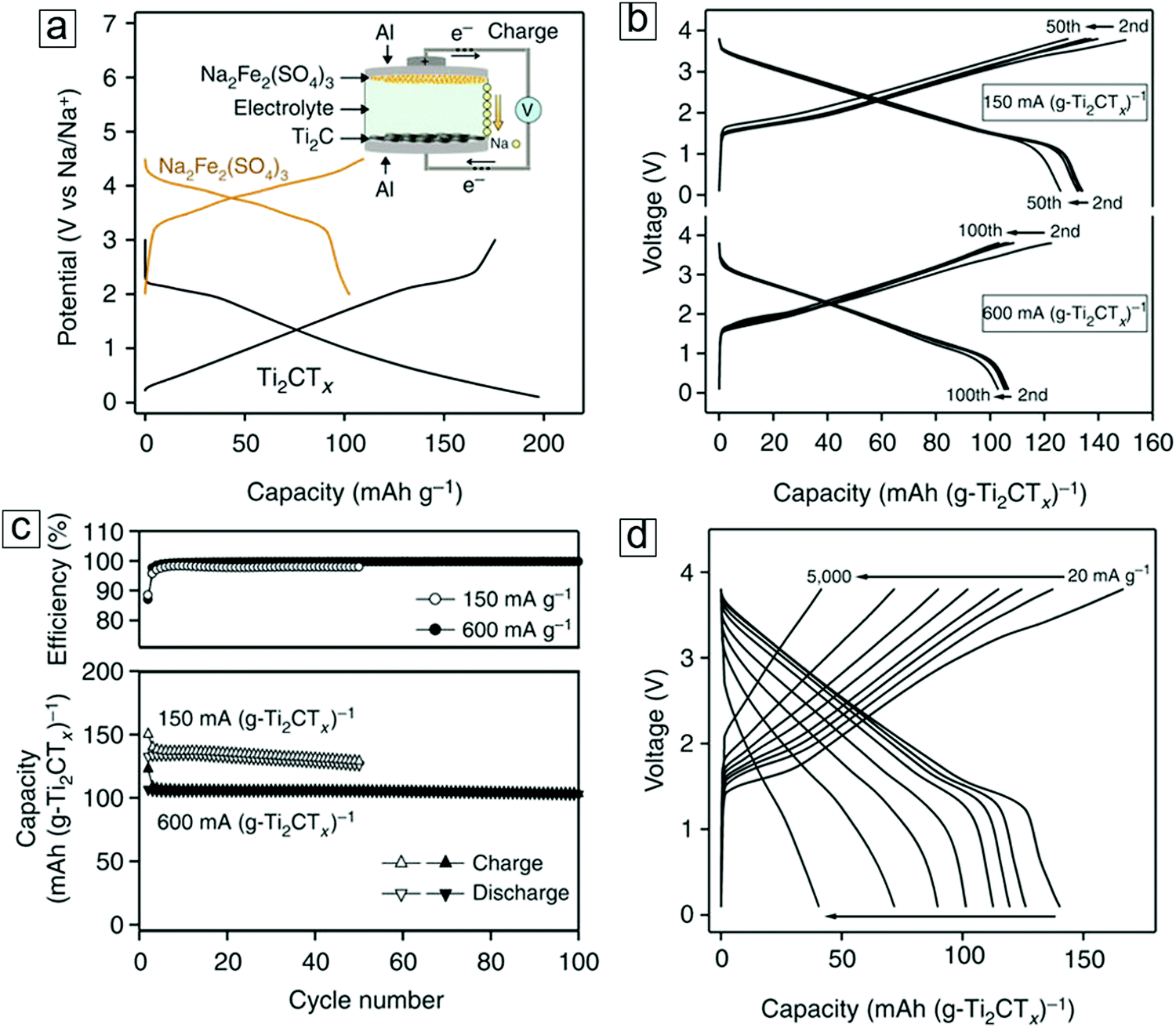 | ||
| Fig. 24 Electrochemical performance of Na-ion hybrid capacitors in full cells. (a) Charge/discharge curves of MXene Ti2CTx and Na2Fe2(SO4)3 alluaudite versus Na/Na+. The inset is a full cell with MXene and Na2Fe2(SO4)3 electrodes. (b) Voltage profiles of the MXene and alluaudite full cell at two specific currents. The specific currents are normalized with the weight of MXene, e.g., 150 and 600 mA (g-Ti2CTx)−1. (c) Coulombic efficiency and cycle stability of the full cell. (d) Charge/discharge profiles at different currents. Reprinted by permission of Macmillan Publishers Ltd: [Nature Communications] ref. 20 copyright (2015). | ||
The charging/discharging profiles of the full cell exhibit high-power performance. Reversible capacities of 90 mA h g−1 and 40 mA h g−1 at respective rates of 1000 mA g−1 and 5000 mA g−1 were observed.
We will now examine the mechanisms of sodiation and desodiation.
The MXene Ti3C2Tx anode in an organic Na-ion electrolyte exhibits a dominant intercalation and deintercalation mechanism during the charging/discharging processes.464 Before sodiation, pristine Ti3C2Tx has an interlayer distance of 0.97 nm (top panel in Fig. 25a). After the first sodiation cycle, the interlayer distance of the fresh sodiated MXene expands to 1.25 nm (middle panel in Fig. 25a). Upon vacuum annealing treatment, the interlayer spacing of the dried sodiated MXene decreases to 1.1 nm (Fig. 25b). The 0.15 nm difference between the dried and fresh sodiated MXene sheets is attributed to evacuation of the solvent molecules from the organic electrolyte. Thus, the 0.13 nm difference between the pristine and dried sodiated MXene probably stems from Na ions trapped between the MXene sheets, which play the role of a supporting pillar.
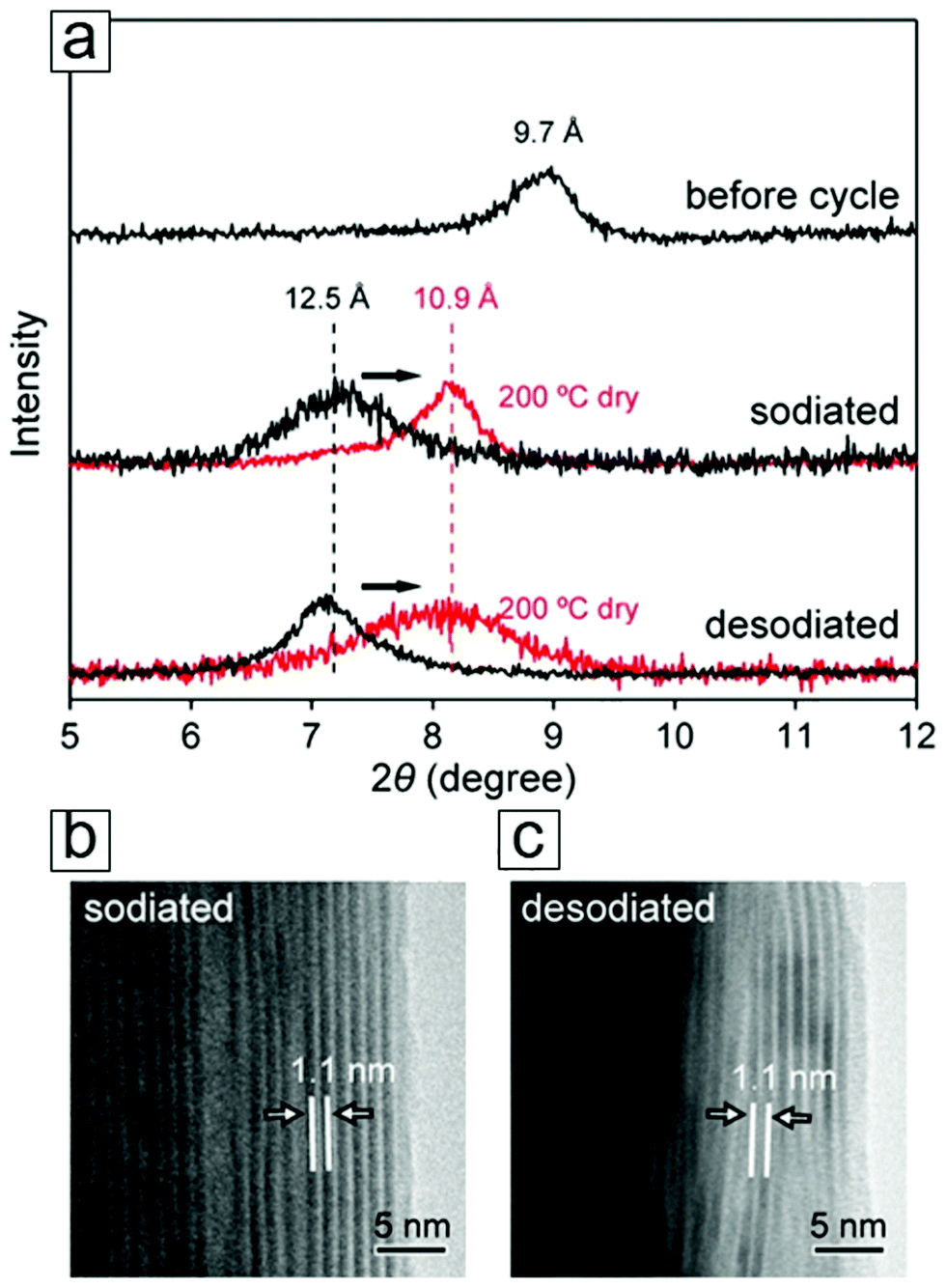 | ||
| Fig. 25 Sodium-ion reaction mechanism in electrochemical charging/discharging. (a) X-ray diffraction (XRD) curves of the pristine (upper), fully sodiated (middle), and fully desodiated (bottom) MXene Ti3C2Tx. The red curves represent the samples after vacuum annealing treatment at 200 °C. (b) Transmission electron microscopy (TEM) micrograph of fully sodiated MXene after the annealing treatment. (c) TEM micrograph of the fully desodiated MXene after the annealing process. Reprinted with permission from ref. 464. Copyright 2016 American Chemical Society. | ||
The desodiation reaction does not change the interlayer spacing (1.25 nm) derived from XRD data (bottom panel in Fig. 25a). Vacuum drying treatment at 200 °C also eliminates the organic solvents between the MXene sheets, as revealed by TEM analysis (Fig. 25c). These data show an identical layer distance upon sodiation, confirming that the interlayer spacing is preserved along with both the Na pillars and organic electrolyte molecules during the desodiation process. Following the irreversible capacity in the first cycle of the charging/discharging test, the equivalent of a 120 mA h g−1 capacity or 0.15 Na per formula unit of Ti3C2O2, is attributed to the inactive Na species that is either trapped between the MXene sheets (pillaring) or deactivated in the solid electrolyte interphase (SEI). This also explains the low coulombic efficiency of the initial cycle (∼60%).
In greater detail, the dominant contribution of the intercalation/deintercalation mechanism was also confirmed by NMR spectroscopy. This technique can determine the chemical environment of the sodium ions. Thus, a peak at σ = −9 ppm indicates the presence of Na ions in a freestanding electrolyte;465–467 Na ions adsorbed at the surface and edges of the MXene sheets are observed at σ = −8 ppm, and Na ions intercalated between the MXene sheets are observed at σ = +5 ppm.24 At the lowest chemical shift (−8 ppm; Fig. 26a), there is a strong shielding effect from the electrons donated from the counter anions and the solvent molecules from the organic Na electrolyte.466
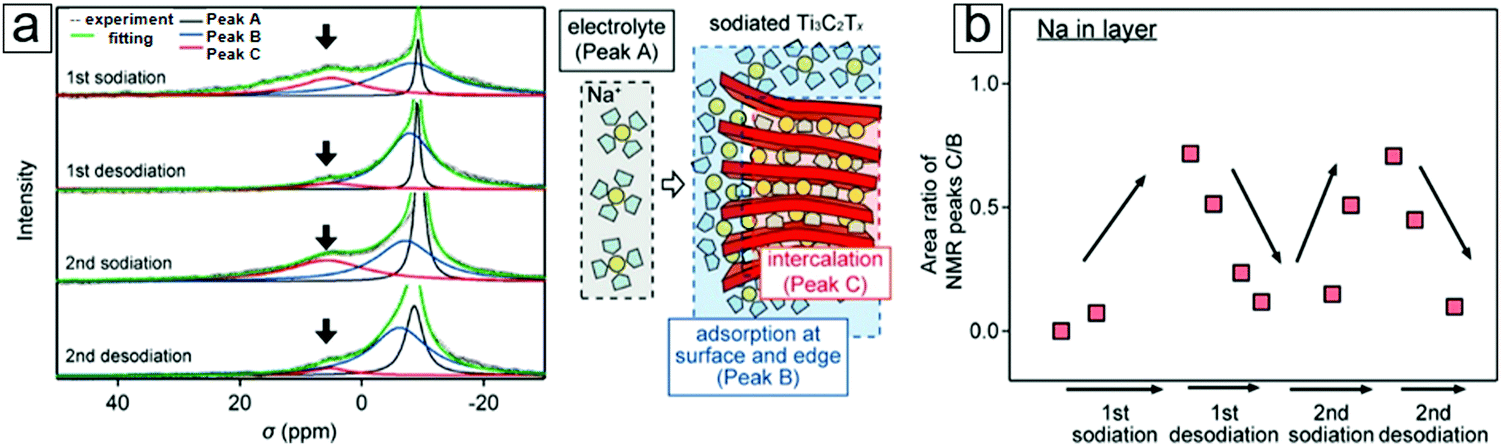 | ||
| Fig. 26 Nuclear magnetic resonance (NMR) spectra acquired to determine the mechanism of Na incorporation into the MXene Ti3C2Tx. (a) NMR spectra of 23Na for the MXene electrodes during the initial two cycles. Three-peak fittings are deconvoluted for the three types of chemical environments with Na-ion sites illustrated in the schematic in the middle panel. (b) Area ratios of the NMR peaks of intercalated Na ions with respect to the adsorbed Na ions. These data can be used to estimate the variation in the number of Na ions during the charging/discharging cycles. Reprinted with permission from ref. 464. Copyright 2016 American Chemical Society. | ||
At an analogous negative frequency of −8 ppm, the broad line width of the peak indicates fixation of the solvated Na ion. This is partially due to entrapment in the SEI layer and partially due to capacitive Na-ion incorporation. The MXene Ti3C2Tx electrode possesses both electric double-layer capacitance and pseudocapacitance. At a positive high frequency of +5 ppm, this peak represents the desolvated Na ion undergoing intercalation into the MXene sheets,24 in which the Knight shift is triggered by the Pauli magnetic susceptibility.
During the first two sodiation and desodiation processes, the intercalated Na ions undergo significant changes, while the Na ions in the freestanding electrolyte and the adsorbed Na ions do not exhibit any variations (Fig. 26a). This again proves that intercalation is the dominant mechanism in sodium-ion batteries. Moreover, the relative peak area ratio of Na intercalation with respect to Na adsorption shows high reversibility (Fig. 26b). This is reflected in the high cycling performance observed for MXene electrodes. XRD, TEM, and NMR data confirm the superior structural and chemical stabilities of MXene electrodes for Na storage. These afford zero strain and enable a large capacity of 70 mA h g−1 at a high charge/discharge rate of 500 mA g−1.464
We will now examine the atomic resolution view of the MXene Ti3C2Tx to understand its surface structure and intercalation chemistry. Aberration-corrected STEM revealed structural variations in MXenes and Na atomic occupation upon Na-ion intercalation during the discharge cycle.468 First, partial intercalation of the MXene electrode was achieved with a cutoff discharge potential of 0.5 V during the galvanostatic cycle. High-angle annular dark-field (HAADF) micrographs illustrate the partial occupation of Na ions between the MXene sheets (yellow arrows in Fig. 27a). A compensatory annular dark-field (ADF) TEM phase-contrast image confirmed the assignment of Na ions (yellow arrows in Fig. 27d). In this process, the Na ions are initially intercalated from the edge of the material and subsequently diffuse to the bulk, as indicated by the gradually increasing contrast of the interlayer points (Na ions; from left to right in Fig. 27d). Secondly, full Na intercalation was realized by tuning the cutoff discharge potential to 0.0 V. A Na-ion monolayer fully occupies the interlayer spacing (Fig. 27b and e). Thereafter, excessive Na intercalation was experimentally observed to form a Na-ion double-layer between the MXene sheets (double yellow arrows in Fig. 27c and f). Careful study of the ADF micrographs indicated the locations of the Na ions in the MXene sheets. These micrographs revealed that Na ions selectively reside on top of the C atoms rather than over the surface or the bulk Ti atoms (red lines in Fig. 27d and e). These experimental results agree well with theoretical reports.24,324 Double-layer Na ions occupy the same relative positions on top of the C atoms and are 0.23 nm apart (Fig. 27f). These double-layer Na ions are believed to occur in the solid solution reaction as a final intercalation step.
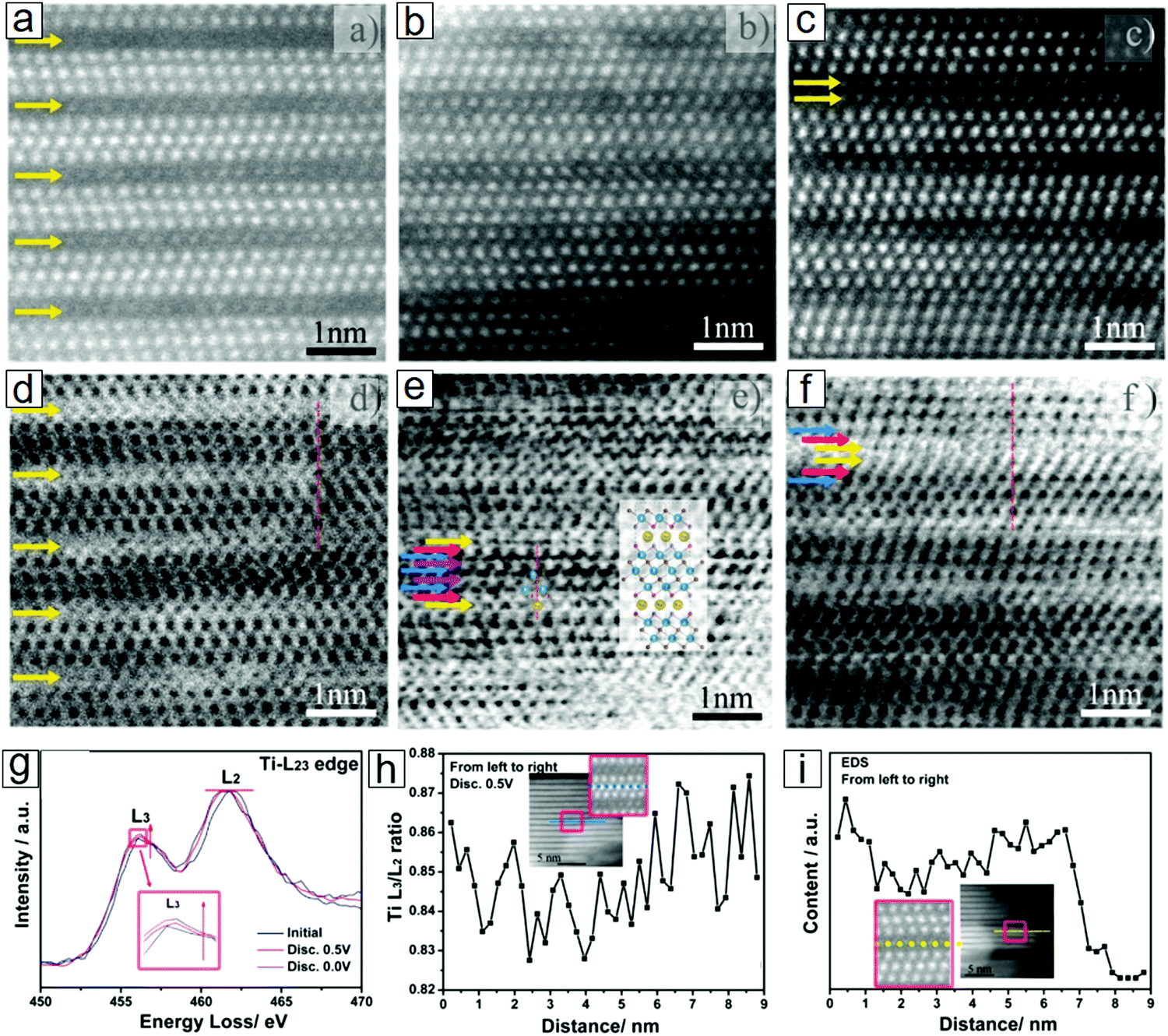 | ||
| Fig. 27 Sodium ions partially and fully occupying the interlayers of MXenes in the sodiation process. (a–c) High-angle annular dark-field (HAADF) images. Double-layered sodium-atom layers, represented in (c) as double arrows, indicate a fully sodiated state. (d–f) Annular bright-field micrographs; sodium ions remain on top of the C atoms rather than the surface or center of the Ti atoms in (e). (g) Electron energy loss spectroscopy (EELS) spectra. (h) Changes in the Ti L3/L2 (energy loss peaks) intensity ratio extracted from (g) from the MXene center to the surface. (i) Changes in sodium content from the MXene center to the surface. Sodium ions occupy the interlayers upon sodium intercalation partially at a cutoff potential of 0.5 V (a, d, h and i) and fully at 0.0 V (b, c, e and f). The insets in (h and i) are the regions where the EELS and energy dispersion spectra were collected. Reprinted with permission from ref. 468. Copyright 2015 American Chemical Society. | ||
The chemical environment of the Ti atoms in the Na-intercalated MXenes can be determined at the atomic scale using EELS. The data reveals two edges of Ti in the EELS spectrum, i.e., an L2 edge at 456.4 eV and an L3 edge at 461.7 eV (Fig. 27g). The relative L3/L2 intensity ratio reflects the chemical states of the Ti atoms in Ti3C2Tx.469,470 Upon Na intercalation, the L3/L2 intensity ratio increases because of the decrease in Ti atoms through the acceptance of electrons from the intercalated Na (electron donors). The atomic Na diffusion mechanism is confirmed by the gradual decrease of the L3/L2 ratio (Fig. 27h) as well as the Na content (Fig. 27i) in a line scan from the surface to the bulk of the MXene. Theoretical studies provide insight into intercalation chemistry and surface geometry. An energetically favorable atomic configuration is displayed with F and OH terminations in the side view (Fig. 28a). This is analogous to LiMn2 adsorption at the Mn surface in Li2MnO3.471 The functional groups prefer to reside on top of the bulk Ti atoms in the top view (Fig. 28a). The Na ions are shown to diffuse into the MXene interlayer spacing and occupy sites on the top of the C atoms (Fig. 28b).
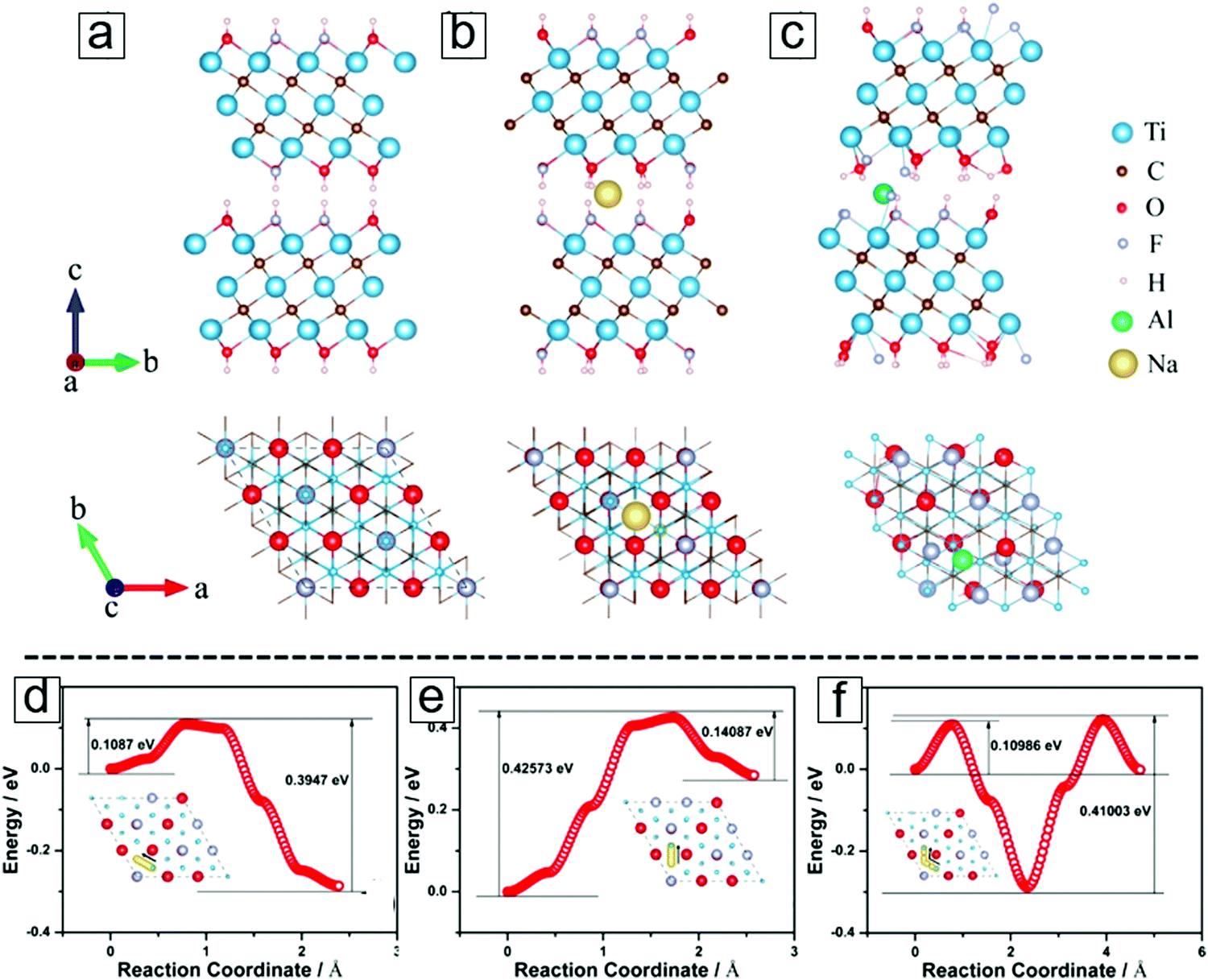 | ||
| Fig. 28 Equilibrium geometries of MXene Ti3C2X after intercalation of Na and Al ions. (a) Pristine MXene, (b) Na-intercalated MXene, and (c) Al-intercalated MXene; upper panel: side view, lower panel: top view. (d–f) Diffusion energies of a Na ion with respect to its predesigned pathways. The insets correspond to three types of migration paths for Na-ion diffusion along the Ti3C2X surface. The density functional theory (DFT) calculations were based on a modified model and experimental results. Low diffusion energy barriers are predicted for facile Na diffusion. Reprinted with permission from ref. 468. Copyright 2015 American Chemical Society. | ||
Density-of-states analysis has revealed that Ti3C2Tx is metallic, accounting for its high electrical conductivity. In addition, the Na-ion diffusion path and energy barrier were computed from DFT calculations and first principles molecular dynamics. Three low diffusion energy pathways are proposed with diffusion from the top C atom to the top surface Ti atom as well as direct hopping from the top C atom to a neighboring top C atom (Fig. 28d–f). Eventually, the low diffusion barrier (0.41 eV) allows Na to diffuse easily into Ti3C2Tx (Fig. 28f).
To sum up, the high electrical and ionic conductivities of Ti3C2Tx result in its extraordinary rate performance in sodium-ion batteries. Other theoretical and experimental work has provided further insight. For example, Al-ion intercalation has been demonstrated with atomic resolution STEM by immersing MXene sheets in an Al(OH)3 aqueous solution.468 Upon Al-ion intercalation, horizontal sliding of a Ti3C2Tx monolayer sheet relative to the two nearest neighboring top and bottom MXene sheets was observed. Calculations revealed that the intercalated Al ions can reside on top of the C atoms at the top of the MXene sheet and remain close to the surface Ti atoms of the bottom MXene sheet (Fig. 28c). This strain caused by Al-ion intercalation raises the possibility of using MXenes for Al-ion storage.
4.4 Sulfur immobilizers in lithium–sulfur batteries
Cathode materials in rechargeable batteries are also important to couple with the anode to increase the total energy capacity to accommodate lithium. Conventional lithium–transition metal oxides, e.g. LiCoO2, possess layered structures and display long cycle life; however, they have limited theoretical capacities472,473 (280 mA h g−1) and energy densities.474,475 This does not satisfy the requirements of low cost and high energy density for use in electric vehicles and electrified transportation in general. Earth-abundant sulfur has a large gravimetric capacity and high energy density and represents a highly promising cathode material for next-generation batteries. A typical lithium–sulfur battery (Fig. 26a) consists of a Li–metal anode, sulfur cathode, polymeric separator, and organic electrolyte. The solid electrolyte or ionic conducting liquid delivers Li ions. In its most energetically stable form, sulfur exhibits a stacked octagon ring (S8) with a density of 2.07 g cm−3, which is optimal for low weight storage cells. To address the insulator disadvantage of sulfur and its lithium compound species, highly electrically conducting porous carbon and nanosized metals with large surface areas have been explored to implement sulfur as a hosting matrix.476–479 With the highest theoretical capacity (1675 mA h g−1) among solid cathode materials, sulfur has a high theoretical energy density of 2500 W h kg−1.480–483 In the discharge process, the sulfur–sulfur bonds are broken and sulfur reacts with the Li ions to produce lithium polysulfide, i.e., Li2Sx, 2 < x < 8. These highly soluble lithium polysulfide discharge products cause mass loss at the cathode and anode surfaces via a sulfur shuttling mechanism (sulfur crosses the separator).484–488 Currently, this is the greatest disadvantage that is hindering commercialization of lithium–sulfur batteries. To overcome this limitation, strategies are being developed involving additives and surface modification. Examples include porous oxide additives for the physical absorption of soluble polysulfides474 and polymer coatings over the cathode surface to physically retard polysulfides.489 Surface-coating-modified separators have shown effective restriction of polysulfide migration across the separators. This is reflected in the high cycling life and rate capacity performance found in such systems.490–492Ti3C2Tx was deposited as a thin surface layer over a common separator to immobilize soluble polysulfides. A conformal MXene layer over the separator improved both the electrical conductivity and polysulfide entrapment without causing structural degradation (Fig. 29a). This MXene surface coating enhanced the initial discharge capacity (1246.3 mA h g−1) and capacity retention (860.7 mA h g−1) after 30 cycles. Additionally, excellent coulombic efficiency was observed after 500 cycles (Fig. 29b).
 | ||
| Fig. 29 MXene-modified separator for immobilizing polysulfide in Li–S batteries. (a) Schematic of lithium–sulfur batteries using a polypropylene membrane and MXene-modified polypropylene as separators. At the cathode, carbon black is used as the conducting carbon for hosting sulfur. (b) Cycling performance of lithium–sulfur cells with MXene-modified polypropylene separators at various high rates; the black curve represents 0.5C and the red curve represents 1C. High retention ratios of specific capacity and 100% coulombic efficiency were observed after long cycle times. Reprinted with permission from ref. 493. Copyright 2016 American Chemical Society. | ||
Electrochemical charging/discharging was carried out to analyze the impact of MXene modification on the separator. The lithium–sulfur cells were discharged and charged stepwise from 0.2C to 1C for a forward scan and subsequently from 1C to 0.2C as a backward scan. The initial discharge profiles reveal two discharge potential plateaus for both cells, one with a pristine separator (Fig. 30a) and the other with an MXene-modified separator (Fig. 30b). In the discharge reactions, the upper discharge voltage plateau at 2.3 V stems from the reduction of sulfur to form polysulfides, i.e., Li2Sx, 3 < x < 8. At a lower discharge voltage, the plateau is attributed to further sulfur reduction due to Li2S2 and Li2S formation.494 In the charging process, two continuous plateaus at ∼2.2 and 2.4 V correspond to oxidation reactions from Li2S and Li2S2 to Li2S8 and S8, respectively. The charge/discharge profiles of the cells with the MXene-modified separator (Fig. 30b) exhibit more favorable flat plateaus compared to those of the pristine separator (Fig. 30a). In addition, the MXene-modified separator displays small voltage plateau gaps between the charge/discharge plots (Fig. 30c) of 140, 175, and 230 mV at 0.2, 0.5, and 1C, respectively. This is advantageous for improving the redox reaction kinetics and capacity reversibility. A specific capacity of 1046.9 mA h g−1 at a rate of 0.2C was achieved. With increasing current density, high capacity retentions (848.7 mA h g−1 at 0.5C and 743.7 mA h g−1 at 1C) were realized. These results far exceed the values observed for cells with a pristine separator.
 | ||
| Fig. 30 Electrochemistry performance of the MXene-modified separators. Lithium–sulfur batteries comprising (a) a polypropylene separator and (b) an MXene-modified polypropylene separator showing first cycle galvanostatic charge/discharge curves at 0.2, 0.5, and 1C. (c) Potential differences of lithium–sulfur cells with a polypropylene separator and MXene-modified polypropylene separator between the charge/discharge plateaus at different rates. (d) Rate performance of lithium–sulfur batteries with polypropylene and MXene-modified polypropylene separators. Reprinted with permission from ref. 493. Copyright 2016 American Chemical Society. | ||
Moreover, high reversibility of the capacities was retained at 90.7% and 78.5% of the starting discharge capacities with backward scanning (Fig. 30d).
