
Development of organic semiconducting materials for deep-tissue optical imaging, phototherapy and photoactivation
Jingchao
Li
and
Kanyi
Pu
 *
*
School of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, 637457, Singapore. E-mail: kypu@ntu.edu.sg
First published on 2nd November 2018
Abstract
Biophotonics as a highly interdisciplinary frontier often requires the assistance of optical agents to control the light pathways in cells, tissues and living organisms for specific biomedical applications. Organic semiconducting materials (OSMs) composed of π-conjugated building blocks as the optically active components have recently emerged as a promising category of biophotonic agents. OSMs possess common features including excellent optical properties, good photostability and biologically benign composition. This review summarizes the recent progress in the development of OSMs based on small-molecule fluorophores, aggregation-induced emission (AIE) dyes and semiconducting oligomer/polymer nanoparticles (SONs/SPNs) for advanced biophotonic applications. OSMs have been exploited as imaging agents to transduce biomolecular interactions into second near-infrared fluorescence, chemiluminescence, afterglow or photoacoustic signals, enabling deep-tissue ultrasensitive imaging of biological tissues, disease biomarkers and physiological indexes. By fine-tuning the molecular structures, OSMs can also convert light energy into cytotoxic free radicals or heat, allowing for effective cancer phototherapy. Due to their instant light response and efficient light-harvesting properties, precise regulation of biological activities using OSMs as remote transducers has been demonstrated for protein ion channels, gene transcription and protein activation. In addition to highlighting OSMs as a multifunctional platform for a wide range of biomedical applications, current challenges and perspectives of OSMs in biophotonics are discussed.
 Jingchao Li | Jingchao Li received his PhD degree in Materials Science and Engineering from the University of Tsukuba (Japan) in 2017. Then he worked as a Postdoctoral Research Fellow in Prof. Kanyi Pu's group at the School of Chemical and Biomedical Engineering (SCBE), Nanyang Technological University (NTU). His current research focuses on the development of advanced agents for cancer theranostics. |
 Kanyi Pu | Kanyi Pu received his PhD from the National University of Singapore in 2011 followed by a postdoctoral study at Stanford University School of Medicine. He joined the School of Chemical and Biomedical Engineering (SCBE) at Nanyang Technological University (NTU) as an Associate Professor in 2015. His research lies at the intersection of chemistry, nanotechnology, biophotonics and medicine, aiming to develop smart molecular probes and advanced imaging technologies to understand, detect and treat life-threatening diseases. |
1. Introduction
Biophotonics that involves the study of light-activated biological processes at the molecular, cellular or tissue level is an interdisciplinary research frontier encompassing life science, optics and engineering.1 Significant progress in the application of biophotonics in modern medicine has been achieved in the past decade owing to the improvements of photonic technologies.2 For example, biophotonics has allowed us to accurately distinguish early-stage lesions with high spatial resolution ranging from the nanometer to the centimeter scale, longitudinally observe pathological events with an extremely broad temporal range from femtoseconds to days, sensitively detect disease biomarkers at a limit of detection (LOD) down to nanomoles, and precisely control therapeutic processes and biological activities.3,4Biophotonic applications often rely on optical materials that have light absorption and conversion capabilities. Advancement of chemistry and nanotechnology has led to many biophotonic agents, which include inorganic optical nanomaterials such as semiconducting quantum dots (QDs),5 silica nanoparticles,6 gold nanoparticles,7 carbon nanotubes,8 2D transition metal dichalcogenide nanosheets,9 nanographene oxide,10 and upconversion nanocomposites.11 They have shown the capabilities to transduce biomolecular interactions into optical signals to allow noninvasive disease diagnosis,12 as well as convert light energy into free radicals or thermal effect for localized cancer phototherapy,13 controlled cargo delivery,14 biological stimulation,15 and photochemical tissue binding.16 However, inorganic nanoparticles are excreted slowly after administration and are largely retained in the major organs of the reticuloendothelial system, such as the liver and spleen, potentially resulting in long-term toxicity.17 In sharp contrast, organic optical agents that are made from completely benign and biologically inert components have the potential to circumvent the toxicity concerns while possessing optical advantages equal or even superior to inorganic optical nanomaterials.18
As an emerging class of organic optical agents, organic semiconducting materials (OSMs) have been proven effective in biophotonics. OSMs can be characterized by small molecules, polymers or nanoparticles composed of electron-delocalized π-conjugated components. Typical OSMs include small molecular fluorophores, aggregation-induced emission (AIE) molecules/nanoparticles, and semiconducting oligomer/polymer nanoparticles (SONs/SPNs). The photophysical properties of OSMs are mainly determined by the chemical structures or aggregate states of the electron-delocalized π-conjugated components, and thus can be adjusted through rational molecular engineering. Until now, OSMs have been exploited as contrast agents for cell imaging and tracking,19 targeted tumor imaging,20 vascular imaging,21 lymph node imaging,22 drug release tracking,23 real-time monitoring of physiological indexes and biomarkers,24,25 and phototherapeutic agents for cancer and bacteria killing.26,27
In this review, we summarize the recent progress of OSMs as a class of optical agents for biophotonic applications: (i) deep-tissue molecular optical imaging including second near-infrared (NIR-II) fluorescence, chemiluminescence, afterglow and photoacoustic (PA) imaging, (ii) cancer phototherapy, and (iii) biological photoactivation (Fig. 1). The molecular or nanoparticle design principles of OSMs are discussed along with their key applications in optical molecular imaging with an emphasis on the strategies to amplify brightness, improve tissue penetration depth and enhance signal-to-noise ratios. Next, rational designs and examples of OSMs as therapeutic nanoagents for amplified or combinational cancer phototherapy are discussed. Then, the potential of OSMs as light transducers is revealed using the examples of regulating different biological activities. Finally, a brief summary is given together with the discussion of current challenges and perspectives in this field.
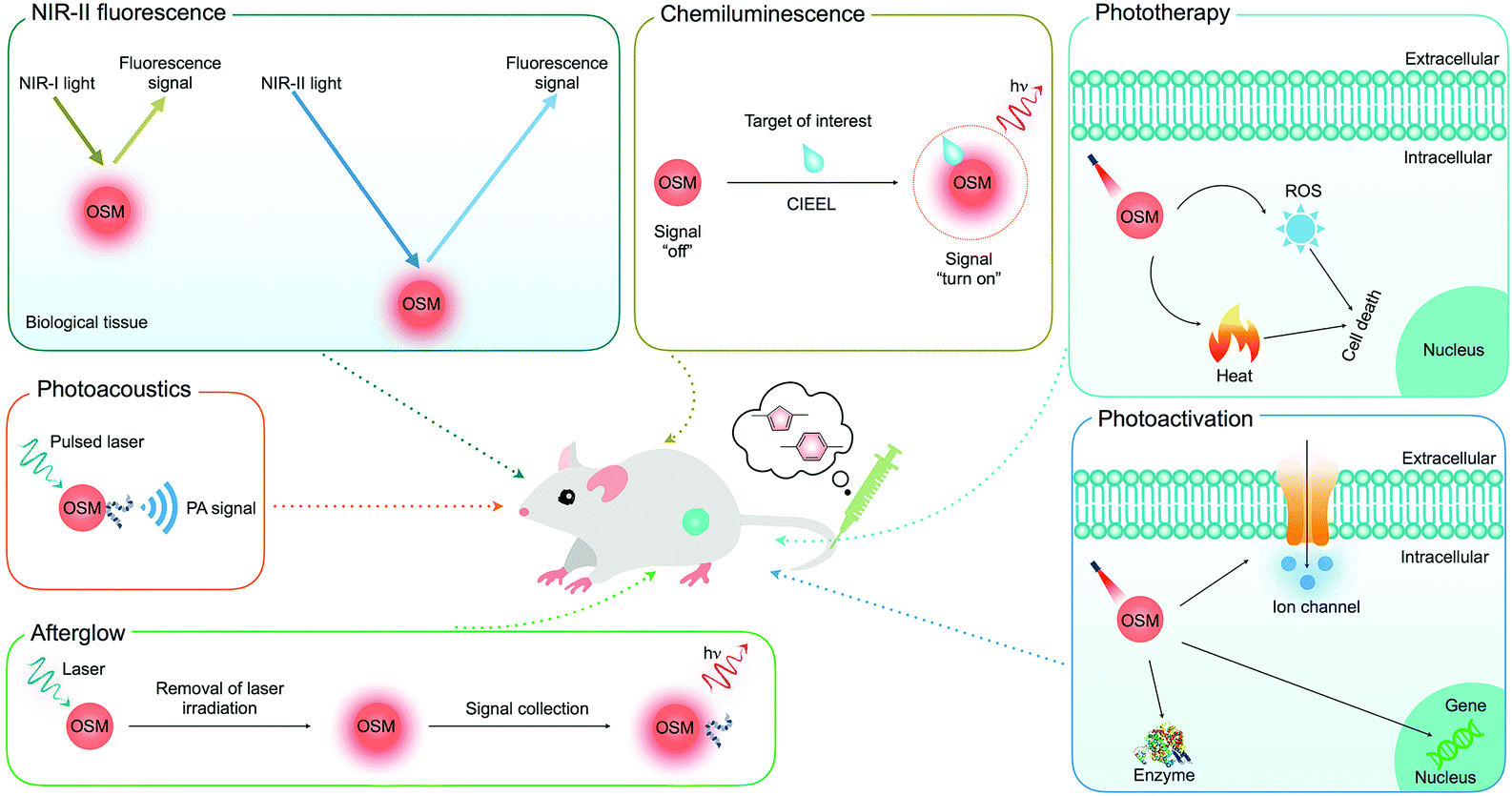 | ||
| Fig. 1 Summary of the applications of OSMs for deep-tissue optical imaging (NIR-II fluorescence, chemiluminescence, afterglow and photoacoustics), phototherapy and biological photoactivation. | ||
2. NIR-II fluorescence imaging
Fluorescence imaging is a convenient and cost-effective method for biological analysis, clinical diagnosis, therapeutic monitoring and drug discovery.28,29In vivo fluorescence imaging is performed with light excitation in different regions, which include the visible region (400–700 nm), the conventional first NIR region (NIR-I, 700–1000 nm) and the second NIR region (NIR-II, 1000–1800 nm).30 Because visible light has shallow tissue penetration depth (∼1 mm) and can be significantly distributed by tissue autofluorescence,31 the general research interest in fluorescence imaging has been focused in the NIR-I region, defined as the “biological transparency NIR window”,32 allowing for a light penetration depth of 1–3.5 mm.33 However, NIR-I fluorescence imaging still has the intrinsic issues of relatively high tissue autofluorescence and unsatisfactory imaging depth. By contrast, NIR-II fluorescence imaging has significantly diminished background noise and improved tissue penetration depth (5–20 mm) due to the minimal photon scattering and low tissue absorption of NIR-II light.34 With these merits, NIR-II fluorescence imaging is able to visualize biological molecules and tissue structures in deep tissue that are otherwise difficult to achieve for NIR-I imaging.35Because not many optical agents can emit light in the NIR-II window,36 Dai's and other groups have made efforts to design and develop NIR-II fluorescent agents, including inorganic nanoparticles such as single-walled carbon nanotubes (SWCNTs),37–39 QDs,40–44 and rare-earth doped nanoparticles,45 and OSMs composed of organic semiconducting nanoparticles and small molecular fluorophores (Table 1).36,46,47 In contrast to inorganic nanoparticles, OSMs are more desirable and optimal candidates for NIR-II fluorescence imaging mainly because their optical properties, pharmacokinetics, and biodistribution can be easily tuned to fulfill diverse applications.
| Probes | Excitation/emission | QY (%) | Imaging applications | Ref. |
|---|---|---|---|---|
| SWCNTs | 785 nm/1100–1400 nm | 0.4 | Vascular, deep-tissue anatomical and cancer metastasis | 37–39 |
| Ag2S QDs | 808 nm/1100–1400 nm | 15.5 | Tumor, lymphatic drainage and vascular networks | 40 and 41 |
| Ag2Se QDs | 785 nm/1300 nm | 29.4 | Deep organs and vascular structures | 42 |
| PbS QDs | 450 nm/1000–1200 nm | 6–16 | Breast tumor and lymph node | 43 and 44 |
| LaF3:Nd nanoparticles | 808 nm/910, 1050 and 1340 nm | 50 | Internal tissue | 45 |
| IR-1061-PEG nanoparticles | 808 or 980 nm/1000–1400 nm | 1.8 | Inner organs and blood vessels | 48 |
| LbL-IR1061 nanoparticles | 808 nm/1100 nm | 1.8 | Evaluation of biodistribution, pharmacokinetics, and toxicities | 30 |
| CQS1000 nanoparticles | 808 nm/1000 nm | — | Circulatory systems and imaging-guided tumor resection and sentinel lymph node biopsy | 49 |
| TQ-BPN AIE nanoparticles | 635 nm/900–1200 nm | 2.8 | Brain capillaries, blood–brain barrier damage and EPR effect in tumor | 51 |
| SPN1 | 808 nm/1050–1350 nm | 1.7 | Cancer cells and arterial blood flow | 52 |
| SCH1100 | 808 nm/1100 nm | 0.2 | Prostate cancers | 46 |
| FD-1080 | 1064 nm/1080 nm | 0.31 | Hindlimb vasculature, brain vessel, respiratory craniocaudal motion of liver | 47 |
| CH1055 | 808 nm/1055 nm | 0.3 | SLN, orthotopic glioblastoma brain tumor, brain vasculature and imaging guided tumor surgery | 56 |
| IR-E1 | 808 nm/1071 nm | 0.7 | TBI-induced hemodynamic abnormalities and cerebrovascular damage | 57 |
| IR-FEP | 808 nm/910 and 1100 nm | 2.0 | Blood flow in hindlimb and tumors | 58 |
| IR-FEPC | 808 nm/1100 nm | 2.6 | Adult female ovary in living mice | 59 |
| IR-FG | 808 nm/1050 nm | 1.9 | Histological tissue staining | 60 |
| IR-FTAP | 808 nm/1048 nm | 5.3 | Blood vessels of a mouse hindlimb | 61 |
| NIR-II@Si | 780 nm/900–1300 nm | 0.37 | H2S in tumor | 64 |
| HISSNPs | 808 nm/900–1500 nm | — | Tumor microenvironment | 65 |
2.1. NIR-II fluorescent nanoparticles
To generate NIR-II fluorescent organic nanoparticles, hydrophobic fluorophores or molecules have been transformed into water-soluble SONs through nanoprecipitation (Fig. 2a). For instance, Dai and co-workers embedded a commercially available water-insoluble IR-1061 dye (Scheme 1) into a poly(acrylic acid) (PAA) matrix with a surface coating of an amphiphilic polymer phospholipid-conjugated polyethylene glycol (DSPE-mPEG) to generate IR-1061-PEG nanoparticles that displayed a major fluorescence emission peak at 920 nm and a shoulder peak at 1064 nm.48 This simple embedding process afforded a relatively high fluorescence quantum yield (QY) of 1.8% for IR-1061-PEG nanoparticles. Intravenous injection of IR-1061-PEG nanoparticles allowed for NIR-II fluorescence imaging of different inner organs including the lungs and kidneys, and subcutaneous arteries in the hindlimb thigh and abdomen of living mice. In another study, Cheng and co-workers subsequently synthesized a NIR-II absorbing small molecule, 5,5′-((1H,5H-benzo[1,2-c:4,5-c′]bis[1,2,5]thiadiazole)-4,8-diyl)bis(N,N-bis(4-(3-((tert-butyldimethylsilyl)oxy)propyl)phenyl)thiophen-2-amine) (CQ1, Fig. 2b), and encapsulated it into phospholipids to yield CQS1000 nanoparticles as imaging probes.49 With a NIR-II fluorescence emission at 1000 nm, CQS1000 nanoparticles allowed for noninvasive and dynamical visualization and monitoring of circulatory systems, including the hindlimb and cerebral blood vessels, angiogenesis of tumors, arterial thrombus formation and incomplete hindlimb ischemia, and lymphatic drainage and routing, as well as imaging-guided precise resection of tumors and sentinel lymph node mapping and biopsy.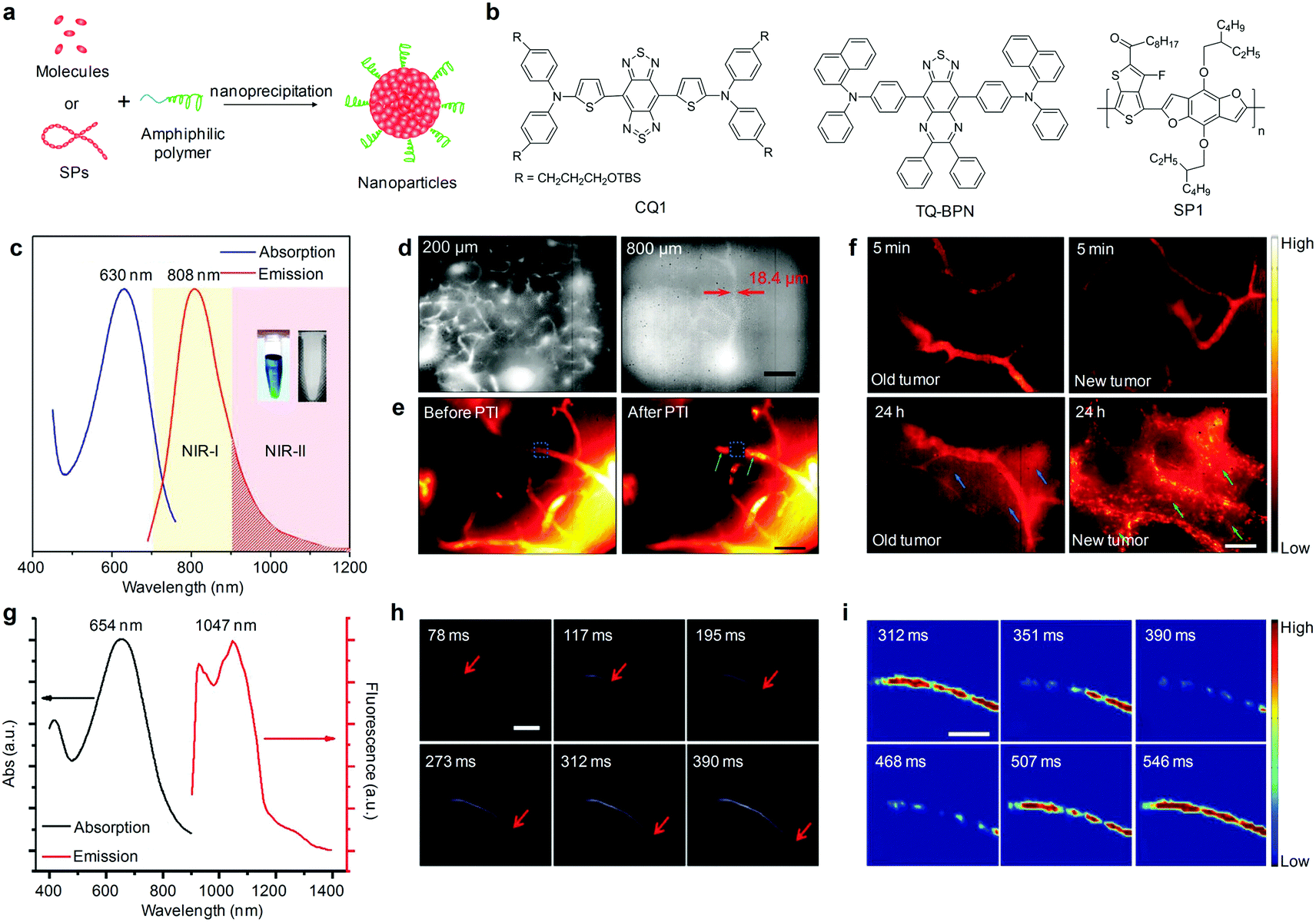 | ||
| Fig. 2 (a) Schematic illustration of the preparation of organic nanoparticles via nanoprecipitation. (b) Chemical structures of CQ1, TQ-BPN and SP1 used for preparation of SONs, AIE nanoparticles and SPNs for NIR-II fluorescence imaging. (c) UV-Vis absorption and photoluminescence (PL) spectra of TQ-BPN AIE nanoparticles dispersed in water. The inset shows the photographs of the bright-field (left) and NIR-II (right) images of the nanoparticles in aqueous dispersion. (d) In vivo real-time fluorescence microscopic imaging of mouse brain vasculature at different depths (200 and 800 μm) after the intravenous injection of TQ-BPN AIE nanoparticles. (e) NIR-II fluorescence microscopic images and corresponding heat maps of brain blood vessels before and after PTI induction using TQ-BPN AIE nanoparticles. (f) NIR-II fluorescence microscopic imaging for visualizing the EPR effect in old (left) and new (right) tumors at different time points after intravenous injection of TQ-BPN AIE nanoparticles. (g) UV-Vis absorption and fluorescence emission spectra of SPN1. (h) A time course of NIR-II fluorescence images of a mouse hindlimb immediately after intravenous injection of SPN1, showing the blood flow front moving inside the femoral artery (indicated by red arrows). (i) Time course NIR-II fluorescence images of a mouse femoral artery after subtracting the background. (c)–(f) were reproduced from ref. 51 with permission from John Wiley & Sons Ltd, copyright 2018. (g)–(i) were reproduced from ref. 52 with permission from Nature Publishing Group, copyright 2014. | ||
 | ||
| Scheme 1 Chemical structures of organic small molecules, fluorophores and semiconducting oligomers (SOs) used for preparation of OSMs. | ||
AIE nanoparticles, a class of organic luminophores showing enhanced brightness upon aggregation,50 have been another candidate for NIR-II fluorescence imaging. Tang's group recently described the use of AIE nanoparticles assembled from a small molecular TQ-BPN (Fig. 2a and b) as contrast agents for angiography in mouse brain and tumor.51 The formed TQ-BPN AIE nanoparticles showed an absorption peak at 630 nm and emitted light in a broad range of 700–1200 nm (Fig. 2c). Owing to the long-wavelength emission and high fluorescence brightness (QY = 2.8%), TQ-BPN AIE nanoparticles allowed direct visualization of the brain vasculature with high temporal resolution (25 frames per second) and spatial resolution (2.6 μm) and deep tissue penetration up to 800 μm (Fig. 2d). In vivo real-time microscopic angiography using TQ-BPN AIE nanoparticles as the NIR-II fluorescent probes enabled monitoring of the photothrombotic ischemia (PTI) induced blood–brain barrier (BBB) damage in a mouse brain (Fig. 2e), and visualization of the enhanced permeability and retention (EPR) effect in tumor sites (Fig. 2f).
In addition to SONs and AIE nanoparticles, SPNs made from hydrophobic π-conjugated semiconducting polymers (SPs) through nanoprecipitation have been used as NIR-II fluorescence imaging agents (Fig. 2a). Utilizing a donor–acceptor (D–A) alternating copolymerization reaction, Dai and co-workers synthesized poly(benzo[1,2-b:3,4-b0]difuran-alt-fluorothieno-[3,4-b]thiophene) (SP1, Fig. 2b) and formulated it into SPN1 with the assistance of DSPE-mPEG.52 SPN1 had a high QY (≈1.7%) and showed an absorption peak at 654 nm and an emission peak at 1048 nm with a large Stokes shift (≈400 nm) (Fig. 2g). The use of SPN1 allowed fluorescence imaging of living cells in the >1000 nm window for the first time, and enabled real-time tracking of arterial blood flow in a mouse hindlimb with an exposure time as short as 20 ms and an ultrafast frame rate of 25 frames per second in the NIR-II window (Fig. 2h), as well as facilitated spatiotemporally resolved imaging of the blood flow pattern in a cardiogram waveform over a single cardiac cycle (∼200 ms) of a mouse (Fig. 2i).
2.2. NIR-II fluorescent small molecular fluorophores
To achieve rapid renal clearance of imaging agents, a significant amount of efforts have been made to synthesize small molecular fluorophores with size below the renal filtration threshold of ∼40 kD (5.5 nm).53–55 The first NIR-II fluorescent small molecular fluorophore was developed by Dai and co-workers and utilized for NIR-II fluorescence imaging guided surgery.56 A donor–acceptor–donor (D–A–D) structured CH1055 composed of benzobisthiadiazole (BBTD) as the acceptor and triphenylamine as the donor was PEGylated to yield water soluble CH1055-PEG (Fig. 3a), showing maximum excitation at ∼750 nm and maximum emission at 1055 nm with a tail extending into the NIR-II region. With a relatively small size (8.9 kD), CH1055-PEG displayed a rapid renal clearance with ∼90% removal through urine within 24 h after intravenous injection, while nearly no accumulation was observed in the liver tissue (Fig. 3b). In striking contrast, the injected SWCNTs had slower pharmacokinetics and were almost retained in the liver and spleen. In addition, CH1055-PEG showed a high signal contrast in sentinel lymph node (SLN) mapping, which enabled selective removal of SLN to alleviate lymphoedema and prevent cancer metastasis (Fig. 3c). NIR-II fluorescence imaging of an orthotopic glioblastoma brain tumor at a depth of ∼4 mm was also performed using CH1055-PEG (Fig. 3d). After 72 h of intravenous injection, the tumor was clearly visualized with a tumor-to-normal tissue (T/NT) ratio of 5.5. Imaging of brain vasculature in living mice was also carried out, showing that the imaging quality of CH1055-PEG in the NIR-II window was obviously higher than that of indocyanine green (ICG) in the NIR-I window (Fig. 3e). CH1055 was attached with a small protein antiepidermal growth factor receptor (anti-EGFR) affibody for passive tumor accumulation (Fig. 3a). Such a chemical bioconjugation not only facilitated targeted NIR-II fluorescence imaging of EGFR-overexpressed squamous cell carcinoma (SAS) tumors with an unprecedentedly high T/NT ratio of up to ∼15 (Fig. 3f), but also permitted the first proof-of-concept NIR-II imaging guided tumor excision surgery (Fig. 3g).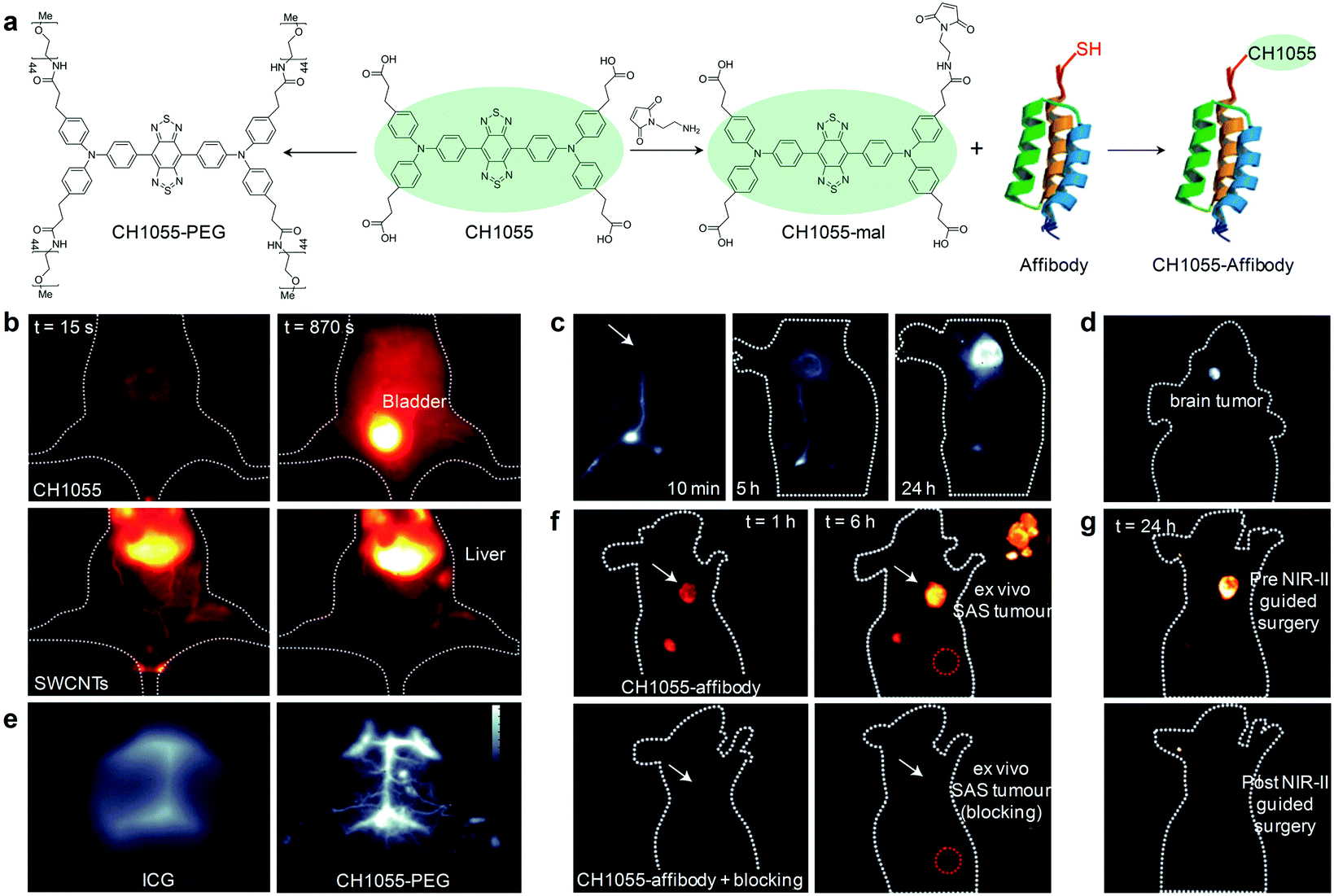 | ||
| Fig. 3 (a) Chemical structure of CH1055 and schematic illustration of the preparation of CH1055-PEG and CH1055-affibody. (b) NIR-II fluorescence imaging (1200 nm long-pass filter, 100 ms) of a mouse in the supine position at 15 and 870 s after the intravenous injection of CH1055-PEG or SWCNTs. (c) NIR-II fluorescence imaging (1200 nm long-pass filter, 200 ms) of a nude mouse with a 4T1 xenograft tumor at 10 min, 5 h and 24 h after the intradermal injection of CH1055-PEG near the base of the tail. White arrow points to the tumor location. (d) NIR-II fluorescence imaging (1200 nm long-pass filter, 800 ms) of glioblastoma brain tumor at 24 h post intravenous injection of CH1055-PEG. (e) NIR-II fluorescence imaging of brain vasculature through the scalp and skull in C57BL/6 mice with shaved heads using either ICG or CH1055-PEG. The greyscale bar corresponds to all NIR-II images. (f) NIR-II fluorescence imaging of SAS tumor at 1 and 6 h after intravenous injection of CH1055-affibody (60 μg). The upper panel shows clear tumor targeting and the bottom panel shows mice injected with a blocking dose consisting of CH1055-affibody plus free affibody concomitantly intravenously injected. The inset shows the SAS tumor ex vivo after excision in both targeted and blocked mice. The red circle corresponds to the normal tissue region used to calculate the T/NT ratio. (g) Representative images of a mouse at 24 h post-injection before (upper) and after (bottom) performing NIR-II fluorescence image-guided surgery to excise the tumors. Reproduced from ref. 56 with permission from Nature Publishing Group, copyright 2016. | ||
Despite the promising example of CH1055, the applications of small molecular fluorophores in NIR-II fluorescence imaging are still limited mainly due to their low fluorescence QYs (0.2–0.31%) in aqueous solutions, attributed to the strong interactions between the π-conjugated segments and environmental water molecules.46,47,56 Thus, rational design of molecular structures has been performed to develop new fluorophores with improved fluorescence brightness. Replacing the triphenylamine in CH1055 with bulky 3,4-ethoxylene dioxythiophene (EDOT) as the donor generated a novel NIR-II fluorophore (IR-E1, Scheme 1).57 Such a replacement reduced the inter- and intra-molecular interactions between the π-conjugated backbones, increasing the QY to 0.7% at 1071 nm in aqueous solution, more than twice as high as that of CH1055 (0.3%). However, this improvement is still insufficient to provide high-quality imaging performances.
