
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Varying oxygen coverage on Cu55 and its effect on CO oxidation†
Li
Ma
 a and
Jaakko
Akola
*ab
a and
Jaakko
Akola
*ab
aComputational Physics Laboratory, Tampere University, P.O. Box 692, FI-33014 Tampere, Finland
bDepartment of Physics, Norwegian University of Science and Technology, NO-7491 Trondheim, Norway. E-mail: jaakko.akola@ntnu.no
First published on 7th May 2019
Abstract
Adsorption of molecular oxygen on a Cu55 cluster and the resulting oxidation effects have been investigated by spin-polarized density functional theory (DFT). The optimal structure for each Cu55O2N (N = 1–20) complex has been obtained via a sequential addition of O2 and systematic screening of the preferable adsorption sites. Upon structural optimization, several O2 molecules dissociate readily on Cu55 at different oxygen coverages, and further DFT molecular dynamics simulations at 300 K confirm the instability (small dissociation barrier) of the remaining O2 and a spontaneous movement of some oxygen atoms from the surface sites towards the cluster interior. The Cu55 cluster and its oxidized derivatives have been placed on a γ-Al2O3(100) surface to study the cluster–support interaction, and furthermore, CO oxidation reactions on both Cu55(O)2N and Cu55(O)2N/γ-Al2O3(100) have been studied as a function of oxygen coverage. The CO oxidation reaction barrier is rather insensitive to the oxygen coverage regardless of the support, indicating a small increase in activity with the number of surface oxygen atoms.
1. Introduction
The application of nanoparticles in heterogeneous catalysis is an active area of research. Noble metals, in particular the platinum group metals (PGMs) such as Pd, Pt, Ru, and Rh, have shown high activities and good stabilities for many reactions.1–4 However, they are classified as critical metals owing to their high cost, limited availability, and socio-economic issues related to mining processes and supply (environment, working conditions, etc.). This has motivated the active search for more abundant metal catalysts with similar and/or improved performance characteristics for PGM replacement. Such catalysts are highly desirable for a wide variety of applications, not only in heterogeneous catalysis (e.g. acetylene hydrogenation5 and ammonia decomposition6) but also in electrochemistry (hydrogen energy and CO2 reduction).7 Here, Cu-based catalysts have attracted considerable attention due to their low cost (earth-abundant) and a variety of industrial applications, including hydrogenation,8 oxidation9 and synthesis of methanol.10 Much work has been performed over a number of years to optimize their performance. However, under industrial conditions, the catalyst surfaces are readily covered by the reactant, intermediate and product species causing issues with stability. The existence of the adsorbed species or layers may significantly affect the adsorption behavior of other species and reaction mechanisms or induce catalyst reconstruction.It is well known that the CO oxidation reaction mechanism on a catalyst is influenced by the reactant gas coverage. Under high O2 pressure, surface oxides have been proposed as the active phases for late-transition and noble metal elements,11–13 which contradicts the well-accepted opinion that the transition or noble metal per se is responsible for CO oxidation. At low CO coverage, dissociated oxygen is the active species for the production of CO2 from CO, whereas at higher CO coverage, molecular O2 can directly react with CO via the formation of an OOCO intermediate.14 Such studies of CO oxidation on transition metal surfaces and supported particles under varying partial pressures are beneficial for understanding the reaction mechanisms under realistic or industrial conditions and also for providing insight on the changing activity on metal surfaces or particles (catalyst stability).
Concerning the adsorption of oxygen on Cu catalysts, most studies have considered only a single O2 molecule.15–17 The effects of several oxygen molecules or atoms on Cu catalysts have been rarely studied,15 especially on the saturation coverage. For the CO oxidation reaction, there is still controversy regarding the oxidation state of Cu catalysts as the correlation between reactivity and oxidation states is not well established. For example, Huang et al. proposed that Cu2O is the most active among the three commercial catalysts, Cu, Cu2O, and CuO powders, under oxygen-lean and -rich conditions, and they attribute this to the high mobility of lattice oxygen.18 In contrast, Fredriksson et al.19 and Jernigan et al.20 found that metallic Cu is significantly more active than Cu oxides. On Cu single-crystal surfaces, it is proposed that high oxygen coverage retards the reaction rate.21,22 On the theoretical side, we recently investigated CO oxidation catalyzed by a Cu20 cluster and found that O2 activation upon adsorption (affected by the cluster charge state) is crucial for the rate of CO oxidation.23 To the best of our knowledge, previous calculations of CO oxidation on a Cu catalyst usually focused on low O2 concentrations, and the effects of varying the oxygen coverage are not clear.
Typically, the Cu nanoparticles used experimentally have an average diameter of ∼30 nm for catalytic CO oxidation,19 while they are in the range of 15–35 nm for copper oxide catalysts.24,25 A smaller copper oxide catalyst with a particle size of 3.6 nm was obtained by specific preparation methods,26 and it displays an enhanced catalytic activity. Moreover, previous studies have revealed that the CO oxidation conversion rate is considerably affected by the crystal size of the catalyst where it generally increases up to an optimal crystal size, after which the conversion rate decreases again.27 In this article, we present the results of density functional theory (DFT) calculations for the icosahedral Cu55 cluster and explore the interaction of several oxygen molecules/atoms on the cluster surface, the subsequent cluster modifications, and the resulting effect on CO oxidation. Similar 55-atom clusters have been previously used as model structures for larger nanoclusters.28 Here, the ideal icosahedral structure is the global minimum of Cu55 where the number of symmetrically inequivalent adsorption sites is limited. This makes it possible to examine all individual sites to find the most favorable adsorption structure, and hence, the icosahedral Cu55 cluster was chosen as the model structure for copper nanoparticles.
It is known that the support effects play a crucial role in the catalytic activity. Alumina is widely used as a basic catalytic support material because of its high chemical inertness, strength and hardness.29 γ-Al2O3 possesses an excellent surface area owing to its small particle size, which results in high activity of the surface as a catalyst support. γ-Al2O3 crystals are terminated by the (100), (110) and (111) planes. The two main orientations (under practical operation conditions) of γ-Al2O3 facets are (100) and (110). The high-resolution images show that the (110) face is not atomically flat but significantly reconstructed. The (100) surface has the lowest surface energy, which is considered to be the most representative surface.30 For these reasons, we have also chosen to study CO oxidation on Cu55/γ-Al2O3(100) at different oxygen coverages. In addition, we note that in CO oxidation over metal-based catalysts, O2 adsorption and activation is often regarded as the rate-limiting step.31 The use of traditional PGM catalysts with inert supports such as SiO2, Al2O3 and zeolites is inefficient.32 In this type of catalyst, there is a serious competition between the adsorption and activation of CO and O2 on the metal sites. The strong adsorption of CO seriously hinders O2 adsorption and activation.
2. Computational methods
All electronic structure calculations were based on DFT using the spin-polarized, gradient-corrected exchange–correlation functional of Perdew, Burke, and Ernzerhof (PBE)33 as implemented in the CP2K program package.34,35 The single-particle Kohn–Sham wave functions were expanded by a combined Gaussian and plane wave (GPW) basis set. A molecularly-optimized double-zeta valence plus polarization (DZVP) basis set was used for the Gaussian expansion,36 where the basis set superposition error (BSSE) has been reduced during optimization. An additional auxiliary plane wave basis with an energy cutoff of 600 Ry was used for operations involving electron density. The Cu and O valence states are 3d104s1 and 2s22p4, respectively, and the valence electron–ion interaction is based on the analytical pseudopotentials derived by Goedecker, Teter, and Hutter (GTH).37The Cu55 cluster with icosahedral structure is shown in Fig. S1(a) (ESI†). For the support [Fig. S1(b) and (c), ESI†], a slab model was used by cleaving the bulk γ-Al2O3 model30,38 in the (100) direction. The Al2O3(100) support was chosen as a four-layered (4 × 3) slab (in total 480 atoms) and a vacuum spacing of 25 Å thickness was used within the simulation box. The top two atomic layers of the support were allowed to relax while the bottom two were fixed throughout the calculations. The Bader algorithm was used for analyzing the spatial charge decomposition among atoms.39 The climbing image nudged elastic band (CI-NEB) method was applied for mapping the reaction pathways.40 No symmetry constraints were imposed during structural relaxations. Vibrational analysis was performed to identify the stability of the obtained structures and zero-point energy (ZPE) corrections were included for energetics. DFT molecular dynamics (MD) calculations were performed at 300 K using a Nose thermostat41 and the Born–Oppenheimer mode with a time step of 1 fs.
The electronic ground state of O2 is a triplet, and correspondingly, the doublet and quartet states were examined for the Cu55 cluster (odd number of electrons), Cu55O2 (molecular adsorption), and Cu55(O)2 (dissociative adsorption). The spin multiplicity of Cu55 is a quartet, but this is only 0.03 eV lower than the doublet in total energy. On the other hand, the spin multiplicities of Cu55O2 and Cu55(O)2 are doublets, i.e. the adsorption complexes prefer the lowest spin state.
3. Results and discussion
3.1. Varying oxygen coverage on Cu55
To study the properties of oxygen coverage on copper, we carried out a set of simulations on the Cu55 cluster for molecular and/or dissociative O2 adsorption. First, the lowest-energy arrangements of Cu55O2N complexes were determined by sequentially adding O2 molecules on the cluster surface (Approach 1). Here N is an integer indicating the total number of O2 molecules adsorbed. An extensive adsorption site sampling was performed in Approach 1, where all symmetrically inequivalent sites on Cu55 were examined to find the most favourable adsorption structure for each step. The detailed description of the stepwise adsorption of O2 molecules for N = 1–20 is shown in the ESI† (Fig. S2–S5). The relaxation process led to a spontaneous O2 dissociation to atomic oxygen in several cases.After the optimal structure was found for Cu55O2N, we computed the adsorption energy of the last molecular or dissociative adsorption as42:
| Ea(N) = E(Cu55O(2N−2)) + E(O2) − E(Cu55O2N) | (1) |
| 〈Ea(N)〉 = [E(Cu55) + N × E(O2) − E(Cu55O2N)]/N | (2) |
By applying Approach 1, the Ea(N) value becomes negative at N = 20 (Fig. S5 and Table S1, ESI†) indicating that the 20th molecule cannot adsorb exothermally (at 0 K). However, this changes if O2 is allowed to dissociate upon adsorption. Taking into account the pronounced instability of adsorbed O2, we decided to apply another method (Approach 2) to generate oxidized Cu55 geometries based on the stepwise O2 adsorptive geometries. The initial structures of Approach 2 were based on the O2 adsorption site scanning in Approach 1. Here, we obtained the corresponding O2 dissociative geometries by splitting each O2 upon adsorption one-by-one (Fig. 1; full set in Fig. S6, ESI†). The corresponding oxygen adsorption energies of Approach 2 are shown in Fig. 2.