4.5 Protection layers for lithium–metal electrodes
Metallic lithium, which has a high theoretical capacity of 3860 mA h g−1 and a low potential of −3.04 V, represents a highly promising anode material for lithium-based batteries. Metallic Li was the first anode material used in a Li-ion battery.495 However, the lithium–metal cell retains a short life cycle, with the drawbacks of large volume expansion and Li dendrite formation. Dendritic Li tends to pierce the separator membrane and contact the cathode, thereby leading to short circuit damage of the cells. Thus, pure Li anode studies have been limited. With advances in nanotechnology, especially in nanosized 2D materials with large specific areas, lithium metals as electrode materials are being revisited and are becoming an important branch of rechargeable lithium–metal cells454 and lithium–sulfur496–499 and lithium–oxygen batteries.500–503 Three strategies have been developed to overcome these limitations. Firstly, the strength of the separators can be enhanced to tolerate Li dendrite penetration. Secondly, the surfaces of the Li metal can be passivated and modified to form a stable SEI coating. Thirdly, the entrapment of Li metals into hosting materials can limit the volume changes and suppress vertical dendrite growth, viz. Li metals are accommodated into porous or nanosized matrix materials with large specific areas. Among these, the incorporation of Li metal into conductive porous structures remains a less explored and focused topic. A few examples have explored the confinement concept by infusing molten lithium into lithiophilic hosting materials, e.g., rGO,504 ZnO-covered polyimide,505 and Si-deposited carbon fiber networks.506 The electrochemical performance of such systems exhibits flat voltage profiles, high cycling performance, and low overpotentials (80 mV at 3 mA cm−2). The weak interactions of Li metal with most of these hosting materials lead to low lithiophilicity, which is not favorable.MXene–lithium lamellar structures have been explored for preserving Li metal in the electrode design through a rolling and folding protocol. In brief, Li metal is placed on the surface of an MXene film. Upon heating, the Li metal becomes molten and is rolled, fully wet, over the MXene surface. The MXene/Li thin films are then folded multiple times to form a stacking structure and subsequently undergo a rolling treatment. After repeating this procedure several times, the MXene/Li composite film displays a lamellar layered structure (Fig. 31a) with Li well-dispersed between the MXene sheets. This composite film shows good mechanical flexibility (Fig. 31b) and fine surface morphologies (Fig. 31c and d).
 | ||
| Fig. 31 Synthesis of an MXene Ti3C2 and metallic lithium composite electrode. (a) Sketch of the synthetic process of MXene Ti3C2–lithium films: first, MXene nanosheets and the lithium plate are rolled together for expansion. Subsequently, these expanded composite films are folded to increase the layer numbers. (b) Photograph of the MXene–lithium composite exhibiting its good flexibility (easily twisted by tweezers). (c) Top view and (d) cross-sectional view of scanning electron microscopy (SEM) images of the MXene–lithium composite displaying a layered lamellar morphology with MXene well-dispersed between the Li layers. Reprinted with permission from ref. 507. Copyright 2017 Elsevier. | ||
In electrochemistry, the overpotential of a half-reaction is the potential difference between the potential at thermodynamic equilibrium and the potential that is necessary for the redox reaction to occur. The overpotential increases with increasing current density by tuning the voltage and is correlated by the Tafel equation. Commonly, the term overpotential refers to the reaction overpotential that enables electron transfer from electrochemical reactions such as electroplating. The overpotential describes the external energy that is required to retain a continuous reaction that eventually transforms into heat. A low overpotential is required to increase the energy conversion efficiency in a lithium–oxygen battery.508 In addition, the low overpotential of an electrode can enhance the sulfur efficiency in a Li–S battery.509 The electrochemical performance of an MXene–Li lamellar structure was examined by fabricating a symmetrical cell with two identical MXene–Li electrodes. Its overpotential (32 mV at 1.0 mA cm−2) enabled facile Li plating. This overpotential over the MXene–Li lamellar structure electrode represents the lowest value among Li-hosting composites using 2D materials, e.g., 190 mV for boron nitride, 106 mV for graphene, and 40 to 00 mV for various other composites.510–514 The Ti3C2TxLi structure shows excellent electrochemical performance,507 including a flat charge/discharge plateau and high rate performance.
MXene–Li anodes provide superior performance for lithium–sulfur cells,507i.e., a high initial specific capacity of 948 mA h g−1 at a current density of 0.5 mA cm−2 and high retention after 100 cycles with a capacity of 841 mA h g−1; this is nearly twice that of a bare Li–sulfur battery. MXene, as a host for Li metal, exhibits high rate performance with a capacity of 360 mA h g−1, which is fourfold that observed for a bare lithium metal–sulfur cell at a high current density of 5 mA cm−2 (equivalent to a rate of 2C in the sulfur cathode). Notably, in a rate performance test for non-lithium anodes with layered structured materials, a gravimetric current density of 1 mA g−1 or 1C (1C = 1000 mA g−1) is employed. On the other hand, an areal current density is used to quantify lithium–metal anode batteries, where 1 mA cm−2 is equivalent to 400 mA h g−1 for a sulfur cathode.
Studies on the electrochemical reaction mechanism of MXene hosting Li electrodes have also been conducted. The evolution of Li plating at the electrode was investigated with in situ TEM (Fig. 32). MXene covers the Li surface (Fig. 32a). Li growth is then confined to the MXene nanocap, while Li dendrite formation occurs over the bare Li surface (Fig. 32b). From 0 s to 14 s, Li encounters wetting inside the MXene nanosheets at a voltage of 0.1 V (Fig. 32c). XPS data reveals the formation of a Li–C bond, which agrees well with the fact that the Li atom is energetically favored to reside on top of a C atom as indicated by DFT calculations. Further reactions lead to Li extrusion at the edges, i.e., the formation of a Li dendrite after 20 s (Fig. 32d) when the MXene nanosheets are fully intercalated with the Li metals.
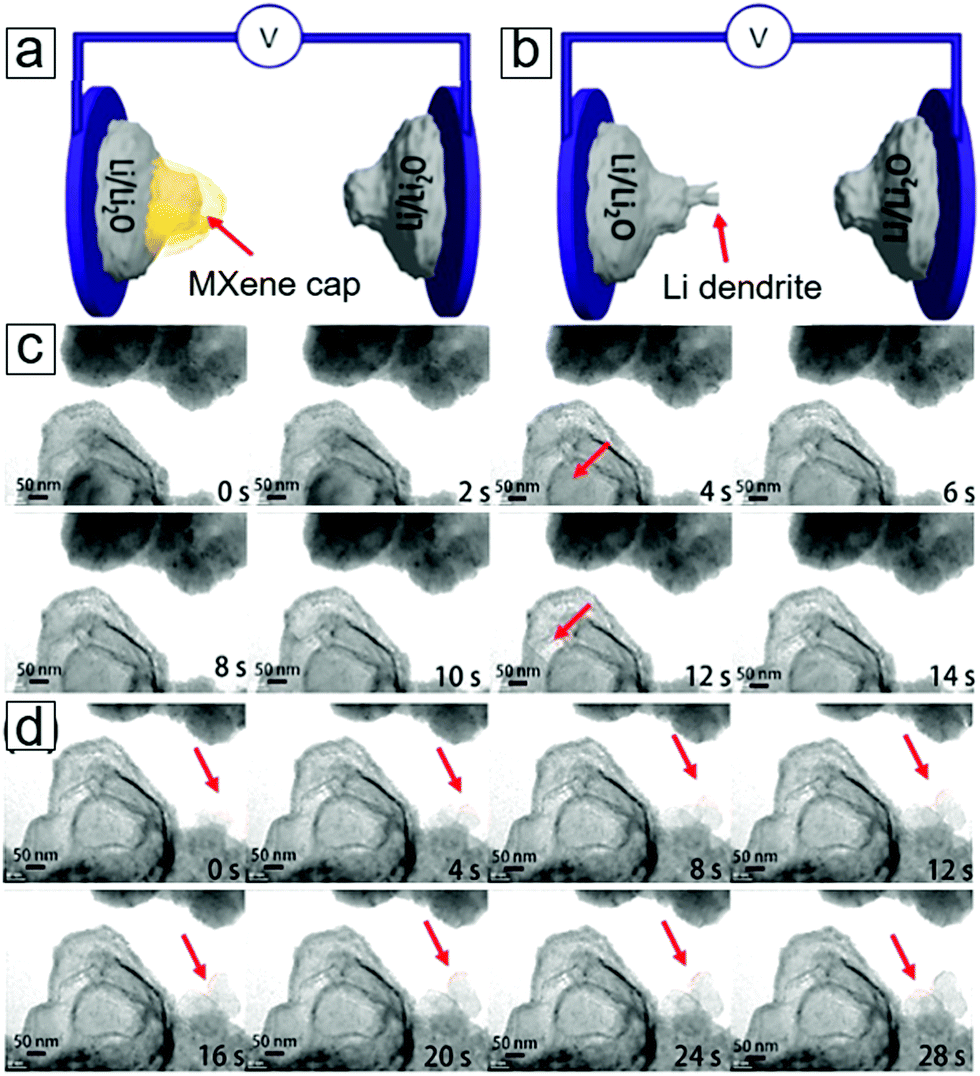 | ||
| Fig. 32 In situ transmission electron microscopy (TEM) investigation of the lithium electrochemical deposition process in MXene-protected Li metal. (a) Schematic of the fabricated microcells with MXene Ti3C2Tx-capped Li as the working electrode and (b) bare Li metal. (c) Time-evolving micrographs of an MXene-covered Li electrode in the Li electroplating process. (d) Time-evolving micrographs of a Li electrode after full filling of Li in an MXene nanocage during the electroplating process. The dendrite of the Li metal forms at the surface frontier of the electrode when the MXene is fully filled with the Li species. A voltage of 100 mV is applied between the two electrodes. Reprinted with permission from ref. 507. Copyright 2017 Elsevier. | ||
In brief, MXene nanosheets act as an SEI film to stabilize Li. The lamellar structures can be maintained during repeated anodic lithium dissolution and cathodic plating due to the strong mechanical properties of the MXene film, which can tolerate high stress.
4.6 Electrocatalysts in zinc–air batteries
Metal–air batteries represent yet another alternative to lithium-ion batteries, promising low cost and large energy density storage. A metal–air battery consists of a metal anode, an oxygen cathode, and an electrolyte. Based on the reactivity of the metal with water, metal–air batteries are divided into two types: an electrochemical cell with an aqueous electrolyte,515–524 including metals such as Ca, Al, Fe, Cd, and Zn, and a battery cell with a non-aqueous electrolyte,525–533e.g., Li or Na. Aqueous electrolyte metal–air batteries have the advantage of easy handling without the need of an inert atmosphere. Among these, Zn is relatively stable in alkaline solution as the aqueous electrolyte. Zn is abundant, inexpensive, environmentally friendly, and exhibits extraordinary electrochemical performance: a low equilibrium potential, flat discharge voltage, and long shelf life.534,535 Used in an open cell configuration, the oxygen required for the cathode reaction can be extracted from air. Such cells have a high theoretical specific energy density, e.g. 1084 W h kg−1 for a Zn–air battery.536Zinc–air batteries are categorized into primary, secondary, and mechanically replaceable cells. The secondary zinc–air battery is of high interest and has received much attention for proof of concept experiments.537,538 Some questions still remain to be fully answered: the chemistry occurring at the zinc anode, bifunctional electrocatalysts for both the OER and ORR, oxygen cathodes with low resistance, separators for confining zinc species, and carbon dioxide management.
MXene heterostructures exhibit outstanding performance when used as an electrocatalyst for the OER in alkaline aqueous solutions. For example, Ti3C2 interacts with graphitic carbon nitrides (g-C3N4) to form a porous matrix.539 This results in strong coupling between the nitrogen-rich network and transition metal centers, in which the Ti–Nx motifs provide electroactive sites to enhance the oxygen electrochemistry activity and stability.
MXene/g-C3N4 heterostructures readily function as working electrodes for the OER. MXene-based heterostructures exhibit a high OER anodic current with a sharp onset potential of 1.44 V versus a reversible hydrogen electrode. On the other hand, individual MXenes and g-C3N4 display low OER yields under the recorded anodic current (Fig. 33a), indicating that the coupling of these two compounds can enhance the catalytic activity. Additionally, the MXene heterostructure exceeds RuO2 in OER activity at potentials >1.54 V. Moreover, it requires a low potential of 1.65 V to provide a current density of 10 mA cm−2, which is comparable to or slightly better than that required by IrO2 (1.7 V) and many state-of-the-art electrocatalysts in 0.1 M KOH aqueous solution.540–545 A Tafel slope can be used to identify the reaction kinetics. The Tafel slope revealed 74.6 mV decade−1 for the MXene/g-C3N4 heterostructures, which is lower than those of g-C3N4 (136.2 mV decade−1), MXene (119.7 mV decade−1), and IrO2/C (93.2 mV decade−1; Fig. 33b). Electrochemical impedance spectroscopy showed low contact resistance and charge transfer impedance for MXene/g-C3N4 heterostructures that were significantly smaller than those of the control set.
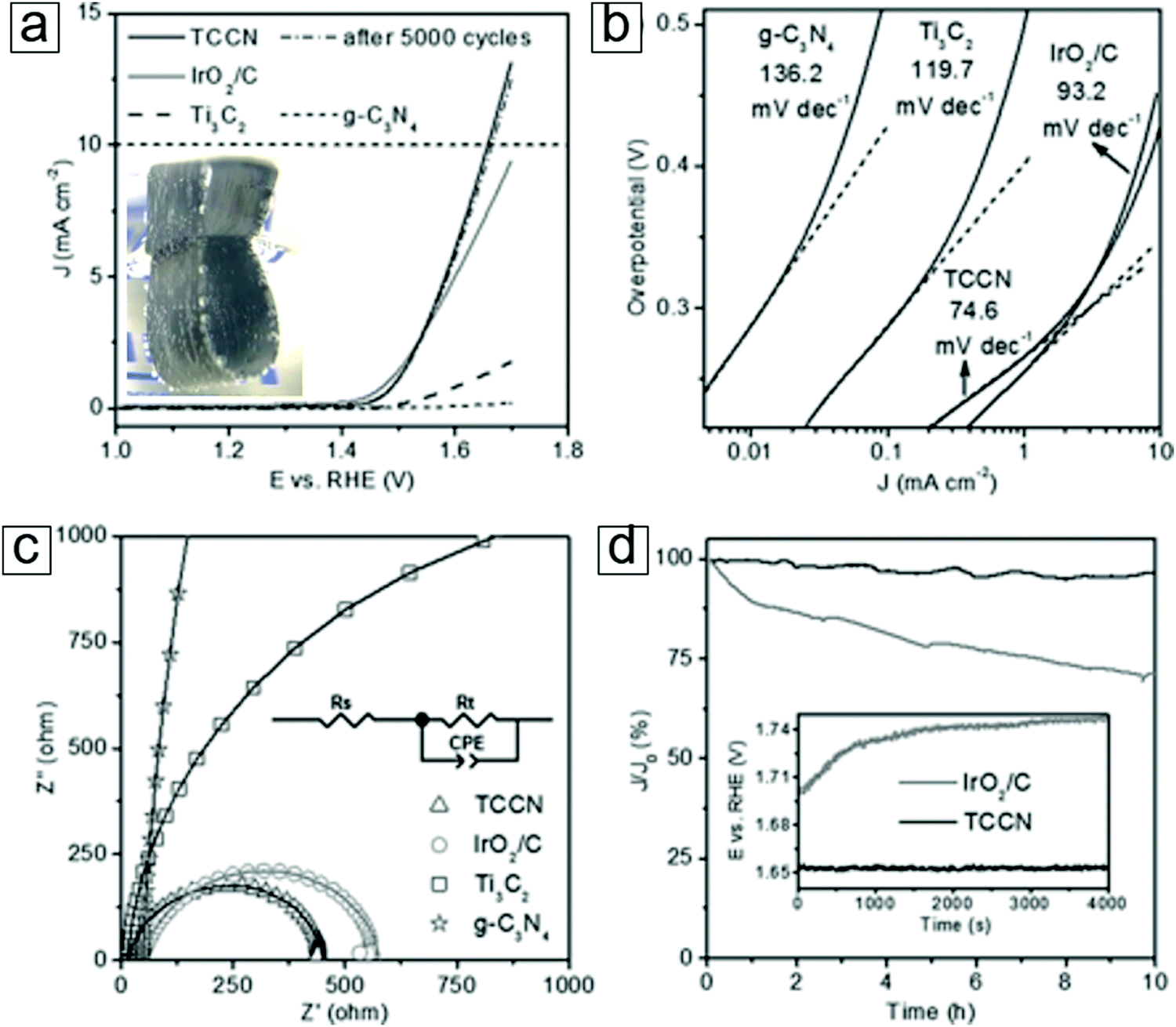 | ||
| Fig. 33 Oxygen evolution reaction (OER) over MXene–g-C3N4 hybrid electrocatalysts. (a) Polarization curves, (b) Tafel plots, (c) electrochemical impedance spectra of MXene Ti3C2, g-C3N4, MXene–g-C3N4 hybrid TCCN, and other electrocatalysts collected at 1.6 V with an inset equivalent circuit diagram. The inset of (a) is a photograph of the TCCN hybrid generating oxygen bubbles when working as an electrode at 1.8 V. (d) Chronoamperometric response of TCCN at a constant potential of 1.65 V. The inset in (a) displays the chronopotentiometric response of TCCN and IrO2/C at a constant current density of 10.0 mAcm−2. The performance after 5000 cycles shows excellent cycling stability. MXene and g-C3N4 coupling lead to high performance electrocatalysis, in which similar two-dimensional (2D) geometries and metal–nitrogen interactions are employed. From ref. 539. Copyright © [2015] by [John Wiley & Sons, Inc.]. Reprinted by permission of [John Wiley & Sons, Inc.]. | ||
Furthermore, the rotating ring-disk electrode technique confirmed a high faradaic efficiency of 99.5% and a preferred four-electron pathway (4OH− → O2 + 2H2O + 4e−) with negligible peroxide formation. The chronoamperometric curve reveals high electrocatalyst stability with a high anodic current retention of 95.7% after 10 h of operation (Fig. 33d). In addition, the MXene heterostructure displays a high anodic current retention of 95.6% after 5000 continuous cycles (Fig. 33a) with a stepwise increasing scanning rate of 100 mV s−1. The freestanding 2D nanosheet architecture accounts for the structural stability over long cycling. Moreover, accelerated scanning voltages exhibit advantages over post-deposited powder catalysts on electrode surfaces, which tend to peel off during the evolution of oxygen gas.546–548 The coupling of nitrogen ligands with the Ti in MXenes results in superior electrocatalytic performance. Indeed, a hybrid MXene/graphene film does not achieve this reactivity, with an onset potential of 1.47 V and a Tafel slope of 108.9 mV decade−1 (Fig. 34a). The XPS spectra reveal that the binding energy of the Ti 2p3/2 profile upshifts to 455.9 eV for MXene/g-C3N4 heterostructures compared to 455.0 eV for the graphene/MXene hybrid and pure MXene (Fig. 34b). The binding energy shift is attributed to the interaction of Ti with g-C3N4, which impairs the electron density of Ti atoms. This enhances the electron transfer, with enhanced hydroxide affinity for faster O2 evolution.539 When coupling with Ti3C2 occurs, a new shoulder peak at 397.4 eV, assigned to the triazine rings (CN–C) in g-C3N4, is observed in the N 1s profile XPS spectrum (Fig. 34c). Analogously, X-ray absorption spectroscopy reveals the near-edge fine structure of pristine g-C3N4 and its MXene heterostructure (Fig. 34d). Two dominant π* resonances for N at 399.8 eV in the CN–C moiety and 402.2 eV in the N(–C)3 moiety are also present. On hybridization with MXene, the CN–C coordination peak shifts to 399.5 eV, indicating perturbation of the N atoms549–551 due to strong interfacial Ti–Nx interactions (inset of Fig. 34d). These highly dispersed active sites552 enable superior OER activity.
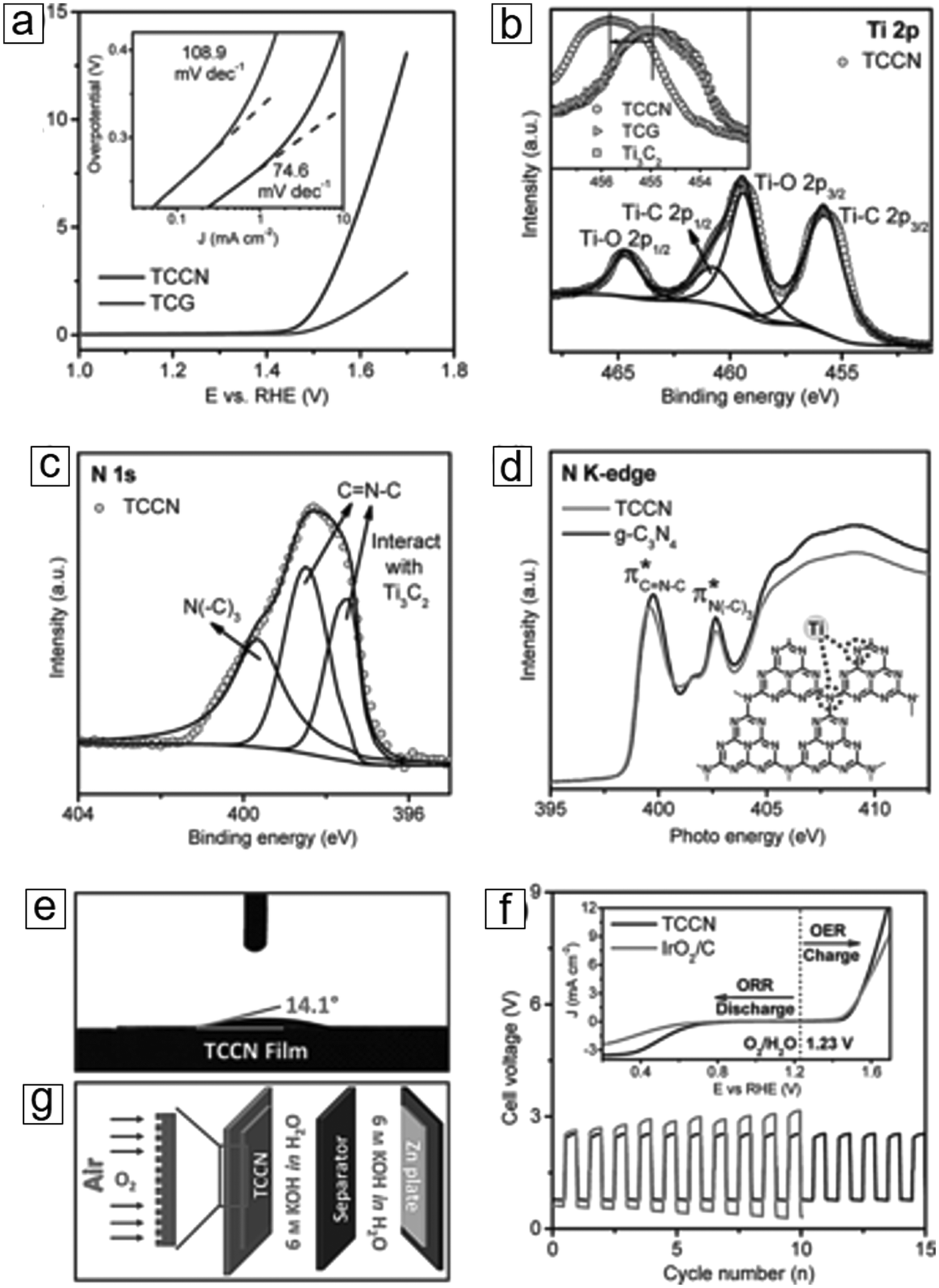 | ||
| Fig. 34 Oxygen evolution reactions (OER) and Zn–air battery performance of MXene–g-C3N4 hybrid electrocatalysts. (a) Polarization curves and Tafel plots as insets. TCCN represents the MXene–g-C3N4 hybrid. (b and c) X-ray photoelectron spectroscopy (XPS) spectra of Ti 2p and N 1s core levels in the MXene–g-C3N4 hybrid. TCG represents the MXene–graphene hybrid. (d) Near edge X-ray absorption fine structure spectra of the N K-edge for the MXene–g-C3N4 hybrid and g-C3N4. The insets show the active N sites. (e) Contact angle of the MXene–g-C3N4 hybrid film. (f) MXene–g-C3N4 hybrid as an electrode in a Zn–air battery. (g) Charge–discharge cycling performance of Zn–air batteries with MXene–g-C3N4 hybrid loading gas diffusion layers as air cathodes. IrO2/C is also shown for comparison. The inset displays the polarization curves of the MXene–g-C3N4 hybrid film and IrO2/C powder in a three-electrode system in the whole OER/oxygen reduction reaction (ORR) regions in an oxygen-saturated 0.1M KOH solution. From ref. 539. Copyright © [2015] by [John Wiley & Sons, Inc.]. Reprinted with permission from [John Wiley & Sons, Inc.]. | ||
The MXene/g-C3N4 heterostructure has a small contact angle of 14.1° (Fig. 34e) due to the oxygen- and hydroxyl-terminated surfaces of MXene, while its hydrophilicity favors the aqueous electrolytes used in electrochemical energy storage.2,176 These advantages are reflected in practical rechargeable Zn–air batteries (Fig. 34f). The MXene/g-C3N4 heterostructure as an air cathode shows a current density of 20 mA cm−2 at 0.87 V for the discharging ORR process and 2.55 V for the charging OER process. These values exceed those from IrO2/C-coated gas diffusion layers used as cathodes, which present potentials of 0.61 and 2.70 V, respectively. Moreover, the heterostructure shows stability over 15 pulse cycles (Fig. 34g). In contrast, the control sample (IrO2/C) displays a significant overpotential increase after five cycles because of three main factors: mass loss of the noble metal over its support, degradation of the carbonaceous gas diffusion layer, and the polymeric binder used to fix the powders. This proof of concept report shows the promising application of the MXene/g-C3N4 heterostructure as a flexible oxygen cathode with high durability and strong activity.
5 Energy utilization and conversion
5.1 Photocatalysts for energy conversion
Photocatalysis, which converts solar light to chemical energy, has been extensively investigated since the first photocatalytic H2 production was reported in 1972.553 Photocatalysts utilize solar light to drive many photocatalytic reactions, including hydrogen production from water splitting,554–556 carbon dioxide fixation,557,558 transformation,559,560 organic synthesis,561–563 and photodegradation of organic pollutants564–567 in water and air.568–574The different types of photocatalysts575–577 can be categorized into conventional transition metal oxides, including TiO2, BiOCl, BiOBr, and WO3, the latest metal organic frameworks, graphene-like carbon nitride, and band gap tunable 2D materials such as MXene and phosphorene.
In this section, we introduce the fundamentals of photocatalysts and the role of MXenes in several processes, including water amelioration, hydrogen evolution production, and fuel conversion from carbon dioxide. We first examine the basics of photocatalysis, taking TiO2 as an example.
Indeed, several efforts have been explored to address these limitations, including band gap tuning via nonmetal doping,578,587–590 metal doping,584,591–595 heterostructure formation with other semiconductors,596–602 photosensitization with organic molecules,589,603,604 and composite formation605,606 with 2D materials that can absorb visible light. Among these, one of the most successful solutions has been the hybridization of TiO2 with 2D materials. This technique takes advantage of the extraordinary electrical conductivities and tunable band gaps of 2D materials.
MXenes have also shown extraordinary potential when forming TiO2 composites for photocatalysis.607 MXenes can enhance the electrical transport in the composite materials.608–610 MXene, especially in the presence of Ti metal, represents an exciting material for hybridization with TiO2 not only because of its oxygen surface terminal groups, which can interact with Ti atoms, but also because of the facile partial oxidation of Ti3C2 and Ti4C3. Indeed, TiO2 is favorably formed at the surface of Ti3C2 in a hydrothermal process (Fig. 35a and b). From a closer perspective, TiO2 can grow along the Ti3C2 lattice fringes (Fig. 35c and d) as well as perpendicular to the [001] direction (Fig. 35e and f).
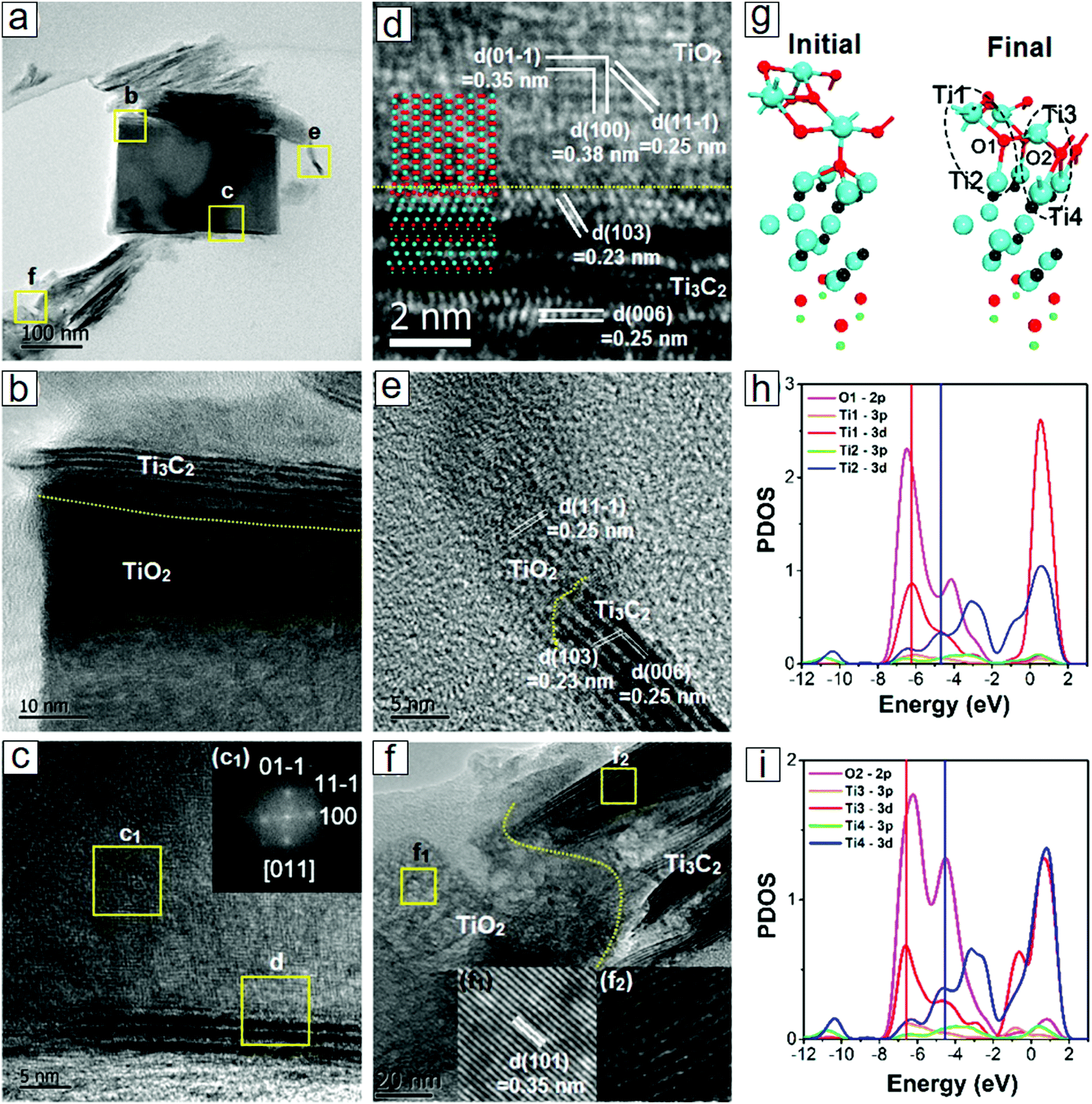 | ||
| Fig. 35 Interfaces of MXene Ti3C2 and TiO2. (a) Low magnification transmission electron microscopy (TEM) graphs. (b and c) Intimate contact of Ti3C2 with a TiO2 nanocrystal. The inset of (c) is a corresponding fast Fourier transform (FFT) image. (d) High resolution TEM micrograph displaying the interface with both layers parallel to each other. (e) TiO2 grown perpendicular to the [001] direction of the MXene Ti3C2 layered structure. (f) TiO2 forming at the edge of an MXene sheet. The insets display the lattice fringes. (g) Initial and final atomic configuration structures of the interface from density functional theory (DFT) results. Cyan, black, red, and green balls represent Ti, C, O, and H atoms, respectively. (h) Projected density of states of the Ti–O–Ti bonds of Ti1–O1–Ti2. (i) Projected density of states of the Ti3–O2–Ti4 bonds. Reprinted with permission from ref. 607. Copyright 2016 American Chemical Society. | ||
DFT calculations show an equilibrium atomic configuration of TiO2 residing over the Ti3C2 surfaces (Fig. 35g) with two types of Ti–O–Ti bonds at the interface. The corresponding densities of states for these bonds have been theoretically predicted (Fig. 35h and i).
