
Alumina: discriminative analysis using 3D correlation of solid-state NMR parameters†
C. Vinod
Chandran
 ,
Christine E. A.
Kirschhock
,
Sambhu
Radhakrishnan
,
Francis
Taulelle
,
Johan A.
Martens
and
Eric
Breynaert
*
,
Christine E. A.
Kirschhock
,
Sambhu
Radhakrishnan
,
Francis
Taulelle
,
Johan A.
Martens
and
Eric
Breynaert
*
Center for Surface Chemistry and Catalysis, Celestijnenlaan 200 F – box 2461, KU Leuven, 3001 Heverlee, Belgium. E-mail: eric.breynaert@kuleuven.be
First published on 16th November 2018
Abstract
Synthetic transition aluminas (χ, κ, θ, γ, δ, η, ρ) exhibit unique adsorptive and catalytic properties leading to numerous practical applications. Generated by thermal transformation of aluminium (oxy)hydroxides, structural differences between them arise from the variability of aluminium coordination numbers and degree of dehydroxylation. Unequivocal identification of these phases using X-ray diffraction has proven to be very difficult. Quadrupolar interactions of 27Al nuclei, highly sensitive to each site symmetry, render advanced 27Al solid-state NMR a unique spectroscopic tool to fingerprint and identify the different phases. In this paper, 27Al NMR spectroscopic data on alumina reported in literature are collected in a comprehensive library. Based on this dataset, a new 3D correlative method of NMR parameters is presented, enabling fingerprinting and identification of such phases. Providing a gold standard from crystalline samples, this approach demonstrates that any sort of crystalline, ill crystallized or amorphous, mixed periodic or aperiodically ordered transition alumina can now be assessed beyond the current limitations of characterisation. Adopting the presented approach as a standard characterisation of alumina samples will readily reveal NMR parameter–structure–property relations suitable to develop new or improved applications of alumina. Methodological guidance is provided to assist consistent implementation of this characterisation throughout the fields involved.
 From left to right: Dr C. Vinod Chandran, Professor Christine E. A. Kirschhock, Dr Sambhu Radhakrishnan, Prof. Francis Taulelle, Prof. Johan A. Martens and Dr Eric Breynaert | All authors are affiliated with the NMR center at Leuven Chem&Tech (KU Leuven). This facility was founded in 2015 to support rational synthesis of advanced materials and converge with the X-ray core to a multidisciplinary NMR/X-ray platform for structural and functional characterisation of materials and processes. The mission of the NMR center is to perform convergent research across physics, chemistry, materials science, health and food science. The center focusses on investigating crystallization processes, confined molecular water, in situ adsorption studies, para-H2 based sensitivity enhancement and NMR crystallography. A particular strength is multi-receiver solid-state NMR for sensitivity enhancement and structure determination. Dr C. Vinod Chandran (official name: Vinodchandran Chandrasekharan) was born in 1982 in Chirayinkeezhu, Kerala, India. After obtaining his PhD from the Max-Planck-Institute for Solid state Research (Prof. M. Jansen, Stuttgart, Germany), he has worked as post doc in Radboud University (Nijmegen, Netherlands) and Leibniz University (Hannover, Germany). At present, he is employed as a post doc at the NMR Center of KU Leuven Chem&Tech with Prof. J. A. Martens and Dr E. Breynaert. In his research career, he mainly worked on solid-state NMR investigations of hydrogen storage materials, lithium-ion battery materials and solid catalysts. Professor Christine E. A. Kirschhock is a full professor at the KU Leuven (Belgium). Her main interest lies in the formation, structure and guest–host interaction of porous materials, especially zeolites. Being an expert in powder crystallography, she refines crystal structure models by combining Rietveld analysis with structural information obtained from complementary methods like SAXS, NMR, EXAFS and modelling. The resulting structure description on atomic scale reveals insight in fundamental mechanisms of zeolite genesis and function, which allows optimization of existing, and design of new functional porous materials. Prof. Kirschhock is heading the X-ray core of Leuven Chem&Tech and participates to combined XRD/NMR methods in NMR Crystallography. She is a member of the structure commission of the International Zeolite Association (IZA) and designs scientific art to bring complex scientific concepts into visual display as graphics or movies. Dr Sambhu Radhakrishnan was born in 1987 in Kerala, India. He obtained Master of Science in Chemistry from Mahatma Gandhi University in 2010 and he started PhD research in the group of Prof. Johan Martens in the field of catalysts for bio-derived fuel additives. During his PhD research, he demonstrated first spectroscopic proof of pore mouth catalysis by in situ solid state NMR studies. Presently he is working as a post doc at the NMR center of KU Leuven under the guidance of Prof. J. A. Martens, Prof. F. Taulelle and Dr Eric Breynaert. His research includes physicochemical characterization of catalytic materials (zeolites, aluminas, silicas), competitive adsorption and in situ catalytic studies studies by NMR. Professor Francis Taulelle is a visiting professor at KU Leuven and scientific advisor of the NMR Center. After investigating structures and (thermo-)dynamics of inorganic ionic liquids, his research focused on NMR crystallography, NMR methodology and instrumentation related to crystallogenesis of nanoporous materials. He developed methods for studying pharmaceutical polymorphs and pushed in situ NMR methods to investigate nanoporous materials as adsorbent, heterogeneous catalyst and as energy storage medium. He implemented NMR facilities in several laboratories (ESPCI Paris, University Pierre & Marie Curie, Paris, CNRS Orléans, University of Strasbourg, University of Versailles and KU Leuven) and leads the development of NMR Crystallography. He is member of the structure commission of IZA, and president of the NMR Crystallography commission of International Union of Crystallography (IUCr). Prof. Taulelle embodies convergent research across physics, chemistry, materials science to medicinal research and food science. Professor Johan A. Martens is a full professor at the KU Leuven (Belgium). As expert in nanoporous materials, he contributes to the advancement of catalysis science and technology. His original synthesis approaches yielded twenty COK-x materials, including ordered mesoporous silicate COK-12, OKO zeolite type material COK-14, metalorganic frameworks COK-15 and COK-18, spherosilicates and Posisils. Martens' research provides a fundamental basis for catalytic processes in (bio)refinery, renewable chemical intermediates, solar fuels, and environmental protection. He embodies innovation with several patents and spin off companies. His group pioneered the use of ordered mesoporous silica for enhancing bioavailability of poorly soluble drug molecules (FORMAC Pharmaceuticals). Other highlights are the use of magnetohydrodynamic (MHD) technology for cosmetic, food and chemical industries (Magnets for Emulsions) and a new aquaculture concept involving adsorptive-photocatalytic systems to eliminate off flavor compounds (AQUA4C, 2013). Dr Eric Breynaert is a research expert at KU Leuven (Belgium) and NMR/X-ray Facility Manager at Leuven Chem&Tech. He is an expert in physicochemical characterization and design of nanoporous materials, focussing on investigating the structure and surface properties of periodic and non-periodic materials (zeolites, ordered mesoporous materials, silica/alumina, clays, layered double hydroxides, etc.), their rational synthesis and post-synthetic modification. Multi-disciplinary research combining molecular level information derived from (i) magnetic resonance, core and valence electron spectroscopy (NMR, XAS, EPR, UV-VIS, etc.) (ii) meso-scale structure determination (SAXS, SANS, HEXS) and (iii) integration of this information in a multi-scale model via chemical and functional characterisation and chemical, molecular and spectroscopic modelling is a key aspect of his work. This involves large-scale infrastructure research (synchrotron and neutron sources) and the development of experimental strategies for new facilities such as the European XFEL. |
1. Introduction
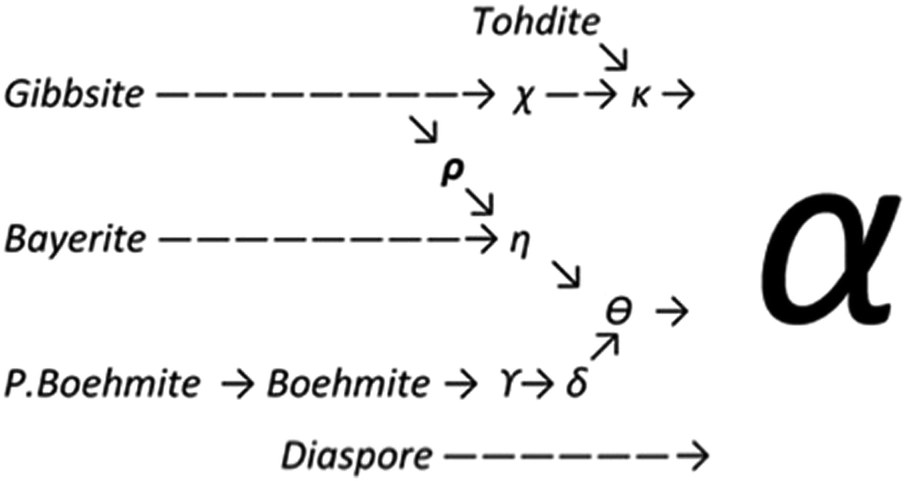
Upon heating, aluminium (oxy)hydroxides (AlOOH or Al(OH)3) pass through a series of phase transitions before converting to the entirely dehydroxylated α-alumina (Al2O3) at high temperature.1–3 The structural transformation from the (oxy)hydroxides to alpha-alumina involves successive loss of water and significant densification. Thermal treatment irreversibly removes structural hydroxyl groups, successively transforming the initial (oxy)hydroxides in a sequence of more and more dense transition phases between 300 and 900 °C (Fig. 1). These so-called transition alumina's are stable at ambient conditions and are distinguished by Greek letters (χ, κ, θ, γ, δ, η, ρ), according to the moment of their discovery and first description. | ||
| Fig. 1 Schematic representation of the thermal evolution (black arrows) of transition alumina phases in air, from the hydroxide and oxyhydroxide precursors, gibbsite, boehmite, bayerite and diaspore.1–3,5,6 The non-thermal transformations are shown with blue arrows. | ||
The transition aluminas exhibit a high surface-area and both Brønsted and Lewis type acidity. Typically these phases are applied as dessicant, adsorbent, catalyst, catalyst support and as ceramics precursor. Abrasion and attrition resistant extrudates of transition alumina are obtained by calcination of extruded precursor aluminium hydroxide or oxyhydroxide. They are suitable sources of Al for manufacturing aluminium metal through the Bayer process. Gamma, theta and eta alumina are popular in catalytic applications such as dehydration of alcohols, isomerisation of olefins, production of sulphur from H2S, etc.4 Other important alumina-catalysed reactions include chlorination of methanol, de-hydrofluorination of organic fluorides, alkylation of phenols, de-oxygenation of bio-oils and decomposition of triglycerides. Large scale applications as catalyst support for metals include hydrodesulfurization in petroleum refinery using cobalt and nickel promoted molybdenum sulfide, NOx reduction, CO and hydrocarbon oxidation reactions on supported noble metals. Wash coated monolithic exhaust gas purification catalysts are further examples.4
The transformation sequences from (oxy)hydroxide up to alpha-alumina (Fig. 1), are highly influenced by starting material, crystallite size, relative humidity, alkalinity, heating rate, pressure and bed depth.1–3,5,6
Clear assignment of structural models to the different phases severely was, and still is, hampered by the occurrence of complex phase assemblies along the calcination path in combination with their typical micro and nanocrystalline nature. Whereas the latter is a direct consequence of their formation by release of structural water and consequent, densification, it results in broadening of the X-ray diffraction lines, rendering PXRD patterns very difficult or impossible to analyse (see ESI,† Discussion S1). Alumina phases such as ρ or χ alumina, for example, inherently exhibit a degree of disorder too high to allow identification from X-ray diffraction.7,8 Comparing the structures proposed in literature revealed these phases can clearly be distinguished by three parameters only: (1) nominal water content, comprising structural hydroxyls and adsorbed water, (2) the order of the anionic sublattice, and (3) the distribution of aluminium cations therein (Fig. 2). As the thermally induced transformations involve both dehydration and dehydroxylation, the atomic ratio of H versus Al is a rather insensitive parameter to globally describe the progress of the transformation. Distinguishing phases with a similar degree of hydration requires additional parameters.9,10 As the condensation of hydrogen-bonded oxygen layers implies successive re-ordering of the oxygen sublattice, this order can serve as second parameter for phase identification. Indeed, all phases under consideration, with the exception of the fully amorphous alumina gels and maybe the extremely disordered rho alumina, can be described as dense packings of oxygen planes, with aluminium cations occupying the vacancies in between the planes. In dense stacking sequences, the ratio of tetrahedral and octahedral vacancies is always 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
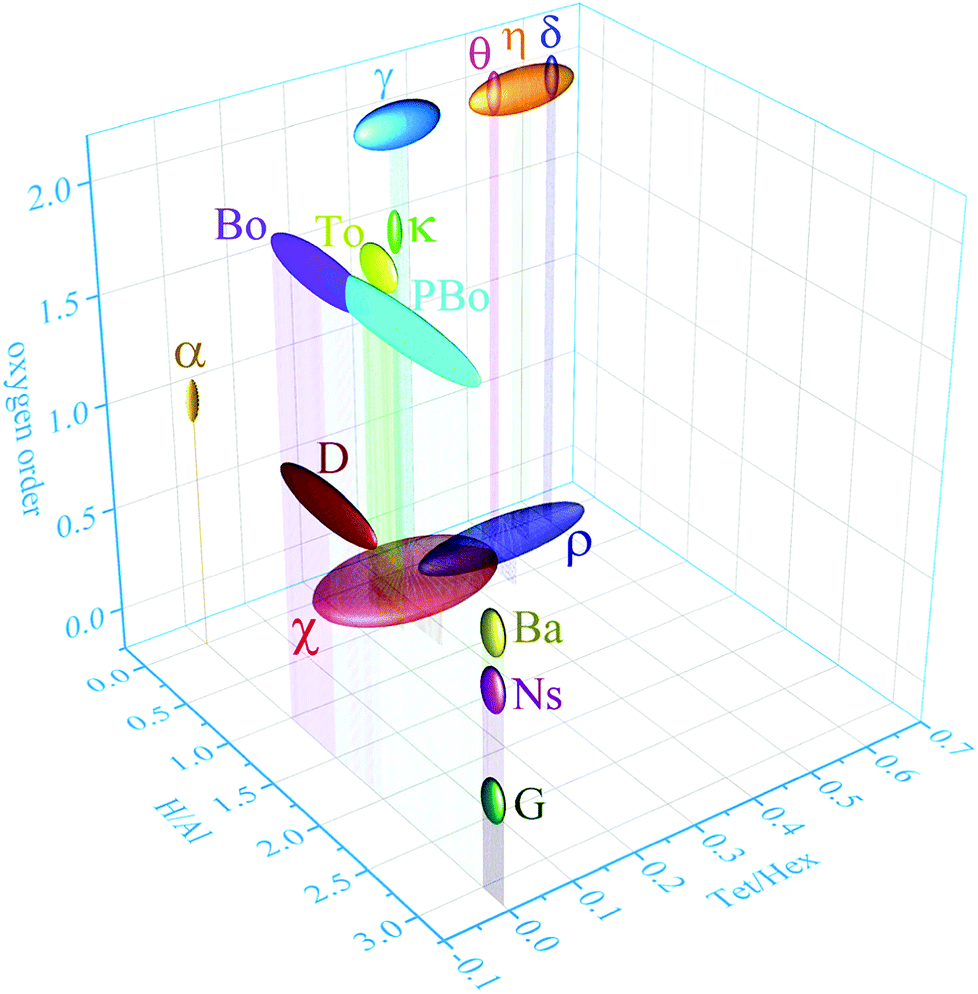 | ||