 | ||
| Fig. 1 Geometries of Cu55(O)2N (N = 13, 16, 18, 20) with increasing number of endohedral oxygen atoms (blue color), as generated by Approach 2. The last structure [Cu55(O)40] is obtained after MD annealing at 300 K. | ||
 | ||
| Fig. 2 Adsorption energy Ea(N) of O2 upon molecular (O2) or dissociative (O)2 adsorption on Cu55 and the corresponding average adsorption energy 〈Ea(N)〉 of added O2 molecules (Approach 2, top). The separate point is the Ea(N) of MD simulation for N = 20. The structures of the initial state (IS), transition state (TS), and final state (FS) of the reaction pathways for O2 → O + O on Cu55O2N (N = 1, 10, 20) together with the energy changes with respect to the IS (below). The bond lengths of the reactive O2 molecules are included. | ||
Beginning with the single molecular adsorption configuration Cu55O2, dissociative adsorption structures Cu55(O)2 were obtained by placing 2 × O atoms at different sites on the triangular facet of the 55-atom icosahedron (Approach 1). Here, two O atoms located at the nearby three-fold hollow sites is the most stable configuration [(O)2 in Fig. S6, ESI†]. The second O2 was added to Cu55(O)2 on the same triangular facet as the second O2 in Cu55(O2)2 (Approach 1, Fig. S3, ESI†), and the dissociated adsorption geometry was obtained in the same way as that of the first step of Approach 2 [(O)4 in Fig. S6, ESI†]. The procedure was repeated with the subsequent O2 molecules step by step until N = 20. We note that Cu55 undergoes a significant distortion with increasing oxygen coverage. It is possible that the high-coverage Cu55(O)2N structures in Fig. S6 (ESI†) may not correspond to the lowest energy isomers anymore as they are based on the icosahedral Cu55 structure. For practical reasons, it is difficult to consider other Cu55(O)2N structures as each composition would constitute a substantial ground-state search problem itself, and we have confined the geometry search within the icosahedral Cu55 motif.
Most of the dissociated O atoms are located on convex adsorption sites binding with three surface Cu atoms. However, the adsorption sites of several O atoms gradually move from convex, to surface, and to concave sites as the number of O atoms increases. As shown in Fig. 1, one O atom completely falls into an endohedral site in Cu55 at N = 13, forming bonds with surface and core Cu atoms. Furthermore, the endohedral O atoms increase to two at N = 16 and up to five at N = 20.
As mentioned, the structural optimizations are performed at 0 K. To see how the adsorption structure changes under more realistic reaction conditions, we carried out a DFT-MD simulation of 20 ps for Cu55(O)40 at room temperature (300 K). During this short period, the number of endohedral oxygen atoms increases up to eight (Fig. 1), which confirms the low-barrier movement of oxygen from the cluster surface towards the interior. In addition, we tested the insertion of a single oxygen atom into a clean Cu55 cluster. In this case, O always bounces back to the surface after optimization regardless of whether it is placed initially on a sub-surface or at the central interstitial site. This indicates that surface oxidation has to take place first before oxygen can move to the interior sites. Experimentally,13 the interaction of oxygen with Pd nanoparticles has been studied by temperature-programmed desorption (TPD), temperature-programmed low-energy ion scattering (TP-LEIS), and X-ray photoelectron spectroscopy (XPS). A low oxygen exposure leads to surface oxygen adatoms on Pd nanoparticles. Surface O adatoms on the Pd nanoparticles move to subsurface sites starting at 400 K, and they almost all move to the sub-surface at ∼750 K, desorbing only at higher temperature. High-pressure oxygen exposure readily converts small Pd nanoparticles to PdO-like species.13
Using eqn (1) and (2), the adsorption energies Ea(N) and 〈Ea(N)〉 for Approach 2 have been calculated for the geometries in Fig. 1 and Fig. S6 (ESI†). The corresponding energies and properties are listed in Table S2 (ESI†). To see the energy changing trends clearly, Ea(N) and 〈Ea(N)〉 are plotted in Fig. 2 for both molecular and atomic adsorption. The dissociative O2 adsorption is energetically much more favorable than molecular adsorption: Ea(N) is above 2.5 eV for dissociative adsorption until N = 16, which is much higher than 1.4 eV for the highest value of O2 molecular adsorption. For N = 17–20, Ea(N) decreases down to 1.6 eV for (O)2 adsorption, which is still significantly higher than O2 adsorption (<0.7 eV). The decrease of Ea(N) for N ≥ 17 is consistent with the energy changing trend in Fig. S5 (Approach 1, ESI†), and it is due to a large oxygen concentration on the cluster surface. Our MD simulations for N = 20 reveal that oxygen diffusion to endohedral sites causes an increase in the Ea(N) value to 2.58 eV, i.e. back to the original level (Fig. 2). However, one cannot observe any saturation at N = 20 in Approach 2 as Ea(N) does not change the sign. For the corresponding geometry of Cu55(O)40, each triangular facet of Cu55 has two oxygen atoms. Evidently, it is possible to add even more oxygen to the cluster but we did not pursue further towards the fully saturated Cu55(O)2N due to the computational demands.
For the average adsorption energy 〈Ea(N)〉, the fully dissociated adsorption (O)2N is also higher than the adsorption with a molecule, i.e. O2(O)2(N−1), and they approach each other with N. At the same time, the net charge transfer increases with the number of dissociated oxygen atoms (Table S2, ESI†). The average 〈Cu–Cu〉 bond length in Cu55(O)2N is larger than that in Cu55O2(O)2(N−1) for each number of N (Table S2, ESI†), and the Cu binding O atoms distort the Cu55 geometry severely.
Obviously, O2 is very unstable on Cu55, and we have simulated the O–O bond cleavage with respect to the geometries in Fig. S6 (ESI†). The geometries of the reactant, the transition state (TS), and the product of the reaction paths are shown in Fig. 2 where N = 1, 10 and 20 have been chosen as the representative cases. The initial O2 adsorption results in activation (elongation) of the O–O bond to the super-oxo state, and the calculated dissociative reaction barriers (TS) are small on all clusters as shown in Fig. 2; the highest barrier at N = 20 is only 0.25 eV. The barrier height correlates inversely with the O–O bond elongation and charge transfer from Cu55 to O2. These results show that O2 can be effectively activated on Cu55 regardless of the oxygen coverage (within the studied range).
3.2. CO oxidation at different oxygen coverages
Our previous study23 has demonstrated that CO oxidation proceeds efficiently on Cu20 when O2 is pre-adsorbed in a dissociative manner. The results in Section 3.1 also show that O2 dissociates easily on the Cu55 cluster at different oxygen coverages. In addition, experimental studies20–22 have suggested that CO oxidation takes place between adsorbed CO molecules and O atoms, following the Langmuir–Hinshelwood (L–H) mechanism. To check the effect of oxygen coverage, we have studied CO oxidation in the presence of fully dissociated O2 atoms.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is hardly influenced upon adsorption (isolated 1.14 Å, max. 1.16 Å on the cluster). Again N = 1, 10 and 20 are chosen to represent the oxygen coverage from low to high. For N = 20, the annealed geometry from the DFT-MD simulation at 300 K is used which is 0.63 eV more stable than the relaxed starting structure at 0 K.
O bond is hardly influenced upon adsorption (isolated 1.14 Å, max. 1.16 Å on the cluster). Again N = 1, 10 and 20 are chosen to represent the oxygen coverage from low to high. For N = 20, the annealed geometry from the DFT-MD simulation at 300 K is used which is 0.63 eV more stable than the relaxed starting structure at 0 K.
The catalytic reaction pathways for CO oxidation on Cu55(O)2N are shown in Fig. 3. Upon the reaction, CO and the nearest O move closer to each other, followed by a crossing of the energy barrier (TS) to form an intermediate (IM) complex. Subsequent to the IM complex, there is a barrier-free release of the formed CO2 molecule. The activation energy barriers decrease as the oxygen coverage increases, while at the same time, the adsorption energies of CO in the corresponding IS also decrease (1.09 eV, 0.83 eV, and 0.78 eV, respectively). Moreover, the binding energies of O in the neighboring site of CO exhibit a similar changing trend to CO adsorption (Table S2, ESI†). Correspondingly, at high oxygen coverage, the adsorbed CO and neighboring O are more weakly bound to Cu55 giving rise to a more efficient reaction rate. According to CO oxidation experiments on Ru(0001),11 it has been identified that the highest rates of CO2 production occurred for high oxygen partial pressures and concomitantly high oxygen coverages.
 | ||
| Fig. 3 Reaction pathways of CO oxidation on Cu55(O)2N (N = 1, 10, and 20). The corresponding structures of the initial state (IS), transition state (TS), intermediate (IM), and final state (FS) are shown. The energy changes are with respect to the IS. | ||
In addition, the d-band centers (Cd) of the selected Cu55(O)2N clusters in Fig. 3 were calculated from the electronic densities of states, DOS (Fig. S7, ESI†). The Cd values are −2.44, −2.66, and −2.78 eV, respectively, displaying a downward shift with oxygen coverage due to the increased charge transfer from Cu55. From the above results, we can see that CO oxidation benefits from increasing oxygen coverage, while the d-band center and the CO and O binding energies are reduced.