For example, strong interfacial binding and good grain contact has led to successful photocatalytic photodegradation of organic pollutants.
An example of this extraordinary photocatalytic degradation performance is organic methyl orange dyes (Fig. 36). A concentration decay profile of the dye plotted against processing time is illustrated in Fig. 36a. The synthetic conditions for the composite (e.g., hydrothermal temperature) can influence the photodegradation performance. In this case, a temperature of 160 °C provided the most efficient rate.607 Furthermore, the duration of the TiO2/MXene composite synthetic process also affects the photocatalytic performance (Fig. 36b). Thus, a 12 h hydrothermal synthetic procedure exhibits an optimal photodegradation rate. This TiO2/MXene hybrid far exceeded the photocatalytic performance of a commercial TiO2-based photocatalyst, Degussa P25.611,612
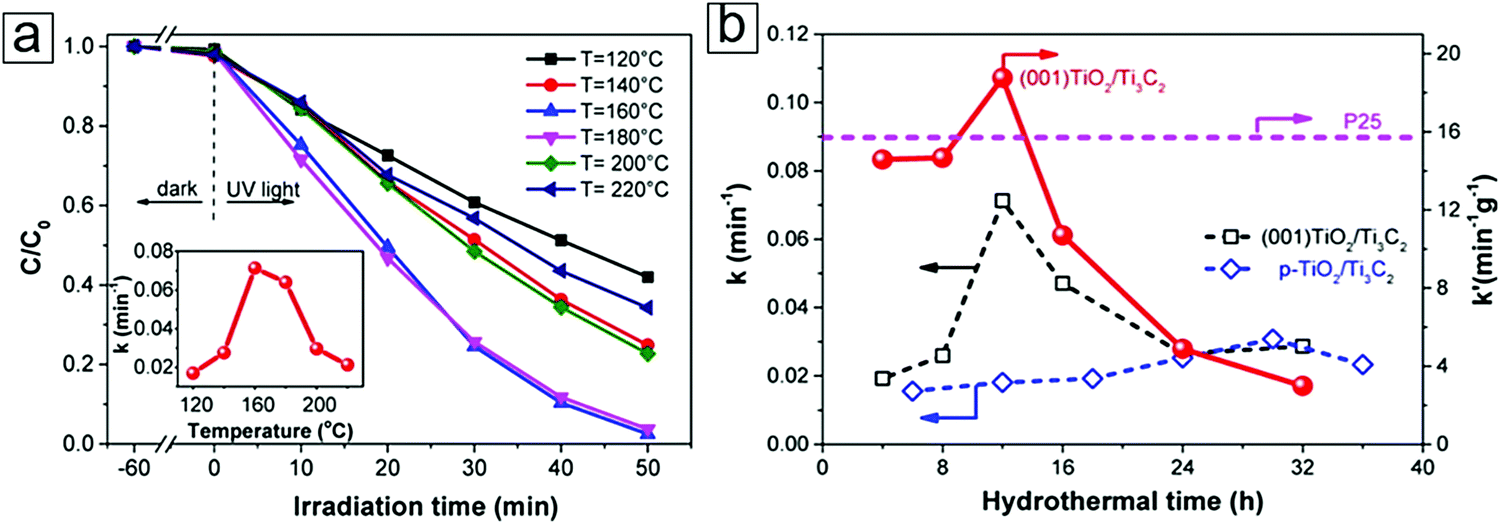 | ||
| Fig. 36 Photocatalytic degradation of methyl orange dye on the interfaces of MXene Ti3C2 and TiO2 hybrids. (a) Concentration decay of methyl orange dye with time depending on the hydrothermal temperature for the synthesized MXene/(001)TiO2. Inset: Rate constants for temperature-dependent synthesized hybrid materials. (b) Rate constants (k) for MXene/(001)TiO2 and MXene/particulate TiO2 dependent on the duration of the hydrothermal synthetic process. The rate constants were normalized by the mass of TiO2. The rate constant of commercial Degussa P25 (aeroxide TiO2) is also illustrated. Reprinted with permission from ref. 607. Copyright 2016 American Chemical Society. | ||
CO2 conversion consists of a multistep electroreduction through the incorporation of proton and electron pairs. First, CO2 fixation requires external energy and pressure to allow chemisorption over a catalyst surface. Second, the initial hydrogenation step through a single electron process, i.e., CO2 + e− → CO2*−, requires a negative reduction potential of −1.9 V versus normal hydrogen electrode.614 Based on the total number of proton and electron pairs transferred after the complete reaction, various types of hydrocarbon (green) fuels, including CO, HCOOH, H2CO, CH3OH, and CH4, can be synthesized. A thorough understanding of the reaction pathway is essential in developing novel catalysts for targeting specific products. In a computational study, various MXenes (Ti2CO2, V2CO2, and Ti3C2O2) were investigated. The data revealed that the catalytic reduction of CO2 preferentially occurs at the oxygen vacancies,615 which can be generated in the presence of CO and H2. Among these MXenes, Ti2CO2 exhibits the best catalytic activity for the hydrogenation of CO2. A typical reaction pathway is illustrated in Fig. 37. In step I, CO2 molecules chemisorb vertically in an oxygen vacancy, while a proton chemisorbs in a neighboring site. Upon chemisorption, a proton migrates towards the carbon atom after overcoming an energy barrier of 0.45 eV. Subsequently, an HCOO intermediate forms with an energy release of 1.48 eV.
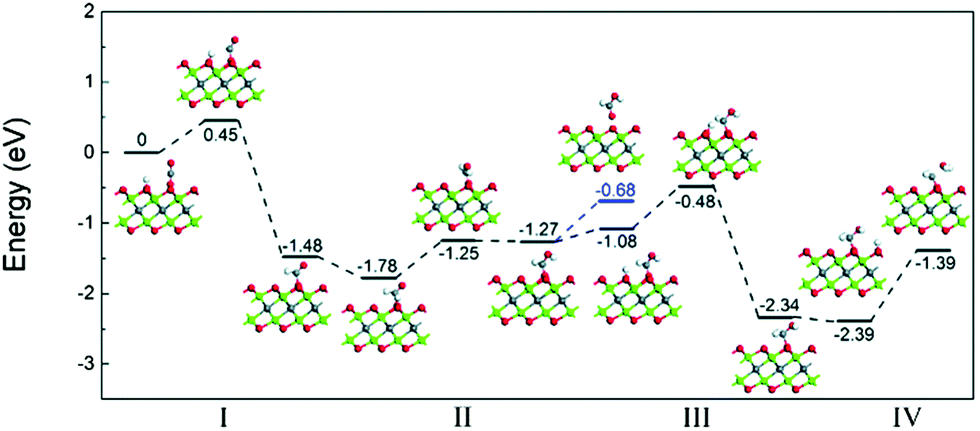 | ||
| Fig. 37 Hydrogenation of CO2 for fuel production in an MXene Ti2CO2 monolayer sheet. Thermodynamic pathways are shown when the reactions occur at an oxygen vacancy of MXene. Step I: CO2 forms HCOO; step II: HCOO is converted into HCOOH (blue lines represent the desorption of HCOOH acid); step III: HCOOH forms H2COOH; step IV: H2COOH decomposes to release HCHO and H2O. Reprinted with permission from ref. 615. Copyright 2017 The Royal Society of Chemistry. | ||
In step II, a second proton is incorporated in the oxygen site to yield HCOOH with an energy barrier of 0.53 eV. This rate limiting step requires much lower energy than that necessary for anatase TiO2 (0.87 eV).616 Thermodynamically, the hydrogenation of HCOO is exothermic (0.51 eV), as is the desorption of formic acid (HCOOH, 0.59 eV). The formation of HCOOH requires a lower energy barrier than other types of carbon species, which enables highly selective production of this hydrocarbon. Thus, due to its band gap of 0.91 eV, efficient visible light absorption, and practical hole and electron separation, MXene Ti2CO2 is an ideal photocatalyst for CO2 reduction.
The most probable reaction pathway for CH4 synthesis from CO2 over an MXene was determined from DFT calculations. Cr3C2 and Mo3C2 are the two most promising catalysts for the selective formation of methane from carbon dioxide.617 A side view of the energetically minimal energy reveals the chemisorption of a CH4 molecule over the Mo3C2 surface (Fig. 38).
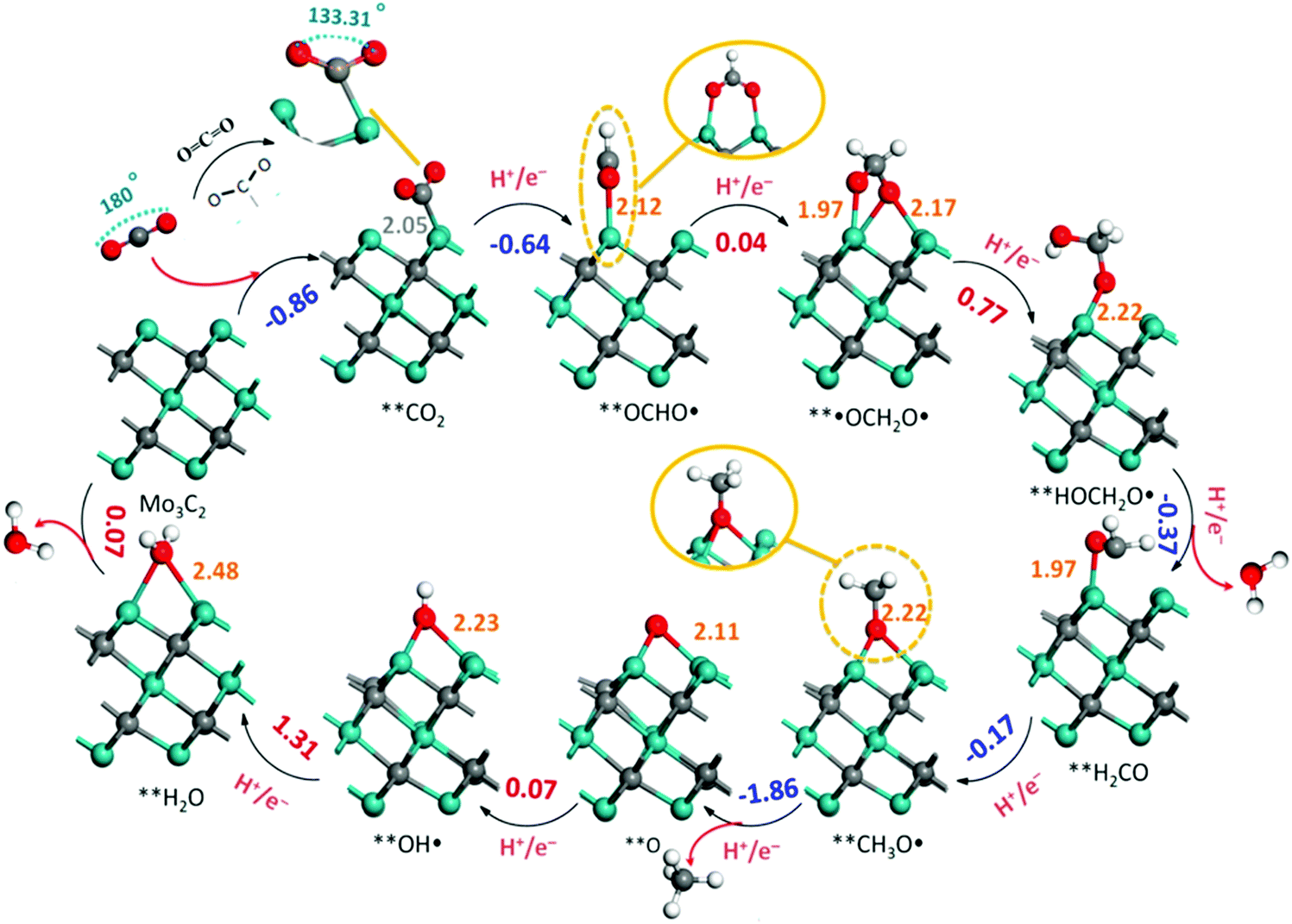 | ||
| Fig. 38 Mechanism for the conversion of CO2 into fuel methane over an MXene Mo3C2 catalyst. The minimum energy pathway is established from Perdew, Burke, and Ernzerhof DFT calculations. Mo, C, O, and H atoms are represented by lilac, grey, red, and white balls, respectively; ** indicates chemisorbed species. Grey and orange numbers refer to the Mo–C and Mo–O bond lengths (Å), respectively. Blue numbers represent the energy released spontaneously in the reaction. Red numbers denote the energy (eV) required to initialize the reaction path. Reprinted with permission from ref. 617. Copyright 2017 American Chemical Society. | ||
In more depth, after a six-step hydrogenation reaction, chemisorbed CH4 is formed by the inclusion of four proton–electron pairs over the Mo3C2 surface of the chosen MXene. This reaction has a total energy barrier of 1.31 eV. Cr3C2 requires a minimum energy input of 1.05 eV to convert CO2 to methane. When selecting an O- and OH-terminated MXene (Mo3C2), the predicted energy barriers are even lower (0.35 and 0.54 eV, respectively). This suggests that a moist environment may be practical.
MXenes such as M2XTx exhibit superior activity for catalysis of the HER. A screening computational study on Mo2CTx predicted that these compounds should exhibit high HER activities. Experimental work has confirmed that MXenes comprise active basal planes that enhance the HER. An MXene thin film drop cast over glassy carbon electrode with a Nafion binder was used in electrochemical tests comprising 0.5 M H2SO4 aqueous solution in a three-electrode cell with a rotating disk electrode. Fig. 39a presents anodic iR-corrected linear sweep voltammograms (LSVs) for pristine glassy carbon, Mo2CTx, and Ti2CTx. Mo2CTx exhibited high HER activity, delivering 10 mA cm−2 at an overpotential of 283 mV. After 30 cycles, the HER activity was maintained without any significant decrease and required an overpotential of 305 mV to reach a current density of 10 mA cm−2. In contrast, the glassy carbon displayed no activity, while Ti2CTx exhibited low catalytic performance with a high overpotential of 609 mV to reach 10 mA cm−2 and presented fast degradation upon 20 cycles.
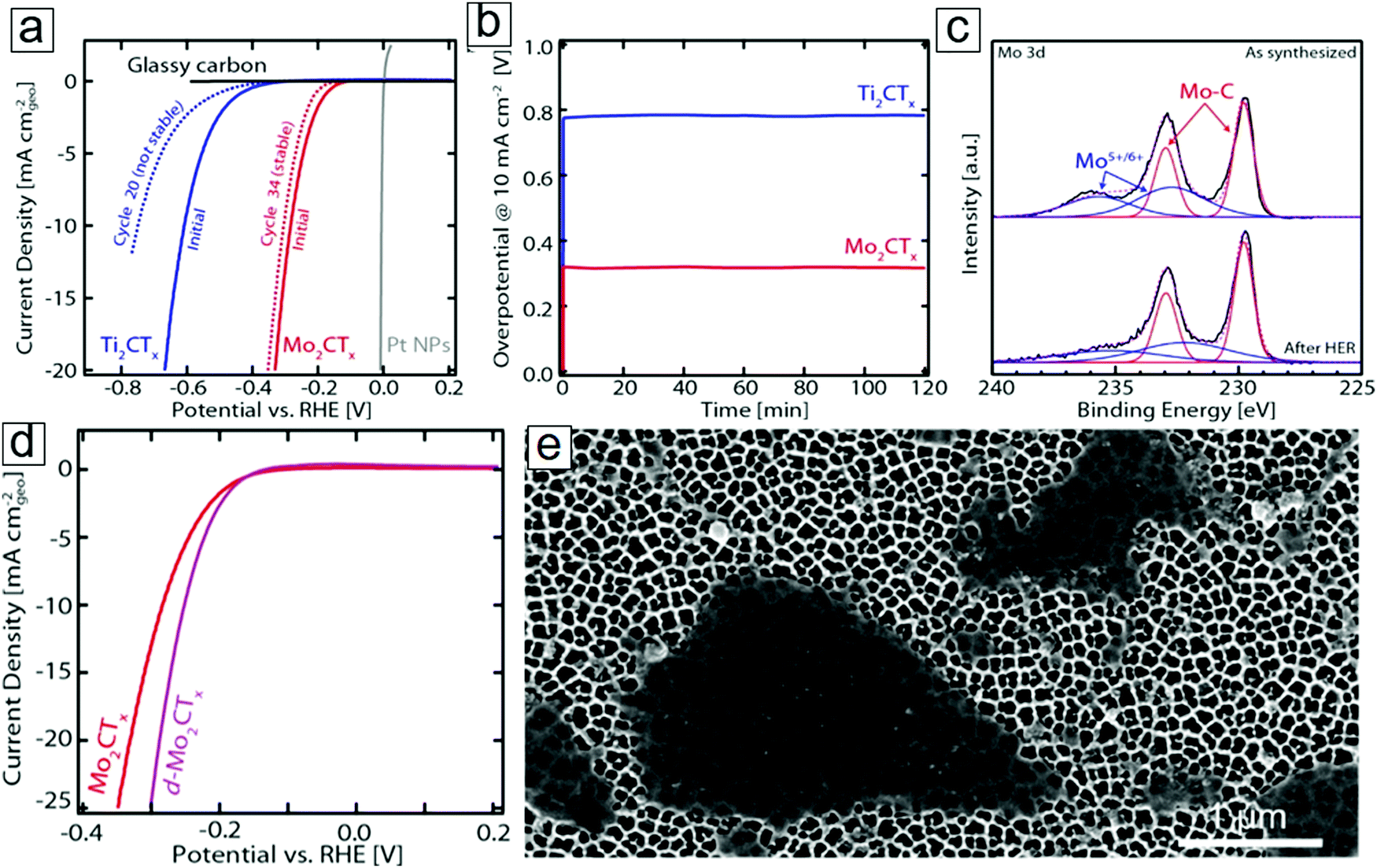 | ||
| Fig. 39 Activity and stability of MXene electrocatalysts in the hydrogen evolution reaction (HER). (a) Positive-going IR-corrected linear sweep voltammetry of MXenes over glassy carbon and bare glassy carbon. MXenes (Mo2CTx and Ti2CTx) prepared from HF etching methods with a mass loading of 0.1 mg cm−2; Pt nanoparticles are shown for comparison (Pt mass loading: 25 μg cm−2). (b) Temporal evolution of the overpotential for retaining a current density of 10 mA cm−2. (c) X-ray photoelectron spectroscopy (XPS) spectra of Mo 3d for as-synthesized Mo2CTx (top) and after HER testing (bottom). (d) Positive-going iR-corrected linear sweep voltammetry of Mo2CTx from the HF etching method and delaminated MXene (d-Mo2CTx) from the LiF/HCl etching method. (e) Scanning electron microscopy (SEM) micrograph of Mo2CTx over an alumina filter. The MXene displays sharp contrast and facilitates transparent observation of the bottom of the alumina particles (15 kV electron beam) due to its few atomic layers and high electrical conductance. Reprinted with permission from ref. 653. Copyright 2016 American Chemical Society. | ||
The temporal evolution of the overpotential654 can be used to evaluate the stability of a catalyst in the HER while maintaining a galvanostatic hold of 10 mA cm−2. There were no significant changes in the overpotentials of either Mo2CTx or Ti2CTx after a 2 h evolution period (Fig. 39b). Following an LSV test after the galvanostatic hold, no change was observed for Mo2CTx, while a fast decay was observed for Ti2CTx. This highlights the stability of Mo2CTx for long-duration HER performance. XPS was employed to determine the change in chemical states for Mo in Mo2CTx during the HER. Before the HER, the Mo2CTx presented four peaks with fitting, two doublets for the Mo–C bonds at 233.0 eV for 2d3/2 and 229.8 eV for 2d5/2 and two doublets for Mo–O bonds (Mo5+ and Mo6+) at 235.7 eV and 232.7 eV, respectively (Fig. 39c). After the HER, the Mo–C bonds remained unchanged, demonstrating the stability of the compound during HER activity. The Mo–O bonds decreased slightly in intensity. This was attributed to the reduction of the oxidized species under the cathodic potentials due to their dissolution in the presence of an acid. Further delamination resulted in the exposure of a larger portion of basal plane surfaces. This is reflected by an enhancement in the HER activity (Fig. 39d), e.g., a lower overpotential in the electrode geometry activity.
The HER activity of MXenes depends on the surface terminal groups and high charge transfer kinetics with surfaces terminated with mixed hydroxyl and oxygen species and fully terminated with oxygen atoms. Oxygen atoms residing on the top and bottom layers of the MXene sheets are active sites for HER reactions because of their interaction strength with H radicals. The Gibbs free energies for atomic hydrogen adsorption over oxygen-terminated MXenes were calculated for varying hydrogen coverages (Fig. 40). Ti2CO2 with a H coverage of 1/8 shows a Gibbs free energy of −0.04 eV; meanwhile, at a H coverage of 2/8, a free energy of −0.13 eV was observed (Fig. 40a). This negligible free energy suggests that Ti2CO2 is a promising HER catalyst with a wide range of H coverage. For Ti3C2O2 and V2CO2, the interactions between the oxygen atoms and H* enable the liberation of hydrogen from the surface, resulting in small negative free energies of −0.02 eV (Fig. 40b) and −0.05 eV (Fig. 40d) for 3/8 hydrogen high coverage, respectively. In contrast, Nb2CO2 and Nb4C3O2 display HER activities with low free energies of 0.11 eV (Fig. 40c) and 0.14 eV (Fig. 40e), respectively, at relatively low 2/8 H coverage.
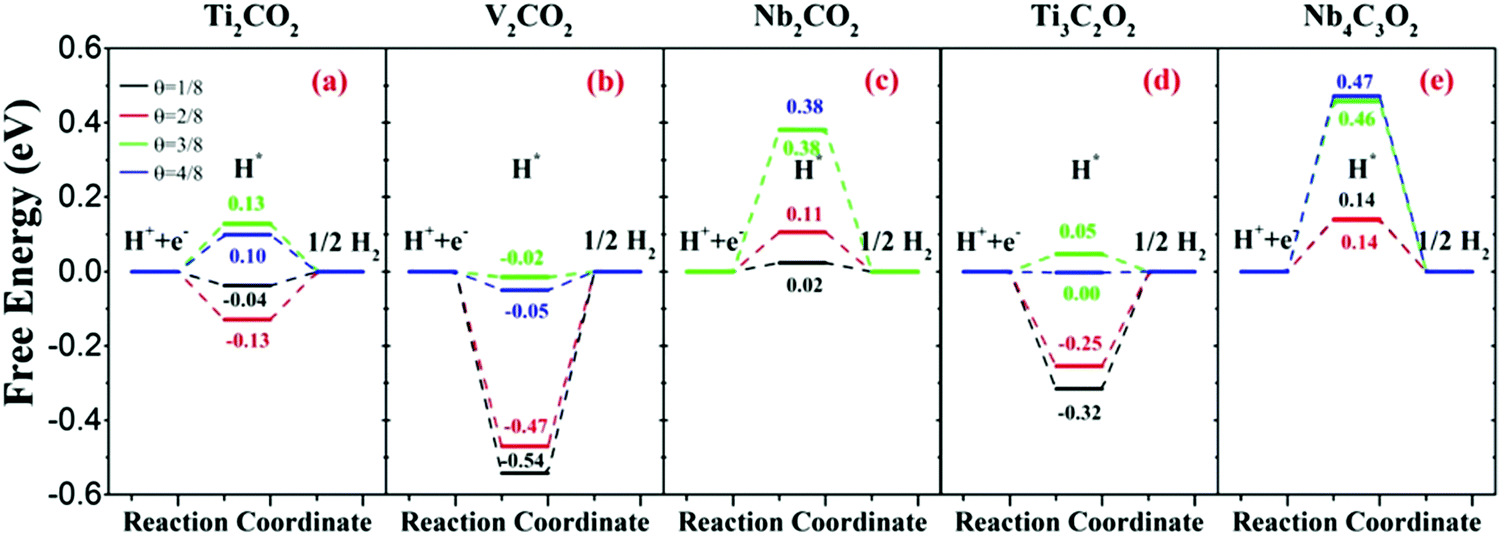 | ||
| Fig. 40 The hydrogen evolution reaction (HER) catalyzed over MXenes. The reactions proceed from H ions through H* radicals to H2 molecules. MXenes of Ti2CO2, V2CO2, Nb2CO2, Ti3C2O2, and Nb4C3O2 were employed for calculating the Gibbs free energy diagrams of these reactions under standard conditions. The lines represent the coverage ratio θ of H radicals over the surfaces of the MXenes. Reprinted with permission from ref. 655. Copyright 2017 American Chemical Society. | ||
The HER activities of various MXenes were compared with and without oxygen termination. HER performance can be quantitatively characterized with a volcano peak, viz., the nearest plots to the volcano peak represent the best catalytic activity.656 A volcano plot is presented for the theoretical exchange current, i0, versus the Gibbs free energy in Fig. 41. The pristine MXenes are far from the volcano peak, with a free energy of −0.73 eV, and show very low exchange currents. This implies insufficient catalyst activity because of the strong interactions between the pristine MXenes and H*. With oxygen termination, the weak interaction enables the liberation of hydrogen and leads to a high exchange current for enhanced HER performance. Indeed, the Gibbs free energy of Ti2CO2 is 0.01 eV and the exchange current, which is predicted to exceed the HER performance of Nb2CO2 and Ti3C2O2, is located at the maximum of the volcano plot.
 | ||
| Fig. 41 Volcano plot of the exchange current (i0) as a function of the average Gibbs free energy of hydrogen adsorption. The H* radicals form during the hydrogen evolution reaction (HER) over MXene with and without oxygen surface groups. Oxygen surface groups provide more redox reaction sites. Reprinted with permission from ref. 655. Copyright 2017 American Chemical Society. | ||
Further calculations show the reaction pathways based on the Volmer–Heyrovsky and Volmer–Tafel mechanisms.657 The Ti2CO2 supercell, with H* covering one surface, exhibits energy barriers of 0.29 eV and 0.99 eV for the Heyrovsky and Tafel pathways, respectively.
5.2 Catalysts for synthetic reactions
MXenes display promising catalyst activity for N2 capture and reduction. This catalytic activity depends on the transition metal incorporated,664i.e., d2: Ti, Zr, and Hf; d3: V, Nb, and Ta; or d4: Cr and Mo. Analogous to CO2 and H2 capture, the initial step of this process is N2 capture, i.e., the interaction of N2 with the catalytic surface. DFT calculations have revealed that the N2 molecules spontaneously transition from a gas phase to a chemisorbed state without an intermediate physisorption step, viz. through a negative change in the Gibbs free energy. V3C2 and Nb3C2 show large binding energies (−2.17 eV and −2.44 eV, respectively), indicating strong N2-philicity. The rate limiting step of the Haber–Bosch process was determined to be the N2 dissociation step in both theoretical665 and experimental666 reports. During MXene catalysis, the weakening of the triple bond in a N2 molecule and the subsequent activation occur spontaneously.
For MXene-catalyzed N2 reduction mechanisms, five possible pathways are proposed with multistep reactions, e.g., proton–electron pair inclusion and incorporation of additional pairs (Fig. 42a). These routes667 include single-double routes prior to and after the third hydrogenation reaction, leading to the formation of HN–NH2* or N* radicals. The d2 M3C2 MXenes prefer path 4, the second hydrogenated HN–NH and the third hydrogenated HN–NH2*. On the other hand, the d3 and d4 MXenes host path 2 reactions, viz. the N–NH2 and N* species form along their minimal energy pathways.
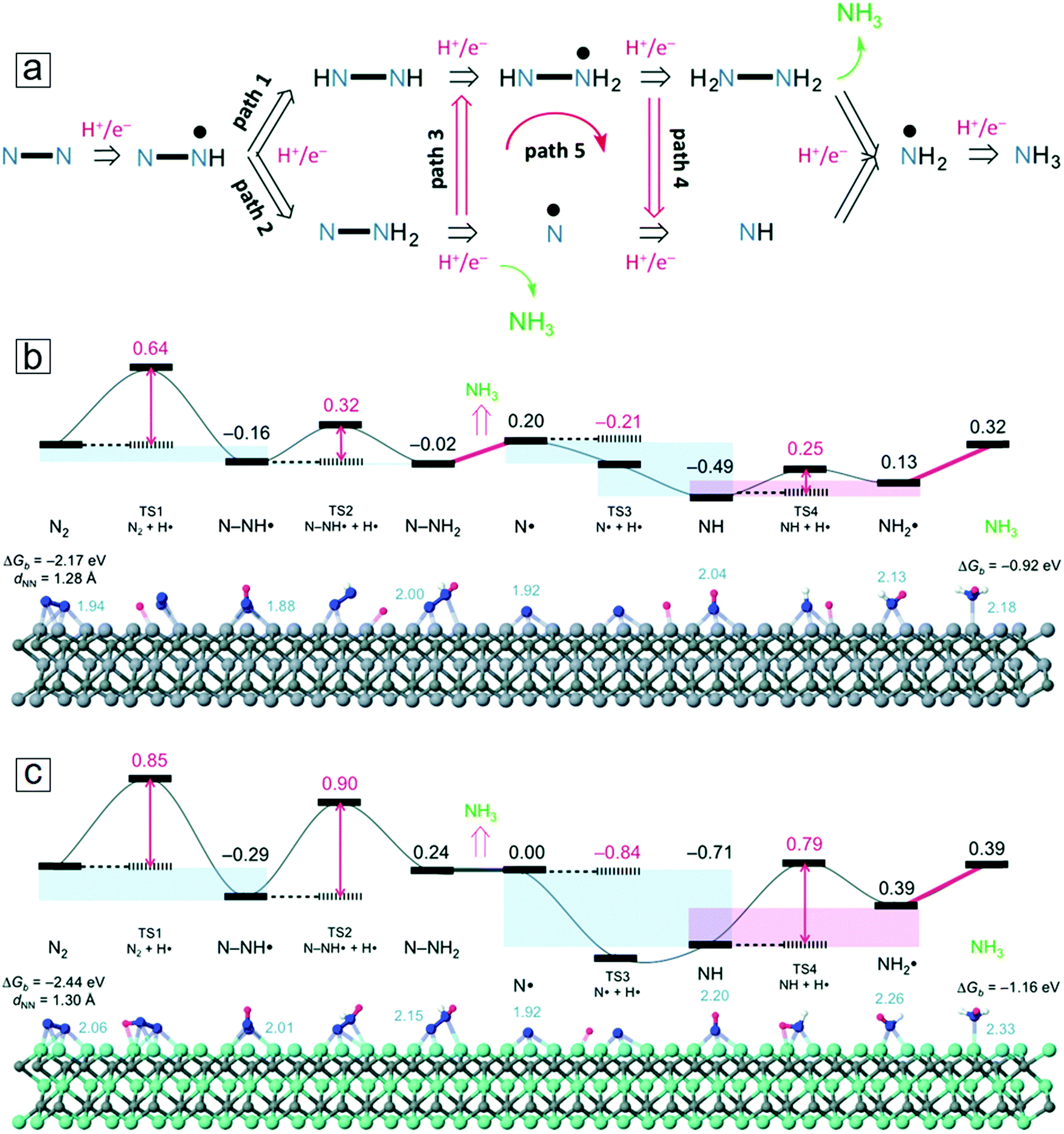 | ||
| Fig. 42 Ammonia synthesized from N2 over MXene catalysts. (a) Proposed reaction pathway for N2 conversion into NH3 fertilizer. (b) Minimum energy path for NH3 formation from N2 conversion over the MXene V3C2. Included are the atomic structures and energies of the transition states (TS). (c) Minimum energy path for NH3 synthesis over the MXene Nb3C2. These results were calculated at the DFT+D3 level. Gibbs free energies (black) and activation energies (red) are provided in eV at room temperature vs. standard hydrogen electrode (SHE; pH 0, H2 pressure = 1 bar, voltage = 0 V). Notes: dMN is the distance between the metal ions and N atoms. Blue and pink spheres represent N and H atoms, respectively. Blue shading indicates spontaneous steps, while red represents non-spontaneous routes. Reprinted with permission from ref. 664. Copyright 2016 The Royal Society of Chemistry. | ||
Among the investigated MXenes, V3C2 and Nb3C2 proved to be the most promising catalysts for the electrochemical synthesis of NH3. The largest energies for single step reactions are the six proton–electron pairs gained from the NH2* species (Fig. 42b and c), namely 0.32 eV for V3C2 and 0.39 eV for Nb3C2versus standard hydrogen electrode (SHE). The first N2 hydrogenation requires a substantial amount of energy input to form the N–NH* radical, with electron affinities for N2* generation of −0.16 eV and −0.29 eV for V3C2 and Nb3C2, respectively. The kinetics can be evaluated by considering the effects of the transition states. The activation energy barriers for the first transition state of N2 + H*, which bridges N2 and N–NH*, are 0.64 eV for V3C2 and 0.85 eV for Nb3C2. When N2 is spontaneously captured, the barriers of the rate-limiting reactions are only 0.64 eV for V3C2 and 0.90 eV for Nb3C2 during the total electrochemical synthesis of NH3. In addition, the reaction profile is smoother for V3C2 than for Nb3C2. Eventually, the liberation of chemisorbed NH3 requires energy injections of 0.92 eV and 1.16 eV, respectively, for these two MXenes.