To significantly improve the fluorescence brightness, shielding units have been introduced to construct shielding unit–donor–acceptor–donor–shielding unit (S–D–A–D–S) type NIR-II small molecular fluorophores. For instance, Dai and co-workers synthesized a S–D–A–D–S structured fluorophore (IR-FE, Scheme 1) with BBTD, EDOT and dialkyl-fluorene serving as the acceptor, donor and shielding unit, respectively.58,59 The introduction of dialkyl-fluorene as a shielding unit protected the π-conjugated backbone from intermolecular interactions and reduced aggregation, resulting in an improved QY of 2.0–2.6% in aqueous solution. In addition to the introduction of dialkyl-fluorene as the shielding unit, incorporation of tert(ethylene glycol) (TEG)-substituted thiophene as the bridging donor unit afforded another S–D–A–D–S structured NIR-II fluorophore (IR-FG, Scheme 1).60 In such a molecule, TEG substitution could distort the conjugated backbone and increase the dihedral angle between the acceptor and donor, reducing the molecular interactions and increasing the QY to 1.9%.
The QYs of small molecular fluorophores could be further increased by introducing two donor units into the π-conjugated backbones to have a S–D2–D1–A–D1–D2–S structure.61 This molecular engineering on the donor units has allowed the generation of a novel NIR-II molecular fluorophore IR-FTAP (Scheme 1), in which the alkyl thiophene and thiophene were employed as the first and second donors to connect the acceptor unit (BBTD) and the shielding unit (dialkyl-fluorene), respectively. Such a structure afforded a larger distortion of the π-conjugated backbone and further reduced intermolecular interactions, resulting in an enhanced QY of 5.3% for IR-FTAP in aqueous solution.
In addition to these chemical strategies, simple binding of small molecular fluorophores with proteins can improve the QYs.47 This should be due to the fact that the aggregates of fluorophores are hindered by the protein binding and therefore a rigid fluorophore conformation can be maintained to minimize torsion and consequently minimize non-radiative decay.47 For example, the QYs of FD-1080 (Scheme 1) could be increased from 0.31% to 5.94% after binding with fetal bovine serum (FBS).47 Besides the high-resolution deep-tissue NIR-II fluorescence imaging of hindlimb vasculature and brain vessel, the FD-1080-FBS complexes could be utilized for NIR-II fluorescence dynamic imaging of the liver in living mice to quantify the respiratory rate. In another study, FBS binding afforded an around 6-fold increment in the QYs for a cyanine dye that displayed tail emission in the 1000–1300 nm range.62 The NIR-II fluorescence imaging of this cyanine dye was used to image the liver and intestine clearly in a CD-1 mouse after administration, while it was not possible using NIR-I imaging.
2.3. NIR-II fluorescent activatable probes
NIR-II fluorescence imaging has exhibited unprecedented advantages over the traditional NIR-I fluorescence imaging, but the aforementioned agents fail to provide specific and sensitive signals toward targets of interest, leading to poor signal-to-background ratios (SBRs).63 Thus, activatable NIR-II fluorescent probes that can produce signals only in response to specific disease biomarkers or biological events have been recently proposed.63 As an example, Tian and co-workers devised an activatable organic nanoprobe that displayed hydrogen sulphide (H2S)-activated ratiometric fluorescence and NIR-II emission for the deep-tissue imaging of H2S-rich colorectal cancers.64 Such a nanoprobe (termed as NIR-II@Si) was fabricated by trapping a H2S-responsive boron-dipyrromethene (ZX-NIR) dye and a H2S inert aza-BODIPY (aza-BOD) dye into the hydrophobic interior of silica coated micelles (Fig. 4a and b). Upon reaction with H2S, the fluorescence emission at 600 nm was quenched, while that at 700 nm showed no appreciable change under excitation at 520 nm, allowing for ratiometric fluorescence detection (I700/I600) of H2S with a LOD of 37 nM. Moreover, this H2S specific response triggered a NIR-II fluorescence “turn-on” under NIR laser irradiation (Fig. 4c and d). The H2S-activated NIR-II fluorescence signals were clearly detected even at a depth of 10 mm, while the NIR-I fluorescence signals were only visible below 3 mm. By using NIR-II@Si as the activatable and target specific nanoprobe, deep-tissue NIR-II fluorescence imaging of H2S-rich colorectal HCT116 tumors in animal models was achieved with a NIR-II signal ratio between the tumor and normal site (T/N) as high as 5.7 (Fig. 4e–g). In sharp contrast, no NIR-II fluorescence signals could be detected in the HepG2 tumors because of the low level of H2S, even though NIR-I fluorescence signals were observed (Fig. 4f).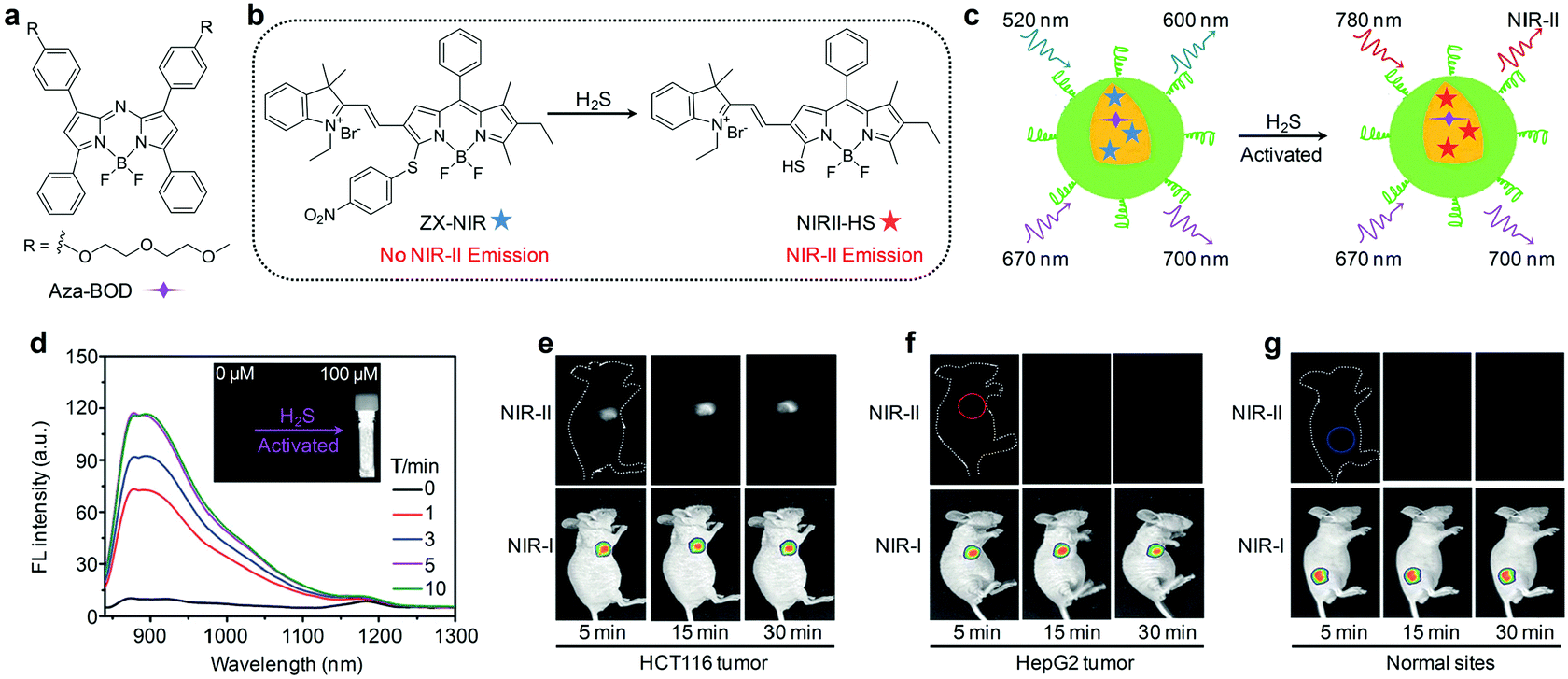 | ||
| Fig. 4 (a) Chemical structure of aza-BOD. (b) Chemical structures of ZX-NIR and NIRII-HS. (c) Mechanism of H2S activated NIR-II fluorescence signals. (d) Time-dependent NIR-II fluorescence spectra of NIR-II@Si in the presence of H2S under excitation at 780 nm. The inset shows the photograph of the H2S-activated NIR-II emission. NIR-I and NIR-II fluorescence imaging of HCT116 tumors (e), HepG2 tumors (f) and normal tissues (g) after subcutaneous injection of NIR-II@Si. Reproduced from ref. 64 with permission from John Wiley & Sons Ltd, copyright 2018. | ||
Because most biomarkers are often expressed in multiple types of cells, activatable probes may encounter the issue of nonspecific activation and are unable to accurately identify the complex and dynamic pathological environment.66 To improve the diagnostic specificity and accuracy, Fan and co-workers developed a dual-pathological-parameter cooperatively activatable organic nanoprobe by using a “dual lock-and-key” strategy.65 IR-1061 was conjugated with hyaluronic acid (HA) to generate amphiphilic IR-1061-pendent HA polymers that consequently self-assembled into nanoparticles (“single lock-and-key”-controlled HINPs) in aqueous solution. Further crosslinking of the HA on the surface of HINPs using disulfide formed the final nanoprobes (“dual lock-and-key”-controlled HISSNPs). Both HINPs and HISSNPs had very weak fluorescence owing to the fluorescence-quenched aggregation state of IR-1061. Overexpressed hyaluronidase and thiols in tumor microenvironments specifically cleaved HA chains and disulfide bonds, respectively, resulting in the dissociation of nanoparticles and the decrease of the aggregation degree of IR-1061, which led to fluorescence “turn-on”. With HA-mediated targeting and the EPR effect, HISSNPs effectively accumulated into tumor tissues, and allowed such a cooperatively activatable fluorescence resonance property for NIR-II fluorescence imaging of specific tumors. As compared to HINPs, HISSNPs not only significantly reduced the nonspecific activation in vivo, but also achieved a higher T/NT ratio of up to 15.4 (3.3 for HINPs) and higher tumor-to-liver ratio of 5.87 (1.1 for HINPs) at 24 h post-injection.
3. Chemiluminescence imaging
Chemiluminescence imaging that relies on the energy release from a chemical reaction has appeared to be an ideal technology for different biological analysis and disease diagnosis.67–69 The major advantages of chemiluminescence imaging are the negligible photo-damage and the absence of background noise from biological tissues attributed to the elimination of light excitation, permitting deep-tissue imaging with a relatively high SBR.70–72 At present, a great number of OSMs including organic semiconducting nanoparticles and dioxetane-based small molecules have been utilized as probes for chemiluminescence imaging.3.1. Chemiluminescent nanoprobes
The chemiluminescence imaging of targets of interest with organic semiconducting nanoprobes is generally achieved through a two-step process (Fig. 5a). First, the targets of interest react with the responsive substrates in the nanoprobes to afford high-energy intermediates. Second, intermolecular electron transfer from the fluorophores or SPs to the high-energy intermediates occurred through the chemically initiated electron-exchange luminescence (CIEEL) mechanism, leading to the generation of chemiluminescence signals.72 Since reactive oxygen species (ROS) have high activities to react with responsive substrates,73–77 chemiluminescent organic semiconducting nanoprobes have been widely utilized to detect ROS in living systems.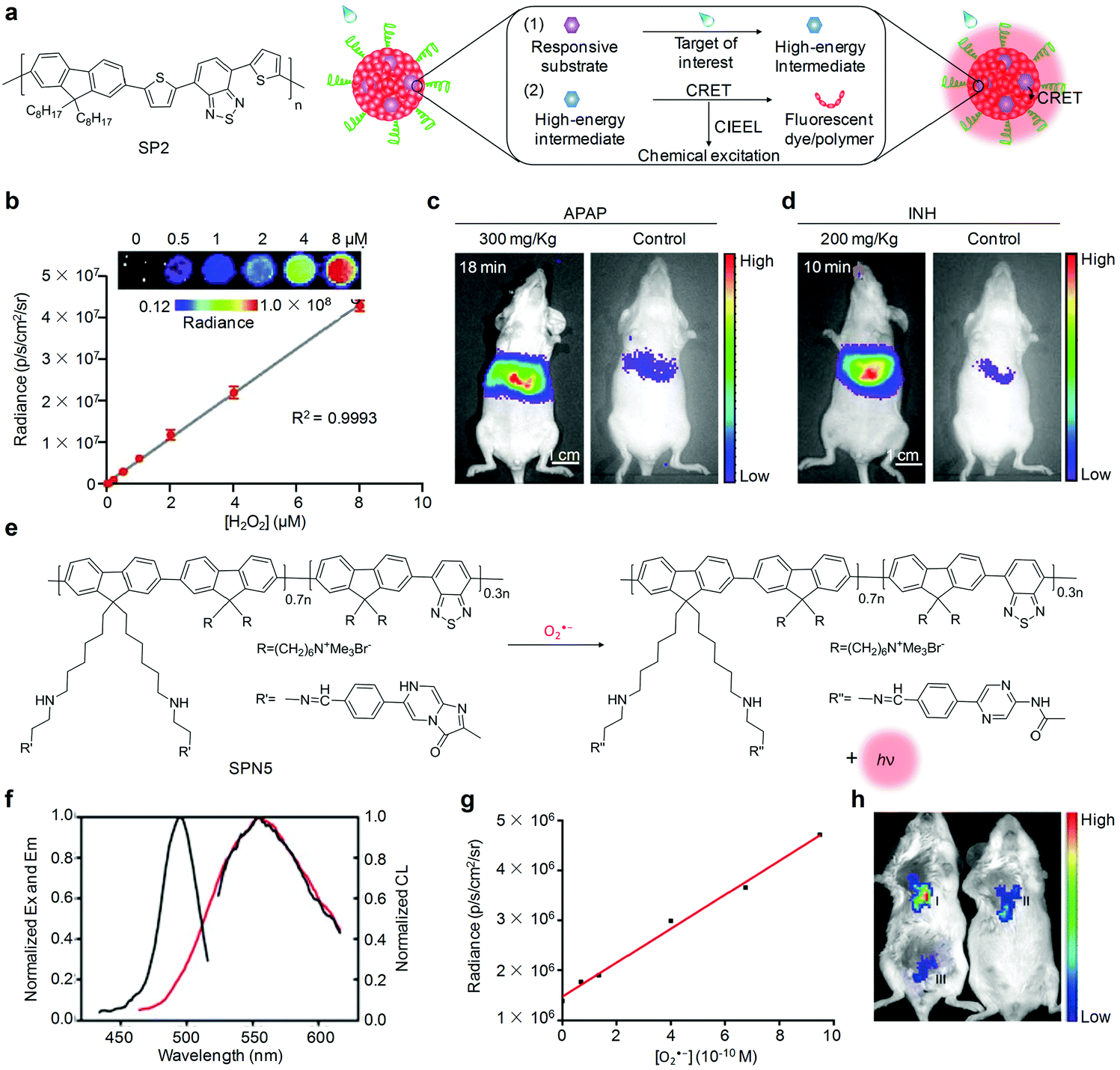 | ||
| Fig. 5 (a) Chemical structure of SP2 and schematic illustration of the mechanism of chemiluminescent nanoprobes for imaging of target of interest. (b) Chemiluminescence response of CF-SPN2 to H2O2 at different concentrations (inset, representative chemiluminescence images of CF-SPN2 response to indicated concentrations of H2O2). (c) Representative chemiluminescence images of mice receiving an intraperitoneal injection of APAP or saline (control), followed by an intravenous injection of CF-SPN2 at 18 min. (d) Representative chemiluminescence images of mice receiving an intraperitoneal injection of INH or saline (control), followed by an intravenous injection of CF-SPN2 at 10 min. (e) Schematic illustration of the molecular structure of SP5 and O2˙− sensing using SPN5. (f) Excitation/emission spectra (black lines) and chemiluminescence spectrum (red line) of SPN5 in the presence of 10 μM O2˙−. (g) Linear relationship between the chemiluminescence intensity of SPN5 and O2˙− concentration. (h) Representative chemiluminescence images of tumor (I), tumor + Tiron (II) and normal tissue (III) in mice followed by a local injection of SPN5. (b)–(d) were reproduced from ref. 87 with permission from Nature Publishing Group, copyright 2014. (f)–(h) were reproduced from ref. 94 with permission from the American Chemical Society, copyright 2016. | ||
Murthy and co-workers reported the utilization of fluorescent dye (pentacene) encapsulated peroxalate nanoparticles as contrast agents for in vivo chemiluminescence imaging of hydrogen peroxide (H2O2),72 a major ROS in biological systems that modulated redox-dependent cellular signaling transductions.78–80 Upon interaction with H2O2, the peroxalate ester groups within nanoparticles were specifically oxidized to generate high-energy dioxetanediones that could chemically excite the nearby fluorescent dye, resulting in chemiluminescence at an emission wavelength of 630 nm. Such peroxalate nanoparticles were able to detect H2O2 at a concentration as low as 250 nM, while this LOD was far above the in vivo concentration (∼100 nM) under normal conditions.81 Soon afterwards, several other chemiluminescent organic semiconducting nanoprobes with the encapsulation of peroxalate compounds have been designed and developed for H2O2 imaging (Table 2), but failed to provide high detection sensitivity in living subjects, largely because their LODs were still slightly high (10–100 nM).82–86
| Probes | Imaging mode | Biomarker | LOD | Imaging applications | Ref. |
|---|---|---|---|---|---|
| Dye@peroxalate nanoparticles | Chemiluminescence | H2O2 | 250 nM | Exogenous H2O2 and LPS-induced acute inflammation | 72 |
| CPPO@AIE nanoparticles | Chemiluminescence | H2O2 | 80 nM | LPS-induced arthritis | 82 |
| CPPO@DSA nanoparticles | Chemiluminescence | H2O2 | 100 nM | LPS-induced arthritis and Duox2-derived viral infection in a nasal mucosa | 83 and 84 |
| HPOX nanoparticles | Chemiluminescence | H2O2 | 100 nM | LPS-induced inflammation | 85 |
| CPPO@Cy5 nanoparticles | Chemiluminescence | H2O2 | 10 nM | Exogenous H2O2, LPS-induced acute inflammation and blood sugar level | 86 |
| CF-SPN | Chemiluminescence/fluorescence | H2O2/ONOO− | 5 nM/10 nM | APAP and INH induced hepatotoxicity | 87 |
| SPN3 | Chemiluminescence | H2O2 | 5 nM | LPS induced peritonitis and neuroinflammation | 88 |
| SPN4 | Chemiluminescence | H2O2 | 1 nM | Inflammation in murine models of arthritis and peritonitis | 89 |
| TBD-AIE nanoparticles | Chemiluminescence | H2O2 | 2 nM | Primary and peritoneal metastatic tumors | 90 |
| SPN5 | Chemiluminescence | O2˙− | 19.3 pM | Visualization of O2˙− level differences between normal and tumor tissues | 94 |
| NIR-II@Si | NIR-II fluorescence | H2S | 37 nM | H2S-rich colorectal tumors | 64 |
| CHS-3 | Chemiluminescence | H2S | 5.4 μM | H2S in living animals | 106 |
| HyCL-1, HyCL-2 | Chemiluminescence | Hypoxia | — | Nitroreductase, tissue oxygenation and hypoxia in tumors | 107 |
| Probe 3 | Chemiluminescence | β-Gal | — | β-Galactosidase activity in tumor models | 108 |
| Probe 7 | Chemiluminescence | H2O2 | 30 nM | Endogenous H2O2 in LPS induced inflammatory mice | 98 |
| CFAP540, CFAP700 | Chemiluminescence | Formaldehyde | 10 μM | Endogenous formaldehyde released from folate metabolism in living mice | 105 |
| SPN6-thiol | Afterglow | Biothiols | 0.6 μM | Drug-induced hepatotoxicity | 118 |
| SPN8 | Photoacoustic | ONOO−/ClO− | ∼50 nM | ROS in zymosan-induced mouse models of acute edema | 131 |
| PCBP | Photoacoustic | ONOO−/H2O2 | 0.15 mM | Drug-induced ROS in tumor tissues | 145 |
| SON-BT | Photoacoustic | ONOO− | 100 nM | ONOO− levels in xenograft tumor models after drug treatment | 146 |
| SON-N | Photoacoustic | ClO− | 0.70 μM | ClO− in tumor models | 147 |
| SON-BDP | Photoacoustic | pH | 7.4–5.5 | Tumor pH | 152 |
| HAS-BOPx-IR825 | Photoacoustic | pH | 5.0–7.0 | Tumor pH and pH variation | 122 |
| CyGal-P | Photoacoustic | β-Gal | — | β-Galactosidase activated imaging and PTT of tumors | 159 |
| HyP-1 | Photoacoustic | Hypoxic | — | Hypoxic in a mouse tumor and hindlimb ischemia model | 160 |
For ultrasensitive imaging of H2O2, Rao and co-workers contributed a SPN-based sensor that was able to simultaneously but differentially detect drug-induced ROS and reactive nitrogen species (RNS) using chemiluminescence and fluorescence independent optical channels.87 In such a system, poly(2,7-(9,9-dioctylfluorene)-alt-4,7-bis(thiophen-2-yl)benzo-2,1,3-thiadiazole) (SP2, Fig. 5a) was used as both the chemiluminescence resonance energy transfer (CRET) energy acceptor and the fluorescence resonance energy transfer (FRET) energy donor. A cyanine dye IR775S (Scheme 1) and bis-(2,4,5-trichloro-6-(pentyloxycarbonyl)phenyl)oxalate (CPPO, Scheme 1) were encapsulated into the SP2 matrix, acting as the fluorescence-based responsive sensor and H2O2 reactive chemiluminescent substrate, respectively. Combining CRET and FRET into a single SPN2 afforded CRET–FRET-SPN2 (CF-SPN2). To impart the ability to target hepatocytes for hepatotoxic evaluation, an amphiphilic PEG-grafted poly(styrene) copolymer with a conjugation of galactose (PS-g-PEG-Gal) was used to prepare CF-SPN2. As the FRET acceptor, IR775S within CF-SPN2 decomposed after being oxidized by both peroxynitrite (ONOO−) and hypochlorite (ClO−), leading to FRET abolishment and thereby ratiometric fluorescence signals. However, only a chemiluminescence response of CF-SPN2 to H2O2 occurred without external light excitation, allowing for highly selective and sensitive imaging of H2O2 with a LOD as low as 5 nM (Fig. 5b). The improved detection sensitivity of this approach enabled early-stage in vivo detection of anti-pyretic acetaminophen (APAP) and isoniazid (INH) induced hepatotoxicity (Fig. 5c and d).
Pu's group have reported another optimized approach, intraparticle energy level alignment, to amplify the chemiluminescence signals of SPNs for ultrasensitive detection of H2O2.88 Five polyfluorene derived SPs with different molecular orbitals were chosen as chemiluminescence reporters to respectively pair with a chemiluminescent substrate peroxalate bis(2,4,6-trichlorophenyl) oxalate (TCPO, Scheme 1), affording a series of SPNs that selectively emitted chemiluminescence signals in response to H2O2. Among these SPNs, the energy interval between the highest occupied molecular orbital (HOMO) of poly[(9,9′-dioctyl-2,7-divinylene-fluorenylene)-alt-(2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylene)] (SP3, Scheme 2) and the lowest unoccupied molecular orbital (LUMO) of 1,2-dioxetanedione was the smallest, and thereby the intermolecular electron transfer between them was most facilitated. As a result, SPN3 had the highest chemiluminescence efficiency and enabled detection of H2O2 with a LOD down to 5 nM. Further doping of a naphthalocyanine NIR dye (NCBS, Scheme 1) created an intraparticle CRET and endowed SPN3 with the ability to emit luminescence NIR light, allowing for in vivo ultrasensitive imaging of H2O2 in the mouse models of lipopolysaccharide (LPS) induced peritonitis and neuroinflammation.
 | ||
| Scheme 2 Chemical structures of some SPs used for preparation of SPNs. | ||
To improve the NIR chemiluminescence efficacy of SPNs for H2O2 detection, Kim and co-workers subsequently proposed a simple nanophotonic energy relay pathway.89 SPN4 was constructed by nanoprecipitation of a low-bandgap conjugated polymer (SP4, Scheme 2), CPPO and BODIPY dye (Scheme 1). As compared to other probes used for H2O2 detection, the only difference lay in the use of BODIPY dye as an energy gap-bridging photonic molecule to effectively accept the energy from the chemically excited intermediate of peroxalates and then relay it to SP4 in the same nanoscopic space through the consecutive intra-particle energy transfer. Such an energy-relay effect boosted the chemiluminescence emission by 50-fold and thus enabled reliable detection of H2O2 with a LOD down to 1 nM and presented a fairly high tissue penetration depth (>12 mm). Due to these merits, SPN4 facilitated sensitive deep-tissue chemiluminescence imaging of inflammation in the mouse models of arthritis and peritonitis.
In addition to SPNs, AIE nanoparticles have been utilized for chemiluminescence imaging of H2O2.82,90 In one example, a specially designed small molecule TBD (Scheme 1), CPPO and soybean oil were co-precipitated with an amphiphilic triblock copolymer (PEG-b-PPG-b-PEG) to obtain TBD AIE nanoparticles.90 Chemiluminescence of TBD AIE nanoparticles in the presence of H2O2 was around 900-fold higher than that with other ROS, allowing detection of H2O2 at an ultralow concentration of 2 nM. In addition, the encapsulated soybean oil served as a retarder to slow down the reaction rate between H2O2 and CPPO, prolonging the half-life of chemiluminescence to 2.3 h for in vivo imaging applications. Because the H2O2 concentration in tumors was obviously elevated relative to normal tissues,91 TBD AIE nanoparticles were able to accurately distinguish primary and peritoneal metastatic tumors.
In addition to the imaging of H2O2, chemiluminescent OSMs were used to detect superoxide anions (O2˙−), one of the most common ROS generated from the reduction of oxygen molecules.92,93 Tang and co-workers recently synthesized a chemiluminescent probe (SPN5) through nano-precipitation of SP5 comprising two covalently connected segments.94 In such a nanoparticle, the imidazopyrazinone moiety on the side chain of SP5 acted as both the O2˙− responsive group and CRET donor, while SP5 was utilized as both the CRET acceptor and signal amplification matrix. The specific reaction between the imidazopyrazinone moiety and environmental O2˙− produced high energy (Fig. 5e), which was then transferred to SP5 through CRET, leading to an intense luminescence at 560 nm (Fig. 5f). The LOD of SPN5 was estimated to be 19.3 pM (Fig. 5g). Additionally, the covalently linked molecular structure of SP5 contributed to a prolonged chemiluminescence time of more than 25 min, favoring a practical application of in vivo molecular imaging. With these advantages over other existing small molecule compounds, SPN5 was utilized for chemiluminescence imaging of O2˙− in mouse models of inflammation and tumor. After the local injection of SPN5, the chemiluminescence signal in tumor tissues was 3-fold higher than that in normal tissues, while the signal intensity in the tumor tissues reduced after the intratumoral injection of a ROS scavenger (Tiron) (Fig. 5h).