| Eb(N) = E(Cu55O2N) + E(Al2O3) − E(Cu55O2N/Al2O3) | (3) |
| Δρ = ρ(Cu55O2N + Al2O3) − ρ(Cu55O2N) − ρ(Al2O3) | (4) |
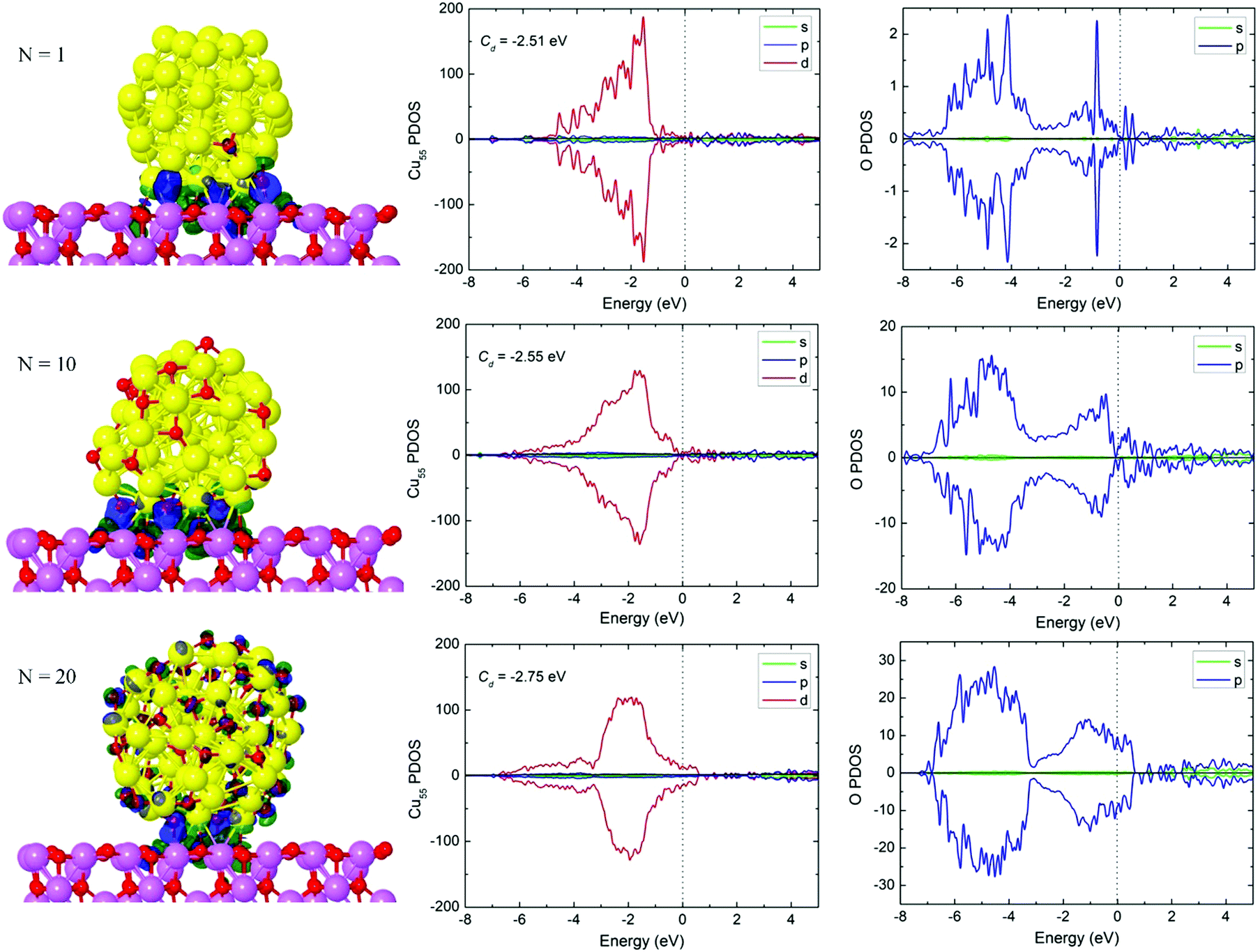 | ||
| Fig. 4 Optimized Cu55(O)2N/γ-Al2O3(100) (N = 1, 10, 20) geometries and charge density differences (CDDs) of the Cu55(O)2N clusters adsorbed on γ-Al2O3(100). Green and blue colors represent charge depletion and accumulation, respectively. The isosurface values are ±0.002 e Å−3 (left column). Spin-polarized projected density of states (PDOS) of Cu55 with (O)2N coverage for the Cu55(O)2N/γ-Al2O3(100) system. The PDOS are projected onto the Cu atoms in Cu55 (middle column) and the O atoms in the coverage (right column). The dotted line represents the Fermi level at 0 eV. | ||
| Cu55(O)2N/γ-Al2O3(100) | E b (eV) | Q (e) | r O–Al (Å) | E ad (eV) | E r (eV) |
|---|---|---|---|---|---|
| N = 1 | 2.87 | 0.22 | 1.83 | 1.06 | 0.57 |
| N = 10 | 2.76 | −0.56 | 1.86 | 1.00 | 0.65 |
| N = 20 | 2.71 | −0.49 | 1.91 | 0.87 | 0.59 |
As expected, the Cu55(O)2N clusters become more distorted on the Al2O3 substrate (Fig. 4). The number of oxygen atoms at the Cu55(O)2N/Al2O3 interface increases from 1 to 3, then to 6 as N increases. The interfacial oxygen atoms are closer to the Al2O3 surface than Cu atoms. The Cu–O bond lengths across the interface are always greater than 2.0 Å, while the O–Al bonds increase from 1.83 Å to 1.91 Å (Table 1), i.e. the distance between the cluster and substrate increases with N. The corresponding binding energies decrease from 2.87 eV to 2.17 eV, and CDDs in Fig. 4 show how the interaction at the cluster–support interface changes as there is more weight on the cluster itself for Cu55(O)40. According to the Bader charge analysis (Table 1), Cu55(O)2 carries a positive net charge, which indicates that the cluster has transferred 0.22 electron to the γ-Al2O3(100) surface. The other two systems carry about 0.5 e negative net charge, each, indicating charge transfer from the support. On the whole, the charge transfer between Cu55(O)2N and Al2O3 is weak, which is consistent with the previous reports43,44 where charge transfers of similar magnitude were reported. For example, the net charge of Cu is 0.08 e and 0.15 e, respectively, for the Cu2 cluster adsorbed on Al2O343 and ZnO44 surfaces. In addition, it has been reported43 that the charge may be locally transferred from Cu to the Al2O3 carrier, and the transfer of electron charge further decreases during Cu deposition.
To understand how the electronic structure is modified due to oxygen coverage, we have performed a detailed analysis of the Kohn–Sham states by examining the projected density of states (PDOS) from the contribution of different atom-centered orbital components (s, p, d). Fig. 4 shows the PDOS of Cu55 with (O)2N (N = 1, 10, and 20) coverage on γ-Al2O3(100). PDOS is projected with respect to the Cu atoms in the cluster and O atoms in the coverage, and the spin-up and spin-down contributions are plotted separately (paramagnetic system). PDOS shows an interesting overall evolution at the Fermi energy where both Cu55 and oxygen weights gradually increase with oxidation such that the N = 20 case exhibits an enhanced metallic character. The electronic states near the Fermi energy have a major influence on the p states of O atoms, and the underlying valence band PDOS corresponds mainly to the atomic d states of Cu55. Thus, the bonding of O atoms with Cu results in a strong hybridization between the O p-states and the Cu d-states. Moreover, the d-band center (Cd) is also shown in Fig. 4. The d-band center moves downward with an increase in the oxygen coverage, which is similar to the isolated Cu55(O)2N clusters (Fig. S7, ESI†). For N = 1, the Cd value decreases to −2.51 eV compared to the −2.44 eV in the isolated Cu55(O)2 cluster which is related to the charge transfer from Cu55(O)2 to the substrate. In contrast, the Cd values increase for the other two systems (−2.55 eV and −2.75 eV) in comparison with the isolated clusters. The reason is that Cu55(O)2N receive electron density from the support in these cases, which lifts the d-band center closer to the Fermi level.
The catalytic reaction pathways and CO oxidation energetics are shown in Fig. 5. The reaction paths are similar to those on the isolated Cu55(O)2N clusters (Fig. 3). The activation energy barriers are 0.57 eV, 0.65 eV and 0.59 eV for N = 1, 10 and 20, respectively. The CO adsorption energies for the corresponding IS are 1.06 eV, 1.00 eV and 0.87 eV, respectively, showing a gradual decrease versus the number of oxygen atoms. The CO oxidation activation energy barriers are of the same magnitude as that of the isolated Cu55(O)2N clusters. For N = 10 and 20, the reaction barriers are slightly higher (by <0.1 eV) and this correlates with the larger CO adsorption energy for the supported clusters. Here, the charge transfer from Al2O3 to Cu55(O)2N (N = 10 and 20) increases the adsorption strength of CO, which slightly increases the reaction barrier.
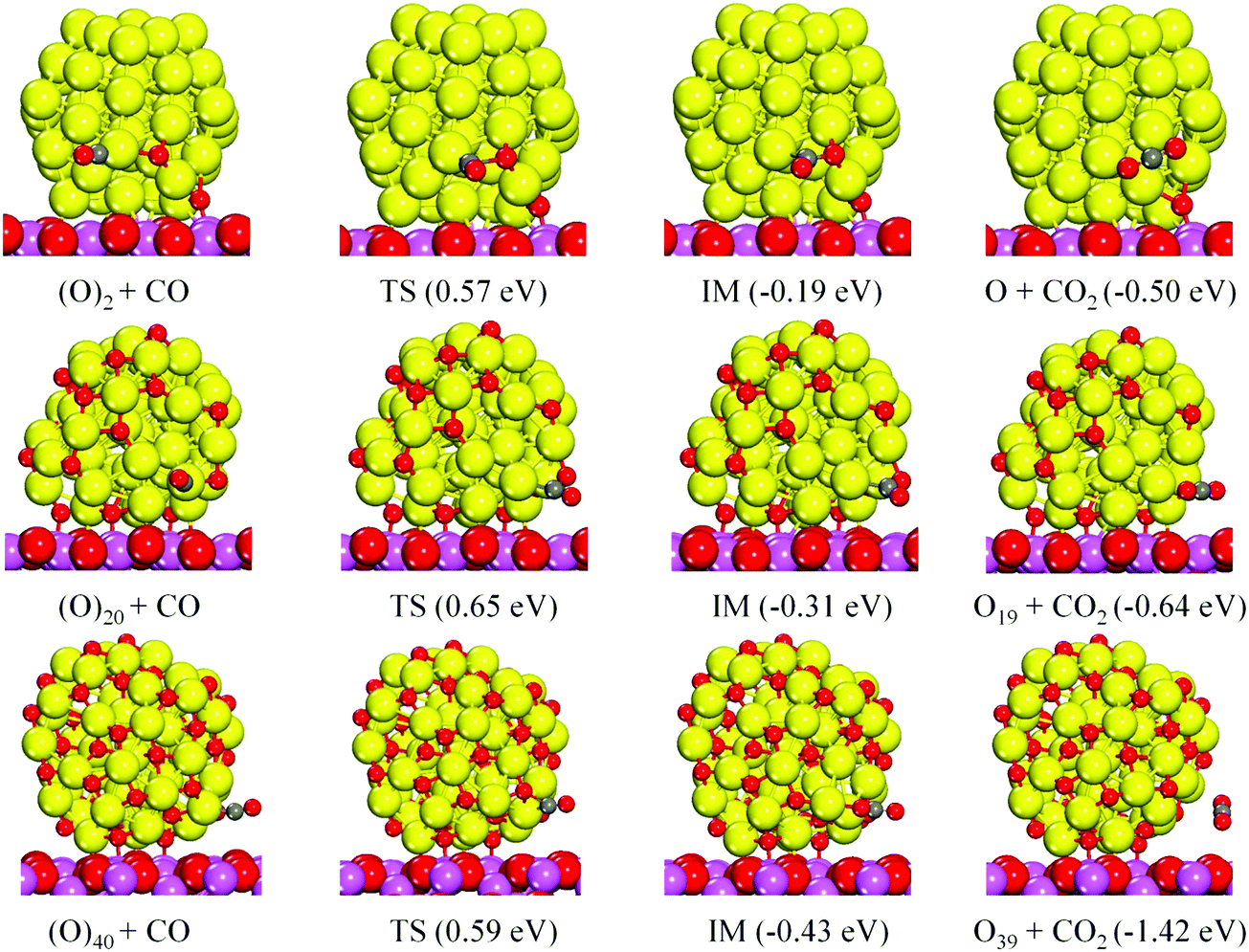 | ||
| Fig. 5 Structures of the initial state (IS), transition state (TS), intermediate (IM), and final state (FS) for the catalytic CO oxidation on Cu55(O)2N/γ-Al2O3(100) (N = 1, 10, 20) and the energy changes with respect to the IS. | ||
Finally, we remark that some experiments20–22 indicate that the reaction order of oxygen is negative over metallic Cu, which is due to the fact that adsorbed atomic O interacts strongly with the metal surface and blocks the active sites so that CO cannot adsorb. However, there is also opposite evidence11 which shows that the reaction with CO2 at high gas pressures occurs both via scattering of gas-phase CO molecules and by CO molecules weakly adsorbed at vacancies in the oxygen adlayer, i.e. the Eley–Rideal (E–R) mechanism contributes to the reaction. Here, we tested the E–R mechanism for Cu55(O)40 and Cu55(O)40/γ-Al2O3(100) (Fig. S8, ESI†), and the reaction barriers are clearly higher than those for the L–H mechanism. In our calculations, as the oxygen coverage increases up to N = 20, the adsorbed CO molecule and the neighboring O atom come close to one another while binding more weakly on Cu55, and this is reflected in the modest reaction barrier height.
4. Conclusion
We have systematically studied the adsorption of several (N = 1–20) oxygen molecules on the icosahedral Cu55 cluster using the spin-polarized DFT calculations. Herein, we must emphasize the representative nature of the Cu55(O)2N clusters obtained, as it is very difficult to obtain the true ground states for such numbers of atoms via DFT calculations. Based on these model structures, we focus on the behavior of oxygen on Cu55, cluster–support interaction, and the effects of oxygen coverage on CO oxidation.As the oxygen coverage increases, several adsorbed O atoms gradually move from convex sites, to surface, and to concave sites. The first O atom that spontaneously relaxes to an endohedral site of Cu55 is observed at N = 13. Furthermore, the endohedral O atoms increase with greater oxygen exposure. The O–O bond cleavage processes on Cu55 with varying numbers of oxygen atoms are analyzed, and it is observed that O2 gets activated upon adsorption and it dissociates either spontaneously or with a very small barrier. A short DFT-MD simulation (20 ps) at 300 K confirms the small dissociation barrier and the active movement of oxygen atoms from the surface to the interior sites at larger oxygen coverages. Furthermore, CO oxidation reactions have been studied on Cu55(O)2N and Cu55(O)2N/γ-Al2O3(100) (N = 1, 10, and 20) systems with fully dissociated oxygen molecules. For Cu55(O)2N/γ-Al2O3(100), the interaction between the cluster and the substrate decreases as the distance between them increases with the number of oxygen atoms, N, and the charge transfer across the Cu55(O)2N/Al2O3 interface is weak. The CO oxidation reaction order is positive over oxygen coverage on both Cu55(O)2N and Cu55(O)2N/γ-Al2O3 systems with similar reaction barrier heights (0.50–0.65 eV). This is due to the inherently small adsorption energies of the reactants and their modest variation as a function of the varying oxygen coverage and the presence of the Al2O3 support.
Finally, it should be noted that the range of adsorbed oxygen is still limited in this work. The Cu55 cluster reaches a maximum content of 40 oxygen atoms (i.e. beyond Cu2O but not yet at CuO), and, as evidenced by the results (small number of endohedral O atoms; modest reaction barriers), it appears that the cluster can host even more oxygen without losing its catalytic activity. In particular, it is likely that the number of endohedral oxygen atoms will increase further with increasing oxygen content.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
DFT calculations were carried out at CSC – the IT Center for Science Ltd, Espoo, Finland. Financial support has been provided by the European Union Horizon 2020 NMP programme through the CritCat Project under Grant Agreement No. 686053.References
- X. Huang, S. Tang, X. Mu, Y. Dai, G. Chen, Z. Zhou, F. Ruan, Z. Yang and N. Zheng, Nat. Nanotechnol., 2011, 6, 28–32 CrossRef CAS PubMed.
- L. Hu, X. Cao, D. Ge, H. Hong, Z. Guo, L. Chen, X. Sun, J. Tang, J. Zheng, J. Lu and H. Gu, Chem. – Eur. J., 2011, 17, 14283–14287 CrossRef CAS PubMed.
- C. Chiu, P. Chung, K. Lao, C. Liao and M. Huang, J. Phys. Chem. C, 2012, 116, 23757–23763 CrossRef CAS.
- N. Iwasa, S. Arai and M. Arai, Appl. Catal., B, 2008, 79, 132–141 CrossRef CAS.
- Y. Chan and J. Chen, Appl. Surf. Sci., 2016, 387, 16–27 CrossRef.
- T. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- S. De, J. Zhang, R. Luque and N. Yan, Energy Environ. Sci., 2016, 9, 3314–3347 RSC.
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC.
- C. Zhou, J. N. Beltramini, C. Lin, Z. Xu, G. Q. Lu and A. Tanksale, Catal. Sci. Technol., 2011, 1, 111–122 RSC.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. Kniep, M. Tovar, R. W. Fischer, J. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- C. Stampfl and M. Scheffler, Surf. Sci., 1997, 377, 808–812 CrossRef.