5.3 Solar steam production
Photothermal conversion is a primitive means of solar energy utilization in which sunlight is converted into heat, e.g., solar cookers and solar water heating.668–670 With advances in nanomaterial processing and high resolution characterization tools, solar-thermal conversion has been revisited for a number of applications, including solar steam production and cancer therapy.671–677 Photothermal performance has been enhanced by the advent of nanomaterials, including carbon nanotubes,678–680 graphene,681,682 metal nanoparticles,683–685 black TiO2,686 Ti2O3,687 and bimetallic plasmonic nanoshells.688MXenes, with high visible light absorption, exhibit extraordinary solar-thermal conversion efficiencies. For example, a Ti3C2 membrane achieved 100% light-to-heat conversion efficiency as measured in a droplet laser heating process.689 Indeed, this extraordinary energy conversion efficiency is reflected by a high solar steam production efficiency of 84% under solar illumination. This value represents the efficiency of the photothermal material.
5.4 Photothermal therapies
The outstanding heat conversion efficiency of MXenes can also be successfully applied to photothermal therapy. Nb2C shows highly efficient in vivo ablation of tumor xenografts in white mice under near-IR illumination.690 Quantitatively, MXenes exhibit extraordinary photothermal efficiencies, i.e., 36.4% in a near-IR light range of 750 to 1000 nm and 45.7% in a range of 1000 to 1350 nm. In addition, the stability of MXenes enables long life under heated conditions. Colloids of MXene/polyvinylpyrrolidone (PVP) show good biocompatibility and non-toxicity. This work may broaden MXene applications to surgery-free cancer therapy.5.5 Photovoltaic devices
A photovoltaic device consists of a heterostructure made from two materials to provide an interface that hosts the separation of photoinduced electron–hole charge pairs and the subsequent charge transfer towards electrodes for photocurrent generation. The heterostructures can be categorized as semiconducting p–n, p–i–n, and metal/semiconductor Schottky junctions. Light absorbing materials in solar cells include bulk silicon, silicon thin films, compound thin films, photo-sensitized dyes, fullerene conjugated polymers, and perovskite hybrid organic–inorganic materials. Photovoltaic devices are usually termed solar cells for solar energy conversion (e.g., thin films as light absorbers691–693) and photodetectors when the photocurrent yield is small with very thin, specifically atom-thin, 2D materials.MXenes have extraordinary properties for photoelectronic energy conversion applications, such as adjustable electronic properties for metallic or semiconducting conductivity,694 tunable work functions, and high optical transmittance.67
The work functions of MXenes are highly dependent on their surface functional groups, e.g., OH groups decrease the work functions, while O groups increase them. MXenes possess a wide range of work functions, from <2.14 eV (the minimum value among all metals)695 to >5.65 eV (the maximum value among metals). A typical example, Ti3C2Tx, has a work function of 4.37 eV. Therefore, the metal/semiconductor contact will lead to a suitable barrier height for photoelectric current generation.696,697 When MXene/Si contact each other in a vertical van der Waals heterostructure, the efficient separation of photogenerated electron hole pairs and their collection can be achieved. Indeed, a photovoltaic device of an MXene/Si Schottky junction cell698 demonstrated an open voltage of 0.34 V and a short circuit current density of 12.9 mA cm−2 under light irradiation (100 mW cm−2). This photoelectric cell can be used as a self-powered photodetector with a high sensitivity of 26.95 mA W−1, a superior ON/OFF ratio of 105, and a high-speed response, with 0.84 ms response time and 1.67 ms recovery time.
Secondly, we will examine photovoltaic devices using semiconducting MXenes. An MXene and blue phosphorene heterostructure was employed in proof-of-concept theoretical computational work.699 In the band alignment diagram, the MXene Zr2CO2 and a blue phosphorene junction presented a conductance band minimum and a valence band maximum from both MXene sides. Strain engineering699 can induce manipulation of the electronic band structure for semiconducting materials and leads to extraordinary photoelectronic applications.700–705 Under moderate strain, the heterostructure displays a band alignment with a valence band minimum from MXene and a conduction band minimum from blue phosphorene. When compressing the junction with tensile strain, the heterostructure displays band alignment between the conduction band minimum from the MXene and valence band minimum from the blue phosphorene. This MXene/semiconductor heterostructure junction may pave the way for new photoelectronic and photonic applications.
5.6 Thermoelectric power generation
A few MXenes, including Sc2CTx, Zr2CTx, Ti2CTx, and Hf2CTx, are predicted to be semiconducting with non-zero band gaps.42,706,707 Calculations have revealed that these MXenes possess high Seebeck coefficients, e.g., 1140 μV K−1 for Ti2CO2 and 2200 μV K−1 for Sc2C(OH)2 at 100 K. Moreover, MoCF2 has a superior thermoelectric power factor.21In an experimental study, Mo-based MXenes in the form of freestanding papers were fabricated by vacuum filtration of their suspensions.708 All the tested MXenes (Mo2CTx, Mo2TiC2Tx, and Mo2Ti2C3Tx) displayed stability and improved electrical conductivity at >500 K. Among them, Mo2TiC2Tx exhibited optimum performance, with a large Seebeck coefficient of −47.3 μV K−1 and a high electric conductivity of 1380 S cm−1. Its thermoelectric power factor reached 3.09 × 10−4 W m−1 K−2 at 803 K. The thermoelectric performance of the MXenes exceeded that of their parent MAX phases and is comparable to the performance of some conventional thermoelectric materials.
The MXene type and edge direction affect their thermal and electronic properties. Thus, in a theoretical study,709 Hf2CO2 also displayed promising thermoelectric properties with a band gap of 1.657 eV. With armchair edge termination, MXene stripes have a thermal conductivity of 86.25 W m−1 K and a conductivity of 36.5 W m−1 K along the zig-zag direction. In terms of the electronic conductivity, the hole-carrier mobility in Hf2CO2 was calculated as 13.5 × 103 cm2 V−1 s−1 in the armchair direction and 17.6 × 103 cm2 V−1 s−1 in the zig-zag direction. These values suggest that Hf2CO2 can be used in nanoelectronic devices, while the use of Ti2CO2 as a building block requires good thermal dispersion out of the device package.
The electron and phonon transport properties of a material also influence its thermoelectric performance. In a large sample with diffusive heat dispersion via n-doped charge carriers,706 the figures of merit of Ti2CO2, Zr2CO2, and Hf2CO2 were calculated as 0.45, 0.35, and 0.29, respectively. Ti2CO2 had the highest figure of merit and the lowest contribution from the lattice to the total thermal conductivity. This occurs because the acoustic phonon branches show strong dispersion. The figure of merit decreases with a lower carrier concentration because of the minority carrier effect. When the sample size of each MXene type was limited to a value below the mean free path of the phonon, i.e., 132, 486, and 850 nm for Ti2CO2, Zr2CO2, and Hf2CO2, respectively, the thermoelectric performance was enhanced by means of ballistic heat transport.
Beyond energy applications, these properties have led to other important applications, such as hydrogen storage media,17,710 transparent conducting films,67,151 field effect transistors,26,711 sensors,712,713 photoluminescent labelling agents for biomedical imaging,714,715 and heavy metal and dye adsorption for environmental water purification.175,676 Moreover, especially in the field of physics, MXenes provide enrichful platforms for new phenomena, e.g., ohmic contact with other semiconducting 2D materials to form field effect transistors,41,716–720 topological insulators in oxygen-terminated MXenes,45,48 and switchable 2D electron hole gas for spin orbitronics.721,722
The progress of MXene research has demonstrated many novel uses with increasing understanding of their continuously explored extraordinary properties. For example, the structures of MXenes have been updated with the presence of Mo1.33C with divacancy ordering723 or carbon vacancy.724 Furthermore, the role of water was investigated during a selective etching protocol.725 Second, computational predications have rapidly increased the understanding of the band gap properties of MXenes.726,727 Other emerging topics include surface-enhanced Raman spectroscopy,728 structure–property correlations,729,730 anisotropic electronic conduction,731 bipolar magnetic properties,732 magnetoelectronic performance,733 magnetic properties,734 multipolar surface plasmons,735 elastic properties,736 thermoelectric properties,737 transport properties738 for understanding free carrier dynamics,739 and thermal conductivity.740
There are also innovative methods for tuning the structures and properties of MXenes. Examples are structural engineering,741 nanopore engineering,742–744 phase transition745–747 by oxidation,748 surface alloying,749 liquid exfoliation,750,751 fluorine-free synthesis,752 and cold sintering.753 Superior performance of MXenes can be achieved by forming heterostructures754,755 and composites.756–762
MXene-based sensors, e.g., glucose biosensors,763 piezoresistive sensors,764–766 and force sensors,767 have also attracted great interest. These sensing materials feature hydrogels for stretching768 and 3D frameworks for high sensing volumes.769 Other electronic applications include transparent conducting films770,771 and electromagnetic interference shielding772,773 by microwave absorption.774–777
MXenes have made variable contributions to renewable energy production. These wide uses include photocatalysts,778–785 water splitting786 for hydrogen production,787–789 and electrocatalysts.790,791 Gas evolution with MXenes has exhibited great promise, where they play multiple roles as catalysts for CO2 abatement,792–794 CO oxidation,795,796 N2 photofixation,797,798 the oxygen reduction reaction,799 and urea decomposition800 while also serving as catalyst host materials.801
Rechargeable ion batteries account for great efforts in MXene energy investigations. These contributions include lithium ion batteries,329,802–810 sodium ion batteries811–817 and their cathodes,818 lithium sulfur batteries,819–824 magnesium ion batteries,825 alkaline hybrid batteries,826–829 and seawater electrolytes.830 The future of battery research is the achievement of high capacity, high rate batteries.831 Further strategies include uniaxial strain for tunable Li diffusion.832
In terms of supercapacitors,833,834 efforts have been made to improve their capacitances835–837 and understand their charge transfer mechanisms.838 The new trends in supercapacitor developments include satisfying the requirements for flexible,839,840 solid,841 stretchable842 and bendable843 materials which are suitable for wearable electronics.844,845 These supercapacitors incorporate yarn fabrication,846 pseudocapactive polymers,847 inorganic capacitive materials,848 on-chip micro-supercapacitors,849,850 flexible arrays,851 and planar structures.852 Also, their performance is being continuously enhanced to achieve the goals of high cycling stability,853 ultrahigh areal capacitance,854 high voltage,855 fast ion storage,856 and high volumetric performance.857
For environmental amelioration, MXenes have been widely employed in water deionization,858–861 water desalination862,863 and antimicrobial water treatment864 as well as gas filtering.865–868 In healthcare fields, MXenes have shown promise in cancer chemotherapy,869 cancer theranostics,870,871 and related biomedical applications.872,873
Most recently, MXenes have been successfully integrated in device systems with the maturation of individual functional devices. First, MXene was employed as an electrode material in a self-powered charging unit874 connecting both interfaces of a triboelectric nanogenerator (TENG)609,875 and supercapacitor.327,876–878
6 Summary and future opportunities
The aim of this review is to introduce the basics of MXenes associated with energy storage and conversion in terms of their performance. Initially, their fundamentals, such as their structures, morphologies, and properties, as well as their rich characterizations were presented. The most recent advances in MXenes have been described in the application sections and the experimental progress in rechargeable batteries and supercapacitors has been updated. The multifunctional capabilities of MXenes show advantageous performance as anode materials in both lithium- and sodium-ion batteries, as passivation materials in lithium–metal batteries, and as sulfur immobilizers in lithium–sulfur batteries. The reported performance of MXene electrodes has been compared in a comprehensive table. Furthermore, supercapacitors have been shown to be enhanced by the porous morphology of the MXene electrodes. Beyond double symmetrical electrodes for ion adsorption, other types of feasible supercapacitors have been fabricated. These include the incorporation of transition metal oxide hybrids serving as pseudocapacitors as well as electrochemical capacitors. Experimental updates on the photodegradation of organic pollutants as well as electrocatalysts for both the HER and OER have also been presented.The photocatalytic and electrocatalytic performance of MXenes for fuel production remains mostly theoretical. Theoretical calculations indicate energetically favorable reaction paths for catalytic fuel production, such as hydrogen generation, carbon dioxide hydrogenation, and ammonia fuel production.
Many aspects of MXenes are still unknown. These include the number of family members and the ultimate theoretical capacity for lithium storage as well as other energy-related applications. In future, theoretical predictions will continue to lead to the expansion of the MXene family through different elemental combinations. As rechargeable lithium-ion battery anodes, the capacities of MXenes will be enhanced through the use of composites with other elements, such as Si, Sn, Ge, and transition metal oxides. Moreover, photocatalytic reactions can be tailored when forming MXene composites with wide band gap semiconductors such as TiO2 and tunable band gap semiconductors such as phosphorene. Experimentally, the design and coupling of both the OER and ORR with MXene electrocatalysts has shown proof-of-concept success in rechargeable Zn–air batteries. The potential of MXenes in energy applications is truly enormous.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly thank the National Science Foundation China (NSFC, Project 51672181), the National Science Center within the frame of the Sonata Program (Grant agreement 2014/13/D/ST5/02853) and the Opus program (Grant agreement 2015/19/B/ST5/03399). J. P. gives his gratitude to the IKM fellowship in the Institut für Komplexe Materialien of IFW Dresden, Germany.Notes and references
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CAS.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- M. W. Barsoum and M. Radovic, Annu. Rev. Mater. Res., 2011, 41, 195–227 CrossRef CAS.
- M. W. Barsoum, in Ceramics Science and Technology, ed. R. Riedel and I.-W. Chen, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, vol. 2, pp. 299–347 Search PubMed.
- Z. M. Sun, Int. Mater. Rev., 2011, 56, 143–166 CrossRef CAS.
- M. W. Barsoum, MAX Phases: Properties of Machinable Ternary Carbides and Nitrides, Wiley-VCH Verlag GmbH & Co. KGaA, Weiheim, Germany, 1st edn, 2013 Search PubMed.
- M. Naguib and Y. Gogotsi, Acc. Chem. Res., 2015, 48, 128–135 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- M. Ghidiu, M. Naguib, C. Shi, O. Mashtalir, L. M. Pan, B. Zhang, J. Yang, Y. Gogotsi, S. J. Billinge and M. W. Barsoum, Chem. Commun., 2014, 50, 9517–9520 RSC.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, J. Am. Chem. Soc., 2013, 135, 15966–15969 CrossRef CAS PubMed.
- M. Kurtoglu, M. Naguib, Y. Gogotsi and M. W. Barsoum, MRS Commun., 2012, 2, 133–137 CrossRef CAS.
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS PubMed.
- J. Chen, K. Chen, D. Tong, Y. Huang, J. Zhang, J. Xue, Q. Huang and T. Chen, Chem. Commun., 2015, 51, 314–317 RSC.
- R. B. Rakhi, B. Ahmed, M. N. Hedhili, D. H. Anjum and H. N. Alshareef, Chem. Mater., 2015, 27, 5314–5323 CrossRef CAS.
- J. C. Lei, X. Zhang and Z. Zhou, Front. Phys., 2015, 10, 276–286 CrossRef.
- Q. Hu, D. Sun, Q. Wu, H. Wang, L. Wang, B. Liu, A. Zhou and J. He, J. Phys. Chem. A, 2013, 117, 14253–14260 CrossRef CAS PubMed.
- Z. Ma, Z. Hu, X. Zhao, Q. Tang, D. Wu, Z. Zhou and L. Zhang, J. Phys. Chem. C, 2014, 118, 5593–5599 CrossRef CAS.
- Z. Y. Li, L. B. Wang, D. D. Sun, Y. D. Zhang, B. Z. Liu, Q. K. Hu and A. G. Zhou, Mater. Sci. Eng., B, 2015, 191, 33–40 CrossRef CAS.
- X. Wang, S. Kajiyama, H. Iinuma, E. Hosono, S. Oro, I. Moriguchi, M. Okubo and A. Yamada, Nat. Commun., 2015, 6, 6544 CrossRef CAS PubMed.
- M. Khazaei, M. Arai, T. Sasaki, M. Estili and Y. Sakka, Phys. Chem. Chem. Phys., 2014, 16, 7841–7849 RSC.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- T. L. Tan, H. M. Jin, M. B. Sullivan, B. Anasori and Y. Gogotsi, ACS Nano, 2017, 11, 4407–4418 CrossRef CAS PubMed.
- Y. Xie, Y. Dall'Agnese, M. Naguib, Y. Gogotsi, M. W. Barsoum, H. L. Zhuang and P. R. Kent, ACS Nano, 2014, 8, 9606–9615 CrossRef CAS PubMed.
- G. R. Berdiyorov, Europhys. Lett., 2015, 111, 67002 CrossRef.
- X. Zhang, X. Zhao, D. Wu, Y. Jing and Z. Zhou, Nanoscale, 2015, 7, 16020–16025 RSC.
- P. Eklund, J. Rosen and P. O. Å. Persson, J. Phys. D: Appl. Phys., 2017, 50, 113001 CrossRef.
- J. Halim, S. Kota, M. R. Lukatskaya, M. Naguib, M. Q. Zhao, E. J. Moon, J. Pitock, J. Nanda, S. J. May, Y. Gogotsi and M. W. Barsoum, Adv. Funct. Mater., 2016, 26, 3118–3127 CrossRef CAS.
- M. Naguib, J. Come, B. Dyatkin, V. Presser, P. L. Taberna, P. Simon, M. W. Barsoum and Y. Gogotsi, Electrochem. Commun., 2012, 16, 61–64 CrossRef CAS.
- J. Come, M. Naguib, P. Rozier, M. W. Barsoum, Y. Gogotsi, P. L. Taberna, M. Morcrette and P. Simon, J. Electrochem. Soc., 2012, 159, A1368–A1373 CrossRef CAS.
- A. Vaughn, J. Ball, T. Heil, D. J. Morgan, G. I. Lampronti, G. Marsalkaite, C. L. Raston, N. P. Power and S. Kellici, Chemistry, 2017, 23, 8128–8133 CrossRef CAS PubMed.
- I. R. Shein and A. L. Ivanovskii, Comput. Mater. Sci., 2012, 65, 104–114 CrossRef CAS.
- H. Qu, Y. Wang, Y. S. Ye, W. Zhou, S. P. Bai, X. P. Zhou, H. Y. Peng, X. L. Xie and Y. W. Mai, J. Mater. Chem. A, 2017, 5, 22361–22371 RSC.
- L. Wang, P. Yang, Y. Liu, X. Fang, X. Shi, S. Wu, L. Huang, H. Li, X. Huang and W. Huang, Nanoscale, 2017, 9, 9997–10001 RSC.
- Y. Wu, P. Nie, H. Dou, J. Jiang, Y. Zhu and X. Zhang, Nano-Struct. Nano-Objects, 2018, 15, 197–204 CrossRef CAS.
- P. Sun, R. Ma and T. Sasaki, Chem. Sci., 2018, 9, 33–43 RSC.
- Q. Zhang, W. Wang, X. Kong, R. G. Mendes, L. Fang, Y. Xue, Y. Xiao, M. H. Rummeli, S. Chen and L. Fu, J. Am. Chem. Soc., 2016, 138, 11101–11104 CrossRef CAS PubMed.
- X. Chen, R. A. Boulos, J. F. Dobson and C. L. Raston, Nanoscale, 2013, 5, 498–502 RSC.
- J. Wang, J. Hao, D. Liu, S. Qin, C. Chen, C. Yang, Y. Liu, T. Yang, Y. Fan, Y. Chen and W. Lei, Nanoscale, 2017, 9, 9787–9791 RSC.
- A. Lipatov, M. Alhabeb, M. R. Lukatskaya, A. Boson, Y. Gogotsi and A. Sinitskii, Adv. Electron. Mater., 2016, 2, 1600255 CrossRef.
- M. Khazaei, A. Ranjbar, M. Arai, T. Sasaki and S. Yunoki, J. Mater. Chem. C, 2017, 5, 2488–2503 RSC.
- M. Khazaei, M. Arai, T. Sasaki, C. Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2013, 23, 2185–2192 CrossRef CAS.
- M. Ashton, K. Mathew, R. G. Hennig and S. B. Sinnoet, J. Phys. Chem. C, 2016, 120, 3550–3556 CrossRef CAS.
- L. H. Li, Comput. Mater. Sci., 2016, 124, 8–14 CrossRef CAS.
- C. Si, J. You, W. Shi, J. Zhou and Z. Sun, J. Mater. Chem. C, 2016, 4, 11524–11529 RSC.
- C. Si, K. H. Jin, J. Zhou, Z. Sun and F. Liu, Nano Lett., 2016, 16, 6584–6591 CrossRef CAS PubMed.
- M. Khazaei, A. Ranjbar, M. Arai and S. Yunoki, Phys. Rev. B, 2016, 94, 125152 CrossRef.
- H. M. Weng, A. Ranjbar, Y. Y. Liang, Z. D. Song, M. Khazaei, S. Yunoki, M. Arai, Y. Kawazoe, Z. Fang and X. Dai, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 075436 CrossRef.
- Y. Lee, S. B. Cho and Y. C. Chung, ACS Appl. Mater. Interfaces, 2014, 6, 14724–14728 CrossRef CAS PubMed.
- X. F. Yu, J. B. Cheng, Z. B. Liu, Q. Z. Li, W. Z. Li, X. Yang and B. Xiao, RSC Adv., 2015, 5, 30438–30444 RSC.
- L. H. Li, J. Phys. Chem. C, 2016, 120, 24857–24865 CrossRef CAS.
- Y. Lee, Y. Hwang, S. B. Cho and Y. C. Chung, Phys. Chem. Chem. Phys., 2014, 16, 26273–26278 RSC.
- R. C. Longo, R. Addou, S. Kc, J.-Y. Noh, C. M. Smyth, D. Barrera, C. Zhang, J. W. P. Hsu, R. M. Wallace and K. Cho, 2D Mater., 2017, 4, 025050 CrossRef.
- X. J. Qiu, Q. Lv and Z. Z. Cao, J. Phys. D: Appl. Phys., 2017, 50, 455106 CrossRef.
- Q.-H. Tan, X. Zhang, X.-D. Luo, J. Zhang and P.-H. Tan, J. Semicond., 2017, 38, 031006 CrossRef.
- Z. Song, Z. Li, H. Wang, X. Bai, W. Wang, H. Du, S. Liu, C. Wang, J. Han, Y. Yang, Z. Liu, J. Lu, Z. Fang and J. Yang, Nano Lett., 2017, 17, 2079–2087 CrossRef CAS PubMed.
- Y. Ye, J. Xiao, H. Wang, Z. Ye, H. Zhu, M. Zhao, Y. Wang, J. Zhao, X. Yin and X. Zhang, Nat. Nanotechnol., 2016, 11, 598–602 CrossRef CAS PubMed.
- X. T. An, J. Xiao, M. W. Tu, H. Yu, V. I. Fal'ko and W. Yao, Phys. Rev. Lett., 2017, 118, 096602 CrossRef PubMed.
- V. H. Nguyen, S. Dechamps, P. Dollfus and J. C. Charlier, Phys. Rev. Lett., 2016, 117, 247702 CrossRef PubMed.
- M. J. Szary, M. T. Michalewicz and M. W. Radny, Phys. Rev. B, 2017, 95, 205421 CrossRef.
- J. Pang, A. Bachmatiuk, I. Ibrahim, L. Fu, D. Placha, G. S. Martynkova, B. Trzebicka, T. Gemming, J. Eckert and M. H. Rümmeli, J. Mater. Sci., 2015, 51, 640–667 CrossRef.
- J. B. Pang, A. Bachmatiuk, L. Fu, C. L. Yan, M. Q. Zeng, J. Wang, B. Trzebicka, T. Gemming, J. Eckert and M. H. Rummeli, J. Phys. Chem. C, 2015, 119, 13363–13368 CrossRef.
- B. Xu, M. Zhu, W. Zhang, X. Zhen, Z. Pei, Q. Xue, C. Zhi and P. Shi, Adv. Mater., 2016, 28, 3333–3339 CrossRef CAS PubMed.
- L. Lorencova, T. Bertok, E. Dosekova, A. Holazova, D. Paprckova, A. Vikartovska, V. Sasinkova, J. Filip, P. Kasak, M. Jerigova, D. Velic, K. A. Mahmoud and J. Tkac, Electrochim. Acta, 2017, 235, 471–479 CrossRef CAS PubMed.
- A. Miranda, J. Halim, A. Lorke and M. W. Barsoum, Mater. Res. Lett., 2017, 5, 322–328 CrossRef CAS.
- J. Come, J. M. Black, M. R. Lukatskaya, M. Naguib, M. Beidaghi, A. J. Rondinone, S. V. Kalinin, D. J. Wesolowski, Y. Gogotsi and N. Balke, Nano Energy, 2015, 17, 27–35 CrossRef CAS.
- M. Mariano, O. Mashtalir, F. Q. Antonio, W. H. Ryu, B. Deng, F. Xia, Y. Gogotsi and A. D. Taylor, Nanoscale, 2016, 8, 16371–16378 RSC.
- A. T. Murdock, C. D. van Engers, J. Britton, V. Babenko, S. S. Meysami, H. Bishop, A. Crossley, A. A. Koos and N. Grobert, Carbon, 2017, 122, 207–216 CrossRef CAS.
- A. Castro-Muniz, H. Nishihara, T. Hirota, M. Ohwada, L. X. Li, T. Tsuda, S. Kuwabata, J. Maruyama and T. Kyotani, Chem. Commun., 2017, 53, 13348–13351 RSC.
- N. E. Richey, C. Haines, J. L. Tami and L. McElwee-White, Chem. Commun., 2017, 53, 7728–7731 RSC.
- D. E. Starr, Z. Liu, M. Havecker, A. Knop-Gericke and H. Bluhm, Chem. Soc. Rev., 2013, 42, 5833–5857 RSC.
- C. Ye, M. Q. Wang, G. Chen, Y. H. Deng, L. J. Li, H. Q. Luo and N. B. Li, J. Mater. Chem. A, 2017, 5, 7791–7795 RSC.
- J. Huang, H. Liu, B. Jin, M. Liu, Q. Zhang, L. Luo, S. Chu, S. Chu and R. Peng, Nanotechnology, 2017, 28, 275704 CrossRef PubMed.
- H. P. Steinruck, Phys. Chem. Chem. Phys., 2012, 14, 5010–5029 RSC.
- E. Mazzotta, S. Rella, A. Turco and C. Malitesta, RSC Adv., 2015, 5, 83164–83186 RSC.
- T. O. M. Samuels, A. W. Robertson, H. Kim, M. Pasta and J. H. Warner, J. Mater. Chem. A, 2017, 5, 10457–10469 RSC.
- D. Wu, C. Zhang, S. Xu, Y. Zhu, D. Xiong, P. Guo, Y. Wu, R. Qi, R. Huang, L. Wang and P. K. Chu, J. Mater. Chem. C, 2016, 4, 9251–9260 RSC.
- S. Chen, W. Xiong, Y. S. Zhou, Y. F. Lu and X. C. Zeng, Nanoscale, 2016, 8, 9746–9755 RSC.
- W. Xiong, Y. S. Zhou, W. J. Hou, T. Guillemet, J. F. Silvain, Y. Gao, M. Lahaye, E. Lebraud, S. Xu, X. W. Wang, D. A. Cullen, K. L. More, L. Jiang and Y. F. Lu, RSC Adv., 2015, 5, 99037–99043 RSC.
- W. Chen, J. Zhao, J. Zhang, L. Gu, Z. Yang, X. Li, H. Yu, X. Zhu, R. Yang, D. Shi, X. Lin, J. Guo, X. Bai and G. Zhang, J. Am. Chem. Soc., 2015, 137, 15632–15635 CrossRef CAS PubMed.
- X. Wang, H. Feng, Y. Wu and L. Jiao, J. Am. Chem. Soc., 2013, 135, 5304–5307 CrossRef CAS PubMed.
- S. Umrao, J. Jeon, S. M. Jeon, Y. J. Choi and S. Lee, Nanoscale, 2017, 9, 594–603 RSC.
- L. Zhang, C. Wang, X. L. Liu, T. Xu, M. Long, E. Liu, C. Pan, G. Su, J. Zeng, Y. Fu, Y. Wang, Z. Yan, A. Gao, K. Xu, P. H. Tan, L. Sun, Z. Wang, X. Cui and F. Miao, Nanoscale, 2017, 9, 19124–19130 RSC.
- B. Li, S. Yang, N. Huo, Y. Li, J. Yang, R. Li, C. Fan and F. Lu, RSC Adv., 2014, 4, 26407 RSC.
- J. Halim, K. M. Cook, M. Naguib, P. Eklund, Y. Gogotsi, J. Rosen and M. W. Barsoum, Appl. Surf. Sci., 2016, 362, 406–417 CrossRef CAS.
- S. A. Shah, T. Habib, H. Gao, P. Gao, W. Sun, M. J. Green and M. Radovic, Chem. Commun., 2016, 53, 400–403 RSC.
- W. Sun, S. A. Shah, Y. Chen, Z. Tan, H. Gao, T. Habib, M. Radovic and M. J. Green, J. Mater. Chem. A, 2017, 5, 21663–21668 RSC.
- Y. Wen, T. E. Rufford, X. Chen, N. Li, M. Lyu, L. Dai and L. Wang, Nano Energy, 2017, 38, 368–376 CrossRef CAS.
- H. B. Wang, J. F. Zhang, Y. P. Wu, H. J. Huang, G. Y. Li, X. Zhang and Z. Y. Wang, Appl. Surf. Sci., 2016, 384, 287–293 CrossRef CAS.
- S. Carenco, S. Moldovan, L. Roiban, I. Florea, D. Portehault, K. Valle, P. Belleville, C. Boissiere, L. Rozes, N. Mezailles, M. Drillon, C. Sanchez and O. Ersen, Nanoscale, 2016, 8, 1260–1279 RSC.
- R. Pal, A. K. Sikder, K. Saito, A. M. Funston and J. R. Bellare, Polym. Chem., 2017, 8, 6927–6937 RSC.
- D. B. Williams and C. B. Carter, in Transmission Electron Microscopy: A Textbook for Materials Science, ed. D. B. Williams and C. B. Carter, Springer US, Boston, MA, 2nd edn, 2009, pp. 679–698 DOI:10.1007/978-0-387-76501-3_37.
- C. Wiktor, M. Meledina, S. Turner, O. I. Lebedev and R. A. Fischer, J. Mater. Chem. A, 2017, 5, 14969–14989 RSC.
- W. Zhang and W. T. Zheng, Phys. Chem. Chem. Phys., 2015, 17, 14461–14469 RSC.
- H. Tan, S. Turner, E. Yucelen, J. Verbeeck and G. Van Tendeloo, Phys. Rev. Lett., 2011, 107, 107602 CrossRef PubMed.
- D. Magne, V. Mauchamp, S. Celerier, P. Chartier and T. Cabioc'h, Phys. Chem. Chem. Phys., 2016, 18, 30946–30953 RSC.
- J. R. Jinschek, Chem. Commun., 2014, 50, 2696–2706 RSC.
- X. H. Liu and J. Y. Huang, Energy Environ. Sci., 2011, 4, 3844–3860 RSC.
- R. J. Zou, Z. Cui, Q. Liu, G. Q. Guan, W. L. Zhang, G. J. He, J. M. Yang and J. Q. Hu, J. Mater. Chem. A, 2017, 5, 20072–20094 RSC.
- J. C. Yang, M. W. Small, R. V. Grieshaber and R. G. Nuzzo, Chem. Soc. Rev., 2012, 41, 8179–8194 RSC.
- K. Kimoto, T. Asaka, T. Nagai, M. Saito, Y. Matsui and K. Ishizuka, Nature, 2007, 450, 702–704 CrossRef CAS PubMed.
