
Observation of electrochemically active Fe3+/Fe4+ in LiCo0.8Fe0.2MnO4 by in situ Mössbauer spectroscopy and X-ray absorption spectroscopy†
Christoph
Dräger
 *a,
Florian
Sigel
a,
Ralf
Witte
b,
Robert
Kruk
b,
Lukas
Pfaffmann
a,
Stefan
Mangold
c,
Valeriu
Mereacre
a,
Michael
Knapp
ad,
Helmut
Ehrenberg
ad and
Sylvio
Indris
ad
*a,
Florian
Sigel
a,
Ralf
Witte
b,
Robert
Kruk
b,
Lukas
Pfaffmann
a,
Stefan
Mangold
c,
Valeriu
Mereacre
a,
Michael
Knapp
ad,
Helmut
Ehrenberg
ad and
Sylvio
Indris
ad
aInstitute for Applied Materials (IAM), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany. E-mail: office-ess@iam.kit.edu
bInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
cInstitute for Photon Science and Synchrotron Radiation (IPS), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
dHelmholtz Institute Ulm for Electrochemical Energy Storage (HIU), Helmholtzstrasse 11, 89081 Ulm, Germany
First published on 26th November 2018
Abstract
LiCo0.8Fe0.2MnO4 has been investigated as an active material for the positive electrode in lithium-ion batteries (LIBs) with a discharge potential of around 5 V (vs. Li+|Li). After synthesis by a Pechini based sol–gel route, the structural and morphological properties have been investigated by X-ray diffraction, scanning electron microscopy, 7Li MAS NMR spectroscopy, and 57Fe Mössbauer spectroscopy. With galvanostatic cycling, it was possible to obtain a specific discharge capacity of 117 mA h g−1, which is more than 80% of the theoretical capacity. The lithium extraction/insertion mechanism has been characterized by in situ synchrotron powder diffraction. The reversible oxidation process of Fe3+ to Fe4+ has been observed by in situ Mössbauer spectroscopy and in situ XAS measurements.
Introduction
Lithium-ion batteries are widely used in mobile electronic devices and increasingly in electric vehicles.1 Future applications are aiming at even larger energy storage systems for storage of discontinuously generated forms of energy such as wind energy and solar energy. Improvements of these systems are aiming at increasing the energy density of these batteries and lowering the cost of the battery-materials used. LiMn2O4 is a known active material for lithium-ion batteries (LIBs) and delivers discharge potentials of around 4 V.2–4However, this material suffers from a low capacity and fast capacity fading. To compensate the low capacity in a full cell, it is necessary to increase the discharge voltage of the cell and therefore the energy density of the battery. This can be achieved by substituting Mn3+ with another transition metal with a higher redox potential, like Ni2+, Co3+ or Fe3+.5–9
This substitution also reduces the Jahn–Teller activity, since Mn3+ is a Jahn–Teller active ion and destabilizes the host lattice of the material if it is oxidized to Mn4+ during electrochemical cycling, which leads to a continuous degradation with increasing number of cycles.9,10 The highest discharge potential of up to 5.2 V can be achieved by substituting Mn3+ with Co3+,11–13 but cobalt is toxic and quite expensive. In addition to that, for LiCoMnO4 spinels, more than 75% of their theoretical discharge capacity has not been achieved so far.13–17
Fe3+, on the other hand, can also provide high operating potentials in combination with other transition metals in the spinel structure and is inexpensive, non-toxic, and widely available.18,19 In this study, we replaced 20% of Co3+ with Fe3+ to obtain LiCo0.8Fe0.2MnO4. This material was firstly synthesized by an established Pechini2 based sol–gel method,13 and characterized via synchrotron powder diffraction (XRD), scanning electron microscopy (SEM), 7Li magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy, and Fe Mössbauer spectroscopy. The lithium extraction/insertion mechanism during electrochemical cycling has been investigated via in situ synchrotron diffraction. The reversible oxidation process of Fe3+ to Fe4+ has been characterized by in situ Mössbauer spectroscopy as well as in situ X-ray absorption spectroscopy (XAS).