3.2. Chemiluminescent small-molecular probes
A class of chemiluminescent small molecular probes based on dioxetane have been developed for imaging of different disease biomarkers.95–99 These small-molecular probes do not require any prior oxidation steps to trigger chemiexcitation because the dioxetane compounds are pre-oxidized in a thermally stable form, which is different from most of the other chemiluminescent nanoprobes.100–102 Such chemiluminescence is excited upon removal of the protecting groups (PGs), which results in an electron transfer from a phenolate to the peroxide bond of dioxetane (Fig. 6a).96,103 To optimize the optical properties for in vivo imaging applications, various electron-withdrawing groups (EWGs) have been conjugated at the ortho position of phenol, leading to a 1000-fold increase in the chemiluminescence quantum efficiency and red-shifted emission in the NIR region.98,104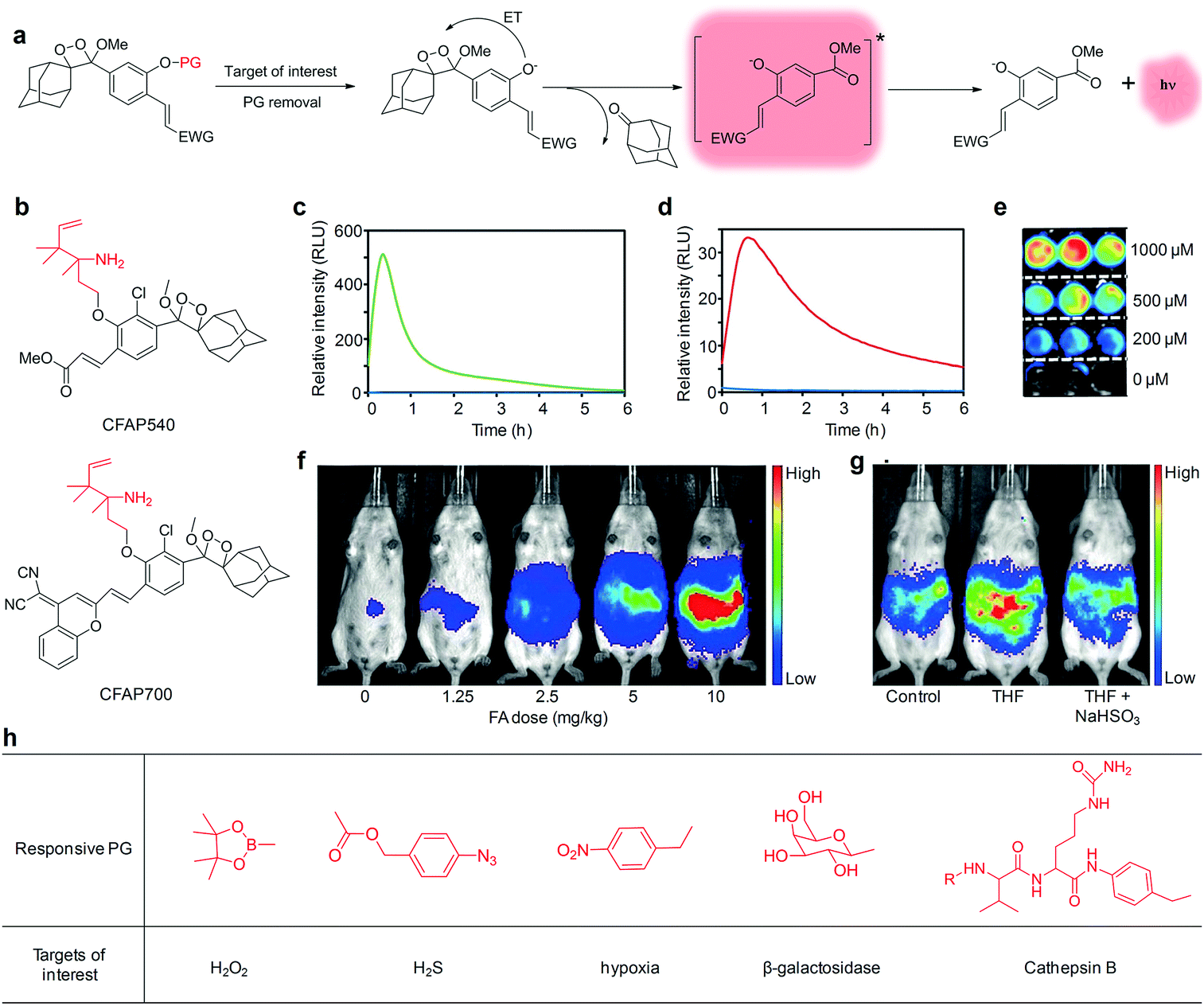 | ||
| Fig. 6 (a) Chemiluminescent activation pathway of dioxetane probes for imaging of target of interest (PG, protecting groups; ET, electron transfer; EWG, electron-withdrawing groups). (b) Chemical structures of CFAP540 and CFAP700. Chemiluminescence kinetic profiles of (c) CFAP540 and (d) CFAP700 to 0 mM FA (blue) and 10 mM FA (green or red). (e) Representative chemiluminescence cell images of HEK293 cells after treatment with CFAP540 and FA at different concentrations. (f) Chemiluminescence imaging of CFAP700 in response to exogenously injected FA at different doses. (g) Chemiluminescence imaging of mice after an intraperitoneal injection of tetrahydrofolate (THF), then aqueous solution of sodium bisulfite (NaHSO3) or water, then CFAP700. (h) Other responsive PG used for chemiluminescence imaging of different targets of interest. (c)–(g) were reproduced from ref. 105 with permission from John Wiley & Sons Ltd, copyright 2018. | ||
Chemiluminescent small molecular probes using a 2-aza-Cope reactive trigger as the PG were developed for activatable detection of formaldehyde (FA) in living animals.105 A specific reaction between FA and the 2-aza-Cope reactive trigger within probes resulted in immolation of the two-carbon linker, generating free phenoxy-dioxetane that subsequently decomposed to produce chemiexcitation. Two different EWGs were conjugated to the dioxetane scaffolds to allow visible (CFAP540 probe) and NIR (CFAP700 probe) emission, respectively (Fig. 6b). These two probes displayed a robust response to FA, with maximum light emission increased by 500-fold for CFAP540 and 33-fold for CFAP700 (Fig. 6c and d). Due to the superior signal-to-noise response, CFAP540 detected FA at the concentration as low as 10 μM, while CFAP700 could detect 25–50 μM levels of FA. Meanwhile, the probes showed a high selectivity for FA with minimal interference from other biologically relevant aldehydes and molecules. In addition, CFAP540 was cell-trappable because of the existence of a methyl ester substituent, making it highly suitable for FA detection in living cells (Fig. 6e), whereas the NIR emission profile of CFAP700 was exploited for imaging of exogenously injected FA (Fig. 6f) and endogenous FA released from folate metabolism in living mice (Fig. 6g). Beyond this example, various other responsive PGs have been applied to trigger the chemiexcitation, allowing for imaging of different targets of interest in living biological systems (Fig. 6h), including H2O2,98 H2S,106 hypoxia,107 β-galactosidase,104,108,109 and Cathepsin B95 (Table 2).
4. Molecular afterglow imaging
Afterglow imaging is a luminescent process that occurs after the removal of the illumination source, providing a promising imaging technique for in vivo disease diagnosis owing to its elimination of real-time excitation and minimum tissue autofluorescence.110–112 Thus afterglow imaging has the advantages of relatively high SBRs and high imaging sensitivity. The current afterglow luminescence in experimental animals largely relies on inorganic nanoparticles. However, these inorganic contrast agents have the concerns of low brightness, short luminescence time and underlying toxicity.113–115 As alternative candidates, a category of biocompatible organic nanoparticles with an ultralong luminescence lifetime and relatively high brightness have been developed, permitting high quality in vivo afterglow imaging applications.116To provide ideal organic afterglow probes, Pu's group used a top-down approach to greatly preserve the aggregates of phosphorescent molecules within the nanoparticles (SONs-T).117 As compared to the nanoparticles made from a bottom-up approach (SONs-B), SONs-T facilitated the formation of H-aggregates more due to their stronger molecular packing. Since H-aggregation provided lower energy levels that served as additional energy traps to effectively stabilize the triplet excited states, SONs-T had 1.75-fold longer phosphorescence lifetime (861 ms) relative to SONs-B (492 ms). Such an ultralong phosphorescence could be repeatedly activated with nearly no loss in the total intensity, allowing for longitudinal afterglow imaging of lymph nodes in living mice with a SBR as high as 40.
Despite the feasibility of using SONs-T for in vivo afterglow imaging, their brightness was low and their emission was in the visible region. To overcome these limitations, Pu's group have further developed a class of biodegradable SPNs showing a more than 100-fold higher brightness than inorganic probes and emitting long NIR luminescence for molecular afterglow imaging in living mice.118 We demonstrated that the chemical structures of SPs, rather than the nanoparticle structures played an indispensable role in producing the afterglow luminescence, and only phenylenevinylene (PPV)-based SPs with strong electron-donating substituents could afford detectable afterglow signals. The mechanism of afterglow luminescence was described as follows (Fig. 7a): light irradiation of PPVs generated 1O2 that reacted with the vinylene bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) in PPVs to form a dioxetane intermediate; the unstable intermediate then spontaneously degraded into a PPV-aldehyde to produce luminescence. Among all PPV-based SPs, MEHPPV (SP6) displayed the highest afterglow intensity and was thus transformed into nanoparticles doped with NCBS at a weight percentage of 5% (SPN6-NCBS5). NCBS doping contributed to an amplified afterglow owing to the increased generation of 1O2 by NCBS, and modulation of the emission into the NIR optical window through the energy transfer (Fig. 7b and c). Such an amplified SPN6-NCBS5 allowed an afterglow imaging penetration depth of 4 cm in vitro and 1.7 cm through a body of mouse, as well as permitted fast and high-contrast afterglow imaging of lymph nodes and tumors in living mice with a SBR more than 127-fold higher than that obtained by NIR fluorescence imaging (Fig. 7d and e).
C) in PPVs to form a dioxetane intermediate; the unstable intermediate then spontaneously degraded into a PPV-aldehyde to produce luminescence. Among all PPV-based SPs, MEHPPV (SP6) displayed the highest afterglow intensity and was thus transformed into nanoparticles doped with NCBS at a weight percentage of 5% (SPN6-NCBS5). NCBS doping contributed to an amplified afterglow owing to the increased generation of 1O2 by NCBS, and modulation of the emission into the NIR optical window through the energy transfer (Fig. 7b and c). Such an amplified SPN6-NCBS5 allowed an afterglow imaging penetration depth of 4 cm in vitro and 1.7 cm through a body of mouse, as well as permitted fast and high-contrast afterglow imaging of lymph nodes and tumors in living mice with a SBR more than 127-fold higher than that obtained by NIR fluorescence imaging (Fig. 7d and e).
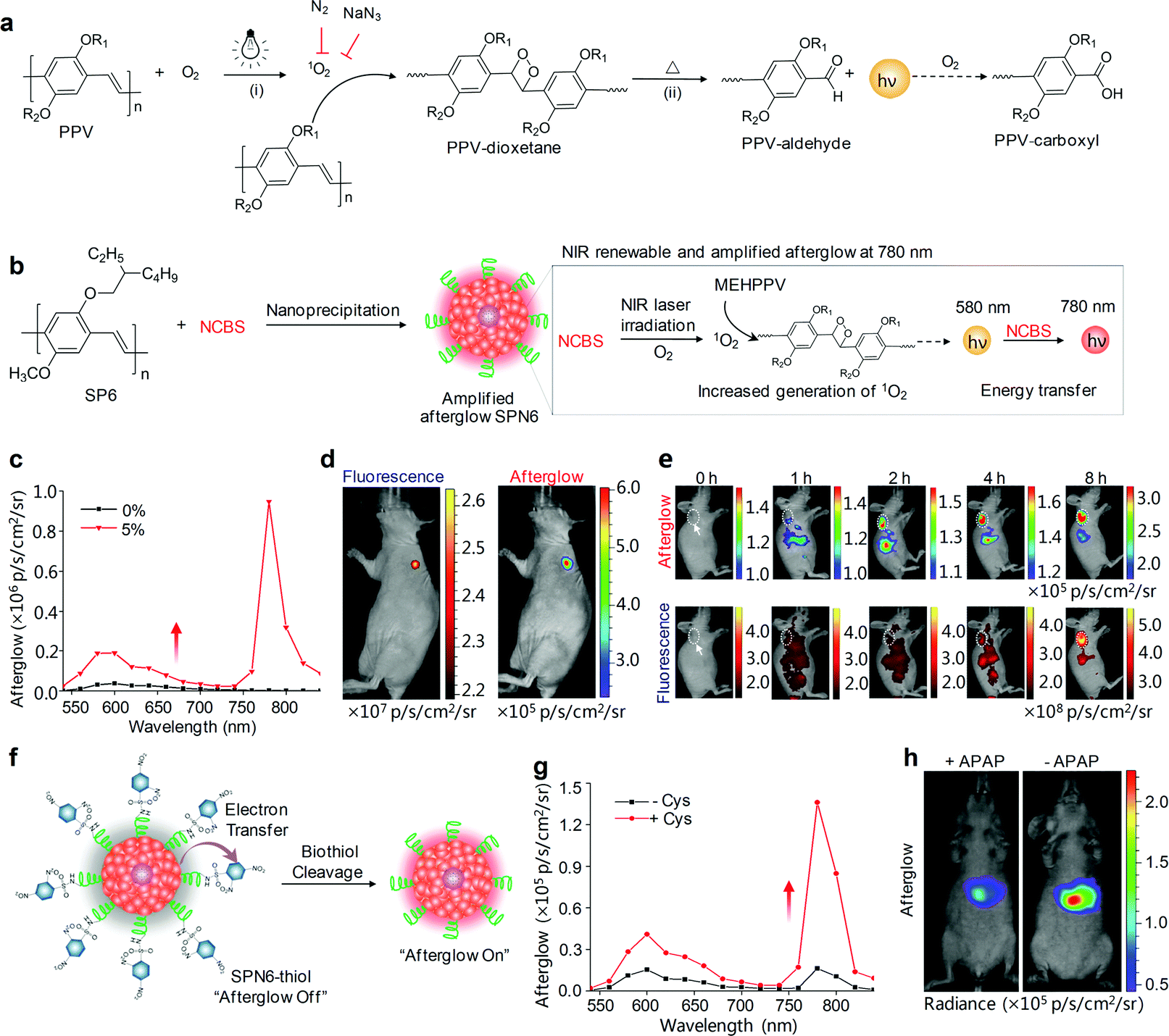 | ||
| Fig. 7 (a) Proposed mechanism for the afterglow luminescence of PPV-based SPs. R1 and R2 are alkyl chains. (b) Schematic illustration of the proposed mechanism for 1O2-sensitizer-amplified NIR afterglow of SPN6-NCBS5. (c) NIR-induced afterglow luminescence spectra of SPN6 without the NCBS doping and SPN6-NCBS5 dispersed in 1× PBS buffer. (d) Fluorescence (left) and afterglow luminescence (right) imaging of a lymph node in a living mouse at t = 65 min after intradermal injection of SPN6-NCBS5 into the forepaw of the mouse. (e) Representative afterglow images (upper panel) and fluorescence images (lower panel) of a tumor in a living mouse at representative time points after systemic administration of SPN6-NCBS5 via tail vein injection. The tumor was on the right shoulder as indicated by the white dashed circles and arrows. (f) Schematic illustration of the design and turn-on mechanism of a biothiol activatable afterglow probe (SPN6-thiol). (g) Afterglow luminescence spectra of SPN6-thiol in the absence or presence of Cys. (h) Representative afterglow luminescence images of mice with or without intraperitoneal injection of APAP, followed by the administration of SPN6-thiol via tail vein at t = 20 min later. Afterglow luminescence images were acquired for 180 s after irradiation at 808 nm (1 W cm−2) for 1 min. Reproduced from ref. 118 with permission from Nature Publishing Group, copyright 2017. | ||
SPN6-NCBS5 could be developed into a smart activatable afterglow probe (termed as SPN6-thiol) after a covalent conjugation with an electron-withdrawing quencher (2,4-dinitrophenylsulfonyl, DNBS) through sulfonamide bonds for specific and sensitive imaging of biothiols. Biothiols including glutathione (GSH), homocysteine (Hcy) and cysteine (Cys) are the major portions of antioxidants that defend against oxidative stress in living body. The luminescence of SPN6-thiol was substantially quenched at its initial afterglow “off” state due to the intra-particle efficient electron transfer, while the sulfonamide bonds on the surface of SPN6-thiol, when activated by biothiols, could be cleaved to release DNBS from nanoparticles (Fig. 7f). As a result, the electron transfer was abolished to turn on afterglow signals (Fig. 7g), allowing for high-sensitive early-stage imaging of APAP-induced hepatotoxicity (Fig. 7h). Because the APAP treatment induced oxidative and nitrosative stress and thus consumed biothiols, the afterglow signals for the APAP-injected mice were 1.99-fold lower than those for the control mice without APAP injection at t = 2 h post-injection of SPN6-thiol.
To optimize the biodistribution of afterglow probes, Pu's group subsequently synthesized an amphiphilic polymer composed of a hydrophobic PPV backbone and hydrophilic PEG brushes (Fig. 8a).119 This PPV-derived amphiphile was able to self-assemble into nanoparticles (SPPVN) that had a smaller size (24 nm), higher energy transfer efficiency and brighter afterglow luminescence as compared with the counterpart SPN6 (34 nm) prepared from nanoprecipitation. Due to the smaller size and higher surface PEG density, SPPVN showed faster and better accumulation into tumors than SPN6, allowing for afterglow imaging of xenograft tumors (≈5 mm3) as early as 40 min post intravenous injection (Fig. 8b). In sharp contrast, the tumors were only detectable with fluorescence imaging at 4 h post-injection for SPPVN, and at 4 h and 8 h post-injection with afterglow imaging and fluorescence imaging for SPN6, respectively. Such enhanced afterglow imaging performance of SPPVN also enabled detection of early-stage tumors with size as small as ≈1 mm3 and tiny peritoneal metastatic tumors that were almost invisible to the naked eye (Fig. 8c).
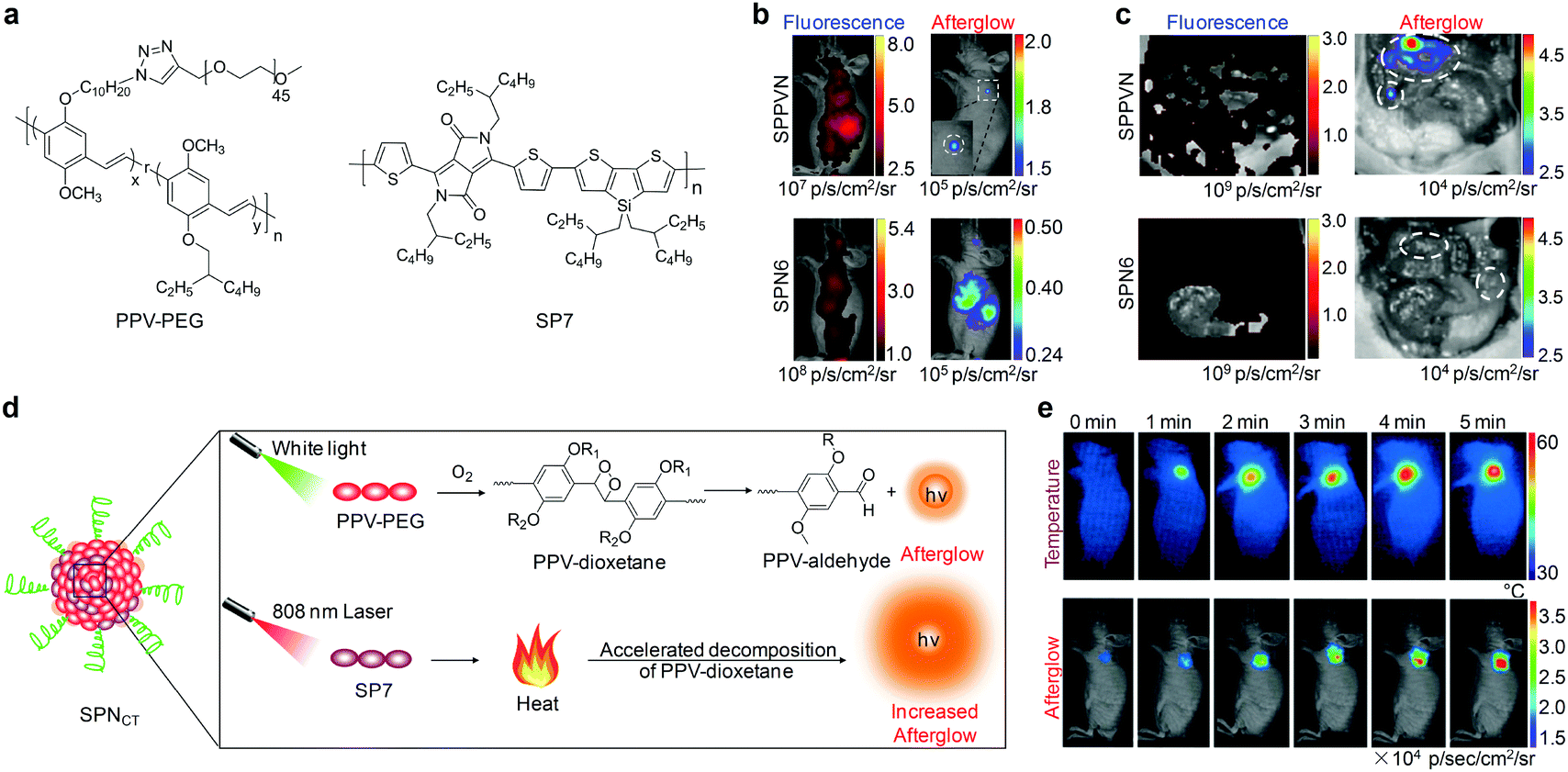 | ||
| Fig. 8 (a) Molecular structures of PPV-PEG and SP7 used for preparation of nanoparticles for afterglow imaging. (b) Representative in vivo fluorescence and afterglow luminescence images of tumors in living mice at 40 min after intravenous injection of SPPVN or SPN6. (c) Fluorescence and afterglow images of mice after skin resection to expose the abdominal cavity at 1.5 h post-injection of SPPVN or SPN6. Tumor regions are marked by white circles. (d) Schematic illustration of the mechanism for photothermally enhanced afterglow luminescence of SPNCT. (e) Representative thermal and afterglow images of tumor in living mice at 24 h after intravenous injection of SPNCT under laser irradiation at 808 nm for different times. (b) and (c) were reproduced from ref. 119 with permission from John Wiley & Sons Ltd, copyright 2018. (d) and (e) were reproduced from ref. 120 with permission from John Wiley & Sons Ltd, copyright 2018. | ||
Because afterglow luminescence from PPVs is dependent on the generation of 1O2, it shows an oxygen-sensitive nature that can be utilized for in vivo imaging of tissue oxygen levels.119 In addition, the temperature-dependent luminescence is another intrinsic characteristic for afterglow of PPVs, because the photo-releasing decomposition of dioxetane intermediate is thermodynamically controlled.118 Therefore, Pu's group recently developed a semiconducting polymer nanococktail (SPNCT) that had a temperature-correlated afterglow luminescence for cancer photothermal therapy (PTT).120 This SPNCT was made of the amphiphilic PEG grafted PPV and NIR absorbing poly(silolodithiophene-alt-diketopyrrolopyrrole) (PCSD, SP7, Fig. 8a), which served as the temperature-monitoring sensor and the photothermal agent, respectively (Fig. 8d). NIR laser irradiation of SPNCT elevated the temperature, which in turn accelerated decomposition of the PPV-dioxetane, affording an increased afterglow signal. Such an afterglow imaging of SPNCT facilitated real-time temperature monitoring during PTT without light excitation (Fig. 8e).
5. PA imaging
PA imaging has emerged as a promising imaging modality for providing three-dimensional (3D) images with real-time correlation, deep imaging penetration depth of up to 5–7 cm and relatively high spatial resolution.121–124 During PA imaging, the optical absorbers (imaging agents) in biological tissues of interest are irradiated with a short-pulsed laser and they convert the photon energy into heat, leading to the generation of transient thermo-elastic expansion and thus wideband acoustic waves. These ultrasonic waves can be recorded by ultrasonic transducers to produce PA images. Therefore, PA imaging possesses the merits of optical and acoustic methods including sensitive optical absorption contrast and minimized acoustic scattering.1255.1. “Always on” PA probes
To provide detectable variations for PA imaging in deep tissue, a broad range of exogenous imaging contrast agents have been designed and developed.126–128 Owing to the good photo-stability, high mass absorption coefficient and excellent photothermal conversion efficiency, OSMs have been exploited as a class of “always on” PA probes, allowing for in vivo imaging of blood vessels,129 lymph nodes,130,131 protein sulfenic acids,75 thrombus,132 tumors133–135 and so on.PA imaging of blood vessels has emerged as a challenging undertaking on the molecular imaging front, in which blood generates an interferential PA signal because of its strong light-absorption properties, leading to poor imaging performance, sensitivity and SBR.129 Thereby, there is a demand to develop PA imaging probes exhibiting higher PA signals relative to the surrounding vasculatures. In this regard, an organic PA contrast agent with a broad and strong optical absorption in the visible-light region has been developed through the nanoprecipitation of a newly designed PIID-DTBT (SP8, Scheme 2) with functional poly(styrene-co-maleic anhydride) (PSMA).129 The higher PA intensity than vasculatures under 700 nm illumination made SPN8 a highly effective contrast agent for PA imaging of blood vessels. In another study, Liu and co-workers synthesized a SP with strong absorption in the NIR window, poly[9,9-bis(4-(2-ethylhexyl)phenyl)fluorene-alt-co-6,7-bis(4-(hexyloxy)phenyl)-4,9-di(thiophen-2-yl)-thiadiazoloquinoxaline] (PFTTQ, SP9, Fig. 9a), and formulated it into SPN9, which displayed a high PA signal benefited from its large absorption coefficient and high non-radiative quantum yield of almost 100%.21 The authors demonstrated the feasibility of utilizing SPN9 as a PA probe for brain vasculature imaging with a ∼3-fold signal enhancement.