- K. Sohlberg, T. J. Pennycook, W. Zhou and S. J. Pennycook, Phys. Chem. Chem. Phys., 2015, 17, 3982–4006 RSC.
- K. Jarausch, P. Thomas, D. N. Leonard, R. Twesten and C. R. Booth, Ultramicroscopy, 2009, 109, 326–337 CrossRef CAS PubMed.
- G. Servanton, R. Pantel, M. Juhel and F. Bertin, Micron, 2009, 40, 543–551 CrossRef CAS PubMed.
- G. Möbus, R. C. Doole and B. J. Inkson, Ultramicroscopy, 2003, 96, 433–451 CrossRef.
- L. Zhao, H. Q. Ta, A. Dianat, A. Soni, A. Fediai, W. Yin, T. Gemming, B. Trzebicka, G. Cuniberti, Z. Liu, A. Bachmatiuk and M. H. Rummeli, Nano Lett., 2017, 17, 4725–4732 CrossRef CAS PubMed.
- J. Zhao, Q. Deng, A. Bachmatiuk, G. Sandeep, A. Popov, J. Eckert and M. H. Rummeli, Science, 2014, 343, 1228–1232 CrossRef CAS PubMed.
- M. A. Hope, A. C. Forse, K. J. Griffith, M. R. Lukatskaya, M. Ghidiu, Y. Gogotsi and C. P. Grey, Phys. Chem. Chem. Phys., 2016, 18, 5099–5102 RSC.
- V. Vales, P. Kovaricek, M. Fridrichova, X. Ji, X. Ling, J. Kong, M. S. Dresselhaus and M. Kalbac, 2D Mater., 2017, 4, 025087 CrossRef.
- J. Pang, R. G. Mendes, P. S. Wrobel, M. D. Wlodarski, H. Q. Ta, L. Zhao, L. Giebeler, B. Trzebicka, T. Gemming, L. Fu, Z. Liu, J. Eckert, A. Bachmatiuk and M. H. Rummeli, ACS Nano, 2017, 11, 1946–1956 CrossRef CAS PubMed.
- Y. Y. Shin, M. Lozada-Hidalgo, J. L. Sambricio, I. V. Grigorieva, A. K. Geim and C. Casiraghi, Appl. Phys. Lett., 2016, 108, 221907 CrossRef.
- I. A. Verzhbitskiy, M. D. Corato, A. Ruini, E. Molinari, A. Narita, Y. Hu, M. G. Schwab, M. Bruna, D. Yoon, S. Milana, X. Feng, K. Mullen, A. C. Ferrari, C. Casiraghi and D. Prezzi, Nano Lett., 2016, 16, 3442–3447 CrossRef CAS PubMed.
- J. B. Pang, A. Bachmatiuk, L. Fu, R. G. Mendes, M. Libera, D. Placha, G. S. Martynkova, B. Trzebicka, T. Gemming, J. Eckert and M. H. Rummeli, RSC Adv., 2015, 5, 60884–60891 RSC.
- M. Akhtar, G. Anderson, R. Zhao, A. Alruqi, J. E. Mroczkowska, G. Sumanasekera and J. B. Jasinski, npj 2D Mater. Appl., 2017, 1, 5 CrossRef.
- M. Baibarac, A. Nila and I. Baltog, RSC Adv., 2016, 6, 58003–58009 RSC.
- S. K. Pandey, N. Izquierdo, R. Liptak and S. A. Campbell, RSC Adv., 2017, 7, 46201–46207 RSC.
- F. Alsaffar, S. Alodan, A. Alrasheed, A. Alhussain, N. Alrubaiq, A. Abbas and M. R. Amer, Sci. Rep., 2017, 7, 44540 CrossRef CAS PubMed.
- J. Quereda, J. J. Palacios, N. Agrait, A. Castellanos-Gomez and G. Rubio-Bollinger, 2D Mater., 2017, 4, 021006 CrossRef.
- W. T. Su, Y. C. Wang, W. W. Wu, H. Y. Qin, K. X. Song, X. W. Huang, L. J. Zhang and D. Q. Chen, J. Mater. Chem. C, 2017, 5, 6352–6358 RSC.
- E. Mercado, A. Goodyear, J. Moffat, M. Cooke and R. S. Sundaram, J. Phys. D: Appl. Phys., 2017, 50, 184005 CrossRef.
- A. Bera, D. V. S. Muthu and A. K. Sood, J. Raman Spectrosc., 2018, 49, 100–105 CrossRef CAS.
- A. Bera and A. K. Sood, in MoS2: Materials, Physics, and Devices, ed. Z. M. Wang, Springer International Publishing, Cham, 1st edn, 2014, pp. 155–215 DOI:10.1007/978-3-319-02850-7_7.
- A. Zafar, H. Y. Nan, Z. Zafar, Z. T. Wu, J. Jiang, Y. M. You and Z. H. Ni, Nano Res., 2017, 10, 1608–1617 CrossRef CAS.
- B. R. Carvalho, Y. Wang, S. Mignuzzi, D. Roy, M. Terrones, C. Fantini, V. H. Crespi, L. M. Malard and M. A. Pimenta, Nat. Commun., 2017, 8, 14670 CrossRef PubMed.
- A. V. Kolobov and J. Tominaga, in Two-Dimensional Transition-Metal Dichalcogenides, ed. A. V. Kolobov and J. Tominaga, Springer International Publishing, Cham, 1st edn, 2016, pp. 227–294 DOI:10.1007/978-3-319-31450-1_7.
- X. Zhang, X. F. Qiao, W. Shi, J. B. Wu, D. S. Jiang and P. H. Tan, Chem. Soc. Rev., 2015, 44, 2757–2785 RSC.
- R. Saito, Y. Tatsumi, S. Huang, X. Ling and M. S. Dresselhaus, J. Phys.: Condens. Matter, 2016, 28, 353002 CrossRef CAS PubMed.
- T. Hu, J. Wang, H. Zhang, Z. Li, M. Hu and X. Wang, Phys. Chem. Chem. Phys., 2015, 17, 9997–10003 RSC.
- J. Bartelmess, S. J. Quinn and S. Giordani, Chem. Soc. Rev., 2015, 44, 4672–4698 RSC.
- X. Zhang, Q. H. Tan, J. B. Wu, W. Shi and P. H. Tan, Nanoscale, 2016, 8, 6435–6450 RSC.
- A. Jorio and L. G. Cancado, Phys. Chem. Chem. Phys., 2012, 14, 15246–15256 RSC.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
- A. V. Ewing and S. G. Kazarian, Analyst, 2017, 142, 257–272 RSC.
- M. Manley, Chem. Soc. Rev., 2014, 43, 8200–8214 RSC.
- B. L. Mojet, S. D. Ebbesen and L. Lefferts, Chem. Soc. Rev., 2010, 39, 4643–4655 RSC.
- M. K. Kuimova, W. Z. Alsindi, J. Dyer, D. C. Grills, O. S. Jina, P. Matousek, A. W. Parker, P. Portius, X. Z. Sun, M. Towrie, C. Wilson, J. X. Yang and M. W. George, Dalton Trans., 2003, 3996–4006, 10.1039/b306303h.
- K. Olszowska, J. Pang, P. S. Wrobel, L. Zhao, H. Q. Ta, Z. Liu, B. Trzebicka, A. Bachmatiuk and M. H. Rummeli, Synth. Met., 2017, 234, 53–85 CrossRef CAS.
- O. H. Wheeler, Chem. Rev., 1959, 59, 629–666 CrossRef CAS.
- M. Zheng, R. Guo, Z. Liu, B. Wang, L. Meng, F. Li, T. Li and Y. Luo, J. Alloys Compd., 2018, 735, 1262–1270 CrossRef CAS.
- Y. Wang, H. Dou, J. Wang, B. Ding, Y. L. Xu, Z. Chang and X. D. Hao, J. Power Sources, 2016, 327, 221–228 CrossRef CAS.
- B. Tyagi, C. D. Chudasama and R. V. Jasra, Spectrochim. Acta, Part A, 2006, 64, 273–278 CrossRef PubMed.
- M. Beekes, P. Lasch and D. Naumann, Vet. Microbiol., 2007, 123, 305–319 CrossRef CAS PubMed.
- V. Kanokkantapong, T. F. Marhaba, B. Panyapinyophol and P. Pavasant, J. Hazard. Mater., 2006, 136, 188–196 CrossRef CAS PubMed.
- G. A. Olah, I. Bucsi, C. Lambert, R. Aniszfeld, N. J. Trivedi, D. K. Sensharma and G. K. S. Prakash, J. Am. Chem. Soc., 1991, 113, 9385–9387 CrossRef CAS.
- H. Vigué, P. Quintard, T. Merle-Méjean and V. Lorenzelli, J. Eur. Ceram. Soc., 1998, 18, 305–309 CrossRef.
- Q. Xue, H. Zhang, M. Zhu, Z. Pei, H. Li, Z. Wang, Y. Huang, Y. Huang, Q. Deng, J. Zhou, S. Du, Q. Huang and C. Zhi, Adv. Mater., 2017, 29, 1604847 CrossRef PubMed.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- A. D. Dillon, M. J. Ghidiu, A. L. Krick, J. Griggs, S. J. May, Y. Gogotsi, M. W. Barsoum and A. T. Fafarman, Adv. Funct. Mater., 2016, 26, 4162–4168 CrossRef CAS.
- R. M. Pasquarelli, D. S. Ginley and R. O'Hayre, Chem. Soc. Rev., 2011, 40, 5406–5441 RSC.
- S. B. Yang, B. S. Kong, D. H. Jung, Y. K. Baek, C. S. Han, S. K. Oh and H. T. Jung, Nanoscale, 2011, 3, 1361–1373 RSC.
- K. Hantanasirisakul, M. Q. Zhao, P. Urbankowski, J. Halim, B. Anasori, S. Kota, C. E. Ren, M. W. Barsoum and Y. Gogotsi, Adv. Electron. Mater., 2016, 2, 1600050 CrossRef.
- D. Wang, F. You and G.-H. Hu, in Graphene-Based Polymer Nanocomposites in Electronics, ed. K. K. Sadasivuni, D. Ponnamma, J. Kim and S. Thomas, Springer International Publishing, Cham, 2015, pp. 49–65 DOI:10.1007/978-3-319-13875-6_3.
- V. N. Borysiuk, V. N. Mochalin and Y. Gogotsi, Nanotechnology, 2015, 26, 265705 CrossRef PubMed.
- H. Q. Ta, L. Zhao, D. Pohl, J. B. Pang, B. Trzebicka, B. Rellinghaus, D. Pribat, T. Gemming, Z. F. Liu, A. Bachmatiuk and M. H. Rummeli, Crystals, 2016, 6, 100 CrossRef.
- Z. Guo, J. Zhou, C. Si and Z. Sun, Phys. Chem. Chem. Phys., 2015, 17, 15348–15354 RSC.
- Z. Ling, C. E. Ren, M. Q. Zhao, J. Yang, J. M. Giammarco, J. Qiu, M. W. Barsoum and Y. Gogotsi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16676–16681 CrossRef CAS PubMed.
- H. Sadeghzadeh and A. Morsali, CrystEngComm, 2010, 12, 370–372 RSC.
- J. Yan, C. E. Ren, K. Maleski, C. B. Hatter, B. Anasori, P. Urbankowski, A. Sarycheva and Y. Gogotsi, Adv. Funct. Mater., 2017, 27, 1701264 CrossRef.
- L. Wang, W. Tao, L. Yuan, Z. Liu, Q. Huang, Z. Chai, J. K. Gibson and W. Shi, Chem. Commun., 2017, 53, 12084–12087 RSC.
- K. Maleski, V. N. Mochalin and Y. Gogotsi, Chem. Mater., 2017, 29, 1632–1640 CrossRef CAS.
- Q. Gao, J. Come, M. Naguib, S. Jesse, Y. Gogotsi and N. Balke, Faraday Discuss., 2017, 199, 393–403 RSC.
- V. M. Hong, Ng, H. Huang, K. Zhou, P. S. Lee, W. Que, J. Z. Xu and L. B. Kong, J. Mater. Chem. A, 2017, 5, 3039–3068 RSC.
- S. K. Xu, G. D. Wei, J. Z. Li, W. Han and Y. Gogotsi, J. Mater. Chem. A, 2017, 5, 17442–17451 RSC.
- J. Halim, M. R. Lukatskaya, K. M. Cook, J. Lu, C. R. Smith, L. A. Naslund, S. J. May, L. Hultman, Y. Gogotsi, P. Eklund and M. W. Barsoum, Chem. Mater., 2014, 26, 2374–2381 CrossRef CAS PubMed.
- R. H. Friend and A. D. Yoffe, Adv. Phys., 1987, 36, 1–94 CrossRef CAS.
- A. Lerf, Dalton Trans., 2014, 43, 10276–10291 RSC.
- M. J. Crane, M. B. Lim, X. Zhou and P. J. Pauzauskie, Microsyst. Nanoeng., 2017, 3, 17032 CrossRef.
- R. Zhang, J. Waters, A. K. Geim and I. V. Grigorieva, Nat. Commun., 2017, 8, 15036 CrossRef CAS PubMed.
- A. U. Liyanage and M. M. Lerner, RSC Adv., 2014, 4, 47121–47128 RSC.
- L. Bernard, M. Mckelvy, W. Glaunsinger and P. Colombet, Solid State Ionics, 1985, 15, 301–310 CrossRef CAS.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall'Agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, Nat. Commun., 2013, 4, 1716 CrossRef PubMed.
- O. Mashtalir, M. R. Lukatskaya, M. Q. Zhao, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2015, 27, 3501–3506 CrossRef CAS PubMed.
- M. Naguib, R. R. Unocic, B. L. Armstrong and J. Nanda, Dalton Trans., 2015, 44, 9353–9358 RSC.
- Y. Xie, M. Naguib, V. N. Mochalin, M. W. Barsoum, Y. Gogotsi, X. Yu, K. W. Nam, X. Q. Yang, A. I. Kolesnikov and P. R. Kent, J. Am. Chem. Soc., 2014, 136, 6385–6394 CrossRef CAS PubMed.
- O. Mashtalir, K. M. Cook, V. N. Mochalin, M. Crowe, M. W. Barsoum and Y. Gogotsi, J. Mater. Chem. A, 2014, 2, 14334–14338 RSC.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
- X. M. Feng, N. N. Chen, Y. Zhang, Z. Z. Yan, X. F. Liu, Y. W. Ma, Q. M. Shen, L. H. Wang and W. Huang, J. Mater. Chem. A, 2014, 2, 9178–9184 RSC.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- L. Li, A. R. Raji and J. M. Tour, Adv. Mater., 2013, 25, 6298–6302 CrossRef CAS PubMed.
- X. M. Feng, Z. Z. Yan, N. N. Chen, Y. Zhang, Y. W. Ma, X. F. Liu, Q. L. Fan, L. H. Wang and W. Huang, J. Mater. Chem. A, 2013, 1, 12818–12825 RSC.
- O. Ghodbane, F. Ataherian, N. L. Wu and F. Favier, J. Power Sources, 2012, 206, 454–462 CrossRef CAS.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- C. C. Lai, R. Meshkian, M. Dahlqvist, J. Lu, L. A. Naslund, O. Rivin, E. N. Caspi, O. Ozeri, L. Hultman, P. Eklund, M. W. Barsoum and J. Rosen, Acta Mater., 2015, 99, 157–164 CrossRef CAS.
- C. Hu, C. C. Lai, Q. Tao, J. Lu, J. Halim, L. Sun, J. Zhang, J. Yang, B. Anasori, J. Wang, Y. Sakka, L. Hultman, P. Eklund, J. Rosen and M. W. Barsoum, Chem. Commun., 2015, 51, 6560–6563 RSC.
- C. C. Lai, H. Fashandi, J. Lu, J. Palisaitis, P. O. A. Persson, L. Hultman, P. Eklund and J. Rosen, Nanoscale, 2017, 9, 17681–17687 RSC.
- R. Meshkian, L. A. Naslund, J. Halim, J. Lu, M. W. Barsoum and J. Rosen, Scr. Mater., 2015, 108, 147–150 CrossRef CAS.
- Z. Liu, C. Xu, N. Kang, L. Wang, Y. Jiang, J. Du, Y. Liu, X. L. Ma, H. M. Cheng and W. Ren, Nano Lett., 2016, 16, 4243–4250 CrossRef CAS PubMed.
- L. Wang, C. Xu, Z. Liu, L. Chen, X. Ma, H. M. Cheng, W. Ren and N. Kang, ACS Nano, 2016, 10, 4504–4510 CrossRef CAS PubMed.
- C. Xu, L. Wang, Z. Liu, L. Chen, J. Guo, N. Kang, X. L. Ma, H. M. Cheng and W. Ren, Nat. Mater., 2015, 14, 1135–1141 CrossRef CAS PubMed.
- J. Jia, T. Xiong, L. Zhao, F. Wang, H. Liu, R. Hu, J. Zhou, W. Zhou and S. Chen, ACS Nano, 2017, 11, 12509–12518 CrossRef CAS PubMed.
- H. Kim, G. Yoon, K. Lim and K. Kang, Chem. Commun., 2016, 52, 12618–12621 RSC.
- Y. Zhao and A. Manthiram, Chem. Commun., 2015, 51, 13205–13208 RSC.
- Z. Q. Zhu, F. Y. Cheng, Z. Hu, Z. Q. Niu and J. Chen, J. Power Sources, 2015, 293, 626–634 CrossRef CAS.
- R. Zhang, F. Mizuno and C. Ling, Chem. Commun., 2015, 51, 1108–1111 RSC.
- P. Sood, K. C. Kim and S. S. Jang, J. Energy Chem., 2018, 27, 528–534 CrossRef.
- P. Bairi, R. G. Shrestha, J. P. Hill, T. Nishimura, K. Ariga and L. K. Shrestha, J. Mater. Chem. A, 2016, 4, 13899–13906 RSC.
- Y. Fu, Y. S. Su and A. Manthiram, Angew. Chem., Int. Ed., 2013, 52, 6930–6935 CrossRef CAS PubMed.
- D. Kong, X. Li, Y. Zhang, X. Hai, B. Wang, X. Qiu, Q. Song, Q.-H. Yang and L. Zhi, Energy Environ. Sci., 2016, 9, 906–911 RSC.
- M. Xie, X. Sun, H. T. Sun, T. Porcelli, S. M. George, Y. Zhou and J. Lian, J. Mater. Chem. A, 2016, 4, 537–544 RSC.
- T. Kowase, K. Hori, K. Hasegawa, T. Momma and S. Noda, J. Power Sources, 2017, 363, 450–459 CrossRef CAS.
- M. P. Yu, J. S. Ma, H. Q. Song, A. J. Wang, F. Y. Tian, Y. S. Wang, H. Qiu and R. M. Wang, Energy Environ. Sci., 2016, 9, 1495–1503 RSC.
- D. D. Li, L. Zhang, H. B. Chen, J. Wang, L. X. Ding, S. Q. Wang, P. J. Ashman and H. H. Wang, J. Mater. Chem. A, 2016, 4, 8630–8635 RSC.
- V. Ivanistsev, T. Mendez-Morales, R. M. Lynden-Bell, O. Cabeza, L. J. Gallego, L. M. Varela and M. V. Fedorov, Phys. Chem. Chem. Phys., 2016, 18, 1302–1310 RSC.
- K. Y. Zhou, G. Y. Chen, J. A. Liu, Z. P. Zhang, P. Sun, W. Z. Zhang, F. Niu, W. X. Zhang and J. C. Liang, RSC Adv., 2016, 6, 90069–90075 RSC.
- X. Wang, Q. Weng, Y. Yang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 4042–4073 RSC.
- Y. P. Gao, X. Wu, K. J. Huang, L. L. Xing, Y. Y. Zhang and L. Liu, CrystEngComm, 2017, 19, 404–418 RSC.
- S. K. Balasingam, J. S. Lee and Y. Jun, Dalton Trans., 2016, 45, 9646–9653 RSC.
- A. Eftekhari, J. Mater. Chem. A, 2017, 5, 18299–18325 RSC.
- Z. Sofer, D. Bousa, J. Luxa, V. Mazanek and M. Pumera, Chem. Commun., 2016, 52, 1563–1566 RSC.
- J. X. Zhao, Y. G. Yang, R. S. Katiyar and Z. F. Chen, J. Mater. Chem. A, 2016, 4, 6124–6130 RSC.
- Q. Xia, W. Li, Z. Miao, S. Chou and H. Liu, Nano Res., 2017, 10, 4055–4081 CrossRef CAS.
- M. Qiu, Z. T. Sun, D. K. Sang, X. G. Han, H. Zhang and C. M. Niu, Nanoscale, 2017, 9, 13384–13403 RSC.
- J. Liu, A. X. Wei, M. Chen and X. Xia, J. Mater. Chem. A, 2018, 6, 3857–3863 RSC.
- L. Kong, C. C. Xie, H. Gu, C. P. Wang, X. Zhou, J. Liu, Z. Zhou, Z. Y. Li, J. Zhu and X. H. Bu, Small, 2018, 14, e1800639 CrossRef PubMed.
- X. Xia, J. Zhan, Y. Zhong, X. Wang, J. Tu and H. J. Fan, Small, 2017, 13, 1602742 CrossRef PubMed.
- D. Xie, X. Xia, Y. Zhong, Y. Wang, D. Wang, X. Wang and J. Tu, Adv. Energy Mater., 2017, 7, 1601804 CrossRef.
- Y. Zhong, X. Xia, S. Deng, J. Zhan, R. Fang, Y. Xia, X. Wang, Q. Zhang and J. Tu, Adv. Energy Mater., 2018, 8, 1701110 CrossRef.
- J. Zhan, S. Deng, Y. Zhong, Y. Wang, X. Wang, Y. Yu, X. Xia and J. Tu, Nano Energy, 2018, 44, 265–271 CrossRef CAS.
- M. Yu, S. Zhou, Z. Wang, J. Zhao and J. Qiu, Nano Energy, 2018, 44, 181–190 CrossRef CAS.
- C. J. Zhang, M. P. Kremer, A. Seral-Ascaso, S.-H. Park, N. McEvoy, B. Anasori, Y. Gogotsi and V. Nicolosi, Adv. Funct. Mater., 2018, 28, 1705506 CrossRef.
- X. Zhang, Z. Zhang and Z. Zhou, J. Energy Chem., 2018, 27, 73–85 CrossRef.
- S. Sarkar, P. Howli, B. Das, N. S. Das, M. Samanta, G. C. Das and K. K. Chattopadhyay, ACS Appl. Mater. Interfaces, 2017, 9, 22652–22664 CrossRef CAS PubMed.
- B. K. Deka, A. Hazarika, J. Kim, Y. B. Park and H. W. Park, Int. J. Energy Res., 2017, 41, 1397–1411 CrossRef.
- S. Bag and C. R. Raj, J. Mater. Chem. A, 2016, 4, 587–595 RSC.
- M. Jana, J. S. Kumar, P. Khanra, P. Samanta, H. Koo, N. C. Murmu and T. Kuila, J. Power Sources, 2016, 303, 222–233 CrossRef CAS.
- Y. Y. Li, Z. M. Chen, J. N. Zhang and Q. Xu, RSC Adv., 2016, 6, 9180–9185 RSC.
- T. M. Masikhwa, M. J. Madito, D. Y. Momodu, J. K. Dangbegnon, O. Guellati, A. Harat, M. Guerioune, F. Barzegar and N. Manyala, RSC Adv., 2016, 6, 46723–46732 RSC.
- A. Kafy, A. Akther, L. D. Zhai, H. C. Kim and J. Kim, Synth. Met., 2017, 223, 94–100 CrossRef CAS.
- W. Long, B. Fang, A. Ignaszak, Z. Wu, Y. J. Wang and D. Wilkinson, Chem. Soc. Rev., 2017, 46, 7176–7190 RSC.
- H. S. Zhou, Energy Environ. Sci., 2013, 6, 2256 RSC.
- Y. F. Deng, C. C. Fang and G. H. Chen, J. Power Sources, 2016, 304, 81–101 CrossRef CAS.
- F. Hu and T. Song, RSC Adv., 2017, 7, 54203–54212 RSC.
- M. Cai, R. A. Outlaw, R. A. Quinlan, D. Premathilake, S. M. Butler and J. R. Miller, ACS Nano, 2014, 8, 5873–5882 CrossRef CAS PubMed.
- Y. H. Lu, F. Zhang, T. F. Zhang, K. Leng, L. Zhang, X. Yang, Y. F. Ma, Y. Huang, M. J. Zhang and Y. S. Chen, Carbon, 2013, 63, 508–516 CrossRef CAS.
- L. Zhang, X. Yang, F. Zhang, G. Long, T. Zhang, K. Leng, Y. Zhang, Y. Huang, Y. Ma, M. Zhang and Y. Chen, J. Am. Chem. Soc., 2013, 135, 5921–5929 CrossRef CAS PubMed.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103–13107 CrossRef CAS.
- L. Zhang, F. Zhang, X. Yang, K. Leng, Y. Huang and Y. Chen, Small, 2013, 9, 1342–1347 CrossRef CAS PubMed.
- W. Xiong, Y. Gao, X. Wu, X. Hu, D. Lan, Y. Chen, X. Pu, Y. Zeng, J. Su and Z. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 19416–19423 CrossRef CAS PubMed.
- Z. L. Jian, V. Raju, Z. F. Li, Z. Y. Xing, Y. S. Hu and X. L. Ji, Adv. Funct. Mater., 2015, 25, 5778–5785 CrossRef CAS.
- B. P. Bastakoti, H. S. Huang, L. C. Chen, K. C. Wu and Y. Yamauchi, Chem. Commun., 2012, 48, 9150–9152 RSC.
- Y. Wang, S. F. Yu, C. Y. Sun, T. J. Zhu and H. Y. Yang, J. Mater. Chem., 2012, 22, 17584–17588 RSC.
- H. Chen, J. Jiang, L. Zhang, H. Wan, T. Qi and D. Xia, Nanoscale, 2013, 5, 8879–8883 RSC.
- J. Li, M. Yang, J. Wei and Z. Zhou, Nanoscale, 2012, 4, 4498–4503 RSC.
- M. Yang, J. X. Li, H. H. Li, L. W. Su, J. P. Wei and Z. Zhou, Phys. Chem. Chem. Phys., 2012, 14, 11048–11052 RSC.
- Z. S. Wu, W. Ren, D. W. Wang, F. Li, B. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS PubMed.
- Y. Huang, J. Liang and Y. Chen, Small, 2012, 8, 1805–1834 CrossRef CAS PubMed.
- F. Zhang, T. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 1623 RSC.
- K. Leng, F. Zhang, L. Zhang, T. F. Zhang, Y. P. Wu, Y. H. Lu, Y. Huang and Y. S. Chen, Nano Res., 2013, 6, 581–592 CrossRef CAS.
- A. Burke, Electrochim. Acta, 2007, 53, 1083–1091 CrossRef CAS.
- J. H. Warner, F. Schäffel, A. Bachmatiuk and M. H. Rümmeli, Graphene Fundamentals and Emergent Applications, Elsevier, Waltham, USA, 1st edn, 2013, pp. 333–437 DOI:10.1016/B978-0-12-394593-8.00006-0.
- G. K. Ramesha and S. Sampath, in Graphene Synthesis, Properties and Phenomena, ed. C. N. R. Rao and A. K. Sood, Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 269–301 DOI:10.1002/9783527651122.ch9.
- Y. Q. Sun, Q. O. Wu, Y. X. Xu, H. Bai, C. Li and G. Q. Shi, J. Mater. Chem., 2011, 21, 7154–7160 RSC.
- Z. S. Wu, K. Parvez, S. Li, S. Yang, Z. Liu, S. Liu, X. Feng and K. Mullen, Adv. Mater., 2015, 27, 4054–4061 CrossRef CAS PubMed.
- Z. Wang, P. Tammela, M. Stromme and L. Nyholm, Nanoscale, 2015, 7, 3418–3423 RSC.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L. C. Qin, Phys. Chem. Chem. Phys., 2011, 13, 17615–17624 RSC.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- A. P. Yu, I. Roes, A. Davies and Z. W. Chen, Appl. Phys. Lett., 2010, 96, 253105 CrossRef.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS PubMed.
- Q. Jiang, N. Kurra, M. Alhabeb, Y. Gogotsi and H. N. Alshareef, Adv. Energy Mater., 2018, 8, 1703043 CrossRef.
- C. Couly, M. Alhabeb, K. L. Van Aken, N. Kurra, L. Gomes, A. M. Navarro-Suárez, B. Anasori, H. N. Alshareef and Y. Gogotsi, Adv. Electron. Mater., 2017, 1700339, DOI:10.1002/aelm.201700339.
- Y. P. Tian, C. H. Yang, W. X. Que, X. B. Liu, X. T. Yin and L. B. Kong, J. Power Sources, 2017, 359, 332–339 CrossRef CAS.
- K. Zhu, H. Zhang, K. Ye, W. Zhao, J. Yan, K. Cheng, G. Wang, B. Yang and D. Cao, ChemElectroChem, 2017, 4, 3018–3025 CrossRef CAS.
- R. B. Rakhi, B. Ahmed, D. Anjum and H. N. Alshareef, ACS Appl. Mater. Interfaces, 2016, 8, 18806–18814 CrossRef CAS PubMed.
- J. F. Zhu, X. Lu and L. Wang, RSC Adv., 2016, 6, 98506–98513 RSC.
- J. F. Zhu, Y. Tang, C. H. Yang, F. Wang and M. J. Cao, J. Electrochem. Soc., 2016, 163, A785–A791 CrossRef CAS.
- W. Zheng, P. Zhang, W. Tian, Y. Wang, Y. Zhang, J. Chen and Z. Sun, Mater. Lett., 2017, 209, 122–125 CrossRef CAS.
- F. Wang, M. J. Cao, Y. Qin, J. F. Zhu, L. Wang and Y. Tang, RSC Adv., 2016, 6, 88934–88942 RSC.
- L. Qin, Q. Tao, A. El Ghazaly, J. Fernandez-Rodriguez, P. O. Å. Persson, J. Rosen and F. Zhang, Adv. Funct. Mater., 2017, 1703808, DOI:10.1002/adfm.201703808.
- M. Boota, B. Anasori, C. Voigt, M. Q. Zhao, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2016, 28, 1517–1522 CrossRef CAS PubMed.
- M. S. Zhu, Y. Huang, Q. H. Deng, J. Zhou, Z. X. Pei, Q. Xue, Y. Huang, Z. F. Wang, H. F. Li, Q. Huang and C. Y. Zhi, Adv. Energy Mater., 2016, 6, 1600969 CrossRef.
- M. Boota, M. Pasini, F. Galeotti, W. Porzio, M. Q. Zhao, J. Halim and Y. Gogotsi, Chem. Mater., 2017, 29, 2731–2738 CrossRef CAS.
- M. Hu, Z. Li, G. Li, T. Hu, C. Zhang and X. Wang, Adv. Mater. Technol., 2017, 2, 1700143 CrossRef.
- Y. Ren, J. Zhu, L. Wang, H. Liu, Y. Liu, W. Wu and F. Wang, Mater. Lett., 2018, 214, 84–87 CrossRef CAS.
- S. Y. Lin and X. T. Zhang, J. Power Sources, 2015, 294, 354–359 CrossRef CAS.
- J. Zhang, S. Seyedin, Z. Gu, W. Yang, X. Wang and J. M. Razal, Nanoscale, 2017, 9, 18604–18608 RSC.
- C. H. Yang, W. X. Que, X. T. Yin, Y. P. Tian, Y. W. Yang and M. D. Que, Electrochim. Acta, 2017, 225, 416–424 CrossRef CAS.
- S. Seyedin, E. R. S. Yanza and J. M. Razal, J. Mater. Chem. A, 2017, 5, 24076–24082 RSC.
- L. Li, F. Wang, J. Zhu and W. Wu, Dalton Trans., 2017, 46, 14880–14887 RSC.
- M. Hu, Z. Li, H. Zhang, T. Hu, C. Zhang, Z. Wu and X. Wang, Chem. Commun., 2015, 51, 13531–13533 RSC.
- Y. P. Tian, C. H. Yang, W. X. Que, Y. C. He, X. B. Liu, Y. Y. Luo, X. T. Yin and L. B. Kong, J. Power Sources, 2017, 369, 78–86 CrossRef CAS.
- Q. Y. Yang, Z. Xu, B. Fang, T. Q. Huang, S. Y. Cai, H. Chen, Y. J. Liu, K. Gopalsamy, W. W. Gao and C. Gao, J. Mater. Chem. A, 2017, 5, 22113–22119 RSC.
- A. M. Navarro-Suárez, K. L. Van Aken, T. Mathis, T. Makaryan, J. Yan, J. Carretero-González, T. Rojo and Y. Gogotsi, Electrochim. Acta, 2018, 259, 752–761 CrossRef.