57Fe Mössbauer spectroscopy is the prime method to observe changes of the oxidation state of Fe ions and also the changes of the local environment around these ions.20,21 Since Fe4+ is hard to be formed via common synthesis techniques, especially in oxide compounds, only very few Mössbauer investigations on Fe4+ have been reported in the literature.19,20,22–24 Mössbauer spectroscopy has often been used to confirm electrochemical reactions including redox reactions of Fe2+/Fe3+. In electrochemical cells, the formation of Fe4+ in some materials has been claimed in order to explain the electrochemical behavior, but clear evidence has not been often provided,22,25,26 since Fe4+ is not stable outside electrochemical cells, and therefore ex situ investigations do not give clear results. So far, only a few in situ Mössbauer investigations have been published with respect to this matter.22
Experimental
LiCo0.8Fe0.2MnO4 was synthesized by a citric acid based sol–gel method. A detailed description of the procedure (for LiCoMnO4) can be found elsewhere,13 but briefly it can be described as follows: equivalent amounts of metal acetates for Li, Co and Mn (LiCH3COO·2H2O, Mn(CH3COO)2·4H2O, Co(CH3COO)2·4H2O, all were obtained from Sigma Aldrich, and of reagent grade) were mixed with citric acid (5 eq. with respect to Mn and Li) and ethylene glycol (20 eq.). For the standard experiments, Fe(III) citrate (C6H5FeO7·H2O, Fluka) was used as the Fe-source. For the in situ Mössbauer experiments, isotopically enriched [57Fe3O(O2CC9H19)6(H2O)3]NO3 was used as the Fe-source.After mixing the metal salts, the solution was stirred for 12 h at 90 °C, followed by evaporation of the excess ethylene glycol and water residues at 180 °C, leading to a dark violet gel. This gel was pyrolized at 350 °C for 3 h in a muffle furnace (Nabertherm L5/12) in air, yielding a dark brown/violet precursor powder. This powder was ground in a mortar and calcined under an oxygen atmosphere for 24 h at 600 °C, directly followed by 12 h at 750 °C in a tube furnace (Carbolite CTF 18/300) at heating and cooling rates of 3 °C min−1.
In order to perform electrochemical tests, pristine LiCo0.8Fe0.2MnO4 powder (80 w/w) was mixed with carbon black (10 w/w, TIMCAL, Super C65), PVDF (10 w/w, Solef 6020, Solvay) and NMP (Sigma-Aldrich) to obtain a slurry, which was stirred overnight and then coated on aluminium foil (20 μm) with a wet thickness of 250 μm.
The resulting electrode film was then dried for 24 h at 80 °C and cut into discs with a diameter of 14 mm for coin cells and 12 mm for the Mössbauer experiments. All cells were assembled in an argon filled glove box (MBraun) with lithium foil (MTI, 15.6 mm diameter, 250 μm thickness) as the anode, 180 μL electrolyte (1 M LiPF6 (ABCR) in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (wt) mixture of bis(2,2,2-trifluoroethyl)carbonate and 4-fluoro-1,3-dioxolan-2-one (TCI)) and two layers of Celgard 2325 as the separator. The cells were cycled using a VMP3 multi-channel potentiostat (Bio-Logic, France) at 25 °C.
:50 (wt) mixture of bis(2,2,2-trifluoroethyl)carbonate and 4-fluoro-1,3-dioxolan-2-one (TCI)) and two layers of Celgard 2325 as the separator. The cells were cycled using a VMP3 multi-channel potentiostat (Bio-Logic, France) at 25 °C.
The morphology of the particles was analyzed using a Zeiss MERLIN SEM with a primary electron energy of 10 keV and an in-lens detector. Ex situ and in situ synchrotron powder diffraction experiments were performed at the Materials Science and Powder Diffraction Beam Line (MSPD) at ALBA at 30 keV (λ = 0.413250 Å) using a MYTHEN 6 K position sensitive detector27 and a dedicated coin cell setup28 with an exposure time of 45 s per pattern. Rietveld refinements were performed using the FullProf software package.29 Mössbauer spectroscopy was performed in the transmission mode with a 57Co(Rh) source. All isomer shifts are given with respect to that of metallic α-Fe at room temperature. In order to perform in situ Mössbauer spectroscopy on a complete Li battery during operation, a cathode sample with enriched 57Fe was synthesized from an 57Fe containing precursor and an Fe Mössbauer source (30 mCi) was used. Measurements were performed on a flat stainless steel cell with Kapton windows.
For 7Li MAS NMR, a Bruker Avance 200 MHz spectrometer (4.7 T) with 1.3 mm zirconia rotors and a spinning frequency of 60 kHz was used. Spectra were recorded with a rotor-synchronized Hahn-echo pulse sequence and are referenced to an aqueous 1 M LiCl solution (0 ppm).
Results and discussion
Structural and morphological characterization of LiCo0.8Fe0.2MnO4
The results of the Rietveld refinement are shown in Fig. 1 (left). The pattern can be indexed to the space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, origin choice 2. The tetrahedral 8a site is mostly occupied by lithium, but the intensity of the 220 reflection indicates that there is also some transition metal contribution on this site. Similar results were found for the LiCoMnO4 spinel.13
m, origin choice 2. The tetrahedral 8a site is mostly occupied by lithium, but the intensity of the 220 reflection indicates that there is also some transition metal contribution on this site. Similar results were found for the LiCoMnO4 spinel.13
 | ||
| Fig. 1 Rietveld refinement against synchrotron powder diffraction data of LiCo0.8Fe0.2MnO4 (left) and a SEM image of pristine LiCo0.8Fe0.2MnO4 (right). | ||
The transition metals Co, Mn, and Fe are distributed homogenously on the octahedral 16d site, and oxygen occupies one 32e site. The results of the refinement are listed in Table S1 (ESI†). A small contribution (less than 1% w/w) of the second spinel phase with slightly increased lattice parameters can also be found, which could be interpreted as an Fe3O4 phase.
Fig. 1 (right) shows a SEM image of the as prepared sample. The particle sizes range from ∼80 nm up to 500 nm, but most particles have a size of around 100 nm. The shape of the particles can be described as a truncated octahedron. Additional SEM images can be found in the ESI† (Fig. S1).
Electrochemical characterization
The charge and discharge curves of the first, second, and 100th cycles of LiCo0.8Fe0.2MnO4 at a C-rate of C/2 (i.e. complete charge/discharge within 2 h) in the voltage range of 5.4–4.5 V are shown in Fig. 2a. The resulting specific charge and discharge capacities are displayed in Fig. 2b. It can be seen that the first charge process starts around 4.6 V, followed by a plateau at 5.05 V and ends around 5.4 V. The first discharge process starts at 5.1 V and shows a plateau down to 4.6 V, resulting in a first cycle coulombic efficiency of 75.6%. The plateau of the second charge process starts at a lower potential, which reveals that there is some kind of formation process involved in the first cycle. | ||
| Fig. 2 Electrochemical cycling of LiCo0.8Fe0.2MnO4vs. Li+/Li in a voltage range of 5.4–4.5 V. (a) Resulting charge and discharge potentials and (b) specific capacities over 100 cycles (b). | ||
The second discharge process also differs slightly from the first one and starts at a higher potential of around 5.15 V, i.e. the electrode polarization is lower than that in the first cycle. On the other hand, the electrode polarization is strongly increased after 100 cycles, accompanied by a loss of capacity. The discharge process starts even below 5.0 V.
The maximum discharge capacity is reached in cycles 2 and 3, both with 117 mA h g−1. This corresponds to 80% of the theoretical capacity (146 mA h g−1). After 100 cycles, a specific discharge capacity of 73 mA h g−1 is left. In order to investigate the lithium extraction/insertion mechanism, in situ synchrotron powder diffraction experiments were conducted at a C-rate of C/2. The evolution of the diffraction patterns is shown in Fig. 3a. Starting with delithiation, the Bragg reflections are shifting to higher angles, corresponding to the smaller lattice parameters (Fig. 3b). The shift stops when a cut-off potential of 5.4 V is reached, which suggests that the charging process has not been completed and the material might not be fully delithiated. The point of full delithiation is therefore not within the usable voltage-range of the used electrolyte. The maximum change of the lattice parameters from the start to the end of the charging process is just 1.2%.
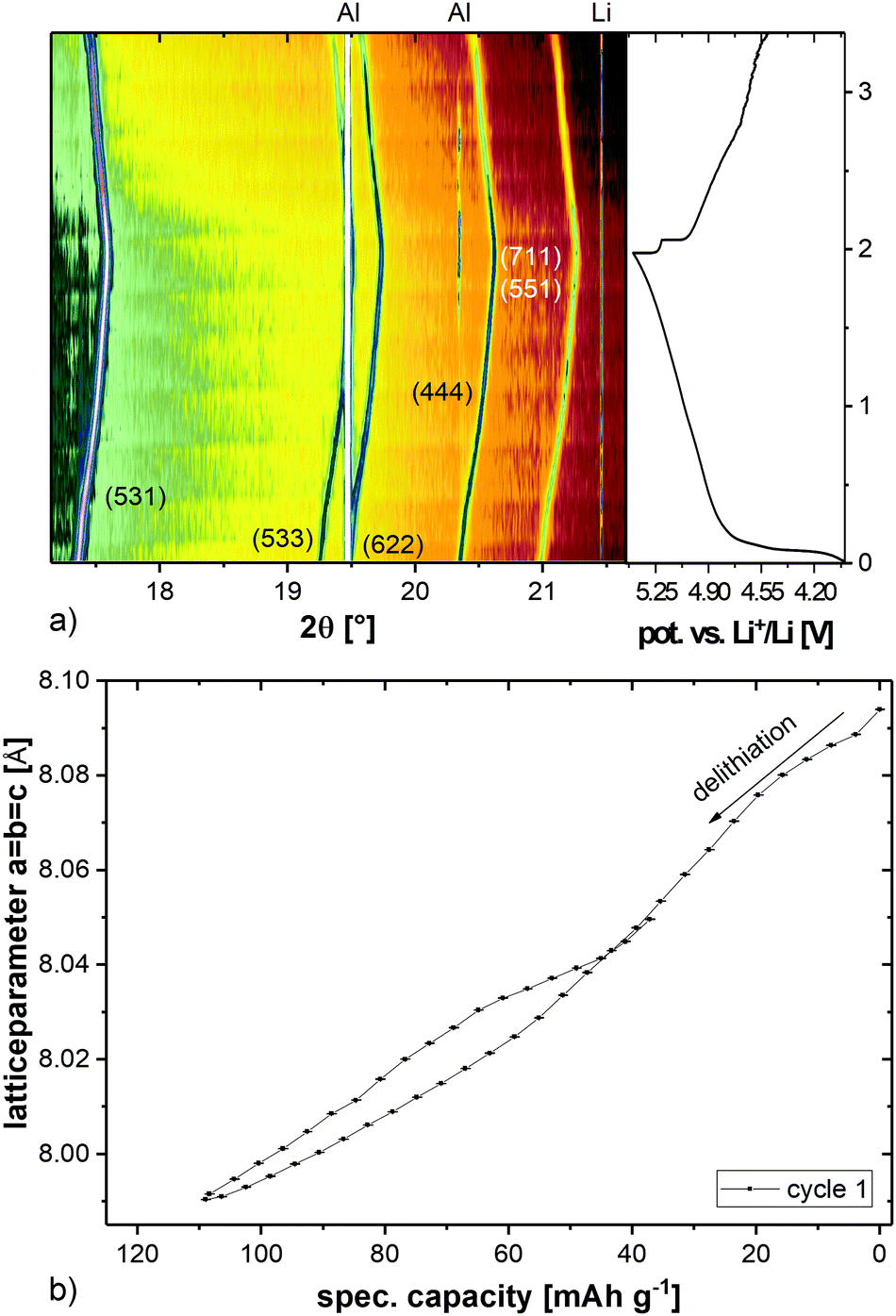 | ||
| Fig. 3 In situ synchrotron powder diffraction of LiCo0.8Fe0.2MnO4vs. Li+/Li in a voltage range of 5.4–4.5 V (a) and evolution of the unit cell parameter a = b = c of the spinel phase (b). | ||
The discharge process of the cell is accompanied by a shift of the reflections to smaller angles. This reveals that the lattice parameters decrease again, but they do not reach the initial values. The discharge process is therefore also not complete at 4.5 V and limits the usable discharge capacity of the material even further. It is also noticeable that there is no second phase (apart from the minor impurity with weight fraction <1% (w/w), cf.Fig. 1), either inactive or active, and the material shows, therefore, a solid-solution behavior over the whole voltage range.
7Li MAS NMR spectroscopy
The 7Li MAS NMR spectrum of pristine LiCo0.8Fe0.2MnO4 is shown in Fig. 4. The minor contribution at 730 ppm can be ascribed to the presence of a small amount of Li2MnO3.13 The broad peak around 595 ppm represents a large variety of different environments around the lithium on the tetrahedral site (8a) of the spinel phase. These environments can be described by a kind of random occupation of 12 next nearest neighboring octahedral (16d) sites with Fe, Mn and Co. In comparison to the 7Li NMR spectrum of LiCoMnO4,13 the maximum of this broad peak is shifted toward larger NMR shift values. This is caused by the substitution of diamagnetic Co3+ (electron configuration 3d6: t62ge0g) for paramagnetic Fe3+ (3d5: t32ge2g). | ||
| Fig. 4 7Li MAS NMR spectrum of LiCo0.8Fe0.2MnO4. | ||
In situ Mössbauer spectroscopy
Fig. 5 shows the ex situ Mössbauer spectra of LiCo0.8Fe0.2MnO4 in the pristine state and after charging in a battery to 5.3 V. The pristine state can be well described by a doublet with isomer shift IS = (0.330 ± 0.001) mm s−1 and quadrupole splitting QS = (0.722 ± 0.001) mm s−1. These values are characteristic of Fe3+ with a high-spin configuration on the octahedral sites of the spinel structure. The spectrum obtained after charging to 5.3 V also shows this doublet, but a second component has been formed. Due to the strong overlap of these two components, the fitting is ambiguous and the formation of Fe4+ cannot be clearly derived from this spectrum. Therefore, the changes in the oxidation state of the Fe ions and also their local environments were investigated by in situ Mössbauer spectroscopy on a complete battery cell during operation, with LiCo0.8Fe0.2MnO4 as the positive electrode and Li metal as the negative electrode. The voltage of the cell was increased/decreased stepwise by 0.1 V in the range between 4.3 V and 5.4 V and then, during a 1 h long potentiostatic step, the Mössbauer spectrum was acquired. The spectra acquired during the first two cycles are shown in Fig. 6. As described above, the pristine state (top spectrum) can be well fitted with one doublet. During the initial charging of the battery, the isomer shift does not show any changes, while the quadrupole splitting shows a slight increase from 0.72 mm s−1 to about 0.8 mm s−1. This reveals an increasing electric field gradient at the site of the Fe nuclei, i.e. an increased asymmetry of the local environment. Subsequently, this Fe3+ doublet successively disappears and is replaced almost completely by a new doublet (see Fig. 8). This new doublet has an isomer shift IS = (−0.134 ± 0.006) mm s−1 and a quadrupole splitting QS = (0.300 ± 0.008) mm s−1. The negative value for IS clearly reveals that Fe4+ is formed. During further cycling, the Fe4+ is reduced again to Fe3+ and in the second cycle an overall similar behavior is observed. A comparison of the Mössbauer spectra with the voltage profile acquired during the stepwise potentiostatic cycling reveals that the oxidation/reduction of Fe3+/Fe4+ is highly reversible, but some time-shift reveals sluggish kinetics and thus a retarded oxidation and reduction of Fe. The fit–parameters used in Fig. 7 can be found in Table 1. In contrast to the ex situ Mössbauer measurements, the in situ experiments show an almost complete conversion of Fe3+ to Fe4+ during charging and back to Fe3+ during discharging. The fact that this process could not be observed ex situ shows that the Fe4+ is not stable outside the battery and that its formation can only be observed by the in situ measurements on a complete battery cell. | ||
| Fig. 5 Ex situ Mössbauer spectra of LiCo0.8Fe0.2MnO4 acquired on the pristine powder (top) and on the electrode after charging to 5.3 V (bottom). The experimental spectra are shown as red spheres, the fit as black lines, and the subspectra as blue and purple doublets. | ||
 | ||
| Fig. 6 In situ Mössbauer spectroscopy results over 2 cycles (left) and the applied voltage profile (right). | ||
 | ||
| Fig. 7 Selected in situ Mössbauer spectra of LiCo0.8Fe0.2MnO4 acquired at different potentials as indicated in the figure. The used fit–parameters can be found in Table 1. | ||
| Potential [V] | Species | IS [mm s−1] | QS [mm s−1] | Γ [mm s−1] | Area fraction [%] |
|---|---|---|---|---|---|
| 4.0 | Fe3+ | 0.330(1) | 0.722(1) | 0.316(1) | 100 |
| 5.0 | Fe3+ | 0.312(3) | 0.788(6) | 0.364(9) | 100 |
| 4.9 | Fe3+ | 0.52(2) | 0.42(2) | 0.37(4) | 26 |
| Fe4+ | −0.134(6) | 0.300(8) | 0.33(1) | 74 | |
| 4.5 | Fe3+ | 0.326(5) | 0.82(1) | 0.29(1) | 63 |
| Fe4+ | 0.07(3) | 0.49(5) | 0.6(1) | 37 |
In situ XAS measurements
The results of in situ XAS measurements on the Fe-K-edge of LiCo0.8Fe0.2MnO4 are shown in Fig. 8. The data concerning the Co-K-edge and the Mn-K-edge can be found in the ESI† (Fig. S2 and S3). Only selected spectra are shown for better visibility. During the charging process, the shift of the absorption edge to higher energies is in good agreement with an oxidation state change of +1. Since the oxidation state of the pristine material was verified to be Fe3+, the reaction to Fe4+ can be demonstrated by XAS. During the discharge process, Fe4+ is reduced again to Fe3+, revealing a reversible redox process of Fe3+ to Fe4+ and back, and thus confirming the Mössbauer results. | ||
| Fig. 8 Results of the in situ XAS measurements on the Fe-K-edge of LiCo0.8Fe0.2MnO4. (a) The voltage profile acquired during these galvanostatic measurements. The markers represent the times where an XAS spectrum was acquired. Only selected spectra are shown for better readability (red markers). (b) The XAS spectra acquired during the charge process (oxidation) and (c) the spectra acquired during the discharge process (reduction). The reference-spectrum of Fe3+ (Fe2O3) is plotted as a dashed line. | ||
Conclusions
In this paper, we have presented the synthesis and the structural as well as electrochemical characterization of LiCo0.8Fe0.2MnO4. The delithiation mechanism has been revealed as solid-solution behavior by in situ synchrotron diffraction. In addition to that, we demonstrated the reversible oxidation/reduction of Fe4+/Fe3+ in a 5 V battery cell by in situ Mössbauer spectroscopy. This enables the use of non-toxic and highly abundant transition metal elements in cathode materials for rechargeable high-voltage Li-ion batteries and at least partial substitution of expensive and toxic cobalt. In turn, this might lead to higher energy densities and reduced costs of energy storage devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the German Federal Ministry of Education and Research, as part of the DESIREE project (grant no. 03SF0477B). Financial support for the Zeiss MERLIN SEM was provided by the Federal Ministry of Economics and Technology on the basis of a decision by the German Bundestag within the BMWi grant no. 03ET6017. This work has benefitted from beamtime allocation by the Materials Science and Powder Diffraction Beam Line (MSPD) at ALBA.Notes and references
- P. Kurzweil and O. K. Dietlmeier, Elektrochemische Speicher, Springer Fachmedien Wiesbaden, Wiesbaden, 2015 Search PubMed.
- W. Liu, Synthesis and Electrochemical Studies of Spinel Phase LiMn2O4 Cathode Materials Prepared by the Pechini Process, J. Electrochem. Soc., 1996, 143, 879 CrossRef CAS.
- R. J. Gummow, A. de Kock and M. M. Thackeray, Improved capacity retention in rechargeable 4 V lithium/lithium-manganese oxide (spinel) cells, Solid State Ionics, 1994, 69, 59–67 CrossRef CAS.
- M. M. Thackeray, Manganese oxides for lithium batteries, Prog. Solid State Chem., 1997, 25, 1–71 CrossRef CAS.
- C. M. Julien and A. Mauger, Review of 5 V electrodes for Li-ion batteries: status and trends, Ionics, 2013, 19, 951–988 CrossRef CAS.
- L. Guohua, The Spinel Phases LiMyMn2−yO4 (M = Co, Cr, Ni) as the Cathode for Rechargeable Lithium Batteries, J. Electrochem. Soc., 1996, 143, 178 CrossRef.
- Q. Zhong, Synthesis and Electrochemistry of LiNixMn2−xO4, J. Electrochem. Soc., 1997, 144, 205 CrossRef CAS.
- N. Amdouni, F. Gendron, A. Mauger, H. Zarrouk and C. M. Julien, LiMn2−yCoyO4 (0 ≤ y ≤ 1) intercalation compounds synthesized from wet-chemical route, Mater. Sci. Eng., B, 2006, 129, 64–75 CrossRef CAS.
- A. Bhaskar, D. Mikhailova, N. Kiziltas-Yavuz, K. Nikolowski, S. Oswald, N. N. Bramnik and H. Ehrenberg, 3d-Transition metal doped spinels as high-voltage cathode materials for rechargeable lithium-ion batteries, Prog. Solid State Chem., 2014, 42, 128–148 CrossRef CAS.
- L. Xiong, Y. Xu, T. Tao, J. Song and J. B. Goodenough, Excellent stability of spinel LiMn2O4-based composites for lithium ion batteries, J. Mater. Chem., 2012, 22, 24563–24568 RSC.
- M. Hu, Y. Tian, J. Wei, D. Wang and Z. Zhou, Porous hollow LiCoMnO4 microspheres as cathode materials for 5 V lithium ion batteries, J. Power Sources, 2014, 247, 794–798 CrossRef CAS.
- H. Kawai, M. Nagata, M. Tabuchi, H. Tukamoto and A. R. West, Novel 5 V Spinel Cathode Li2FeMn3O8 for Lithium Ion Batteries, Chem. Mater., 1998, 10, 3266–3268 CrossRef CAS.
- C. Dräger, F. Sigel, S. Indris, D. Mikhailova, L. Pfaffmann, M. Knapp and H. Ehrenberg, Delithiation/relithiation process of LiCoMnO4 spinel as 5 V electrode material, J. Power Sources, 2017, 371, 55–64 CrossRef.
- H. Kawai, M. Nagata, H. Kageyama, H. Tukamoto and A. R. West, 5 V lithium cathodes based on spinel solid solutions Li2Co1+XMn3−XO8: −1 ≤ X ≤ 1, Electrochim. Acta, 1999, 45, 315–327 CrossRef CAS.
- H. Kawai, A New Lithium Cathode LiCoMnO4: Toward Practical 5 V Lithium Batteries, Electrochem. Solid-State Lett., 1999, 1, 212 CrossRef.
- N. Kuwata, S. Kudo, Y. Matsuda and J. Kawamura, Fabrication of thin-film lithium batteries with 5-V-class LiCoMnO4 cathodes, Solid State Ionics, 2014, 262, 165–169 CrossRef CAS.
- H. Kawai, M. Nagata, H. Tukamoto and A. R. West, High-voltage lithium cathode materials, J. Power Sources, 1999, 81–82, 67–72 CrossRef CAS.
- R. Alcántara, M. Jaraba, P. Lavela and J. L. Tirado, Electrochemical, 6Li MAS NMR, and X-ray and Neutron Diffraction Study of LiCoxFeyMn2−(x+y)O4 Spinel Oxides for High-Voltage Cathode Materials, Chem. Mater., 2003, 15, 1210–1216 CrossRef.
- T. Ohzuku, K. Ariyoshi, S. Takeda and Y. Sakai, Synthesis and characterization of 5 V insertion material of LiFeyMn2−yO4 for lithium-ion batteries, Electrochim. Acta, 2001, 46, 2327–2336 CrossRef CAS.
- F. Menil, Systematic trends of the 57Fe Mössbauer isomer shifts in (FeOn) and (FeFn) polyhedra. Evidence of a new correlation between the isomer shift and the inductive effect of the competing bond T–X (→Fe) (where X is O or F and T any element with a formal positive charge), J. Phys. Chem. Solids, 1985, 46, 763–789 CrossRef CAS.
- P. Gütlich, Physikalische Methoden in der Chemie: Mößbauer-Spektroskopie I, Chem. Unserer Zeit, 1970, 4, 133–144 CrossRef.
- E. McCalla, M. T. Sougrati, G. Rousse, E. J. Berg, A. Abakumov, N. Recham, K. Ramesha, M. Sathiya, R. Dominko, G. Van Tendeloo, P. Novák and J. M. Tarascon, Understanding the Roles of Anionic Redox and Oxygen Release during Electrochemical Cycling of Lithium-Rich Layered Li4FeSbO6, J. Am. Chem. Soc., 2015, 137, 4804–4814 CrossRef CAS.
- R. Rajagopalan, B. Chen, Z. Zhang, X.-L. Wu, Y. Du, Y. Huang, B. Li, Y. Zong, J. Wang, G.-H. Nam, M. Sindoro, S. X. Dou, H. K. Liu and H. Zhang, Improved Reversibility of Fe3+/Fe4+ Redox Couple in Sodium Super Ion Conductor Type Na3Fe2(PO4)3for Sodium-Ion Batteries, Adv. Mater., 2017, 29, 1605694 CrossRef.
- E. Lee, D. E. Brown, E. E. Alp, Y. Ren, J. Lu, J. J. Woo and C. S. Johnson, New Insights into the Performance Degradation of Fe-Based Layered Oxides in Sodium-Ion Batteries: Instability of Fe3+/Fe4+ Redox in α-NaFeO2, Chem. Mater., 2015, 27, 6755–6764 CrossRef CAS.
- R. Chen, M. Knapp, M. Yavuz, S. Ren, R. Witte, R. Heinzmann, H. Hahn, H. Ehrenberg and S. Indris, Nanoscale spinel LiFeTiO4 for intercalation pseudocapacitive Li+ storage, Phys. Chem. Chem. Phys., 2015, 17, 1482–1488 RSC.
- R. Chen, R. Heinzmann, S. Mangold, V. S. K. Chakravadhanula, H. Hahn and S. Indris, Structural Evolution of Li2Fe1−yMnySiO4 (y = 0, 0.2, 0.5, 1) Cathode Materials for Li-Ion Batteries upon Electrochemical Cycling, J. Phys. Chem. C, 2013, 117, 884–893 CrossRef CAS.
- F. Fauth, I. Peral, C. Popescu and M. Knapp, The new Material Science Powder Diffraction beamline at ALBA Synchrotron, Powder Diffr., 2013, 28, S360–S370 CrossRef CAS.
- M. Herklotz, J. Weiß, E. Ahrens, M. Yavuz, L. Mereacre, N. Kiziltas-Yavuz, C. Dräger, H. Ehrenberg, J. Eckert, F. Fauth, L. Giebeler and M. Knapp, A novel high-throughput setup for in situ powder diffraction on coin cell batteries, J. Appl. Crystallogr., 2016, 49, 1–6 CrossRef.
- T. Roisnel and J. Rodríquez-Carvajal, WinPLOTR: A Windows Tool for Powder Diffraction Pattern Analysis, Mater. Sci. Forum, 2001, 378–381, 118–123 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06177g |
| This journal is © the Owner Societies 2019 |