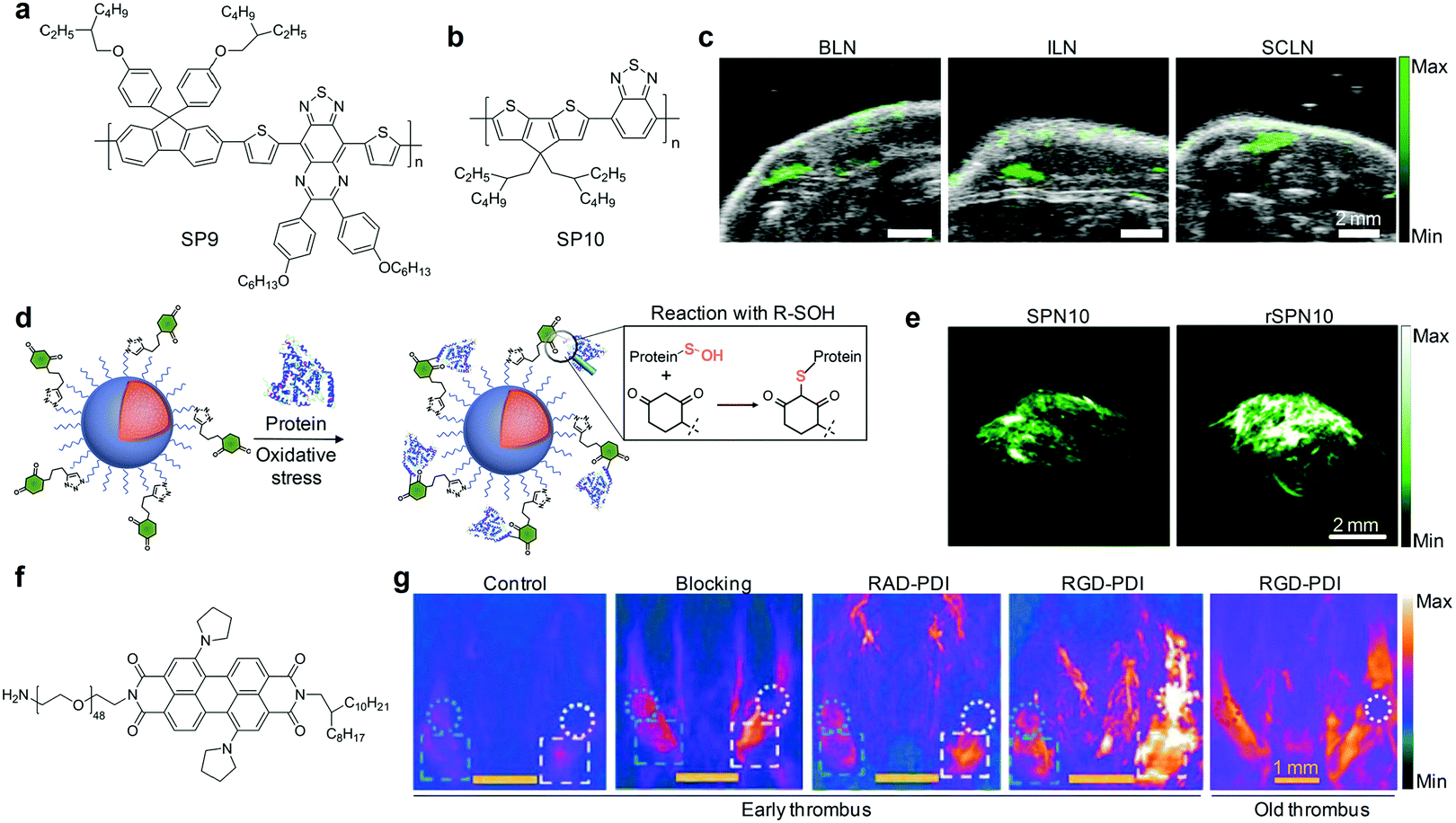 | ||
| Fig. 9 (a) Chemical structure of SP9 used for preparation of SPN9 for PA imaging of blood vessels. (b) Chemical structure of SP10 used for preparation of SPN10. (c) PA images of mouse lymph nodes following tail vein injection of SPN10. Images represent transverse slices through the lymph nodes. BLN, brachial lymph node; ILN, inguinal lymph node; SCLN, superficial cervical lymph node. (d) Illustration of the mechanism of the specific reaction between rSPN10 and BSA-SOH. (e) PA images with axial view of tumors at 24 h time-point after systemic administration of rSPN10 or SPN10 via tail vein injection. (f) Chemical structure of PDI used for preparation of RGD-PDI nanoparticles. (g) PA images of the mouse jugular veins with early or old thrombus after an intravenous injection with RGD-PDI nanoparticles, RAD-PDI (non-targeting) nanoparticles, blocking group, and PBS (Control) at 24 h post-injection treatment time. (c) was reproduced from ref. 131 with permission from Nature Publishing Group, copyright 2014. (d) and (e) were reproduced from ref. 75 with permission from the American Chemical Society, copyright 2016. (g) was reproduced from ref. 132 with permission from the American Chemical Society, copyright 2017. | ||
Noninvasive PA imaging of lymph nodes is highly desirable for the detection of cancer metastasis and lymph node biopsy.130 To fully utilize the potential of SPNs for PA imaging of lymph nodes, SP10 (Fig. 9b) with strong absorption in the NIR window was transformed into SPN10, which produced a higher PA signal and showed better photo-stability relative to the most commonly used SWNTs and gold nanorods on a per mass basis.131 Systemic administration of SPN10 permitted whole-body PA imaging of lymph nodes in living mice with a high SBR of 13.3, at a low injection mass (50 μg) (Fig. 9c). Similarly, the utilization of newly designed SONs that show strong NIR absorption, excellent imaging contrast and quick body clearance for effective PA imaging of sentinel lymph nodes has also been reported.130
Pu's group recently reported a reaction-based nanoprobe (rSPN10) for imaging of protein sulfenic acids,75 a class of proteins whose cysteines were oxidized into sulfenic acid (SOH) by ROS.136,137 The surface of rSPN10 was modified with 1,3-cyclohexanedione groups that could covalently bind to SOH sites (Fig. 9d).75 Such a nanoparticle reaction enabled monitoring of the dynamic changes in the concentration of protein sulfenic acids under elevated oxidative stress in cancer cells, and real-time PA imaging of protein sulfenic acids in the tumor microenvironment of living mice (Fig. 9e).
In addition, the feasibility of utilizing NIR absorbing nanoprobes, self-assembled from amphiphilic perylene-3,4,9,10-tetracarboxylic diimide (PDI, Fig. 9f) derivatives, for selective PA imaging of early thrombus in living mice was demonstrated.132 Surface modification of these nanoparticles with RGD peptides enabled their specific bindings to glycoprotein IIb/IIIa overexpressed in early thrombus. In combination with other advantages, such as relatively high PA intensity, good colloidal stability and long blood circulation half-life (around 22 h), RGD-PDI nanoparticles enabled the visualization of a FeCl3-induced mouse model of jugular vein thrombus, affording a nearly 4-fold higher PA signal relative to that in the control and old thrombus groups (Fig. 9g).
Beyond the abovementioned imaging applications, a large variety of OSMs have been developed and used for PA imaging of tumors.138–140 In particular, a series of SPNs composed of low-bandgap diketopyrrolopyrrole (DPP)-based SPs were synthesized to systematically study and compare their PA properties.133 SPNs self-assembled from amphiphilic SPs attached with PEG side chains often have a small size and PEG-passivated surface, and thus can efficiently accumulate into tumors of living mice, permitting PA imaging of tumors with a high SBR.134,135 Moreover, rational designs of molecular or nanoparticle structures contributed to the generation of OSMs exhibiting amplified PA brightness, leading to sensitive tumor imaging.
For instance, intra-particle molecular orbital engineering that favored photo-induced electron transfer (PET) resulted in quenched fluorescence and enhanced heat generation for nanoparticles upon laser irradiation, leading to amplified PA signals.141 This approach was demonstrated by doping (6,6)-phenyl-C71-butyric acid methyl ester (PC70BM) that acted as an electron acceptor into SPN10, wherein SP10 served as the electron donor through nanoprecipitation (Fig. 10b).141 The HOMO and LUMO of SP10 and PC70BM were ideally aligned to facilitate intra-particle PET. The PA brightness of PC70BM doped SPN10 was increased by 2.6-fold relative to the nondoped SPN10 at the same concentration, and thus afforded 1.8-fold PA signal amplification to clearly delineate tumors.
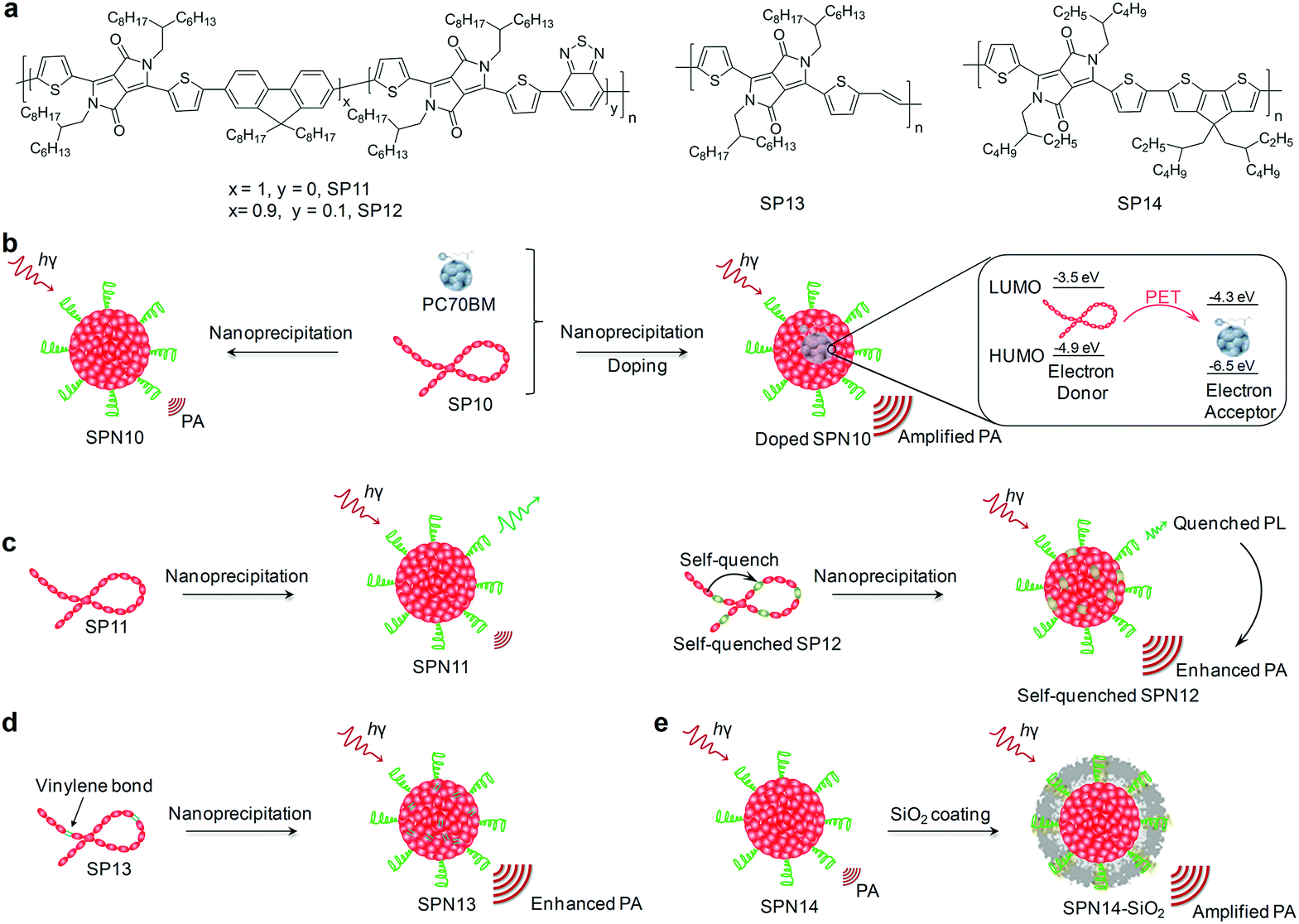 | ||
| Fig. 10 (a) Chemical structures of SP11–14 used for preparation of SPNs for amplified PA imaging of tumors. (b) Schematic illustration of an intra-particle molecular orbital engineering design approach for amplified tumor PA imaging. (c) Schematic illustration of a self-quenched molecular engineering approach to synthesize self-quenched SPN12 via nanoprecipitation for enhanced tumor PA imaging. (d) Schematic illustration of molecular structure engineering to synthesize SPN13 from vinylene bond incorporated SP13 for enhanced tumor PA imaging. (e) Schematic illustration of a surface engineering approach to synthesize SPN14-SiO2 for amplified tumor PA imaging. | ||
As an alternative to intra-particle engineering, a self-quenched molecular engineering approach that incorporated an electron-deficient structure unit into polymer backbones has been proposed to amplify PA brightness of SPNs.142 This was feasible because the electron-deficient units promoted the nonradiative decay, leading to fluorescence quenching and thus enhanced thermal deactivation. Doping of benzothiadiazole (BT) as an electron-deficient structure unit with an amount of 10% into the backbone of PDPPF (SP11) yielded SP12 (Fig. 10a), and its corresponding SPN12 after nanoprecipitation with the amphiphilic triblock copolymer (PEG-b-PPG-b-PEG) showed a 1.7-fold PA signal amplification as compared with the control SPN11 (Fig. 10c). Therefore, SPN12 after surface conjugations of cyclic-RGD was highly promising to effectively delineate the xenografted 4T1 tumors in living mice with the PA intensity increased by 4.7-fold after systemic administration.
Another strategy to obtain SPNs with amplified PA signals is to incorporate a vinylene bond as a structural unit into the backbone of SPs.143 Because of the low molecular weight and proper planar conformation, vinylene bonds facilitated the electron delocalization and increased the mass absorption coefficients, improving the photothermal conversion efficiencies and amplifying PA signals. As demonstrated in a recent study of Pu's group, SP13 (Fig. 10a) with the incorporation of vinylene bonds was nanoprecipitated with an amphiphilic polymer (PLGA–PEG) to generate SPN13, showing a 2.2-fold higher PA signal relative to its nanoparticle counterpart without vinylene bonds (Fig. 10d), which allowed tumor PA imaging in living mice in a more sensitive and effective way.143 Moreover, SPN13 could be efficiently digested in the presence of peroxidase owing to the oxidizable nature of vinylene bonds, representing a class of biodegradable agents for cancer theranostics.
In addition to these approaches relying on the molecular engineering of SPs, Pu et al. have found that surface coating of SPNs with silica layers amplified PA brightness, largely due to the higher heat interfacial conductance of the interface between the silica layer and water relative to that between the SP core and water.144 Both poly(cyclopentadithiophene-alt-diketopyrrolopyrrole) (SP14, Fig. 10a) and SP10 were developed into silica-coated SPNs through nanoprecipitation and surface coating of a thin silica shell. Silica-coated SPNs had a ∼1.4 higher PA amplitude than that of the corresponding uncoated SPNs (Fig. 10e).
5.2. Activatable PA probes
Rational design of molecular structures leads to the generation of a number of organic optical PA contrast agents that have shown promising applications in the visualization of biological structures and disease biomarkers. However, these “always on” probes often suffer from a strong background signal and thus a low SBR, even if with conjugations of a specific targeting ligand.121 In contrast, activatable PA probes that can only be activated by specific biomolecular recognition or interaction serve as exciting candidates for detection of disease-related biomarkers and physiological indexes with improved imaging sensitivity and specificity (Table 2).An activatable PA probe (SPN10) was developed through nanoprecipitation of SP10 and ROS-responsive IR775S (Fig. 11a) for real-time imaging of ONOO− and ClO− in drug-induced mice.131 Upon activation with ONOO− or ClO−, the PA peak of SPN10 at 700 nm corresponding to SP10 remained nearly the same, while the peak at 735 nm decreased significantly and that at 820 nm almost disappeared owing to the ROS-mediated rapid oxidative decomposition of IR775S (Fig. 11b), resulting in ratiometric PA imaging (PA700/PA820) of ONOO−/ClO−. With a high selectivity towards ONOO−/ClO− and low LOD of ∼50 nM, SPN10 was used as an activatable PA contrast agent for in vivo real-time imaging of ROS in the zymosan-induced mouse models of acute edema (Fig. 11c). The ratiometric PA signal (PA700/PA820) gradually increased to 2.7 at 120 min post-injection of SPN10 for zymosan-treated mice, about 2-fold higher relative to the control mice (Fig. 11d).
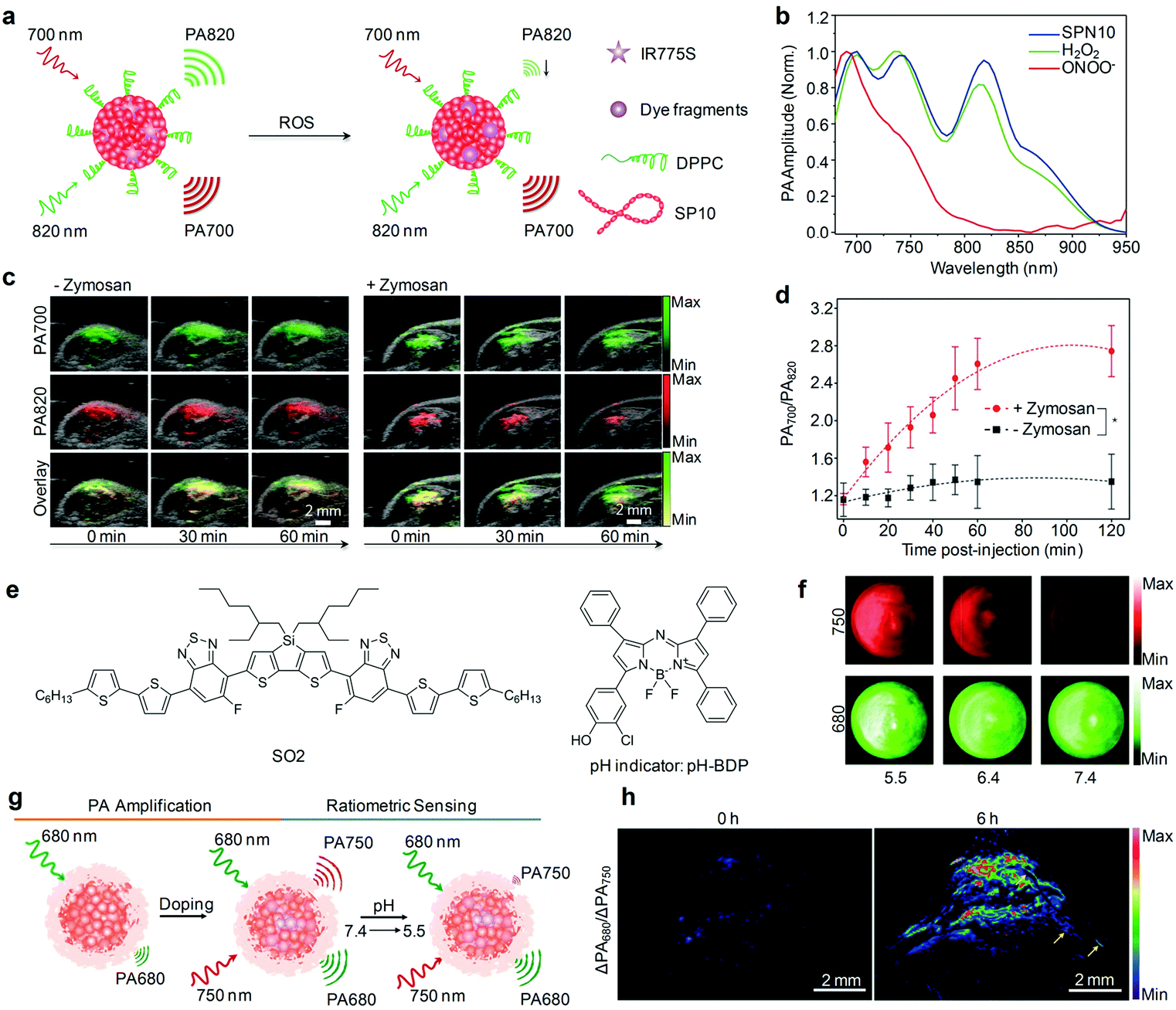 | ||
| Fig. 11 (a) Proposed mechanism for ROS PA imaging. (b) Representative PA spectra of SPN10 in the absence and presence of ROS. (c) Representative PA/US overlaid images of saline-treated (−zymosan) and zymosan-treated (+zymosan) regions in the thigh of living mice. SPN10 was intramuscularly injected into the thigh 20 min after zymosan treatment. (d) Ratio of PA amplitude at 700 nm to that at 820 nm (PA700/PA820) as a function of time post-injection of SPN10. (e) Chemical structures of SO2 and pH-BDP used for pH sensing. (f) PA images of the SON-BDP solution at pH = 7.4, 6.4, or 5.5. A pulsed laser was tuned to 680 or 750 nm for ratiometric imaging. (g) Schematic illustration of the amplification and sensing of a SON-BDP-based activatable probe for pH. (h) Representative ratiometric PA images (ΔPA680/ΔPA750) of tumors in living mice before (0 h) and at 6 h after the intravenous administration of SON-BDP. (b)–(d) were reproduced from ref. 131 with permission from Nature Publishing Group, copyright 2014. (f)–(h) were reproduced from ref. 152 with permission from John Wiley & Sons Ltd, copyright 2016. | ||
To detect drug-induced ROS in tumor models, Pu's group recently developed a semiconducting macromolecular activatable probe based on the nanoparticle regrowth enhanced PA signals.145 This macromolecular probe (PCBP) was composed of NIR absorbing phthalocyanine (Pc, Scheme 1) as the hydrophobic core and four PEG chains as the hydrophilic arms. These two components were covalently linked by responsive phenylboronic acid pinacol ester groups that could be specifically cleaved by ROS including H2O2 and ONOO−. Such a ROS response allowed it to undergo a self-immolative process to release PEG and regrowth of remaining Pc segments into large nanoparticles via increased hydrophobic interactions and π–π stacking. Larger nanoparticles were beneficial for heat transfer, thus displaying an enhanced PA signal for imaging of ROS. After an intravenous injection of PCBP into tumor-bearing mice, the PA signal in the tumors pre-treated with D,L-buthionine-(S,R)-sulphoximine (BSO) to have an elevated ROS level was 2.03 times higher than that of untreated tumors. It should be noted that the small size of PCBP contributed to its effective penetration into the tumors of living mice, while the ROS-activated nanoparticle regrowth could extend the retention time along with an amplified PA signal, allowing for imaging of ROS during chemotherapy.
To increase the specificity of activatable PA probes toward a certain ROS, Pu's group synthesized an activatable NIR-absorbing SON for real-time ratiometric PA imaging of ONOO− in the tumor tissues of living mice.146 Such a probe (termed as SON-BT) was composed of a boronate-caged boron-dipyrromethene dye (BBD, Scheme 1) as the primary component for ONOO− sensing and tris(pentafluorophenyl)borane (TPFB, Scheme 1) as the secondary component. BBD responded to both ONOO− and H2O2, but doping of a large excess of bulky boranes inhibited the reaction between BBD and H2O2, probably owing to the weaker oxidative capability of H2O2 relative to ONOO−, affording the specific response of SON-BT only toward ONOO−. Upon addition of ONOO−, the PA signal at 680 nm (PA680) remained nearly unchanged, while that at 750 nm (PA750) significantly increased due to the rapid oxidative cleavage of the borate ester moiety of BBD by ONOO−, generating an anionic phenoxide product with the characteristic maximum absorption at 745 nm. The ratiometric PA signal (PA750/PA680) of SON-BT increased linearly as the concentration of ONOO−, with a LOD of ≈100 nM, and thus could potentially be used to monitor the variation of ONOO− levels in xenograft tumor models after drug treatment.
Pu's group have also proposed a self-assembly approach to synthesize an activatable and degradable PA probe for in vivo specific imaging of ClO−.147 In this approach, a semiconducting oligomer (SO1, Scheme 1) with the integration of a π-conjugated but ClO− degradable backbone (phenothiazine) was grafted with PEG chains to allow its spontaneous encapsulation of the ROS-inert NCBS dye, providing a SON (SON-N) through π–π stacking and hydrophobic interactions. The presence of ClO− induced the degradation of SON-N, whose PA signal at 680 nm was hence decreased, while its PA signal at 780 nm from NCBS remained nearly unchanged. These spectral changes allowed ratiometric PA imaging (PA780/PA680) specifically toward ClO− with a LOD of 0.70 μM. The feasibility of SON-N to be used as an activatable probe for in vivo imaging of ClO− was demonstrated on subcutaneous xenograft tumor models.
In addition to ROS detection, activatable organic PA nanoprobes can be used for in vivo ratiometric imaging of pH. As a very critical physiological parameter, pH participates in cellular and tissue homeostasis and its aberrance has a distinct influence on the biomolecular structure, protein activity and metabolic rate.148–151 Pu's group recently developed an activatable nanoprobe (termed as SON-BDP) with an amplified PA signal for pH imaging in tumor tissues.152 SON-BDP displayed a quenched fluorescence and in turn enhanced PA brightness, because of the PET between encapsulated SO2 acting as the inert PA matrix and BODIPY dye (pH-BDP) serving as both the PA enhancer and pH indicator (Fig. 11e). The hydroxyl group on the backbone of pH-BDP underwent protonation in an acidic environment to generate signal alteration. In the pH range from 7.4 to 5.5, absorbance of pH-BDP at 750 nm decreased as a function of pH, while that of SO2 at 680 nm barely changed. As a result, the PA signals at 750 and 680 nm changed accordingly (Fig. 11f), permitting ratiometric PA imaging (PA680/PA750) of pH in tumors after an intravenous injection of SON-BDP (Fig. 11g and h), as well as pH estimation of tumors at ∼6.3 according to the calibration curve.
In another work, Liu and his colleagues reported another activatable pH probe via self-assembly of benzo[α]phenoxazine (BPOx) and IR825 (Scheme 1) with human serum albumin (HSA).122 Within this probe (HAS-BPOx-IR825), IR825 was inert to pH change, acting as an internal reference, while BPOx responded to pH, because of its protonation and intramolecular charge-transfer process with pH change, serving as a pH indicator. With the decrease of pH values, the PA signal at 680 nm for BPOx displayed an obvious increase, while that at 825 nm for IR825 remained unchanged, affording good pH-dependent ratiometric PA imaging (PA680/PA825) in the range of 5.0–7.0. This ratiometric response combined with quantitative analysis using HAS-BOPx-IR825 as an activatable probe was used for PA imaging of pH in tumor microenvironments, as well as pH variation after injection of buffer solution.
Some activatable organic PA probes have been utilized for imaging of enzyme activity in living animals, which is a meaningful but challenging task to decipher enzyme function and detect related diseases.153,154 The smart PA response towards enzymes relies on the specific enzymatic cleavage of targeted substrates.155,156 Such an enzymatic cleavage can initiate the self-assembly or induce the separation of fluorophores so as to afford PA signal variations (Fig. 12a). Matrix metalloproteinases (MMPs) are the most prominent family of proteinases associated with tumors and can degrade the extracellular matrix (ECM) components.155 Wang and co-workers developed an activatable PA probe targeted for MMPs (MMP-2 and MMP-9), which consisted of purpurin 18 as the NIR fluorophore, Pro-Leu-Gly-Val-Arg-Gly (PLGVRG) peptide as the enzyme-responsive linker, and RGD peptide as the cancer targeting ligand.157 MMP-2/-9 triggered a specific cleavage of PLGVRG peptide and thereby self-assembly of residual purpurin 18 into nanoparticles, resulting in enhanced PA signals for imaging of overexpressed MMPs in xenografted mouse models. To specifically detect MMP-2 activity in follicular thyroid carcinoma, Gambhir and co-workers developed an activatable organic PA probe by conjugating an Alexa750-fluorophore and a black hole quencher (BHQ3) on each side of a MMP-2 specific cleavable cell-penetrating peptide, CXEEEEXPLGLAGRRRRRXK.156 Due to the presence of BHQ3 and Alexa750, this probe showed two PA peaks at 675 and 750 nm. MMP-2 induced a specific cleavage of the peptide, leading to separation of these two fluorophores. Subsequently, BHQ3 conjugated to the cell-penetrating peptide effectively accumulated into tumors, while the Alexa750 fragment diffused away, resulting in enhanced subtraction PA signals. β-Galactosidase is an important enzyme for living organisms as it breaks down lactose into galactose and glucose, providing energy and a carbon source for metabolism.158 Pu's group developed an activatable PA probe composed of a D-galactose caged NIR hemicyanine dye (CyOH, Fig. 12b) and linked PEG chains for PA imaging of β-galactosidase.159 The designed probe, termed as CyGal-P, was initially in its PA “turn-off” state owing to the diminished electron-donating ability of oxygen atoms in the caged CyOH, while β-galactosidase-mediated specific cleavage of the D-galactose moiety activated the PA signal for imaging of β-galactosidase in associated tumor models (Fig. 12c).
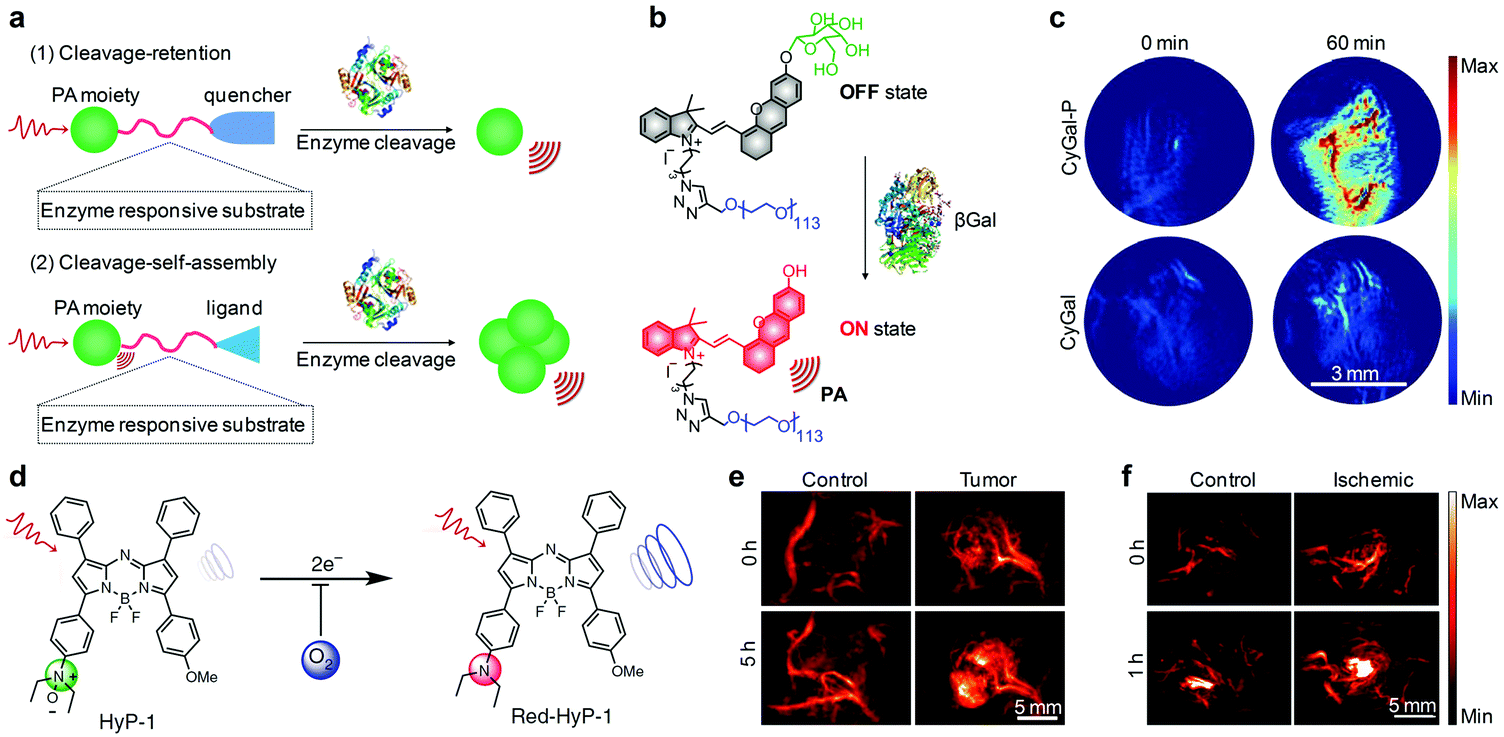 | ||
| Fig. 12 (a) Mechanisms of activatable PA probes for imaging of enzymes. (b) Schematic illustration of the activation mechanism of CyCal-P for β-galactosidase PA imaging. (c) Representative PA images of SKOV3 tumor-bearing mice before (0 min) and at 60 min post-injection time point after an intravenous injection of CyGal-P or CyGal. (d) Chemical structures of HyP-1 and red-HyP-1 and the activation mechanism for hypoxia PA imaging. (e) PA images (770 nm) of the tumor-bearing and control flank before (0 h) and at 5 h after intratumoral or subcutaneous injection of HyP-1. (f) PA images (770 nm) of the ischemic and control leg before (0 h) and at 1 h after intramuscular injection of HyP-1. (b) and (c) were reproduced from ref. 159 with permission from John Wiley & Sons Ltd, copyright 2018. (d)–(f) were reproduced from ref. 160 with permission from Nature Publishing Group, copyright 2017. | ||
Apart from the detection of ROS, pH and enzyme, an activatable PA probe (termed as HyP-1) was developed for imaging of the hypoxic environment.160 Hypoxia refers to a lower tissue oxygen level (<5 mmHg, 0.7%) than the physiological level (30–52 mmHg, 3.9–6.8%) in normal tissues, which is a pathogenic indicator of solid tumors.161,162 HyP-1 was made of unsymmetric aza-BODIPY containing a hypoxia-responsive trigger on one side and a methoxy substituent on the other side (Fig. 12d). The PA response of HyP-1 to hypoxia relied on the conversion of N-oxide to aniline, eliciting a red-shift of its absorbance from 670 to 760 nm for PA signal enhancing. Rapid and highly selective hypoxia-mediated activation of HyP-1 could be observed in cancer cells cultured under hypoxic conditions, even at an oxygen content as low as <0.1%. Thereby, activatable HyP-1 was a promising probe for PA imaging of hypoxia in a tumor-bearing mouse model and a mouse hindlimb ischemia model (Fig. 12e and f).