- M. Q. Zhao, C. E. Ren, Z. Ling, M. R. Lukatskaya, C. Zhang, K. L. Van Aken, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2015, 27, 339–345 CrossRef CAS PubMed.
- X. Q. Xie, M. Q. Zhao, B. Anasori, K. Maleski, C. E. Ren, J. W. Li, B. W. Byles, E. Pomerantseva, G. X. Wang and Y. Gogotsi, Nano Energy, 2016, 26, 513–523 CrossRef CAS.
- Y. Dall’Agnese, P. Rozier, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Power Sources, 2016, 306, 510–515 CrossRef.
- P. T. Yan, R. J. Zhang, J. Jia, C. Wu, A. G. Zhou, J. Xu and X. S. Zhang, J. Power Sources, 2015, 284, 38–43 CrossRef CAS.
- Q. Fu, X. Wang, N. Zhang, J. Wen, L. Li, H. Gao and X. Zhang, J. Colloid Interface Sci., 2018, 511, 128–134 CrossRef CAS PubMed.
- H. Y. Li, Y. Hou, F. X. Wang, M. R. Lohe, X. D. Zhuang, L. Niu and X. L. Feng, Adv. Energy Mater., 2017, 7, 1601847 CrossRef.
- M. Hu, T. Hu, R. Cheng, J. Yang, C. Cui, C. Zhang and X. Wang, J. Energy Chem., 2018, 27, 161–166 CrossRef.
- C. Zhao, Q. Wang, H. Zhang, S. Passerini and X. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 15661–15667 CrossRef CAS PubMed.
- M. R. Lukatskaya, S. Kota, Z. Lin, M.-Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P.-L. Taberna, M. W. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 6, 17105 CrossRef.
- J. Luo, W. Zhang, H. Yuan, C. Jin, L. Zhang, H. Huang, C. Liang, Y. Xia, J. Zhang, Y. Gan and X. Tao, ACS Nano, 2017, 11, 2459–2469 CrossRef CAS PubMed.
- S. K. Xu, G. D. Wei, J. Z. Li, Y. Ji, N. Klyui, V. Izotov and W. Han, Chem. Eng. J., 2017, 317, 1026–1036 CrossRef CAS.
- Y. Tang, J. F. Zhu, C. H. Yang and F. Wang, J. Alloys Compd., 2016, 685, 194–201 CrossRef CAS.
- H. B. Wang, J. F. Zhang, Y. P. Wu, H. J. Huang and Q. G. Jiang, Electrochem. Commun., 2017, 81, 48–51 CrossRef CAS.
- Y. Tang, J. F. Zhu, C. H. Yang and F. Wang, J. Electrochem. Soc., 2016, 163, A1975–A1982 CrossRef CAS.
- Z. F. Lin, D. Barbara, P. L. Taberna, K. L. Van Aken, B. Anasori, Y. Gogotsi and P. Simon, J. Power Sources, 2016, 326, 575–579 CrossRef CAS.
- C. J. Zhang, B. Anasori, A. Seral-Ascaso, S. H. Park, N. McEvoy, A. Shmeliov, G. S. Duesberg, J. N. Coleman, Y. Gogotsi and V. Nicolosi, Adv. Mater., 2017, 29, 1702678 CrossRef PubMed.
- Y. Dall'Agnese, M. R. Lukatskaya, K. M. Cook, P. L. Taberna, Y. Gogotsi and P. Simon, Electrochem. Commun., 2014, 48, 118–122 CrossRef.
- L. Li, M. Y. Zhang, X. T. Zhang and Z. G. Zhang, J. Power Sources, 2017, 364, 234–241 CrossRef CAS.
- W. Y. Wu, S. Y. Lin, T. T. Chen, L. Li, Y. Pan, M. Y. Zhang, L. L. Wu, H. Gao and X. T. Zhang, J. Alloys Compd., 2017, 729, 1165–1171 CrossRef CAS.
- Q. X. Xia, J. J. Fu, J. M. Yun, R. S. Mane and K. H. Kim, RSC Adv., 2017, 7, 11000–11011 RSC.
- Y. Y. Peng, B. Akuzum, N. Kurra, M. Q. Zhao, M. Alhabe, B. Anasori, E. C. Kumbur, H. N. Alshareef, M. D. Ger and Y. Gogotsi, Energy Environ. Sci., 2016, 9, 2847–2854 RSC.
- N. Kurra, B. Ahmed, Y. Gogotsi and H. N. Alshareef, Adv. Energy Mater., 2016, 6, 1601372 CrossRef.
- H. B. Hu and T. Hua, J. Mater. Chem. A, 2017, 5, 19639–19648 RSC.
- K. Krishnamoorthy, P. Pazhamalai, S. Sahoo and S. J. Kim, J. Mater. Chem. A, 2017, 5, 5726–5736 RSC.
- Z. S. Wu, D. W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H. M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- S. Cho, J. Kim, Y. Jo, A. T. A. Ahmed, H. S. Chavan, H. Woo, A. I. Inamdar, J. L. Gunjakar, S. M. Pawar, Y. Park, H. Kim and H. Im, J. Alloys Compd., 2017, 725, 108–114 CrossRef CAS.
- I. Ryu, M. Yang, H. Kwon, H. K. Park, Y. R. Do, S. B. Lee and S. Yim, Langmuir, 2014, 30, 1704–1709 CrossRef CAS PubMed.
- C. C. Hu, K. H. Chang, M. C. Lin and Y. T. Wu, Nano Lett., 2006, 6, 2690–2695 CrossRef CAS PubMed.
- H. Ma, D. Kong, Y. Xu, X. Xie, Y. Tao, Z. Xiao, W. Lv, H. D. Jang, J. Huang and Q. H. Yang, Small, 2017, 13, 1701026 CrossRef PubMed.
- M. W. Barsoum, Prog. Solid State Chem., 2000, 28, 201–281 CrossRef CAS.
- J. Li, X. T. Yuan, C. Lin, Y. Q. Yang, L. Xu, X. Du, J. L. Xie, J. H. Lin and J. L. Sun, Adv. Energy Mater., 2017, 7, 1602725 CrossRef.
- L. Bai, H. Yin, L. Wu and X. Zhang, Comput. Mater. Sci., 2018, 143, 225–231 CrossRef CAS.
- Y. Xin and Y. X. Yu, Mater. Des., 2017, 130, 512–520 CrossRef CAS.
- B. Xu, S. Yue, Z. Sui, X. Zhang, S. Hou, G. Cao and Y. Yang, Energy Environ. Sci., 2011, 4, 2826–2830 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- J. Han, D. Kim and M. Sunwoo, J. Power Sources, 2009, 188, 606–612 CrossRef CAS.
- P. Kurzweil, J. Power Sources, 2010, 195, 4424–4434 CrossRef CAS.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112 RSC.
- Y. Nishi, J. Power Sources, 2001, 100, 101–106 CrossRef CAS.
- T. Ohzuku and R. J. Brodd, J. Power Sources, 2007, 174, 449–456 CrossRef CAS.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- C. Zhan, M. Naguib, M. Lukatskaya, P. R. C. Kent, Y. Gogotsi and D. E. Jiang, J. Phys. Chem. Lett., 2018, 9, 1223–1228 CrossRef CAS PubMed.
- N. Shpigel, S. Sigalov, M. D. Levi, T. Mathis, L. Daikhin, A. Janes, E. Lust, Y. Gogotsi and D. Aurbach, Joule, 2018, 2, 988–1003 CrossRef CAS.
- C. Chen, M. Boota, P. Urbankowski, B. Anasori, L. Miao, J. Jiang and Y. Gogotsi, J. Mater. Chem. A, 2018, 6, 4617–4622 RSC.
- X. Xie, K. Kretschmer, B. Anasori, B. Sun, G. Wang and Y. Gogotsi, ACS Appl. Nano Mater., 2018, 1, 505–511 CrossRef CAS.
- D. Zhao, M. Clites, G. Ying, S. Kota, J. Wang, V. Natu, X. Wang, E. Pomerantseva, M. Cao and M. W. Barsoum, Chem. Commun., 2018, 54, 4533–4536 RSC.
- J. Muldoon, C. B. Bucur, A. G. Oliver, T. Sugimoto, M. Matsui, H. S. Kim, G. D. Allred, J. Zajicek and Y. Kotani, Energy Environ. Sci., 2012, 5, 5941–5950 RSC.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martinez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 RSC.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC.
- Y. Niu, M. Xu, C. Dai, B. Shen and C. M. Li, Phys. Chem. Chem. Phys., 2017, 19, 17270–17277 RSC.
- J. Song, M. Noked, E. Gillette, J. Duay, G. Rubloff and S. B. Lee, Phys. Chem. Chem. Phys., 2015, 17, 5256–5264 RSC.
- M. Sawicki and L. L. Shaw, RSC Adv., 2015, 5, 53129–53154 RSC.
- C. B. Bucur, M. Jones, M. Kopylov, J. Spear and J. Muldoon, Energy Environ. Sci., 2017, 10, 905–911 RSC.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- L. Carbone, S. G. Greenbaum and J. Hassoun, Sustainable Energy Fuels, 2017, 1, 228–247 RSC.
- Q. C. Liu, J. J. Xu, S. Yuan, Z. W. Chang, D. Xu, Y. B. Yin, L. Li, H. X. Zhong, Y. S. Jiang, J. M. Yan and X. B. Zhang, Adv. Mater., 2015, 27, 5241–5247 CrossRef CAS PubMed.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- A. A. Arie and J. K. Lee, J. Nanosci. Nanotechnol., 2011, 11, 6569–6574 CrossRef CAS PubMed.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J. G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- N. Mahne, O. Fontaine, M. O. Thotiyl, M. Wilkening and S. A. Freunberger, Chem. Sci., 2017, 8, 6716–6729 RSC.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- F. X. Wang, X. W. Wu, C. Y. Li, Y. S. Zhu, L. J. Fu, Y. P. Wu and X. Liu, Energy Environ. Sci., 2016, 9, 3570–3611 RSC.
- K. N. Jung, J. Kim, Y. Yamauchi, M. S. Park, J. W. Lee and J. H. Kim, J. Mater. Chem. A, 2016, 4, 14050–14068 RSC.
- D. Higgins, P. Zamani, A. P. Yu and Z. W. Chen, Energy Environ. Sci., 2016, 9, 357–390 RSC.
- S. M. Xu, S. Lau and L. A. Archer, Inorg. Chem. Front., 2015, 2, 1070–1079 RSC.
- A. Eftekhari and B. Ramanujam, J. Mater. Chem. A, 2017, 5, 7710–7731 RSC.
- D. U. Lee, P. Xu, Z. P. Cano, A. G. Kashkooli, M. G. Park and Z. W. Chen, J. Mater. Chem. A, 2016, 4, 7107–7134 RSC.
- C. R. Raj, A. Samanta, S. H. Noh, S. Mondal, T. Okajima and T. Ohsaka, J. Mater. Chem. A, 2016, 4, 11156–11178 RSC.
- C. Tang and Q. Zhang, J. Mater. Chem. A, 2016, 4, 4998–5001 RSC.
- Z. Z. Yang, L. Gu, Y. S. Hu and H. Li, Annu. Rev. Mater. Res., 2017, 47, 175–198 CrossRef CAS.
- M. A. Hannan, M. S. H. Lipu, A. Hussain and A. Mohamed, Renewable Sustainable Energy Rev., 2017, 78, 834–854 CrossRef.
- Y. Abu-Lebdeh, in Nanotechnology for Lithium-Ion Batteries, ed. Y. Abu-Lebdeh and I. Davidson, Springer US, Boston, MA, 1st edn, 2013, pp. 1–11 DOI:10.1007/978-1-4614-4605-7_1.
- T. Wei, R. Zeng, Y. Sun, Y. Huang and K. Huang, Chem. Commun., 2014, 50, 1962–1964 RSC.
- M. Okubo, E. Hosono, J. Kim, M. Enomoto, N. Kojima, T. Kudo, H. Zhou and I. Honma, J. Am. Chem. Soc., 2007, 129, 7444–7452 CrossRef CAS PubMed.
- J. Geder, H. E. Hoster, A. Jossen, J. Garche and D. Y. W. Yu, J. Power Sources, 2014, 257, 286–292 CrossRef CAS.
- C. F. Liu, Z. G. Neale and G. Z. Cao, Mater. Today, 2016, 19, 109–123 CrossRef CAS.
- B. K. Lesel, J. B. Cook, Y. Yang, T. C. Lin and S. H. Tolbert, ACS Energy Lett., 2017, 2, 2293–2298 CrossRef CAS.
- X. Yang, F. Y. Qu, H. Niu, Q. Wang, J. Yan and Z. J. Fan, Electrochim. Acta, 2015, 180, 287–294 CrossRef CAS.
- H. Jiang, Y. Fu, Y. Hu, C. Yan, L. Zhang, P. S. Lee and C. Li, Small, 2014, 10, 1096–1100 CrossRef CAS PubMed.
- J. T. Hu, Y. Jiang, S. H. Cui, Y. D. Duan, T. C. Liu, H. Guo, L. P. Lin, Y. Lin, J. X. Zheng, K. Amine and F. Pan, Adv. Energy Mater., 2016, 6, 1600856 CrossRef.
- J. L. Li, M. W. Xiang, Y. Wang, J. H. Wu, H. Zhao and H. Liu, J. Mater. Chem. A, 2017, 5, 7952–7960 RSC.
- J. K. Kim, R. Vijaya, L. K. Zhu and Y. Kim, J. Power Sources, 2015, 275, 106–110 CrossRef CAS.
- B. Z. Li, Y. Wang, L. Xue, X. P. Li and W. S. Li, J. Power Sources, 2013, 232, 12–16 CrossRef CAS.
- K. Naoi, K. Kisu, E. Iwama, S. Nakashima, Y. Sakai, Y. Orikasa, P. Leone, N. Dupre, T. Brousse, P. Rozier, W. Naoi and P. Simon, Energy Environ. Sci., 2016, 9, 2143–2151 RSC.
- B. Das, A. Pohl, V. S. K. Chakravadhanula, C. Kubel and M. Fichtner, J. Power Sources, 2014, 267, 203–211 CrossRef CAS.
- L. M. Shang, H. Li, H. W. Lai, D. Q. Li, Q. Wu, L. J. Yang, X. Z. Wang and Z. Hu, J. Power Sources, 2016, 326, 279–284 CrossRef CAS.
- Y. C. Zhang and C. Y. Wang, J. Electrochem. Soc., 2009, 156, A527–A535 CrossRef CAS.
- S. Xu, C. M. Hessel, H. Ren, R. Yu, Q. Jin, M. Yang, H. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632–637 RSC.
- J. Kong, W. A. Yee, Y. Wei, L. Yang, J. M. Ang, S. L. Phua, S. Y. Wong, R. Zhou, Y. Dong, X. Li and X. Lu, Nanoscale, 2013, 5, 2967–2973 RSC.
- H. Tian, F. Xin, X. Wang, W. He and W. Han, J. Materiomics, 2015, 1, 153–169 CrossRef.
- N. Liu, Z. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- F. Zhang, X. Yang, Y. Q. Xie, N. B. Yi, Y. Huang and Y. S. Chen, Carbon, 2015, 82, 161–167 CrossRef CAS.
- M. H. Rummeli, S. Gorantla, A. Bachmatiuk, J. Phieler, N. Geissler, I. Ibrahim, J. B. Pang and J. Eckert, Chem. Mater., 2013, 25, 4861–4866 CrossRef CAS.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 RSC.
- W. P. Si, X. L. Sun, X. H. Liu, L. X. Xi, Y. D. Jia, C. L. Yan and O. G. Schmidt, J. Power Sources, 2014, 267, 629–634 CrossRef CAS.
- Y. L. Qin, F. Li, X. B. Bai, X. L. Sun, D. Q. Liu and D. Y. He, Mater. Lett., 2015, 138, 104–106 CrossRef CAS.
- A. M. Chockla, K. C. Klavetter, C. B. Mullins and B. A. Korgel, ACS Appl. Mater. Interfaces, 2012, 4, 4658–4664 CrossRef CAS PubMed.
- X. Sun, X. Lu, S. Huang, L. Xi, L. Liu, B. Liu, Q. Weng, L. Zhang and O. G. Schmidt, ACS Appl. Mater. Interfaces, 2017, 9, 38556–38566 CrossRef CAS PubMed.
- X. L. Sun, W. P. Si, L. X. Xi, B. Liu, X. J. Liu, C. L. Yan and O. G. Schmidt, ChemElectroChem, 2015, 2, 737–742 CrossRef CAS.
- N. G. Rudawski, B. R. Yates, M. R. Holzworth, K. S. Jones, R. G. Elliman and A. A. Volinsky, J. Power Sources, 2013, 223, 336–340 CrossRef CAS.
- L. Qiao, L. Qiao, X. W. Li, X. L. Sun, H. W. Yue and D. Y. He, J. Mater. Sci., 2017, 52, 6969–6975 CrossRef CAS.
- X. L. Sun, X. H. Wang, L. Qiao, D. K. Hu, N. Feng, X. W. Li, Y. Q. Liu and D. Y. He, Electrochim. Acta, 2012, 66, 204–209 CrossRef CAS.
- X. L. Sun, L. Qiao, L. Qiao, H. Pang and D. Li, Ionics, 2017, 23, 3177–3185 CrossRef CAS.
- X. L. Sun, X. H. Wang, Y. L. Qin, X. W. Li, L. Qiao, N. Feng, D. K. Hu and D. Y. He, Mater. Lett., 2012, 66, 193–195 CrossRef CAS.
- X. Liu, W. Si, J. Zhang, X. Sun, J. Deng, S. Baunack, S. Oswald, L. Liu, C. Yan and O. G. Schmidt, Sci. Rep., 2014, 4, 7452 CrossRef CAS PubMed.
- W. H. Xie, L. L. Gu, X. L. Sun, M. T. Liu, S. Y. Li, Q. Wang, D. Q. Liu and D. Y. He, Electrochim. Acta, 2016, 220, 107–113 CrossRef CAS.
- X. Sun, C. Yan, Y. Chen, W. Si, J. Deng, S. Oswald, L. Liu and O. G. Schmidt, Adv. Energy Mater., 2014, 4, 1300912 CrossRef.
- H. Liu, G. X. Wang, J. Liu, S. Z. Qiao and H. J. Ahn, J. Mater. Chem., 2011, 21, 3046–3052 RSC.
- X. H. Wang, X. W. Li, X. L. Sun, F. Li, Q. M. Liu, Q. Wang and D. Y. He, J. Mater. Chem., 2011, 21, 3571–3573 RSC.
- X. H. Wang, Z. B. Yang, X. L. Sun, X. W. Li, D. S. Wang, P. Wang and D. Y. He, J. Mater. Chem., 2011, 21, 9988–9990 RSC.
- X. H. Wang, L. M. Sun, X. L. Sun, X. W. Li and D. Y. He, Mater. Res. Bull., 2017, 96, 533–537 CrossRef CAS.
- X. L. Sun, W. P. Si, X. H. Liu, J. W. Deng, L. X. Xi, L. F. Liu, C. L. Yan and O. G. Schmidt, Nano Energy, 2014, 9, 168–175 CrossRef CAS.
- N. Feng, X. L. Sun, H. W. Yue and D. Y. He, RSC Adv., 2016, 6, 72008–72014 RSC.
- X. L. Sun, G. P. Hao, X. Y. Lu, L. X. Xi, B. Liu, W. P. Si, C. S. Ma, Q. M. Liu, Q. Zhang, S. Kaskel and O. G. Schmidt, J. Mater. Chem. A, 2016, 4, 10166–10173 RSC.
- S. Huang, L. Zhang, X. Lu, L. Liu, L. Liu, X. Sun, Y. Yin, S. Oswald, Z. Zou, F. Ding and O. G. Schmidt, ACS Nano, 2017, 11, 821–830 CrossRef CAS PubMed.
- M. Zhou, T. Cai, F. Pu, H. Chen, Z. Wang, H. Zhang and S. Guan, ACS Appl. Mater. Interfaces, 2013, 5, 3449–3455 CrossRef CAS PubMed.
- Q. Z. Xiao, Y. Fan, X. H. Wang, R. A. Susantyoko and Q. Zhang, Energy Environ. Sci., 2014, 7, 655–661 RSC.
- K. Yan, H. W. Lee, T. Gao, G. Zheng, H. Yao, H. Wang, Z. Lu, Y. Zhou, Z. Liang, Z. Liu, S. Chu and Y. Cui, Nano Lett., 2014, 14, 6016–6022 CrossRef CAS PubMed.
- I. H. Son, J. Hwan Park, S. Kwon, S. Park, M. H. Rummeli, A. Bachmatiuk, H. J. Song, J. Ku, J. W. Choi, J. M. Choi, S. G. Doo and H. Chang, Nat. Commun., 2015, 6, 7393 CrossRef CAS PubMed.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS PubMed.
- H. Li, G. Richter and J. Maier, Adv. Mater., 2003, 15, 736–739 CrossRef CAS.
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13, 621–625 CrossRef CAS.
- S. Suresh, Z. P. Wu, S. F. Bartolucci, S. Basu, R. Mukherjee, T. Gupta, P. Hundekar, Y. Shi, T. M. Lu and N. Koratkar, ACS Nano, 2017, 11, 5051–5061 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- S. Goriparti, E. Miele, F. De Angelis, E. Di Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- S. Choi, T. W. Kwon, A. Coskun and J. W. Choi, Science, 2017, 357, 279–283 CrossRef CAS PubMed.
- L. Liu, Q. Weng, X. Lu, X. Sun, L. Zhang and O. G. Schmidt, Small, 2017, 13, 1701847 CrossRef PubMed.
- J. J. Zhu and U. Schwingenschlogl, 2D Mater., 2017, 4, 025073 CrossRef.
- J. Zhu, A. Chroneos, J. Eppinger and U. Schwingenschlögl, Appl. Mater. Today, 2016, 5, 19–24 CrossRef.
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef PubMed.
- F. Feng, J. Wu, C. Wu and Y. Xie, Small, 2015, 11, 654–666 CrossRef CAS PubMed.
- C. Eames and M. S. Islam, J. Am. Chem. Soc., 2014, 136, 16270–16276 CrossRef CAS PubMed.
- D. Sun, Q. Hu, J. Chen, X. Zhang, L. Wang, Q. Wu and A. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 74–81 CrossRef CAS PubMed.
- J. Luo, X. Tao, J. Zhang, Y. Xia, H. Huang, L. Zhang, Y. Gan, C. Liang and W. Zhang, ACS Nano, 2016, 10, 2491–2499 CrossRef CAS PubMed.
- C. E. Ren, M. Q. Zhao, T. Makaryan, J. Halim, M. Boota, S. Kota, B. Anasori, M. W. Barsoum and Y. Gogotsi, ChemElectroChem, 2016, 3, 689–693 CrossRef CAS.
- X. Wu, Z. Wang, M. Yu, L. Xiu and J. Qiu, Adv. Mater., 2017, 29, 1607017 CrossRef PubMed.
- F. Wang, Z. J. Wang, J. F. Zhu, H. B. Yang, X. J. Chen, L. Wang and C. H. Yang, J. Mater. Sci., 2017, 52, 3556–3565 CrossRef CAS.
- B. Ahmed, D. H. Anjum, Y. Gogotsi and H. N. Alshareef, Nano Energy, 2017, 34, 249–256 CrossRef CAS.
- B. Ahmed, D. H. Anjum, M. N. Hedhili, Y. Gogotsi and H. N. Alshareef, Nanoscale, 2016, 8, 7580–7587 RSC.
- C. Shen, L. Wang, A. Zhou, H. Zhang, Z. Chen, Q. Hu and G. Qin, J. Electrochem. Soc., 2017, 164, A2654–A2659 CrossRef CAS.
- Z. Y. Lin, D. F. Sun, Q. Huang, J. Yang, M. W. Barsoum and X. B. Yan, J. Mater. Chem. A, 2015, 3, 14096–14100 RSC.
- G. Zou, Z. Zhang, J. Guo, B. Liu, Q. Zhang, C. Fernandez and Q. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 22280–22286 CrossRef CAS PubMed.
- S. Zhao, X. Meng, K. Zhu, F. Du, G. Chen, Y. Wei, Y. Gogotsi and Y. Gao, Energy Storage Mater., 2017, 8, 42–48 CrossRef.
- F. F. Liu, J. Zhou, S. W. Wang, B. X. Wang, C. Shen, L. B. Wang, Q. K. Hu, Q. Huang and A. G. Zhou, J. Electrochem. Soc., 2017, 164, A709–A713 CrossRef CAS.
- C. Shen, Y. Cao, A. Zhou, Q. Hu, G. Qin, Z. Yang, X. Liu and L. Wang, J. Alloys Compd., 2018, 735, 530–535 CrossRef CAS.
- C. F. Zhang, S. J. Kim, M. Ghidiu, M. Q. Zhao, M. W. Barsoum, V. Nicolosi and Y. Gogotsi, Adv. Funct. Mater., 2016, 26, 4143–4151 CrossRef CAS.
- J. Zhou, S. H. Gao, Z. L. Guo and Z. M. Sun, Ceram. Int., 2017, 43, 11450–11454 CrossRef CAS.
- F. Du, H. Tang, L. M. Pan, T. Zhang, H. M. Lu, J. Xiong, J. Yang and C. Zhang, Electrochim. Acta, 2017, 235, 690–699 CrossRef CAS.
- D. D. Sun, M. S. Wang, Z. Y. Li, G. X. Fan, L. Z. Fan and A. G. Zhou, Electrochem. Commun., 2014, 47, 80–83 CrossRef CAS.
- S. J. Kim, M. Naguib, M. Q. Zhao, C. F. Zhang, H. T. Jung, M. W. Barsoum and Y. Gogotsi, Electrochim. Acta, 2015, 163, 246–251 CrossRef CAS.
- M. R. Gao, Y. F. Xu, J. Jiang and S. H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209–231 RSC.
- D. Chen, W. Chen, L. Ma, G. Ji, K. Chang and J. Y. Lee, Mater. Today, 2014, 17, 184–193 CrossRef CAS.
- H. Wang, H. Feng and J. Li, Small, 2014, 10, 2165–2181 CrossRef CAS PubMed.
- Y. Pan, J. Zhang and H. Lu, Chemistry, 2017, 23, 9937–9945 CrossRef CAS PubMed.
- J. Hu, B. Xu, C. Ouyang, S. A. Yang and Y. Yao, J. Phys. Chem. C, 2014, 118, 24274–24281 CrossRef CAS.
- Z. Xu, X. Lv, J. Chen, L. Jiang, Y. Lai and J. Li, Phys. Chem. Chem. Phys., 2017, 19, 7807–7819 RSC.
- Y. G. Zhou and X. T. Zu, Electrochim. Acta, 2017, 235, 167–174 CrossRef CAS.
- D. Ccedilakir, C. Sevik, O. Gulseren and F. M. Peeters, J. Mater. Chem. A, 2016, 4, 6029–6035 RSC.
- J. P. Hu, B. Xu, C. Y. Ouyang, Y. Zhang and S. Y. A. Yang, RSC Adv., 2016, 6, 27467–27474 RSC.
- Q. Sun, Y. Dai, Y. Ma, T. Jing, W. Wei and B. Huang, J. Phys. Chem. Lett., 2016, 7, 937–943 CrossRef CAS PubMed.
- D. Wang, Y. Gao, Y. Liu, D. Jin, Y. Gogotsi, X. Meng, F. Du, G. Chen and Y. Wei, J. Phys. Chem. C, 2017, 121, 13025–13034 CrossRef CAS.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 11173–11179 CrossRef CAS PubMed.
- X. Chen, Z. Kong, N. Li, X. Zhao and C. Sun, Phys. Chem. Chem. Phys., 2016, 18, 32937–32943 RSC.
- M. Ashton, R. G. Hennig and S. B. Sinnott, Appl. Phys. Lett., 2016, 108, 023901 CrossRef.
- T. Yu, S. Zhang, F. Li, Z. Zhao, L. Liu, H. Xu and G. Yang, J. Mater. Chem. A, 2017, 5, 18698–18706 RSC.
- X. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Chem. Commun., 2012, 48, 2198–2200 RSC.
- M. Wu, J. E. Sabisch, X. Song, A. M. Minor, V. S. Battaglia and G. Liu, Nano Lett., 2013, 13, 5397–5402 CrossRef CAS PubMed.
- N. Lin, L. B. Wang, J. B. Zhou, J. Zhou, Y. Han, Y. C. Zhu, Y. T. Qian and C. H. Cao, J. Mater. Chem. A, 2015, 3, 11199–11202 RSC.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- J. Qian, D. Qiao, X. Ai, Y. Cao and H. Yang, Chem. Commun., 2012, 48, 8931–8933 RSC.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633–4636 CrossRef CAS PubMed.
- H. Liu, G. Wang, J. Park, J. Wang, H. Liu and C. Zhang, Electrochim. Acta, 2009, 54, 1733–1736 CrossRef CAS.
- Z. Wang, L. Zhou and X. W. Lou, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS PubMed.
- D. C. Bock, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, Chem. Commun., 2017, 53, 13145–13148 RSC.
- M. Y. Cheng and B. J. Hwang, J. Power Sources, 2010, 195, 4977–4983 CrossRef CAS.
- J. Zhu, T. Zhu, X. Zhou, Y. Zhang, X. W. Lou, X. Chen, H. Zhang, H. H. Hng and Q. Yan, Nanoscale, 2011, 3, 1084–1089 RSC.
- F. D. Wu and Y. Wang, J. Mater. Chem., 2011, 21, 6636–6641 RSC.
- C. Peng, B. Chen, Y. Qin, S. Yang, C. Li, Y. Zuo, S. Liu and J. Yang, ACS Nano, 2012, 6, 1074–1081 CrossRef CAS PubMed.
- S. Kajiyama, L. Szabova, K. Sodeyama, H. Iinuma, R. Morita, K. Gotoh, Y. Tateyama, M. Okubo and A. Yamada, ACS Nano, 2016, 10, 3334–3341 CrossRef CAS PubMed.
- K. Gotoh, M. Izuka, J. Arai, Y. Okada, T. Sugiyama, K. Takeda and H. Ishida, Carbon, 2014, 79, 380–387 CrossRef CAS.
- R. Alcantara, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Solid-State Lett., 2005, 8, A222–A225 CrossRef CAS.
- L. Sawers, Solid State Ionics, 1998, 107, 13–23 CrossRef CAS.
- X. Wang, X. Shen, Y. Gao, Z. Wang, R. Yu and L. Chen, J. Am. Chem. Soc., 2015, 137, 2715–2721 CrossRef CAS PubMed.
- E. Stoyanov, F. Langenhorst and G. Steinle-Neumann, Am. Mineral., 2007, 92, 577–586 CrossRef CAS.
- D. H. Pearson, C. C. Ahn and B. Fultz, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 8471–8478 CrossRef CAS.
- R. Wang, X. Q. He, L. H. He, F. W. Wang, R. J. Xiao, L. Gu, H. Li and L. Q. Chen, Adv. Energy Mater., 2013, 3, 1358–1367 CrossRef CAS.
- S.-T. Myung, K. Izumi, S. Komaba, H. Yashiro, H. J. Bang, Y.-K. Sun and N. Kumagai, J. Phys. Chem. C, 2007, 111, 4061–4067 CrossRef CAS.
- A. Sakuda, H. Kitaura, A. Hayashi, K. Tadanaga and M. Tatsumisago, J. Power Sources, 2009, 189, 527–530 CrossRef CAS.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135–1143 CrossRef CAS PubMed.
- J. X. Song, T. Xu, M. L. Gordin, P. Y. Zhu, D. P. Lv, Y. B. Jiang, Y. S. Chen, Y. H. Duan and D. H. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS.
- J. Cao, C. Chen, Q. Zhao, N. Zhang, Q. Lu, X. Wang, Z. Niu and J. Chen, Adv. Mater., 2016, 28, 9629–9636 CrossRef CAS PubMed.
- H. J. Peng, T. Z. Hou, Q. Zhang, J. Q. Huang, X. B. Cheng, M. Q. Guo, Z. Yuan, L. Y. He and F. Wei, Adv. Mater. Interfaces, 2014, 1, 1400227 CrossRef.
- Q. Lu, X. Wang, J. Cao, C. Chen, K. Chen, Z. Zhao, Z. Niu and J. Chen, Energy Storage Mater., 2017, 8, 77–84 CrossRef.
- K. Chen, J. Cao, Q. Lu, Q. Wang, M. Yao, M. Han, Z. Niu and J. Chen, Nano Res., 2018, 11, 1345–1357 CrossRef CAS.
- J. Q. Huang, T. Z. Zhuang, Q. Zhang, H. J. Peng, C. M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS PubMed.
- H. J. Peng, J. Liang, L. Zhu, J. Q. Huang, X. B. Cheng, X. Guo, W. Ding, W. Zhu and Q. Zhang, ACS Nano, 2014, 8, 11280–11289 CrossRef CAS PubMed.
- X. Yang, L. Zhang, F. Zhang, Y. Huang and Y. Chen, ACS Nano, 2014, 8, 5208–5215 CrossRef CAS PubMed.
- P. Y. Zhu, J. X. Song, D. P. Lv, D. H. Wang, C. Jaye, D. A. Fischer, T. P. Wu and Y. S. Chen, J. Phys. Chem. C, 2014, 118, 7765–7771 CrossRef CAS.
- R. Fang, S. Zhao, P. Hou, M. Cheng, S. Wang, H. M. Cheng, C. Liu and F. Li, Adv. Mater., 2016, 28, 3374–3382 CrossRef CAS PubMed.
- H. J. Peng, D. W. Wang, J. Q. Huang, X. B. Cheng, Z. Yuan, F. Wei and Q. Zhang, Adv. Sci., 2016, 3, 1500268 CrossRef PubMed.
- J.-Q. Huang, Q. Zhang and F. Wei, Energy Storage Mater., 2015, 1, 127–145 CrossRef.
- J. Liang, Z.-H. Sun, F. Li and H.-M. Cheng, Energy Storage Mater., 2016, 2, 76–106 CrossRef.
- D. W. Wang, Q. C. Zeng, G. M. Zhou, L. C. Yin, F. Li, H. M. Cheng, I. R. Gentle and G. Q. M. Lu, J. Mater. Chem. A, 2013, 1, 9382–9394 RSC.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS PubMed.
- J. Balach, H. K. Singh, S. Gomoll, T. Jaumann, M. Klose, S. Oswald, M. Richter, J. Eckert and L. Giebeler, ACS Appl. Mater. Interfaces, 2016, 8, 14586–14595 CrossRef CAS PubMed.
- J. Balach, T. Jaumann, M. Klose, S. Oswald, J. Eckert and L. Giebeler, Adv. Funct. Mater., 2015, 25, 5285–5291 CrossRef CAS.
- J. Balach, T. Jaumann, S. Muhlenhoff, J. Eckert and L. Giebeler, Chem. Commun., 2016, 52, 8134–8137 RSC.
- J. Song, D. Su, X. Xie, X. Guo, W. Bao, G. Shao and G. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 29427–29433 CrossRef CAS PubMed.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed.
- L. F. Nazar and O. Crosnier, in Lithium Batteries: Science and Technology, ed. G.-A. Nazri and G. Pistoia, Springer US, Boston, MA, 1st edn, 2003, pp. 112–143 DOI:10.1007/978-0-387-92675-9_4.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- X. Tao, Y. Liu, W. Liu, G. Zhou, J. Zhao, D. Lin, C. Zu, O. Sheng, W. Zhang, H. W. Lee and Y. Cui, Nano Lett., 2017, 17, 2967–2972 CrossRef CAS PubMed.
- X. Tao, J. Wang, C. Liu, H. Wang, H. Yao, G. Zheng, Z. W. Seh, Q. Cai, W. Li, G. Zhou, C. Zu and Y. Cui, Nat. Commun., 2016, 7, 11203 CrossRef CAS PubMed.
- Z. Wei Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. McDowell, P. C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef PubMed.
- X. Lu, J. Deng, W. Si, X. Sun, X. Liu, B. Liu, L. Liu, S. Oswald, S. Baunack, H. J. Grafe, C. Yan and O. G. Schmidt, Adv. Sci., 2015, 2, 1500113 CrossRef PubMed.
- X. Lu, L. Zhang, X. Sun, W. Si, C. Yan and O. G. Schmidt, J. Mater. Chem. A, 2016, 4, 4155–4160 RSC.
- X. Lu, W. Si, X. Sun, B. Liu, L. Zhang, C. Yan and O. G. Schmidt, Nano Energy, 2016, 19, 428–436 CrossRef CAS.
- X. Lu, Y. Yin, L. Zhang, L. Xi, S. Oswald, J. Deng and O. G. Schmidt, Nano Energy, 2016, 30, 69–76 CrossRef CAS.
- D. Lin, Y. Liu, Z. Liang, H. W. Lee, J. Sun, H. Wang, K. Yan, J. Xie and Y. Cui, Nat. Nanotechnol., 2016, 11, 626–632 CrossRef CAS PubMed.
- Y. Liu, D. Lin, Z. Liang, J. Zhao, K. Yan and Y. Cui, Nat. Commun., 2016, 7, 10992 CrossRef CAS PubMed.
- Z. Liang, D. Lin, J. Zhao, Z. Lu, Y. Liu, C. Liu, Y. Lu, H. Wang, K. Yan, X. Tao and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2862–2867 CrossRef CAS PubMed.
- B. Li, D. Zhang, Y. Liu, Y. Yu, S. Li and S. Yang, Nano Energy, 2017, 39, 654–661 CrossRef CAS.
- J. Lu, Y. Lei, K. C. Lau, X. Luo, P. Du, J. Wen, R. S. Assary, U. Das, D. J. Miller, J. W. Elam, H. M. Albishri, D. A. El-Hady, Y. K. Sun, L. A. Curtiss and K. Amine, Nat. Commun., 2013, 4, 2383 CrossRef PubMed.
- S. E. Cheon, K. S. Ko, J. H. Cho, S. W. Kim, E. Y. Chin and H. T. Kim, J. Electrochem. Soc., 2003, 150, A800–A805 CrossRef CAS.
- B. C. Yu, K. Park, J. H. Jang and J. B. Goodenough, ACS Energy Lett., 2016, 1, 633–637 CrossRef CAS.
- Q. Yun, Y. B. He, W. Lv, Y. Zhao, B. Li, F. Kang and Q. H. Yang, Adv. Mater., 2016, 28, 6932–6939 CrossRef CAS PubMed.
- D. Zhou, R. L. Liu, Y. B. He, F. Y. Li, M. Liu, B. H. Li, Q. H. Yang, Q. Cai and F. Y. Kang, Adv. Energy Mater., 2016, 6, 1502214 CrossRef.
- C. P. Yang, Y. X. Yin, S. F. Zhang, N. W. Li and Y. G. Guo, Nat. Commun., 2015, 6, 8058 CrossRef CAS PubMed.
- Z. Tu, Y. Lu and L. Archer, Small, 2015, 11, 2631–2635 CrossRef CAS PubMed.
- X. Chen, B. Liu, C. Zhong, Z. Liu, J. Liu, L. Ma, Y. D. Deng, X. P. Han, T. P. Wu, W. B. Hu and J. Lu, Adv. Energy Mater., 2017, 7, 1700779 CrossRef.
- C. Y. Su, H. Cheng, W. Li, Z. Q. Liu, N. Li, Z. F. Hou, F. Q. Bai, H. X. Zhang and T. Y. Ma, Adv. Energy Mater., 2017, 7, 1602420 CrossRef.
- P. Moni, S. Hyun, A. Vignesh and S. Shanmugam, Chem. Commun., 2017, 53, 7836–7839 RSC.
- S. Clark, A. Latz and B. Horstmann, ChemSusChem, 2017, 10, 4735–4747 CrossRef CAS PubMed.
- J. Park, M. Risch, G. Nam, M. Park, T. J. Shin, S. Park, M. G. Kim, Y. Shao-Horn and J. Cho, Energy Environ. Sci., 2017, 10, 129–136 RSC.
- S. Z. Liu, W. Han, B. C. Cui, X. J. Liu, F. L. Zhao, J. Stuart and S. Licht, J. Power Sources, 2017, 342, 435–441 CrossRef CAS.
- B. Li, S.-W. Chien, X. Ge, J. Chai, X.-Y. Goh, K.-T. Nai, T. S. Andy Hor, Z. Liu and Y. Zong, Mater. Chem. Front., 2017, 1, 677–682 RSC.
- S. X. Qu, Z. S. Song, J. Liu, Y. B. Li, Y. Kou, C. Ma, X. P. Han, Y. D. Deng, N. Q. Zhao, W. B. Hu and C. Zhong, Nano Energy, 2017, 39, 101–110 CrossRef CAS.
- Y. Fan, S. Ida, A. Staykov, T. Akbay, H. Hagiwara, J. Matsuda, K. Kaneko and T. Ishihara, Small, 2017, 13, 1700099 CrossRef PubMed.
- N. Xu, Q. Nie, Y. Wei, H. Xu, Y.-D. Wang, X.-D. Zhou and J. Qiao, Sustainable Energy Fuels, 2018, 2, 91–95 RSC.
- R. Morasch, D. G. Kwabi, M. Tulodziecki, M. Risch, S. Zhang and Y. Shao-Horn, ACS Appl. Mater. Interfaces, 2017, 9, 4374–4381 CrossRef CAS PubMed.
- S. A. Cho, Y. J. Jang, H.-D. Lim, J.-E. Lee, Y. H. Jang, T.-T. H. Nguyen, F. M. Mota, D. P. Fenning, K. Kang, Y. Shao-Horn and D. H. Kim, Adv. Energy Mater., 2017, 7, 1700391 CrossRef.
- D. G. Kwabi, V. S. Bryantsev, T. P. Batcho, D. M. Itkis, C. V. Thompson and Y. Shao-Horn, Angew. Chem., Int. Ed., 2016, 55, 3129–3134 CrossRef CAS PubMed.
- S. Y. Sayed, K. P. Yao, D. G. Kwabi, T. P. Batcho, C. V. Amanchukwu, S. Feng, C. V. Thompson and Y. Shao-Horn, Chem. Commun., 2016, 52, 9691–9694 RSC.
- M. Tulodziecki, G. M. Leverick, C. V. Amanchukwu, Y. Katayama, D. G. Kwabi, F. Barde, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 1828–1842 RSC.
- C. V. Amanchukwu, M. Gauthier, T. P. Batcho, C. Symister, Y. Shao-Horn, J. M. D'Arcy and P. T. Hammond, J. Phys. Chem. Lett., 2016, 7, 3770–3775 CrossRef CAS PubMed.
- D. G. Kwabi, M. Tulodziecki, N. Pour, D. M. Itkis, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2016, 7, 1204–1212 CrossRef CAS PubMed.
- K. P. C. Yao, J. T. Frith, S. Y. Sayed, F. Bardé, J. R. Owen, Y. Shao-Horn and N. Garcia-Araez, J. Phys. Chem. C, 2016, 120, 16290–16297 CrossRef CAS.
- D. G. Kwabi, T. P. Batcho, S. Feng, L. Giordano, C. V. Thompson and Y. Shao-Horn, Phys. Chem. Chem. Phys., 2016, 18, 24944–24953 RSC.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS PubMed.
- F. R. McLarnon, J. Electrochem. Soc., 1991, 138, 645 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Y. Z. Su, Z. Q. Yao, F. Zhang, H. Wang, Z. Mics, E. Canovas, M. Bonn, X. D. Zhuang and X. L. Feng, Adv. Funct. Mater., 2016, 26, 5893–5902 CrossRef CAS.
- D. U. Lee, J. Fu, M. G. Park, H. Liu, A. Ghorbani Kashkooli and Z. Chen, Nano Lett., 2016, 16, 1794–1802 CrossRef CAS PubMed.
- T. Y. Ma, J. L. Cao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 1138–1142 CrossRef CAS PubMed.
- S. Chen and S. Z. Qiao, ACS Nano, 2013, 7, 10190–10196 CrossRef CAS PubMed.
- S. Chen, J. Duan, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2014, 26, 2925–2930 CrossRef CAS PubMed.
- S. Chen, J. Duan, J. Ran and S. Z. Qiao, Adv. Sci., 2015, 2, 1400015 CrossRef PubMed.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grutzke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- M. R. Gao, Y. F. Xu, J. Jiang, Y. R. Zheng and S. H. Yu, J. Am. Chem. Soc., 2012, 134, 2930–2933 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- H. Over, Chem. Rev., 2012, 112, 3356–3426 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- W. Fan, Q. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2632–2649 RSC.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- M. Xing, B. Qiu, X. Li and J. Zhang, in Nanostructured Photocatalysts: Advanced Functional Materials, ed. H. Yamashita and H. Li, Springer International Publishing, Cham, 1st edn, 2016, pp. 23–67 DOI:10.1007/978-3-319-26079-2_2.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. B. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. L. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- M. Z. Rahman, J. R. Ran, Y. H. Tang, M. Jaroniec and S. Z. Qiao, J. Mater. Chem. A, 2016, 4, 2445–2452 RSC.
- A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander Iii, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem Rev, 2015, 115, 12888–12935 Search PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- X. Li, J. Wen, J. Low, Y. Fang and J. Yu, Sci. China Mater., 2014, 57, 70–100 CrossRef.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188–7216 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- S. Pal, S. Maiti, U. N. Maiti and K. K. Chattopadhyay, CrystEngComm, 2015, 17, 1464–1476 RSC.
- R. C. Pawar, V. Khare and C. S. Lee, Dalton Trans., 2014, 43, 12514–12527 RSC.
- H. Y. Jing, T. Wen, C. M. Fan, G. Q. Gao, S. L. Zhong and A. W. Xu, J. Mater. Chem. A, 2014, 2, 14563–14570 RSC.
- S. E. Mousavinia, S. Hajati, M. Ghaedi and K. Dashtian, Phys. Chem. Chem. Phys., 2016, 18, 11278–11287 RSC.
- G. Li, B. Jiang, S. Xiao, Z. Lian, D. Zhang, J. C. Yu and H. Li, Environ. Sci.: Processes Impacts, 2014, 16, 1975–1980 RSC.
- J. Di, J. X. Xia, S. Yin, H. Xu, L. Xu, Y. G. Xu, M. Q. He and H. M. Li, J. Mater. Chem. A, 2014, 2, 5340–5351 RSC.
- Y. Zhou, L. Zhang, J. Liu, X. Fan, B. Wang, M. Wang, W. Ren, J. Wang, M. Li and J. Shi, J. Mater. Chem. A, 2015, 3, 3862–3867 RSC.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- N. M. Flores, U. Pal, R. Galeazzi and A. Sandoval, RSC Adv., 2014, 4, 41099–41110 RSC.
- B. Li, J. Q. Zhao, J. Liu, X. Y. Shen, S. B. Mo and H. Tong, RSC Adv., 2015, 5, 15572–15578 RSC.
- Z. W. Zhao, Y. J. Sun, F. Dong, Y. X. Zhang and H. Zhao, RSC Adv., 2015, 5, 39549–39556 RSC.
- P. Kumar, A. Kumar, C. Joshi, R. Boukherroub and S. L. Jain, Advanced 2D Materials, John Wiley & Sons, Inc., 2016, pp. 353–386 DOI:10.1002/9781119242635.ch9.
- S. Deng, Y. Zhong, Y. Zeng, Y. Wang, Z. Yao, F. Yang, S. Lin, X. Wang, X. Lu, X. Xia and J. Tu, Adv. Mater., 2017, 29, 1700748 CrossRef PubMed.
- S. Deng, Y. Zhong, Y. Zeng, Y. Wang, X. Wang, X. Lu, X. Xia and J. Tu, Adv. Sci., 2018, 5, 1700772 CrossRef PubMed.
- M. Y. Xing, J. L. Zhang and F. Chen, Appl. Catal., B, 2009, 89, 563–569 CrossRef CAS.
- M. Xing, J. Zhang, F. Chen and B. Tian, Chem. Commun., 2011, 47, 4947–4949 RSC.
- M. Y. Xing, D. Y. Qi, J. L. Zhang and F. Chen, Chemistry, 2011, 17, 11432–11436 CrossRef CAS PubMed.
- M. Y. Xing, J. L. Zhang and F. Chen, J. Phys. Chem. C, 2009, 113, 12848–12853 CrossRef CAS.
- M. Xing, Y. Wu, J. Zhang and F. Chen, Nanoscale, 2010, 2, 1233–1239 RSC.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- M. Y. Xing, W. Z. Fang, M. Nasir, Y. F. Ma, J. L. Zhang and M. Anpo, J. Catal., 2013, 297, 236–243 CrossRef CAS.
- M. Y. Xing, D. Y. Qi, J. L. Zhang, F. Chen, B. Z. Tian, S. Bagwas and M. Anpo, J. Catal., 2012, 294, 37–46 CrossRef CAS.
- M. Y. Xing, B. X. Yang, H. Yu, B. Z. Tian, S. Bagwasi, J. L. Zhang and X. Q. Gongs, J. Phys. Chem. Lett., 2013, 4, 3910–3917 CrossRef CAS.
- Y. M. Wu, M. Y. Xing, B. Z. Tian, J. L. Zhang and F. Chen, Chem. Eng. J., 2010, 162, 710–717 CrossRef CAS.
- Y. Cong, F. Chen, J. L. Zhang and M. Anpo, Chem. Lett., 2006, 35, 800–801 CrossRef CAS.
- Y. Wu, M. Xing and J. Zhang, J. Hazard. Mater., 2011, 192, 368–373 CAS.
- D. Y. Qi, M. Y. Xing and J. L. Zhang, J. Phys. Chem. C, 2014, 118, 7329–7336 CrossRef CAS.
- Y. Cong, J. L. Zhang, F. Chen, M. Anpo and D. N. He, J. Phys. Chem. C, 2007, 111, 10618–10623 CrossRef CAS.
- P. Yang, C. Lu, N. P. Hua and Y. K. Du, Mater. Lett., 2002, 57, 794–801 CrossRef CAS.
- L. Yu, S. Yuan, L. Y. Shi, Y. Zhao and J. H. Fang, Microporous Mesoporous Mater., 2010, 134, 108–114 CrossRef CAS.
- X. L. Yuan, J. L. Zhang, M. Anpo and D. N. He, Res. Chem. Intermed., 2010, 36, 83–93 CrossRef CAS.
- M. Xing, X. Li and J. Zhang, Sci. Rep., 2014, 4, 5493 CrossRef CAS PubMed.
- W. Li, M. Zhang, Z. Du, Q. Ma, H. Jameel and H.-m. Chang, BioResources, 2015, 10, 1245–1259 CAS.
- R. F. Dong, B. Z. Tian, J. L. Zhang, T. T. Wang, Q. S. Tao, S. Y. Bao, F. Yang and C. Y. Zeng, Catal. Commun., 2013, 38, 16–20 CrossRef CAS.
- X. Du, Y. Wang, Y. Mu, L. Gui, P. Wang and Y. Tang, Chem. Mater., 2002, 14, 3953–3957 CrossRef CAS.
- F. Dong, Y. J. Sun and M. Fu, Int. J. Photoenergy, 2012, 569716, DOI:10.1155/2012/569716.
- C. He, B. Tian and J. Zhang, J. Colloid Interface Sci., 2010, 344, 382–389 CrossRef CAS PubMed.
- M. D. Driessen, A. L. Goodman, T. M. Miller, G. A. Zaharias and V. H. Grassian, J. Phys. Chem. B, 1998, 102, 549–556 CrossRef CAS.
- C. Cheng, A. Amini, C. Zhu, Z. Xu, H. Song and N. Wang, Sci. Rep., 2014, 4, 4181 CrossRef PubMed.
- W. Z. Fang, M. Y. Xing and J. L. Zhang, Appl. Catal., B, 2014, 160, 240–246 CrossRef.
- R. Marschall and L. Z. Wang, Catal. Today, 2014, 225, 111–135 CrossRef CAS.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- Y. Y. Liang, H. L. Wang, H. S. Casalongue, Z. Chen and H. J. Dai, Nano Res., 2010, 3, 701–705 CrossRef CAS.
- C. Peng, X. Yang, Y. Li, H. Yu, H. Wang and F. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 6051–6060 CrossRef CAS PubMed.
- I. Persson, A. El Ghazaly, Q. Tao, J. Halim, S. Kota, V. Darakchieva, J. Palisaitis, M. W. Barsoum, J. Rosen and P. O. A. Persson, Small, 2018, 14, e1703676 CrossRef PubMed.
- Y. Dong, S. S. K. Mallineni, K. Maleski, H. Behlow, V. N. Mochalin, A. M. Rao, Y. Gogotsi and R. Podila, Nano Energy, 2018, 44, 103–110 CrossRef CAS.
- A. D. Handoko, K. D. Fredrickson, B. Anasori, K. W. Convey, L. R. Johnson, Y. Gogotsi, A. Vojvodic and Z. W. Seh, ACS Appl. Energy Mater., 2017, 1, 173–180 CrossRef.
- K. Nagaveni, G. Sivalingam, M. S. Hegde and G. Madras, Environ. Sci. Technol., 2004, 38, 1600–1604 CrossRef CAS PubMed.
- H. Zhang, R. L. Zong, J. C. Zhao and Y. F. Zhu, Environ. Sci. Technol., 2008, 42, 3803–3807 CrossRef CAS PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal and J. Perez-Ramirez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- W. H. Koppenol and J. D. Rush, J. Phys. Chem., 1987, 91, 4429–4430 CrossRef CAS.
- X. Zhang, Z. H. Zhang, J. L. Li, X. D. Zhao, D. H. Wu and Z. Zhou, J. Mater. Chem. A, 2017, 5, 12899–12903 RSC.
- H. Y. He, P. Zapol and L. A. Curtiss, Energy Environ. Sci., 2012, 5, 6196–6205 RSC.
- N. Li, X. Chen, W. J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- C. Ma, J. Sheng, N. Brandon, C. Zhang and G. Li, Int. J. Hydrogen Energy, 2007, 32, 2824–2829 CrossRef CAS.
- S. A. Grigoriev, P. Millet and V. N. Fateev, J. Power Sources, 2008, 177, 281–285 CrossRef CAS.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 12703–12706 CrossRef CAS PubMed.
- W. F. Chen, C. H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. T. Muckerman, Y. Zhu and R. R. Adzic, Energy Environ. Sci., 2013, 6, 943–951 RSC.
- L. Liao, S. N. Wang, J. J. Xiao, X. J. Bian, Y. H. Zhang, M. D. Scanlon, X. L. Hu, Y. Tang, B. H. Liu and H. H. Girault, Energy Environ. Sci., 2014, 7, 387–392 RSC.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- J. Kibsgaard, C. Tsai, K. Chan, J. D. Benck, J. K. Norskov, F. Abild-Pedersen and T. F. Jaramillo, Energy Environ. Sci., 2015, 8, 3022–3029 RSC.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- J. Tian, Q. Liu, A. M. Asiri and X. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jorgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Norskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- D. Kong, H. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Norskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- J. Kibsgaard and T. F. Jaramillo, Angew. Chem., Int. Ed., 2014, 53, 14433–14437 CrossRef CAS PubMed.
- M. Caban-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. C. Chang, M. L. Tsai, J. H. He and S. Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef CAS PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 2230–2234 CrossRef CAS PubMed.
- W. Cui, Q. Liu, N. Cheng, A. M. Asiri and X. Sun, Chem. Commun., 2014, 50, 9340–9342 RSC.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- J. Pang, A. Bachmatiuk, Y. Yin, B. Trzebicka, L. Zhao, L. Fu, R. G. Mendes, T. Gemming, Z. Liu and M. H. Rummeli, Adv. Energy Mater., 2017, 1702093, DOI:10.1002/aenm.201702093.
- J. Ran, B. Zhu and S. Z. Qiao, Angew. Chem., Int. Ed., 2017, 56, 10373–10377 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, J. Chen, J. Liu, J. Liang, A. Du, W. Zhang, Z. Zhu, S. C. Smith, M. Jaroniec, G. Q. Lu and S. Z. Qiao, J. Am. Chem. Soc., 2011, 133, 20116–20119 CrossRef CAS PubMed.
- G. P. Gao, Y. Jiao, F. X. Ma, Y. L. Jiao, E. Waclawik and A. J. Du, J. Catal., 2015, 332, 149–155 CrossRef CAS.
- G. Gao, Y. Jiao, F. Ma, Y. Jiao, E. Waclawik and A. Du, Phys. Chem. Chem. Phys., 2015, 17, 31140–31144 RSC.
- G. P. Gao, G. Sun and A. J. Du, J. Phys. Chem. C, 2016, 120, 16761–16766 CrossRef CAS.
- H. Fei, J. Dong, M. J. Arellano-Jimenez, G. Ye, N. Dong Kim, E. L. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. Fernandez Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- O. K. Varghese, M. Paulose, T. J. Latempa and C. A. Grimes, Nano Lett., 2009, 9, 731–737 CrossRef CAS PubMed.
- G. H. Liu, N. Hoivik, K. Y. Wang and H. Jakobsen, Sol. Energy Mater. Sol. Cells, 2012, 105, 53–68 CrossRef CAS.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- G. P. Gao, A. P. O'Mullane and A. J. Dui, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- T. Kandemir, M. E. Schuster, A. Senyshyn, M. Behrens and R. Schlogl, Angew. Chem., Int. Ed., 2013, 52, 12723–12726 CrossRef CAS PubMed.
- R. Schlogl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- A. Vojvodic, A. J. Medford, F. Studt, F. Abild-Pedersen, T. S. Khan, T. Bligaard and J. K. Norskov, Chem. Phys. Lett., 2014, 598, 108–112 CrossRef CAS.
- V. Smil, Nature, 1999, 400, 415 CrossRef CAS.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636 CrossRef CAS.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Norskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- L. M. Azofra, N. Li, D. R. MacFarlane and C. Sun, Energy Environ. Sci., 2016, 9, 2545–2549 RSC.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Norskov, Science, 2005, 307, 555–558 CrossRef CAS PubMed.
- S. Dahl, P. A. Taylor, E. Tornqvist and I. Chorkendorff, J. Catal., 1998, 178, 679–686 CrossRef CAS.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- A. E. Kabeel and S. A. El-Agouz, Desalination, 2011, 276, 1–12 CrossRef CAS.
- V. Velmurugana and K. Srithar, Renewable Sustainable Energy Rev., 2008, 12, 2253–2263 CrossRef.
- R. D. Jackson and C. H. Van Bavel, Science, 1965, 149, 1377–1379 CrossRef CAS PubMed.
- O. Neumann, A. S. Urban, J. Day, S. Lal, P. Nordlander and N. J. Halas, ACS Nano, 2013, 7, 42–49 CrossRef CAS PubMed.
- Y. Liu, S. Yu, R. Feng, A. Bernard, Y. Liu, Y. Zhang, H. Duan, W. Shang, P. Tao, C. Song and T. Deng, Adv. Mater., 2015, 27, 2768–2774 CrossRef CAS PubMed.
- J. Xuan, Z. Wang, Y. Chen, D. Liang, L. Cheng, X. Yang, Z. Liu, R. Ma, T. Sasaki and F. Geng, Angew. Chem., Int. Ed., 2016, 55, 14569–14574 CrossRef CAS PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- D. Jaque, L. Martinez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martin Rodriguez and J. Garcia Sole, Nanoscale, 2014, 6, 9494–9530 RSC.
- R. Li, L. Zhang and P. Wang, Nanoscale, 2015, 7, 17167–17194 RSC.
- D. Boyer, P. Tamarat, A. Maali, B. Lounis and M. Orrit, Science, 2002, 297, 1160–1163 CrossRef CAS PubMed.
- N. T. Panagiotopoulos, E. K. Diamanti, L. E. Koutsokeras, M. Baikousi, E. Kordatos, T. E. Matikas, D. Gournis and P. Patsalas, ACS Nano, 2012, 6, 10475–10485 CrossRef CAS PubMed.
- Y. C. Wang, L. B. Zhang and P. Wang, ACS Sustainable Chem. Eng., 2016, 4, 1223–1230 CrossRef CAS.
- I. Ibrahim, J. Kalbacova, V. Engemaier, J. B. Pang, R. D. Rodriguez, D. Grimm, T. Gemming, D. R. T. Zahn, O. G. Schmidt, J. Eckert and M. H. Rummeli, Chem. Mater., 2015, 27, 5964–5973 CrossRef CAS.
- Y. Ito, Y. Tanabe, J. Han, T. Fujita, K. Tanigaki and M. Chen, Adv. Mater., 2015, 27, 4302–4307 CrossRef CAS PubMed.
- X. Li, W. Xu, M. Tang, L. Zhou, B. Zhu, S. Zhu and J. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13953–13958 CrossRef CAS PubMed.
- L. Zhou, Y. L. Tan, J. Y. Wang, W. C. Xu, Y. Yuan, W. S. Cai, S. N. Zhu and J. Zhu, Nat. Photonics, 2016, 10, 393–399 CrossRef CAS.
- L. Zhou, Y. Tan, D. Ji, B. Zhu, P. Zhang, J. Xu, Q. Gan, Z. Yu and J. Zhu, Sci. Adv., 2016, 2, e1501227 CrossRef PubMed.
- Z. Wang, Y. Liu, P. Tao, Q. Shen, N. Yi, F. Zhang, Q. Liu, C. Song, D. Zhang, W. Shang and T. Deng, Small, 2014, 10, 3234–3239 CrossRef CAS PubMed.
- W. Ren, Y. Yan, L. Zeng, Z. Shi, A. Gong, P. Schaaf, D. Wang, J. Zhao, B. Zou, H. Yu, G. Chen, E. M. Brown and A. Wu, Adv. Healthcare Mater., 2015, 4, 1526–1536 CrossRef CAS PubMed.
- J. Wang, Y. Li, L. Deng, N. Wei, Y. Weng, S. Dong, D. Qi, J. Qiu, X. Chen and T. Wu, Adv. Mater., 2017, 29, 1603730 CrossRef PubMed.
- M. S. Zielinski, J. W. Choi, T. La Grange, M. Modestino, S. M. Hashemi, Y. Pu, S. Birkhold, J. A. Hubbell and D. Psaltis, Nano Lett., 2016, 16, 2159–2167 CrossRef CAS PubMed.
- R. Li, L. Zhang, L. Shi and P. Wang, ACS Nano, 2017, 11, 3752–3759 CrossRef CAS PubMed.
- H. Lin, S. Gao, C. Dai, Y. Chen and J. Shi, J. Am. Chem. Soc., 2017, 139, 16235–16247 CrossRef CAS PubMed.
- W. Liu, J. J. He, Z. G. Li, W. L. Jiang, J. B. Pang, Y. Zhang and Y. Sun, Phys. Scr., 2012, 85, 055806 CrossRef.
- J. B. Pang, Y. A. Cai, Q. He, H. Wang, W. L. Jiang, J. J. He, T. Yu, W. Liu, Y. Zhang and Y. Sun, Phys. Procedia, 2012, 32, 372–378 CrossRef CAS.
- H. Wang, Y. Zhang, X. L. Kou, Y. A. Cai, W. Liu, T. Yu, J. B. Pang, C. J. Li and Y. Sun, Semicond. Sci. Technol., 2010, 25, 055007 CrossRef.
- Z. L. Guo, J. Zhou, L. G. Zhu and Z. M. Sun, J. Mater. Chem. A, 2016, 4, 11446–11452 RSC.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729–4733 CrossRef CAS.
- X. Li, Z. Lv and H. Zhu, Adv. Mater., 2015, 27, 6549–6574 CrossRef CAS PubMed.
- X. M. Li and H. W. Zhu, Phys. Today, 2016, 69, 47–51 Search PubMed.
- Z. Kang, Y. Ma, X. Tan, M. Zhu, Z. Zheng, N. Liu, L. Li, Z. Zou, X. Jiang, T. Zhai and Y. Gao, Adv. Electron. Mater., 2017, 3, 1700165 CrossRef.
- Z. L. Guo, N. H. Miao, J. Zhou, B. S. Sa and Z. Sun, J. Mater. Chem. C, 2017, 5, 978–984 RSC.
- J. Zhou and P. Jena, J. Phys. Chem. Lett., 2017, 8, 5764–5770 CrossRef CAS PubMed.
- Q. Yang, S. L. Zhang, C. J. Tan, H. Y. Ye, X. Ming, S. Ingebrandt and X. P. Chen, J. Mater. Chem. C, 2017, 5, 9412–9420 RSC.
- G. W. Jones, D. A. Bahamon, A. H. Castro Neto and V. M. Pereira, Nano Lett., 2017, 17, 5304–5313 CrossRef CAS PubMed.
- M. Kang, B. Kim, S. H. Ryu, S. W. Jung, J. Kim, L. Moreschini, C. Jozwiak, E. Rotenberg, A. Bostwick and K. S. Kim, Nano Lett., 2017, 17, 1610–1615 CrossRef CAS PubMed.
- Z. Wang, L. Zhao, K. F. Mak and J. Shan, Nano Lett., 2017, 17, 740–746 CrossRef CAS PubMed.
- C. W. Niu, P. M. Buhl, G. Bihlmayer, D. Wortmann, Y. Dai, S. Blugel and Y. Mokrousov, Phys. Rev. B, 2017, 95, 075404 CrossRef.
- A. N. Gandi, H. N. Alshareef and U. Schwingenschlögl, Chem. Mater., 2016, 28, 1647–1652 CrossRef CAS.
- S. Kumar and U. Schwingenschlögl, Phys. Rev. B, 2016, 94, 035405 CrossRef.
- H. Kim, B. Anasori, Y. Gogotsi and H. N. Alshareef, Chem. Mater., 2017, 29, 6472–6479 CrossRef CAS.
- X. H. Zha, Q. Huang, J. He, H. He, J. Zhai, J. S. Francisco and S. Du, Sci. Rep., 2016, 6, 27971 CrossRef PubMed.
- Y. Liu, H. Du, X. Zhang, Y. Yang, M. Gao and H. Pan, Chem. Commun., 2016, 52, 705–708 RSC.
- S. Lai, J. Jeon, S. K. Jang, J. Xu, Y. J. Choi, J. H. Park, E. Hwang and S. Lee, Nanoscale, 2015, 7, 19390–19396 RSC.
- X. Chen, X. Sun, W. Xu, G. Pan, D. Zhou, J. Zhu, H. Wang, X. Bai, B. Dong and H. Song, Nanoscale, 2018, 10, 1111–1118 RSC.
- Y. I. Jhon, M. Seo and Y. M. Jhon, Nanoscale, 2017, 10, 69–75 RSC.
- Y. Xu, X. Wang, W. L. Zhang, F. Lv and S. Guo, Chem. Soc. Rev., 2018, 47, 586–625 RSC.
- X. Yu, X. Cai, H. Cui, S. W. Lee, X. F. Yu and B. Liu, Nanoscale, 2017, 9, 17859–17864 RSC.
- Y. Liu, H. Xiao and W. A. Goddard, 3rd, J. Am. Chem. Soc., 2016, 138, 15853–15856 CrossRef CAS PubMed.
- H. Fashandi, M. Dahlqvist, J. Lu, J. Palisaitis, S. I. Simak, I. A. Abrikosov, J. Rosen, L. Hultman, M. Andersson, A. Lloyd Spetz and P. Eklund, Nat. Mater., 2017, 16, 814–818 CrossRef CAS PubMed.
- N. Gao, S. Zhou, N. Liu, Y. Bai and J. Zhao, J. Mater. Chem. C, 2018, 6, 6764–6770 RSC.
- Q. Li, J. Yang, R. Quhe, Q. Zhang, L. Xu, Y. Pan, M. Lei and J. Lu, Carbon, 2018, 135, 125–133 CrossRef CAS.
- S. Lai, S. K. Jang, J. H. Cho and S. Lee, Nanoscale, 2018, 10, 5191–5197 RSC.
- J. J. He, P. B. Lyu and P. Nachtigall, J. Mater. Chem. C, 2016, 4, 11143–11149 RSC.
- A. Chandrasekaran, A. Mishra and A. K. Singh, Nano Lett., 2017, 17, 3290–3296 CrossRef CAS PubMed.
- Q. Tao, M. Dahlqvist, J. Lu, S. Kota, R. Meshkian, J. Halim, J. Palisaitis, L. Hultman, M. W. Barsoum, P. O. A. Persson and J. Rosen, Nat. Commun., 2017, 8, 14949 CrossRef PubMed.
- T. Hu, J. Yang and X. Wang, Phys. Chem. Chem. Phys., 2017, 19, 31773–31780 RSC.
- M. Ghidiu, S. Kota, V. Drozd and M. W. Barsoum, Sci. Adv., 2018, 4, eaao6850 CrossRef PubMed.
- A. C. Rajan, A. Mishra, S. Satsangi, R. Vaish, H. Mizuseki, K.-R. Lee and A. K. Singh, Chem. Mater., 2018, 30, 4031–4038 CrossRef CAS.
- W. Jiang, X. Zou, H. Du, L. Gan, C. Xu, F. Kang, W. Duan and J. Li, Chem. Mater., 2018, 30, 2687–2693 CrossRef CAS.
- E. Satheeshkumar, T. Makaryan, A. Melikyan, H. Minassian, Y. Gogotsi and M. Yoshimura, Sci. Rep., 2016, 6, 32049 CrossRef CAS PubMed.
- N. M. Caffrey, Nanoscale, 2018, 10, 13520–13530 RSC.
- G. Rehman, S. A. Khan, B. Amin, I. Ahmad, L.-Y. Gan and M. Maqbool, J. Mater. Chem. C, 2018, 6, 2830–2839 RSC.
- T. Hu, H. Zhang, J. Wang, Z. Li, M. Hu, J. Tan, P. Hou, F. Li and X. Wang, Sci. Rep., 2015, 5, 16329 CrossRef CAS PubMed.
- X. H. Zha, J. C. Ren, L. Feng, X. Bai, K. Luo, Y. Zhang, J. He, Q. Huang, J. S. Francisco and S. Du, Nanoscale, 2018, 10, 8763–8771 RSC.
- A. Bandyopadhyay, D. Ghosh and S. K. Pati, Phys. Chem. Chem. Phys., 2018, 20, 4012–4019 RSC.
- W. Sun, Y. Xie and P. R. C. Kent, Nanoscale, 2018, 10, 11962–11968 RSC.
- J. K. El-Demellawi, S. Lopatin, J. Yin, O. F. Mohammed and H. N. Alshareef, ACS Nano, 2018, 12, 8485–8493 CrossRef CAS PubMed.
- A. Lipatov, H. Lu, M. Alhabeb, B. Anasori, A. Gruverman, Y. Gogotsi and A. Sinitskii, Sci. Adv., 2018, 4, eaat0491 CrossRef PubMed.
- S. Sarikurt, D. Cakir, M. Keceli and C. Sevik, Nanoscale, 2018, 10, 8859–8868 RSC.
- Y. Zhou, G. Zhai, T. Yan, J. S. Francisco, H. Tian, Q. Huang and S. Du, J. Phys. Chem. C, 2018, 122, 14908–14917 CrossRef CAS.
- G. Li, K. Kushnir, Y. Dong, S. Chertopalov, A. M. Rao, V. N. Mochalin, R. Podila and L. V. Titova, 2D Mater., 2018, 5, 035043 CrossRef.
- P. Yasaei, Z. Hemmat, C. J. Foss, S. J. Li, L. Hong, A. Behranginia, L. Majidi, R. F. Klie, M. W. Barsoum, Z. Aksamija and A. Salehi-Khojin, Adv. Mater., 2018, e1801629, DOI:10.1002/adma.201801629.
- Y. Zhu, L. Peng, Z. Fang, C. Yan, X. Zhang and G. Yu, Adv. Mater., 2018, 30, e1706347 CrossRef PubMed.
- S. Kondrat, P. Wu, R. Qiao and A. A. Kornyshev, Nat. Mater., 2014, 13, 387–393 CrossRef CAS PubMed.
- C. Liu, X. Yan, F. Hu, G. Gao, G. Wu and X. Yang, Adv. Mater., 2018, 30, e1705713 CrossRef PubMed.
- Z. Li, H. Zhang, J. Han, Y. Chen, H. Lin and T. Yang, Adv. Mater., 2018, 30, e1706981 CrossRef PubMed.
- J. J. Zhang, L. Lin, Y. Zhang, M. Wu, B. I. Yakobson and S. Dong, J. Am. Chem. Soc., 2018, 140, 9768–9773 CrossRef CAS PubMed.
- X. Sang, Y. Xie, D. E. Yilmaz, R. Lotfi, M. Alhabeb, A. Ostadhossein, B. Anasori, W. Sun, X. Li, K. Xiao, P. R. C. Kent, A. C. T. van Duin, Y. Gogotsi and R. R. Unocic, Nat. Commun., 2018, 9, 2266 CrossRef PubMed.
- N. Shpigel, M. D. Levi, S. Sigalov, T. S. Mathis, Y. Gogotsi and D. Aurbach, J. Am. Chem. Soc., 2018, 140, 8910–8917 CrossRef CAS PubMed.
- R. Lotfi, M. Naguib, D. E. Yilmaz, J. Nanda and A. C. T. van Duin, J. Mater. Chem. A, 2018, 6, 12733–12743 RSC.
- W. T. Tysoe, Nat. Catal., 2018, 1, 316–317 CrossRef.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- M. Khazaei, A. Ranjbar, K. Esfarjani, D. Bogdanovski, R. Dronskowski and S. Yunoki, Phys. Chem. Chem. Phys., 2018, 20, 8579–8592 RSC.
- T. Li, L. Yao, Q. Liu, J. Gu, R. Luo, J. Li, X. Yan, W. Wang, P. Liu, B. Chen, W. Zhang, W. Abbas, R. Naz and D. Zhang, Angew. Chem., 2018, 130, 6223–6227 CrossRef.
- J. Guo, B. Legum, B. Anasori, K. Wang, P. Lelyukh, Y. Gogotsi and C. A. Randall, Adv. Mater., 2018, 30, e1801846 CrossRef PubMed.
- E. Pomerantseva and Y. Gogotsi, Nat. Energy, 2017, 2, 17089 CrossRef CAS.
- S. Zhou, X. Yang, W. Pei, N. Liu and J. Zhao, Nanoscale, 2018, 10, 10876–10883 RSC.
- R. Han, X. Ma, Y. Xie, D. Teng and S. Zhang, RSC Adv., 2017, 7, 56204–56210 RSC.
- Q. Xue, Z. Pei, Y. Huang, M. Zhu, Z. Tang, H. Li, Y. Huang, N. Li, H. Zhang and C. Zhi, J. Mater. Chem. A, 2017, 5, 20818–20823 RSC.
- W. Zheng, P. Zhang, J. Chen, W. B. Tian, Y. M. Zhang and Z. M. Sun, J. Mater. Chem. A, 2018, 6, 3543–3551 RSC.
- G.-M. Weng, J. Li, M. Alhabeb, C. Karpovich, H. Wang, J. Lipton, K. Maleski, J. Kong, E. Shaulsky, M. Elimelech, Y. Gogotsi and A. D. Taylor, Adv. Funct. Mater., 2018, 1803360, DOI:10.1002/adfm.201803360.
- Y. Sun, Z. Xu, Y. Zhuang, G. Liu, W. Jin, G. Liu and W. Jing, 2D Mater., 2018, 5, 045003 CrossRef.
- K. Li, T. Jiao, R. Xing, G. Zou, J. Zhou, L. Zhang and Q. Peng, Sci. China Mater., 2018, 61, 728–736 CrossRef.
- W. Shao, M. Tebyetekerwa, I. Marriam, W. Li, Y. Wu, S. Peng, S. Ramakrishna, S. Yang and M. Zhu, J. Power Sources, 2018, 396, 683–690 CrossRef CAS.
- R. B. Rakhi, P. Nayuk, C. Xia and H. N. Alshareef, Sci. Rep., 2016, 6, 36422 CrossRef CAS PubMed.
- Y. Ma, N. Liu, L. Li, X. Hu, Z. Zou, J. Wang, S. Luo and Y. Gao, Nat. Commun., 2017, 8, 1207 CrossRef PubMed.
- Y. Yue, N. Liu, W. Liu, M. Li, Y. Ma, C. Luo, S. Wang, J. Rao, X. Hu, J. Su, Z. Zhang, Q. Huang and Y. Gao, Nano Energy, 2018, 50, 79–87 CrossRef CAS.
- Y. Ma, Y. Yue, H. Zhang, F. Cheng, W. Zhao, J. Rao, S. Luo, J. Wang, X. Jiang, Z. Liu, N. Liu and Y. Gao, ACS Nano, 2018, 12, 3209–3216 CrossRef CAS PubMed.
- S. Xu, Y. Dall’Agnese, G. Wei, C. Zhang, Y. Gogotsi and W. Han, Nano Energy, 2018, 50, 479–488 CrossRef CAS.
- Y. Z. Zhang, K. H. Lee, D. H. Anjum, R. Sougrat, Q. Jiang, H. Kim and H. N. Alshareef, Sci. Adv., 2018, 4, eaat0098 CrossRef PubMed.
- W. Yuan, K. Yang, H. Peng, F. Li and F. Yin, J. Mater. Chem. A, 2018, 6, 18116–18124 RSC.
- G. Ying, S. Kota, A. D. Dillon, A. T. Fafarman and M. W. Barsoum, FlatChem, 2018, 8, 25–30 CrossRef CAS.
- C. Zhang and V. Nicolosi, Energy Storage Mater., 2019, 16, 102–125 CrossRef.
- X. Li, X. Yin, C. Song, M. Han, H. Xu, W. Duan, L. Cheng and L. Zhang, Adv. Funct. Mater., 2018, 1803938, DOI:10.1002/adfm.201803938.
- Y. Jiang, X. Xie, Y. Chen, Y. Liu, R. Yang and G. Sui, J. Mater. Chem. C, 2018, 6, 8679–8687 RSC.
- B. Dai, B. Zhao, X. Xie, T. Su, B. Fan, R. Zhang and R. Yang, J. Mater. Chem. C, 2018, 6, 5690–5697 RSC.
- W. T. Cao, F. F. Chen, Y. J. Zhu, Y. G. Zhang, Y. Y. Jiang, M. G. Ma and F. Chen, ACS Nano, 2018, 12, 4583–4593 CrossRef CAS PubMed.
- G. Choi, F. Shahzad, Y.-M. Bahk, Y. M. Jhon, H. Park, M. Alhabeb, B. Anasori, D.-S. Kim, C. M. Koo, Y. Gogotsi and M. Seo, Adv. Opt. Mater., 2018, 6, 1701076 CrossRef.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. Man Hong, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137–1140 CrossRef CAS PubMed.
- C.-F. Fu, X. Li, Q. Luo and J. Yang, J. Mater. Chem. A, 2017, 5, 24972–24980 RSC.
- D. Wang, Y. Bao, T. Wu, S. Gan, D. Han and L. Niu, Carbon, 2018, 134, 22–28 CrossRef CAS.
- W. Yuan, L. Cheng, Y. An, S. Lv, H. Wu, X. Fan, Y. Zhang, X. Guo and J. Tang, Adv. Sci., 2018, 5, 1700870 CrossRef PubMed.
- M. H. Tran, T. Schäfer, A. Shahraei, M. Dürrschnabel, L. Molina-Luna, U. I. Kramm and C. S. Birkel, ACS Appl. Energy Mater., 2018, 1, 3908–3914 CrossRef CAS.
- P. Li, J. Zhu, A. D. Handoko, R. Zhang, H. Wang, D. Legut, X. Wen, Z. Fu, Z. W. Seh and Q. Zhang, J. Mater. Chem. A, 2018, 6, 4271–4278 RSC.
- X. An, W. Wang, J. Wang, H. Duan, J. Shi and X. Yu, Phys. Chem. Chem. Phys., 2018, 20, 11405–11411 RSC.
- X. Zhang, Z. Zhang, D. Wu, X. Zhang, X. Zhao and Z. Zhou, Small Methods, 2018, 2, 1700359 CrossRef.
- J. Ran, G. Gao, F. T. Li, T. Y. Ma, A. Du and S. Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS PubMed.
- C.-F. Du, K. N. Dinh, Q. Liang, Y. Zheng, Y. Luo, J. Zhang and Q. Yan, Adv. Energy Mater., 2018, 8, 1801127 CrossRef.
- C. Peng, P. Wei, X. Li, Y. Liu, Y. Cao, H. Wang, H. Yu, F. Peng, L. Zhang, B. Zhang and K. Lv, Nano Energy, 2018, 53, 97–107 CrossRef CAS.
- Y. Sun, D. Jin, Y. Sun, X. Meng, Y. Gao, Y. Dall’Agnese, G. Chen and X.-F. Wang, J. Mater. Chem. A, 2018, 6, 9124–9131 RSC.
- N. H. Attanayake, S. C. Abeyweera, A. C. Thenuwara, B. Anasori, Y. Gogotsi, Y. Sun and D. R. Strongin, J. Mater. Chem. A, 2018, 6, 16882–16889 RSC.
- L. Arnarson, P. S. Schmidt, M. Pandey, A. Bagger, K. S. Thygesen, I. E. L. Stephens and J. Rossmeisl, Phys. Chem. Chem. Phys., 2018, 20, 11152–11159 RSC.
- H. Wang, Y. Wu, X. Yuan, G. Zeng, J. Zhou, X. Wang and J. W. Chew, Adv. Mater., 2018, 30, e1704561 CrossRef PubMed.
- Y. Xu, S. Wang, J. Yang, B. Han, R. Nie, J. Wang, Y. Dong, X. Yu, J. Wang and H. Jing, J. Mater. Chem. A, 2018, 6, 15213–15220 RSC.
- R. Morales-Salvador, A. Morales-Garcia, F. Vines and F. Illas, Phys. Chem. Chem. Phys., 2018, 20, 17117–17124 RSC.
- Á. Morales-García, A. Fernández-Fernández, F. Viñes and F. Illas, J. Mater. Chem. A, 2018, 6, 3381–3385 RSC.
- C. Cheng, X. Zhang, Z. Yang and Z. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 32903–32912 CrossRef CAS PubMed.
- C. Cheng, X. Zhang, M. Wang, S. Wang and Z. Yang, Phys. Chem. Chem. Phys., 2018, 20, 3504–3513 RSC.
- M. Shao, Y. Shao, W. Chen, K. L. Ao, R. Tong, Q. Zhu, I. N. Chan, W. F. Ip, X. Shi and H. Pan, Phys. Chem. Chem. Phys., 2018, 20, 14504–14512 RSC.
- Q. Liu, L. Ai and J. Jiang, J. Mater. Chem. A, 2018, 6, 4102–4110 RSC.
- Z. Li, Z. Zhuang, F. Lv, H. Zhu, L. Zhou, M. Luo, J. Zhu, Z. Lang, S. Feng, W. Chen, L. Mai and S. Guo, Adv. Mater., 2018, 1803220, DOI:10.1002/adma.201803220.
- S. H. Overbury, A. I. Kolesnikov, G. M. Brown, Z. Zhang, G. S. Nair, R. L. Sacci, R. Lotfi, A. C. T. van Duin and M. Naguib, J. Am. Chem. Soc., 2018, 140, 10305–10314 CrossRef CAS PubMed.
- Z. Li, Y. Cui, Z. Wu, C. Milligan, L. Zhou, G. Mitchell, B. Xu, E. Shi, J. T. Miller, F. H. Ribeiro and Y. Wu, Nat. Catal., 2018, 1, 349–355 CrossRef.
- A. Ali, K. Hantanasirisakul, A. Abdala, P. Urbankowski, M. Q. Zhao, B. Anasori, Y. Gogotsi, B. Aissa and K. A. Mahmoud, Langmuir, 2018, 34, 11325–11334 CrossRef CAS PubMed.
- C. Wang, H. Xie, S. Chen, B. Ge, D. Liu, C. Wu, W. Xu, W. Chu, G. Babu, P. M. Ajayan and L. Song, Adv. Mater., 2018, 30, e1802525 CrossRef PubMed.
- Y. T. Du, X. Kan, F. Yang, L. Y. Gan and U. Schwingenschlogl, ACS Appl. Mater. Interfaces, 2018, 10, 32867–32873 CrossRef CAS PubMed.
- R. Meng, J. Huang, Y. Feng, L. Zu, C. Peng, L. Zheng, L. Zheng, Z. Chen, G. Liu, B. Chen, Y. Mi and J. Yang, Adv. Energy Mater., 2018, 8, 1801514 CrossRef.
- M. Lu, W. Han, H. Li, W. Shi, J. Wang, B. Zhang, Y. Zhou, H. Li, W. Zhang and W. Zheng, Energy Storage Mater., 2019, 16, 163–168 CrossRef.
- Y. T. Liu, P. Zhang, N. Sun, B. Anasori, Q. Z. Zhu, H. Liu, Y. Gogotsi and B. Xu, Adv. Mater., 2018, 30, e1707334 CrossRef PubMed.
- Y. Aierken, C. Sevik, O. Gülseren, F. M. Peeters and D. Çakır, J. Mater. Chem. A, 2018, 6, 2337–2345 RSC.
- Y. Wang, Y. Li, Z. Qiu, X. Wu, P. Zhou, T. Zhou, J. Zhao, Z. Miao, J. Zhou and S. Zhuo, J. Mater. Chem. A, 2018, 6, 11189–11197 RSC.
- P. Yu, G. Cao, S. Yi, X. Zhang, C. Li, X. Sun, K. Wang and Y. Ma, Nanoscale, 2018, 10, 5906–5913 RSC.
- Z. Chen, T. Yuan, X. Pu, H. Yang, X. Ai, Y. Xia and Y. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 11689–11698 CrossRef CAS PubMed.
- X. Guo, J. Zhang, J. Song, W. Wu, H. Liu and G. Wang, Energy Storage Mater., 2018, 14, 306–313 CrossRef.
- J. Li, D. Yan, S. Hou, Y. Li, T. Lu, Y. Yao and L. Pan, J. Mater. Chem. A, 2018, 6, 1234–1243 RSC.
- Q. Meng, J. Ma, Y. Zhang, Z. Li, A. Hu, J.-J. Kai and J. Fan, J. Mater. Chem. A, 2018, 6, 13652–13660 RSC.
- Q. Meng, J. Ma, Y. Zhang, Z. Li, C. Zhi, A. Hu and J. Fan, Nanoscale, 2018, 10, 3385–3392 RSC.
- Y. Wu, P. Nie, L. Wu, H. Dou and X. Zhang, Chem. Eng. J., 2018, 334, 932–938 CrossRef CAS.
- J. Luo, C. Fang, C. Jin, H. Yuan, O. Sheng, R. Fang, W. Zhang, H. Huang, Y. Gan, Y. Xia, C. Liang, J. Zhang, W. Li and X. Tao, J. Mater. Chem. A, 2018, 6, 7794–7806 RSC.
- X. Li, Y. Qian, T. Liu, F. Cao, Z. Zang, X. Sun, S. Sun, Q. Niu and J. Wu, J. Mater. Sci., 2018, 53, 11078–11090 CrossRef CAS.
- H. Tang, W. Li, L. Pan, C. P. Cullen, Y. Liu, A. Pakdel, D. Long, J. Yang, N. McEvoy, G. S. Duesberg, V. Nicolosi and C. J. Zhang, Adv. Sci., 2018, 5, 1800502 CrossRef PubMed.
- Z.-W. Zhang, H.-J. Peng, M. Zhao and J.-Q. Huang, Adv. Funct. Mater., 2018, 28, 1707536 CrossRef.
- X. T. Gao, Y. Xie, X. D. Zhu, K. N. Sun, X. M. Xie, Y. T. Liu, J. Y. Yu and B. Ding, Small, 2018, e1802443, DOI:10.1002/smll.201802443.
- W. Cai, G. Li, K. Zhang, G. Xiao, C. Wang, K. Ye, Z. Chen, Y. Zhu and Y. Qian, Adv. Funct. Mater., 2018, 28, 1704865 CrossRef.
- Y. Dong, S. Zheng, J. Qin, X. Zhao, H. Shi, X. Wang, J. Chen and Z. S. Wu, ACS Nano, 2018, 12, 2381–2388 CrossRef CAS PubMed.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- M. Xu, S. Lei, J. Qi, Q. Dou, L. Liu, Y. Lu, Q. Huang, S. Shi and X. Yan, ACS Nano, 2018, 12, 3733–3740 CrossRef CAS PubMed.
- C. Han, H. Li, R. Shi, L. Xu, J. Li, F. Kang and B. Li, Energy Environ. Mater., 2018, 1, 75–87 CrossRef.
- V. Aravindan and Y. S. Lee, J. Phys. Chem. Lett., 2018, 9, 3946–3958 CrossRef CAS PubMed.
- N. Kurra, M. Alhabeb, K. Maleski, C.-H. Wang, H. N. Alshareef and Y. Gogotsi, ACS Energy Lett., 2018, 3, 2094–2100 CrossRef CAS.
- X. Dong, Y. Zhang, B. Ding, X. Hao, H. Dou and X. Zhang, J. Power Sources, 2018, 390, 208–214 CrossRef CAS.
- Q. X. Xia, N. M. Shinde, T. Zhang, J. M. Yun, A. Zhou, R. S. Mane, S. Mathur and K. H. Kim, Dalton Trans., 2018, 47, 8676–8682 RSC.
- D. Fan, S. Lu, Y. Guo and X. Hu, J. Phys. Chem. C, 2018, 122, 15118–15124 CrossRef CAS.
- S. Chen, Z. Fu, H. Zhang, D. Legut, T. C. Germann, Q. Zhang, S. Du, J. S. Francisco and R. Zhang, Adv. Funct. Mater., 2018, 1804867, DOI:10.1002/adfm.201804867.
- K. S. Kumar, N. Choudhary, Y. Jung and J. Thomas, ACS Energy Lett., 2018, 3, 482–495 CrossRef CAS.
- Z. Fan, Y. Wang, Z. Xie, D. Wang, Y. Yuan, H. Kang, B. Su, Z. Cheng and Y. Liu, Adv. Sci., 2018, 1800750, DOI:10.1002/advs.201800750.
- C. Cui, M. Hu, C. Zhang, R. Cheng, J. Yang and X. Wang, Chem. Commun., 2018, 54, 8132–8135 RSC.
- M. Hu, T. Hu, Z. Li, Y. Yang, R. Cheng, J. Yang, C. Cui and X. Wang, ACS Nano, 2018, 12, 3578–3586 CrossRef CAS PubMed.
- A. Jain, Y. Shin and K. A. Persson, Nat. Rev. Mater., 2016, 1, 15004 CrossRef CAS.
- P. Nayak, Q. Jiang, R. Mohanraman, D. Anjum, M. N. Hedhili and H. N. Alshareef, Nanoscale, 2018, 10, 17030–17037 RSC.
- L. Yu, L. Hu, B. Anasori, Y.-T. Liu, Q. Zhu, P. Zhang, Y. Gogotsi and B. Xu, ACS Energy Lett., 2018, 3, 1597–1603 CrossRef CAS.
- Z. Zhou, W. Panatdasirisuk, T. S. Mathis, B. Anasori, C. Lu, X. Zhang, Z. Liao, Y. Gogotsi and S. Yang, Nanoscale, 2018, 10, 6005–6013 RSC.
- P. Li, W. Shi, W. Liu, Y. Chen, X. Xu, S. Ye, R. Yin, L. Zhang, L. Xu and X. Cao, Nanotechnology, 2018, 29, 445401 CrossRef PubMed.
- T. H. Chang, T. Zhang, H. Yang, K. Li, Y. Tian, J. Y. Lee and P. Y. Chen, ACS Nano, 2018, 12, 8048–8059 CrossRef CAS PubMed.
- H. An, T. Habib, S. Shah, H. Gao, M. Radovic, M. J. Green and J. L. Lutkenhaus, Sci. Adv., 2018, 4, eaaq0118 CrossRef PubMed.
- D. P. Dubal, N. R. Chodankar, D. H. Kim and P. Gomez-Romero, Chem. Soc. Rev., 2018, 47, 2065–2129 RSC.
- M. Ha, S. Lim and H. Ko, J. Mater. Chem. B, 2018, 6, 4043–4064 RSC.
- Z. Wang, S. Qin, S. Seyedin, J. Zhang, J. Wang, A. Levitt, N. Li, C. Haines, R. Ovalle-Robles, W. Lei, Y. Gogotsi, R. H. Baughman and J. M. Razal, Small, 2018, e1802225, DOI:10.1002/smll.201802225.
- F. Deng, X. Li, F. Ding, B. Niu and J. Li, J. Phys. Chem. C, 2018, 122, 5325–5333 CrossRef CAS.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef PubMed.
- H. Hu, Z. Bai, B. Niu, M. Wu and T. Hua, J. Mater. Chem. A, 2018, 6, 14876–14884 RSC.
- Y. Yue, N. Liu, Y. Ma, S. Wang, W. Liu, C. Luo, H. Zhang, F. Cheng, J. Rao, X. Hu, J. Su and Y. Gao, ACS Nano, 2018, 12, 4224–4232 CrossRef CAS PubMed.
- B. D. Boruah, A. Maji and A. Misra, ACS Appl. Mater. Interfaces, 2018, 10, 15864–15872 CrossRef CAS PubMed.
- S. Zheng, W. Lei, J. Qin, Z.-S. Wu, F. Zhou, S. Wang, X. Shi, C. Sun, Y. Chen and X. Bao, Energy Storage Mater., 2018, 10, 24–31 CrossRef.
- W. Yuan, L. Cheng, H. Wu, Y. Zhang, S. Lv and X. Guo, Chem. Commun., 2018, 54, 2755–2758 RSC.
- M. Guo, C. Liu, Z. Zhang, J. Zhou, Y. Tang and S. Luo, Adv. Funct. Mater., 2018, 28, 1803196 CrossRef.
- S. A. Melchior, K. Raju, I. S. Ike, R. M. Erasmus, G. Kabongo, I. Sigalas, S. E. Iyuke and K. I. Ozoemena, J. Electrochem. Soc., 2018, 165, A501–A511 CrossRef CAS.
- R. Wang, S. Wang, Y. Zhang, D. Jin, X. Tao and L. Zhang, J. Mater. Chem. A, 2018, 6, 1017–1027 RSC.
- C. Yang, Y. Tang, Y. Tian, Y. Luo, Y. He, X. Yin and W. Que, Adv. Funct. Mater., 2018, 28, 1705487 CrossRef.
- M. E. Suss and V. Presser, Joule, 2018, 2, 10–15 CrossRef.
- Y. Xia, T. S. Mathis, M. Q. Zhao, B. Anasori, A. Dang, Z. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409–412 CrossRef CAS PubMed.
- W. Bao, X. Tang, X. Guo, S. Choi, C. Wang, Y. Gogotsi and G. Wang, Joule, 2018, 2, 778–787 CrossRef CAS.
- R. P. Pandey, K. Rasool, V. E. Madhavan, B. Aïssa, Y. Gogotsi and K. A. Mahmoud, J. Mater. Chem. A, 2018, 6, 3522–3533 RSC.
- J. Zhao, Y. Yang, C. Yang, Y. Tian, Y. Han, J. Liu, X. Yin and W. Que, J. Mater. Chem. A, 2018, 6, 16196–16204 RSC.
- L. Guo, X. Wang, Z. Y. Leong, R. Mo, L. Sun and H. Y. Yang, FlatChem, 2018, 8, 17–24 CrossRef CAS.
- F. Alimohammadi, M. Sharifian Gh, N. H. Attanayake, A. C. Thenuwara, Y. Gogotsi, B. Anasori and D. R. Strongin, Langmuir, 2018, 34, 7192–7200 CrossRef CAS PubMed.
- J. Shen, G. Liu, Y. Ji, Q. Liu, L. Cheng, K. Guan, M. Zhang, G. Liu, J. Xiong, J. Yang and W. Jin, Adv. Funct. Mater., 2018, 28, 1801511 CrossRef.
- A. Junkaew and R. Arroyave, Phys. Chem. Chem. Phys., 2018, 20, 6073–6082 RSC.
- L. Li, T. Zhang, Y. Duan, Y. Wei, C. Dong, L. Ding, Z. Qiao and H. Wang, J. Mater. Chem. A, 2018, 6, 11734–11742 RSC.
- L. Ding, Y. Wei, L. Li, T. Zhang, H. Wang, J. Xue, L. X. Ding, S. Wang, J. Caro and Y. Gogotsi, Nat. Commun., 2018, 9, 155 CrossRef PubMed.
- X. Han, J. Huang, H. Lin, Z. Wang, P. Li and Y. Chen, Adv. Healthcare Mater., 2018, 7, e1701394 CrossRef PubMed.
- L. Zong, H. Wu, H. Lin and Y. Chen, Nano Res., 2018, 11, 4149–4168 CrossRef CAS.
- Z. Liu, M. Zhao, H. Lin, C. Dai, C. Ren, S. Zhang, W. Peng and Y. Chen, J. Mater. Chem. B, 2018, 6, 3541–3548 RSC.
- H. Lin, Y. Chen and J. Shi, Adv. Sci., 2018, 1800518, DOI:10.1002/advs.201800518.
- K. Huang, Z. Li, J. Lin, G. Han and P. Huang, Chem. Soc. Rev., 2018, 47, 5109–5124 RSC.
- Q. Jiang, C. Wu, Z. Wang, A. C. Wang, J.-H. He, Z. L. Wang and H. N. Alshareef, Nano Energy, 2018, 45, 266–272 CrossRef CAS.
- F. Yi, H. Ren, J. Shan, X. Sun, D. Wei and Z. Liu, Chem. Soc. Rev., 2018, 47, 3152–3188 RSC.
- C. Yu, Y. Gong, R. Chen, M. Zhang, J. Zhou, J. An, F. Lv, S. Guo and G. Sun, Small, 2018, e1801203, DOI:10.1002/smll.201801203.
- Z. Fan, Y. Wang, Z. Xie, X. Xu, Y. Yuan, Z. Cheng and Y. Liu, Nanoscale, 2018, 10, 9642–9652 RSC.
- E. Kayali, A. VahidMohammadi, J. Orangi and M. Beidaghi, ACS Appl. Mater. Interfaces, 2018, 10, 25949–25954 CrossRef CAS PubMed.
Footnote |
| † J. P. and R. G. M. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |