
Distance-dependent formation of electronic charge-transfer states in the ground states of anthracene and pyrene covalently linked to a TEMPO free radical†
Alok Kumar
Tripathi
 ,
Sushma
Kundu
and
Ranjan
Das
*
,
Sushma
Kundu
and
Ranjan
Das
*
Tata Institute of Fundamental Research, Homi Bhabha Road, Mumbai 400005, India. E-mail: ranjan@tifr.res.in
First published on 5th December 2018
Abstract
Charge-transfer (CT) electronic states are generally seen in molecules involving interactions between species of low ionization potential and high electron affinity. In this context, the 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) free radical is not considered to be a typical molecule to form charge transfer states with aromatic hydrocarbons. Nevertheless, involvement of such CT states has been invoked in rationalising the spin-dependent photophysical quenching of excited states of aromatic systems by TEMPO during bimolecular collisions. Direct observation of such CT states, however, has been elusive until recently, with our first report on the observation of CT states involving naphthalene and TEMPO moieties covalently linked through a spacer group (Rane et al., J. Fluoresc., 2015, 25, 1351–1361). With a view to demonstrating more systems of CT states involving a TEMPO donor, and establishing a possible dependence on its distance from an acceptor chromophore, we have now extended our investigation to anthracene (An) and pyrene (Py) moieties linked to TEMPO, using two different spacer groups of different lengths. The molecules are An–CH2–O–TEMPO, Py–CH2–O–TEMPO, Py–(CH2)2–O–TEMPO, Py–(CH2)4–O–TEMPO, Py–CH2–CO–O–TEMPO and Py–(CH2)3–CO–O–TEMPO, where a linear alkyl chain containing an ether or an ester moiety constitutes the spacer group. We established the formation of CT states in their ground states by comparing their electronic absorption spectra, steady-state fluorescence spectra and time-resolved fluorescence signals with those of the parent molecules An–CH2–OH, Py–CH2–OH and Py–CH2–COOH. CT bands of appreciable intensity were seen only with An–CH2–O–TEMPO, Py–CH2–O–TEMPO and Py–CH2–CO–O–TEMPO, the molecules with the shortest spacer group. Approximate shapes of the absorption and emission bands of the CT states have been determined. For the rest, very weak bands were seen. Similar trends were seen in their fluorescence lifetimes also. Absorption intensities of the CT bands were found to decrease exponentially with the length of the spacer group. The presence of the ether or the ester moiety in the spacer groups showed little influence on the intensities of the CT bands. Our results are probably the first experimental demonstration of the expected exponential dependence of the efficiency of the formation of CT states on the length of the spacer groups of chromophore–TEMPO linked molecules.
Introduction
When a species of low ionization potential (IP) meets another species of high electron affinity (EA), a partial or total transfer of electronic charge between them may take place, leading to the formation of ‘charge-transfer’ (CT) electronic states, which are distinct from the electronic states of the individual species. Though, in general, the CT states can form when both the species are in their ground states or one of them is in its electronic excited state, it is in the latter case that CT states in organic molecules are more readily seen. This is because a molecule in an electronic excited state has a lower IP and a higher EA than when it is in its ground state, making the excited molecule a good electron donor as well as a good electron acceptor [ref. 1, page 249]. Some of the extensively studied CT complexes involve naphthalene, anthracene and pyrene as electron donors, and iodine, p-chloranil, 1,3,5-trinitrobenzene and tetracyanoethylene as electron acceptors.2 Amines are also known to act as a good electron donor. CT complexes of different amines with pyrene as an electron acceptor have been well studied [ref. 1, page 256]. In this context, CT complexes involving a nitroxyl radical are not as ubiquitous.2 Nevertheless, nitroxyl radicals are known to quench both excited singlet and triplet states, and involvement of intermediate CT states during their quenching process has been invoked occasionally.3–6When a doublet free radical meets another species in an excited singlet or a triplet state, the overall spin state of the pair can be a doublet or a quartet. As a result, an otherwise spin-forbidden intersystem crossing (ISC) process between the singlet and the triplet states becomes spin-allowed. The quenching of the excited state often takes place through this spin-allowed enhanced intersystem crossing (EISC) process. Spin-dependent magnetic interactions arising from the zero-field splitting of the triplet or the hyperfine interaction of the radical can cause mixing of the sublevels of the electronic states to different extent, and consequently, during the quenching process, the spin distribution of the radical usually deviates from its Boltzmann distribution. Observation of such electron spin polarization (ESP) and its evolution gives rich insight into the dynamics of radical-induced photophysical quenching of excited states.
The doublet and quartet spin states of the radical-excited molecule pair, as alluded to earlier, are generally a linear combination of the doublet radical in the ground state and the locally excited singlet or triplet state of the chromophore of a molecule. Such a linear combination of a doublet state and a triplet state can produce a doublet and a quartet state. To specifically indicate the overall spin states of the pair in terms of the local spin states of the two entities, these states are generally called sing-doublet (the chromophore in excited singlet state, the radical in ground doublet state, and the overall spin is a doublet), trip-doublet (the chromophore in excited triplet state, the radical in ground doublet state, and the overall spin is a doublet), or trip-quartet (the chromophore in excited triplet state, the radical in ground doublet state, and the overall spin is a quartet).7
Even though the quenching dynamics and ESP generation in a nitroxyl radical participating in photophysical quenching of the excited state of a large number of molecules can be well-understood in terms of the overall spin states of the pair mentioned above, there are instances when additional electronic states of charge-transfer type have become necessary to rationalize the observation.4–6 When the chromophore and the free radical are independent entities freely diffusing in the solution, however, it is extremely difficult to detect the presence of any charge-transfer state. It is because such CT states have only transient existence, as only when the two species are in close proximity, allowing significant overlaps of their electronic orbitals, can such states develop. Until recently, CT states involving the 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) free radical and its derivatives have been reported only with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (TCNQF4) and 2,3-dichloro-5,6-dicyanobenzo-1,4-quinone (DDQ), which are very strong electron acceptors (standard reduction potential: +0.57 V (TCNQF4) and +0.47 V (DDQ)).8 Salts of these donor–acceptor complexes are realized in the solid state. These salts are, however, not representatives of the scenario of the TEMPO radical quenching an excited chromophore by random encounters while diffusing freely in solutions. When the TEMPO radical and the chromophore are covalently linked together, their random diffusive motion is severely restricted. Mutual electronic interactions through the covalent bonds joining the two moieties may facilitate formation of persistent CT states. The first demonstration of this was in the case of a naphthalene (Nap) moiety covalently linked to a TEMPO moiety through a spacer group of different lengths.9 By a combined study of electronic absorption and fluorescence spectroscopy, formation of a weak CT state in the ground state of Nap–CH2–O–TEMPO has been reported. The intensity of the CT band diminishes on increasing the length of the spacer group in the series Nap–CH2–O–TEMPO, Nap–(CH2)2–O–TEMPO and Nap–CH2–O–(CH2)4–O–TEMPO.
The present work was undertaken with a view to finding more systems forming CT states involving a TEMPO moiety, and establishing the dependence of the efficiency of their formation on the distance of separation between the chromophore and the radical. Here we chose pyrene (Py) and anthracene (An) as two acceptor moieties and covalently linked them to TEMPO through a spacer group of different lengths, with the expectation that CT states would form more prominently in them than in the naphthalene–TEMPO linked systems. The basis of such an expectation is that the EA of these two acceptors is higher than that of naphthalene (reduction potentials with respect to Ag/Ag+: naphthalene, −2.53 V; anthracene, −2.04 V; pyrene −2.29 V),10 and hence, they should form CT states more easily. Along with changing the length of the spacer group, we also changed the nature of the spacer by incorporating either an ether or an ester moiety in the linear alkyl chains of the spacer groups. Here we report our findings on the observation of CT states in these different sets of linked molecules, and their dependence on the length and nature of the spacer groups.
Materials and methods
Molecular structures and the symbolic names of the molecular systems studied here are given in Chart 1. The systems belong to three classes: anthracene linked to TEMPO through an alkyl–ether linkage (An–CH2–O–TEMPO), pyrene linked to TEMPO through an alkyl–ether linkage (Py–CH2–O–TEMPO, Py–(CH2)2–O–TEMPO and Py–(CH2)4–O–TEMPO), and pyrene linked to TEMPO through an alkyl–ester linkage (Py–CH2–(CO)–O–TEMPO and Py–(CH2)3–(CO)–O–TEMPO). In all these molecules, there is at least one –CH2– group separating the aromatic moiety from other functional groups. This was done to minimize perturbation to the electronic states of the aromatic group on forming the linked molecules. Different lengths of the spacer groups should reveal the distance dependence of the efficiency of the formation of CT states. In addition, the two sets of molecules with spacer groups containing the ether or the ester group could be used to examine the influence of the spacer group on the formation of the CT states. We used the term “unlinked system” to mean a homogeneous solution containing the chromophore molecule (An–CH2–OH, Py–CH2–OH or Py–CH2–COOH) and 4-hydroxy-TEMPO (TEMPOL) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio (the three pairs in the bottom row of Chart 1). These aromatic molecules too have a –CH2– group separating the functional groups from the chromophores. This should ensure that the chromophores of both linked molecules and their “unlinked” partners have similar electronic states, and any observed difference could be ascribed only to the interaction between the chromophore and the TEMPO moieties because of their being in close proximity.
:1 mole ratio (the three pairs in the bottom row of Chart 1). These aromatic molecules too have a –CH2– group separating the functional groups from the chromophores. This should ensure that the chromophores of both linked molecules and their “unlinked” partners have similar electronic states, and any observed difference could be ascribed only to the interaction between the chromophore and the TEMPO moieties because of their being in close proximity.
 | ||
| Chart 1 The structure of the molecules and their symbolic names, used in this study. In this report, the pairs of molecules, in the bottom row, are called “unlinked systems”, which contain equimolar concentrations of the aromatic molecule and TEMPOL. | ||
The detailed synthetic procedures and characterization of all the linked molecules have been described in the ESI.† The purity of these newly synthesized molecules was checked by NMR and fluorescence spectroscopy. 9-Anthracenemethanol, 1-pyrenemethanol, 1-pyreneacetic acid, 1-pyrenebutyric acid, 4-hydroxy-TEMPO, sodium hydride and all other chemicals were purchased from Sigma-Aldrich (USA) and used without further purification. The ground-state electronic absorption experiments were done by using a Lambda 25 UV/vis spectrophotometer (Perkin Elmer, USA). Steady-state fluorescence measurements were done by using a JASCO Spectro-fluorometer FP-8300. Fluorescence lifetimes were recorded by using a picosecond spectrometer using the time-correlated single-photon counting technique (TCSPC), built in the laboratory. The excitation source for these measurements was either a frequency-doubled output of a mode-locked picosecond Ti-Sapphire laser (Tsunami, SpectraPhysics, USA) (720–900 nm) pumped by a diode-pumped CW Nd:Yttrium-vanadate laser (532 nm) (Millennia X, Spectra-Physics, USA) for anthracene systems, or a frequency-doubled output of a rhodamine 6G dye laser (Spectra Physics, USA) pumped by a passively mode-locked, frequency-doubled Nd:YAG laser (Vanguard, Spectra Physics) for pyrene systems. For recording the TCSPC experiments we used deoxygenated samples. Deoxygenation of the samples was done by bubbling pure and dry nitrogen gas. The solvents used in this study were of spectroscopic grade. All measurements were carried out at room temperature of 21 °C.
Results and discussion
Steady-state UV-vis absorption spectroscopic measurements
Anthracene and pyrene chromophores have a strong absorption band around 200–400 nm (molar absorptivity ∼7200 dm3 mol−1 cm−1, at 340 nm) and TEMPO has a relatively weak band (molar absorptivity ε ∼ 10 dm3 mol−1 cm−1 at 475 nm) around 400–550 nm. Therefore, to record these two bands with modest absorbance, spectra were recorded at concentrations of 20 μM and 10 mM, respectively (Fig. 1). There was no measurable difference in the intensities of the chromophore band of An–CH2–O–TEMPO, An–CH2–OH and the “unlinked system” of An–CH2–OH + TEMPOL (Fig. 1a). This shows that there was little interaction between the anthracene and the TEMPO moieties of An–CH2–O–TEMPO to influence the local excitation of anthracene. Similarly, little change in the intensity of absorption was seen in the red-edge region (>∼450 nm) of TEMPO in these systems (Fig. 1b). In the 420 nm region, however, extra absorbance was noted only for An–CH2–O–TEMPO. Specifically, while the intensity of the “unlinked system” matched with the sum of the spectra of An–CH2–OH and TEMPOL (each 10 mM), indicating no bimolecular electronic interaction between them at this concentration, the enhanced absorption around 400 nm in the linked molecule An–CH2–O–TEMPO was prominently seen. | ||
| Fig. 1 Absorption spectra of An–CH2–OH, An–CH2–OH + TEMPOL (1:1) and An–CH2–O–TEMPO (a), TEMPOL, An–CH2–OH, An–CH2–OH + TEMPOL (1:1) and An–CH2–O–TEMPO (b), TEMPOL, Py–CH2–OH, Py–CH2–OH + TEMPOL (1:1), Py–(CH2)4–O–TEMPO, Py–(CH2)2–O–TEMPO and Py–CH2–O–TEMPO (c), and TEMPOL, Py–CH2–COOH, Py–CH2–COOH + TEMPOL (1:1), Py–(CH2)3–CO–O–TEMPO and Py–CH2–CO–O–TEMPO (d) in acetonitrile. In (b), the sum of the spectra of TEMPOL (black) and An–CH2–OH (red) exactly matched with the spectrum of the “unlinked system” An–CH2–OH + TEMPOL (blue). Concentrations of the samples: 20 μM in (a), and 10 mM in (b), (c) and (d). | ||
The absorption spectra of the pyrene systems showed qualitatively similar features as those of their anthracene analogs, when compared with the corresponding “unlinked system” of Py–CH2–OH + TEMPOL or Py–CH2–COOH + TEMPOL (Fig. 1c and d). The spectra in the UV region of the pyrene moiety alone of these two systems are shown in ESI,† Fig. S5 and S6.
From the intensities of this extra absorption around 420 nm, we concluded that the intensity of this band was maximum when pyrene and –O–TEMPO moieties were separated by a single methylene group (–CH2–), and it progressively decreased with the separation between them, becoming non-discernible when this separation became longer than two carbon atoms (either –CH2–CH2– or –CH2–(CO)–). We obtained an approximate shape of this extra absorption band around 420 nm, by subtracting the absorbance of the “unlinked system” from that of the linked molecules, recorded under identical conditions (ESI,† Fig. S1). We attributed this band to a CT band in the ground state, which is similar to what has been reported in naphthalene–TEMPO linked molecules.9
Steady-state fluorescence measurements
 | ||
| Fig. 2 Fluorescence emission spectra of An–CH2–OH, An–CH2–OH + TEMPOL and An–CH2–O–TEMPOL at 20 μM in acetonitrile (a) at λex = 340 nm and (c) at λex = 440 nm. Fluorescence excitation spectra of An–CH2–OH, An–CH2–OH + TEMPOL and An–CH2–O–TEMPO at 20 μM in acetonitrile (b) at λem = 420 nm and (d) at λem = 550 nm. The sharp peaks around 475–525 nm in (c) are Raman lines of acetonitrile. | ||
Fluorescence emission spectra were also recorded by exciting at λex = 440 nm (approximately in the CT absorption band shown in Fig. 1a). Here, the emission spectra were much weaker than those in Fig. 2a, and also contaminated by three prominent Raman lines of solvent acetonitrile (Fig. 2c). Despite that, the overall intensity pattern around 500–650 nm of An–CH2–O–TEMPO was found to be perceptively different from that of An–CH2–OH or the “unlinked system” An–CH2–OH + TEMPOL, indicating a possible small contribution from the CT state in the former.
The fluorescence excitation spectra of this system were recorded by monitoring the emission at λem = 440 nm (emission from the anthracene moiety (Fig. 2b)). Once again, whereas the spectra of An–CH2–OH and the “unlinked system” An–CH2–OH + TEMPOL were indistinguishable, the spectrum of An–CH2–O–TEMPO was somewhat different. This difference was more pronounced when the excitation spectra were recorded at the emission wavelength λem = 550 nm (emission from the possible CT state) (Fig. 2d). This observation thus reinforced our earlier inference from the electronic absorption spectra (Fig. 1b) that, in the linked molecule An–CH2–O–TEMPO, there was a weak absorption band lying slightly red-shifted with respect to the absorption band of the anthracene moiety. We therefore concluded that there was a CT state of An–CH2–O–TEMPO in its ground state.
When the solvent was changed from polar acetonitrile to non-polar n-hexane, very similar spectral properties were seen. The extra absorption band (data not shown here) similar to that in Fig. 1a, pertaining to the CT absorption, and also similar fluorescence and excitation spectra were seen (Fig. 3). The fluorescence intensity of the chromophore was substantially reduced in An–CH2–O–TEMPO, compared to that of An–CH2–OH and the “unlinked system” An–CH2–OH + TEMPOL (Fig. 3a). Enhanced extra emission from the CT state (Fig. 3c) and a distinct difference in the fluorescence excitation spectra of An–CH2–O–TEMPO, compared to An–CH2–OH and the “unlinked system” (Fig. 3b and d), were seen.
 | ||
| Fig. 3 Emission spectra of An–CH2–OH, An–CH2–OH + TEMPOL and An–CH2–O–TEMPO in n-hexane (a) at λex = 340 nm and (c) at λex = 440 nm. Excitation spectra of An–CH2–OH, An–CH2–OH + TEMPO and An–CH2–O–TEMPO in n-hexane (b) at λem = 420 nm and (d) at λem = 550 nm. Conc. of all samples: 20 μM. The sharp peak near 500 nm in (c) is a Raman line of n-hexane. | ||
The effect of solvent polarity on the spectra of An–CH2–O–TEMPO could be more readily seen, when the background signal from the solvent along with its Raman lines was subtracted from the spectra of the molecules recorded under identical conditions (Fig. 4). The fluorescence intensity of the linked molecule was higher than that of the “unlinked system” (Fig. 4a and b), indicating the extra intensity to arise from the possible CT states. This is consistent with the enhanced intensity of the excitation spectrum in polar acetonitrile compared to that in non-polar n-hexane (Fig. 4c).
 | ||
| Fig. 4 Fluorescence emission spectra of An–CH2–OH, An–CH2–OH + TEMPOL and An–CH2–O–TEMPOL at λex = 450 nm in acetonitrile (a) and in n-hexane (b), after subtraction of Raman lines. Fluorescence excitation spectra of An–CH2–O–TEMPOL in n-hexane and acetonitrile at λem = 450 nm (c). | ||
 | ||
| Fig. 5 Fluorescence emission spectra of Py–CH2–OH, Py–CH2–OH + TEMPOL, Py–(CH2)4–O–TEMPO, Py–(CH2)2–O–TEMPO and Py–CH2–O–TEMPO at 20 μM in acetonitrile at λex = 340 nm (a) and at λex = 440 nm (c). Fluorescence excitation spectra of Py–CH2–OH, Py–CH2–OH + TEMPOL, Py–(CH2)4–O–TEMPO, Py–(CH2)2–O–TEMPO and Py–CH2–O–TEMPO at 20 μM in acetonitrile at λem = 420 nm (b) and at λem = 550 nm (d). The sharp peaks around 475–525 nm in (c) are Raman lines of acetonitrile. | ||
Fluorescence spectra were also recorded by exciting the molecules at λex = 440 nm (the CT absorption band) (Fig. 5c). The spectra had Raman lines of acetonitrile overlapping with the fluorescence spectra. Despite that, a fluorescence band of good intensity around the 500–600 nm region was seen for Py–CH2–O–TEMPO. This intensity decreased steadily from Py–(CH2)2–O–TEMPO to Py–(CH2)4–O–TEMPO. Under similar conditions, Py–CH2–OH or the “unlinked system” gave fluorescence signals of negligible intensity in this region. This excess intensity observed in the linked molecules may be attributed to their CT emission.
The fluorescence excitation spectra of these systems were recorded by monitoring the emission at λem = 420 nm (emission from the pyrene moiety) (Fig. 5b). The spectra of Py–CH2–OH and the unlinked system Py–CH2–OH + TEMPOL overlapped, but that of the linked molecules Py–(CH2)2–O–TEMPO and Py–(CH2)4–O–TEMPO showed smaller intensity, with little change in their shape. The excitation spectrum of Py–CH2–O–TEMPO, however, was distinctly different from that of the “unlinked system”; it had a distinct red-edge tail, which was more easily seen when the spectra were normalized to the same peak intensity. These similarities and differences became more pronounced when the excitation spectra were recorded by monitoring the emission at λem = 550 nm (emission from the CT state) (Fig. 5d).
When the solvent was changed from polar acetonitrile to non-polar n-hexane, the spectral properties were seen to be qualitatively very similar to what were seen in acetonitrile (Fig. 6): Steady decrease in the fluorescence intensity of the pyrene chromophore from Py–(CH2)4–O–TEMPO to Py–(CH2)2–O–TEMPO to Py–CH2–O–TEMPO, compared to Py–CH2–OH and the “unlinked system” Py–CH2–OH + TEMPOL (Fig. 6a); distinct extra emission from the CT state (Fig. 6c); distinct difference in the excitation spectra of Py–CH2–O–TEMPO and Py–(CH2)4–O–TEMPO, compared to An–CH2–OH or the “unlinked system” (Fig. 6b and d). Little measurable difference in the absorption or the fluorescence spectrum could be detected in Py–CH2–OH and the “unlinked system” Py–CH2–OH + TEMPOL, indicating negligible quenching and formation of CT state at the concentration of TEMPOL used here.
 | ||
| Fig. 6 Fluorescence emission spectra of Py–CH2–OH, Py–CH2–OH + TEMPOL, Py–(CH2)4–O–TEMPO, Py–(CH2)2–O–TEMPO and Py–CH2–O–TEMPO at 20 μM in n-hexane at λex = 320 nm (a) and at λex = 440 nm (c). Fluorescence excitation spectra of Py–CH2–OH, Py–CH2–OH + TEMPOL, Py–(CH2)4–O–TEMPO, Py–(CH2)2–O–TEMPO and Py–CH2–O–TEMPO at 20 μM in n-hexane at λem = 420 nm (b) and at λem = 550 nm (d). The sharp peak near 500 nm in (c) is a Raman line of n-hexane. | ||
All the above studies showed that the TEMPO moiety forms CT electronic states with a pyrene moiety, when linked by an alkyl–ether spacer group of –(CH2)n–O– (n = 1, 2, 4), giving characteristic absorption and emission spectra.
In order to examine the role of the spacer group in the formation of the CT state, we next studied the pyrene–TEMPO linked molecules with an alkyl-ester group of the type –(CH2)n–(CO)–O– (n = 1, 3) as a linker. Evidence of the formation of CT states in these molecules has been seen in their electronic absorption spectra (Fig. 1c).
The fluorescence emission spectra of these linked molecules and the “unlinked system” Py–CH2–COOH + TEMPOL, in acetonitrile solution, are shown in Fig. 7. When excited at 320 nm, corresponding to the absorption of the pyrene moiety, the intensity of the unlinked system was almost identical to that of Py–CH2–COOH (Fig. 7a), showing negligible bimolecular quenching of excited pyrene by the TEMPOL free radical at the concentration used here. The intensities of the linked molecules, at the same concentration, were, however, considerably reduced. The reduction in intensity was most for Py–CH2–CO–O–TEMPO and a little less in Py–(CH2)3–CO–O–TEMPO, indicating the distance-dependent efficiency of intramolecular quenching. No discernible change in the shape of the spectra was seen. These spectra showed the emission from the pyrene moiety. When the molecules were excited at 440 nm, corresponding to the CT absorption band (Fig. 1d), an emission band of good intensity around 500–600 nm could be seen only in Py–CH2–CO–O–TEMPO (Fig. 7c). Fluorescence excitation spectra, when monitored at 420 nm (Fig. 7b) gave the absorption profile of the pyrene moiety in all the systems. But when monitored at 550 nm (Fig. 7d), the linked molecule Py–CH2–CO–O–TEMPO gave a broad featureless band around 300–450 nm, which could be ascribed to its CT absorption band. With the increase in the length of the spacer group, the intensity of this band decreased considerably in Py–(CH2)3–CO–O–TEMPO.
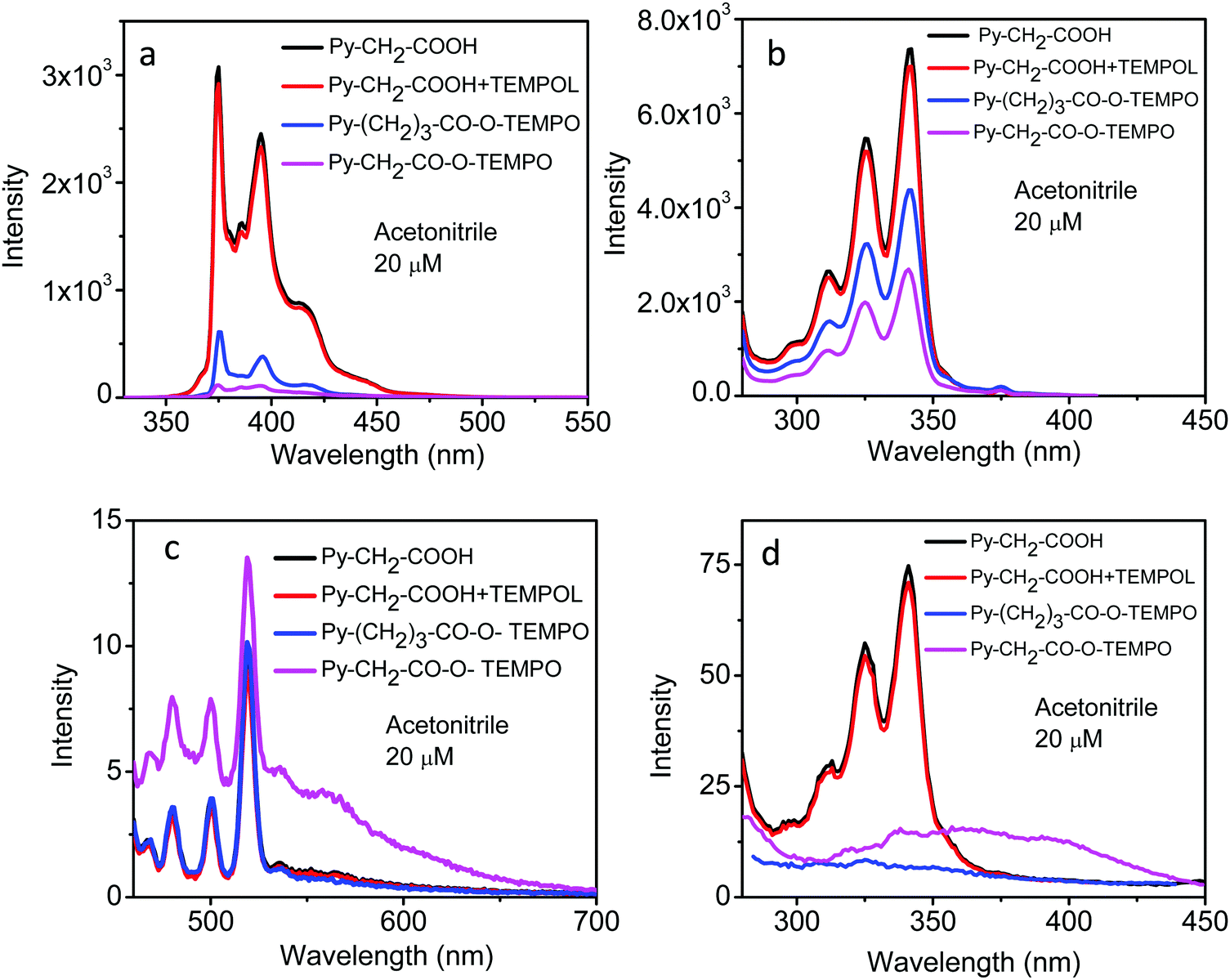 | ||
| Fig. 7 Fluorescence emission spectra of Py–CH2–COOH, Py–CH2–COOH + TEMPOL, Py–(CH2)3–CO–O–TEMPO and Py–CH2–CO–O–TEMPO at 20 μM in acetonitrile at λex = 320 nm (a) and at λex = 440 nm (c). Fluorescence excitation spectra of Py–CH2–COOH, Py–CH2–COOH + TEMPOL, Py–(CH2)4–CO–O–TEMPO and Py–CH2–CO–O–TEMPO at 20 μM in acetonitrile at λem = 420 nm (b) and at λem = 550 nm (d). The sharp peaks around 475–525 nm in (c) are Raman lines of acetonitrile. | ||
When the solvent was changed from polar acetonitrile to non-polar n-hexane, qualitatively very similar spectral properties were seen (Fig. 8): Decrease in the quenching efficiency from Py–CH2–CO–O–TEMPO to Py–(CH2)3–CO–O–TEMPO, with negligible quenching in the “unlinked system” (Fig. 8a); CT emission band for Py–(CH2)–CO–O–TEMPO (Fig. 8c); little change in the shape of the excitation spectra when monitored at 320 nm (Fig. 8b); and a distinctly different excitation spectrum of Py–CH2–CO–O–TEMPO when monitored at 550 nm and attributable to a CT state (Fig. 8d).
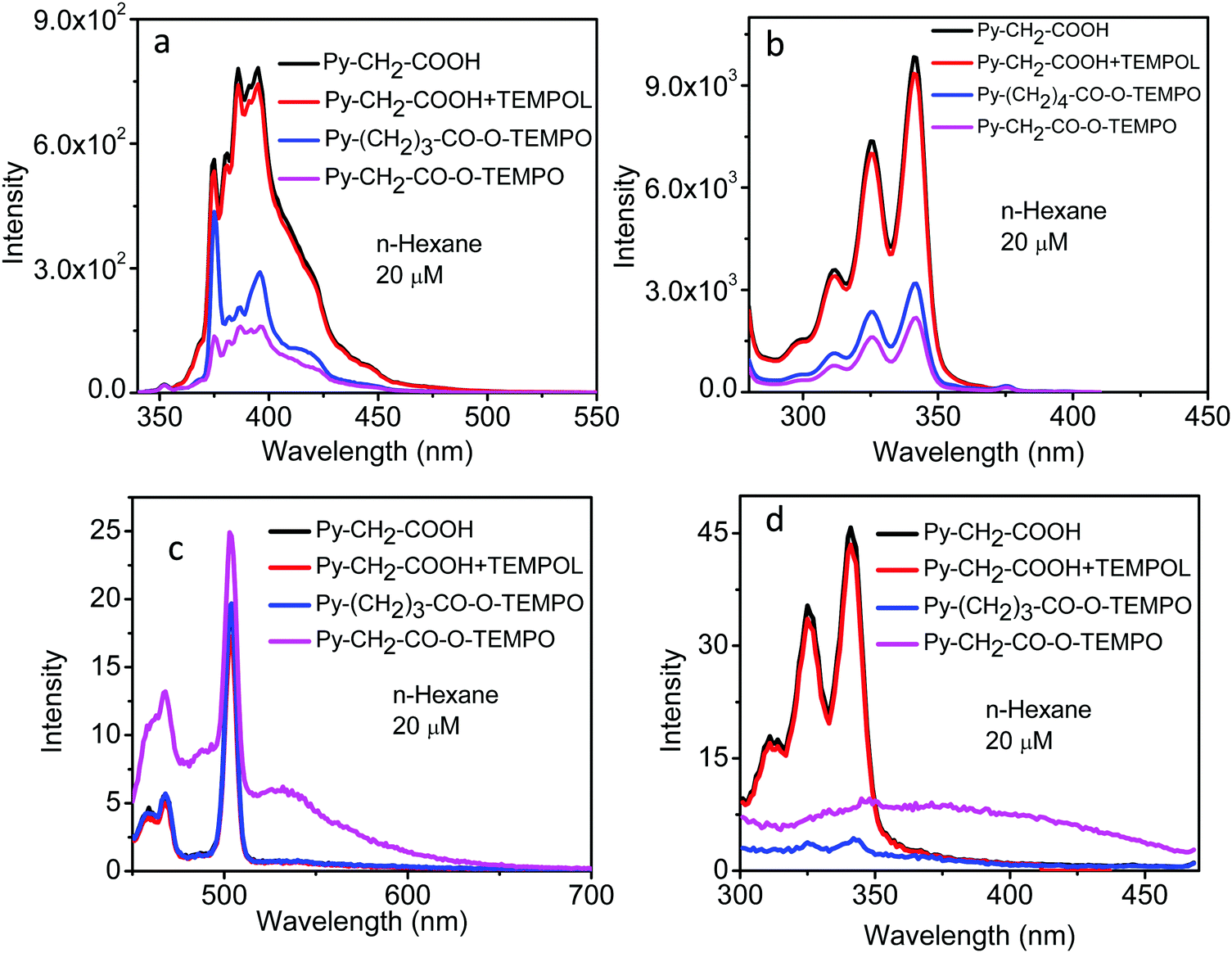 | ||
| Fig. 8 Fluorescence emission spectra of Py–CH2–COOH, Py–CH2–COOH + TEMPOL, Py–(CH2)3–CO–O–TEMPO and Py–CH2–CO–O–TEMPO at 20 μM in n-hexane (a) at λex = 320 nm and (c) at λex = 440 nm. Fluorescence excitation spectra of Py–CH2–COOH, Py–CH2–COOH + TEMPOL, Py–(CH2)4–CO–O–TEMPO and Py–CH2–CO–O–TEMPO at 20 μM in n-hexane at λem = 420 nm (b) and at λem = 550 nm (d). The sharp peak at 500 nm in (c) is a Raman line of n-hexane. | ||
Approximate spectral shapes of the CT states
In order to obtain an approximate shape of the absorption spectra of the charge transfer states of the linked molecules studied here, we subtracted the absorption spectra of the “unlinked systems” from that of the corresponding linked systems, after recording the spectra at the same concentration and under identical experimental conditions. As the absorbance of the chromophore moieties below ∼400 nm was too high to be measured accurately, the difference spectra of the linked molecules could not be obtained below the same wavelength. Therefore, only partial absorption bands of the CT states are shown in ESI,† Fig. S1.The fluorescence emission spectra of these systems have overlapping Raman lines of the solvents (Fig. 5c, 6c, 7c and 8c). To obtain the spectra of the CT states free from these Raman lines, we subtracted from these spectra the spectrum of the neat solvent, recorded under identical experimental conditions. The results are shown in ESI,† Fig. S2 and S3. In a similar manner, the fluorescence excitation spectra of the CT states of the linked molecules were also obtained and are shown in ESI,† Fig. S4. In all cases, the intensities in acetonitrile were found to be higher than that in n-hexane. This is consistent with the polar nature of the CT states, which are more stabilized in the polar solvent.
Time-resolved fluorescence measurements
Having established the presence of CT states of linked molecules containing a TEMPO moiety and anthracene or pyrene as a chromophore moiety, we measured their fluorescence lifetimes using the TCSPC technique. The observed fluorescence decay signals were fit to a sum of exponentials of the type to obtain different lifetimes (τk) and their contributions (αk) to the total intensity. Representative decay signals and multiexponential fits to them are shown in ESI,† Fig. S7. The data are summarized in Table 1. We have not investigated in detail the origin of the small components (α ≤ 5%) that were obtained by the multiexponential fits. As the count rates of fluorescence photons were rather low, particularly in the TEMPO-linked molecules, the TCSPC measurements had to be carried out for about 15 to 30 minutes, for each recording of the time-resolved signal. Because of such prolonged exposure of the samples to the excitation laser, a small amount of decomposition of the molecules is likely. The small components with long lifetimes could be due to such decomposed products. For all the major components, the measured lifetimes were considerably shorter than those of their corresponding “unlinked systems”.
to obtain different lifetimes (τk) and their contributions (αk) to the total intensity. Representative decay signals and multiexponential fits to them are shown in ESI,† Fig. S7. The data are summarized in Table 1. We have not investigated in detail the origin of the small components (α ≤ 5%) that were obtained by the multiexponential fits. As the count rates of fluorescence photons were rather low, particularly in the TEMPO-linked molecules, the TCSPC measurements had to be carried out for about 15 to 30 minutes, for each recording of the time-resolved signal. Because of such prolonged exposure of the samples to the excitation laser, a small amount of decomposition of the molecules is likely. The small components with long lifetimes could be due to such decomposed products. For all the major components, the measured lifetimes were considerably shorter than those of their corresponding “unlinked systems”.
| Molecules | Ex (nm) | Em (nm) | τ 1 (α1) | τ 2 (α2) | τ 3 (α3) | χ 2 |
|---|---|---|---|---|---|---|
| a The values shown here are those obtained in one measurement. We repeated the measurements 3 times, and found that the lifetimes (τ) were accurate within 10%, and the contributions (α) within 5%. | ||||||
| An–CH2–OH | 386 | 420 | 0.85 (0.99) | 63.8 (0.01) | — | 1.2 |
| An–CH2–OH + TEMPOL | 386 | 420 | 0.78 (0.98) | 68.26 (0.02) | — | 1.13 |
| An–CH2–O–TEMPO | 386 | 420 | 0.08 (0.90) | 1.03 (0.09) | 6.35 (0.01) | 1.12 |
| Py–CH2–OH | 324 | 410 | 35.58 (1.0) | — | — | 1.2 |
| Py–CH2–OH + TEMPOL | 324 | 410 | 33.50 (1.0) | — | — | 1.13 |
| Py–CH2–O–TEMPO | 324 | 410 | 0.15 (0.90) | 5.04 (0.07) | 36.74 (0.03) | 1.10 |
| Py–(CH2)2–O–TEMPO | 324 | 410 | 0.43 (0.89) | 4.62 (0.08) | 32.62 (0.02) | 1.20 |
| Py–(CH2)4–O–TEMPO | 0.64 (0.94) | 52.18 (0.06) | — | 1.10 | ||
| Py–CH2–COOH | 324 | 410 | 33.63 (1.0) | — | — | 1.13 |
| Py–CH2–COOH + TEMPOL | 324 | 410 | 30.69 (1.0) | — | — | 1.12 |
| Py–CH2–CO–O–TEMPO | 324 | 410 | 0.74 (0.88) | 1.01 (0.11) | 41.14 (0.01) | 1.20 |
| Py–(CH2)3–CO–O–TEMPO | 324 | 410 | 0.82 (0.74) | 47.70 (0.26) | — | 1.01 |
Time-resolved fluorescence signals of An–CH2–OH showed a predominantly single exponential decay with a lifetime of ≈0.8 ns in nitrogen saturated solutions. In the presence of 20 μM TEMPOL in the same solution, this lifetime showed only a slight decrease (0.7 ns), indicating little bimolecular quenching of the excited state at the concentrations used there. This is consistent with the reported bimolecular quenching constant kQ = 1.55 × 1010 M−1 s−1 of singlet excited state of anthracene by TEMPO.11 For An–CH2–O–TEMPO, the fluorescence decay was predominantly biexponential. The first component of lifetime τ1 ≈ 0.08 ns was attributed to the decay of the locally excited (LE) state of the anthracene moiety, whose lifetime was significantly shortened by the presence of the TEMPO moiety. The second component of lifetime τ2 ≈ 1.0 ns was attributed to the emission from the CT state of An–CH2–O–TEMPO.
The fluorescence lifetime of pyrene is known to be long, 650 ns in cyclohexane.12 Consequently, its measured value is often sensitive to the presence of trace amounts of impurities and dissolved oxygen. Our measured values of the fluorescence lifetime of Py–CH2–OH in nitrogen saturated solutions showed a large variation of about 20% among different samples prepared in a similar manner. Within this variation, the measured lifetime of Py–CH2–OH was found to be ≈35 ns. Similar values were seen, when 20 μM TEMPOL was added to the same sample, indicating negligible bimolecular quenching of the excited singlet state of pyrene at that concentration of the radical. This is again consistent with the reported bimolecular quenching constant of pyrene singlet state by TEMPO of 0.86 × 1010 M−1 s−1.11 Py–CH2–O–TEMPO, in contrast, showed two predominant fluorescence lifetimes. The short component of τ1 ≈ 0.15 ns was attributed to the LE emission of the pyrene moiety, and the long component of τ2 ≈ 4.9 ns was attributed to the emission from the CT state. In Py–(CH2)4–O–TEMPO, with its much longer spacer group, τ1 increased to about ≈0.64 ns, showing reduced effectiveness of quenching of the excited singlet state of pyrene by TEMPO. In this linked molecule, emission from the CT state could not be detected unequivocally, though a long component lifetime τ2 ≈ 52 ns, of small contribution, could be seen.
A similar trend was also seen in the linked molecules Py–CH2–CO–O–TEMPO and Py–(CH2)3–O–TEMPO. The single exponential lifetime of ∼33 ns, in nitrogen saturated solutions of Py–CH2–COOH, changed little (lifetime ∼30 ns) in the presence of 20 μM TEMPOL, again showing negligible bimolecular quenching at that concentration. For Py–CH2–CO–O–TEMPO, a predominantly biexponential decay was seen. The component of τ1 ≈ 0.74 ns was considerably shorter than the lifetime of Py–CH2–COOH (τ ≈ 33 ns), and was assigned to the LE emission. The other component of τ2 ≈ 1 ns was assigned to the CT emission. This component was undetectable in Py–(CH2)3–CO–O–TEMPO.
The time-resolved emission signals from the CT states, as reported above, are consistent with the observation from the characteristic steady-state absorption, fluorescence and excitation spectra, all of which indicate that these states are intramolecular charge-transfer states present in the ground states of the linked molecules.
Charge-transfer states and the distance of separation between the donor and the acceptor moieties
Electron transfer efficiency between a donor and an acceptor molecule depends on the electronic coupling between the two. The magnitude of this coupling generally goes down exponentially with the distance of separation (RDA), since the overlap of their wavefunctions follows the same. For a long-distance electron transfer, where RDA is so large that a direct overlap of the wavefunctions of D and A is negligible, a “superexchange” or “through-bond” coupling mechanism13 has been developed. In this mechanism, the electronic coupling between the donor moiety and the acceptor moiety is mediated via the intervening orbitals of the σ bonds of the spacer group. The electron transfer rate constants (ket) for such systems have been shown to follow the relation| ket = |V0|2ρexp(−β(N − 1)), |
Formation of electronic charge-transfer states may be considered as a “partial” electron transfer, and therefore, its efficiency is expected to follow a similar distance dependence. The efficiency of the formation of CT states associated with the electronic coupling between a specific pair of D and A may be taken to be proportional to exp(−β(N − 1)). We assigned this efficiency to the intensity of the CT states. In our linked molecules, the major part of the spacer group is a linear alkyl group. We assumed that our linked molecules would take extended conformations in the solution. With this assumption, we attempted to correlate the efficiency of formation of the CT state to the length separating the two moieties. The absorption intensity of the CT states of Py–TEMPO linked molecules was calculated by taking the difference between the absorbance values, at 440 nm, of a given linked molecule and that of the “unlinked system”, recorded under identical concentrations. These values are plotted against the number of σ bonds separating the pyrene moiety from the oxygen atom of the 4-oxo group of TEMPO. Fig. 9 shows the results. A small difference could be noted when the spacer group changed from an ether linkage to an ester. Despite the limited number of points, the trend was highly nonlinear. An empirical fit to an exponential function, also shown there, may be considered satisfactory. From this empirical exponential fit, the value β was found to be ≈2.0. This shows that the formation efficiency of charge transfer states in the ground state depends more strongly on the distance of separation between the donor and the acceptor than the rate constant of excited state electron transfer does.
 | ||
| Fig. 9 Absorbance of the charge transfer state, determined at 440 nm, of pyrene–TEMPO linked molecules in acetonitrile at 10 mM and plotted against the number of σ bonds separating the pyrene moiety of the linked molecules from the oxygen atom of the 4-oxo group of TEMPO. The blue curve is a fit of an exponential function to the data. | ||
Conclusions
In this study, we have shown the presence of charge-transfer states, in their ground states, between the TEMPO free radical and anthracene or pyrene covalently linked through a spacer group of different lengths. The spacer group is a linear alkyl chain with an ether or an ester group. The efficiency of the formation of the CT states shows no appreciable difference with regard to the nature of these two groups, but shows a strong dependence on the overall length of the spacer group. For the few molecules of the pyrene–TEMPO system studied here, the efficiency decreases approximately exponentially with the length. Similar distance-dependent quenching of the singlet excited states is also seen in their time-dependent fluorescence signals. Of all the linked molecules, Ay–CH2–O–TEMPO, Py–CH2–O–TEMPO and Py–CH2–CO–O–TEMPO—the ones with the shortest spacer groups—show the most prominent CT states. But even for these molecules, the distance between the chromophore and the nitroxyl group of TEMPO moieties is 6–7 bonds long. Therefore, when TEMPO and the chromophore are independent entities freely moving in solutions, during their collisions the transient formation of CT states is expected to be much more efficient than what has been seen in these linked molecules, because of their extremely close proximity during the collision. Such transient states can therefore play prominent roles in influencing their photophysical dynamics, as has been invoked in the generation of ESP in several cases.4–6Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors thank Dr Vinayak Rane for fruitful discussions, Ms Mamata Kallianpur for TCSPC measurements and Mr Dyaneshwar P. Avhad for various types of technical help.References
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, Viva Books, New Delhi, 2015 Search PubMed.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970, ch. 9 Search PubMed.
- S. A. Green, D. J. Simpson, S. G. Zhou, P. S. Ho and N. V. Blough, Intramolecular quenching of excited singlet states by stable nitroxyl radicals, J. Am. Chem. Soc., 1990, 112, 7337–7346 CrossRef CAS.
- A. Kawai, K. Shibuya and K. Obi, Effect of charge transfer state on the exchange interaction of radical-triplet encounter pairs, Appl. Magn. Reson., 2000, 18, 343–350 CrossRef CAS.
- A. Kawai and K. Shibuya, Charge-Transfer Controlled Exchange Interaction in Radical-Triplet Encounter Pairs as Studied by FT-EPR Spectroscopy, J. Phys. Chem. A, 2007, 111, 4890–4910 CrossRef CAS PubMed.
- Y. Kobori, S. Sekiguchi, K. Akiyama and S. Tero-Kubota, Chemically induced dynamic electron polarization study on the mechanism of exchange interaction in radical ion pairs generated by photoinduced electron transfer reactions, J. Phys. Chem. A, 1999, 103, 5416–5424 CrossRef CAS.
- M. Gouterman, R. A. Mathies, B. E. Smith and W. S. Caughey, Porphyrins. XIX. Tripdoublet and Quartet Luminescence in Cu and VO Complexes, J. Chem. Phys., 1970, 52, 3795 CrossRef CAS.
- S. Nakatsuji, A. Takai, K. Nishikawa, Y. Morimoto, N. Yasuoka, K. Suzuki, T. Enoki and H. Anzai, CT complexes based on TEMPO radicals, J. Mater. Chem., 1999, 9, 1747–1754 RSC.
- V. Rane, S. Kundu and R. Das, Photophysical studies on covalently-linked naphthalene and TEMPO free radical systems: observation of a charge transfer state in the ground state, J. Fluoresc., 2015, 25, 1351–1361 CrossRef CAS PubMed.
- T. Fuchigami, M. Atobe and S. Inagi, Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, John Wiley & Sons, 1st edn, 2015 Search PubMed.
- A. R. Watkins, Quenching of Electronically Excited States by the Free Radical Tetramethylpiperidene Nitroxide, Chem. Phys. Lett., 1974, 29, 526–528 CrossRef CAS.
- J. F. Delouis, J. A. Delaire and N. Ivanoff, Pyrene Fluorescence Quenching and Triplet-state Formation in the Presence of DABCO (1,4-diazobicyclo[2-2-2]octane): A Laser Photolysis Study, Chem. Phys. Lett., 1979, 61, 343–346 CrossRef CAS.
- R. Hoffman, Interaction of Orbitals through Space and through Bonds, Acc. Chem. Res., 1971, 4, 1–9 CrossRef.
- O. H. Oevering, M. N. Paddon-Row, M. Heppener, A. M. Oliver, E. Cotsaris, J. W. Verhoeven and N. S. Hush, Long-Range Photoinduced Through-Bond Electron Transfer and Radiative Recombination via Rigid Nonconjugated Bridges: Distance and Solvent Dependence, J. Am. Chem. Soc., 1987, 109, 3258–3269 CrossRef.
- G. L. Closs, P. Piotrowiak, J. M. Maclnnis and G. R. Fleming, Determination of long distance intramolecular triplet energy transfer rates. A quantitative comparison with electron transfer, J. Am. Chem. Soc., 1988, 110, 2652–2653 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06722h |
| This journal is © the Owner Societies 2019 |