5.3. NIR-II PA probes
In view of the advantages of NIR-II light, it is envisioned that PA imaging in the NIR-II window can give a further improved tissue penetration depth and enhanced imaging contrast, regardless of the slightly higher endogenous absorption in this window.163 A desired imaging depth of 11.6 cm in chicken breast tissues has been reached through PA imaging at 1064 nm using a phosphorus phthalocyanine-based organic probe.164 However, a very small number of NIR-II PA imaging applications have been demonstrated, which is largely attributed to the lack of available contrast agents.Pu's group synthesized an organic imaging probe (SPN15) with a broadband absorption in both the NIR-I and NIR-II windows and applied it for NIR-II PA imaging (Fig. 13a and b).165 Rational molecular engineering afforded a D–A1–D–A2 structured SP15 (Fig. 13a), which exhibited a lower bandgap and subsequently strong absorption in the NIR-II window. After nanoprecipitation of SP15 with amphiphilic PEG-b-PPG-b-PEG, the formed SPN15 facilitated PA imaging at 1064 nm with a 1.4-times higher signal-to-noise ratio (SNR) than that at 750 nm at a tissue depth of 3 cm. Such an excellent NIR-II PA imaging probe was applied for in vivo brain vasculature imaging on living rats. At 70 min post-injection of SPN15 via the tail vein, NIR-II PA imaging at 1064 nm provided a higher contrast and clearer branch vessels relative to NIR-I PA imaging at 750 nm, showing a 1.5-times increase in SNR (Fig. 13c and d).
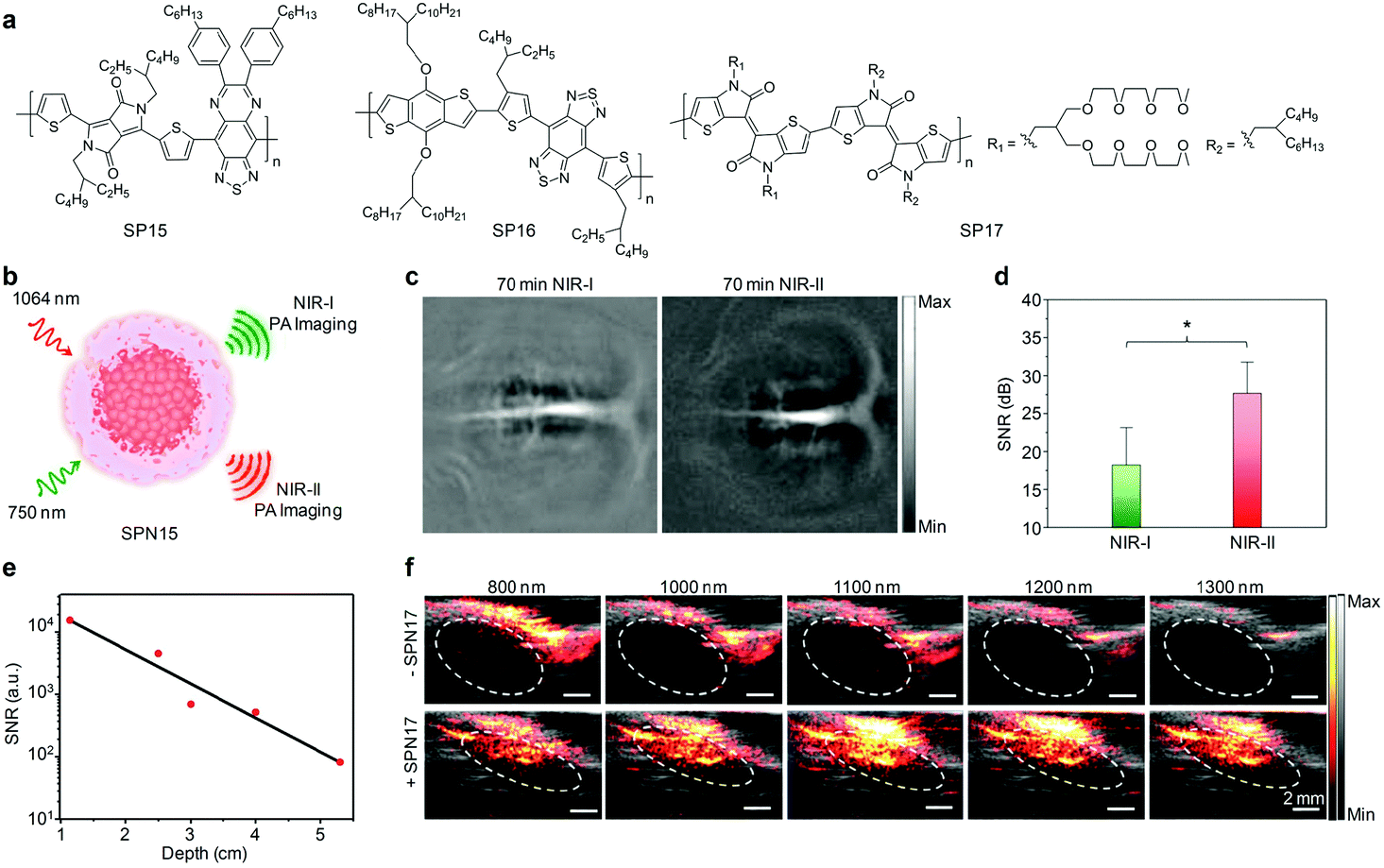 | ||
| Fig. 13 (a) Chemical structures of SP15, SP16 and SP17 used for PA imaging in the NIR-II window. (b) Schematic illustration of SPN15 for NIR-I and NIR-II PA imaging. (c) Representative PA images of rat cortex at 70 min post-injection of SPN15 at 750 (NIR-I) and 1064 nm (NIR-II). (d) SNR in decibels of brain cortex at 70 min post-injection of SPN15 at 750 and 1064 nm. (e) SNR of SPN17 at a concentration of 40 μg mL−1 contained in a transparent plastic tube as a function of depth from the illuminated tissue surface at a laser wavelength of 1064 nm. (f) In vivo PA/US imaging of a mouse tumor (dashed circles) with or without the intratumoral injection of SPN17. (b)–(d) were reproduced from ref. 165 with permission from the American Chemical Society, copyright 2017. (e) and (f) were reproduced from ref. 167 with permission from John Wiley & Sons Ltd, copyright 2017. | ||
Using suitable contrast agents, NIR-II PA imaging has been extended to visualize a mouse brain tumor model, as demonstrated by Liu and co-workers.166 In their work, a well-known electron donor, benzodithiophene, was copolymerized with a strongly electron-deficient acceptor, benzobisthiadiazole, to yield SP16 (Fig. 13a) that exhibited a red-shifted absorption in the NIR-II window with a relatively high extinction coefficient (22.6 L g−1 cm−1). The corresponding SPN16 generated strong PA signals at 1064 nm that could be detectable even at a very low concentration of 6 μg mL−1. In addition, PA imaging of orthotopic brain tumors in living mice was successfully performed at 1064 nm using intravenously injected SPN16 as a contrast agent. The tumors started to be clearly delineated at 3 h post-injection and the tumor regions as deep as 3.4 mm were clearly imaged at 24 h with a SBR of 59.
In another study, Mei and co-workers synthesized a completely new thienoisoindigo (TII)-based SP17 (Fig. 13a) with a strong absorption in the 1000–1350 nm range and transformed it into SPN17 for tumor PA imaging in the NIR-II region.167 Benefiting from the electron deficiency and high planarity of TII in SP17, the corresponding nanoparticles (SPN17) generated a strong PA signal in the NIR-II optical window, affording an imaging depth of up to 5.3 cm with a SBR of 82 in chicken-breast tissue under 1064 nm excitation (Fig. 13e). With a local injection of SPN17, both the skin tissue of rats and tumors in living mice could be clearly depicted by PA imaging conducted at multiple wavelengths ranging from 800 to 1300 nm. As compared to the images from the control tumors, the PA signals in the tumor regions were enhanced by approximately 7.0- and 13.3-fold at 1100 and 1300 nm after an intratumoral injection of SPN17, respectively (Fig. 13f).
6. Phototherapy
Phototherapy has been considered as an emerging cancer therapeutic modality and has attracted considerable attention, which is much different from conventional chemotherapy, radiotherapy, and gene therapy owing to its features including noninvasiveness, simple operation, negligible drug resistance, high temporal-spatial resolution and minimized adverse side effects.168–170 In general, phototherapy relies on the conversion of light into hyperthermia for photothermal therapy (PTT) or ROS for photodynamic therapy (PDT), thereby requiring light-absorbing materials as therapeutic agents.171–174 Nowadays, a large amount of nanomaterials are being extensively exploited as agents for cancer phototherapy, in particular, OSMs featuring strong light absorbance, tunable optical properties and good photostability have become a recent addition to this class of therapeutic agents.1756.1. PTT
A broad range of OSMs with diverse optical cores, particle architectures and surface properties have been designed and used as photothermal therapeutic agents for cancer ablation.176–181 Their photothermal conversion efficiencies have been reported in the range of 33.3–65.0% (Table 3), which are equivalent to or even higher than those of the most commonly used inorganic nanoparticles, such as SWCNTs (38.3%),182 UCNPs (38.1%),183 black phosphorus QDs (28.4%),184 CuS nanocrystals (25.7%),185 Au nanorods (21%),186 and so on. Moreover, the PTT performance of OSMs can be further improved by modulating their chemical structures or components.187| OSMs | Surface property | Laser irradiation | PTT/PDT effect | Ref. |
|---|---|---|---|---|
| PFTTQ SPNs | DSPE-PEG2000 | 808 nm (0.75 W cm−2, 10 min) | ΔT = 30 °C (0.5 mg mL−1) | 176 |
| Polypyrrole nanoparticles | Poly(vinyl alcohol) | 808 nm (0.5 W cm−2, 5 min) | ΔT = ∼34 °C (1 mg mL−1) | 177 |
| BDT-IID SPNs | PSMA | 660 nm (0.2 W cm−2, 10 min) | ΔT = 30 °C (0.1 mg mL−1), η = ∼45% | 178 |
| TDI nanoparticles | Poly(acrylic acid) | 660 nm (1 W cm−2, 10 min) | ΔT = 36.8 °C (50 μM), η = 41% | 179 |
| DPP SPNs | PSMA | 808 nm (0.5 W cm−2, 5 min) | ΔT = 36.4 °C (50 μg mL−1), η = ∼65% | 180 |
| Porphyrin conjugates | Peptide | 635 nm (1.2 W cm−2, 10 min) | ΔT = 35.3 °C (0.8 mg mL−1), η = 54.2% | 181 |
| PDPP-TT SPNs | mPEG-b-PHEP | 808 nm (2 W cm−2, 15 min) | ΔT = 29.3 °C (30 μg mL−1) | 187 |
| Porphyrin SPNs | DSPE-PEG, cell penetrating peptide | 808 nm (0.75 W cm−2, 4 min) | ΔT = 42 °C (50 μg mL−1), η = 63.8% | 188 |
| TPA-T-TQ SONs | DSPE-PEG2000 | 808 nm (0.8 W cm−2, 5 min) | ΔT = ∼53 °C (100 μM) | 189 |
| SPN19 | DSPE-PEG2000 | 808 nm (0.8 W cm−2, 3 min) | ΔT = 44 °C (12.5 μg mL−1) | 191 |
| SPN20 | PMHC18-mPEG, HOOC-PEG-COOH | 635 nm (1.4 W cm−2, 5 min) | ΔT = 33 °C (400 μg mL−1), η = 62.3% | 192 |
| DPPCN-Fc nanoparticles | — | 730 nm (1.0 W cm−2, 10 min) | ΔT = 33.4 °C (100 μg mL−1), η = 59.1% | 190 |
| SPN10-F20 | PEG-b-PPG-b-PEG | 808 nm (0.5 W cm−2, 5 min) | ΔT = ∼34 °C (15 μg mL−1) | 141 |
| SPN13 | PLGA-PEG | 808 nm (0.3 W cm−2, 5 min) | ΔT = ∼30 °C (60 μg mL−1), η = 71% | 143 |
| SPN21 | PEG-b-PPG-b-PEG | 1064 nm (1.0 W cm−2, 6 min) | ΔT = ∼50 °C (20 μg mL−1), η = 43.4% | 196 |
| SPN22 | mPEG-b-PHEP | 1064 nm (2.0 W cm−2, 5 min) | ΔT = 31.4 °C (30 μg mL−1), η = 66.4% | 197 |
| TBDOPV-DT SPNs | mPEG-PDLLA | 1064 nm (0.9 W cm−2, 10 min) | ΔT = 25.7 °C (10 μg mL−1), η = 50% | 198 |
| PT2 SPNs | DSPE-PEG | 808 nm (5.0 W cm−2, 20 min) | ΦΔ = 0.61 | 200 |
| TTD AIE nanoparticles | DSPE-mPEG, DSPE-PEG-cRGD | 530 nm (0.25 W cm−2, 15 min) | ΦΔ = 0.51 (1 μg mL−1) | 201 |
| PFBT-TPP SPNs | PSMA | 460 nm (100 mW cm−2, 20 min) | ΦΔ = 0.35 (20 μg mL−1) | 202 |
| Ce6-PFDTBT SPNs | PSMA | 520 nm (0.1 W cm−2, 10 min) | ΦΔ = ∼0.4 (5 μg mL−1) | 203 |
| TPP-PFBT SPNs | PSMA | 460 nm (50 mW cm−2, 8 min) | ΦΔ = 0.5 (20 μg mL−1) | 204 |
| RET-BDP SPNs | PEG-b-PPG-b-PEG | 645 nm (10 mW cm−2, 20 min) | ΦΔ = 0.53 (5 μM) | 205 |
| DPPBDPI SPNs | — | Xenon lamp (8 min) | ΦΔ = ∼0.8 (5 μM) | 206 |
| TPETCAQ AIE nanoparticles | DSPE-PEG, HIV-1 Tat | 400–1000 nm (60 mW cm−2, 6 min) | ΦΔ = 0.81 | 208 |
| SPN10 | PS-b-PAA, silica, MnO2, PEG-b-PPG-b-PEG | 808 nm (0.44 W cm−2, 6 min) | ΦΔ = 0.073 (10 μg mL−1) | 210 and 211 |
| SPN23–24 | DSPE-PEG, anti-HER2 | 808 nm (1 W cm−2, 10 min) | ΔT = 40 °C (100 nM), η = 47.6%, ΦΔ = 0.604 | 218 |
| SPN25 | PEG-PCL | 785 nm (1.5 W cm−2, 10 min) | ΔT = 65.2 °C (20 μg mL−1), η = 34.7%, ΦΔ = 0.1 | 219 |
| DPP-TPA nanoparticles | — | 660 nm (1.0 W cm−2, 10 min) | ΔT = ∼34 °C (80 μg mL−1), η = ∼34.5% | 220 |
| ΦΔ = 0.336 | ||||
| Tri-BDP nanoparticles | PEG-PCL | 780 nm (0.5 W cm−2, 5 min) | ΔT = 38 °C (80 μg mL−1), η = 45.2% | 221 |
| ΦΔ = 0.18 |
Quenching the fluorescence emission of OSMs is an effective way to improve the photothermal conversion efficiency. For example, a facile intraparticle molecular orbital engineering approach was used to quench the fluorescence of SPN10 and ultimately enhance the PTT efficiency.141 SPN10 was doped with different amounts of PC70BM to favor the intra-particle PET. With increasing doping amount of PC70BM, the fluorescence intensities of SPN10 decreased rapidly and the temperatures of nanoparticle solutions under laser irradiation increased gradually. As a result, SPN10 with a 20% doping amount (SPN10-F20) showed a 1.3-fold maximum temperature increase as compared to the nondoped nanoparticles (SPN10-F0), affording a better efficiency for cancer PTT.
In addition to the intraparticle molecular orbital engineering approach, a self-quenched molecular engineering approach was used to improve the photothermal properties of SPNs.133 Pu and Rao et al. synthesized a series of D–A structured SPs with DPP as the electron acceptor copolymerized with three different electron-donating monomers and then transformed them into water-soluble SPNs.133 Among these SPNs, SPN18 showed the highest fluorescence quenching efficiency, displaying the best photothermal property. Later on, this approach was used to develop other D–A structured molecules and SPs for enhanced PTT performance.188–191 For instance, Dong and co-workers recently synthesized a DPP derivative (termed as DPPCN-Fc, Scheme 1), wherein two ferrocene (Fc) moieties acted as the electron-donating units to quench the fluorescence through a PET process.190 After self-assembly into nanoparticles through π–π stacking interactions and hydrophobic interactions, DPPCN-Fc SONs exposed to 730 nm laser irradiation resulted in a temperature elevation of 33.4 °C within 10 min and showed an amplified photothermal conversion efficiency reaching up to 59.1%. In another study, Liu and co-workers conjugated a planar electron donor with the DPP electron acceptor to quench the fluorescence of SP19 (Scheme 2) and thus increased the photothermal heating efficiency of SPN19 for cancer PTT.191
Since the light-harvesting property is directly related to the extinction coefficient that greatly modulates PTT efficiencies of SPNs, integration of light-harvesting units into D–A structured SPs has been recently exploited for further enhancing the light-absorbing ability and in turn improving the photothermal conversion efficiency.192 Lee and co-authors synthesized a D–A structured SP20 (Scheme 2) containing a porphyrin–pyrene pendant acting as the light-harvesting unit.192 In addition, a fluorescence quenching effect occurred during transformation of SP20 into nanoparticles through nanoprecipitation. The fluorescence quenching effect and enhanced light-absorbing ability synergistically improved the photothermal conversion efficiency of SPN20 to 62.3%.
In addition, the PTT performance of SPNs can be significantly improved by incorporating vinylene bonds into the backbone of SPs.143 In a recent study of Pu's group, incorporation of vinylene bonds into SPN13 increased the maximum photothermal temperature and photothermal conversion efficiency by 1.2- and 2.4-fold, respectively, relative to the control SPN18 without vinylene bonds (Fig. 14a and b).143 Such an amplified PTT performance of SPN13 allowed effective ablation of tumors in living mice, while the tumors of SPN18-treated mice grew as fast as those of saline-treated mice (Fig. 14c).
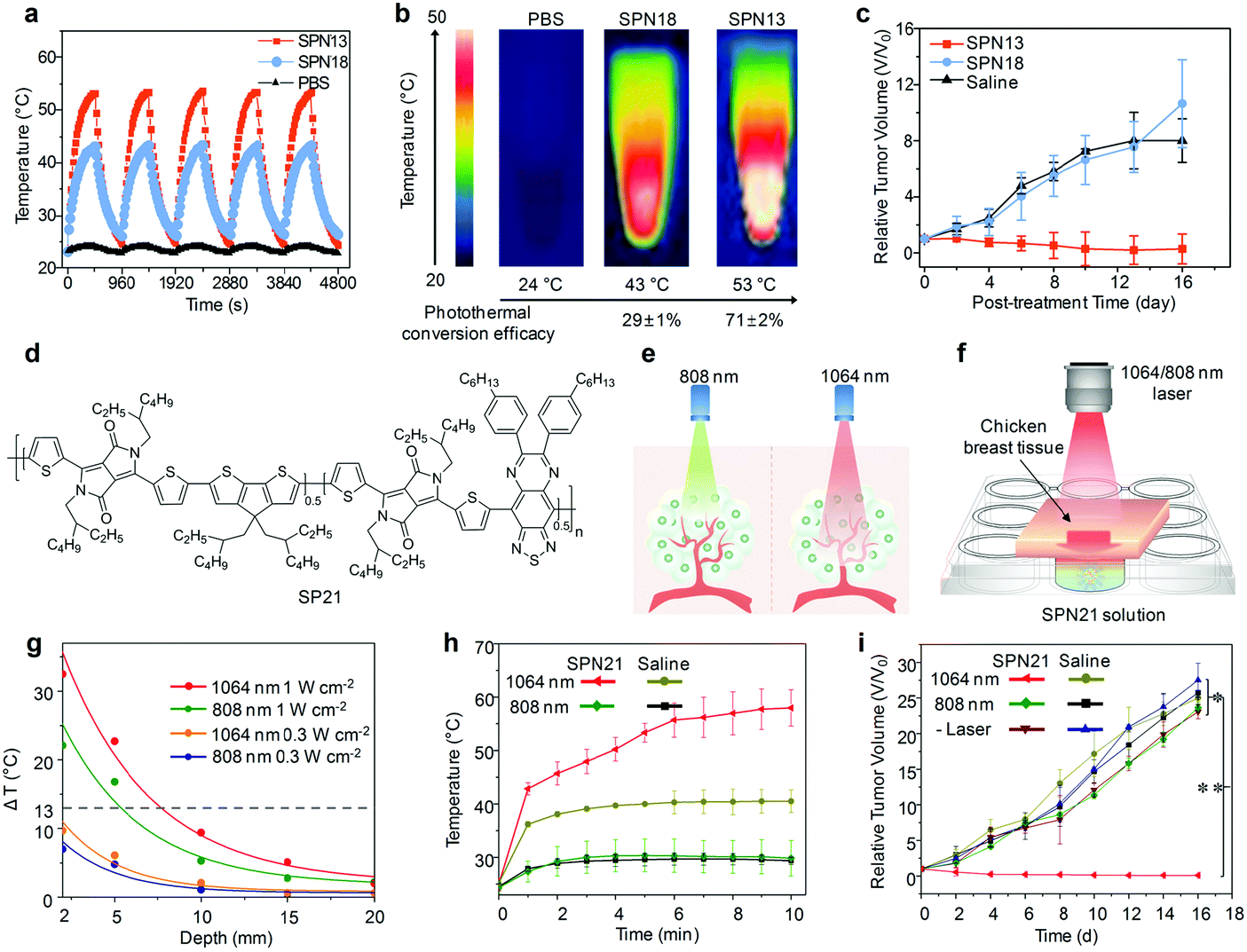 | ||
| Fig. 14 (a) Photothermal heating and natural cooling cycles of SPN13 and SPN18 under 808 nm laser irradiation with a power density of 0.3 W cm−2. (b) Thermal images of PBS, SPN13 and SPN18 at their respective maximum temperatures (both the corresponding maximum temperature values and photothermal conversion efficacies are indicated below the respective images). (c) Tumor growth curves of mice after systemic injection of saline, SPN13 or SPN18 with 808 nm laser irradiation. (d) Chemical structure of SP21 used for deep-tissue NIR-II PTT. (e) Scheme illustrating the deeper tissue penetration of the 1064 nm laser relative to that of the 808 nm laser. (f) Schematic illustration of the photothermal study of the SPN21 solution at different tissue penetration depths. (g) Fitted exponential decay of temperature change of the SPN21 solution at different depths of chicken breast tissue under different laser conditions. (h) Mean tumor temperature as a function of laser irradiation time after intratumoral administration of SPN21. (i) Tumor growth curves of mice after intratumoral injection of SPN21 or saline with and without irradiation of the 808 or 1064 nm laser. (a)–(c) were reproduced from ref. 143 with permission from the American Chemical Society, copyright 2018. (e)–(i) were reproduced from ref. 196 with permission from John Wiley & Sons Ltd, copyright 2018. | ||
Despite great efforts being devoted to increase the photothermal conversion efficiencies of therapeutic agents, current PTT applications are only suited for superficial tumors due to the limited penetration depth of lasers.193 To achieve effective ablation of deep-seated tumors, NIR-II PTT has been recently used because of the intrinsic advantages of NIR-II light, including deeper tissue penetration and larger maximum permissible exposure (MPE) for skin as compared to the NIR-I light.193–195 To reveal the practical superiority of NIR-II over NIR-I light in terms of cancer PTT, Pu's group have recently synthesized a semiconducting copolymer SP21 (Fig. 14d) that showed dual-peak absorption in both the NIR-I and NIR-II windows and formulated it into a nanoparticle (SPN21) through nanoprecipitation with PEG-b-PPG-b-PEG.196 The deeper tissue penetration in combination with a higher MPE limit of the 1064 nm laser (1 W cm−2) relative to that of the 808 nm laser (0.3 W cm−2) allowed higher heat generation for SPN21 solution covered with chicken breast tissue of different thicknesses (Fig. 14e–g), even though the photothermal conversion efficiency of SPN21 at 1064 nm (43.4%) was nearly the same as that at 808 nm (44.9%). In a mimic deep-tissue tumor model at a tissue depth of 5 mm, intratumoral injected SPN21 with 1064 nm laser irradiation generated more heat and totally ablated tumors, while the 808 nm laser failed to do so (Fig. 14h and i). Such a deep-tissue heating capability also allowed effective PTT of xenograft tumors after systemic administration of SPN21.
In addition to SPN21, some other SPNs have been subsequently developed and used for NIR-II PTT of cancers so far.197,198 For instance, a thieno-isoindigo derivative-based D–A structured polymer (SP22, Scheme 2) with a maximum absorption peak at approximately 1107 nm was synthesized and formulated into nanoparticles.197 The obtained SPN22 showed superior photothermal conversion efficiency (66.4%) under laser irradiation at 1064 nm. Similarly, another SP (termed as TBDOPV-DT) possessing the same polymer backbone as SP22 was transformed into nanoparticles to have a narrow and strong absorbance in the NIR-II window and a photothermal conversion efficiency of 50%.198
6.2. PDT
Due to the characteristic absorbance that can be utilized to produce ROS under laser irradiation, a number of organic semiconducting components have been formulated into nanoparticles for cancer PDT.199,200 The formed nanoparticles can well overcome the drawbacks of hydrophobicity and poor photostability, as well as afford targeted delivery into tumors through surface modifications, leading to effective PDT performance.201 Singlet oxygen (1O2) is the major ROS type generated during PDT with OSMs as agents.201 However, PDT inherently requires O2 to generate 1O2 and thus its therapeutic efficacy is generally limited due to the hypoxic microenvironment of solid tumors.161 Therefore, there is an urgent demand to develop effective strategies to amplify or modulate the PDT performances.Incorporation of photosensitizers into SPNs has been considered as a good method to enhance the PDT efficiency (Table 3).202 Wu and co-workers have synthesized a class of photodynamic agents with amplified generation of 1O2 by doping tetraphenylporphyrin (TPP) and chlorin e6 (Ce6) into SPNs.203,204 These designs allowed efficient resonance energy transfer (RET) from SPs to photosensitizer molecules, resulting in a 3.5–5-fold improvement in 1O2 quantum yield, which led to better outcomes of cancer PDT in living mice. By utilizing the same RET mechanism, Han et al. subsequently constructed a novel small dyad molecule (termed as RET-BDP, Scheme 1) by connecting a fluorophore (distyryl-BODIPY) as the energy donor moiety to a photosensitizer (diiodo-distyryl-BODIPY) as the energy acceptor.205 After being encapsulated by folic acid modified PEG-b-PPG-b-PEG, RET-BDP molecules were formulated into SOMs that exhibited a 1.8-fold higher 1O2 quantum yield relative to the nanoparticle counterparts of the photosensitizer alone under NIR laser irradiation, which enabled a better therapeutic PDT effect against cancer cells both in vitro and in vivo. In addition to the RET mechanism, photosensitizer incorporation has induced a synergistic effect that can be utilized to enhance PDT efficiency.206 For instance, Dong and co-workers synthesized a novel D–A–D structured organic photosensitizer (termed as DPPBDPI, Scheme 1) by linking DPP and BODIPY with a benzene ring as a π bridge, wherein the synergistic effect of DPP and BODIPY elevated the 1O2 quantum yield to 80%.206
The fact that some AIE photosensitizers display efficient 1O2 generation in the aggregate state makes AIE based nanoparticles alternative candidates for cancer PDT, but most of these photosensitizers often suffer from the drawbacks of absorption and emission at short wavelengths, and low efficiency of 1O2 generation.207 To address these issues, Liu's group used a new molecular design strategy to synthesize a novel AIE photosensitizer (termed as TPETCAQ, Scheme 1).208 Introduction of a phenyl ring between the dicyanovinyl (DC) group and methoxy-substituted tetraphenylethylene (TPE) within TPETCAQ led to increased 1O2 generation efficiency while addition of another DC group to the acceptor red-shifted the absorption and emission to the NIR region. As such, the corresponding AIE nanoparticles after surface modification with a cell membrane penetrating peptide enabled image-guided PDT of tumors.
In addition to the molecular engineering approaches, O2-evolving nanoparticles that can continuously generate O2 have been developed and used to overcome the hypoxic microenvironment of tumors, representing a class of promising candidates for amplified PDT.209 Pu's group utilized a one-pot straightforward method to in situ grow manganese dioxide (MnO2) nanosheets on the surface of SiO2-coated SPN10, yielding a hybrid core–shell nanoparticle (SPN10-M1).210 The surface coated MnO2 nanosheets acted as a sacrificing component to convert H2O2 to O2 under hypoxic and acidic tumor microenvironments, resulting in a 2.68-fold higher 1O2 generation under NIR laser irradiation at 808 nm (Fig. 15a–c). With such an oxygen-evolution capability, SPN10-M1 displayed better efficiency in eradicating cancer cells both in vitro and in vivo as compared with its uncoated counterpart (SPN10-M0).
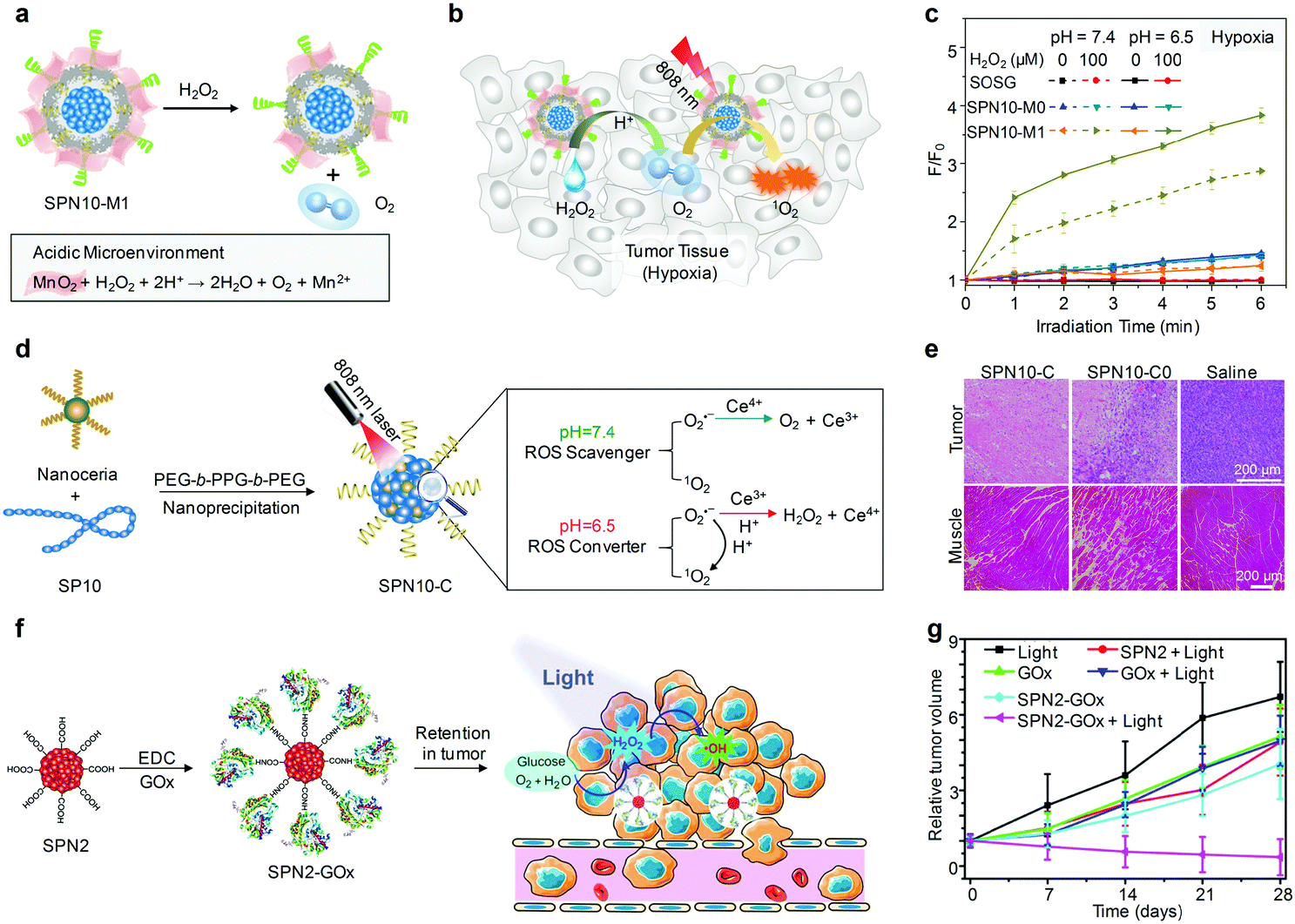 | ||
| Fig. 15 (a) Schematic illustration for the H2O2-responsive mechanism of SPN10-M1 and (b) the detailed mechanism of SPN10-M1 for amplified PDT in tumor. (c) The generation of 1O2 determined by the increased fluorescence of SOSG for SPN10-M0 and SPN10-M1 with or without addition of H2O2 in a hypoxic environment under NIR laser irradiation at 808 nm. (d) Schematic illustration of the synthesis of SPN10-C and the self-regulated photodynamic properties under physiologically neutral and pathologically acidic conditions. (e) Histological H&E staining for mouse tumor and muscle at 24 h after treatment with saline, SPN10-C or SPN10-C0 under 808 nm laser irradiation. (f) Schematic illustration of SPN2-GOx for generation and photolysis of H2O2 in tumors for therapy. (g) Tumor growth curves in xenograft-bearing mice after different treatments. (a)–(c) were reproduced from ref. 210 with permission from the American Chemical Society, copyright 2017. (d) and (e) were reproduced from ref. 211 with permission from the American Chemical Society, copyright 2017. (f) and (g) were reproduced from ref. 212 with permission from the American Chemical Society, copyright 2017. | ||
To increase the therapeutic efficiency and selectivity for cancers but reduce the risk of damage to normal tissues of PDT, Pu's group developed a nanoceria doped hybrid SPN10 that could self-regulate the PDT properties according to the tumor microenvironment for optimized therapy.211 The nanoceria functioned as a smart intraparticle modulator to decrease the ROS generation from SPN10 under 808 nm laser irradiation in physiologically neutral environments, but to increase the ROS generation under pathologically acidic conditions, respectively (Fig. 15d). Under laser irradiation, the nanoceria doped hybrid SPN10 (SPN10-C) showed a 2.9-fold improvement in killing cancer cells relative to that for SPN10 without nanoceria doping (SPN10-C0). Moreover, the self-regulated properties of SPN10-C not only afforded a better PDT performance for cancer ablation under NIR laser irradiation, but also resulted in reduced nonspecific damage to normal tissue (Fig. 15e).
In addition, a novel strategy called enzyme-enhanced phototherapy (EEPT) has been proposed for cancer treatment, showing a significantly enhanced therapeutic efficiency as compared to typical PDT.212 This nanoparticle platform was constructed by covalently conjugating glucose oxidase (GOx) onto the surface of SPN2 to afford SPN2-GOx, which efficiently catalyzed the conversion of glucose in tumor tissues into gluconic acid and H2O2. Under laser irradiation, the in situ generated H2O2 was photolyzed to produce hydroxyl radicals (˙OH), the most reactive ROS, to kill cancer cells (Fig. 15f). In view of the persistent immobilization of SPN2-GOx in tumors, a conspicuous inhibition of tumor growth was realized in xenograft-bearing mice (Fig. 15g), suggesting the potential for effective cancer treatment.
6.3. Combinational phototherapy
Owing to the limitations of monotherapy, cancer recurrence often occurs after treatment.213 To further improve the therapeutic efficiency, combinational cancer therapy has been widely used.214,215 The combination of PTT and PDT can induce a synergistic effect to amplify the PDT efficacy, as photothermal effects accelerate the blood flow to allow more oxygen transport into tumors.216,217 To realize such a combinational phototherapy of cancer, Liu and co-workers co-encapsulated PFVBT (SP23, Scheme 2) with efficient ROS generating capability and PIDTTTQ (SP24) with an excellent photothermal effect into a single SPN, which displayed a high 1O2 quantum yield of 60.4% and photothermal conversion efficiency of 47.6%.218 The co-loaded SPNs with conjugation of anti-HER2 affibody allowed for targeted PTT and PDT of HER2 over-expressing tumors with ∼53% of tumor suppressive efficiency. In addition to the co-doping of two different SPs, rational molecular design of D–A structured SPs contributed to generate light-triggered photoconversion behaviors with both photothermal and photodynamic performance. As demonstrated in a recent study of Chen's group, a low-bandgap D–A type SP25 (Fig. 16a) with BT as the electron donor and BIBDF as the electron acceptor has been synthesized and transformed into SPN25.219 Laser irradiation of SPN25 allowed singlet-to-triplet transition triggered by the charge transfer in the excited D–A system to produce 1O2, as well as nonradiative decay induced by the high electron-deficiency of the electron acceptor to generate a thermal effect (Fig. 16b). Furthermore, SPN25 exhibited an enhanced cellular uptake efficiency and effective tumor accumulation, accelerating the synergistic PDT/PTT effects for severe cell damage and tumor ablation. | ||
| Fig. 16 (a) Chemical structure of SP25 used for preparation of SPN25 for combinational cancer PTT and PDT. (b) Photophysical mechanism of SPN25 for photoactive cancer therapy (Ex., excitation; NRR, nonradiative recombination; Phos., phosphorescence; ISC, intersystem crossing). (c) Schematic of the preparation and light-activated hypoxia-responsive drug delivery system of DSPN26. (a) and (b) were reproduced from ref. 219 with permission from John Wiley & Sons Ltd, copyright 2017. (c) was reproduced from ref. 232 with permission from John Wiley & Sons Ltd, copyright 2016. | ||
In addition to SPNs, molecular engineering of organic small molecules has led to the generation of SONs for synergistic PTT/PDT. For instance, triphenylamine (TPA) was conjugated to the DPP backbone for absorption red-shift and charge transport capacity enhancement, obtaining a D–A–D structured DPP-TPA (Scheme 1).220 After self-assembly into SONs, the absorption of DPP-TPA further red-shifted from 630 nm to 660 nm and the D–A–D structure was further enhanced, which facilitated the charge transport to generate more heat under laser irradiation. The obtained DPP-TPA nanoparticles exhibited a high photothermal conversion efficiency of 34.5% and 1O2 quantum yield of 33.6%, and thus effectively ablated tumors under laser irradiation at 660 nm. In another study, conjugated coupling of BDP monomers into trimeric BDP (tri-BDP) could trigger photoconversions from fluorescence to singlet-to-triplet or nonradiative transitions and red-shift of the absorption into the NIR region.221 Therefore, tri-BDP based nanoparticles not only had a 2-fold enhanced singlet oxygen generation efficiency (18%), but also showed a preferable photothermal conversion efficiency (45.2%) under NIR light exposure, realizing complete tumor ablation through their cooperative anticancer outcome.
Photothermal-chemotherapy that combines thermal effect and drug-induced toxicity has been considered as a promising treatment for solid tumors to overcome the drug resistance of conventional chemotherapy.222 In such a therapeutic system, the photothermal effect can induce cancer cell death and also facilitate the drug release, and thus enhance chemotherapeutic efficiency.9,223,224 Pu's group recently utilized amphiphilic SP10 attached with PEG as both photothermal agents and nanocarriers to encapsulate an anti-cancer drug doxorubicin (DOX), obtaining multifunctional DSPN10 for imaging guided chemo-photothermal therapy.225 After intravenous injection into tumor-bearing living mice, DSPN10 exhibited superior antitumor efficacy over SPN10-PEG and free DOX because of the synergistic effect of PTT and chemotherapy. Similarly, Yang and Li et al. contributed another two related studies, in which anticancer drugs were loaded into SPNs with high photothermal conversion efficiency to allow light-triggered enhanced drug release for synergistic photothermal-chemotherapy of xenograft tumor models in living subjects.226,227 In addition, polyaniline (PANI)-based SPNs in combination with rapamycin (RAPA), an antiangiogenic drug that can destroy the tumor vessels and inhibit the tumor growth, have been developed for cancer therapy.228 NIR laser irradiation of these PANI SPNs resulted in thermal generation and burst release of RAPA, which enabled enhanced photothermal and antiangiogenic combination therapy.
The combination of chemotherapy and PDT has also been used to achieve high therapeutic outcomes, in particular for the ROS-responsive drug delivery systems.229,230 Liu's group reported a novel therapeutic SPN based on a conjugated-polyelectrolyte (CPE) polyprodrug for cancer targeted PDT and chemotherapy.231 In this platform, PEGylated CPE serving as both the photosensitizer and nanocarrier was covalently conjugated with DOX through a ROS-cleavable linker (thioketal). Upon laser irradiation, the generated ROS from the photosensitizer not only could be used for PDT, but also caused on-demand drug release due to the cleavage of the linker, allowing for combinational and enhanced cancer therapeutic efficiency. In another study, Gu and co-workers reported the combination of PDT with a hypoxia-responsive drug-release system based on SPNs for enhanced cancer therapy.232 A ROS-generating and hypoxia-sensitive 2-nitroimidazole-grafted polymer (SP26) was utilized to encapsulate DOX through a double-emulsion method to generate DSPN26. Laser irradiation of DSPN26 produced 1O2 for PDT and rapidly consumed the dissolved oxygen, leading to a local hypoxic environment. Therefore, the hydrophobic 2-nitroimidazole (NI) groups were reduced to hydrophilic 2-aminoimidazoles (AI) under the bioreductive conditions of cells, resulting in the dissociation of DSPN26 for drug release (Fig. 16c). Such a light-activated hypoxia-responsive drug delivery system allowed for combinational photodynamic-chemotherapy of tumors with an enhanced therapeutic efficiency.
7. Photoactivation
Remote regulation of biomolecular activities in living systems at designated locations and times is of fundamental necessity for better understanding of physiological processes and pathogenesis, and potentially leads to innovative therapeutic medicine.233 On this front, magnetic fields,234 genetic engineering,235 chemical modification,236 ultrasounds,237 electric fields,238 radio waves239 and light irradiation240 are mainly used approaches that can control cellular behaviors, protein activities, transgene systems and so on. Among these approaches, photoactivation that utilizes light stimulus to trigger biological activation has gained increasing popularity and interest owing to its advantages, such as easy production, good controllability, simple operability, high spatiotemporal resolution and noninvasive nature.241,2427.1. Photoactivation of ion channels
Rapid thermal stimulation in the innoxious temperature range can open the Ca2+ channels in plasma membranes, eliciting the activation of neurons.243 Gold nanorods and carbon nanomaterials have been used to convert photon energy into local heat for remote photoactivation of the temperature-sensitive transient receptor potential cation channel subfamily V member 1 (TRPV1, a typical Ca2+ channel protein) intrinsically overexpressed on the plasma membrane of neurons.244–246 However, these agents are short of the specificity and have the risk of damaging the cellular membrane. To overcome these limitations, Pu's group synthesized a TRPV1 targeted SPN14 for specific photothermal activation of neurons.247 In this study, SP14 was transformed into SPN14 through nanocoprecipitation to have a faster heating capability under NIR laser irradiation at 808 nm as compared to both SPN10 and gold nanorods. Anti-TRPV1 antibodies were conjugated onto SPN14 to generate semiconducting polymer nanobioconjugates (SPN14bc) that could precisely target TRPV1, ensuring quick diffusion of locally generated heat from SPN14bc to the TRPV1 ion channels under NIR laser irradiation (Fig. 17a). Thereby, SPN14bc worked as a wireless remote nanomodulator to rapidly and specifically activate the TRPV1 ion channels, inducing intracellular Ca2+ influx of neurons within milliseconds in a safe and reversible manner (Fig. 17b). | ||
| Fig. 17 (a) Schematic illustration of SPNbc controlled photothermal activation of Ca2+ channels in neurons. The intracellular concentration of Ca2+ was monitored in real-time by using Fluo-8 as the indicator, which turned on its fluorescence upon binding with Ca2+. (b) Representative confocal fluorescence images of mouse neuroblastoma/DRG neuron hybrid ND7/23 or HeLa cells treated with SPN14bc or SPN10bc before and after laser irradiation at 808 nm for 2 s. (c) Schematic illustration of the synthesis of SPN7-C. (d) Schematic illustration of the proposed apoptosis mechanism induced by SPN7-C under NIR laser irradiation. (a) and (b) were reproduced from ref. 247 with permission from the American Chemical Society, copyright 2016. (c) and (d) were reproduced from ref. 250 with permission from the American Chemical Society, copyright 2018. | ||
TRPV1 is also highly over-expressed in many types of cancer cells,248 and thereby activation of TRPV1 ion channels represents a promising approach for cell-specific therapy.249 For instance, use of a temperature-responsive semiconducting photothermal nanoagonist enabled remote and specific apoptosis of cancer cells from the cell membrane.250 To make the nanoagonist responsive to NIR light, SP7 with silicon in the backbone to red-shift the absorption was designed. Self-assembly of SP7, capsaicin (Cap, a TRPV1 agonist) and lipid layers led to the generation of a semiconducting photothermal nanoagonist (SPN7-C, Fig. 17c). Under NIR laser irradiation at the time scale of seconds, photothermal conversion of SPN7 increased the temperature and melted the lipid layers, allowing for the controlled release of Cap to active TRPV1 Ca2+ channels on the cellular membrane. As a cumulative consequence of multiple photoactivation of TRPV1 ion channels, over-influx of Ca2+ into mitochondria occurred, inducing cell apoptosis specifically for TRPV1-postive cancer cells (Fig. 17d).
7.2. Photoactivation of gene expression
Photoactivation can also be used to remotely switch on gene expression for treating inherited genetic diseases and cancers.251,252 A proof-of-concept application of photothermal semiconducting polymer nanocarriers for remote photoactivation of gene expression in living cells and animals was recently demonstrated by Pu's group.253 A NIR light absorbing dendronized semiconducting polymer (DSP, Fig. 18a) was synthesized by grafting polyamidoamine (PAMAM) side chains and PEG blocks onto SP10, serving as a nanocarrier to load plasmids with the gene of interest cloned downstream of a heat shock promoter (HSP70). After cellular internalization, the plasmids were released from DSP and entered into cellular nucleus, while DSP was retained in the cytoplasm to transform photons into heat under NIR laser irradiation. This mild photothermal effect did not cause obvious cytotoxicity, but effectively triggered a heat-inducible promoter to activate the intracellular downstream targeting gene transcription and protein expression (Fig. 18b and c). Using the luciferase gene (pSV40-Luc) as a model gene, such DSP-mediated photoactivation could rapidly and safely result in 25- and 4.5-fold increases in the luciferase expression in living cells and mice, respectively (Fig. 18d and e). | ||
Fig. 18 (a) Chemical structure of DSP. (b) Illustration of the DSP-mediated gene delivery and remote photoactivation of intracellular gene expression under 808 nm laser irradiation. (c) Representative confocal fluorescence images of HeLa cells transfected with DSP/pHSP70-EGFP nanocomplexes with a N/P ratio of 7.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with or without laser irradiation at 808 nm (0.75 W cm−2) for 30 min. Red indicates the DSP, blue indicates the cell nucleus and green indicates the expressed EGFP. (d) Illustration of laser irradiation in living nude mice and (e) whole-animal BL images at representative times with (left side of animal) and without (right side of animal) laser irradiation at 808 nm (0.42 W cm−2). The red/white dashed circles indicate the respective regions subcutaneously implanted with HeLa cell pellets transfected with DSP/pHSP70-Luc nanocomplexes and treated with/without laser irradiation. (b)–(e) were reproduced from ref. 253 with permission from John Wiley & Sons Ltd, copyright 2017. :1 with or without laser irradiation at 808 nm (0.75 W cm−2) for 30 min. Red indicates the DSP, blue indicates the cell nucleus and green indicates the expressed EGFP. (d) Illustration of laser irradiation in living nude mice and (e) whole-animal BL images at representative times with (left side of animal) and without (right side of animal) laser irradiation at 808 nm (0.42 W cm−2). The red/white dashed circles indicate the respective regions subcutaneously implanted with HeLa cell pellets transfected with DSP/pHSP70-Luc nanocomplexes and treated with/without laser irradiation. (b)–(e) were reproduced from ref. 253 with permission from John Wiley & Sons Ltd, copyright 2017. | ||
Wang and co-workers subsequently reported a similar strategy for remote photoactivation of intracellular gene expression using SPNs as the photothermal nanotransducers under NIR laser irradiation.254 To improve the specificity and efficiency, they conjugated a cell-penetrating peptide (Tat) onto SPN27 to allow its location on the surface of the cell membrane after incubation. Upon irradiation with a 808 nm laser, SPN27-Tat generated localized heat in plasmid transfected cancer cells. As a result, the heat-inducible HSP-70 promoter could start transcription of any downstream gene of interest (for example, enhanced green fluorescence protein, EGFP) inside cells.
7.3. Photoactivation of enzyme activity
Remote controlling of enzyme activity is no longer just a method to regulate cell fates, but also provides new opportunities for monitoring health and treating diseases.255–257 Because the activity of most enzymes is temperature dependent, photothermal effect mediated photoactivation can be used to dynamically regulate enzyme activity.258,259 In this regard, Pu's group recently described the design of a semiconducting polymer nanoenzyme, a SPN/enzyme complex, that could be activated by a NIR laser for enhanced cancer PTT.260 To improve the coupling efficiency of the enzyme, SP10 grafted with short-chain methoxy-PEG (Mw = 1000) and long-chain carboxyl-PEG (Mw = 2000) was designed and synthesized (Fig. 19a). Bromelain (Bro), a temperature-sensitive enzyme that could proficiently digest collagen, was covalently conjugated to SPN10 via a carbodiimide coupling reaction, affording SPN10-Bro. SPN10-Bro could convert photon energy into heat under NIR laser irradiation at 808 nm, leading to increased local temperature. As the optimal activity of Bro was at ∼45 °C, the local heat resulted in more than 2-fold enhancement in the enzymatic activity of SPN10-Bro (Fig. 19b and c). Such a photothermally triggered remote activation of enzyme enabled an improved penetration depth of SPN10-Bro in 3D tumor spheroids and an enhanced nanoparticle accumulation in tumors (Fig. 19d), which should be attributed to the in situ digestion of collagen type I (Fig. 19e), the most abundant ECM proteins in the tumor microenvironment. Therefore, SPN10-Bro induced higher heat generation in the tumor regions under 808 nm laser irradiation and eventually afforded better anticancer efficiency relative to control SPN10.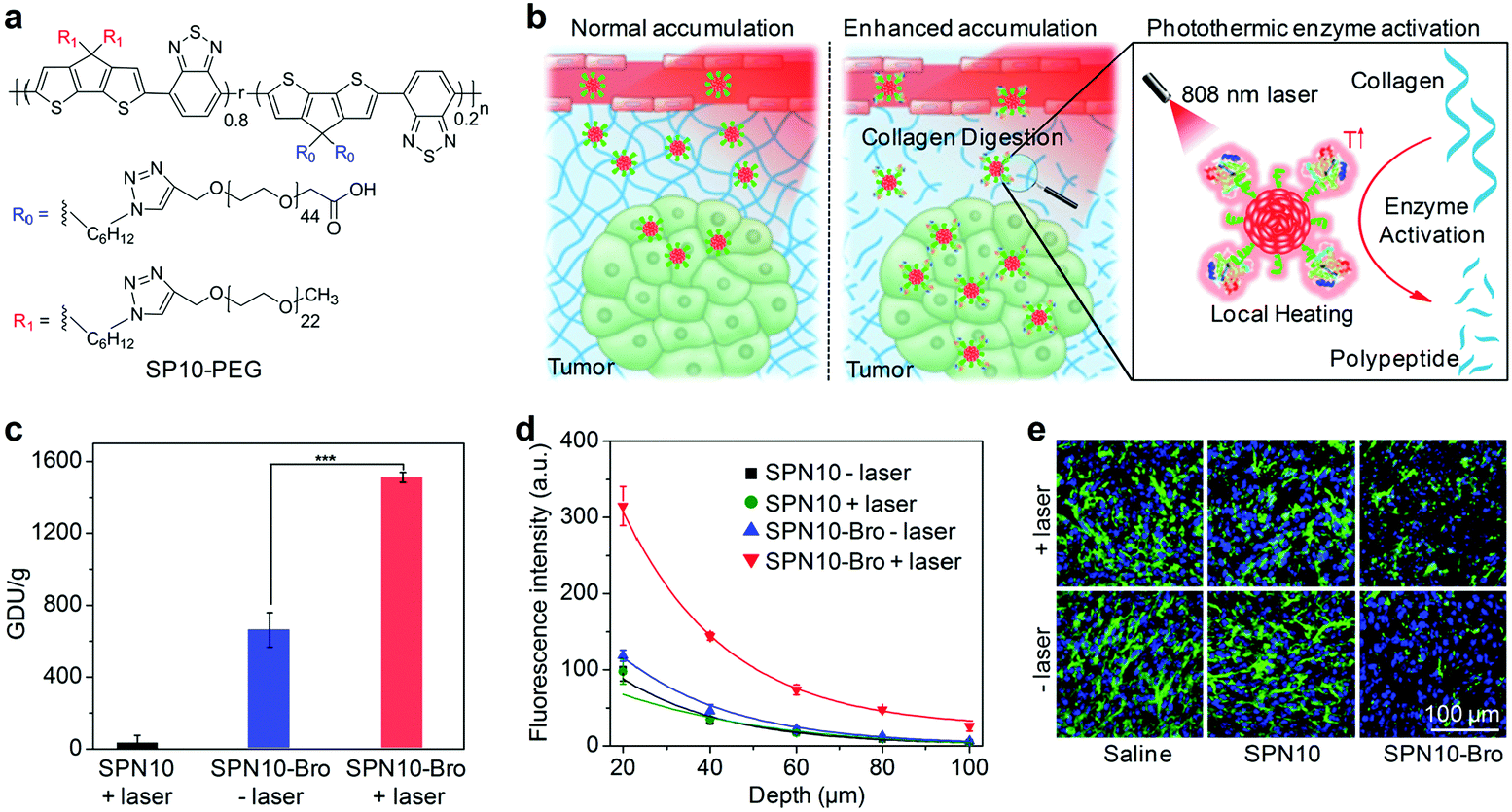 | ||
| Fig. 19 (a) Chemical structure of SP10-PEG. (b) Illustration of the photothermally triggered enzyme activation of SPN10-Bro towards collagen I digestion for enhanced accumulation of nanoparticles in tumors. (c) Enzymatic activity of SPN10-Bro with or without NIR laser irradiation using gelatin as a substrate; the enzymatic activity was defined as gelatin digestion unit (GDU). (d) Fluorescence intensities of 3D tumor spheroids after treatments of SPN10 or SPN10-Bro with or without 808 nm laser irradiation at different depths. (e) Immunofluorescence collagen I staining images of 4T1 tumors after intratumoral injection of saline, SPN10 or SPN10-Bro with or without NIR laser irradiation. Cell nuclei (blue color) were stained with 4′,6-diamidino-2-phenylindole (DAPI) and collagen I (green color) was stained with Alexa Fluor 488 conjugated anti-collagen I antibody. (b)–(e) were reproduced from ref. 260 with permission from John Wiley & Sons Ltd, copyright 2018. | ||
7.4. Photoactivation of chloroplast photosynthesis
In addition to the photothermal effect of OSMs, their unique light harvesting ability can allow activation of chloroplast photosynthesis (Fig. 20a).261 Two typical light-harvesting SPs (SP28 and SP29) with a backbone structure containing fluorene units were utilized to nanoprecipitate into SPN28 and SPN29, respectively, which were then coated onto the surface of chloroplasts. In such a chloroplast/SPNs system, SPNs could strongly absorb the irradiated light, especially UV light, and emitted visible light for chloroplast absorption. As a result, photosystem II (PS II), a critical protein complex in chloroplasts, captured more visible light to oxidize water, which produced more electrons in the electron-transport chain, leading to augmented photosynthetic activity (Fig. 20b) and more ATP generation (Fig. 20c). This work presented the potential to utilize light-harvesting OSMs for reforming chloroplasts to increase solar energy conversion.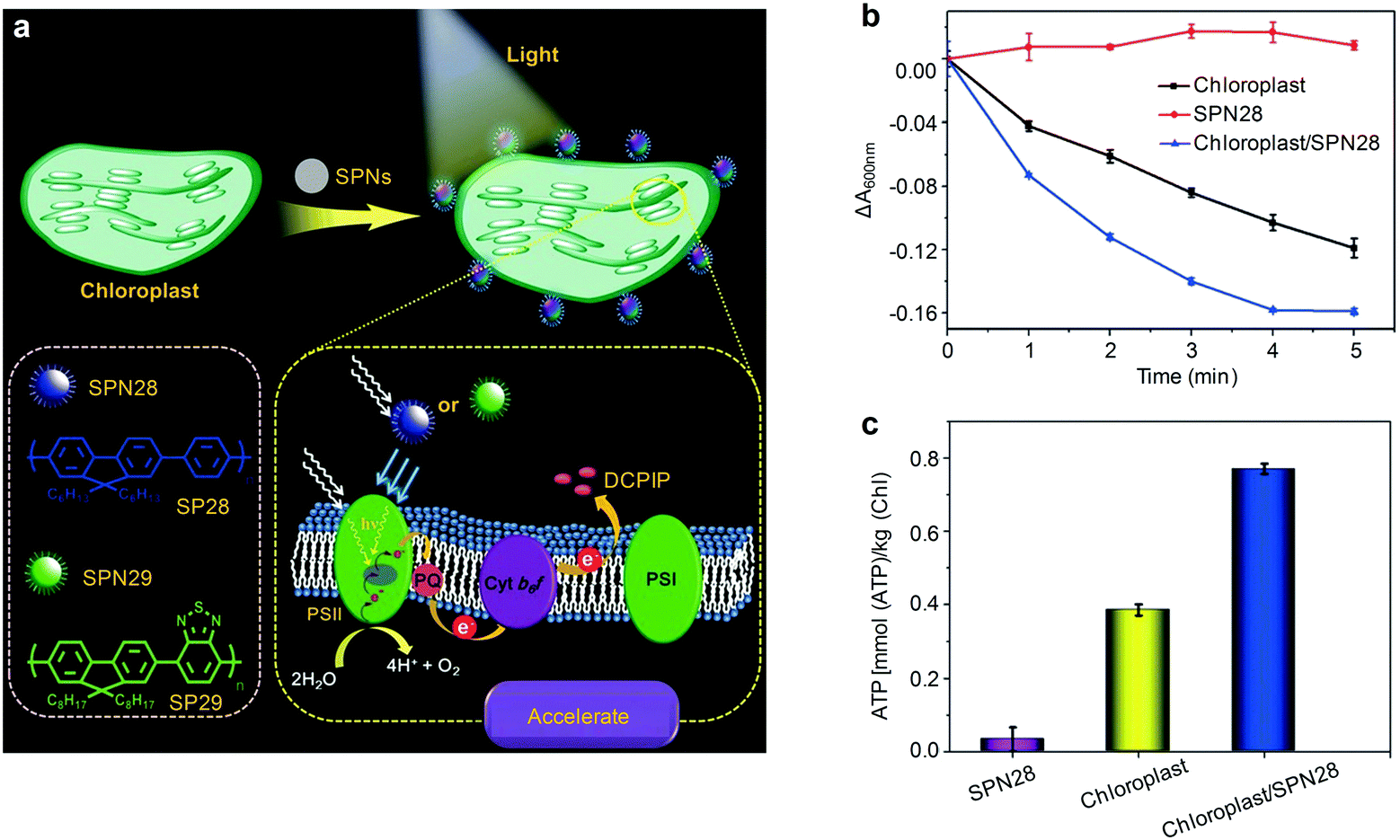 | ||
| Fig. 20 (a) Illustration of the strategy for augmenting the light reaction of isolated chloroplasts utilizing SPNs. The chemical structures of SP28 and SP29 are shown. PS = photosystem, PQ = plastoquinone, DCPIP= 2,6-dichlorophenolindophenol dye. (b) The absorption changes of DCPIP at 600 nm for chloroplast (black line), SPN28 (red line), and chloroplast/SPN28 (blue line) upon irradiation with an intensity of 4 mW cm−2 for 5 min. (c) ATP production of SPN28, chloroplast, and chloroplast/SPN28 complex under light irradiation. (a)–(c) were reproduced from ref. 261 with permission from John Wiley & Sons Ltd, copyright 2018. | ||
8. Summary and outlook
The convergence of chemistry, materials science, photology and medicine has facilitated the development of a large number of OSMs with tunable optical properties, excellent photo-stability and good cytocompatibility for versatile biophotonic applications. The emergence of OSMs has provided new opportunities for in vivo deep-tissue optical imaging with improved tissue penetration depths, spatial resolutions, detection sensitivities and SBRs as compared with the traditional NIR-I fluorescence imaging. Moreover, by incorporating molecular sensors or target-responsive components, OSMs have been developed into smart activatable optical probes to correlate signals with the level or activity of disease-specific biomarkers. NIR-II fluorescent OSMs have enabled deep-tissue visualization of vascular circulation, cardiac cycle, tumors and brain tissues, imaging-guided precise resection of tumors and sentinel lymph node biopsy, as well as molecular detection of H2S, GSH and enzymes in tumor microenvironments. Chemiluminescent OSMs in both nanoparticle and small-molecular forms have been used to detect biomarkers, such as ROS, hypoxia and enzymes. OSM-based afterglow agents have been utilized to delineate lymph nodes, xenograft/metastatic tumors, tissue oxygen levels and drug-induced hepatotoxicity, as well as real-time temperature monitoring during PTT. PA OSMs have permitted in vivo imaging of blood vessels, lymph nodes, tumors, thrombus and protein sulfenic acids and detection of ROS, pH, enzyme activities and hypoxic conditions.Owing to their highly efficient light conversion capabilities under NIR laser irradiation, OSMs can serve as phototherapeutic agents for cancer PTT and PDT. Rational alternation of molecular structures or nanoparticle components has been an effective way to improve the photothermal conversion efficiencies and 1O2 generation efficiencies, optimize phototherapeutic properties responding to the tumor microenvironment, and combine phototherapy with other therapeutic modes, enabling high treatment outcomes. Taking advantage of the photothermal effects, OSMs can be used as heat transducers to remotely regulate ion channels, enzyme activity and gene transcription.