| Fig. 2 Three-dimensional graph correlating the ratio H/Al, the ratio of tetra-coordinated and hexa-coordinated Al and the oxygen order of α, η, δ, θ, γ, κ, χ, ρ-alumina and bayerite (Ba), boehmite (Bo), tohdite (To), pseudoboehmite (PBo), diaspore (D), nordstrandite (Ns) and gibbsite (G). The chemical formulae are listed in the ESI,† Table S1. | ||
Within a layer, octahedra share edges with neighbouring octahedra and faces with tetrahedra, and opposite. In stacking sequence ABA, octahedra share faces with octahedra in the upper and lower layers, whereas in the ABC sequence, they only share edges between layers (Fig. 3). As charge distribution forbids occupation of tetrahedra sharing a face with either octahedra or tetrahedra, the stacking sequence of the anion sublattice limits the possibilities to distribute cations over the vacancies. Furthermore, early transition aluminas systematically show disorder in their cation occupation. With increasing temperature, order in cation as well as oxygen sublattice is increasing. Hence, as criterion for phase identification the layer sequence of the anionic sublattice and the ratio of occupied tetrahedra versus octahedra can serve to fully identify a transition alumina and an aluminium (oxy)hydroxide. However, direct identification of some alumina phases (e.g., ρ or χ alumina) from X-ray diffraction alone is not possible owing to the degree of disorder in the systems.
 | ||
| Fig. 3 ABC stacking sequence with octahedra and tetrahedra sharing edges. | ||
Solid-state NMR is a powerful characterization technique for materials.11–1327Al NMR spectroscopy is very often used, as aluminium is omnipresent in materials necessary for science and technology. The parameters defining the structure of the transition alumina phases have direct or indirect influence on the local environment of the 27Al nucleus, and should be reflected in its chemical shift and quadrupole parameters. As will be demonstrated below, 27Al MQMAS is the ideal tool to measure these parameters, providing a straightforward spectroscopic identification of any transition alumina by a single experiment. Although very important 1H and 17O NMR experiments have been carried out to investigate the structure of transition aluminas,14–1727Al is definitely the most suitable NMR probe for these phases. Solid-state 27Al NMR is able to give highly resolved NMR signatures even for those phases which cannot be identified by X-ray diffraction.
1.1. 27Al NMR spectroscopy in materials science
The NMR properties of 27Al (see Table 1) provide a good NMR receptivity, at frequencies accessible by common NMR probe-heads. Consequently, 27Al NMR is an obvious choice to assist structure elucidation of aluminium containing materials.18–21 The nuclear spin 5/2 of 27Al results in a nuclear quadrupole moment interacting with local electric field gradients (EFG). Although this interaction can severely broaden the spectra, it provides an extremely sensitive probe to the local chemical environment of a quadrupolar nucleus in solids.| Properties | Magnitude | Unit |
|---|---|---|
| Spin number (I) | 5/2 | |
| Nuclear magnetic moment (μ) | 4.30869 | μN |
| Gyromagnetic ratio (γ) | 6.976 × 107 | rad T−1 s−1 |
| Resonance frequency (at 11.4 T) | 130.284 | MHz |
| Quadrupole moment (Q) | 146.6 | mb |
| Relaxation times (T1) | <10 | s |
| Natural abundance | 100 | % |
| Receptivity (relative to 1H) | 0.207 | |
| Receptivity (relative to 13C) | 1220 | |
| Isotropic chemical shift (δiso) range (for non-metallics) | >−25 to <+110 | ppm |
| Quadrupole coupling constant (CQ) | >1 to <20 | MHz |
In case of 27Al, the magnitude of this quadrupole interaction is typically in the MHz range and can be expressed as a quadrupole coupling constant (CQ), relating the quadrupole moment Q with the local EFG tensor as:
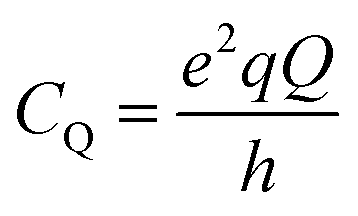 | (1) |
In liquid state NMR, first and second order quadrupole interactions are averaged out. In solids only the first-order quadrupole interaction can be fully averaged by very fast magic-angle spinning (MAS). However, second-order quadrupole interaction is not averaged with single-angle sample spinning. This not only results in anisotropically broadened solid state spectra of the central 27Al transition (m+1/2 ↔ m−1/2), it also gives rise to a quadrupole-induced chemical shift:
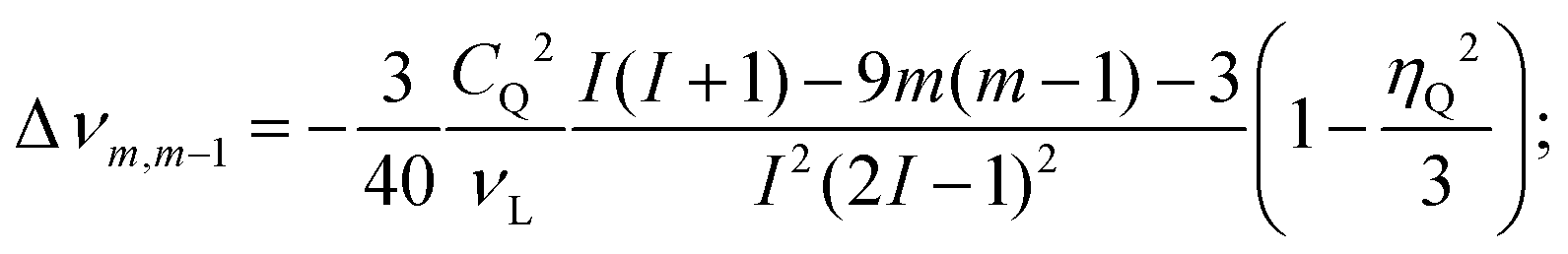 | (2) |
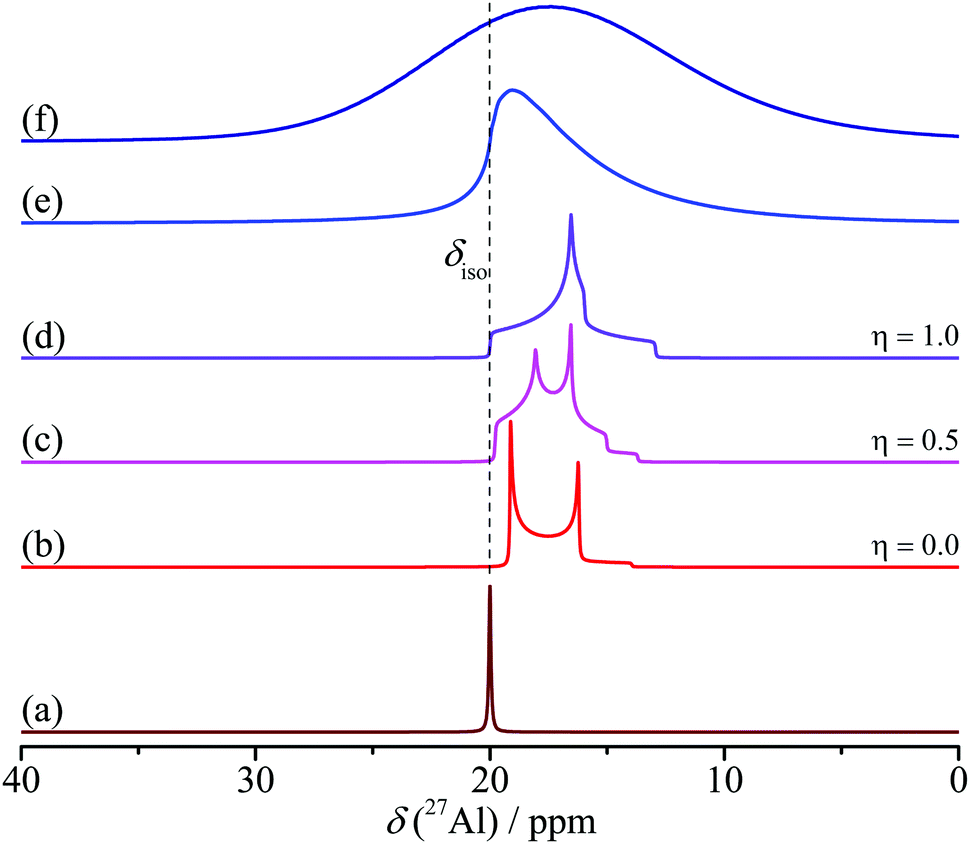 | ||
| Fig. 4 Simulated 27Al central transition spectra under MAS of (a) without quadrupole coupling and (b)–(f) with a quadrupole coupling (CQ) of 2 MHz. The asymmetry parameter (η) varies from 0.0 to 0.5 and to 1.0 in (b)–(d). A distribution of CQ is simulated in (e) and a distribution of CQ and chemical shift are simulated in (f). | ||
In case of 27Al, IUPAC recommends to use a fresh aqueous (D2O) solution of Al(NO3)3 (1.1 M) as the 27Al primary chemical shift reference standard at 0 ppm.12 An aqueous solution of 0.5/0.5 M Al(NO3)3/HNO3 is usually used in case long shelf-life is desired. The range of 27Al isotropic chemical shifts (δiso) for different Al–Ox coordinations is usually between −20 to +90 ppm. The hexa coordination (Al–O6) exhibits 27Al isotropic chemical shifts in the range between −21 and +37 ppm,27–33 while for the tetra-coordinated species (Al–O4) it is between +39 and +85 ppm.27–31 The penta-coordinated aluminium (Al–O5) usually shows shifts in the range between +20 and +52 ppm.24,34–38 Since each coordination exhibits a typical chemical shift, 27Al NMR provides an easy way to identify and quantify Al structural units in materials.23,39 For most materials, the 27Al NMR resolution is sufficient to distinguish between different coordinations. Especially in the case of Al(V), quadrupole broadening is less than that in Al(IV) and Al(VI).
In addition to chemical shift referencing, also pulse calibration is essential for 27Al characterisation of materials. The nutation of half-integer spin quadrupoles (like 27Al) in solution state is different from that in solid-state. The 90° pulse calibrated for a solution standard should be divided by a factor of I + 1/2 to obtain that for solid-state systems when the quadrupole frequency (νQ = CQ/[2I(2I − 1)]) is higher than the nutation frequency. At infinite rotation frequency only the second-order quadrupole broadened central transition survives. The nutation of the quadrupole central transition in solid-state heavily depends on the quadrupole interaction. Therefore, very small flip angles (π/12 or lower) are recommended to remain in the quantitative regime for the central transition signals with different quadrupole couplings.
Once correct chemical shift referencing and pulse calibration are ensured, separation of overlapping resonance contributions broadened by the second-order quadrupole coupling becomes experimentally accessible using high-resolution NMR methods such as double rotation (DOR),40,41 dynamic-angle spinning (DAS),42,43 multiple-quantum magic-angle spinning (MQMAS)44,45 and satellite-transition magic-angle spinning (STMAS).46,47 While in case of 27Al, DOR or DAS experiments don’t provide benefit, as the MAS frequencies which can be used for these experiments cannot average the 27Al–27Al dipolar interactions. MQMAS or STMAS experiments do provide an advantage, as they can have very high MAS frequencies. Using correctly acquired experimental data, distributions of quadrupole parameters can be extracted by simulating them with Gaussian distributions of each of the EFG tensor components, using the Czjzek model,48,49 a feature that has been incorporated in one of the more popular NMR spectral analysis programs, DMFIT.50,51
MQMAS, a 2D experiment correlating single and multiple quantum transitions (3Q and 5Q), is the most popular and convenient choice to achieve high resolution in the case of spin 1/2 quadrupolar nuclei (e.g.27Al) exhibiting central transitions broadened by a second-order quadrupole interaction.52 Isotropic chemical shifts can be directly obtained from the indirect dimension of the experiment. Quadrupole parameters (CQ and ηQ) and their distributions can be extracted by fitting resolved single quantum (horizontal) slices of the 2D experiment.
As the quadrupole coupling constant CQ is impacted by the local symmetry, this parameter reveals information on the coordination of Al atoms. Distortions from perfect octahedron or tetrahedron usually result in large quadrupole interactions (of the order of MHz), especially in the case of Al–O coordinations in alumina. Tetra-coordinated species exhibit larger quadrupole coupling compared to hexa-coordinated species; penta-coordinated Al typically exhibit a broad distribution in quadrupole coupling and/or chemical shift, due to their preferential occurrences at surfaces of the solids and at grain boundaries. 3D correlating δiso, CQ and ηQ consequently yields a fingerprint of the local environment of an 27Al nucleus.
This has been done for a set of transition alumina samples with the help of values reported in literature (infra). 3D ellipsoid representations are a suitable method to present such correlations, as they allow to also include the spread of the reported values by plotting an average value of each parameter (ellipsoid centre) along with its spread (ellipsoid axis). In case of alumina, such correlations have to be built for Al(IV), Al(V) and Al(VI) coordinations separately. In case of transition alumina such 3D ellipsoids were found to be extremely useful in the identification of all alumina phases as they have minimal overlap (infra). Additionally, 1H MAS NMR can be used for absolute quantification of the nominal water content (structural hydroxyls and adsorbed water),157 while 1H–27Al correlation NMR methods are useful to provide extra information on the structure of alumina and other aluminium containing solid systems. In case of surface studies, dynamic nuclear polarization (DNP) enhanced NMR detection has provided crucial information on 1H–27Al connectivity.53–59
2. Aluminium hydroxides and oxyhydroxides with H/Al ≥ 1
Aluminium (oxy)hydroxides are common precursors to generate transition alumina phases by thermal treatment (Fig. 1). Typical for all structures is the presence of Al-bonded hydroxyls which take part in hydrogen bonding. The idealized structure formula for the hydroxides and oxyhydroxides is Al(OH)3 and AlO(OH), respectively, although real materials may contain extra physisorbed water molecules, leading to an Al/H ratio for the hydroxides in the range of 3–3.2, and for oxyhydroxides roughly in the range of 1–2. Furthermore, all these phases exclusively contain hexa-coordinated Al-ions leading to a ratio Altet/Alhex equalling zero. These water-rich phases occur naturally in bauxite, containing the hydroxide gibbsite and the oxyhydroxides boehmite and diaspore as main aluminous components, next to traces of the other three hydroxides, viz. bayerite, nordstandite and doyleite. The Bayer process, introduced in 1888, still today is the most important pathway to refine bauxite.3 Leaching of aluminate from the bauxite mineral followed by precipitation leads to gibbsite, currently the most used precursor for alumina and aluminium synthesis.2.1. Aluminium hydroxides
Only four Al-hydroxides are known: gibbsite, bayerite, doyleite and nordstrandite. Whereas gibbsite and bayerite are common minerals found naturally and produced synthetically, nordstrandite and doyleite are rare. Among the latter two, only nordstrandite can be obtained synthetically. Hence, reports on these two hydroxides, especially on doyleite are limited.Al-Hydroxides are built up from octahedral (6-coordinated Al3+), edge-linked into hexagonal sheets, isomorphic to brucite type sheets with 2/3 occupancy of the metal positions, resulting in the typical honeycomb-like appearance. Each sheet is composed of two densely packed oxygen planes, with AB sequence (Fig. 5). The four hydroxide structures differ in their vertical stacking, leading to different vertical Al distributions and to different hydrogen bonding within and between the hexagonal layers. This not only results in different Al-environments, but also in different sequences of transition alumina upon calcination. Upon thermal treatment, only nordstrandite and bayerite are reported to follow a same transformation sequence.
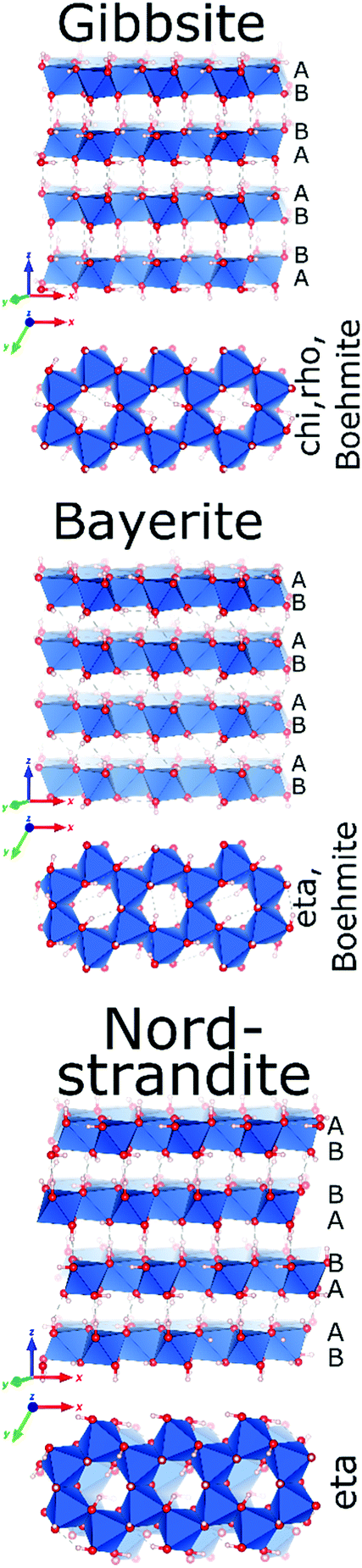 | ||
| Fig. 5 The crystal structures of (from top to bottom) gibbsite,60 bayerite and nordstrandite (Al in blue polyhedra, O in red and H in grey).67 | ||
Gibbsite, the end-product of the commercial Bayer process, is produced on large scale by alkaline precipitation of Al3+. It is widely used as fire retardant, additive in polymers and paper, toothpaste fillers, for stabilization of titania and in the production of artificial marble.