- J. Gustafson, S. Blomberg, N. M. Martin, V. Fernandes, A. Borg, Z. Liu, R. Chang and E. Lundgren, J. Phys.: Condens. Matter, 2014, 26, 055003 CrossRef CAS PubMed.
- S. Penner, P. Bera, S. Pedersen, L. T. Ngo, J. J. W. Harris and C. T. Campbell, J. Phys. Chem. B, 2006, 110, 24577–24584 CrossRef CAS PubMed.
- H. Zhou, X. Chen and J. Wang, Int. J. Quantum Chem., 2016, 116, 939–944 CrossRef CAS.
- X. Yuan, L. Liu, X. Wang, M. Yang, K. Jackson and J. Jellinek, J. Phys. Chem. A, 2011, 115, 8705–8712 CrossRef CAS PubMed.
- E. Fernández, M. Boronat and A. Corma, J. Phys. Chem. C, 2015, 119, 19832–19846 CrossRef.
- E. Florez, W. Tiznado, F. Mondragón and P. Fuentealba, J. Phys. Chem. A, 2005, 109, 7815–7821 CrossRef CAS PubMed.
- T. Huang and D. Tsai, Catal. Lett., 2003, 87, 173–178 CrossRef CAS.
- Y. Bu, J. Niemantsverdriet and H. Fredriksson, ACS Catal., 2016, 6, 2867–2876 CrossRef CAS.
- G. Jernigan and G. Somorjai, J. Catal., 1994, 147, 567–577 CrossRef CAS.
- M. Domagala and C. Campbell, Catal. Lett., 1991, 9, 65–70 CrossRef CAS.
- J. Szanyi and D. Goodman, Catal. Lett., 1993, 21, 165–174 CrossRef CAS.
- L. Ma, M. Melander, K. Laasonen and J. Akola, Phys. Chem. Chem. Phys., 2015, 17, 7067–7076 RSC.
- A. Zedan, A. Mohamed, M. El-Shall, S. AlQaradawi and A. AlJaber, RSC Adv., 2018, 8, 19499–19511 RSC.
- H. Elazab, Biointerface Res. Appl. Chem., 2018, 8, 3278–3281 Search PubMed.
- L. Gong, L. Luo, R. Wang and N. Zhang, J. Chil. Chem. Soc., 2012, 57, 1048–1053 CrossRef CAS.
- N. Soliman, J. Mater. Res. Technol., 2019 DOI:10.1016/j.jmrt.2018.12.012.
- D. Tang and J. Zhang, RSC Adv., 2013, 3, 15225–15236 RSC.
- M. Trueba and S. Trasatti, Eur. J. Inorg. Chem., 2005, 3393–3403 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- D. Widmann and R. Behm, Acc. Chem. Res., 2014, 47, 740–749 CrossRef CAS PubMed.
- J. Lin, X. Wang and T. Zhang, Chin. J. Catal., 2016, 37, 1805–1813 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- CP2K version 2.5. CP2K is freely available from http://www.cp2k.org.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703 CrossRef CAS.
- M. Honkanen, J. Wang, M. Kärkkäinen, M. Huuhtanen, H. Jiang, K. Kallinen, R. L. Keiski, J. Akola and M. Vippola, J. Catal., 2018, 358, 253–265 CrossRef CAS.
- M. Yu and D. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- G. Henkelman, B. Uberuage and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. A. Nosé, J. Chem. Phys., 1984, 81, 511 CrossRef.
- A. Pelzer, J. Jellinek and K. Jackson, J. Phys. Chem. A, 2015, 119, 3594–3603 CrossRef CAS PubMed.
- Z. Zuo, L. Wang, P. Han and W. Huang, Appl. Surf. Sci., 2014, 290, 398–404 CrossRef CAS.
- Z. Zuo, P. Han, Z. Li, J. Hu and W. Huang, Appl. Surf. Sci., 2012, 261, 640–646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of stepwise adsorption of O2 molecules; optimized geometries of Cu55 and Cu55/γ-Al2O3(100) (Fig. S1); geometries of Cu55O2N with the number of O2 dissociation increasing from zero to seven (Fig. S2); The full set of geometries for the stepwise adsorption of O2 molecules (N = 1–20) on Cu55 (Approach 1) (Fig. S3); geometries of Cu55O2N (N = 10) at 0 K and after 20 ps of MD at 300 K (Fig. S4); adsorption energy Ea(N) of each O2 and average adsorption energy 〈Ea(N)〉 of added O2 molecules versus the number of O2 added on the Cu55 cluster (Approach 1) (Fig. S5); properties of the stepwise O2 adsorption complexes Cu55O2N (N = 1–20, Approach 1) (Table S1); the full set of geometries for stepwise adsorption and dissociation of O2 molecules (N = 1–20) on Cu55 (Approach 2) (Fig. S6); spin-polarized Cu55 d-projected density of states (d-PDOS) with (O)2N (N = 1, 10, and 20) coverage (Fig. S7); properties of the stepwise O2 adsorption and dissociation complexes Cu55O2N (N = 1–20, Approach 2) (Table S2); structures and energetics of the catalytic CO oxidation on Cu55(O)40 and Cu55(O)40/γ-Al2O3(100) by the Eley–Rideal (ER) mechanism (Fig. S8). See DOI: 10.1039/c9cp00974d |
| This journal is © the Owner Societies 2019 |