With these promising achievements, OSMs have the potential to be translated from basic research in experimental animal models to daily clinical trials, but several critical concerns need to be addressed. First, OSMs possess relatively good biocompatibility and high biosafety, as well as desirable blood circulation time after administration in living animals. However, they often encounter the main problems of high accumulation in the liver and slow excretion from the body because of their large sizes. To address this concern, other designs of OSMs are needed to enhance their biodegradability or to reduce their dimension to the level below the renal filtration threshold (∼5 nm) for rapid clearance via urine excretion. In this regard, we and others have recently shown that imine bonds,262 imidazole units,263 thiophene moieties,147 and vinylene bonds143 can serve as the cleavable linker in the conjugated backbone of SPs to facilitate the bio-degradation of SPNs, and these molecular designs should be applicable to other OSMs. Alternatively, nanoprecipitation of semiconducting oligomers should be a potential method to prepare ultrasmall nanoparticles with diameters smaller than 5 nm. Second, there are still only a few activatable agents that have been reported for NIR-II fluorescence/PA imaging and phototherapy, which is largely due to the challenges in chemical modifications of NIR-II light responsive agents. With the emergence of new NIR-II dyes or polymers, it is expected that more and more activatable NIR-II OSMs will be developed for disease detection and treatment in photomedicine. Third, although the tissue penetration depth of molecular optical imaging in living subjects has been improved, it is still shallower than clinically available computed tomography (CT), positron emission tomography (PET) and magnetic resonance (MR) imaging that are able to ‘see through’ the entire human body with almost limitless depth in soft tissues. Integration of OSMs with endoscopic diagnostic technologies should be a solution.
By virtue of the facile preparation, OSMs can be further multi-functionalized to broaden their biomedical applications. For example, encapsulation of different semiconducting components potentially can lead to both deep-tissue imaging properties and phototherapeutic effects. Moreover, other imaging components such as radioisotopes and iron oxide nanoparticles can be incorporated into OSMs, allowing for multimodal molecular imaging of diverse biological systems. The versatile surface and structure characteristics of OSMs enable the attachment of genes or antigens or loading of drugs to produce new-generation of therapeutic agents with gene therapy, immunotherapy or chemotherapy capabilities. All the merits of OSMs will allow them to be used as a multi-functional platform to investigate, detect and treat diseases other than cancer, such as autoimmune diseases, cardiovascular diseases and neurodegenerative diseases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. P. thanks Nanyang Technological University (Start-up grant: NTU-SUG: M4081627.120) and Singapore Ministry of Education, Academic Research Fund Tier 1 (RG133/15 M4011559 and 2017-T1-002-134-RG147/17) and Academic Research Fund Tier 2 (MOE2016-T2-1-098) for financial support.Notes and references
- Y. Jiang and K. Pu, Acc. Chem. Res., 2018, 51, 1840–1849 CrossRef CAS PubMed.
- K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012–11042 CrossRef CAS PubMed.
- S.-m. Park, A. Aalipour, O. Vermesh, J. H. Yu and S. S. Gambhir, Nat. Rev. Mater., 2017, 2, 17014 CrossRef CAS PubMed.
- J. Lee, D. G. Udugamasooriya, H.-S. Lim and T. Kodadek, Nat. Chem. Biol., 2010, 6, 258 CrossRef CAS PubMed.
- O. T. Bruns, T. S. Bischof, D. K. Harris, D. Franke, Y. Shi, L. Riedemann, A. Bartelt, F. B. Jaworski, J. A. Carr and C. J. Rowlands, Nat. Biomed. Eng., 2017, 1, 0056 CrossRef PubMed.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- J. Song, X. Yang, Z. Yang, L. Lin, Y. Liu, Z. Zhou, Z. Shen, G. Yu, Y. Dai and O. Jacobson, ACS Nano, 2017, 11, 6102–6113 CrossRef CAS PubMed.
- C. F. Chiu, W. A. Saidi, V. E. Kagan and A. Star, J. Am. Chem. Soc., 2017, 139, 4859–4865 CrossRef CAS PubMed.
- S. Wang, Y. Chen, X. Li, W. Gao, L. Zhang, J. Liu, Y. Zheng, H. Chen and J. Shi, Adv. Mater., 2015, 27, 7117–7122 CrossRef CAS PubMed.
- Z. Sheng, L. Song, J. Zheng, D. Hu, M. He, M. Zheng, G. Gao, P. Gong, P. Zhang, Y. Ma and L. Cai, Biomaterials, 2013, 34, 5236–5243 CrossRef CAS PubMed.
- L. Rao, L. L. Bu, B. Cai, J. H. Xu, A. Li, W. F. Zhang, Z. J. Sun, S. S. Guo, W. Liu and T. H. Wang, Adv. Mater., 2016, 28, 3460–3466 CrossRef CAS PubMed.
- S. K. Maji, S. Sreejith, J. Joseph, M. Lin, T. He, Y. Tong, H. Sun, S. W. K. Yu and Y. Zhao, Adv. Mater., 2014, 26, 5633–5638 CrossRef CAS PubMed.
- H. Moon, D. Kumar, H. Kim, C. Sim, J.-H. Chang, J.-M. Kim, H. Kim and D.-K. Lim, ACS Nano, 2015, 9, 2711–2719 CrossRef CAS PubMed.
- J. Shi, L. Wang, J. Zhang, R. Ma, J. Gao, Y. Liu, C. Zhang and Z. Zhang, Biomaterials, 2014, 35, 5847–5861 CrossRef CAS PubMed.
- J. Li, J. Liu and C. Chen, ACS Nano, 2017, 11, 2403–2409 CrossRef CAS PubMed.
- S. Han, B. W. Hwang, E. Y. Jeon, D. Jung, G. H. Lee, D. H. Keum, K. S. Kim, S. H. Yun, H. J. Cha and S. K. Hahn, ACS Nano, 2017, 11, 9979–9988 CrossRef CAS PubMed.
- S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323–2343 RSC.
- K. Pu, N. Chattopadhyay and J. Rao, J. Controlled Release, 2016, 240, 312–322 CrossRef CAS PubMed.
- C.-S. Ke, C.-C. Fang, J.-Y. Yan, P.-J. Tseng, J. R. Pyle, C.-P. Chen, S.-Y. Lin, J. Chen, X. Zhang and Y.-H. Chan, ACS Nano, 2017, 11, 3166–3177 CrossRef CAS PubMed.
- E. Ahmed, S. W. Morton, P. T. Hammond and T. M. Swager, Adv. Mater., 2013, 25, 4504–4510 CrossRef CAS PubMed.
- J. Liu, J. Geng, L.-D. Liao, N. Thakor, X. Gao and B. Liu, Polym. Chem., 2014, 5, 2854–2862 RSC.
- H. Zhu, Y. Fang, X. Zhen, N. Wei, Y. Gao, K. Q. Luo, C. Xu, H. Duan, D. Ding, P. Chen and K. Pu, Chem. Sci., 2016, 7, 5118–5125 RSC.
- J.-C. Yu, Y.-L. Chen, Y.-Q. Zhang, X.-K. Yao, C.-G. Qian, J. Huang, S. Zhu, X.-Q. Jiang, Q.-D. Shen and Z. Gu, Chem. Commun., 2014, 50, 4699–4702 RSC.
- K. Pu, A. J. Shuhendler and J. Rao, Angew. Chem., Int. Ed., 2013, 52, 10325–10329 CrossRef CAS PubMed.
- Q. Miao and K. Pu, Adv. Mater., 2018, 1801778 CrossRef PubMed.
- L. Cheng, W. He, H. Gong, C. Wang, Q. Chen, Z. Cheng and Z. Liu, Adv. Funct. Mater., 2013, 23, 5893–5902 CrossRef CAS.
- H. Yuan, B. Wang, F. Lv, L. Liu and S. Wang, Adv. Mater., 2014, 26, 6978–6982 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- X. Dang, L. Gu, J. Qi, S. Correa, G. Zhang, A. M. Belcher and P. T. Hammond, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5179–5184 CrossRef CAS PubMed.
- D. Kobat, N. G. Horton and C. Xu, J. Biomed. Opt., 2011, 16, 106014 CrossRef PubMed.
- M.-F. Tsai, S.-H. G. Chang, F.-Y. Cheng, V. Shanmugam, Y.-S. Cheng, C.-H. Su and C.-S. Yeh, ACS Nano, 2013, 7, 5330–5342 CrossRef CAS PubMed.
- C. E. Tedford, S. DeLapp, S. Jacques and J. Anders, Lasers Surg. Med., 2015, 47, 312–322 CrossRef PubMed.
- R. Wang, X. Li, L. Zhou and F. Zhang, Angew. Chem., Int. Ed., 2014, 53, 12086–12090 CrossRef CAS PubMed.
- Y. Tsukasaki, M. Morimatsu, G. Nishimura, T. Sakata, H. Yasuda, A. Komatsuzaki, T. M. Watanabe and T. Jin, RSC Adv., 2014, 4, 41164–41171 RSC.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 0010 CrossRef.
- C. Liang, S. Diao, C. Wang, H. Gong, T. Liu, G. Hong, X. Shi, H. Dai and Z. Liu, Adv. Mater., 2014, 26, 5646–5652 CrossRef CAS PubMed.
- G. Hong, J. C. Lee, J. T. Robinson, U. Raaz, L. Xie, N. F. Huang, J. P. Cooke and H. Dai, Nat. Med., 2012, 18, 1841–1846 CrossRef CAS PubMed.
- K. Welsher, S. P. Sherlock and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8943–8948 CrossRef CAS PubMed.
- C. Li, Y. Zhang, M. Wang, Y. Zhang, G. Chen, L. Li, D. Wu and Q. Wang, Biomaterials, 2014, 35, 393–400 CrossRef CAS PubMed.
- G. Hong, J. T. Robinson, Y. Zhang, S. Diao, A. L. Antaris, Q. Wang and H. Dai, Angew. Chem., Int. Ed., 2012, 51, 9818–9821 CrossRef CAS PubMed.
- B. Dong, C. Li, G. Chen, Y. Zhang, Y. Zhang, M. Deng and Q. Wang, Chem. Mater., 2013, 25, 2503–2509 CrossRef CAS.
- A. Sasaki, Y. Tsukasaki, A. Komatsuzaki, T. Sakata, H. Yasuda and T. Jin, Nanoscale, 2015, 7, 5115–5119 RSC.
- Y. Nakane, Y. Tsukasaki, T. Sakata, H. Yasuda and T. Jin, Chem. Commun., 2013, 49, 7584–7586 RSC.
- U. Rocha, K. U. Kumar, C. Jacinto, I. Villa, F. Sanz-Rodriguez, A. Juarranz, E. Carrasco, F. C. van Veggel, E. Bovero, J. G. Sole and J. Daniel, Small, 2014, 10, 1141–1154 CrossRef CAS.
- Y. Sun, C. Qu, H. Chen, M. He, C. Tang, K. Shou, S. Hong, M. Yang, Y. Jiang and B. Ding, Chem. Sci., 2016, 7, 6203–6207 RSC.
- B. Li, L. Lu, M. Zhao, Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7483–7487 CrossRef CAS PubMed.
- Z. Tao, G. Hong, C. Shinji, C. Chen, S. Diao, A. L. Antaris, B. Zhang, Y. Zou and H. Dai, Angew. Chem., Int. Ed., 2013, 52, 13002–13006 CrossRef CAS PubMed.
- K. Shou, C. Qu, Y. Sun, H. Chen, S. Chen, L. Zhang, H. Xu, X. Hong, A. Yu and Z. Cheng, Adv. Funct. Mater., 2017, 27, 1700995 CrossRef PubMed.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Qi, C. Sun, A. Zebibula, H. Zhang, R. T. Kwok, X. Zhao, W. Xi, J. W. Lam, J. Qian and B. Z. Tang, Adv. Mater., 2018, 30, 1706856 CrossRef.
- G. Hong, Y. Zou, A. L. Antaris, S. Diao, D. Wu, K. Cheng, X. Zhang, C. Chen, B. Liu, Y. He, J. Z. Wu, J. Yuan, B. Zhang, Z. Tao, C. Fukunaga and H. Dai, Nat. Commun., 2014, 4206 CrossRef CAS PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS.
- J.-H. Park, L. Gu, G. Von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed.
- H. Kang, J. Gravier, K. Bao, H. Wada, J. H. Lee, Y. Baek, G. El Fakhri, S. Gioux, B. P. Rubin, J. L. Coll and H. S. Choi, Adv. Mater., 2016, 28, 8162–8168 CrossRef CAS PubMed.
- A. L. Antaris, H. Chen, K. Cheng, Y. Sun, G. Hong, C. Qu, S. Diao, Z. Deng, X. Hu, B. Zhang and H. Dai, Nat. Mater., 2016, 15, 235 CrossRef CAS PubMed.
- X. D. Zhang, H. Wang, A. L. Antaris, L. Li, S. Diao, R. Ma, A. Nguyen, G. Hong, Z. Ma, J. Wang, S. Zhu, J. M. Castellano, T. Wyss-Coray, Y. Liang, J. Luo and H. Dai, Adv. Mater., 2016, 28, 6872–6879 CrossRef CAS PubMed.
- Q. Yang, Z. Ma, H. Wang, B. Zhou, S. Zhu, Y. Zhong, J. Wang, H. Wan, A. Antaris, R. Ma, X. Zhang, J. Yang, X. Zhang, H. Sun, W. Liu, Y. Liang and H. Dai, Adv. Mater., 2017, 29, 1605497 CrossRef PubMed.
- S. Zhu, S. Herraiz, J. Yue, M. Zhang, H. Wan, Q. Yang, Z. Ma, Y. Wang, J. He, A. L. Antaris, Y. Zhong, S. Diao, Y. Feng, Y. Zhou, Y. Kuai, G. Hong, Y. Liang, A. J. Hsueh and H. Dai, Adv. Mater., 2018, 30, 1705799 CrossRef PubMed.
- S. Zhu, Q. Yang, A. L. Antaris, J. Yue, Z. Ma, H. Wang, W. Huang, H. Wan, J. Wang and S. Diao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 962–967 CrossRef CAS PubMed.
- Q. Yang, Z. Hu, S. Zhu, R. Ma, H. Ma, Z. Ma, H. Wan, T. Zhu, Z. Jiang and W. Liu, J. Am. Chem. Soc., 2018, 140, 1715–1724 CrossRef CAS PubMed.
- S. Zhu, Z. Hu, R. Tian, B. C. Yung, Q. Yang, S. Zhao, D. O. Kiesewetter, G. Niu, H. Sun, A. L. Antaris and X. Chen, Adv. Mater., 2018, 30, 1802546 CrossRef PubMed.
- S. Jeong, J. Song, W. Lee, Y. M. Ryu, Y. Jung, S.-Y. Kim, K. Kim, S. C. Hong, S. J. Myung and S. Kim, Nano Lett., 2017, 17, 1378–1386 CrossRef CAS PubMed.
- G. Xu, Q. Yan, X. Lv, Y. Zhu, K. Xin, B. Shi, R. Wang, J. Chen, W. Gao, P. Shi, C. Fan, C. Zhao and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 3626–3630 CrossRef CAS PubMed.
- Y. Tang, Y. Li, X. Hu, H. Zhao, Y. Ji, L. Chen, W. Hu, W. Zhang, X. Li, X. Lu, W. Huang and Q. Fan, Adv. Mater., 2018, 1801140 CrossRef PubMed.
- I. Takashima, R. Kawagoe, I. Hamachi and A. Ojida, Chem. – Eur. J., 2015, 21, 2038–2044 CrossRef CAS PubMed.
- L. J. Kricka, Clin. Chem., 1991, 37, 1472–1481 CAS.
- E. S. Lee, V. Deepagan, D. G. You, J. Jeon, G.-R. Yi, J. Y. Lee, D. S. Lee, Y. D. Suh and J. H. Park, Chem. Commun., 2016, 52, 4132–4135 RSC.
- A. Liu, F. Zhao, Y. Zhao, L. Shangguan and S. Liu, Biosens. Bioelectron., 2016, 81, 97–102 CrossRef CAS PubMed.
- S. Bi, B. Ji, Z. Zhang and S. Zhang, Chem. Commun., 2013, 49, 3452–3454 RSC.
- Z. Wang, J. Li, B. Liu, J. Hu, X. Yao and J. Li, J. Phys. Chem. B, 2005, 109, 23304–23311 CrossRef CAS PubMed.
- D. Lee, S. Khaja, J. C. Velasquez-Castano, M. Dasari, C. Sun, J. Petros, W. R. Taylor and N. Murthy, Nat. Mater., 2007, 6, 765 CrossRef CAS PubMed.
- J. Li, Y. Chen, Y. Yang, N. Kawazoe and G. Chen, J. Mater. Chem. B, 2017, 5, 1353–1362 RSC.
- C. R. Reczek and N. S. Chandel, Curr. Opin. Cell Biol., 2015, 33, 8–13 CrossRef CAS PubMed.
- Y. Lyu, X. Zhen, Y. Miao and K. Pu, ACS Nano, 2016, 11, 358–367 CrossRef PubMed.
- Z. Chen, Z. Liu, Z. Li, E. Ju, N. Gao, L. Zhou, J. Ren and X. Qu, Biomaterials, 2015, 39, 15–22 CrossRef CAS PubMed.
- X. Chen, F. Wang, J. Y. Hyun, T. Wei, J. Qiang, X. Ren, I. Shin and J. Yoon, Chem. Soc. Rev., 2016, 45, 2976–3016 RSC.
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS PubMed.
- J. Li, J. Zhang, Y. Chen, N. Kawazoe and G. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 35683–35692 CrossRef CAS PubMed.
- Y. Zhou, W. Pei, X. Zhang, W. Chen, J. Wu, C. Yao, L. Huang, H. Zhang, W. Huang and J. S. C. Loo, Biomaterials, 2015, 54, 34–43 CrossRef CAS PubMed.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722–728 CrossRef CAS PubMed.
- J. Geng, K. Li, W. Qin, B. Z. Tang and B. Liu, Part. Part. Syst. Charact., 2014, 31, 1238–1243 CrossRef CAS.
- Y.-D. Lee, C.-K. Lim, A. Singh, J. Koh, J. Kim, I. C. Kwon and S. Kim, ACS Nano, 2012, 6, 6759–6766 CrossRef CAS PubMed.
- H. J. Kim, Y. H. Seo, S. An, A. Jo, I. C. Kwon and S. Kim, Theranostics, 2018, 8, 1798–1807 CrossRef PubMed.
- S. Cho, O. Hwang, I. Lee, G. Lee, D. Yoo, G. Khang, P. M. Kang and D. Lee, Adv. Funct. Mater., 2012, 22, 4038–4043 CrossRef CAS.
- C. K. Lim, Y. D. Lee, J. Na, J. M. Oh, S. Her, K. Kim, K. Choi, S. Kim and I. C. Kwon, Adv. Funct. Mater., 2010, 20, 2644–2648 CrossRef CAS.
- A. J. Shuhendler, K. Pu, L. Cui, J. P. Uetrecht and J. Rao, Nat. Biotechnol., 2014, 32, 373–380 CrossRef CAS PubMed.
- X. Zhen, C. Zhang, C. Xie, Q. Miao, K. L. Lim and K. Pu, ACS Nano, 2016, 10, 6400–6409 CrossRef CAS PubMed.
- Y. H. Seo, A. Singh, H.-J. Cho, Y. Kim, J. Heo, C.-K. Lim, S. Y. Park, W.-D. Jang and S. Kim, Biomaterials, 2016, 84, 111–118 CrossRef CAS PubMed.
- D. Mao, W. Wu, S. Ji, C. Chen, F. Hu, D. Kong, D. Ding and B. Liu, Chem, 2017, 3, 991–1007 CAS.
- J. Liu, Q. Chen, W. Zhu, X. Yi, Y. Yang, Z. Dong and Z. Liu, Adv. Funct. Mater., 2017, 27, 1605926 CrossRef.
- K. Teranishi and T. Nishiguchi, Anal. Biochem., 2004, 325, 185–195 CrossRef CAS PubMed.
- M. Sekiya, K. Umezawa, A. Sato, D. Citterio and K. Suzuki, Chem. Commun., 2009, 3047–3049 RSC.
- P. Li, L. Liu, H. Xiao, W. Zhang, L. Wang and B. Tang, J. Am. Chem. Soc., 2016, 138, 2893–2896 CrossRef CAS PubMed.
- M. E. Roth-Konforti, C. R. Bauer and D. Shabat, Angew. Chem., Int. Ed., 2017, 56, 15633–15638 CrossRef CAS PubMed.
- L. S. Ryan and A. R. Lippert, Angew. Chem., Int. Ed., 2018, 57, 622–624 CrossRef CAS PubMed.
- J. Cao, W. An, A. G. Reeves and A. R. Lippert, Chem. Sci., 2018, 9, 2552–2558 RSC.
- O. Green, T. Eilon, N. Hananya, S. Gutkin, C. R. Bauer and D. Shabat, ACS Cent. Sci., 2017, 3, 349–358 CrossRef CAS PubMed.
- D. Shabat, N. Hananya, O. Green, R. Blau and R. Satchi-Fainaro, Angew. Chem., Int. Ed., 2017, 56, 11793–11796 CrossRef PubMed.
- S. Gnaim, O. Green and D. Shabat, Chem. Commun., 2018, 54, 2073–2085 RSC.
- A. P. Schaap, R. S. Handley and B. P. Giri, Tetrahedron Lett., 1987, 28, 935–938 CrossRef CAS.
- N. Hananya and D. Shabat, Angew. Chem., Int. Ed., 2017, 56, 16454–16463 CrossRef CAS PubMed.
- O. Seven, F. Sozmen and I. S. Turan, Sens. Actuators, B, 2017, 239, 1318–1324 CrossRef CAS.
- O. Green, S. Gnaim, R. Blau, A. Eldar-Boock, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2017, 139, 13243–13248 CrossRef CAS PubMed.
- K. J. Bruemmer, O. Green, T. A. Su, D. Shabat and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 7508–7512 CrossRef CAS PubMed.
- J. Cao, R. Lopez, J. Thacker, J. Moon, C. Jiang, S. Morris, J. Bauer, P. Tao, R. Mason and A. Lippert, Chem. Sci., 2015, 6, 1979–1985 RSC.
- J. Cao, J. Campbell, L. Liu, R. P. Mason and A. R. Lippert, Anal. Chem., 2016, 88, 4995–5002 CrossRef CAS PubMed.
- N. Hananya, A. Eldar Boock, C. R. Bauer, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2016, 138, 13438–13446 CrossRef CAS PubMed.
- T. Eilon-Shaffer, M. Roth-Konforti, A. Eldar-Boock, R. Satchi-Fainaro and D. Shabat, Org. Biomol. Chem., 2018, 16, 1708–1712 RSC.
- J. Shi, X. Sun, J. Li, H. Man, J. Shen, Y. Yu and H. Zhang, Biomaterials, 2015, 37, 260–270 CrossRef CAS PubMed.