Gibbsite (γ-Al(OH)3) crystallizes as a monoclinic structure (space group P21/b)60 where consecutive hexagonal layers of Al-octahedra are systematically mirrored. This results in an overall stacking sequence of AB BA AB BA in the oxygen sublattice (Fig. 5). Aluminium ions in different layers are vertically aligned. The intra- and interlayer hydrogen bonding occurring in gibbsite results in two crystallographically different Al environments with a ratio of 1:1.61 While their chemical shift differences are small, the two Al-sites experience significantly different EFGs, reflected in their quadrupole coupling constants.
Like most of Al-containing materials, the direct excitation 27Al MAS NMR spectrum of gibbsite contains a very wide spinning side-band manifold, owing to its large quadrupole interaction. The 27Al CT spectrum of gibbsite under MAS shows two overlapping Al resonances (see simulated spectrum in Table 2) which differ in their second-order quadrupole coupling pattern assigned to two different sites.7,9,62–64 Al occupying the first site is surrounded by hydroxyl groups taking part in four intra-layer and two inter-layer hydrogen bonds with an isotropic chemical shift of 13.6 ppm, CQ of 4.6 MHz and ηQ of 0.4. The second Al site exhibits a similar isotropic shift (11.3 ppm), but a smaller CQ (2.2 MHz) and a larger ηQ (0.7). In this case, four hydroxyls point towards the neighbouring layers, while the other 2 engage in intra-layer interaction.61 Spectral decomposition of the 27Al NMR resonance of the central transition, recorded quantitatively with a π/12 pulse, revealed two equally occupied octahedral Al-sites in accordance with the structural model. Also high-resolution 1H decoupled 27Al 2D 3QMAS spectra showed two isotropic resonances in corresponding to the two Al sites.63,64 Proton decoupling is highly recommended for quadrupolar NMR, especially for MQ experiments in hydroxides and oxyhydroxides.65
| Material | 27Al MAS | 27Al MQMAS | Coordination (occupancy) | δ iso/ppm | C Q/MHz | η Q | Ref. |
|---|---|---|---|---|---|---|---|
| a Assuming the reported values in ref. 123 are quadrupole frequencies νQ. b Estimated from the spectra shown in ref. 7. c Predicted by ab initio calculations139 and deduced from the experimental spectra.123 d Al2 site having five regular and one elongated Al–O bond. | |||||||
|
|
|
Alhex(1) (50%) | 11.3 | 2.2 | 0.7 | 61 |
| Alhex(2) (50%) | 13.6 | 4.6 | 0.4 | ||||

|
|
|
Alhex(1) (50%) | 9.1 | 1.9 | 0.25 | 64 |
| Alhex(2) (50%) | 13.1 | 1.4 | 0.80 | ||||
|
|
|
Alhex (100%) | 12.6 | 1.8–2.8 | 0.5–1.0 | 64 |
|
|

|
Alhex (100%) | 17.0 | 3.4 | 0.8 | 83 |
|
|
|
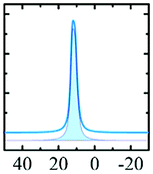
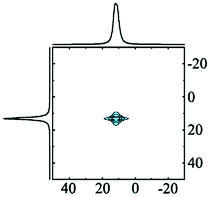
Alhex (100%) | 13.5 | 2.38 | 0.0 | 131 |
| 16.0 | 2.40 | 0.05 | 138 | ||||

|
|
|
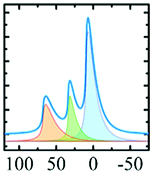
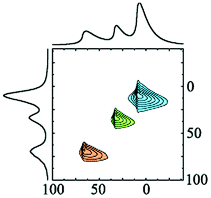
Alhex (30–55%) | 9.8b | 4.8 | 0.6b | 7 |
| Alpent (20–50%) | 34.0b | 4.5b | 0.6b | ||||
| Altet (25–15%) | 68.0b | 5.6b | 0.6b | ||||
|

|
|
Alhex (70–75%) | 11.5 | 4.5 | 0.3 | 8 |
| Alpent (5–10%) | 38.5 | 2.7 | 0.3 | ||||
| Altet (20%) | 71.5 | 5.0 | 0.3 | ||||
|
|
|
Alhex (70–40%) | 10.0 | 5.1 | 0.31 | 120 |
| Alpent (0–20%) | 44.0 | 3.55 | 0.00 | ||||
| Altet (30–60%) | 71.5 | 5.1 | 0.19 | ||||

|
|

|
Alhex (70–55%) | 15.2 | 3.6 | 0.4 | 71 |
| Altet (30–45%) | 78.5 | 4.7 | 0.7 | ||||
|
|

|
Alhex (50%) | 10.5 | 3.5 | 0.0 | 131 |
| Altet (50%) | 80.0 | 6.4 | 0.65 | ||||

|

|

|
Alhex (80–50%) | 14.5–15.1 | 3.9 | 0.4 | 71 |
| Altet (20–50%) | 76.5–77.5 | 4.5–4.7 | 0.7 | ||||
|
|
|
Alhex(1) (25%) | 0.0 | 3.33a | 0.33c | 123 |
| Alhex(2) (25%) | 5.0 | 5.07a | 0.33c | ||||
| AlV+Id (25%) | 4.4c | >10a | 0.77c | ||||
| Altet (25%) | 68.5 | 5.67a | 0.3 | ||||
Dehydroxylation of gibbsite follows different routes (sequence of transition alumina species) when performed in wet air, in dry air, and in vacuum. Heating in presence of excess water vapour (60–300 °C), transforms gibbsite to boehmite,66 being the main route for synthesis of the latter. In dry air, it transforms to χ-alumina (300–500 °C) and κ-alumina (800–1150 °C) before turning into α-alumina.6 In this sequence,9 the population of the 4-coordinated Al (Al–O4) increases gradually and reaches a maximum of Altet/Alhex ≈ 30% around 1000 °C in the kappa phase (Fig. 6). Under vacuum, gibbsite transforms to ρ-alumina (100–400 °C), which then turns into η-alumina (270–500 °C) and θ-alumina (870–1150 °C), before reaching α-alumina (above 1200 °C). Also in this series, the percentage of tetrahedral Al increases and reaches values of 50% in the theta phase. Unlike most other discussed alumina phases, the highly disordered ρ-alumina with strongly fractured, nanosized particles contains a significant fraction of five-fold coordinated aluminium in addition to the tetra and hexa-coordinated Al species.
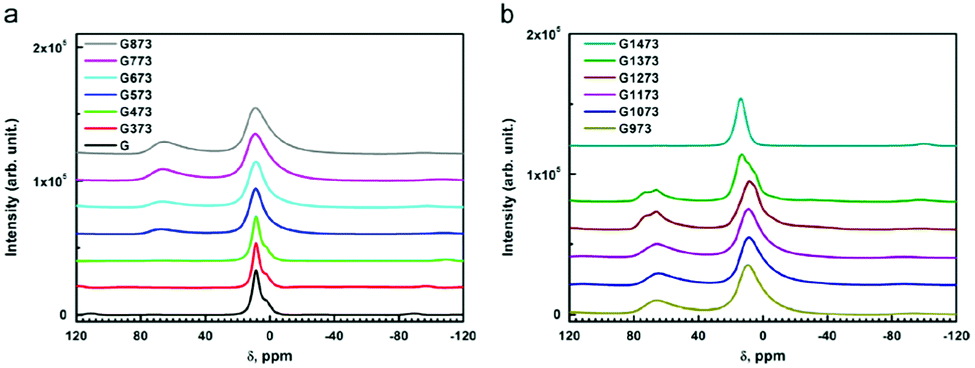 | ||
| Fig. 6 27Al MAS NMR spectra of untreated and thermally treated gibbsite in air from 100 to 1200 °C (373 to 1473 K in the inserted legend). Reproduced from ref. 7 with permission from Elsevier, copyright 2018. | ||
Grinding has significant influence on the gibbsite properties as confirmed with 27Al NMR by MacKenzie et al.66 The mechanically activated surfaces of the fractured gibbsite particles are postulated to adsorb water, resulting in five-fold coordinated Al-species. Ground gibbsite, showed a small fraction (∼10%) of AlO5, which remained present on heating from room temperature up to 600 °C, before diminishing. These observations demonstrate the generally observed sensitivity of alumina transformation to crystallite size, moisture, heating rate, etc.
The aluminium hydroxide bayerite (α-Al(OH)3) crystallizes in monoclinic structure in the space group P21/a.67 Similar to all crystalline Al(OH)3 phases it consists of layers of edge sharing AlO6-octahedra with hexagonal symmetry. Like in gibbsite, the sheets are congruently aligned. Unlike gibbsite, however, the stacking of the oxygen ions in the anionic sublattice follows a sequence AB AB AB…, i.e. each octahedral layer can be generated by simple vertical translation from the layer below. Also like gibbsite, bayerite has two crystallographically distinct Al-sites in a ratio 1:1, owing to the hydrogen bonding in and between the layers.
It was long thought that 27Al NMR could not resolve the two Al sites, since they are very similar from a crystallographic point of view resulting in isotropic chemical shifts differing only slightly (9.1 and 13.1 ppm). In contrast with gibbsite, also the CQ values of the two Al sites of bayerite are almost identical (1.9 and 1.4 MHz), leaving only the ηQ values (0.25, 0.80) to distinguish the two sites. While in double rotation (DOR) NMR experiments, no resolution of the 2 sites was achieved,68 exploiting 27Al 3QMAS NMR, Damodaran et al.64 could resolve the two 27Al NMR resonances. Quantification succeeded with quadrupolar parameters extracted from MQMAS.64
Thermal treatment of bayerite converts it into η-alumina at 300–600 °C, followed by the formation of θ-alumina at 800–1000 °C, reaching α-alumina above 1100 °C (Fig. 1). However, a complete transformation to α-alumina without co-existing additional phases has rarely been observed by 27Al NMR (as in Fig. 6).69 This may be related to the ready conversion of bayerite into boehmite, which follows a different transition route.70
The changes of the 27Al MAS NMR spectrum upon calcination of bayerite are illustrated in Fig. 7. During bayerite calcination, 6-coordinated Al is converted into AlO4, reaching a maximum Altet/Alhex ratio of 50:50 at 800–900 °C. Further heating decreases the AlO4 content again. The broadening of the AlO627Al MAS NMR resonance increases with temperature (from 300 °C to 800 °C), indicating an increased CQ and its distribution. On calcination from 800 °C to 1200 °C, the mean CQ of the 4-coordinated Al increases from 4.7 MHz to 5.5 MHz, while the CQ of the 6-coordinated Al decreased from about 3.7 MHz to 3.0 MHz.71 In the transition sequence, the illustrated de-convolution of the spectra suggests the presence of five-coordinated Al, but NMR parameters were not reported. Ashbrook et al. reported 27Al NMR of bayerite mechanically mixed with silicic acid.63 Two-dimensional 3QMAS experiments were carried out to resolve a third Al site at around 60 ppm corresponding to Al(IV) coordination indicating the formation of Al–O–Si bonds in tetrahedral coordination. Longer grinding also produced five-coordinated Al at the surface as shown by the 27Al NMR peak at around 32 ppm.
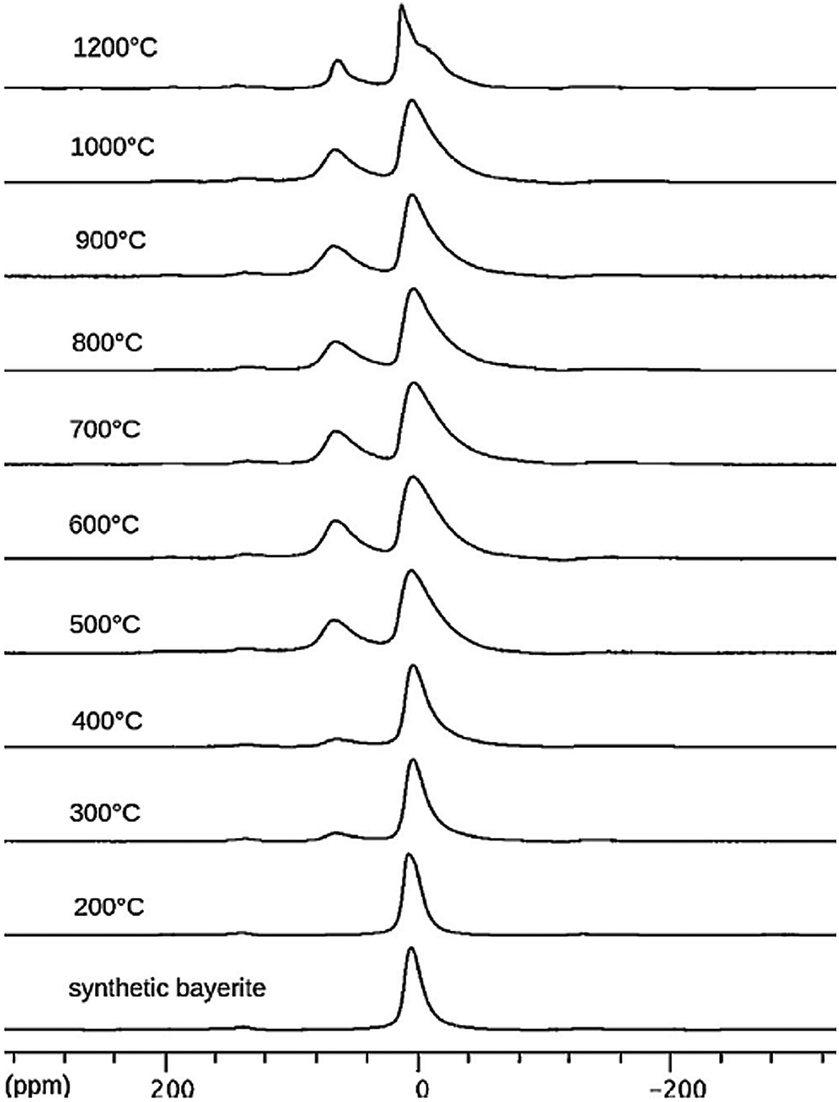 | ||
| Fig. 7 27Al NMR spectra of synthetic bayerite and derivatives obtained by heating at the indicated temperatures. Reproduced from ref. 69 with permission from Elsevier, copyright 2018. | ||
2.2. Aluminium oxyhydroxides
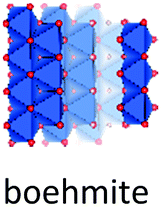
Despite of their quite different structures, the aluminium oxide monohydroxides diaspore and boehmite exhibit a very similar stability,72 with slight preference for boehmite. Both are built-up from edge and corner sharing Al octahedra, but while diaspore is a truly 3D connected octahedral framework, boehmite displays hydrogen-bonded octahedral layers (Fig. 7).Used in the production of ceramics, as catalyst binder, catalyst support and as a flame retardant, boehmite (γ-AlOOH) is the aluminum oxyhydroxide of greatest practical interest. It is produced by hydrothermal synthesis starting from gibbsite and can also be obtained by controlled calcination of bayerite. Boehmite crystallizes in the orthorhombic space group Amam, with one crystallographic site for Al and two for oxygen. It is built up from edge-sharing octahedra arranged in stepped layers, cross-linked by hydrogen bonds.73 Within the octahedral layers the oxygen planes follow an ABC ABC… arrangement. However, H-bonding between Al-octahedra, causes systematic offsets within oxygen planes spanning neighbouring Al-layers (Fig. 8). Boehmite exhibits severe variation of hydration level and particle size.74,75 With increasing water content, layer distance is slightly increased and crystallites tend to decrease in size. The resulting compound is called pseudoboehmite.76 This flaky material has grown in only two crystallographic directions and exhibits a poorly defined X-ray powder pattern. Boehmite-like arrangement of aluminium octahedra persists within the layers.

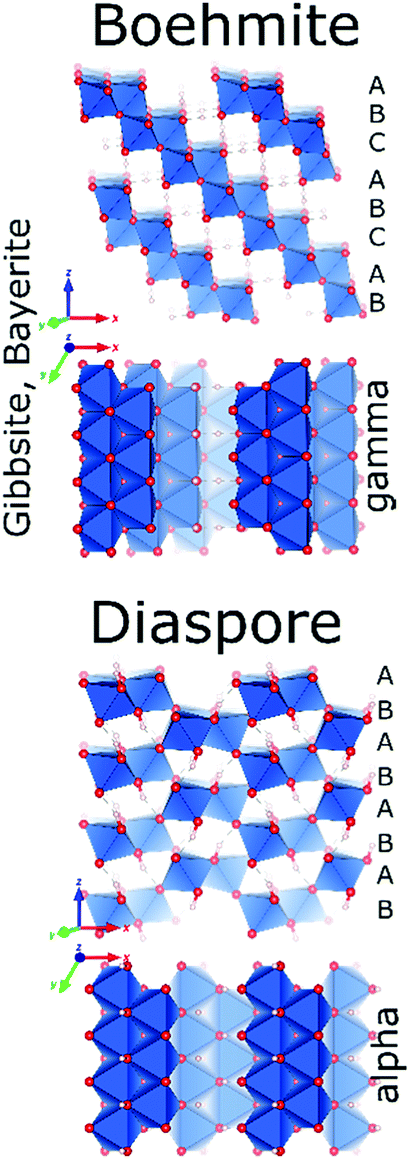 | ||
| Fig. 8 Structure of boehmite and diaspore. | ||
Boehmite and pseudoboehmite show a single 27Al MAS NMR resonance with isotropic chemical shift at 12.6 ppm (Fig. 9).69,71,77–79 From the combined analysis of the spinning side-band manifold and the 3QMAS spectrum, the range of CQ and ηQ for boehmite were deduced as 1.8–2.8 MHz and 0.5–1.0, respectively. While the boehmite 27Al CT resonance exhibits a small distribution in quadrupole couplings and chemical shifts, the pseudoboehmite resonance is significantly broadened. This renders the estimation of CQ and chemical shift difficult. Grinding of boehmite, similar as in the case of gibbsite, has been observed to generate five-fold coordinated Al, a feature which quite often is reported also for pseudoboehmites.80 Upon heating, boehmite sequentially transforms into γ-alumina (500–700 °C), δ-alumina (800–900 °C), θ-alumina (∼1000 °C) and α-alumina (above 1100 °C) (Fig. 1).6 In the delta phase, the ratio Altet/Alhex reaches a maximum of up to 0.6. Calcination of pseudoboehmite follows the same sequence, but phase transformations occur in broader temperature ranges. For the highly disperse and partially disordered pseudoboehmites, commonly complex mixtures of transition aluminas are obtained and, unlike boehmite, five-fold coordinated Al could be observed.77 As boehmite can form from bayerite as well as from gibbsite by calcination, the occurrence of varying amounts of transition aluminas assigned to the boehmite pathway upon thermal transformation of both hydroxides is not surprising.
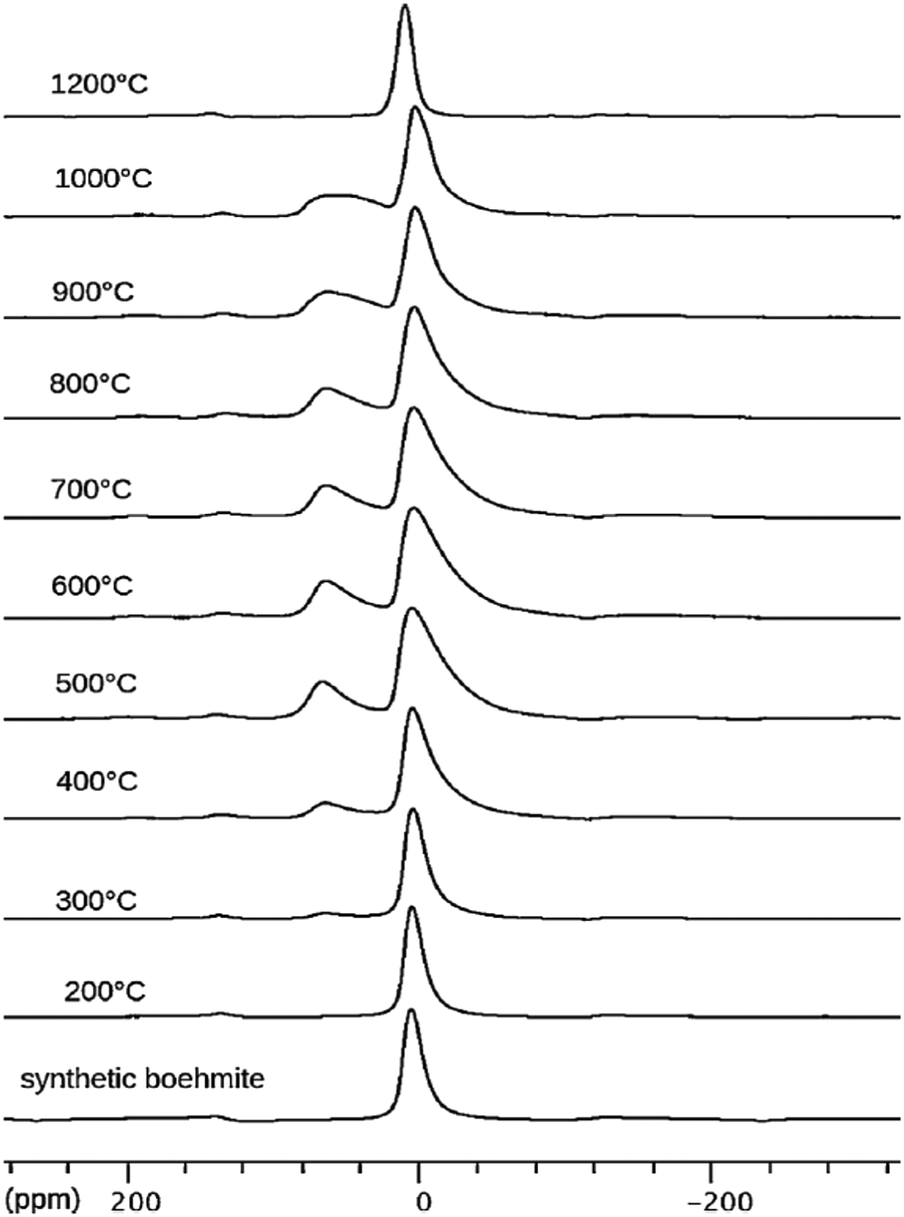 | ||
| Fig. 9 27Al NMR spectra of synthetic boehmite and derivatives obtained by heating at the indicated temperatures. Reproduced from ref. 69 with permission from Elsevier, copyright 2018. | ||
As shown in Fig. 9, 27Al NMR is ideally suited to follow the evolution of the AlO4/AlO6 ratio as function of the calcination temperature.71,77–79 The development of 4-coordinated Al centers (δiso 70–80 ppm) starts at 300 °C. As the fraction of AlO4 increases, the resonance of the residual AlO6 broadens due to an increase in the distribution of its CQ.69 Decomposition of the spectra at even higher temperatures might even reveal the presence of five-coordinated Al, especially when pseudoboehmite is used as starting material.69 From 1200 °C onwards, only the octahedral Al of α-alumina remains.
Diaspore (α-AlOOH) is a naturally occurring mineral that can be obtained synthetically by hydrothermal synthesis at high temperature and pressure.81 It crystallises in the orthorhombic space group Pbnm with a single Al and two O atoms per asymmetric unit.82 The structure consists of a hexagonally ordered, close packed oxygen sublattice with AB AB… sequence and Al occupying octahedral sites in such a way as to generate edge-sharing ribbons which connect via corners to a 3D network. The reported 27Al NMR parameters for the octahedral Al in diaspore are δiso = 17 ppm, CQ = 3.4 MHz and ηQ = 0.8.83

Diaspore topotactically converts to α-alumina when heated in air above 500 °C, without passing through any transition phases. The structural similarity of the anionic sub lattices of diaspore and α-alumina facilitates this transformation. Tsuchida81 observed the transformation using 27Al MAS NMR. The peak maximum of the AlO6 resonance shifted about 5 ppm down-field on calcination in the temperature range 450–500 °C. In contrast to boehmite, grinding of diaspore did not result in any additional species.81 Upon vacuum dehydration, however, formation of a transition alumina phase containing up to 20% of tetra-coordinated Al is reported.84 This α′ alumina phase was detected with 27Al NMR and exhibits peak maxima of the hexa- and tetra-coordinated peaks at 0 and 60 ppm respectively. No other NMR parameters were reported. Interestingly, transformation from α′-alumina to corundum is catalysed by water vapour. Moist air-catalysed transformation of diaspore to α-alumina has been suggested as the reason for the absence of transition alumina's in the diaspore series.
3. Transition alumina phases
Transition alumina's typically are microcrystalline phases exhibiting a certain degree of disorder in their structure. Their complexity and disordered nature defines the reactivity of activated or partially calcined alumina hydrates, which are known to be excellent catalyst supports. Phases like χ, ρ, η and γ have ill-defined structures. This structural diversity manifests in badly resolved X-ray diffraction patterns and 27Al NMR spectra making the structural interpretation complex. The NMR resonance of the six-coordinated Al sites in all the transition alumina's shows a tailing towards smaller chemical shifts. Distributions of NMR parameters like the quadrupole coupling and chemical shifts are the reason for this asymmetry. Surface species of transition alumina's can be considered as highly dynamic. Proton motion on the surface (from chemisorbed or physisorbed water) takes place at great speeds compared to bulk diffusion. This fast re-orientation of the protons randomly creates and annihilates large electric field gradients on the surface, resulting in an efficient quadrupole relaxation.3.1. Early transition aluminas and tohdite with H/Al ≥ 0.2
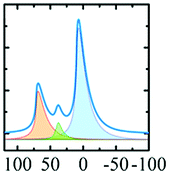
In vacuo calcination of gibbsite at 200 °C generates a highly amorphous and disperse phase called ρ-alumina. It has rarely been characterized owing to its amorphous nature according to X-ray diffraction62 and its high sensitivity towards moisture. Rho-alumina often is referred to as hydratable alumina, since contact with water converts it to a mixture of hydroxides and oxyhydroxides. In construction and building industries rho-alumina is a common additive for cements and as bonding agent, also known as Alphabond.85 Phenomenologically it may be described as the borderline case between a disordered oxyhydroxo compound and early transition alumina, occurring in a broad range of H/Al ratios roughly between 0.6–0.2. The 27Al MAS NMR spectrum reveals three types of overlapping resonances corresponding to different Al coordinations (AlO6, AlO5 and AlO4).7,62 The disorder typical for ρ-alumina can clearly be seen from the large distribution of quadrupole couplings (see Fig. 10). The intensities of the AlO4, AlO5 and AlO6 resonances are approximately 25
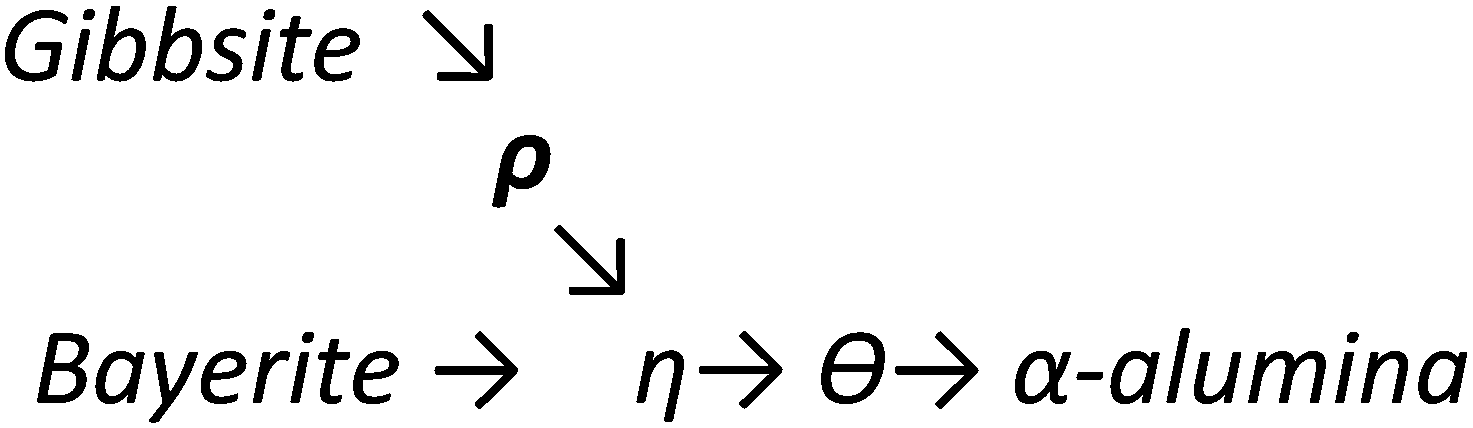 :20:55, respectively (Fig. 10). The corresponding isotropic chemical shifts occur around 68, 34 and 9.8 ppm.
:20:55, respectively (Fig. 10). The corresponding isotropic chemical shifts occur around 68, 34 and 9.8 ppm.
 | ||
| Fig. 10 27Al MAS spectra (and decomposition: grey) of ρ-alumina simulated using the NMR parameters from ref. 7. | ||
Meinhold et al.7 proposed a model with a random distribution of EFG's to fit the experimental line shapes exhibiting unusually long tailings, typical for disordered alumina phases. However, a detailed analysis with a Gaussian isotropic model with Czjzek distribution is essential for the proper analysis of such spectra.48 Slade et al. and Meinhold et al.,7,62 did not carry out the extraction of the mean quadrupole couplings of ρ-alumina, except for those of the AlO6 site (4.8 MHz). Rho-alumina obtained from gibbsite has been reported to convert to eta-alumina upon heating in air, which then follows the bayerite path.3
Amorphous alumina3 thin films produced by atomic layer deposition often exhibit 27Al NMR spectra86–92 similar to that of ρ-alumina, but with a population of five-coordinated Al varying with the layer thickness. If the layer thickness is of the order of a few nanometers, the 27Al NMR spectra resemble ρ-alumina. For sample-thickness of the order of tens of nanometers, the AlO5 population dominates.90 The relative occupancy of different Al–O sites in films has been reported to evolve with temperature92,93 similar as in the gibbsite series, finally reaching α-alumina.90
Tohdite cannot be classified as transition alumina, as it can only be synthesised hydrothermally72 or found in nature as the mineral akdalaite.94 Still it will be briefly discussed in this section as it displays the typical H/Al ratio of 0.2 associated with the early transition alumina and because it transforms into kappa-alumina before alpha alumina is formed, similar as observed for chi-alumina (vide infra). Tohdite, also known as akdalaite is well crystalline, shows a defined Altet/Alhex ratio of 0.25 and displays the same specific stacking of oxygen planes (ABCB…) encountered in kappa alumina. Though lacking disorder in site occupation and having different order of oxygen planes as the spinel-based transition aluminas eta and gamma, it also consists of alternating layers of octahedral and mixed octahedral/tetrahedral aluminium ions. No 27Al study of tohdite is known.

Chi alumina is the first member of the gibbsite calcination series. It forms between 250 and 650 °C and spans a range of H/Al values between 0.9 and 0.1. Despite several efforts since the 1950's,1,95–99 the crystal structure of χ-alumina remains uncertain. This poorly characterized transition alumina eluded most crystallographic studies. It has a very diffuse powder X-ray diffraction pattern with few broad lines, and appears to have stacking faults or fully disordered domains. One cubic1 and two hexagonal98 structures have been proposed. Recently, a study combining ab initio calculations, IR and X-ray diffraction99 suggested the probability of an average hexagonal structure. Also high resolution TEM analysis supports a hexagonal arrangement.100 However, all authors agree the oxygen sub-lattice is best described as a dense packing of oxygen planes with either a fully random stacking sequence one that is ordered only locally. Therein, tetrahedral and octahedral Al sites are both considered to be disordered, and typically in a ratio of roughly 0.3. Dimensions of one of the suggested hexagonal unit cells closely resembles tohdite and kappa-alumina, both with layer sequence ABCB… (Fig. 11).

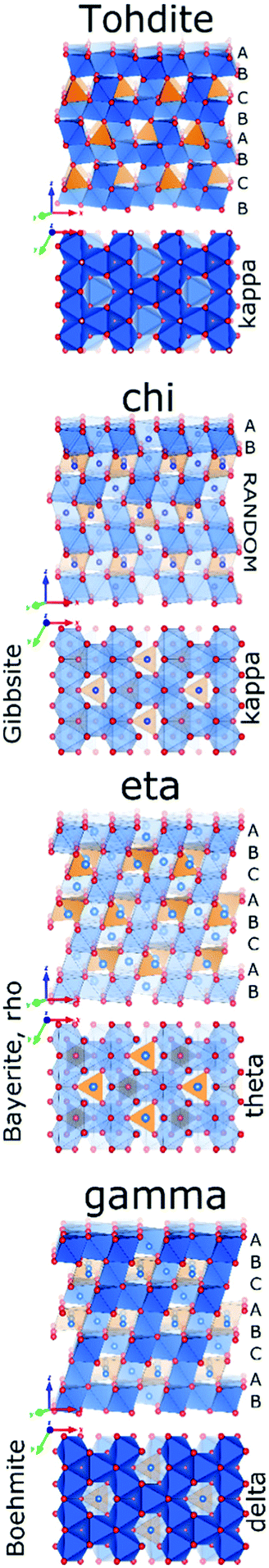 | ||
| Fig. 11 Structure comparison of tohdite, chi, eta and gamma alumina. | ||
Slade et al.79 reported the 27Al MAS spectrum of chi-alumina prepared by calcination of gibbsite. They observed two main resonances (peak maxima at 5.7 and 63.8 ppm), corresponding to the hexa- and the tetra-coordinated Al sites. In addition, there was a small shoulder around 40 ppm, the region where five-coordinated Al is expected. Consequently this peak could be representing species at stacking faults. The high-resolution 27Al NMR spectrum of a χ-alumina phase was reported later, along with line-shape simulations using distributions in quadrupole parameters (Fig. 12).8 The isotropic chemical shifts of AlO6, AlO5 and AlO4 resonances were 11.5, 38.0 and 71.5 ppm, with intensity ratios of 70.0, 7.5 and 22.5 percent respectively. The mean of the distributions of quadrupole couplings are 4.5, 2.7 and 5.0 MHz, respectively. All three resonances were simulated with an ηQ of 0.3, although small differences in ηQ did not affect the simulated line shape significantly.
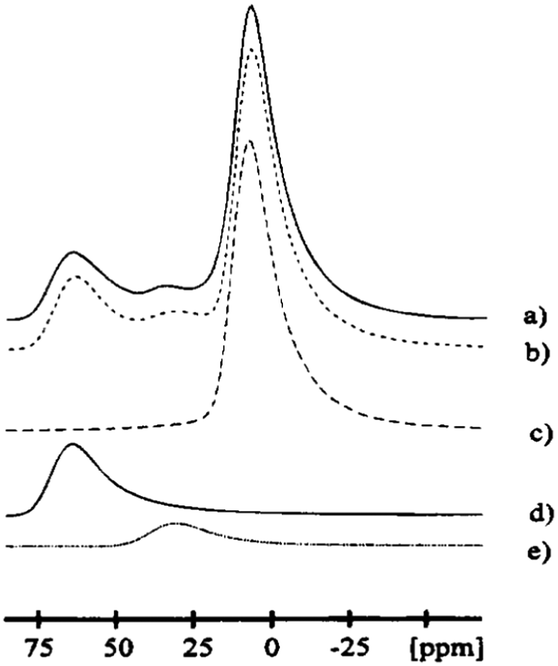 | ||
| Fig. 12 27Al MAS NMR centre-band (right) of eta alumina showing (a) the experimental data, (b) complete simulation and the individual components (c) AlO6, (d) AlO4, and (e) AlO5. Reproduced from ref. 91 with permission from ACS, copyright 2018. | ||
3.2. Intermediate transition aluminas
Though derived from different precursors, eta and gamma alumina show some similarities,2 and even have been considered identical at some point.101 Both are based on an anionic sublattice of ABC… sequence, and both display a tetragonal distortion with respect to the ideal cubic fcc symmetry. Both also show disorder in the cation occupation, though with differing emphasis on tetrahedral and octahedral sites. This results in diffraction patterns, which for both are poorly resolved, and quite similar. Nonetheless, both structures are best described by defective spinel phases with vacancies owing to their stoichiometry,102 and both may contain additional protons up to a H/Al ratio of 0.2. The archetype spinel with formula MgAl2O4 contains hexa-coordinated Al3+ and tetra-coordinated Mg2+ in such a way as to minimise electrostatic repulsion. This results in layers of edge-sharing Al octahedra, which are connected to layers of mixed octahedra, sharing edges with upper or lower layer, and tetrahedra, which strictly are corner-sharing. In eta and gamma alumina, tetrahedral and octahedral sites are both occupied by aluminium, which results in a defective occupation owing to stoichiometry. For both phases, some occupation of nominally ‘forbidden’ sites is observed, which also may arise from the lower occupation of cation sites or from local twinning, i.e. local disorder in the ideal ABC stacking of oxygen planes. Owing to the similarity of the phases, it is extremely difficult to distinguish them based on diffraction, unless the history of the material is known.Calcination of bayerite around 300 °C produces η-alumina. On calcination, η-alumina transforms to θ-alumina, before restructuring to alpha. In the case of η-alumina, the occupation of the four-coordinated positions is fairly high, with a Altet/Alhex ratio of maximally 0.6. In other words, eta-alumina can be described by an idealised spinel formula of Altet□1/3Alhex5/3O4, with □ indicating vacancies on octahedral sites. The higher number of tetrahedral aluminium also has been postulated to explain the higher Lewis acidity compared to gamma alumina,103 an important feature of η-alumina for catalytic applications.103–105

27Al NMR reports on pure-phase η-alumina are scarce, though almost all 27Al NMR studies of the bayerite transition series report the η-alumina phase, which exists over a fairly large temperature range (300–600 °C). One of the initial reports was from John et al.,2 in which the authors determined a higher Al(IV) concentration in η-alumina as compared to γ-alumina. Pecharroman et al.71 observed hexa- and tetra-coordinated Al resonances with isotropic chemical shifts of around 15 and 77 ppm, respectively. The corresponding mean CQ values were 3.9 and 4.6 MHz, with a higher ηQ for AlO4 (0.7) than for AlO6 (0.4). Chagas et al.69 reported the occurrence of the η-phase co-existing with θ-alumina in an even larger temperature range (300–1000 °C) when slow heating rates are applied. In this case, the intensity of the tetra-coordinated Al resonance continuously increased from 300 to 700 °C and then remained almost constant. The same authors also confirmed the often observed co-existence of eta and gamma aluminas as a result of the possibility of formation of boehmite from bayerite and the consecutive formation of gamma alumina next to the eta phase formed directly from unconverted bayerite.
Gamma alumina is formed by thermal dehydroxylation of boehmite between 500–750 °C and then transforms to the delta phase with increased cation ordering. Gamma is often employed in heterogeneous catalysis and like many other aluminas, its catalytic activity is affected by the crystallite size. The structure of gamma alumina (γ-alumina), the most investigated transition alumina phase, was already characterized decades ago.106,107 Like eta alumina, γ-alumina crystallizes as (non-stoichiometric) spinel with some degree of disorder and may contain protons up to a ratio of H/Al of 0.2. Despite its extensive characterization, there is still some debate on the details of the structure, but most authors agree the defect sites in gamma alumina are mostly found on tetrahedral sites, which leads to an idealised spinel formula of Altet2/3□1/3Alhex2O4, with □ symbolising vacancies on tetrahedral sites. Like for eta-alumina, a tetragonal distortion to space group I41/amd from the ideal cubic fcc packing of the anion sublattice is reported.108–111 In theoretical studies, the difficulty to account for defect sites has occasionally been overcome, by simulating orthorhombic structures with full order on the cation sites.112–114Ab initio27Al NMR calculations found a close match using the spinel-type structure with the space group Fd3m115 and Al distributed in different sites as follows: Al(VI) 64%, Al(IV) 34.4% and Al(V) 1.6%. Currently the most widely accepted description of gamma alumina consists of a spinel model with partial occupation of non-spinel positions and considerable disorder next to presence of surface hydroxyl groups.4
Like most transition aluminas, the surface of γ-alumina is hydroxylated to a large extent, as can be observed with 1H MAS NMR.14,56 A combined theoretical and NMR study revealed that the chemical shift and quadrupole coupling of 27Al in γ-alumina strongly depend on the extent of surface hydroxylation.116 Unlike the related eta-alumina, gamma alumina frequently shows the presence of five-fold coordinated aluminium. In a 27Al NMR study, Slade et al.79 proposed the presence of the penta-coordinated species as reason for the crystallographic distortion in gamma alumina. However, usually Al(V) species are assumed to arise from surface species. Disorder and related presence of ‘non-spinel’ species are considered to provide only a negligible number of five-coordinated Al. This is supported by the observation that mechanical milling, diminishing the particle size, can increase the fraction of 5-coordinated Al up to 20%.117 From this perspective, Al(V) content can be used as a measure of activated surface area or, in other words, a measure for particle size. The 5-coordinated Al was suggested to be responsible for the thermal instability of γ-alumina.118
γ-Alumina can be also produced from the calcination of pseudoboehmite.77 In this case, the material displays higher surface area and a higher fraction of five-coordinated Al. Mechanical grinding of γ-alumina also facilitates its thermal transformation to α-alumina. While α-alumina was formed at 1000 °C from ground γ-alumina, 1200 °C was necessary for the unground sample (Fig. 13).78 High-energy mechanical milling alone is enough to convert γ-alumina to α-alumina, as shown in the 27Al NMR study of Düvel et al.117,119
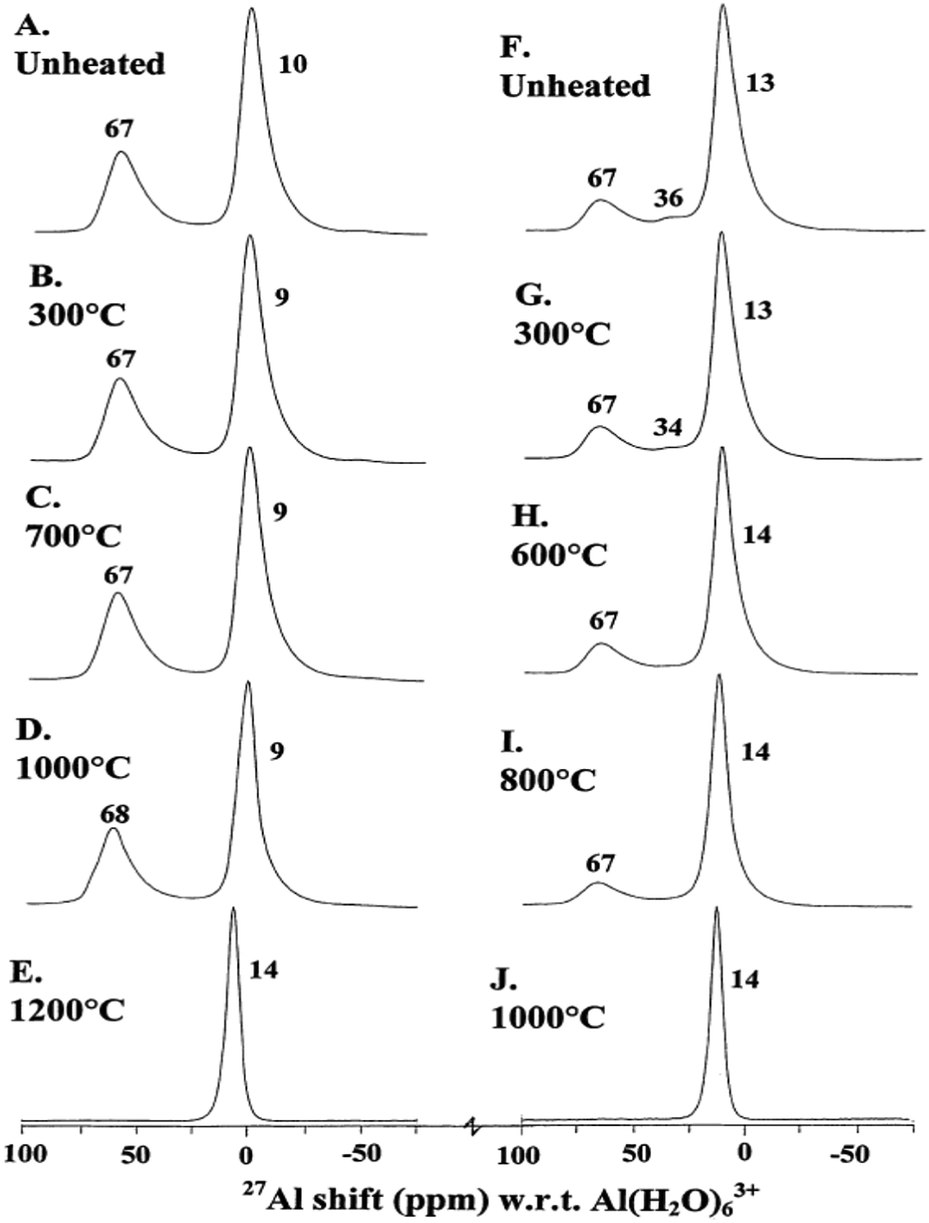 | ||
| Fig. 13 Typical 14.1 T 27Al MAS NMR spectra of unground and ground γ-Al2O3 unheated and heated for 15 min as indicated. (A–E) Unground; (F–J) ground. Reproduced from ref. 67 with permission from Elsevier, copyright 2018. | ||
As evidenced by the above mentioned studies, the detailed history of a given γ-alumina strongly impact the relative populations of its Al-sites and in turn also its properties like surface area, Lewis acidity, and transformation behaviour. For example, an alumina species “Super5” known for its activity and acidity exhibits chemical shifts similar to γ-alumina, but has a very high percentage (∼35%) of Al(V).120,121 A recent paper on the 27Al NMR of highly porous γ-alumina, however, did not observe any significant amount of Al(V).110 Tungsten oxide (WOx) impregnated γ-alumina lacks the penta-coordinated Al sites immediately on increasing the loading. Most probably anchoring of WOx on the surface of γ-alumina occurs preferentially at the penta-coordinated surface sites. In addition, multiplicities of both hexa- and tetra-coordinated Al sites have been observed as a function of WOx concentration, as has been proven by 27Al MAS and 3QMAS experiments.122 In short, in the case of γ-alumina, a regular ratio of Al populations at different Al–O coordination cannot be predicted as it depends too heavily on the synthesis conditions.
Gamma alumina is the most studied transition alumina. So it remains unclear if the manifestation of so varying properties is typical for gamma alumina, or if the other, less studied early transition aluminas like eta and chi show similar diversity.
3.3. Late transition aluminas with H/Al < 0.1
Progressing the calcination to higher temperatures, the X-ray diffraction signatures observed along all different pathways towards alpha alumina show increasing resolution. While the early transition aluminas all show disorder in Al distribution and in the case of chi-alumina also in the oxygen sublattice, the late transition aluminas all can be satisfactorily described by ordered structure models. All late transition aluminas have an increased amount of tetrahedral Al-species compared to their early transition counterparts, before the Altet/Alhex decreases again to reach zero during final reconstruction to the alpha phase (Fig. 14). | ||
| Fig. 14 Structure comparison of kappa, delta and theta alumina. | ||
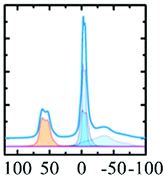
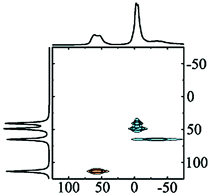
Kappa alumina is obtained by thermal treatment of χ-alumina or of tohdite and is observed in a temperature range between 650 and 1000 °C. With tohdite, kappa alumina shares the same anionic sublattice with ABCB… sequence of oxygen planes, but with different cation distributions. Ollivier et al. determined the crystal structure with X-ray diffraction, electron microscopy and 27Al NMR,123 and described it in the orthorhombic space group Pna21.123,124 Four non-equivalent Al and six non-equivalent O atoms construct the unit cell. Of the four Al-sites, one is in tetrahedral coordination (Al3), the other three (Al1, Al2, Al4) reside in distorted octahedral environment, which arises from slight vertical displacement of oxygen in the anion sublattice. In the case of kappa alumina, Al1 is quite regular, while Al2 and Al4 show severe distortion. Based on the shortest bond length in each octahedron, this results in nominal coordination numbers of roughly 6 for Al1, 5 + 1 for Al4, and 4 + 2 for Al2. All three sites have been resolved by MAS and 3QMAS NMR (Fig. 15).123 Compared to other transition alumina phases, reported CQ values for κ-alumina appear very small, possibly owing to discrepancies in reporting quadrupole parameters in ref. 123. However, if the reported values were quadrupole frequencies (ηQ) instead of CQ's, then they are in agreement with the ab initio calculations reported in ref. 115. While Al1 and Al4 showed CQ values of 3.33 and 5.67 MHz, respectively, the Al2 site was estimated to exhibit values higher than 6 but less than 10 MHz. The tetra-coordinated Al resonance was analysed with a CQ of 5.07 MHz and an ηQ of 0.3. In a combined XRD, XPS and NMR study, Malki et al.9 have estimated the ratio of Altet/Alhex in κ-alumina to be 27%, which is below the ideal ratio according to the proposed structure (33%). The same authors also pointed out that XPS analysis of kappa as well as chi alumina revealed a nonstoichiometric excess in Al versus oxygen and related both observations to the large surface area of the highly dispersed materials.
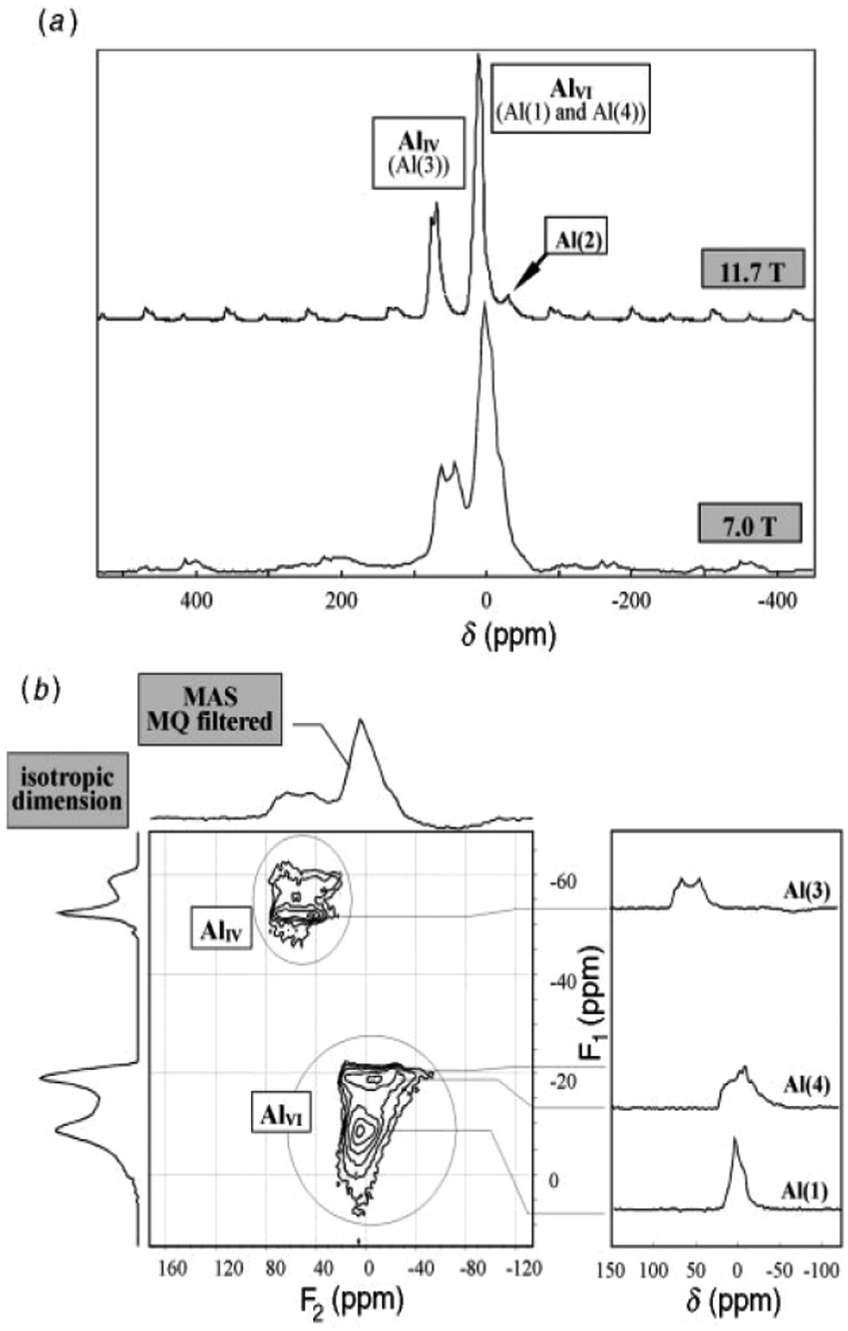 | ||
| Fig. 15 27Al NMR spectra of κ-Al2O3: (a) MAS spectra at 7.0 and 11.7 T; (b) 3Q MQMAS spectra at 7.0 T. The very distorted site Al(2) is only visible at the higher field in MAS while the Al(1), Al(3) and Al(4) sites are fully resolved in the MQMAS spectrum. Reproduced from ref. 114 with permission from RSC, copyright 2018. | ||
In a similar temperature range where chi alumina turns into the more ordered kappa form, also the diffraction pattern of gamma alumina sharpens, indicating increasing regularity. This new phase, called delta-alumina, emerges around 800 °C before further transforming to the theta phase around 950 °C.107,125 In delta alumina, the ABC… stacking of oxygen planes already present in gamma alumina is retained. The cations show significantly higher degree of order compared to its gamma predecessor. All structure models for delta alumina are based on superstructures of the spinel type. Originally a tetragonal structure was proposed in 1961,126 but nowadays intergrowths of at least two structurally related (pseudo)orthorhombic forms, are accepted as good description of delta alumina.127 A recent study combining XRD, TEM and DFT calculations used two representative models, described in space groups P212121 (delta1) and P21 (delta2; pseudo-orthorhombic). Similar as in gamma alumina, both structures show layers of edge-sharing octahedra, alternating with mixed hexa/tetra-coordinated layers in a densely packed oxygen sublattice with ABC… sequence. Occupation of cation sites is explicit within the ordered domains and without vacancies. Combination of both structure models described the TEM observation well and suggested a Altet/Alhex ratio as high as 0.6, which is in rough agreement with experimental data. The study also demonstrated the enthalpy of formation of these variants are very similar to θ-alumina, which explains the common observation of delta next to the theta phase in the boehmite calcination series.127
There are only a few 27Al NMR spectra of pure δ-alumina available in literature. MacKenzie et al.78 followed the formation of δ-alumina (1000 °C) in the boehmite series. They observed a clear δ-alumina XRD pattern and reported two 27Al MAS NMR resonances, corresponding to hexa- and tetra-coordinated Al, with intensity maxima at 8 and 68 ppm, respectively. The corresponding Al population ratio was estimated to be approximately 60:30. Similar NMR characteristics were displayed by δ-alumina produced by directly heating γ-alumina at 1000 °C. However, the δ-alumina phase according to XRD was completely absent when ground (1 h) boehmite or γ-alumina was calcined,78 although this may have been due to an extremely small particle size.7827Al MAS NMR spectra of a gel of hydrolysed Al(OPh)3 fired at 900 °C was interpreted as δ-alumina, based on the high content of Al tetrahedra.128 Pecharroman et al.71 also observed the high 27Al tetra/hexa coordination intensity ratio upon calcination of boehmite calcination at 950 °C, and estimated the corresponding isotropic chemical shifts (14, 77 ppm), CQ's (5.1, 3.3 MHz) and ηQ's (0.7, 0.4). The relative Al populations in the reported δ-alumina spectra seem to be consistent, although the unambiguous synthesis appears to be dependent on optimum crystallite sizes of the precursor. A recent paper reported 27Al 3QMAS NMR experiments (Fig. 16) on nano-crystalline alumina which has mixed γ-, δ- and θ-phases. In combination with calculated shift predictions and 1D line-shape decompositions, the 3QMAS NMR experiments helped to resolve the different species evolved at different temperatures, for varying crystallite sizes. In the mixture, however, the transformation to α-alumina did not occur at 1100–1200 °C, but took place later, at 1300 °C.129
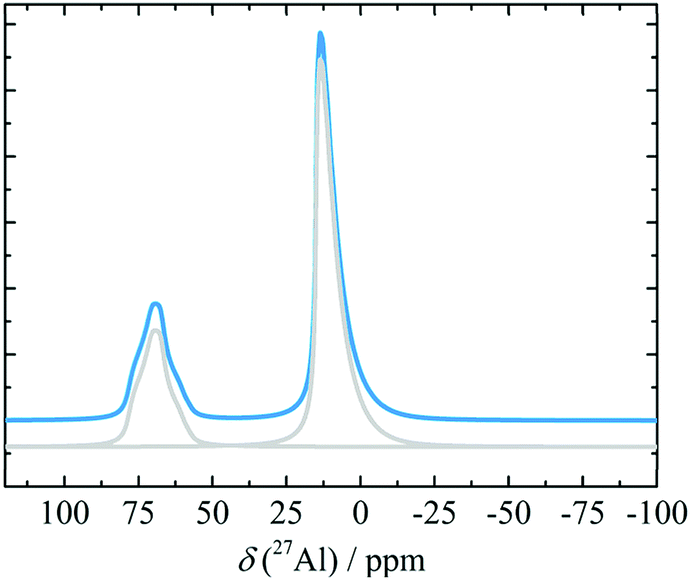 | ||
| Fig. 16 Simulated 27Al NMR spectrum (and decomposition: grey) of δ-alumina using the NMR parameters from ref. 129. | ||
Theta alumina is the common late transition alumina in the series starting from bayerite and boehmite. In the former, it arises from eta, in the latter from delta alumina, in temperature ranges of 800–100 °C and 900–1000 °C, respectively. Like its precursors, it shows ABC… ordering of the oxygen planes and it clearly follows the trend of increasing order with increasing temperature, observed for all calcination pathways. Owing to its high crystallinity and clear X-ray diffraction pattern, its structure was reported as early as 1957130 and found isostructural to beta-Ga2O3. Theta crystallises in monoclinic symmetry with space group C2/m, and contains alternating layers of hexa- and tetra-coordinated Al in a ratio Altet/Alhex = 1.

Syntheses of theta-alumina from bayerite or boehmite both rarely yield pure-phase θ-alumina. Similar enthalpies of delta alumina make it a common side-phase at temperatures up to 950 °C and small amounts of α-alumina phase may be present as an impurity already below 1000 °C. Slade et al. observed this effect with 27Al MAS NMR.79 Overlap between six-coordinated Al resonances from α- and θ-alumina, prevented confirmation of the expected equality of Al(IV) and Al(VI) content without detailed spectral decomposition. In the bayerite series, however, the θ-alumina-phase starts to form already at lower temperatures (900–1000 °C). Consequently, bayerite-derived θ-alumina provides a higher chance to allow observation of its 1:1 distribution of AlO6 and AlO4 with NMR. This effect can be seen in the 27Al NMR spectra of the transition alumina series reported by Pecharroman et al.71 The decomposition and the quantification of the θ-alumina spectra at different magnetic fields were carried out by O'Dell et al. (Fig. 17).131CQ values obtained from the deconvolution were 6.4 and 3.5 MHz for AlO4 and AlO6 species, respectively, while the corresponding ηQ values were 0.65 and 0.00. Isotropic shifts were 80 and 10.5 ppm for AlO4 and AlO6, respectively. These parameters are in very good agreement with the parameters obtained from ab initio calculations.115
 | ||
| Fig. 17 27Al MAS NMR simulations of the sol–gel prepared alumina sample annealed at 1200 °C recorded at (a) 8.45 T and (b) 14.1 T. The top lines show the experimentally obtained spectra, the middle lines are the simulated spectra, and the bottom lines show the individual simulated peaks. Reproduced from ref. 123 with permission from Elsevier, copyright 2018. | ||
4. Alpha-alumina
The final outcome of a calcination of any precursor consisting of Al, O, and H beyond 1350 °C is α-alumina. Alpha-alumina has the same structure as naturally occurring corundum. Many precious stones like rubies or sapphires exhibiting bright colours are corundum single crystals with isomorphic substitution of Al with Fe or Cr. Synthetic single crystals of corundum are used as substrates for epitaxial growth of superconductors and semiconductors. The microcrystalline alpha-alumina obtained by calcination of aluminium (oxy)hydroxides is widely used as catalyst supports, porcelain, advanced ceramics manufacture, refractories and abrasives and also as source for aluminium metal.132Alpha alumina is the most stable Al2O3 phase and its structure has first been reported by Linus Pauling in 1925.133 The structure differs from phases discussed above, in the presence of face-sharing octahedra and shows the highest symmetry in space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c, with one Al and one O site. The structure can be described as being composed of layers of octahedra, similar as encountered in the Al(OH)3 phases, but stacked in such a way that alternating pairs of face sharing octahedra are formed where the layers are merged. The oxygen sublattice follows hcp packing ABAB (Fig. 18). This implies that upon transformation of late transition aluminas derived from the gibbsite, bayerite, and boehmite series, i.e. kappa, and theta alumina, the materials not only have to change cation arrangement but also need to re-construct the anion sublattice. Transformation of these occurs at quite high temperatures usually above 1000 °C. Calcined diaspore, which shares the same oxygen arrangement as alpha alumina has been observed to already show the presence of alpha alumina at temperatures as low as 600 °C.84
c, with one Al and one O site. The structure can be described as being composed of layers of octahedra, similar as encountered in the Al(OH)3 phases, but stacked in such a way that alternating pairs of face sharing octahedra are formed where the layers are merged. The oxygen sublattice follows hcp packing ABAB (Fig. 18). This implies that upon transformation of late transition aluminas derived from the gibbsite, bayerite, and boehmite series, i.e. kappa, and theta alumina, the materials not only have to change cation arrangement but also need to re-construct the anion sublattice. Transformation of these occurs at quite high temperatures usually above 1000 °C. Calcined diaspore, which shares the same oxygen arrangement as alpha alumina has been observed to already show the presence of alpha alumina at temperatures as low as 600 °C.84
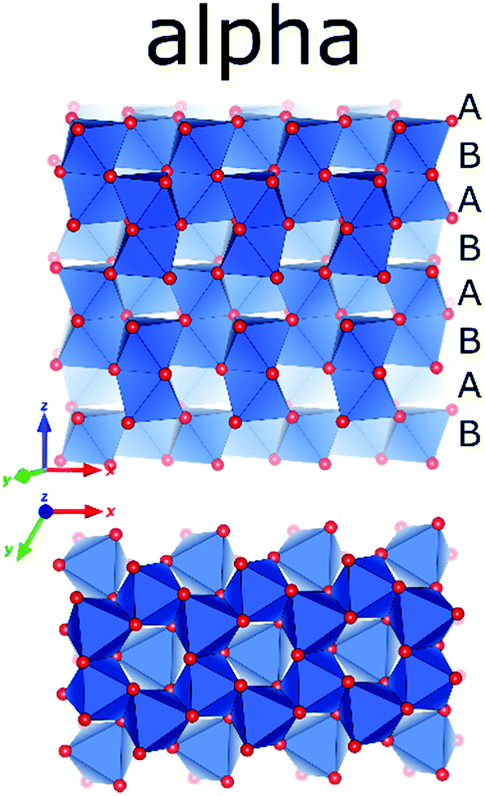 | ||
| Fig. 18 The crystal structure of α-alumina (Al in blue polygons and O in red).156 | ||
As the crystal structure contains a single crystallographic Al site, α-alumina is expected to have a single resonance in its 27Al NMR spectrum. The first 27Al NMR spectrum of α-alumina was reported by Pound,134 who deduced a CQ of 2.39 MHz from single crystal NMR investigation. Huggins and Ellis put forward the structural models for the surface of several alumina materials including α-alumina in a 27Al static NMR study. However, one of the first reports on quantitative 27Al MAS NMR spectroscopy of α-Al2O3 together with line shape simulations was from Jakobsen et al.135 They observed the central (m+1/2 ↔ m−1/2) transition and the satellite transitions (m±3/2 ↔ m±1/2 and m±5/2 ↔ m±3/2) corresponding to a single octahedral Al-site. Accurate estimations of CQ (2.38 MHz) and ηQ (0.00) were carried out for a rather symmetric octahedral environment. Even though a broadening due to the second-order quadrupole interaction was proposed, the central transition (CT) resonance was fairly featureless, lacking the characteristic pattern for an axially symmetric system. An isotropic chemical shift of 12.7 ppm was determined after considering the quadrupole-induced shift correction. These values were later confirmed by Skibsted et al.136 in a study mainly focused on the simulation of the satellite transition (ST) manifold in the 27Al spectrum of α-alumina (Fig. 19).
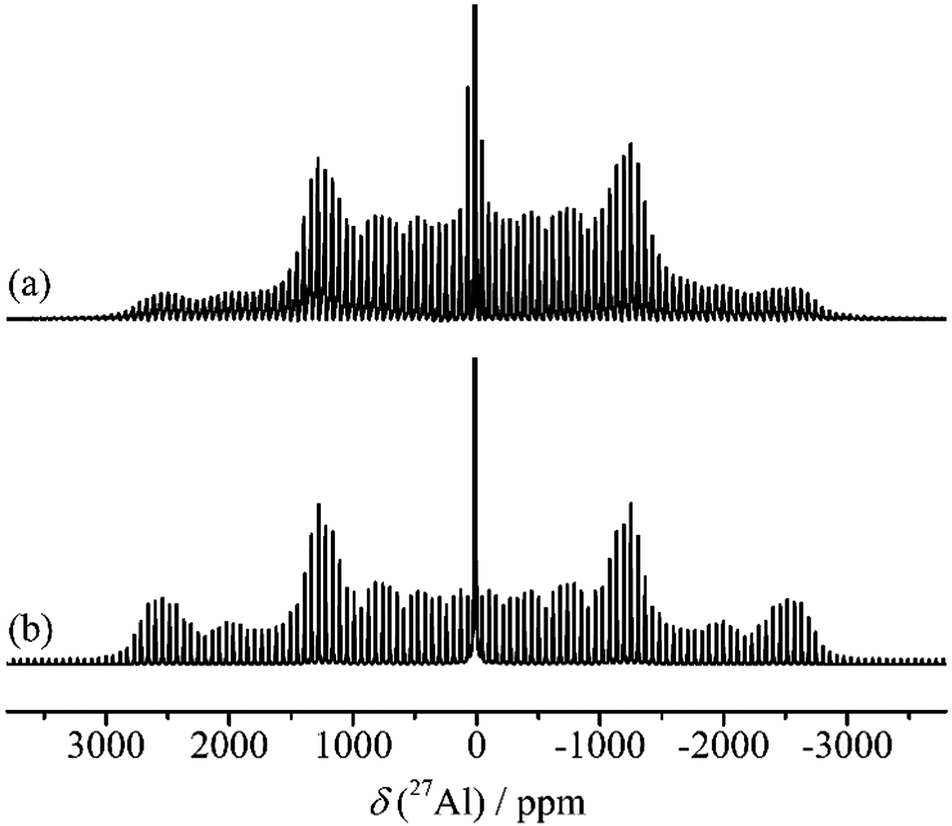 | ||
| Fig. 19 27Al (130.3 MHz) MAS NMR spectra of α-alumina, νr = 7500 Hz. (a) Experimental spectrum. (b) Simulated spectrum using the experimental parameters CQ = 2.38 MHz and η = 0.00. | ||
The 27Al spin–lattice relaxation (SLR) behaviour of α-alumina and the quantitative excitation using radio-frequency (RF) pulses were carefully studied by Kentgens et al.137 Crushed synthetic single crystal samples showed extremely long SLR times compared to natural corundum, which might contain paramagnetic impurities. To measure accurate SLR times, an adiabatic passage was realized by frequency stepping, ensuring proper inversion of the CT and ST magnetization. The influence of the length of the RF pulses on the selective excitation of CT or ST was also discussed. Most literature mentioned in this section showed some disagreements on the 27Al isotopic chemical shift of α-alumina, mostly due to the difficulty to correct for quadrupole-induced shifts. However, by now it has been generally accepted to be in the range of 13.5–16 ppm. Mechanical crystallite size reduction in α-alumina was reported to increase distributions in electric field gradients with the help of 27Al MAS and 3QMAS NMR experiments. Upon size reduction from 52 nm to 12 nm, an up-field shift of about 4 ppm was observed at the peak maximum, but no additional Al coordination beside the octahedral one could be detected.138
5. Tabulation and correlation of NMR parameters
The structural transformation in transition alumina series occurs with severe reorganization of atoms driven by the thermal dehydroxylation process. The most stable alumina phase, alpha has a hexagonal-rhombohedral structure. The phases gamma and eta have similar (cubic disordered spinel) structures. Other stable alumina or precursor phases are either monoclinic or orthorhombic. The rest of the phases are with either amorphous or with disputed structures (Fig. 20). | ||
| Fig. 20 Tree plot of alumina structural transformations and associated changes in the NMR spectra from the hydroxide and oxyhydroxide precursors to α-alumina phase. The monoclinic and orthorhombic alumina species were identified relatively easily, while the others have either a disordered structure or an amorphous nature. | ||
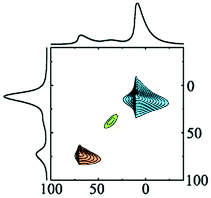
The experimentally determined NMR parameters of aluminium hydroxides and oxyhydroxides and transition alumina phases reported in most recent and reliable literature are collected in Table 2. Some phases possess site multiplicity with same coordination number, as explained in the presentation of the different phases above. Moreover, there is a spread of these NMR parameters due to the physical conditions of the investigated samples (particle size, humidity, pH etc.). Ellipsoid graphs presented in Fig. 21 are more suited to include the distributions in reported 27Al NMR parameters of transition alumina phases.
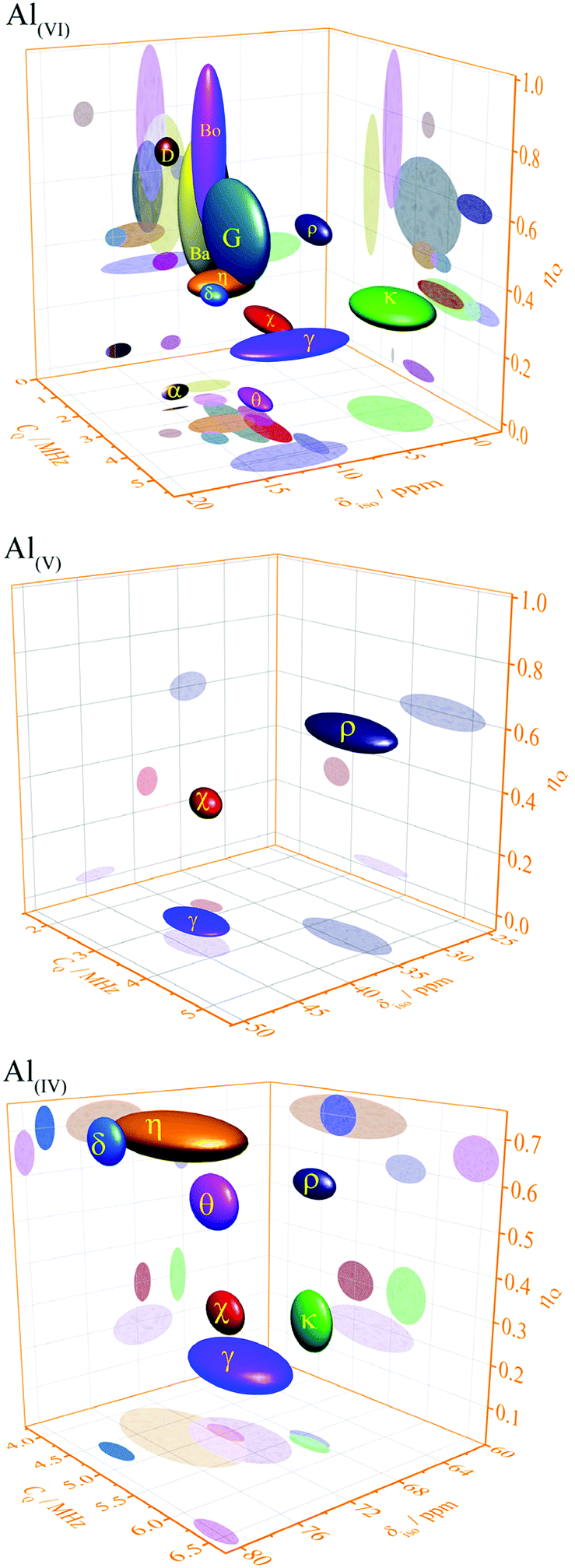 | ||
| Fig. 21 Plots of 27Al NMR parameters (isotropic chemical shift, quadrupole coupling constant and asymmetry parameter) of tetra-, penta- and the hexa-Al–O coordinations in transition alumina phases (α, χ, κ, θ, γ, δ, η, ρ) and their precursors (bayerite (Ba), boehmite (Bo) and gibbsite (G) and diaspore (D)).158 2D projections of all graphs are available in the ESI† (Fig. S1–S3). The phase κ also has an additional highly distorted V + 1-coordinated site with a high quadrupole coupling of around 10 MHz (see ESI,† Fig. S4). | ||
The three-dimensional ellipsoid plots (Fig. 21) correlate the most important NMR parameters of 27Al NMR of alumina, namely the isotropic chemical shift (δiso), quadrupole coupling constant (CQ) and the asymmetry parameter (ηQ). All the three Al–O coordination types (tetra-, penta- and hexa-) were considered for these graphs. These 3D-plots provide important information, which eventually leads to easy identification of an alumina phase under analysis.
The following strategy is proposed to use the 3D ellipsoid plots for identifying an alumina phase. All transition alumina phases have tetra-coordinated Al. The left plot in Fig. 21 provides the NMR parameter correlations of the tetra-coordinated Al. As the first step, one may explore one parameter at a time, like chemical shifts alone. Additional screening through quadrupole coupling constants and asymmetry parameters gives clear indication of the phase. These parameters are useful to add extra-resolution to the 1D 27Al spectrum under analysis. The 2D-projections of the 3-D figures (see ESI†) will be helpful in detailed investigations. The same strategy can also be applied to the hexa-coordinated Al and further on to penta-coordinated Al. The Al(OH)3 and AlOOH precursors have large distributions of NMR parameters of hexa-coordinated Al. Gibbsite and bayerite have two hexa-coordinated Al sites each, adding to the spread of the ellipsoid. Similarly, reports on the boehmite quadrupole parameters show distributions. Transition alumina phases show a smaller spread of NMR parameters of hexa-coordinated Al. None of them have tetra-coordinated Al. Therefore, one can concentrate entirely on the Al(VI) plot for the phase identification of transition aluminas, which show high-resolution in the 3D-representation. An exception is κ-alumina, with multiple AlO6 sites. However, the lower chemical shifts of these sites separate it from the other alumina phases. Only three alumina phases show five-coordinated Al, namely γ, χ and ρ-alumina. The observation of this Al species in the alumina under analysis brings in an additional advantage in the identification process.
This 3D-ellipsoid representation of NMR parameters is a much more powerful tool for phase identification as compared to diffraction methods, especially for complex and rather X-ray amorphous transition alumina phases like ρ, χ or η-alumina. All those phases are sufficiently resolved in the correlation plots of NMR parameters. The three ellipsoid plots in Fig. 21 introduce significant resolution to the NMR data to enable unique identification. This makes the proposed method equivalent to other application fields of NMR spectroscopy for analytical purposes, like high-throughput 1H or 13C NMR spectroscopy of organic compounds where signature peaks are uniquely assigned to chemical functions.
6. Quantum chemical calculations of NMR parameters of alumina
Although 27Al NMR is highly sensitive, elucidating chemical and physical nature of the local crystal structures of aluminium-containing solids, requires an updated state of the art of coupling quantum calculations to NMR data. NMR data are average over the sample in time and space, with different time windows than diffraction methods. Therefore modelling can assess the plausibility of a structural set of models by producing computed NMR observables as a way to validate the most probable model. By considering both the quality of the agreement and the number of parameters required to reach such an agreement, a Bayesian probability can be attached to the model selection. A deeper understanding of the system can be obtained from this additional input from quantum structure calculations. The recent developments in first-principles calculations of NMR parameters are found to be adequately complementing NMR experimental data. The chemical shielding and quadrupole interaction parameters derived from such calculations establish the unique relationship between the NMR data, quantum chemistry and structural environment. Density functional theory (DFT) remains as the basis for shielding and EFG tensor calculations for solids. A DFT based algorithm known as gauge-including projector augmented wave (GIPAW)140 is one of the most successful methods to predict NMR parameters. There are several examples in literature which use DFT-GIPAW with the generalization of plane wave basis sets and pseudopotentials to calculate 27Al NMR parameters and simulate NMR spectra.141–144 This approach is highly useful for exploring the NMR characterization of transition alumina systems, unlike aluminosilicates where periodic molecular origin of NMR parameters make more sense, starting from different alumino siloxy cluster units.145 However, studies comprising quantum chemical NMR calculations on all alumina phases are still missing in literature.Force field calculations are also employed to obtain NMR parameters.146 Mostly they are used for molecular systems, along with molecular modelling, predicting potential energy landscapes in molecular dynamics (MD) simulations. NMR chemical shift of bonded systems is the parameter which is usually estimated in such exercises. However, a modification known as Clay force field (ClayFF) has been developed to extend the applications to non-bonded systems, like in binding molecules (like water) or ions.147 Using ClayFF calculations, largely hydrogen-bonded systems like gibbsite and boehmite were studied with MD simulations.148,149 But the extraction of NMR parameters is still preferred with the help of DFT calculations. Nowadays, different DFT methods are implemented in popular software packages such as WIEN2K150 and CASTEP151 which yield extremely accurate NMR chemical shielding and quadrupole interaction parameters.
One of the initial investigations was from Choi et al.152 exploring α-alumina and θ-alumina structures and predicting NMR shielding and EFG tensors using ab initio calculations. The calculations for the α-alumina model obtained 27Al δiso of 16.66 ppm, CQ of 2.09 MHz and ηQ of 0.00. For θ-alumina, the four and six coordinated Al sites were predicted to have δiso of 79.46 and 11.3 ppm, CQ of 6.03 and 3.15 MHz and ηQ of 0.37 and 0.23, respectively. These values were close to the experimental NMR parameters. Also, gibbsite and bayerite structures were subjected to ab initio calculations obtaining shielding and quadrupole coupling values close to experimental data.64 However, improvements in the field of DFT-GIPAW calculations facilitated data with better agreements to experimental data. Generalized gradient approximation (GGA) was implemented using Perdew–Burke–Ernzerhof (PBE)153 exchange–correlation functional for the studies of alumina structures. Lizarraga et al.139 calculated NMR parameters for some of the most ordered alumina phases (α, θ, κ and γ alumina) with high accuracy. A comprehensive study by Ferreira et al.115 used PBE, revised PBE154 and van der Waals density19 functionals to extract precise values for NMR parameters of α, θ, κ and γ alumina, gibbsite and boehmite. These are the results which have the best agreements with the experimental data until now. For κ-alumina, the chemical shift and quadrupole coupling values obtained in this study remain as the most important reference, as the NMR experimental efforts did not analyze the results properly.
Furthermore, the DFT studies of 27Al NMR parameters focused on the surface of alumina phases, especially of γ-alumina. Modelling the surface of alumina was not trivial, with the presence of penta-coordinated Al, dangling bonds, surface-hydroxyls and hydrogen bonds. However, the effect of surface on the NMR spectra was mostly limited to the intensity of the AlO5 resonance showing up in between 20 and 50 ppm. Recently, Wischert et al.116 have shown that the selective cross-polarization from the hydroxyl protons in the surface can improve the sensitivity of the surface aluminium species, with the support of first principles calculations in periodic boundary conditions. It was shown that very high 27Al quadrupole couplings (>30 MHz) are possible with very low concentration of hydroxyls on the surface, as it leads to highly asymmetric surface structures. In another effort to model different γ-alumina surfaces to do DFT calculations, the chemical shift of penta-coordinated Al was predicted to be between 32 and 39 ppm.122 But in a DFT study involving χ and η alumina, the experimental and theoretical correlation of 1H and 27Al chemical shifts showed that the surface terminal hydroxyls of these phases are linked to tetra-coordinated Al.155
7. Conclusions
A literature overview on 27Al NMR studies of known transition alumina phases reveals the strength of this spectroscopy for disentangling the different phases and their transitions. A library of NMR spectroscopic fingerprints of the different phases is provided. Some structural disorder is always present in transition alumina phases. For disordered materials, NMR can further structural characterization compared to diffraction methods. The different kinds Al–O coordination can be sorted with high accuracy and in quantitative mode. Analysis of the nuclear quadrupole interaction provides insight in deviations from spherical symmetry and their distribution across the crystal structure. The nature and relevance of alumina surface and the dependence of physical properties of the material on the surface is reflected in the NMR spectra. A high contrast between disordered, amorphous and periodic and aperiodic parts of samples allows to characterize on the same footing periodic, amorphous or ill-crystalline, as well as intrinsic coexistence of periodic and disordered situations.Most of the NMR investigations date a few decades back. There have been remarkable improvement on solid-state NMR since, both methodologically and technologically. There are still several transition alumina phases with poorly described structure, the elucidation of which will benefit from the application of recent NMR advances. A complete NMR characterization of all transition alumina phases with high-resolution methods and extraction of parameters using a unified model which takes the inherent disorder into account, is now feasible. 27Al NMR is obviously the most suited method for studies of transition alumina synthesis and transformation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude concerning financial support by Research Foundation Flanders (FWO) via project funding (G.0885.14N). NMR equipment was acquired with financial support by Hercules Foundation (AKUL/13/21), and by the Flemish Government, department EWI via the Hermes Fund (AH.2016.134). J. A. M., F. T. and CEAK acknowledge the Flemish Government for long-term structural funding (Methusalem).References
- H. C. Stumpf, A. S. Russell, J. W. Newsome and C. M. Tucker, Ind. Eng. Chem., 1950, 42, 1398–1403 CrossRef CAS.
- C. S. John, N. C. M. Alma and G. R. Hays, Appl. Catal., 1983, 6, 341–346 CrossRef CAS.
- K. Wefers and C. Misra, Alcoa Tech. Pap., 1987.
- G. Busca, Structural, Surface, and Catalytic Properties of Aluminas, Elsevier Inc., 1st edn, 2014, vol. 57 Search PubMed.
- B. a. Huggins and P. D. Ellis, J. Am. Chem. Soc., 1992, 114, 2098–2108 CrossRef CAS.
- P. S. Santos, H. S. Santos and S. P. Toledo, Mater. Res., 2000, 3, 104–114 CrossRef CAS.
- R. H. Meinhold, R. C. T. Slade and R. H. Newman, Appl. Magn. Reson., 1993, 4, 121–140 CrossRef CAS.
- G. Kunath-Fandrei and T. Bastow, J. Phys. Chem., 1995, 99, 15138–15141 CrossRef CAS.
- A. Malki, Z. Mekhalif, S. Detriche, G. Fonder, A. Boumaza and A. Djelloul, J. Solid State Chem., 2014, 215, 8–15 CrossRef CAS.
- I. Levin and D. Brandon, J. Am. Ceram. Soc., 2005, 81, 1995–2012 CrossRef.
- A. Jerschow, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 63–78 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral De Menezes, R. Goodfellow and P. Granger, Concepts Magn. Reson., 2002, 14, 326–346 CrossRef CAS.
- C. Martineau, J. Senker and F. Taulelle, Annu. Rep. NMR Spectrosc., 2014, 82, 1–57 CrossRef.
- E. C. Decanio, J. C. Edwards and J. W. Bruno, J. Catal., 1994, 148, 76–83 CrossRef CAS.
- G. Piedra, J. J. Fitzgerald, N. Dando, S. F. Dec and G. E. Maciel, Inorg. Chem., 1996, 35, 3474–3478 CrossRef CAS.
- T. H. Walter and E. Oldfield, J. Phys. Chem., 1989, 93, 6744–6751 CrossRef CAS.
- J. Haase and E. Oldfield, Solid State Nucl. Magn. Reson., 1994, 3, 171–175 CrossRef CAS PubMed.
- M. Edén, Annu. Rep. NMR Spectrosc., 2015, 86, 237–331 CrossRef.
- M. Haouas, F. Taulelle and C. Martineau, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 94–95, 11–36 CrossRef CAS PubMed.
- J. Brus, S. Abbrent, L. Kobera, M. Urbanova and P. Cuba, Annu. Rep. NMR Spectrosc., 2016, 88, 79–147 CrossRef CAS.
- C. Martineau, F. Taulelle and M. Haouas, PATAI’S Chemistry of Functional Groups, John Wiley & Sons, Ltd, Chichester, UK, 2016, pp. 1–51 Search PubMed.
- M. H. Levitt, Spin Dynamics: Basics of Nuclear Magnetic Resonance, Wiley, 2008 Search PubMed.
- T. Bräuniger, C. V. Chandran, U. Wedig and M. Jansen, Z. Anorg. Allg. Chem., 2011, 637, 530–535 CrossRef.
- K. Harindranath, K. Anusree Viswanath, C. Vinod Chandran, T. Bräuniger, P. K. Madhu, T. G. Ajithkumar and P. A. Joy, Solid State Commun., 2010, 150, 262–266 CrossRef CAS.
- H. Koller, G. Engelhardt, A. P. M. Kentgens and J. Sauer, J. Phys. Chem., 1994, 98, 1544–1551 CrossRef CAS.
- A. P. M. Kentgens, Geoderma, 1997, 80, 271–306 CrossRef CAS.
- D. Freude and H.-J. Behrens, Krist. Tech., 1981, 16, K36–K38 CrossRef CAS.
- D. Müller, W. Gessner, H. J. Behrens and G. Scheler, Chem. Phys. Lett., 1981, 79, 59–62 CrossRef.
- D. Müller, I. Grunze, E. Hallas and G. Ladwig, Z. Anorg. Allg. Chem., 1983, 500, 80–88 CrossRef.
- D. Müller, W. Gessner, A. Samoson, E. Lippmaa and G. Scheler, Polyhedron, 1986, 5, 779–785 CrossRef.
- H. D. Morris and P. D. Ellis, J. Am. Chem. Soc., 1989, 111, 6045–6049 CrossRef CAS.
- R. W. Schurko, R. E. Wasylishen and H. Foerster, J. Phys. Chem. A, 1998, 102, 9750–9760 CrossRef CAS.
- L. van Wüllen and M. Kalwei, J. Magn. Reson., 1999, 139, 250–257 CrossRef PubMed.
- E. Lippmaa, A. Samoson and M. Magi, J. Am. Chem. Soc., 1986, 108, 1730–1735 CrossRef CAS.
- L. B. Alemany and G. W. Kirker, J. Am. Chem. Soc., 1986, 108, 6158–6162 CrossRef CAS.
- M. C. Cruickshank, L. S. D. Glasser, S. A. I. Barri and I. J. F. Poplett, J. Chem. Soc., Chem. Commun., 1986, 23b–24 RSC.
- L.-S. Du and J. F. Stebbins, J. Phys. Chem. B, 2004, 108, 3681–3685 CrossRef CAS.
- M. Dressler, M. Nofz, F. Malz, J. Pauli, C. Jäger, S. Reinsch and G. Scholz, J. Solid State Chem., 2007, 180, 2409–2419 CrossRef CAS.
- T. Bräuniger and M. Jansen, Z. Anorg. Allg. Chem., 2013, 639, 857–879 CrossRef.
- A. Samoson, E. Lippmaa and A. Pines, Mol. Phys., 1988, 65, 1013–1018 CrossRef CAS.
- B. F. Chmelka, K. T. Mueller, A. Pines, J. Stebbins, Y. Wu and J. W. Zwanziger, Nature, 1989, 339, 42–43 CrossRef CAS.
- A. Llor and J. Virlet, Chem. Phys. Lett., 1988, 152, 248–253 CrossRef CAS.
- K. Mueller, B. Sun, G. Chingas, J. Zwanziger, T. Terao and A. Pines, J. Magn. Reson., 1990, 86, 470–487 CAS.
- A. Medek, J. S. Harwood and L. Frydman, J. Am. Chem. Soc., 1995, 117, 12779–12787 CrossRef CAS.
- G. Wu, D. Rovnyank, B. Sun and R. G. Griffin, Chem. Phys. Lett., 1996, 249, 210–217 CrossRef CAS.
- Z. Gan, J. Am. Chem. Soc., 2000, 122, 3242–3243 CrossRef CAS.
- H.-T. Kwak and Z. Gan, J. Magn. Reson., 2003, 164, 369–372 CrossRef CAS PubMed.
- G. Czjzek, J. Fink, F. Götz, H. Schmidt, J. M. D. Coey, J. P. Rebouillat and A. Liénard, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 2513–2530 CrossRef CAS.
- D. R. Neuville, L. Cormier and D. Massiot, Geochim. Cosmochim. Acta, 2004, 68, 5071–5079 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J. O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- See ESI,† Discussion S2.
- A. Goldbourt and P. K. Madhu, Annu. Rep. NMR Spectrosc., 2004, 54, 81–153 CrossRef.
- X. Xue and M. Kanzaki, J. Phys. Chem. B, 2007, 111, 13156–13166 CrossRef CAS PubMed.
- D. Lee, H. Takahashi, A. S. L. Thankamony, J.-P. Dacquin, M. Bardet, O. Lafon and G. De Paëpe, J. Am. Chem. Soc., 2012, 134, 18491–18494 CrossRef CAS PubMed.
- D. Lee, N. T. Duong, O. Lafon and G. De Paëpe, J. Phys. Chem. C, 2014, 118, 25065–25076 CrossRef CAS.
- M. Taoufik, K. C. Szeto, N. Merle, I. Del Rosal, L. Maron, J. Trébosc, G. Tricot, R. M. Gauvin and L. Delevoye, Chem. – Eur. J., 2014, 20, 4038–4046 CrossRef CAS PubMed.
- Z. Wang, Y. Jiang, O. Lafon, J. Trébosc, K. Duk Kim, C. Stampfl, A. Baiker, J.-P. Amoureux and J. Huang, Nat. Commun., 2016, 7, 13820 CrossRef CAS PubMed.
- B. N. S. Barrow, A. Scullard and N. Collis, Johnson Matthey Technol. Rev., 2016, 60, 90–97 CrossRef.
- A. Venkatesh, M. P. Hanrahan and A. J. Rossini, Solid State Nucl. Magn. Reson., 2017, 84, 171–181 CrossRef CAS PubMed.
- H. Saalfeld and M. Wedde, Z. Kristallogr., 1974, 139, 129–135 CrossRef CAS.
- A. Vyalikh, K. Zesewitz and U. Scheler, Magn. Reson. Chem., 2010, 48, 877–881 CrossRef CAS PubMed.
- R. C. T. Slade, J. C. Southern and I. M. Thompson, J. Mater. Chem., 1991, 1, 563–568 RSC.
- S. E. Ashbrook, J. McManus, K. J. D. MacKenzie and S. Wimperis, J. Phys. Chem. B, 2000, 104, 6408–6416 CrossRef CAS.
- K. Damodaran, P. R. Rajamohanan, D. Chakrabarty, U. S. Racherla, V. Manohar, C. Fernandez, J.-P. Amoureux and S. Ganapathy, J. Am. Chem. Soc., 2002, 124, 3200–3201 CrossRef CAS PubMed.
- C. V. Chandran, G. Hempel and T. Bräuniger, Solid State Nucl. Magn. Reson., 2011, 40, 84–87 CrossRef CAS PubMed.
- K. J. D. MacKenzie, J. Temuujin and K. Okada, Thermochim. Acta, 1999, 327, 103–108 CrossRef CAS.
- F. Zigan, W. Joswig and N. Burger, Z. Kristallogr. – Cryst. Mater., 1978, 148, 255–274 CrossRef CAS.
- T. J. Bastow, J. S. Hall, M. E. Smith and S. Steuernagel, Mater. Lett., 1994, 18, 197–200 CrossRef CAS.
- L. H. Chagas, G. S. G. De Carvalho, R. A. S. San Gil, S. S. X. Chiaro, A. A. Leitão and R. Diniz, Mater. Res. Bull., 2014, 49, 216–222 CrossRef CAS.
- J. Q. Wang, J. L. Liu, X. Y. Liu, M. H. Qiao, Y. Pei and K. N. Fan, Sci. Adv. Mater., 2009, 1, 77–85 CrossRef CAS.
- C. Pecharromán, I. Sobrados, J. E. Iglesias, T. González-Carreño and J. Sanz, J. Phys. Chem. B, 1999, 103, 6160–6170 CrossRef.
- M. Digne, P. Sautet, P. Raybaud, H. Toulhoat and E. Artacho, J. Phys. Chem. B, 2002, 106, 5155–5162 CrossRef CAS.
- R. J. Hill, Clays Clay Miner., 1981, 29, 435–445 CrossRef CAS.
- D. Papee, R. Tertian and R. Biais, Bull. Soc. Chim. Fr., 1958, 5, 1301–1310 Search PubMed.
- R. Tettenhorst and D. A. Hofmann, Clays Clay Miner., 1980, 28, 373–380 CrossRef CAS.
- J. J. Fitzgerald, G. Piedra, S. F. Dec, M. Seger and G. E. Maciel, J. Am. Chem. Soc., 1997, 119, 7832–7842 CrossRef CAS.
- L. B. Alemany, Appl. Magn. Reson., 1993, 4, 179–201 CrossRef CAS.
- K. J. MacKenzie, J. Temuujin, M. Smith, P. Angerer and Y. Kameshima, Thermochim. Acta, 2000, 359, 87–94 CrossRef CAS.
- R. C. T. Slade, J. C. Southern and I. M. Thompson, J. Mater. Chem., 1991, 1, 875–879 RSC.
- F. R. Chen, J. G. Davis and J. J. Fripiat, J. Catal., 1992, 133, 263–278 CrossRef CAS.
- T. Tsuchida and K. Horigome, Thermochim. Acta, 1995, 254, 359–370 CrossRef CAS.
- R. J. Hill, Phys. Chem. Miner., 1979, 5, 179–200 CrossRef CAS.
- X. Xue, M. Kanzaki and H. Fukui, Am. Mineral., 2010, 95, 1276–1293 CrossRef CAS.
- A. H. Carim, G. S. Rohrer, N. R. Dando, S.-Y. Tzeng, C. L. Rohrer and A. J. Perrotta, J. Am. Ceram. Soc., 2005, 80, 2677–2680 CrossRef.
- A. R. Studart, U. T. Gonzenbach, E. Tervoort and L. J. Gauckler, J. Am. Ceram. Soc., 2006, 89, 1771–1789 CrossRef CAS.
- S.-M. Lee, E. Pippel, U. Gösele, C. Dresbach, Y. Qin, C. V. Chandran, T. Bräuniger, G. Hause and M. Knez, Science, 2009, 324, 488–492 CrossRef CAS PubMed.
- S. K. Lee, S. B. Lee, S. Y. Park, Y. S. Yi and C. W. Ahn, Phys. Rev. Lett., 2009, 103, 4–7 Search PubMed.
- S. Keun Lee, S. Young Park, Y. Soo Yi and J. Moon, J. Phys. Chem. C, 2010, 114, 13890–13894 CrossRef.
- C. Detavernier, J. Dendooven, S. P. Sree, K. F. Ludwig and J. a Martens, Chem. Soc. Rev., 2011, 40, 5242–5253 RSC.
- S. K. Lee and C. W. Ahn, Sci. Rep., 2014, 4, 4200 CrossRef PubMed.
- S. P. Sree, J. Dendooven, P. C. M. M. Magusin, K. Thomas, J.-P. Gilson, F. Taulelle, C. Detavernier and J. A. Martens, Catal. Sci. Technol., 2016, 6, 6177–6186 RSC.
- L. Baggetto, V. Sarou-Kanian, P. Florian, A. N. Gleizes, D. Massiot and C. Vahlas, Phys. Chem. Chem. Phys., 2017, 19, 8101–8110 RSC.
- V. Sarou-kanian, A. N. Gleizes, P. Florian, D. Same, D. Massiot and C. Vahlas, J. Phys. Chem. C, 2013, 117, 21965–21971 CrossRef CAS.
- S.-L. Hwang, P. Shen, H.-T. Chu and T.-F. Yui, Int. Geol. Rev., 2006, 48, 754–764 CrossRef.
- B. Whittington and D. Ilievski, Chem. Eng. J., 2004, 98, 89–97 CrossRef CAS.
- T. Kogure, J. Am. Ceram. Soc., 1999, 82, 716–720 CrossRef CAS.
- G. W. Brindley and J. O. Choe, Am. Mineral., 1961, 46, 771–785 Search PubMed.
- H. Saalfeld, Neues Jahrb. Mineral., Abh., 1960, 95, 1–87 CAS.
- L. Favaro, A. Boumaza, P. Roy, J. Lédion, G. Sattonnay, J. B. Brubach, A. M. Huntz and R. Tétot, J. Solid State Chem., 2010, 183, 901–908 CrossRef CAS.
- A. C. Vieira Coelho, H. de Souza Santos, P. K. Kiyohara, K. N. P. Marcos and P. de Souza Santos, Mater. Res., 2007, 10, 183–189 CrossRef.
- A. L. Dragoo and J. J. Diamond, J. Am. Ceram. Soc., 1967, 50, 568–574 CrossRef CAS.
- R. S. Zhou and R. L. Snyder, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 617–630 CrossRef.
- D. S. Maciver, H. H. Tobin and R. T. Barth, J. Catal., 1963, 2, 485–497 CrossRef CAS.
- F. Stone and L. Whalley, J. Catal., 1967, 8, 173–182 CrossRef CAS.
- G. Gatta, B. Fubini, G. Ghiotti and C. Morterra, J. Catal., 1976, 43, 90–98 CrossRef.
- E. J. W. Verwey, Z. Kristallogr. – Cryst. Mater., 1935, 91, 65–69 CAS.
- B. C. Lippens and J. H. de Boer, Acta Crystallogr., 1964, 17, 1312–1321 CrossRef CAS.
- G. Paglia, A. L. Rohl, C. E. Buckley and J. D. Gale, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 224115 CrossRef.
- Y. Rozita, R. Brydson, T. P. Comyn, A. J. Scott, C. Hammond, A. Brown, S. Chauruka, A. Hassanpour, N. P. Young, A. I. Kirkland, H. Sawada and R. I. Smith, ChemCatChem, 2013, 5, 2695–2706 CrossRef CAS.
- L. Samain, A. Jaworski, M. Edén, D. M. Ladd, D.-K. Seo, F. Javier Garcia-Garcia and U. Häussermann, J. Solid State Chem., 2014, 217, 1–8 CrossRef CAS.
- M. F. Peintinger, M. J. Kratz and T. Bredow, J. Phys. Chem. A, 2014, 2, 13143–13158 CAS.
- X. Krokidis, P. Raybaud, A.-E. Gobichon, B. Rebours, P. Euzen and H. Toulhoat, J. Phys. Chem. B, 2001, 105, 5121–5130 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2002, 211, 1–5 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- A. R. Ferreira, E. Küçükbenli, A. A. Leitão and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 235119 CrossRef.
- R. Wischert, P. Florian, C. Copéret, D. Massiot and P. Sautet, J. Phys. Chem. C, 2014, 118, 15292–15299 CrossRef CAS.
- A. Düvel, E. Romanova, M. Sharifi, D. Freude, M. Wark, P. Heitjans and M. Wilkening, J. Phys. Chem. C, 2011, 115, 22770–22780 CrossRef.
- J. H. Kwak, J. Hu, A. Lukaski, D. H. Kim, J. Szanyi and C. H. F. Peden, J. Phys. Chem. C, 2008, 112, 9486–9492 CrossRef CAS.
- M. Wilkening, A. Düvel, F. Preishuber-Pflügl, K. da Silva, S. Breuer, V. Šepelák and P. Heitjans, Z. Kristallogr. – Cryst. Mater., 2017, 232 Search PubMed.
- D. Coster, A. L. Blumenfeld and J. J. Fripiat, J. Phys. Chem., 1994, 98, 6201–6211 CrossRef CAS.
- A. L. Blumenfeld and J. J. Fripiat, Top. Catal., 1997, 4, 119–129 CrossRef CAS.
- C. Wan, M. Y. Hu, N. R. Jaegers, D. Shi, H. Wang, F. Gao, Z. Qin, Y. Wang and J. Z. Hu, J. Phys. Chem. C, 2016, 120, 23093–23103 CrossRef CAS.
- B. Ollivier, R. Retoux, P. Lacorre, D. Massiot and G. Ferey, J. Mater. Chem., 1997, 7, 1049–1056 RSC.
- P. Liu and J. Skogsmo, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 425–433 CrossRef.
- S. J. Wilson and J. D. C. Mc Connell, J. Solid State Chem., 1980, 34, 315–322 CrossRef CAS.
- H. P. Rooksby and C. J. M. Rooymans, Clay Miner. Bull., 1961, 4, 237–238 Search PubMed.
- L. Kovarik, M. Bowden, A. Genc, J. Szanyi, C. H. F. Peden and J. H. Kwak, J. Phys. Chem. C, 2014, 118, 18051–18058 CrossRef CAS.
- E. Ghanti, N. Tomar, P. Gautam and R. Nagarajan, J. Sol-Gel Sci. Technol., 2011, 57, 12–15 CrossRef CAS.
- H. N. Kim and S. K. Lee, Am. Mineral., 2013, 98, 1198–1210 CrossRef CAS.
- J. D. Kohn, J. A. Katz and G. Broder, Am. Mineral., 1957, 42, 398 CAS.
- L. A. O’Dell, S. L. P. Savin, A. V. Chadwick and M. E. Smith, Solid State Nucl. Magn. Reson., 2007, 31, 169–173 CrossRef PubMed.
- L. D. Hart, Alumina Chemicals: Science and Technology Handbook, American Ceramic Society, Westerville, OH, 1990 Search PubMed.
- L. Pauling and S. B. Hendricks, J. Am. Chem. Soc., 1925, 47, 781–790 CrossRef CAS.
- R. V. Pound, Phys. Rev., 1950, 79, 685–702 CrossRef CAS.
- H. J. Jakobsen, J. R. Skibsted, H. Bildsoe and A. C. H. R. Nielsen, J. Magn. Reson., 1989, 85, 173–180 CAS.
- J. Skibsted, N. C. Nielsen, H. Bildsøe and H. J. Jakobsen, J. Magn. Reson., 1991, 95, 88–117 CAS.
- A. P. M. Kentgens, A. Bos and P. J. Dirken, Solid State Nucl. Magn. Reson., 1994, 3, 315–322 CrossRef CAS PubMed.
- V. Sabarinathan, S. Ramasamy and S. Ganapathy, J. Phys. Chem. B, 2010, 114, 1775–1781 CrossRef CAS PubMed.
- R. Lizárraga, E. Holmström, S. C. Parker and C. Arrouvel, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 094201 CrossRef.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- C. Gervais, M. Profeta, F. Babonneau, C. J. Pickard and F. Mauri, J. Phys. Chem. B, 2004, 108, 13249–13253 CrossRef CAS.
- M. Body, C. Legein, J.-Y. Buzaré, G. Silly, P. Blaha, C. Martineau and F. Calvayrac, J. Phys. Chem. A, 2007, 111, 11873–11884 CrossRef CAS PubMed.
- S. E. Ashbrook, M. Cutajar, C. J. Pickard, R. I. Walton and S. Wimperis, Phys. Chem. Chem. Phys., 2008, 10, 5754 RSC.
- B. Tu, X. Liu, H. Wang, W. Wang, P. Zhai and Z. Fu, Inorg. Chem., 2016, 55, 12930–12937 CrossRef CAS PubMed.
- E. Lam, A. Comas-Vives and C. Copéret, J. Phys. Chem. C, 2017, 121, 19946–19957 CrossRef CAS.
- T. A. Halgren, Curr. Opin. Struct. Biol., 1995, 5, 205–210 CrossRef CAS PubMed.
- R. T. Cygan, J.-J. Liang and A. G. Kalinichev, J. Phys. Chem. B, 2004, 108, 1255–1266 CrossRef CAS.
- Z. Shen, E. S. Ilton, M. P. Prange and S. N. Kerisit, J. Phys. Chem. C, 2017, 121, 13692–13700 CrossRef CAS.
- M. Pouvreau, J. A. Greathouse, R. T. Cygan and A. G. Kalinichev, J. Phys. Chem. C, 2017, 121, 14757–14771 CrossRef CAS.
- P. Blaha, K. Schwarz, P. Sorantin and S. B. Trickey, Comput. Phys. Commun., 1990, 59, 399–415 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. – Cryst. Mater., 2005, 220 Search PubMed.
- M. Choi, K. Matsunaga, F. Oba and I. Tanaka, J. Phys. Chem. C, 2009, 113, 3869–3873 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- D. F. Khabibulin, E. Papulovskiy, A. S. Andreev, A. A. Shubin, A. M. Volodin, G. A. Zenkovets, D. A. Yatsenko, S. V. Tsybulya and O. B. Lapina, Z. Phys. Chem., 2017, 231 Search PubMed.
- N. Ishizawa, T. Miyata, I. Minato, F. Marumo and S. Iwai, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 228–230 CrossRef.
- M. Houlleberghs, A. Hoffmann, D. Dom, C. E. A. Kirschhock, F. Taulelle, J. A. Martens and E. Breynaert, Anal. Chem., 2017, 89, 6940–6943 CrossRef CAS PubMed.
- E. Breynaert and C. V. Chandran, 3D Correlation of Solid-State NMR Parameters for Alumina - 3D interactive chart, Harvard Dataverse, V1, 2018, DOI:10.7910/DVN/FRZEGB.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cs00321a |
| This journal is © The Royal Society of Chemistry 2019 |