- L. Zhan-Jun, Z. Hong-Wu, S. Meng, S. Jiang-Shan and F. Hai-Xia, J. Mater. Chem., 2012, 22, 24713–24720 RSC.
- T. Maldiney, G. Sraiki, B. Viana, D. Gourier, C. Richard, D. Scherman, M. Bessodes, K. Van den Eeckhout, D. Poelman and P. Smet, Opt. Mater. Express, 2012, 2, 261–268 CrossRef CAS.
- Z. Li, Y. Zhang, X. Wu, L. Huang, D. Li, W. Fan and G. Han, J. Am. Chem. Soc., 2015, 137, 5304–5307 CrossRef CAS PubMed.
- T. Maldiney, A. Bessière, J. Seguin, E. Teston, S. K. Sharma, B. Viana, A. J. Bos, P. Dorenbos, M. Bessodes and D. Gourier, Nat. Mater., 2014, 13, 418–426 CrossRef CAS PubMed.
- A. Abdukayum, J.-T. Chen, Q. Zhao and X.-P. Yan, J. Am. Chem. Soc., 2013, 135, 14125–14133 CrossRef CAS PubMed.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef PubMed.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102 CAS.
- C. Xie, X. Zhen, Q. Miao, Y. Lyu and K. Pu, Adv. Mater., 2018, 30, 1801331 CrossRef PubMed.
- X. Zhen, C. Xie and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3938–3942 CrossRef CAS PubMed.
- Q. Miao and K. Pu, Bioconjugate Chem., 2016, 27, 2808–2823 CrossRef CAS PubMed.
- Q. Chen, X. Liu, J. Chen, J. Zeng, Z. Cheng and Z. Liu, Adv. Mater., 2015, 27, 6820–6827 CrossRef CAS PubMed.
- C. Kim, T. N. Erpelding, L. Jankovic, M. D. Pashley and L. V. Wang, Biomed. Opt. Express, 2010, 1, 278–284 CrossRef PubMed.
- J. Li, F. Cheng, H. Huang, L. Li and J.-J. Zhu, Chem. Soc. Rev., 2015, 44, 7855–7880 RSC.
- Y. Jiang and K. Pu, Small, 2017, 13, 1700710 CrossRef PubMed.
- J. Weber, P. C. Beard and S. E. Bohndiek, Nat. Methods, 2016, 13, 639–650 CrossRef CAS PubMed.
- W. Li and X. Chen, Nanomedicine, 2015, 10, 299–320 CrossRef CAS PubMed.
- L. Nie and X. Chen, Chem. Soc. Rev., 2014, 43, 7132–7170 RSC.
- J. Zhang, H. Chen, T. Zhou, L. Wang, D. Gao, X. Zhang, Y. Liu, C. Wu and Z. Yuan, Nano Res., 2017, 10, 64–76 CrossRef CAS.
- X. Cai, X. Liu, L. D. Liao, A. Bandla, J. M. Ling, Y. H. Liu, N. Thakor, G. C. Bazan and B. Liu, Small, 2016, 12, 4873–4880 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- C. Cui, Z. Yang, X. Hu, J. Wu, K. Shou, H. Ma, C. Jian, Y. Zhao, B. Qi and X. Hu, ACS Nano, 2017, 11, 3298–3310 CrossRef CAS PubMed.
- K. Pu, J. Mei, J. V. Jokerst, G. Hong, A. L. Antaris, N. Chattopadhyay, A. J. Shuhendler, T. Kurosawa, Y. Zhou, S. S. Gambhir, Z. Bao and J. Rao, Adv. Mater., 2015, 27, 5184–5190 CrossRef CAS PubMed.
- C. Xie, X. Zhen, Q. Lei, R. Ni and K. Pu, Adv. Funct. Mater., 2017, 27, 1605397 CrossRef.
- C. Yin, X. Zhen, H. Zhao, Y. Tang, Y. Ji, Y. Lyu, Q. Fan, W. Huang and K. Pu, ACS Appl. Mater. Interfaces, 2017, 9, 12332–12339 CrossRef CAS PubMed.
- C. M. Furdui and L. B. Poole, Mass Spectrom. Rev., 2014, 33, 126–146 CrossRef CAS PubMed.
- Y. H. Seo and K. S. Carroll, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16163–16168 CrossRef CAS PubMed.
- X. Liang, Y. Li, X. Li, L. Jing, Z. Deng, X. Yue, C. Li and Z. Dai, Adv. Funct. Mater., 2015, 25, 1451–1462 CrossRef CAS.
- Q. Fan, K. Cheng, Z. Yang, R. Zhang, M. Yang, X. Hu, X. Ma, L. Bu, X. Lu and X. Xiong, Adv. Mater., 2015, 27, 843–847 CrossRef CAS PubMed.
- H. Chen, J. Zhang, K. Chang, X. Men, X. Fang, L. Zhou, D. Li, D. Gao, S. Yin, X. Zhang, Z. Yuan and C. Wu, Biomaterials, 2017, 144, 42–52 CrossRef CAS PubMed.
- Y. Lyu, Y. Fang, Q. Miao, X. Zhen, D. Ding and K. Pu, ACS Nano, 2016, 10, 4472–4481 CrossRef CAS PubMed.
- C. Xie, P. K. Upputuri, X. Zhen, M. Pramanik and K. Pu, Biomaterials, 2017, 119, 1–8 CrossRef CAS PubMed.
- Y. Lyu, J. Zeng, Y. Jiang, X. Zhen, T. Wang, S. Qiu, X. Lou, M. Gao and K. Pu, ACS Nano, 2018, 12, 1801–1810 CrossRef CAS PubMed.
- X. Zhen, X. Feng, C. Xie, Y. Zheng and K. Pu, Biomaterials, 2017, 127, 97–106 CrossRef CAS PubMed.
- C. Xie, X. Zhen, Y. Lyu and K. Pu, Adv. Mater., 2017, 29, 1703693 CrossRef PubMed.
- J. Zhang, X. Zhen, P. K. Upputuri, M. Pramanik, P. Chen and K. Pu, Adv. Mater., 2017, 29, 1604764 CrossRef PubMed.
- C. Yin, X. Zhen, Q. Fan, W. Huang and K. Pu, ACS Nano, 2017, 11, 4174–4182 CrossRef CAS PubMed.
- L. Frullano, C. Catana, T. Benner, A. D. Sherry and P. Caravan, Angew. Chem., Int. Ed., 2010, 49, 2382–2384 CrossRef CAS PubMed.
- S. A. Hilderbrand, K. A. Kelly, M. Niedre and R. Weissleder, Bioconjugate Chem., 2008, 19, 1635–1639 CrossRef CAS PubMed.
- S. Schreml, R. J. Meier, O. S. Wolfbeis, M. Landthaler, R.-M. Szeimies and P. Babilas, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2432–2437 CrossRef CAS PubMed.
- J. Y. Ko, S. Park, H. Lee, H. Koo, M. S. Kim, K. Choi, I. C. Kwon, S. Y. Jeong, K. Kim and D. S. Lee, Small, 2010, 6, 2539–2544 CrossRef CAS PubMed.
- Q. Miao, Y. Lyu, D. Ding and K. Pu, Adv. Mater., 2016, 28, 3662–3668 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, S.-R. Kothapalli, G. A. Tikhomirov, J. Rao and S. S. Gambhir, J. Am. Chem. Soc., 2013, 135, 11015–11022 CrossRef CAS PubMed.
- K. Yang, L. Zhu, L. Nie, X. Sun, L. Cheng, C. Wu, G. Niu, X. Chen and Z. Liu, Theranostics, 2014, 4, 134 CrossRef CAS PubMed.
- K. Kessenbrock, V. Plaks and Z. Werb, Cell, 2010, 141, 52–67 CrossRef CAS PubMed.
- J. Levi, S.-R. Kothapalli, S. Bohndiek, J.-K. Yoon, A. Dragulescu-Andrasi, C. Nielsen, A. Tisma, S. Bodapati, G. Gowrishankar and X. Yan, Clin. Cancer Res., 2013, 19, 1494–1502 CrossRef CAS PubMed.
- D. Zhang, G. B. Qi, Y. X. Zhao, S. L. Qiao, C. Yang and H. Wang, Adv. Mater., 2015, 27, 6125–6130 CrossRef CAS PubMed.
- N. Albayrak and S. T. Yang, Biotechnol. Bioeng., 2002, 77, 8–19 CrossRef CAS PubMed.
- X. Zhen, J. Zhang, J. Huang, C. Xie, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 7804–7808 CrossRef CAS PubMed.
- H. J. Knox, J. Hedhli, T. W. Kim, K. Khalili, L. W. Dobrucki and J. Chan, Nat. Commun., 2017, 8, 1794 CrossRef PubMed.
- Y. Dai, C. Xu, X. Sun and X. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS PubMed.
- Y. Jiang and K. Pu, Adv. Biosyst., 2018, 1700262 CrossRef.
- Y. Zhou, D. Wang, Y. Zhang, U. Chitgupi, J. Geng, Y. Wang, Y. Zhang, T. R. Cook, J. Xia and J. F. Lovell, Theranostics, 2016, 6, 688 CrossRef CAS PubMed.
- Y. Jiang, P. K. Upputuri, C. Xie, Y. Lyu, L. Zhang, Q. Xiong, M. Pramanik and K. Pu, Nano Lett., 2017, 17, 4964–4969 CrossRef CAS PubMed.
- B. Guo, Z. Sheng, D. Hu, X. Lin, S. Xu, C. Liu, H. Zheng and B. Liu, Mater. Horiz., 2017, 4, 1151–1156 RSC.
- J. Wu, L. You, L. Lan, H. J. Lee, S. T. Chaudhry, R. Li, J. X. Cheng and J. Mei, Adv. Mater., 2017, 29, 1703403 CrossRef PubMed.
- J. Li, Y. Hu, J. Yang, P. Wei, W. Sun, M. Shen, G. Zhang and X. Shi, Biomaterials, 2015, 38, 10–21 CrossRef CAS PubMed.
- C. Liang, L. Xu, G. Song and Z. Liu, Chem. Soc. Rev., 2016, 45, 6250–6269 RSC.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- J. Li, R. Cai, N. Kawazoe and G. Chen, J. Mater. Chem. B, 2015, 3, 5806–5814 RSC.
- P. Wang, X. Li, C. Yao, W. Wang, M. Zhao, A. M. El-Toni and F. Zhang, Biomaterials, 2017, 125, 90–100 CrossRef CAS PubMed.
- C. S. Jin, J. F. Lovell, J. Chen and G. Zheng, ACS Nano, 2013, 7, 2541–2550 CrossRef CAS PubMed.
- S. Wang, J. Zhao, H. Yang, C. Wu, F. Hu, H. Chang, G. Li, D. Ma, D. Zou and M. Huang, Acta Biomater., 2017, 58, 442–454 CrossRef CAS PubMed.
- J. Li, J. Rao and K. Pu, Biomaterials, 2018, 155, 217–235 CrossRef CAS PubMed.
- J. Geng, C. Sun, J. Liu, L. D. Liao, Y. Yuan, N. Thakor, J. Wang and B. Liu, Small, 2015, 11, 1603–1610 CrossRef CAS PubMed.
- K. Yang, H. Xu, L. Cheng, C. Sun, J. Wang and Z. Liu, Adv. Mater., 2012, 24, 5586–5592 CrossRef CAS PubMed.
- K. Chang, Y. Liu, D. Hu, Q. Qi, D. Gao, Y. Wang, D. Li, X. Zhang, H. Zheng, Z. Sheng and Z. Yuan, ACS Appl. Mater. Interfaces, 2018, 10, 7012–7021 CrossRef CAS PubMed.
- S. Zhang, W. Guo, J. Wei, C. Li, X.-J. Liang and M. Yin, ACS Nano, 2017, 11, 3797–3805 CrossRef CAS PubMed.
- S. Li, X. Wang, R. Hu, H. Chen, M. Li, J. Wang, Y. Wang, L. Liu, F. Lv and X.-J. Liang, Chem. Mater., 2016, 28, 8669–8675 CrossRef CAS.
- Q. Zou, M. Abbas, L. Zhao, S. Li, G. Shen and X. Yan, J. Am. Chem. Soc., 2017, 139, 1921–1927 CrossRef CAS PubMed.
- P. Zhang, H. Huang, J. Huang, H. Chen, J. Wang, K. Qiu, D. Zhao, L. Ji and H. Chao, ACS Appl. Mater. Interfaces, 2015, 7, 23278–23290 CrossRef CAS PubMed.
- X. Zhu, W. Feng, J. Chang, Y.-W. Tan, J. Li, M. Chen, Y. Sun and F. Li, Nat. Commun., 2016, 7, 10437 CrossRef CAS PubMed.
- Z. Sun, H. Xie, S. Tang, X. F. Yu, Z. Guo, J. Shao, H. Zhang, H. Huang, H. Wang and P. K. Chu, Angew. Chem., Int. Ed., 2015, 54, 11526–11530 CrossRef CAS PubMed.
- Q. Tian, F. Jiang, R. Zou, Q. Liu, Z. Chen, M. Zhu, S. Yang, J. Wang, J. Wang and J. Hu, ACS Nano, 2011, 5, 9761–9771 CrossRef CAS PubMed.
- D. K. Roper, W. Ahn and M. Hoepfner, J. Phys. Chem. C, 2007, 111, 3636–3641 CrossRef CAS PubMed.
- D. Li, G. Zhang, W. Xu, J. Wang, Y. Wang, L. Qiu, J. Ding and X. Yang, Theranostics, 2017, 7, 4029–4040 CrossRef PubMed.
- B. Guo, G. Feng, P. N. Manghnani, X. Cai, J. Liu, W. Wu, S. Xu, X. Cheng, C. Teh and B. Liu, Small, 2016, 12, 6243–6254 CrossRef CAS PubMed.
- J. Qi, Y. Fang, R. T. Kwok, X. Zhang, X. Hu, J. W. Lam, D. Ding and B. Z. Tang, ACS Nano, 2017, 11, 7177–7188 CrossRef CAS PubMed.
- P. Liang, Q. Tang, Y. Cai, G. Liu, W. Si, J. Shao, W. Huang, Q. Zhang and X. Dong, Chem. Sci., 2017, 8, 7457–7463 RSC.
- B. Guo, Z. Sheng, D. Hu, A. Li, S. Xu, P. N. Manghnani, C. Liu, L. Guo, H. Zheng and B. Liu, ACS Nano, 2017, 11, 10124–10134 CrossRef CAS PubMed.
- J. Zhang, C. Yang, R. Zhang, R. Chen, Z. Zhang, W. Zhang, S. H. Peng, X. Chen, G. Liu, C. S. Hsu and C.-S. Lee, Adv. Funct. Mater., 2017, 27, 1605094 CrossRef PubMed.
- H. Lin, S. Gao, C. Dai, Y. Chen and J. Shi, J. Am. Chem. Soc., 2017, 139, 16235–16247 CrossRef CAS PubMed.
- X. Ding, C. H. Liow, M. Zhang, R. Huang, C. Li, H. Shen, M. Liu, Y. Zou, N. Gao and Z. Zhang, J. Am. Chem. Soc., 2014, 136, 15684–15693 CrossRef CAS PubMed.
- J. Zhou, Y. Jiang, S. Hou, P. K. Upputuri, D. Wu, J. Li, P. Wang, X. Zhen, M. Pramanik, K. Pu and H. Duan, ACS Nano, 2018, 12, 2643–2651 CrossRef CAS PubMed.
- Y. Jiang, J. Li, X. Zhen, C. Xie and K. Pu, Adv. Mater., 2018, 30, 1705980 CrossRef PubMed.
- Z. Cao, L. Feng, G. Zhang, J. Wang, S. Shen, D. Li and X. Yang, Biomaterials, 2018, 155, 103–111 CrossRef CAS PubMed.
- T. Sun, J.-H. Dou, S. Liu, X. Wang, X. Zheng, Y. Wang, J. Pei and Z. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 7919–7926 CrossRef CAS PubMed.
- L. Cheng, D. Jiang, A. Kamkaew, H. F. Valdovinos, H. J. Im, L. Feng, C. G. England, S. Goel, T. E. Barnhart, Z. Liu and W. Cai, Adv. Funct. Mater., 2017, 27, 1702928 CrossRef PubMed.
- L. Guo, J. Ge, Q. Liu, Q. Jia, H. Zhang, W. Liu, G. Niu, S. Liu, J. Gong, S. Hackbarth and P. Wang, Adv. Healthcare Mater., 2017, 6, 1601431 CrossRef PubMed.
- M. Li, Y. Gao, Y. Yuan, Y. Wu, Z. Song, B. Z. Tang, B. Liu and Q. C. Zheng, ACS Nano, 2017, 11, 3922–3932 CrossRef CAS PubMed.
- K. Chang, Y. Tang, X. Fang, S. Yin, H. Xu and C. Wu, Biomacromolecules, 2016, 17, 2128–2136 CrossRef CAS PubMed.
- Y. Tang, H. Chen, K. Chang, Z. Liu, Y. Wang, S. Qu, H. Xu and C. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 3419–3431 CrossRef CAS PubMed.
- S. Li, K. Chang, K. Sun, Y. Tang, N. Cui, Y. Wang, W. Qin, H. Xu and C. Wu, ACS Appl. Mater. Interfaces, 2015, 8, 3624–3634 CrossRef PubMed.
- L. Huang, Z. Li, Y. Zhao, J. Yang, Y. Yang, A. I. Pendharkar, Y. Zhang, S. Kelmar, L. Chen, W. Wu, J. Zhao and G. Han, Adv. Mater., 2017, 29, 1604789 CrossRef PubMed.
- J. Zou, Z. Yin, P. Wang, D. Chen, J. Shao, Q. Zhang, L. Sun, W. Huang and X. Dong, Chem. Sci., 2018, 9, 2188–2194 RSC.
- G. Feng, W. Wu, S. Xu and B. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 21193–21200 CrossRef CAS PubMed.
- W. Wu, D. Mao, F. Hu, S. Xu, C. Chen, C. J. Zhang, X. Cheng, Y. Yuan, D. Ding and D. Kong, Adv. Mater., 2017, 29, 1700548 CrossRef PubMed.
- J. Kim, H. R. Cho, H. Jeon, D. Kim, C. Song, N. Lee, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2017, 139, 10992–10995 CrossRef CAS PubMed.
- H. Zhu, J. Li, X. Qi, P. Chen and K. Pu, Nano Lett., 2018, 18, 586–594 CrossRef CAS PubMed.
- H. Zhu, Y. Fang, Q. Miao, X. Qi, D. Ding, P. Chen and K. Pu, ACS Nano, 2017, 11, 8998–9009 CrossRef CAS PubMed.
- K. Chang, Z. Liu, X. Fang, H. Chen, X. Men, Y. Yuan, K. Sun, X. Zhang, Z. Yuan and C. Wu, Nano Lett., 2017, 17, 4323–4329 CrossRef CAS PubMed.
- C. He, D. Liu and W. Lin, ACS Nano, 2015, 9, 991–1003 CrossRef CAS PubMed.
- J. Xu, L. Xu, C. Wang, R. Yang, Q. Zhuang, X. Han, Z. Dong, W. Zhu, R. Peng and Z. Liu, ACS Nano, 2017, 11, 4463–4474 CrossRef CAS PubMed.
- Z. Guo, Y. Zou, H. He, J. Rao, S. Ji, X. Cui, H. Ke, Y. Deng, H. Yang, C. Chen, Y. Zhao and H. Chen, Adv. Mater., 2016, 28, 10155–10164 CrossRef CAS PubMed.
- Q. Chen, L. Feng, J. Liu, W. Zhu, Z. Dong, Y. Wu and Z. Liu, Adv. Mater., 2016, 28, 7129–7136 CrossRef CAS PubMed.
- C. Chu, H. Lin, H. Liu, X. Wang, J. Wang, P. Zhang, H. Gao, C. Huang, Y. Zeng, Y. Tan, G. Liu and X. Chen, Adv. Mater., 2017, 29, 1605928 CrossRef PubMed.
- G. Feng, Y. Fang, J. Liu, J. Geng, D. Ding and B. Liu, Small, 2017, 13, 1602807 CrossRef PubMed.
- T. Yang, L. Liu, Y. Deng, Z. Guo, G. Zhang, Z. Ge, H. Ke and H. Chen, Adv. Mater., 2017, 29, 1700487 CrossRef PubMed.
- Y. Cai, P. Liang, Q. Tang, X. Yang, W. Si, W. Huang, Q. Zhang and X. Dong, ACS Nano, 2017, 11, 1054–1063 CrossRef CAS PubMed.
- S. Ye, J. Rao, S. Qiu, J. Zhao, H. He, Z. Yan, T. Yang, Y. Deng, H. Ke, H. Yang, Y. Zhao, Z. Guo and H. Chen, Adv. Mater., 2018, 1801216 CrossRef PubMed.
- T. Liu, C. Wang, X. Gu, H. Gong, L. Cheng, X. Shi, L. Feng, B. Sun and Z. Liu, Adv. Mater., 2014, 26, 3433–3440 CrossRef CAS PubMed.
- X. Liu, F. Fu, K. Xu, R. Zou, J. Yang, Q. Wang, Q. Liu, Z. Xiao and J. Hu, J. Mater. Chem. B, 2014, 2, 5358–5367 RSC.
- X. Liu, Q. Wang, C. Li, R. Zou, B. Li, G. Song, K. Xu, Y. Zheng and J. Hu, Nanoscale, 2014, 6, 4361–4370 RSC.
- Y. Jiang, D. Cui, Y. Fang, X. Zhen, P. K. Upputuri, M. Pramanik, D. Ding and K. Pu, Biomaterials, 2017, 145, 168–177 CrossRef CAS PubMed.
- Y. Cao, J. Yi, X. Yang, L. Liu, C. Yu, Y. Huang, L. Sun, Y. Bao and Y. Li, Biomacromolecules, 2017, 18, 2306–2314 CrossRef CAS PubMed.
- D.-D. Li, J.-X. Wang, Y. Ma, H.-S. Qian, D. Wang, L. Wang, G. Zhang, L. Qiu, Y.-C. Wang and X.-Z. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 19312–19320 CrossRef CAS PubMed.
- J. Wang, F. Guo, M. Yu, L. Liu, F. Tan, R. Yan and N. Li, J. Controlled Release, 2016, 237, 23–34 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, J. Li, Z.-H. Peng, Y. Sheinin, J. Zhou and D. Oupicky, ACS Nano, 2017, 11, 2227–2238 CrossRef CAS PubMed.
- L. Feng, L. Cheng, Z. Dong, D. Tao, T. E. Barnhart, W. Cai, M. Chen and Z. Liu, ACS Nano, 2016, 11, 927–937 CrossRef PubMed.
- Y. Yuan, J. Liu and B. Liu, Angew. Chem., Int. Ed., 2014, 53, 7163–7168 CrossRef CAS PubMed.
- C. Qian, J. Yu, Y. Chen, Q. Hu, X. Xiao, W. Sun, C. Wang, P. Feng, Q. D. Shen and Z. Gu, Adv. Mater., 2016, 28, 3313–3320 CrossRef CAS PubMed.
- G. G. Genchi, A. Marino, A. Grillone, I. Pezzini and G. Ciofani, Adv. Healthcare Mater., 2017, 6, 1700002 CrossRef PubMed.
- H.-M. Yun, S.-J. Ahn, K.-R. Park, M.-J. Kim, J.-J. Kim, G.-Z. Jin, H.-W. Kim and E.-C. Kim, Biomaterials, 2016, 85, 88–98 CrossRef CAS PubMed.
- E. Arbely, J. Torres-Kolbus, A. Deiters and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 11912–11915 CrossRef CAS PubMed.
- M. Wang, S. Sun, C. I. Neufeld, B. Perez-Ramirez and Q. Xu, Angew. Chem., Int. Ed., 2014, 53, 13444–13448 CrossRef CAS PubMed.
- B. Hu, Y. Zhang, J. Zhou, J. Li, F. Deng, Z. Wang and J. Song, PLoS One, 2014, 9, e95168 CrossRef PubMed.
- E. D. Kirson, V. Dbalý, F. Tovaryš, J. Vymazal, J. F. Soustiel, A. Itzhaki, D. Mordechovich, S. Steinberg-Shapira, Z. Gurvich and R. Schneiderman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10152–10157 CrossRef CAS PubMed.
- I. B. Leibiger and P.-O. Berggren, Nat. Med., 2015, 21, 14–16 CrossRef CAS PubMed.
- D. Lee, J. H. Hyun, K. Jung, P. Hannan and H.-B. Kwon, Nat. Biotechnol., 2017, 35, 858–863 CrossRef CAS PubMed.
- L. Lin, L. Liu, B. Zhao, R. Xie, W. Lin, H. Li, Y. Li, M. Shi, Y.-G. Chen and T. A. Springer, Nat. Nanotechnol., 2015, 10, 465–471 CrossRef CAS PubMed.
- M. Karimi, P. Sahandi Zangabad, S. Baghaee-Ravari, M. Ghazadeh, H. Mirshekari and M. R. Hamblin, J. Am. Chem. Soc., 2017, 139, 4584–4610 CrossRef CAS PubMed.
- A. Tay and D. Di Carlo, Nano Lett., 2017, 17, 886–892 CrossRef CAS PubMed.
- H. Huang, S. Delikanli, H. Zeng, D. M. Ferkey and A. Pralle, Nat. Nanotechnol., 2010, 5, 602–606 CrossRef CAS PubMed.
- E. Miyako, J. Russier, M. Mauro, C. Cebrian, H. Yawo, C. Menard-Moyon, J. A. Hutchison, M. Yudasaka, S. Iijima and L. De Cola, Angew. Chem., Int. Ed., 2014, 53, 13121–13125 CrossRef CAS PubMed.
- H. Nakatsuji, T. Numata, N. Morone, S. Kaneko, Y. Mori, H. Imahori and T. Murakami, Angew. Chem., Int. Ed., 2015, 54, 11725–11729 CrossRef CAS PubMed.
- Y. Lyu, C. Xie, S. A. Chechetka, E. Miyako and K. Pu, J. Am. Chem. Soc., 2016, 138, 9049–9052 CrossRef CAS PubMed.
- T. T. Wu, A. A. Peters, P. T. Tan, S. J. Roberts-Thomson and G. R. Monteith, Cell Calcium, 2014, 56, 59–67 CrossRef CAS PubMed.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Nat. Rev. Mol. Cell Biol., 2003, 4, 552–565 CrossRef CAS PubMed.
- X. Zhen, C. Xie, Y. Jiang, X. Ai, B. Xing and K. Pu, Nano Lett., 2018, 18, 1498–1505 CrossRef CAS PubMed.
- X. Wu, Y. Zhang, K. Takle, O. Bilsel, Z. Li, H. Lee, Z. Zhang, D. Li, W. Fan, C. Duan, E. M. Chan, C. Lois, Y. Xiang and G. Han, ACS Nano, 2016, 10, 1060–1066 CrossRef CAS PubMed.
- E. Miyako, T. Deguchi, Y. Nakajima, M. Yudasaka, Y. Hagihara, M. Horie, M. Shichiri, Y. Higuchi, F. Yamashita, M. Hashida, Y. Shigeri, Y. Yoshida and S. Lijima, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7523–7528 CrossRef CAS PubMed.
- Y. Lyu, D. Cui, H. Sun, Y. Miao, H. Duan and K. Pu, Angew. Chem., Int. Ed., 2017, 56, 9155–9159 CrossRef CAS PubMed.
- Y. Wang, S. Li, P. Zhang, H. Bai, L. Feng, F. Lv, L. Liu and S. Wang, Adv. Mater., 2018, 30, 1705418 CrossRef PubMed.
- S. Tadepalli, J. Yim, S. Cao, Z. Wang, R. R. Naik and S. Singamaneni, Small, 2018, 14, 1702382 CrossRef PubMed.
- H. Link, K. Kochanowski and U. Sauer, Nat. Biotechnol., 2013, 31, 357 CrossRef CAS PubMed.
- S. H. Kim, K.-R. Kim, D.-R. Ahn, J. E. Lee, E. G. Yang and S. Y. Kim, ACS Nano, 2017, 11, 9352–9359 CrossRef CAS PubMed.
- C. Wang, Q. Zhang, X. Wang, H. Chang, S. Zhang, Y. Tang, J. Xu, R. Qi and Y. Cheng, Angew. Chem., Int. Ed., 2017, 56, 6767–6772 CrossRef CAS PubMed.
- M. D. Blankschien, L. A. Pretzer, R. Huschka, N. J. Halas, R. Gonzalez and M. S. Wong, ACS Nano, 2012, 7, 654–663 CrossRef PubMed.
- J. Li, C. Xie, J. Huang, Y. Jiang, Q. Miao and K. Pu, Angew. Chem., Int. Ed., 2018, 57, 3995–3998 CrossRef CAS PubMed.
- Y. Wang, S. Li, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 5308–5311 CrossRef CAS PubMed.
- T. Lei, M. Guan, J. Liu, H.-C. Lin, R. Pfattner, L. Shaw, A. F. McGuire, T.-C. Huang, L. Shao, K.-T. Cheng, J. B. H. Tok and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5107–5112 CrossRef CAS PubMed.
- T. Repenko, A. Rix, S. Ludwanowski, D. Go, F. Kiessling, W. Lederle and A. J. Kuehne, Nat. Commun., 2017, 8, 470 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |